


Abstract
BRCA1 mutations are associated with increased breast and ovarian cancer risk. BRCA1-mutant tumors are high-grade, recurrent, and often become resistant to standard therapies. Herein, we performed a targeted CRISPR-Cas9 screen and identified MEPCE, a methylphosphate capping enzyme, as a synthetic lethal interactor of BRCA1. Mechanistically, we demonstrate that depletion of MEPCE in a BRCA1-deficient setting led to dysregulated RNA polymerase II (RNAPII) promoter-proximal pausing, R-loop accumulation, and replication stress, contributing to transcription-replication collisions. These collisions compromise genomic integrity resulting in loss of viability of BRCA1-deficient cells. We also extend these findings to another RNAPII-regulating factor, PAF1. This study identifies a new class of synthetic lethal partners of BRCA1 that exploit the RNAPII pausing regulation and highlight the untapped potential of transcription-replication collision-inducing factors as unique potential therapeutic targets for treating cancers associated with BRCA1 mutations.
INTRODUCTION
The breast cancer susceptibility gene 1 (BRCA1) encodes the tumor suppressor BRCA1, which primarily functions to maintain genomic stability. BRCA1 is a critical component of various multi-protein complexes that allow error-free repair of double-stranded DNA breaks through homologous recombination (HR) (1). BRCA1 also plays a crucial role in cell cycle regulation and transcriptional and epigenetic regulation (2,3). Given its role in maintaining genomic integrity, it is not surprising that mutations in the BRCA1 gene increase the risk of developing breast, ovarian, and other cancers (3,4). Additionally, patients suffering from BRCA1-defective cancers are likely to experience poor clinical outcomes due to relapse, metastasis, and the development of a new contralateral tumor in the years following diagnosis (5,6). Therefore, it is critical to identify mechanisms that underlie the viability of these tumors and pinpoint novel therapeutic targets that specifically eliminate tumors that arise from BRCA1 mutations.
Synthetic lethality refers to the loss of viability resulting from the disruption of two genes whose individual loss is not lethal. Notably, synthetic lethality has been used to identify novel therapeutic targets for cancer therapy. For instance, the identification of poly (ADP-ribose) polymerase (PARP) inhibitors being synthetic lethal with mutations in BRCA1 has garnered great interest as PARP1/2 being potential therapeutic targets (7). While the FDA approved PARP inhibitors (PARPi) such as Olaparib as a monotherapy in patients with BRCA1 germline mutations, different studies have reported clinical resistance to PARPi through various mechanisms (8,9). The abrogation of DNA repair pathways through targeting DNA damage signaling and repair proteins such as DNA Polymerase θ (Pol θ), Fanconi anemia group D2 protein (FANCD2), and the recently identified Ring finger 168 (RNF168), might be a viable option to eliminate BRCA1-mutated tumors (10–12). However, synthetic lethal partners for BRCA1 outside of DNA damage repair remain unknown, reflecting an untapped potential to discover and exploit new vulnerabilities of BRCA1-mutant cells (13). CRISPR-Cas9 pooled screening has made it possible to carry out large-scale unbiased dropout genetic screens directly in human cell lines, leading to the identification of genes that are only essential in specific genetic backgrounds (14). In this study, we utilized a targeted CRISPR-Cas9 dropout screen and identified MEPCE as a potential vulnerability in BRCA1-mutant breast tumors.
MEPCE is a methyltransferase that adds a monomethyl cap onto the γ-phosphate at the 5' end of nascent 7SK small nuclear RNA (snRNA) (15–17). MEPCE and La-related protein 7 (LARP7) stabilize 7SK snRNA by binding to its 3' terminal hairpin, thereby protecting it from degradation by exonucleases (15,18). MEPCE and LARP7 constitute the core 7SK small nuclear ribonucleoproteins (snRNPs) and are essential for the stability of 7SK snRNP (15,18). The core 7SK snRNP provides a dynamic scaffold leading to the assembly of larger 7SK snRNPs such as the hexamethylene bisacetamide (HMBA)-inducible protein 1 (HEXIM1) and the positive transcription elongation factor b (P-TEFb), comprised of CDK9 and its cyclin partners (cyclin T1 and T2) (19–21). The 7SK/MEPCE/LARP7/HEXIM1/P-TEFb snRNP sequesters and inhibits P-TEFb activity, which plays a vital role in the release of stalled RNA polymerase II (RNAPII) from promoter-proximal pausing (PPP), leading to productive elongation (22). Thus, the 7SK snRNP complex functions as a negative transcriptional regulatory complex.
Here, we investigate the essentiality of MEPCE in cancers associated with BRCA1 mutations using cell and mouse models and demonstrate that the loss of MEPCE is lethal with BRCA1 loss and mutations of HR genes. We observed that depletion of MEPCE leads to excessive genomic instability and senescence in BRCA1-mutant breast and ovarian cancer cells. Mechanistically, we demonstrate that MEPCE deficiency in BRCA1-mutant cells deregulates RNAPII, leading to the excessive accumulation of DNA–RNA hybrids (R-loops), transcription-replication conflicts (TRCs) (23) and replication stress. Our dropout screen also identified RNAPII-associated factor 1 (PAF1), which regulates promoter-proximal pausing of RNAPII similar to MEPCE (24), as another synthetic lethal interactor of BRCA1. Not surprisingly, depletion of PAF1 in BRCA1-deficient cells also triggered cell death by exacerbating R-loop accumulation and transcription-replication collisions. Our study identifies dysregulated RNAPII promoter-proximal pausing and inevitable transcription-replication conflicts as a unique BRCA1-mutant cancer cell vulnerability that could be exploited to eliminate these tumors. This study expands on current knowledge of BRCA1 synthetic lethal interactions and highlights the potential of MEPCE inhibition for cancer therapy for this class of tumors.
MATERIALS AND METHODS
Cell lines
HEK293FT (RRID:CVCL_6911), MDA-MB-231 (RRID:CVCL_0062), MDA-MB-468 (RRID:CVCL_0419), MDA-MB-436 (RRID:CVCL_0623) and COV362 (RRID:CVCL_2420), cells were cultured in high-glucose Dulbecco's Modified Eagle Medium (DMEM) containing HEPES buffer, penicillin, and streptomycin (Wisent cat: 319-005-CL) supplemented with 10% fetal bovine serum (FBS) (Wisent cat: 080-150). OVCAR8 (RRID:CVCL_1629) cells were cultured in high-glucose Roswell Park Memorial Institute's (RPMI) medium containing HEPES buffer, penicillin, and streptomycin (Wisent cat: 350-700-CL) supplemented with 10% FBS (Wisent cat: 080-150). SUM149PT (RRID:CVCL_3422), cells were cultured in Ham's F-12 (F12) medium mixture (Gibco cat: 31765035) supplemented with 5% FBS, 10 mM HEPES (Gibco cat: 15630080) 1 μg/ml hydrocortisone (Sigma cat: H0888) and 5 μg/ml insulin (Sigma cat: I-1882). MCF10A (RRID:CVCL_0598), cells were grown in a DMEM/F12 medium mixture (Gibco cat: 11320033) supplemented with 5% horse serum (Invitrogen: 16050-122), 20 ng/ml epidermal growth factor (EGF), 100 ng/ml Cholera toxin, 0.5 mg/ml hydrocortisone (Sigma cat: H0888) and 10 μg/ml insulin (Sigma cat: I-1882). Cell lines were passaged using 0.05% Trypsin-EDTA solution (Wisent cat: 325-542-EL) and grown in a humidified atmosphere at 37°C and 5% CO2.
DNA constructs
shRNA construct targeting a control sequence (luciferase or scramble) or MEPCE were designed and cloned into pLKO.1 puro (Addgene #8453). shCtrl shRNA target sequences were: shLuc (5’-CTTCGAAATGTCCGTTCGGTT3’), shCtrl: (5’-CCTAAGGTTAAGTCGCCCTCG-3’); shMEPCE (5’- AAGAACTACTACCGAATCCA-3’), shMEPCE-2: (5’-AGTTCCAGTATGGGAATTATT-3’); shBRCA1 (5’-GAGTATGCAAACAGCTATAAT-3’) was cloned into the pLKO.1-blast (Addgene #26655); shLARP7 (5’-CTTGAGCTGTTCTTGGGAGAT-3’). ShRNA targeting PAF1 was ordered from Sigma (TRCN0000010939). shRNA constructs targeting a control sequence and MEPCE were also cloned into the Tet-pLKO-puro (Addgene #21915) vector. The RNASEH1 overexpression plasmid and its empty vector control (pcDNA3) used for γH2AX immunofluorescence experiments were gifts from Dr. Karim Mekhail at the University of Toronto. The Lenti‐Cas9‐2A‐Blast plasmid and sgRNAs targeting essential genes PSMD1, PSMB2, and eIF3D were gifts from Dr. Troy Ketela and the Princess Margaret Genomics Centre.
Generation of lentivirus and stable cell lines
Transfections for lentivirus generation in HEK293FT cells were performed using GenJet In Vitro DNA Transfection Reagent (SignaGen cat: SL100489). On the day of transfection, HEK293FT cells were at 90% confluency in a 10 cm tissue culture dish. 1 h prior to transfection, the media was changed to 5 ml of DMEM (10% FBS). 7.5 μg of transfer vector, 3.75 μg of psPAX2 (Addgene #12260) and 3.75 μg of pMD2.G (Addgene #12259) were combined in 500 μl of serum-free DMEM. 45 μl GenJet in 500 μl of serum-free DMEM was mixed with the DNA mixture. The resultant mixture was incubated at room temperature for 15 min. Following the incubation, the full mixture was added dropwise to the plate of HEK293FT cells. These cells were incubated overnight. The next day, the supernatant containing lentiviral particles was collected and filtered through a 0.45 μm low binding filter 24 and 48 h post-transfection. Lentiviral transductions were performed in 6 well tissue culture dishes using 1 ml of appropriate culture media and 1 ml of lentiviral supernatant. Transductions were supplemented with polybrene at a concentration of 4–8 μg/ml (cell line-dependent) to improve transduction efficiency. Transductions were allowed to proceed for 2–3 h. 24–48 h post-transduction, selection with the proper antibiotic and concentration was performed. 1-week post-selection, levels of knockdown or overexpression were assessed via western blot and used for assays or frozen.
CRISPR-Cas9 screen and Cas9 overexpression functionality
MDA-MB-231, MDA-MB-436 and SUM149PT cell lines were transduced with the Cas9-2A- Blasticidin vector (Addgene #73310). Cas9 editing efficiency was validated by transducing the cell lines with viral gRNA expressing viruses targeting the essential genes PSMD1 and PSMB2, and a non-targeting LacZ control. Cells were selected in puromycin for 48 h and cell viability was assessed every 2 days by Trypan Blue staining (Gibco cat: 15250061).
Cell lines characterization
The doubling time of the cell lines used in the screening was determined by counting cells using Trypan Blue staining and using the following formula:
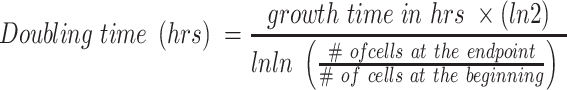 |
Given that the lentiviruses from the library express puromycin resistance, the concentration of puromycin required to kill 100% of uninfected cells 48 h post-treatment was established. In 12 well plates, at ∼70–80% confluency, cells were exposed to a range of concentrations of puromycin. Cell viability was determined using Trypan Blue staining.
Epi/Drug library amplification
The Epi/Drug library (25) contains sgRNAs targeting 334 genes encoding epigenetic regulators and 657 genes with at least one FDA approved drug (10 sgRNAs per gene). In total, the library consists of approximately 12 500 sgRNAs including 400 negative and positive control sgRNAs.
The library was amplified using Stbl4 bacteria cells (Invitrogen cat: 11635018). 20μl Stbl4 cells and 50ng of pooled library plasmids were mixed and placed in an electroporation cuvette (Bio-Rad cat: 1652086). Electroporation was performed at 1.2 kV, 25 μF and 200 Ω using Gene Pulser Xcell™ Total System (Bio-Rad cat: 1652660), then immediately 1 ml of SOC media (Invitrogen cat: 15544034) was added to the cuvette. Bacteria were moved to sterile tubes and shook at 30°C and 225 rpm for 90 min. The bacteria were plated onto 15 cm agar ampicillin (Sigma cat: A0166-5G) plates and incubated at 30°C for 24 h. To determine the number of colonies on the concentrated plate have a good representation of each sgRNA in the pooled library plasmids, a small portion of the transformed bacteria were diluted 40 000-fold and plated it on a separate 15 cm plate. Post-incubation, the number of colonies was counted. An efficiency of >50× colonies per sg plasmid construct was achieved (e.g. for a 12k library, a minimum of 600k colonies is needed). The colonies were scraped into a 50 ml tube using a cell scraper. The pooled library plasmids were purified using the GenEluteTM HP Endotoxin-Free Plasmid Maxiprep Kit (Sigma cat: NA0410-1KT) as per manufacturer's instructions for downstream virus production and the future amplification.
Multiplicity of infection (MOI) determination
Each cell type and virus lot were tested by transducing cells in duplicates with 0, 50, 100, 150 and 200 μl of lentiviral Epi/Drug library virus. 24 h post-transduction, fresh media containing puromycin was added to one duplicate of virus dilution series plates (0, 50, 100, 150 and 200 μl) while the other duplicate received fresh media without puromycin. 48 h post-selection, viable cells from all plates were counted using Trypan Blue staining. The percentage of viable cells post-selection at a given virus volume were determined by comparing plates with and without puromycin. The virus volume that resulted in 25% cell survival post-puromycin selection was calculated. The calculated volume of pooled virus (MOI of 0.25) and the same cell culture conditions were used during the dropout screen.
CRISPR-Cas9 dropout screen
MDA-MB-231, MDA-MB-436 and SUM149PT cells over expressing Cas9 cells were infected with the lentiviral Epi/Drug library at a MOI <0.25. 24 h post-transduction, the media was replaced with fresh medium containing puromycin. 48 h post-selection (72 h post-transduction), cells were split into three technical replicates. Cell pellets from timepoints 0, 7 (SUM149PT) and timepoints 0, 10 (MDA-MB-231 and MDA-MB-436) were harvested for genomic DNA (gDNA) extraction. The average coverage of 400 cells/sgRNA was used for the screen, thus each cell pellet collected contained 5 × 106 cells. The gDNA was isolated using the QIAamp Blood Midi kit (Qiagen cat: 51183) as per manufacturer's instructions. gRNA inserts in the gDNA were subsequently amplified through PCR and Illumina next generation sequencing (HiSeq 2500) was conducted to determine gRNA depletion.
Clonogenic assays
Exponentially growing cell lines were seeded on 6cm dishes in triplicates at equal density, as indicated in figure legends. Cells were allowed to grow under experimental conditions for 14–28 days. In the ataxia telangiectasia and Rad3 related (ATR) inhibitor sensitivity assay, 24 h post-seeding (duplicates), cells were incubated with media containing an ATR inhibitor, Berzosertib (Selleckchem cat: S7102), at various concentrations (0.025, 0.05, 0.1 and 0.2 nM) for 24 h. The following day, the cells received fresh media and were grown for 21 days. Colonies were then fixed in ice-cold methanol for 20–30 min and stained with 0.5% crystal violet solution (Bioshop cat: CRY422), 20% methanol, diluted in PBS for 1 h, then counted. Images of representative dishes were captured using a Cannon LiDE scanner. Images were adjusted using Microsoft PowerPoint. In the ATR sensitivity assay, data from treated samples were normalized to untreated (DMSO) conditions.
Xenotransplantation
All xenotransplantation studies were approved by and performed in compliance with Princess Margaret Cancer Centre (PMCC) Animal Care Committee guidelines. Equal parts of cells (1 × 106 cells for MDA-MB-436 and 3.5 × 105 MDA-MB-231 as indicated in figure legends) and Matrigel (Corning cat: 354248) matrix were injected into inguinal mammary fat pads of 6–7-week-old NOD/SCID/IL2Rγ null (NSG) mice (Jackson Laboratory). Tumor volume was calculated weekly using calliper measurements (length × width2)/2. Mice were euthanized by CO2 inhalation when the tumor size reached the ethical endpoint of 2 cm, in accordance with the rules of PMCC’s Animal Resources Centre.
Senescence-associated β-galactosidase staining
Cells were fixed and stained for senescence-associated β-galactosidase using the senescence β-galactosidase staining kit (Cell Signaling cat: 9860), following the manufacturer's instructions. The cells were visualized using a Leica DM IL inverted contrasting microscope under 10× or 20× magnifications.
RNASEH1 overexpression and cell cycle
500000 shCtrl or shMEPCE expressing MDA-MB-436 cells were transfected with 5 μg of pcDNA3-EV or pcDNA3-RNASEH1 using Lipofectamine 3000 (ThermoFisher) following manufacturer's guidelines. Media was changed after 24 h. 48 h following transfection, cells were trypsinized and fixed in a dropwise manner with 2% PFA while vortexing. Next, cells were permeabilized with 0.5% Triton X-100 (Sigma-Aldrich) and blocked with 10% FBS, 5% BSA (Bioshop cat: ALB001), 1% fish skin gelatin (Sigma cat: G7765), and 0.25% Triton X-100 (Sigma cat: X100-5ML) in PBS for 1 h at room temperature. Next, cells were incubated with anti-RNASEH1 (1:250) (Proteintech Group cat: 15606–1-AP) antibody for 1 h at room temperature. Cells were washed twice with PBS with 0.25% Tween 20 for 15 min/wash. Next, cells were incubated with fluorescent anti-mouse secondary antibody (1:1000) for 1 h at room temperature. Cells were washed twice with PBS with 0.25% Tween 20 for 15 min/wash. Next cells were stained with Hoechst 33342 (ThermoFisher) and subject to flow cytometry using BD LSRFortessa and data was analyzed using FlowJo (version 10).
Immunocytochemistry
Cells were seeded on 18 mm coverslips. 48 h post-seeding, cells were fixed using 4% paraformaldehyde (Bioshop cat: PAR070) for 15 min and permeabilized using 0.1% Triton-X 100 (Sigma cat: X100-5ML) for 10 min at room temperate. To test antibody specificity in the detection of RNA/DNA hybrids, control coverslips were digested using 25 U of exogenous RNASEH (New England Biolabs cat: M0297S) overnight following fixation and permeabilization but prior to addition of primary antibody. Coverslips were incubated using a blocking solution comprising of 10% FBS, 5% BSA (Bioshop cat: ALB001), 1% fish skin gelatin (Sigma cat: G7765), and 0.25% Triton X-100 (Sigma cat: X100-5ML) in PBS for 2 h at room temperature. After blocking, coverslips were incubated with primary antibodies diluted in blocking solution overnight at 4°C with gentle agitation. Following a series of washes with PBS + 0.05% Tween 20 (PBST), slides were stained with secondary antibodies conjugated to Alexa-Fluor 488 or 594 (Invitrogen cat: A11008, A11037, A11001, A11032) at room temperature for 2 h at a concentration of 1:500 while protected from light. Following several washes with PBST, coverslips were counterstained with DAPI (Invitrogen cat: D1306) and mounted using MOWIOL (Sigma cat: 81381). Slides were allowed to dry overnight prior to visualization and analysis.
For RNASEH1 overexpression analysis, cells were seeded in six-well plates were transfected with 2 μg of pcDNA3-Empty or pcDNA3-RNASEH1 using GenJet In Vitro DNA Transfection Reagent (SignaGen cat: SL100489), following manufacturers recommendations. 48 h post-transfection, cells were harvested and re-seeded on 18 mm coverslips, which were processed for immunofluorescence. Anti-γH2AX and anti-RNASEH1 antibodies were used to assess DSBs and confirm successful RNASEH1 overexpression, respectively. For experiments assessing the impact of transcriptional inhibition, cells transduced with shCtrl or shMEPCE were seeded onto coverslips, treated for 2 h with vehicle control or 2 μM flavopiridol (Selleckchem cat: S1230) or 10 μM LDC-000067 (Selleckchem cat: S7461), and then processed for γH2AX immunofluorescence.
Primary antibodies used for immunofluorescence staining were anti-γH2AX (1:500) (Millipore cat: 05-636 JBW301, clone JBW301), anti-RNAPII CTD pSer2 (1:1000) (Abcam cat: ab5095), anti-RNA/DNA hybrid (S9.6) antibody (1:200) (ATCC cat: HB-8730, Hybridoma cell line produces mAb S9.6) and anti-RNASEH1 (1:250) (Proteintech Group cat: 15606-1-AP). All immunofluorescence images were taken using a Lecia DM4000 B fluorescence microscope using 63× or 100× magnification as indicated in figure legends. ImageJ software (FIJI) (National Institutes of Health) was used to process and score S9.6 and γH2AX nuclear staining intensity, by first using the DAPI signal to create masks of nuclei and measuring fluorescence intensity. 10 random fields of view were measured per experiment. Microscope exposure settings were held constant within each experiment.
Immunohistochemistry
Harvested xenograft tumors of MDA-MB-436 cells transduced with shCtrl or shMEPCE were fixed using 10% formalin (Sigma cat: HT501128-4L) and embedded in paraffin sectioning. Sectioned slides were rehydrated utilizing a sequence of washes in Xylene, 100%, 95% ethanol, 90%, 80%, 70% ethanol, distilled H2O and finally PBS. Antigen retrieval was performed by placing the slides in a pressure-cooking containing 10 mM sodium citrate buffer (pH 6.0) for 30 min. After cooling to room temperature, slides were incubated in a blocking solution for 2 h followed by overnight incubation with primary antibodies diluted in blocking solution at 4°C in a humidifying chamber. Following a series of washes with PBST, slides were stained with secondary antibodies at room temperature for 2 h at a concentration of 1:500 while protected from light. Following several washes with PBST, coverslips were counterstained with DAPI and sealed with cover glass using MOWIOL. Slides were allowed to dry overnight before visualization and analysis. Primary antibodies used for immunohistochemistry were anti-Ki67 (1:200) (Novus Biological cat: NB500-170) and anti-Cleaved Caspase-3 (1:500) (Cell Signaling cat: 9664). All immunocytochemistry images were taken using a Lecia DM4000 B fluorescence microscope using 100X magnification. Five xenograft sections in at least five 40× fields were visualized and scored.
Dot blot
Dot blot was performed according to BioRad Bio-Dot Module instruction. Genomic DNA was phenol extracted, ethanol precipitated, and reconstituted in 50 μl Tris–EDTA buffer. Purified genomic DNA were treated accordingly (at 37°C for 3 h) was loaded onto a nylon membrane (GE Healthcare Life Sciences) using BioRad Bio-Dot Module connected to a vacuum, and then the DNA was crossed-linked using UV crosslinker (Stratalinker 2400, Stratagene) at the ‘Auto Crosslink’ setting (1200 μJ × 100). The membrane was blocked in 5% milk in Tris-buffered saline with 0.05% Tween-20 for 1 h and incubated with S9.6 antibody (1:100) overnight at 4°C to detect RNA-DNA hybrids. A duplicate blot was incubated with methylene blue as loading control.
GFP-dRNH1 purification
GFP-tagged RNASEH1 (both WT and D210N, Addgene # 174447 and # 174448) were transformed into Escherichia coli BL21(DE3)/ Rosetta, cultured in media supplemented with kanamycin (50 μg/ml) and overnight induction by IPTG at 15°C. Induced bacterial cells (1L) were harvested and pelleted by centrifugation at 10000 × g for 10 min. Bacterial pellets were resuspended on ice in 100 ml lysis buffer (50 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol, 1mM TCEP and EDTA-free protease inhibitor cocktail tablets) along with benzonase and lysed by sonication. Total cell lysate was centrifuged at 37 000 rpm for 60 min. The cleared supernatant was transferred to a new column containing 2ml of preequilibrated Ni–nitrilotriacetic acid Superflow beads and collected flowthrough. The columns were washed with 20CV wash buffer (25 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol, 1 mM TCEP, 15 mM imidazole, 1 mM PMSF), followed by 20 CV of stringent wash buffer (25 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol, 1 mM TCEP, 30 mM imidazole, 1 mM PMSF) and collected as wash. Samples were eluted five times with 2.5 CV elution buffer (25 mM Tris pH 8.0, 300 mM NaCl, 10% glycerol, 1mM TCEP, 300 mM imidazole, 1 mM PMSF). The eluted samples were flash frozen and stored at –80°C. Purified GFP-RNASEH1 was quantified along with BSA standards and analyzed by Coomassie stained 4–12% SDS-PAGE to determine protein purity and concentrations. The protein stocks were aliquoted, stored at –80°C, and freshly thawed for each experiment.
GFP-dRNH1 immunofluorescence
Cells were seeded on 18mm coverslips. 48 h post seeding, cells were washed twice with 1× PBS and fixed with 4% PFA for 10 min. The coverslips were washed twice with 1× PBS, and permeabilized with 0.3% Triton X-100 in PBS. After two 1 × PBS washes, cells were blocked with 3% BSA in PBS for 30 min. RNASEH (1:50; New England Biolabs cat: M0297S) was diluted in 1× RNASEH buffer and incubated on coverslips for 3 h at 37°C. Cells were washed and blocked with 3% BSA in PBS for 30 min. Coverslips were then incubated with purified GFP-dRNH1 (1:2000; 0.188 mg/ml) in staining buffer for 1.5 h at 37°C. After washing thrice with PBST for 5 min, coverslips were counterstained with DAPI and mounted using MOWIOL. Slides were dried overnight prior to imaging and stored at 4°C in the dark. ImageJ software (FIJI) (National Institutes of Health) was used to analyze GFP-dRNH1 nuclear staining intensity, by first using the DAPI signal to create masks of nuclei and measuring fluorescence intensity. More than 100 cells were quantified per biological replication.
DNA–RNA immunoprecipitation (DRIP)
A 10 cm plate of 80% confluent shCtrl and shMEPCE MDA-MB-436 cells were washed twice with PBS and cells were detached in PBS by scraping and pelleted by centrifugation. Pelleted cells were resuspended in 500 μl of DNA lysis buffer (10 mM Tris pH8.0, 1 mM EDTA and 0.5% SDS) and 8 μl of 10 mg/ml of Proteinase K (Roche cat: 03115887001) and incubated overnight at 37°C. On DRIP day 2, 250μl of 5M NaCl was added to lysed cells and spun at 15 000g. The supernatant was separated and mixed with 0.8 volumes of isopropanol. The precipitate (genomic DNA) was washed with 70% ethanol. The ethanol was removed, and gDNA pellets were dried, followed by gentle resuspension in TE buffer (10 mM Tris and 1 mM EDTA). 50 μg of gDNA was digested in a cocktail of restriction enzymes (HindIII, EcoRI, XbaI and BamHI (New England Biolabs cat: R0104, R0101, R0145, R0136)) and CutSmart buffer (New England Biolabs, B7204) overnight at 37°C with gentle rotation in duplicates. The next day, one of the digested duplicates was incubated with 50U RNASEH (Thermo cat: EN0202) for 4–5 h. The digested gDNA was extracted by phenol:chloroform:isoamyl alcohol extraction. 500 ng of the purified DNA was kept as input while 10 μg of DNA was diluted in IP buffer (0.05% Triton X-100 in PBS) and incubated with 10 μl of anti-(S9.6) antibody (ATCC cat: HB-8730, Hybridoma cell line produces mAb S9.6) or 2 μg control IgG antibody at 4°C overnight with rotation. The following day, 30 μl of Dynabeads (Thermo cat: 10004D) were pre-washed three times with IP buffer. The antibody-DNA mixture was then transferred into tubes containing washed beads and incubated at 4ºC for 2 h. Beads were pelleted and washed three times with IP buffer for 5 min. Beads were treated with 8 μl of Proteinase K and 492 μl of PBS for 3 h at 50°C. Post-incubation, the DNA was extracted by phenol:chloroform:isoamyl alcohol extraction and resuspended in 150 μl of TE buffer. The extracted DNA was mixed with the SYBR Green PCR master mix (Applied Biosystems cat:4 309155) and the indicated forward and reverse primers for each genomic region. Samples were amplified and analyzed using default settings in a 7900HT Fast Real-Time PCR System (Applied Biosystems) with the accompanying SDS v2.3 software. Primers used for RT-PCR are indicated in Supplementary Table S1.
DRIP library preparation and sequencing
DNA–RNA hybrids isolated using the method outlined above were submitted for library preparation and sequencing to Novogene. NEB Next Ultra II DNA Library preparation kit was used to prepare the library. Following quality control, fragments were repaired and dA-tailed. The DNA fragments with A tail were ligated to sequencing adaptors. The final DNA library was obtained by size selection and PCR amplification. After the construction of the library, the initial quantification was done with Qubit 2.0, and the library is diluted to 1 ng/l. Then the insertion size of the library was detected with NGS3K. If meeting the expectation, the accurate concentration of the library was quantified by qPCR (library effective concentration > 2 nM) to ensure the accurate molar amount that will pooled for sequencing. After library quality control, sequencing was performed for different libraries according to the concentration and the demand of data amount on Illumina NovaSeq platform (NovaSeq 6000).
Chromatin immunoprecipitation (ChIP)
For each condition, a 10 cm plate of 80% confluent cells was fixed in 1% formaldehyde for 10 min at room temperature, followed by 125 mM glycine neutralization for 5 min. Following neutralization, the media was removed, and the cells were washed twice with ice-cold PBS, and lysed in 5 ml of lysis buffer (5 mM PIPES, 85 mM KCl, 0.5% NP40 in ddH2O) supplemented with complete Protease Inhibitor Cocktail (Roche cat: 11697498001). Lysed cells were scraped and collected in 15 ml tubes and incubated on ice for 10 min. Nuclei were pelleted by centrifugation for 10 min at 4ºC and then lysed in 500 μl of nuclear lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris–HCl pH 8 in ddH2O) supplemented with protease inhibitor. Chromatin was sonicated using Microson Ultrasonic Cell Disruptor Xl (Misonix) to produce an average fragment length of 250–400 nucleotides, then centrifuged for 10 min at 12 500g at 4ºC. A volume corresponding to a 1% input was recovered and stored overnight at 4ºC. For each IP, 50 μl of chromatin was diluted 10 times in IP dilution buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8, 167 mM NaCl in ddH2O) and incubated with 2 μg anti-RNAPII CTD pSer2 (Abcam cat: ab5095) or 2 μg anti-RNAPII CTD pSer5 (Abcam cat: ab5131) or 2 μg control IgG antibody overnight with rotation at 4ºC. The next day, 30 μl pre-washed Dynabeads (Thermo cat: 10004D) were added to each sample and incubated for 2 h at 4ºC with rotation. Beads were pelleted, resuspended, and washed with 1 ml of a series of cold buffers in the following order: low salt wash, high salt wash, LiCl wash, and TE buffer (twice). Each wash was performed for 5 min with rotation at room temperature. To elute the chromatin, beads were subjected to two rounds of 15 min incubation with 100 μl of elution buffer (1% SDS, 0.1 M NaHCO3 in ddH2O) at room temperature. All tubes (including inputs) were supplemented with 8 μl of 5 M NaCl and subjected to reverse crosslinking overnight in a 65ºC hybridization oven with rotation. The following day, samples were incubated with 3 μl of RNASE A (10 mg/ml) (Thermo cat: EN0531) for 30 min at room temperature followed by a second incubation (2 h at 45ºC) after the addition of 4 μl EDTA (5 M), 8 μl of Tris–HCl (1 M, pH 8.0), and 1 μl of proteinase K (10 mg/ml) to each sample. DNA was then purified using a GeneAid PCR purification kit (GeneAid Biotech cat: DFH300) following the manufacturer's instructions and eluted in a total volume of approximately 200 μl. The eluted DNA was mixed with the SYBR Green PCR master mix and the indicated forward and reverse primers for each genomic region. Samples were amplified and analyzed using default settings in a 7900HT Fast Real-Time PCR System (Applied Biosystems) with the accompanying SDS v2.3 software. Primers used for RT-PCR are indicated in Supplementary Table S1.
DNA fibre spreading assay
shCtrl or shMEPCE cell lines were incubated with media containing either 1 μM doxycycline (DOX) or DMSO for 4 days before DNA fibre analysis. For analysis of untreated cells, shMEPCE or shCtrl cell lines were pulse-labelled with 25 μM CldU for 20 min, washed with medium, before being pulse-labelled with 250 μM IdU for 20 min. Alternatively, shMEPCE or shCtrl cell lines were pulse-labelled with 25 μM CldU for 20 min, and treated with 2 mM hydroxyurea treatment for 2 h, before being washed with media and pulse-labelled with IdU for 20 min. At all times media containing either 1 μM DOX or DMSO was used. Cells were harvested by trypsinization and washed in PBS. 10 000 cells were then lysed directly onto glass microscope slides using spreading buffer (200 mM Tris–HCl, pH 7.5, 50 mM EDTA, 0.5% SDS) and DNA fibres were allowed to spread down the slide by gravity. DNA fibres were fixed in methanol:acetic acid (3:1 ratio), denatured with 2.5 M HCl, and immunostained. CldU was detected using rat anti-BrdU (clone BU1/75, ICR1; Abcam, ab6326; 1:750), and IdU was detected using mouse anti-BrdU (clone B44; BD Biosciences, 347583; 1:750). Slides were fixed in 4% paraformaldehyde and incubated with secondary antibodies conjugated to Alexa Fluor 594 or Alexa Fluor 488 (Life Technologies). Labelled DNA fibres were visualized using a Nikon Eclipse Ni microscope with NIS-Elements software (Nikon Instruments) and images were captured using 60× oil-immersion objectives and analyzed using ImageJ software (US National Institutes of Health; NIH). Tract lengths were measured using ImageJ and arbitrary length values were converted into μM using the scale bars created by the microscope.
Proximity ligation assay (PLA)
Cells were seeded on 8-well chamber slides. 48 h post-seeding, cells were pre-extracted with cold 0.5% NP-40 for 4 min on ice. Cells were then fixed with 4% PFA/PBS for 15 min and washed three times with PBS. Following washes, the slide was blocked for 1 h at room temperate with 2% BSA in PBS. Slides were then incubated with primary antibody overnight at 4ºC (anti-RNAP II CTD 8WG16 (Abcam cat: ab817) alone; anti-PCNA (Santa Cruz cat: sc-7907) alone; anti-γH2AX (Millipore cat: 05-636 JBW301, clone JBW301) alone, or anti-RNAP II CTD 8WG16 with anti-PCNA; anti-γH2AX with anti-RNAP II CTD 8WG16, or anti-γH2AX with anti-PCNA). All antibodies were used at 1:500 dilution. The next day, slides were washed three times with PBS and incubated in a pre-mixed solution of PLA probe anti-mouse minus and PLA probe anti-rabbit plus (Sigma cat: DUO92101) for 1 h at 37ºC. The Duolink In Situ detection reagents (Sigma cat: DUO92101) used to perform the PLA reaction according to the manufacturer's instructions. Slides were counterstained with DAPI and sealed with cover glass using MOWIOL. Slides were allowed to dry overnight before visualization and analysis. All immunofluorescence images were taken and analyzed using a Lecia DM4000 B fluorescence microscope using 100X magnification as indicated in figure legends.
ChIP library preparation
Samples were prepared as outlined by Takara Bio USA Inc's ThruPLEX DNA-seq Kit user guide. Briefly, samples were normalized to 5 ng of DNA input and topped up to 10 μl with nuclease-free water. Samples were then mixed with 3ul of template preparation master mix and incubated at 22°C for 25 min, then 55°C for 20 min in a Veriti 96-well thermocycler (Thermo Fisher). Following template preparation, samples were mixed with 2 μl of library synthesis master mix and incubated at 22°C for 40 min on the same cycler. Samples were prepared for library amplification as outlined in the user guide and indexed individually. From here, PCR cycles were optimized by adding 0.75 μl of 10× SYBR green I nucleic acid gel stain (Thermo Fisher) to each sample. Once mixed, 10 μl from each sample was removed and run on a CFX96 Touch Real-Time PCR cycler (Bio-Rad). Samples were normalized to 1/3 of their amplification curve and amplified on a Veriti 96-well thermocycler. Samples were topped up to 50 μl with nuclease-free water prior to bead cleanup with AMPure XP beads (Beckman Coulter). Final library sizing and QC was evaluated on Agilent's high sensitivity DNA kit run on the 2100 Bioanalyzer (Agilent Technologies).
Sequencing
The libraries were quantified by qPCR then normalized and pooled to 1.25 nM. Each 1.25 nM pool was denatured using 4 μl of 0.2 N NaOH (Sigma) for 8 min at room temperature before being neutralized with 5 μl of 400 mM Tris–HCl (Sigma). Library pools were mixed with illumina's XP master mix and loaded immediately onto a NovaSeq 6000 S1 flow cell. Samples were sequenced with the following run parameters: read 1 – 50 cycles, read 2 – 50 cycles, index 1 – 8 cycles, index 2 – 8 cycles.
ChIP-seq and DRIP-seq data analysis
Paired-end ChIP-seq and DRIP-seq reads were aligned to the human genome hg38 using Bowtie2 (v. 2.2.6) and were then filtered to remove low-quality reads and reads that aligned to multiple loci (MapQ < 40). Duplicates were marked and removed using Picard (v. 2.10.9) and bigwigs were generated using deeptools (v. 3.5.1) bamCoverage normalizing to counts per million (CPM) and calculating the log2 fold-change between shMEPCE and shCtrl using bigwigCompare. Gene annotation and transcript start/termination site information was derived from the TxDb.Hsapiens.hg38.UCSC.knownGene Bioconductor package.
Transcripts considered for analysis have total Pol II coverage totalling at least two CPM in the promoter region (100 bp upstream of TSS to 300 bp downstream of TSS) of the control condition (shCtrl). For genes with multiple transcription start sites, only the transcript with the greatest total PolII occupancy in the promoter was used. Promoter release ratios (PRR) were calculated by taking the total coverage in the gene body (301 bp downstream of the TSS to the TTS) divided by the total coverage in the promoter region (100 bp upstream of the TSS to 300 bp downstream of the TSS). Genes highly regulated by RNAPII PPP were defined as having a fold change increase in PRR >1.5 in shMEPCE compared to shCtrl. Metagene plots and boxplots for both ChIP-seq and DRIP-seq were made in R using coverage matrices generated from deeptools computeMatrix (v.3.5.1) and a custom R script and occupancy heatmaps were generated using deeptools plotHeatmap (v.3.5.1).
Statistical analysis
Statistical analyses were carried out with GraphPad Prism 7, or in the case of ChIP-seq or DRIP-seq analyses, statistical tests were carried out with R. Statistical tests used to determine significance in each figure are outlined in the figure legends. The level of statistical significance is indicated in the figures.
RESULTS
CRISPR-Cas9 screen identifies MEPCE as an essential gene in BRCA1-mutant breast cancers
Given the recent success in targeting chromatin regulators in cancer (26), we performed a CRISPR-Cas9 dropout screen in three triple-negative breast cancer cell lines (MDA-MB-436, SUM149PT, and MDA-MB-231) to identify novel dependencies in BRCA1-mutant cancer cells. Both MDA-MB-436 (5396 + 1G > A) and SUM149PT (2288delT) cell lines carry mutations in the BRCA1 gene (27). We specifically conducted the screening with BRCA1-mutant cell lines providing genetic backgrounds with different magnitudes of HR defects. This approach also promoted the identification of robust synthetic lethal partners of BRCA1 outside of its canonical roles in HR. The MDA-MB-231 cell line, which expresses wild-type BRCA1 and is HR-proficient, served as the screening control (Figure 1A) (27).
Figure 1.

CRISPR-Cas9 dropout screen identifies MEPCE as a potential vulnerability in BRCA1-mutated breast cancers. (A) Schematic of CRISPR screening workflow. (B) Venn diagram representation of high-confidence negatively selected genes in all cell lines. (FDR < 0.1). 2 replicates for each time point were used. (C, D) Dot plot representing gene essentiality in BRCA1 WT cell line compared to BRCA1-mutated cell lines. C:MDA-MB-436 vs. MDA-MB-231. D:SUM149PT vs. MDA-MB-231. Vertical and horizontal dotted lines show 0.1 FDR cut-offs. Control sgRNA construct (sgLacZ) and essential genes PSMD1 and PSMB2 are highlighted. Only MEPCE demonstrates differential essentiality. (E) Scatter plot demonstrating negative correlation between the expression levels of MEPCE and BRCA1 in all TCGA breast cancer samples.
We generated stable Cas9-expressing cells and determined the activity of Cas9 using positive and negative control single guide RNAs (sgRNAs) based on cellular proliferation (Supplementary Figure S1A, B). We infected the Cas9-expressing cells with a sgRNA lentiviral Epi/Drug library targeting epigenetic regulators and druggable proteins with FDA-approved compounds. We used a multiplicity of infection (MOI) of <0.25 before passaging cells for two weeks under puromycin selection (Figure 1A). The sequencing results of the screen were analyzed using the MAGeCK-VISPR pipeline to calculate the depletion score for each gene. We used a false discovery rate (FDR) threshold of 10% in all cell lines to identify high-confidence hits. The screen identified 46 genes as essential in all cell lines for survival (Figure 1B). However, the screen identified only one gene, MEPCE, as essential in both BRCA1-mutant MDA-MB-436 and SUM149PT cells but not BRCA1 proficient MDA-MB-231 cells (Figure 1B–D; Supplementary Figure S1C, D).
To assess the clinical relevance of our findings, we sought to determine the relationship between MEPCE expression and human breast cancer. We observed a significant negative correlation between the expression of MEPCE and BRCA1 in the TCGA breast cancer data set (Figure 1E; provisional TCGA data). While correlative, these data suggest that breast tumors with lower BRCA1 expression tend to highly express MEPCE, underscoring the potential dependence of these tumors on MEPCE expression.
Validation of MEPCE as a synthetic lethal target for BRCA1-mutant tumors
To determine whether MEPCE is specifically required for the viability of BRCA1-mutant cancer cells, we first investigated the effect of depleting MEPCE in a panel of normal and cancer cell lines, each with a different BRCA1 status. Using a short hairpin RNA (shRNA) construct to deplete MEPCE expression, we found that reduced MEPCE levels do not alter the clonogenic growth of BRCA1-WT cells, including the human mammary epithelial cell line MCF10A and the triple-negative breast cancer (TNBC) cell lines MDA-MB-231 and MDA-MB-468 (Figure 2A, C). In contrast, MEPCE depletion drastically attenuated the growth of the BRCA1-mutant breast cancer lines MDA-MB-436 and SUM149 and BRCA1-mutant ovarian cancer cell lines COV362 and OVCAR8 (Figure 2B, C; Supplementary Figure S2A, B; COV362 harbors BRCA1 c.2611fs and c.4095 + 1G > T while OVCAR8 is BRCA1-methylated). To ensure the observed growth defect is dependent on BRCA1 status and not an unforeseen aspect of the genetic or epigenetic heterogeneity of cancer cell lines, we ablated MEPCE alone or in combination with BRCA1 in the same cells using shRNAs. Concomitant depletion of MEPCE and BRCA1 significantly impaired the clonogenic growth of MDA-MB-231 cells compared to the loss of MEPCE or BRCA1 alone (Figure 2D).
Figure 2.

Validation of MEPCE as a synthetic lethal target for BRCA1-mutant tumors. (A) Western blots showing MEPCE expression and representative images of clonogenic growth of indicated BRCA1-WT cells. Cells were fixed after 14–21 days (n = 3). (B) Western blots showing MEPCE expression and representative images of clonogenic growth of indicated BRCA1-mutated cells. Cells were fixed and quantified after 21–28 days (n = 3). (C) Normalized quantification of cells from A and B. (D) Western blot showing MEPCE and BRCA1 expression and representative images of clonogenic growth in indicated cells. Cells were fixed after 14 days of growth (n = 3). (E) In vivo growth curve of indicated xenografts. Tumor volume was measured using external calipers (mean ± SD). Representative images are shown (n = 5 per condition; bar = 1 cm). (F) Quantification of tumor mass post-resection from E (mean ± SEM). (G) Representative images of sectioned tumors from E stained for Ki67 and cleaved CASP3 (bar = 25 μm). Five fields of view were quantified for each tumor (n = 5) (mean ± SEM). Each dot represents a replicate. For A, B and D, 500 cells were seeded in 6 cm dishes. For C, E, F and G, statistical significance was calculated using Student's t-test. For D, statistical significance was calculated using one-way ANOVA (Tukey's multiple comparisons test).
We next examined the effect of MEPCE depletion on the in vivo growth of BRCA1-mutant cancer cells. We orthotopically injected MDA-MB-436 control and MEPCE-depleted cells into the mammary fat pads of nonobese diabetic/severe combined immunodeficiency/gamma (NSG) female mice and monitored tumor growth for 29 days. MEPCE loss significantly reduced tumor growth and tumor mass (Figure 2E, F). Additionally, we stained the resected tumors with antibodies against Ki67 and cleaved CASP3, which denote proliferation and apoptosis, respectively. MEPCE depletion in this xenograft model decreased Ki67-positive and increased cleaved CASP3-marked cells (Figure 2G), indicating reduced proliferation and increased cell death in BRCA1-mutant cells depleted of MEPCE.
We corroborated these findings in our MDA-MB-231 cell line model. Specifically, we generated MDA-MB-231 cells single or double knockdown for MEPCE and BRCA1, injected them into the mammary fat pads of NSG female mice, and monitored tumor growth for 60 days. We observed that the ablation of both MEPCE and BRCA1 in MDA-MB-231 cells significantly attenuated tumor progression compared to the depletion of each protein individually (Supplementary Figure S2C–E). These in vitro and in vivo findings confirm the CRISPR/Cas9 dropout screen results and demonstrate the dependency of BRCA1-mutant cells on MEPCE.
MEPCE suppresses genomic instability and senescence in BRCA1-deficient cells
Depletion of MEPCE in the BRCA1-mutated breast cancer cells MDA-MB-436 and ovarian cancer cells COV362 led to flat and vacuolated cell morphology (Figure 3A), reflecting a potential increase in senescence (28). Indeed, shMEPCE increased the levels of the senescence-associated β-galactosidase (SA-β-gal) activity (29) in MDA-MB-436 cells (Figure 3B). We observed similar results in MDA-MB-231 cells co-depleted of BRCA1 and MEPCE (Figure 3C), further supporting the notion of increased senescence in cells lacking both MEPCE and BRCA1.
Figure 3.

Loss of MEPCE in BRCA1-deficient cells increases genomic instability and induces senescence. (A) Representative images of indicated cells. Arrows indicate large and flat senescent cells. Bar = 50 μm. (B) Quantification of SA-β-gal activity in indicated cells. (C) Representative images and quantification of SA-β-gal activity in indicated cells. Bar = 100 μm. (D) Representative images of γH2AX and DAPI staining in indicated BRCA1-mutant cells. Arrows indicate micronuclei. (Bar = 25 μm). (E) γH2AX and 53BP1 foci quantification in indicated cells. (F) Quantification of micronuclei-positive cells in indicated cell type. (G) Representative images of γH2AX and DAPI staining in indicated cells (bar = 25 μm). (H) γH2AX and 53BP1 foci quantification in indicated cells. (I) Quantification of micronuclei-positive cells in indicated cell type. Each dot represents a replicate. Mean ± SEM is shown for all figures. For B, E and F, statistical significance was calculated using Student's t-test. For C, H and I, statistical significance was calculated using one-way ANOVA (Tukey's multiple comparisons test).
Given the role genomic instability plays in senescence and synthetic lethality (28), we investigated the effect of MEPCE loss in BRCA1-null cells on genomic instability. Loss of MEPCE in MDA-MB-436 and COV362 cells resulted in a significant increase in the percentage of cells with greater than or equal to ten endogenous foci of serine 139 phosphorylated histone variant H2AX (γH2AX) and p53 binding protein 1 (53BP1), markers of DNA damage and DNA double-strand breaks (DSBs) respectively, compared to shRNA control cells (Figure 3D, E; Supplementary Figure S2F, G). Furthermore, these cells had a significantly higher frequency of genomic aberrations, including micronuclei (Figure 3F). Similarly, the absence of both MEPCE and BRCA1 in MDA-MB-231 cells resulted in a significant increase in endogenous γH2AX and 53BP1 foci and micronuclei compared to the single knockdown controls (Figure 3G–I). These findings indicate that the exacerbated genome instability in BRCA1-null cells depleted of MEPCE leads to increased DNA damage and senescence. To determine whether this phenotype could be extended to other HR-deficient settings, we also depleted MEPCE in isogenic PALB2-, FANCD2-, and BRCA2-deficient MDA-MB-231 cells. We observed a significant increase in DNA damage and DSBs as measured by γH2AX and 53BP1 foci respectively following MEPCE depletion in shPALB2 and shFANCD2 cells (Supplementary Figure S2F, G). A significant decrease in survival was only observed in FANCD2-depleted cells (Supplementary Figure S2H).
MEPCE depletion triggers R-loop accumulation in BRCA1-deficient cells
BRCA1 is crucial for the resolution of R-loops through its interaction with SETX, an RNA/DNA helicase (30). R-loops form when a nascent RNA strand reinvades the DNA helix behind an RNA polymerase, resulting in a DNA–RNA hybrid and displaced single-stranded DNA (31,32). These structures are prone to forming G-quadruplexes, impairing replication progression, and promoting genomic instability (31). Thus, we decided to explore the role of MEPCE and aberrant R-loops in the context of BRCA1 deficiency. We employed a multitude of R-loop detecting approaches, including the catalytically-inactive GFP-tagged recombinant RNASEH1 (GFP-dRNH1) purified protein (33) as well as the DNA–RNA hybrid-recognizing S9.6 antibody to detect R-loops via immunofluorescence, in addition to dot blot. We found that depletion of MEPCE in MDA-MB-436 cells resulted in a significant increase in the nuclear GFP-dRNH1 signal (Figure 4A). Immunofluorescence using S9.6 antibody indicated that depletion of MEPCE in MDA-MB-436 cells resulted in a significant increase in nuclear and nucleolar S9.6 staining intensity (Figure 4B). S9.6 dot blot signal was also elevated in MEPCE depleted MDA-MB-436 cells (Supplementary Figure S3A). These results indicated R-loop accumulation in MEPCE deficient MDA-MB-436 cells.
Figure 4.

MEPCE depletion in leads to R-loop accumulation in BRCA1-deficient cells. (A) Representative images and quantification of R-loop levels as detected by GFP-tagged dRHN1 in indicated cells after mock or RNASEH treatment. Bar = 33 μm. (B) Quantification of anti-DNA–RNA hybrid (S9.6) immunofluorescence and quantification of nuclear hybrid levels in response to indicated treatment conditions (Mock, RNASET1, RNASEIII, and/or RNASEH). (C–E) DRIP-qPCR using S9.6 antibody in indicated MDA-MB-436 cells in ACTB gene (C), GAPDH gene (D) and TEFM gene (E). Enrichment is compared to negative control (IgG) followed by normalization to shCtrl treatment (mean ± SEM). (F) Representative images and quantification of co-staining for RNASEH1 and γH2AX in MEPCE-depleted MDA-MB-436 cells transiently transfected with human RNASEH1. Bar = 25 μM. For A, B and F, >100 nuclei were scored per condition, n = 3. Nuclear intensity was quantified using ImageJ. Distribution of all data points as well as the mean ± SEM for each individual replicate is shown. For C, D and E, each dot represents a replicate. C, D, E; Student's t-test. A, B and F; one-way ANOVA (Tukey's multiple comparisons test). a.u.: arbitrary units, TSS: transcription start site, TES: transcription end site, PPP: promoter-proximal pausing site, In1: intron 1, In2: intron 2, In5: intron 5.
Consistent with the reported role of BRCA1 in R-loop resolution, depletion of BRCA1 in the MDA-MB-231 cells resulted in increased S9.6 staining intensity (Supplementary Figure S3B). Interestingly, while knockdown of MEPCE in MDA-MB-231 cells did not affect the level of S9.6 staining, concurrent loss of MEPCE and BRCA1 in these cells further increased S9.6 intensity in these cells (Supplementary Figure S3B). To ensure the S9.6 antibody was not detecting single- and double-strand RNA, the coverslips were subject to pre-treatment with RNASET1 and RNASEIII prior to immunofluorescence.
To corroborate our S9.6 immunofluorescence findings, we performed DNA–RNA immunoprecipitation (DRIP)-quantitative polymerase chain reaction (qPCR) (DRIP-qPCR) in MEPCE-depleted MDA-MB-436 cells and their controls using the S9.6 antibody. We analyzed several genomic DNA sites of three actively transcribed genes, including β-Actin (ACTB), glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and transcription elongation factor mitochondrial (TEFM) (34). Our data indicate that depletion of MEPCE in MDA-MB-436 cells increases the abundance of R-loops proximal to the transcription start site (TSS) of these genes (Figure 4C–E). For instance, the R-loop signal in MEPCE-depleted MDA-MB-436 cells is significantly increased at the 5’ end of the promoter in the ACTB gene and the promoter-proximal pause (PPP) sites in the GAPDH and TEFM genes. These findings indicate that the loss of MEPCE in a BRCA1-null background leads to increased levels of R-loops in highly transcribed genes. Enzymatic treatment of the samples with RNASEH1, an enzyme that preferentially degrades RNA in DNA–RNA hybrids (31), reduced R-loop enrichment at these loci, validating the specificity of the S9.6 antibody in the DRIP assay (Figure 4C–E). We also performed DRIP-qPCR in BRCA1-proficient MDA-MB-231 cells following MEPCE depletion and observed a mild increase in S9.6 enrichment at the ACTB locus; however, statistically significant R-loop enrichment was only observed at intron 1 and this signal was not particularly sensitive to RNASEH treatment (Supplementary Figure S3C). To ensure specificity of the S9.6 immunoprecipitation, we also pulled down DNA–RNA hybrids from three positive (TMEM, CNS and RASA) and two negative (CORT and ANG) genomic loci as control (Supplementary Figure S3D). This suggests that R-loop accumulation following MEPCE depletion is specific to BRCA1-deficient cells.
To determine if R-loop accumulation directly accounts for the observed increase in genomic instability associated with dual loss of MEPCE and BRCA1, we transfected MDA-MB-436 cells with an RNASEH1 expression vector. As already shown, MEPCE-deficient MDA-MB-436 cells displayed increased levels of endogenous γH2AX nuclear staining (Figure 4F). Interestingly, exogenous expression of RNASEH1 significantly reduced endogenous γH2AX nuclear intensity in these cells (Figure 4F). Exogenous expression of catalytically inactive RNASEH1-D210N mutant was unable to rescue endogenous γH2AX nuclear intensity in MEPCE-depleted cells (Supplementary Figure S3E). Furthermore, MDA-MB-436 cells transfected with RNASEH1 had similar transfection efficiencies and a relatively comparable cell cycle profile. Interestingly, transfection of RNASEH1 in MEPCE-depleted MDA-MB-436 cells resulted in a statistically significant reduction in the sub-G1 population (Supplementary Figure S3F). These findings reveal that R-loop accumulation is indeed the causative factor for the elevated genomic instability observed in MEPCE-depleted BRCA1-null cells.
MEPCE depletion leads to increased overall transcription, redistribution of RNAPII from promoters to gene bodies, and increased R-loop accumulation in BRCA1-deficient cells
Since co-transcriptional R-loops can lead to genome instability (35), and MEPCE regulates transcription via P-TEFb control (22), we next examined whether co-transcriptional R-loops contribute to the elevated DNA damage observed in cells deficient in MEPCE and BRCA1. First, we examined transcription levels in shMEPCE and shCtrl MDA-MB-436 cells via 5-ethynyl uridine (EU) incorporation. We observed a significant increase in EU incorporation in the nucleus and the nucleolus following MEPCE depletion in MDA-MB-436 cells (Supplementary Figure S4A). Next, to investigate whether increased DNA damage in these cells could be attributed to increased transcription levels, we treated MEPCE-depleted MDA-MB-436 and control cell lines with the transcription inhibitors flavopiridol and LDC-000067 (36,37). LDC-000067 is a more potent and selective CDK9 inhibitor than flavopiridol (37). We observed a significant decrease in γH2AX nuclear staining intensity in MEPCE-deficient MDA-MB-436 cells following flavopiridol and LDC-000067 treatment (Figure 5A, B). These data suggest that γH2AX accumulation associated with MEPCE and BRCA1 deficiency is due to the deregulation of CDK9 and transcription, resulting in co-transcriptional R-loop accumulation.
Figure 5.

MEPCE depletion leads to redistribution of RNAPII from promoters to gene bodies in BRCA1-deficient cells. (A, B) Representative images and quantification of nuclear γH2AX staining intensity in indicated cell treated with a vehicle control or 2 μM flavopiridol for 2 h and B vehicle control or 10 μM LDC-000067 for 2 h. Bar = 25 μM. (C) Heatmaps of total RNAPII occupancy (scaled CPM) in the –1 kb to +4 kb region around the TSS in indicated cells. Log2 fold change is shown. (D) Schematic representation of parameters used to calculate the RNAPII release ratio (PRR). (E) Boxplots of log2 PRR values of total RNAPII ChIP-seq signal. (F) Heatmaps of total RNAPII occupancy (scaled CPM) in the –1 kb to +4 kb region around the TSS in indicated cells. Regions with PRR >1 are shown. PRR was calculated independently for both shCtrl and shMEPCE cell lines. (G) Scatterplot showing the log10(1 + PRR) in shCtrl (x-axis) and shMEPCE (y-axis). Vertical red dotted line represents PRR in shCtrl >1, lower and upper horizontal red dotted lines represent PRR in shMEPCE >1 and >2, respectively. (H) Venn diagram representing regions with PRR >1 in either shMEPCE (red) or shCtrl (grey) MDA-MB-436 cells. (I) Metagene plot of DRIP signal in all genes for shCtrl and shMEPCE with and without RNASEH treatment as well as DRIP signal in shMEPCE for genes with PRR >1 (green) and PRR >1.5 (purple). (J) Boxplots quantifying DRIP enrichment at PPP sites (DRIP peaks annotated to –100 bp to +300 bp around TSS). Total DRIP signal for shCtrl and shMEPCE as well as DRIP signal for genes with PRR >1 (green) and PRR >1.5 (purple) is shown. For A and B, nuclei were quantified using ImageJ. Median with interquartile ranges are shown. >100 nuclei were scored per condition (n = 3). A and B, one-way ANOVA (Tukey's multiple comparisons test). E and J, Student's t-test (J, Benjamini–Hochberg corrected P-values).
Next, given the CDK9 deregulation associated with MEPCE deficiency, we explored global RNAPII changes by performing RNAPII chromatin immunoprecipitation sequencing (ChIP-seq) following MEPCE depletion in MDA-MB-436 cells. Genome-wide heatmaps of RNAPII occupancy around transcription start site (TSS) regions (total counts per million (CPM) of RNAPII >2 in the promoters of shCtrl cells) demonstrated that RNAPII occupancy at the TSS is decreased and is further diffused into gene bodies in MEPCE-depleted cells (Figure 5C; Supplementary Figure S4B). The observed redistribution of RNAPII into the gene body following MEPCE depletion suggested a potential defect in RNAPII pausing control. To further test this possibility, we calculated the ratio of RNAPII occupancy between the gene body (300 bp to transcription termination site (TTS)) and its promoter (100 bp upstream and 300 bp downstream of the TSS). This ratio is referred to as the RNAPII release ratio (PRR) (Figure 5D). A significant increase in PRR was observed globally, specifically in MEPCE-depleted BRCA1-deficient cells (Figure 5E, F). The observed global changes indicated a differential response to MEPCE depletion. Therefore, we classified a subset of genomic regions most affected by the loss of MEPCE based on a PRR fold change (FC) >1 following MEPCE knockdown. Heatmaps revealed a drastic redistribution of RNAPII from promoters into the gene bodies after MEPCE depletion and apparent RNAPII vacancy was observed in the promoter regions of genes with PRR >1 in shMEPCE cells; however, no apparent differences were noted in promoter regions of genes with PRR > 1 in shCtrl cells (Figure 5F; Supplementary Figure S4C). Overall, 30 702 transcribed regions (CPM of RNAPII > 2) were noted. In shMEPCE cells, we found 7515 regions with a PRR >1 (24.48%) and in shCtrl, 3413 regions with a PRR >1 (11.12%) were identified. 1720 (5.60%) regions had a PRR > 1 in both cell lines (Figure 5G, H).
Attenuation of RNAPII pausing has previously been demonstrated to mitigate BRCA1-associated R-loop accumulation (38). Though our results suggested higher RNAPII release from the PPP site, contrarily we observed a significant increase in DRIP signal via qPCR at the PPP site of ACTB, GAPDH and TEFM genes (Figure 4C–E). Thus, we examined the distribution of DNA–RNA hybrids globally. Through DRIP sequencing (DRIP-seq), we observed a significant increase in S9.6 enrichment in MEPCE-depleted cells compared to control MDA-MB-436 cells (Figure 5I). Importantly, we saw a significant reduction in signal in samples pre-treated with RNASEH. Next, using our RNAPII ChIP-seq data, we examined the distribution of DRIP signal in genes that were determined to have a PRR > 1 and PRR >1.5 (Figure 5I). We also examined DRIP enrichment in PPP site and found that even though shMEPCE cells had an overall increase in DRIP signal at this site compared to shCtrl cells, genes that had a PRR >1 or PRR >1.5 had a statistically significant decrease in DRIP signal in this region (Figure 5J). Overall, decreased total RNAPII and RNAPII phosphorylated Ser2 (pSer2) occupancy was detected at PPP sites in MEPCE-depleted MDA-MB-436 cells compared to shCtrl cells (Supplementary Figure S4D, E).
Next, we aimed to substantiate that the increased RNAPII occupancy in gene bodies in MEPCE knockdown BRCA1-deficient cells is due to the release of paused RNAPII into the elongating form. Using ChIP-seq, we examined the occupancy of pSer2-RNAPII of the carboxy-terminal domain (CTD) in MDA-MB-436 cells transduced with shMEPCE or shCtrl. Measuring the ratio of pSer2-RNAPII to total RNAPII demonstrated that the relative levels of pSer2-RNAPII to total RNAPII increased at promoters globally in the absence of MEPCE (Supplementary Figure S4F). Not surprisingly, these findings are also consistent with the previously reported role of MEPCE as a negative regulator of RNAPII CTD pSer2 (39). Together, our findings demonstrate that the loss of MEPCE leads to a widespread release of the promoter-proximal paused RNAPII into gene bodies and a global increase in R-loop accumulation.
LARP7 is epistatic with MEPCE in maintaining genome stability and tumor viability
Aforementioned, alongside MEPCE, LARP7 forms the core 7SK snRNP and both proteins are essential for the stability of 7SK snRNP (15,18). Given the role of both these proteins in P-TEFb sequestration, we posited that LARP7 would behave similarly to MEPCE in maintaining genome stability in BRCA1-deficient cells, and that dual loss of MEPCE and LARP7 would be epistatic. We first examined cell proliferation following shRNA-mediated knockdown of LARP7 in MDA-MB-436, SUM149PT, and COV362 cells to test our hypothesis. Akin to MEPCE depletion, LARP7 depletion decreased the survival of these BRCA1-mutant cells (Supplementary Figure S5A–C). Also, MDA-MB-436 and COV362 cells depleted of LARP7 exhibited elevated nuclear S9.6 staining and levels of DNA damage (Supplementary Figure S5D–F). Like MEPCE-depleted MDA-MB-436 cells, we were able to substantially decrease the level of DNA damage in LARP7-depleted MDA-MB-436 cells following treatment with the CDK9 inhibitor LDC-000067 (Supplementary Figure S5G). These data suggest that the DNA damage accumulation observed in LARP7 and BRCA1 deficient cells reflects the deregulation of CDK9 function, as observed in MEPCE-depleted MDA-MB-436 cells.
Since we found that LARP7 depletion mimicked MEPCE depletion, we investigated the epistatic relationship between MEPCE and LARP7. We examined proliferation defects following the dual loss of MEPCE and LARP7 in MDA-MB-436 and COV362 cells and found no additional proliferation defects in these cells than controls lacking either MEPCE or LARP7 (Supplementary Figure S6A–D). These data suggest that LARP7 and MEPCE are indeed epistatic. Additionally, any factor that destabilizes the 7SK snRNP complex triggering P-TEFb de-sequestration would likely result in transcription deregulation and contribute to R-loop formation and genomic instability.
The catalytic activity of MEPCE is important for suppressing DNA damage and proliferation in BRCA1-deficient cells
Given our observations following LARP7 depletion and previous studies demonstrating the importance of MEPCE catalytic activity for 7SK snRNP stability (17), we hypothesized that the methyltransferase activity of MEPCE would be important for suppressing DNA damage in BRCA1-deficient cells. Therefore, we reconstituted MEPCE-depleted MDA-MB-436 cells with either full length MEPCE (MEPCE-WT) or catalytically inactive MEPCE-G451/455A (Supplementary Figure S6E). We found that reconstitution of cells with shMEPCE targeting the 3’ UTR with MEPCE-WT leads to a significant, albeit incomplete, rescue of DSB markers γH2AX and 53BP1 in MDA-MB-436 cells (Supplementary Figure S6F, G). Furthermore, a partial but significant rescue was also observed in the percentage of Ki67-positive cells following reconstitution of MEPCE-depleted cells with MEPCE-WT (Supplementary Figure S6H). Reconstitution of shMEPCE cells with MEPCE-G451/455A was unable to significantly rescue DSBs or Ki67 positivity, suggesting that the MEPCE catalytic activity is important for suppressing DNA damage and maintaining proliferation of BRCA1-deficient cancer cells (Supplementary Figure S6F-H).
MEPCE deficiency leads to increased transcription-replication conflicts and replication stress in BRCA1-deficient cells
Given that MEPCE depletion in BRCA1-null cells resulted in the redistribution of RNAPII from promoters into gene bodies, we hypothesized that transcription-replication collision might increase in the absence of MEPCE, contributing to the observed genomic instability. To address whether the increased RNAPII occupancy in gene bodies associated with the loss of MEPCE and BRCA1 could be leading to TRCs, we monitored interactions between the transcription and replication machinery using the proximity ligation assay (PLA). We employed antibodies against the RNAPII CTD and proliferating cell nuclear antigen (PCNA) to mark transcription complexes and replication forks, respectively (Figure 6A). PLA staining of MEPCE-deficient MDA-MB-436 cells revealed a marked increase of cells containing ≥3 PLA foci compared to control cells (Figure 6A), indicating increased co-localization of the transcriptional and replication machinery. Our data suggest that depletion of MEPCE in BRCA1-null cells increases TRCs, potentially contributing to the elevated genomic instability observed in these cells.
Figure 6.

MEPCE deficiency leads to transcription-replication collisions and replication stress in BRCA1-deficient cells. (A) Representative images and quantification of cells with ≥3 PLA foci per nucleus. Bar = 20 μm. More than 100 cells were quantified per biological replication. Three biological replicates were performed. (B) Schematic of conditions for DNA fibre experiments in figures C–G. (C) Quantification of fork track length in indicated cells. (D) Quantification of the fork rate in indicated cells. (E, F) Quantification of the percentage of ongoing forks (E), and stalled forks (F) in indicated cells. (G) Quantification of fork asymmetry. (H) Western blot indicating the expression and phosphorylation levels of the replication stress proteins ATR and RPA32. (I) Representative images and graph of 21-day clonogenic survival of indicated cells treated with DMSO or the ATRi Berzosertib. (mean ± SEM). (J) Nuclear γH2AX quantification in indicated cells treated with vehicle control or 250 nM aphidicolin for 2 h. Median with interquartile range is shown; >100 nuclei were scored per condition. For A, E and F, mean ± SEM is shown. For C and D, median with interquartile range is shown; Kruskal–Wallis test. E and F, multiple Student's t-test. G, Mann–Whitney Rank Sum test. I, two-way ANOVA (Sidak's multiple comparisons test). J, one-way ANOVA (Tukey's multiple comparisons test).
R-loop accumulation and TRCs can stall the replication machinery and/or lead to replication fork collapse (31,40–42). Therefore, we performed the DNA fiber assay to examine whether MEPCE-depleted MDA-MB-436 cells would exhibit elevated replication stress phenotypes. Examination of the replication fork track length revealed a significant decrease in tract length in MDA-MB-436 cells lacking MEPCE (Figure 6B, C). Furthermore, the replication fork rate significantly decreased following the knockdown of MEPCE in untreated MDA-MB-436 cells and post hydroxyurea treatment (Figure 6B, D). Additionally, MEPCE-depleted MDA-MB-436 cells exhibited a significant decrease in ongoing forks, increased fork stalling, and elevated replication fork asymmetry of bi-directional forks (Figure 6E–G). MEPCE-depleted BRCA1-mutant cells displayed high basal levels of phosphorylated ATR and RPA, two replication stress markers (Figure 6H) (42). These cells were also significantly more sensitive to ATR inhibition than their control counterparts, highlighting the increased replication stress and genomic instability when BRCA1-null cells are depleted of MEPCE (Figure 6I). As the MDA-MB-436 cells lacking MEPCE presented with replication stress, a phenomenon known to induce genomic instability (42,43), we sought to examine the levels of DNA damage following treatment with the replication inhibitor aphidicolin. The nuclear intensity of γH2AX in MEPCE-depleted MDA-MB-436 cells decreased significantly following aphidicolin treatment (Figure 6J). Together, these data indicate that the genomic instability observed in MEPCE- and BRCA1-deficient cells is also in part due to the increased replication stress.
To further examine whether TRCs were leading to DNA damage following MEPCE depletion in MDA-MB-436 cells, we also performed PLA using γH2AX antibody. Indeed, we observed a significant increase in γH2AX-PCNA and γH2AX-RNAPII PLA foci in MEPCE-depleted MDA-MB-436 cells (Figure 7A, B). Furthermore, inhibition of transcription and replication using flavopiridol and aphidicolin, respectively, significantly decreased TRCs in MEPCE-depleted MDA-MB-436 cells (Figure 7C). These data, in addition to our previous observation that flavopiridol and aphidicolin treatments significantly reduced nuclear γH2AX intensity to control baseline levels indicate that TRCs are likely the primary source of genomic instability in MEPCE-depleted BRCA1-null cells.
Figure 7.

MEPCE deficiency in BRCA1-mutant cells leads to an increase in transcription-replication conflicts. (A) Representative images and quantification of PCNA-γH2AX PLA foci per nucleus in the indicated cells (mean ± SEM). Quantification for single antibody controls are shown. Bar = 15 μm (n = 3). (B) Representative images and quantification of PolII-γH2AX PLA foci per nucleus in the indicated cells (mean ± SEM). Quantification for single antibody controls are shown. Bar = 15 μm (n = 3). (C) Representative images and quantification of PLA foci per nucleus in the indicated cells (mean ± SEM) treated for 1 h with vehicle control DMSO or aphidicolin (250 nM) or flavopiridol (2 μM). Bar = 20 μm (n = 3). For A–C, statistical significance was calculated using one-way ANOVA (Tukey's multiple comparisons test).
MEPCE is dispensable for replication fork progression and stability in BRCA1-proficient cells
Due to the drastic differences observed at the replication fork following MEPCE depletion in MDA-MB-436 cells, we sought to investigate replication fork stability and progression in isogenic MDA-MB-231 cells. MDA-MB-231 cells depleted for both BRCA1 and MEPCE displayed a significant decrease in the percentage of ongoing forks under HU treatment (Supplementary Figure S7A, B). The percentage of stalled forks in these cells were also significantly higher (Supplementary Figure S7A, C). In addition, these cells also exhibited a significant increase in replication fork asymmetry of bi-directional forks, a significant decrease in replication fork track length, and a significant decrease in replication fork restart (Supplementary Figure S7A, D–G). However, MDA-MB-231 cells depleted of MEPCE alone showed no significant differences at the replication fork when compared to control MDA-MB-231 cells (Supplementary Figure S7A–G). The only significant difference in MEPCE-depleted MDA-MB-231 cells was observed during replication fork restart under HU treated conditions (Supplementary Figure S7A, F–G). These data demonstrate that MEPCE is important for replication fork progression and stability specifically in BRCA1-deficient conditions and is dispensable in BRCA1-proficient cells.
PAF1 deficiency increases R-loop accumulation, genomic instability, and TRCs in BRCA1-deficient cells
Given our data supporting the notion that TRCs due to the deregulation of RNAPII promoter-proximal pausing are the source of genomic instability in MEPCE-depleted BRCA1-null cells, we hypothesized that other RNAPII regulating factors could be similarly essential in BRCA1-mutant cancer cells. Specifically, we aimed to identify additional genes that might be essential for BRCA1-mutant cells and play a role in limiting RNAPII promoter-proximal pausing and transcription-replication collisions. Thus, we reassessed our CRISPR/Cas9 dropout screen results using less stringent cut-offs (FDR < 0.1 in MDA-MB-231 and SUM149PT cells and an FDR < 0.2 in MDA-MB-436 cells). We identified two further hits from our screen, namely PAF1 and ZNF335 (Supplementary Figure S8A). While ZNF335 has no previously reported link to RNAPII regulation, PAF1 plays a role in maintaining promoter-proximal Pol II pausing and is implicated in R-loop control (24,34). PAF1 depletion phenocopies R-loop accumulation after BRCA2 inactivation (34). Therefore, we hypothesized that PAF1 depletion may exacerbate R-loop accumulation and transcription-replication collisions resulting in elevated genomic instability in BRCA1-mutant cells, similar to what we observed with MEPCE depletion. Colony-forming assays revealed that PAF1 depletion decreased colony-forming capability in BRCA1-null MDA-MB-436 cells but not in BRCA1-proficient HEK293T or MDA-MB-231 cells (Supplementary Figure S8B, D). Additionally, we observed higher levels of DNA damage (Supplementary Figure S8E) and S9.6 intensity (Supplementary Figure S8F) in PAF1-depleted MDA-MB-436 cells compared to control cells. Most importantly, higher basal levels of PLA foci for total RNAPII and PCNA were observed in PAF1-deficient cells (Supplementary Figure S8G), suggesting higher levels of TRCs following PAF1 depletion in BRCA1-mutant cancer cells. Together, our data indicate that TRCs due to RNAPII deregulation are indeed a vulnerability of BRCA1-mutant cancer cells.
MEPCE depletion causes non-nucleolar and nucleolar DNA damage
Given that CDK9 is critical for RNAPII transcription elongation and has functions in initiation and termination (44), the primary focus of our mechanism of cell death following MEPCE depletion in BRCA1-mutant cells were TRC events. However, our examination of DNA damage revealed that a subset of γH2AX foci in MEPCE-deficient MDA-MB-436 cells were present in the nucleolus (Supplementary Figure S9A), as indicated by the presence of γH2AX in regions positive for the nucleolar transcription factor UBTF. While RNAPII can operate in the nucleolus, RNA Polymerase I (RNAPI) remains the primary polymerase driving transcription of ribosomal RNA (45,46). To determine which RNA polymerase contributed to the observed accumulation of DNA damage in MEPCE-depleted MDA-MB-436 cells, we examined γH2AX intensity following the inhibition of RNAPI and RNAPII in these cells using low-dose Actinomycin D and Flavopiridol, respectively. As already shown (Figure 5A, B), inhibition of RNAPII significantly reduced nuclear γH2AX intensity in MEPCE-depleted MDA-MB-436 cells (Supplementary Figure S9B, C). Interestingly, inhibition of RNAPI transcription using low-dose Actinomycin D also resulted in significantly decreased DNA damage levels in these cells (Supplementary Figure S9B, C), although not as robustly as RNAPII inhibition. Together, these data reveal that while the mechanism underlying genomic instability in BRCA1-mutant cells following MEPCE loss is primarily PCNA-RNAPII conflicts, collisions between RNAPI and replication machinery can also contribute to the increased genomic instability of cancer cells deficient for BRCA1 and MEPCE.
DISCUSSION
Here, we conducted a CRISPR-Cas9 dropout screen and identified MEPCE as a potential factor necessary for the survival of BRCA1-mutant cancer cells. Our study demonstrates that 7SK snRNP destabilization upon the loss of MEPCE in BRCA1-mutant cells deregulates P-TEFb, compromising the spatiotemporal separation of replication and transcription and promotes TRCs (Figure 8). It has previously been demonstrated that DNA double strand breaks (DSB) in transcriptionally active chromatin recruit RAD51 and are repaired via homologous recombination (HR) (47). Given that the mechanism underlying MEPCE/BRCA1 synthetic lethality is excessive transcription-replication conflicts (TRC), it is likely that loss of MEPCE results in P-TEFb de-sequestration and leads to transcriptional dysregulation, which ultimately lead to TRC. These TRCs lead to an accumulation of R-loops and increased DSB levels (23). Given the role of MEPCE in transcription regulation, it is likely that MEPCE depletion does increase transcription stress in BRCA1-proficient cells; however, it is likely that DNA damage caused during transcription is repaired via RAD51 and HR. Due to the compromised HR repair in BRCA1-mutant cells, they are specifically vulnerable to the loss of MEPCE. The increase in endogenous DSBs in cancer cells deficient for MEPCE and BRCA1 eventually results in DNA damage-induced senescence and thus, promotes synthetic lethality of these cancer cells in vitro and in vivo. Not surprisingly, loss of LARP7, which also destabilizes 7SK snRNP, led to a loss of viability of BRCA1-deficient cells due to increased DNA damage. Preliminary data from this paper suggest that other HR-deficient tumors may also rely on MEPCE for survival; however, this warrants further investigation.
Figure 8.

Proposed model showing consequences of perturbing MEPCE in BRCA1-deficient settings. MEPCE function to stabilize the 7SK snRNP which sequesters P-TEFb to regulate transcription elongation. Following MEPCE depletion, any transcription stress and DNA damage that occurs at transcriptionally active chromatin is repaired via HR. In BRCA1 deficient cells, MEPCE depletion leads to R-loop accumulation, transcription stress, and transcription-replication conflicts, which lead to DNA damage and synthetic lethality in BRCA1-deficient cells.
The link between the P-TEFb subunit CDK9 and RNAPII transcription is well established and therefore was examined in our study. Since we observed a subset of DNA breaks in the nucleolus of MEPCE-depleted BRCA1-mutant cells, we investigated the potential role of RNAPI in our model. Indeed, in addition to inhibition of RNAPII transcription, inhibition of RNAPI transcription also markedly reduced the level of DNA breaks in MEPCE-depleted BRCA1-mutant cells. It is noteworthy that a previous study has demonstrated that CDK9 links RNAPII transcription to ribosomal RNA processing (48). Furthermore, nucleolar RNAPII can drive ribosome biogenesis (45). Thus, MEPCE depletion may alter nucleolar integrity and ribosomal biogenesis through deregulation of RNAPII, resulting in nucleolar DNA damage. Furthermore, despite the widespread use of Actinomycin D as an RNAPI transcription inhibitor, it also affects RNAPII and RNAPIII (49) in a dose-dependent manner, indicating the observed reduction in DNA damage in MEPCE-depleted BRCA1-mutant cells following Actinomycin D could be due to RNAPII inhibition. Nonetheless, our data suggest that MEPCE depletion exacerbates genomic instability in BRCA1-mutant cells through transcription dysregulation and R-loop accumulation, resulting in replication stress, TRCs, and ultimately, genomic instability and loss of cell viability. Additionally, we identify PAF1 as another potential therapeutic target similar to MEPCE, which functions to prevent R-loop accumulation, TRCs, and genomic instability in BRCA1-mutant cancer cells. It should be noted that depletion of any factor that affects transcription, including MEPCE, LARP7, and PAF1, will rewire the transcriptional landscape of a cell, thus also leads to alterations in the replication programme, thereby increasing the cell's reliance on HR and pathways that maintain genome stability. All of the experiments in this manuscript were performed on freshly generated cells approximately 48 h following selection with an antibiotic marker such as puromycin or blasticidin; however, it should be noted that given transcription rewiring caused by MEPCE depletion, it is difficult to separate direct effects from indirect effects. Nonetheless, we observed a significant increase in TRC, replication stress, and genome instability following MEPCE depletion in BRCA1 deficient cells. Moreover, we were able to rescue genome instability phenotypes with WT MEPCE but not with the catalytically inactive MEPCE mutant. Given the excessive amount of DNA damage triggered by MEPCE depletion in HR-deficient cells, it is possible that in a subset of the cell population, such DNA damage events may have impacted other survival genes elsewhere in the genome. In such case, reconstitution with wild-type MEPCE would be unable to fully rescue this damage. In addition, as with any rescue experiment, it would be unlikely that artificial reconstitution would fully recapitulate the molecular and temporal complexity of the mechanisms regulating the expression of endogenously expressed genes. Despite these inherent and general limitations of rescue experiments, we wish to highlight that the partial rescue with wildtype MEPCE was still statistically significant.
In the present study, using inhibitors of P-TEFb, ChIP for RNAPII and RNAPII CTD pSer2, we have demonstrated that the activity of P-TEFb is elevated as a result of MEPCE depletion in BRCA1-mutant cells. However, a prior study revealed that depletion of NELFB, an integral subunit of NELF and an enzymatic target of P-TEFb (50), alleviated RNAPII pausing and suppressed BRCA1-associated R-loop accumulation (38). Based on this study, we were surprised to see a significant increase in R-loops via DRIP-qPCR at the PPP site. Thus, we performed DRIP-seq. Once again, we observed a significant increase in R-loops across the genome in MEPCE-depleted cells compared to control BRCA1-null cells. However, we did see a significant decrease in R-loops in genes that had a PRR >1 as determined through our RNAPII ChIP-seq data. NELFB is known to cooperate with DRB Sensitivity Inducing Factor (DSIF) to repress elongation by RNAPII (50). DSIF is also an enzymatic target of P-TEFb. While NELF phosphorylation by P-TEFb leads to its dissociation from the PPP site, it results in the conversion of DSIF into a positive elongation factor (51). Despite the fact that we did not examine the role of DSIF in our model, we suggest that increased P-TEFb activity due to MEPCE loss is altering the function of NELF and DSIF, thus dysregulation of both of these factors is likely contributing to the observed phenotype. A prior study demonstrated that MEPCE, outside of its functions in stabilizing the 7SK snRNP complex, binds to histone H4 tail to serve as a P-TEFb activator at specific genes important for cellular identity at the chromatin level (52). In the present work, we wanted to explore the global alterations caused by MEPCE depletion; thus, we limited our focus to the canonical function of MEPCE in the sequestration of P-TEFb.
Collectively, our findings demonstrate that MEPCE, and more generally TRCs, are a unique vulnerability in BRCA1-mutant breast and ovarian cancer. Considering that DNA damage in S-phase is primarily repaired via HR and R-loops have been shown to mostly generate DSBs, it is likely that DSBs that accumulate as a result of TRC are left unrepaired, thereby leading to impaired proliferation and cell survival. In addition, cells are continually exposed to transcription-related stress in other phases of the cell cycle due to aberrant P-TEFb activation in MEPCE-depleted cells. Therefore, factors regulating RNAPII promoter-proximal pausing, such as MEPCE and PAF1, highlight a unique vulnerability of BRCA1-mutated tumors and potentially other homologous recombination-deficient cancers. Our findings pave the way to potentially utilize this vulnerability to enhance the benefit from current chemotherapies and PARP-targeted therapies.
DATA AVAILABILITY
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials or have been deposited into a public database (GEO Accession: GSE173223). Additional data related to this paper may be requested from the authors.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all the members of the Hakem, Stewart, He, Prive and Mekhail laboratories for their contributions to this study and helpful discussions. We thank Princess Margaret Genomics Centre and University Health Network Animal Resources Centre for their assistance.
Author contributions: P.S.P., A.A. and R.H. designed the study. P.S.P., A.A., R.K., J.J.R., K.C.J.N., J.H., J.L., Y.F., C.F., T.Y., P.S., F.S., E.L., M.S. and A.H. performed experiments. G.D.B., M.A.P., K.M., H.H.H., G.P., G.S.S., A.H. and R.H. supervised the study. P.S.P., A.A., R.K., A.H. and R.H. wrote and edited the manuscript with feedback from all authors involved in the study.
Contributor Information
Parasvi S Patel, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada; Department of Medical Biophysics, University of Toronto, Ontario, M5G 1L7, Canada.
Arash Algouneh, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada; Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario, M5S 1A8, Canada.
Rehna Krishnan, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada.
John J Reynolds, Institute of Cancer and Genomic Sciences, College of Medical and Dental Sciences, University of Birmingham, Birmingham, UK.
Kevin C J Nixon, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada.
Jun Hao, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada.
Jihoon Lee, Department of Cell and Systems Biology, University of Toronto, Ontario, M5S 3G5, Canada.
Yue Feng, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada; Department of Medical Biophysics, University of Toronto, Ontario, M5G 1L7, Canada.
Chehronai Fozil, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada.
Mia Stanic, Department of Medical Biophysics, University of Toronto, Ontario, M5G 1L7, Canada.
Talya Yerlici, Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario, M5S 1A8, Canada.
Peiran Su, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada; Department of Medical Biophysics, University of Toronto, Ontario, M5G 1L7, Canada.
Fraser Soares, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada.
Elisabeth Liedtke, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada; Department of Medical Biophysics, University of Toronto, Ontario, M5G 1L7, Canada.
Gil Prive, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada.
Gary D Baider, Terrence Donnelly Centre for Cellular and Biomedical Research, University of Toronto, Toronto, ON, Canada.
Miquel Angel Pujana, Program Against Cancer Therapeutic Resistance (ProCURE), Catalan Institute of Oncology (ICO), Bellvitge Institute for Biomedical Research (IDIBELL), L’Hospitalet del Llobregat, Barcelona 08908, Catalonia, Spain.
Karim Mekhail, Department of Medical Biophysics, University of Toronto, Ontario, M5G 1L7, Canada.
Housheng Hansen He, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada; Department of Medical Biophysics, University of Toronto, Ontario, M5G 1L7, Canada.
Anne Hakem, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada.
Grant S Stewart, Institute of Cancer and Genomic Sciences, College of Medical and Dental Sciences, University of Birmingham, Birmingham, UK.
Razqallah Hakem, Princess Margaret Cancer Centre, University Health Network, Toronto, Ontario, M5G 1L7, Canada; Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario, M5S 1A8, Canada.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Lee K. and Margaret Lau Chair in Breast Cancer Research, a joint project with the University of Toronto and the Princess Margaret Hospital (to R.H.); STARS21, Terry Fox Foundation; Princess Margaret Cancer Foundation; Department of Medical Biophysics, University of Toronto; Ontario Graduate Scholarship, Government of Ontario (to P.S.P.); Canada Graduate Scholarships for Master's program; Ontario Graduate Scholarship; Department of Laboratory Medicine and Pathobiology, University of Toronto (to A.A.); Canadian Institute of Health Research [FDN 143214 to R.H.]; Canadian Cancer Society [705367, 706439 to R.H.]; Canadian Institute of Health Research and Cancer Research Society [24418 to R.H.]; Cancer Research UK program [C17183/A23303 to G.S.S.]; University of Birmingham (to J.J.R.); Canadian Institute of Health Research CIHR [PJT-175083 to G.P.]; Samuel Waxman Cancer Research Foundation (to G.P.); Carlos III Institute of Health [PI18/01029] co-funded by European Regional Development Fund (ERDF), a way to build Europe, Generalitat de Catalunya SGR [2017-449]; CERCA Program to IDIBELL (to M.A.P.). Funding for open access charge: CIHR.
Conflict of interest statement. M.A.P. is the recipient of an unrestricted research grant from Roche Pharma for the support of the ProCURE study.
REFERENCES
- 1. Siegel R.L., Miller K.D., Jemal A.. Cancer statistics, 2019. CA A Cancer J. Clin. 2019; 69:7–34. [DOI] [PubMed] [Google Scholar]
- 2. Downs B., Wang S.M.. Epigenetic changes in BRCA1-mutated familial breast cancer. Cancer Genet. 2015; 208:237–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Semmler L., Reiter-Brennan C., Klein A.. BRCA1 and breast cancer: a review of the underlying mechanisms resulting in the tissue-specific tumorigenesis in mutation carriers. J. Breast Cancer. 2019; 22:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mersch J., Jackson M.A., Park M., Nebgen D., Peterson S.K., Singletary C., Arun B.K., Litton J.K.. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015; 121:269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuchenbaecker K.B., Hopper J.L., Barnes D.R., Phillips K.-A., Mooij T.M., Roos-Blom M.-J., Jervis S., van Leeuwen F.E., Milne R.L., Andrieu N.et al.. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017; 317:2402. [DOI] [PubMed] [Google Scholar]
- 6. Krishnan R., Patel P.S., Hakem R.. BRCA1 and metastasis: outcome of defective DNA repair. Cancers (Basel). 2021; 14:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dziadkowiec K.N., Gąsiorowska E., Nowak-Markwitz E., Jankowska A.. PARP inhibitors: review of mechanisms of action and BRCA1/2 mutation targeting. Pharmacogn. Mag. 2016; 4:215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. de Bono J., Mateo J., Fizazi K., Saad F., Shore N., Sandhu S., Chi K.N., Sartor O., Agarwal N., Olmos D.et al.. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2020; 382:2091–2102. [DOI] [PubMed] [Google Scholar]
- 9. Noordermeer S.M., van Attikum H.. PARP inhibitor resistance: a tug-of-war in BRCA-mutated cells. Trends Cell Biol. 2019; 29:820–834. [DOI] [PubMed] [Google Scholar]
- 10. Ceccaldi R., Liu J.C., Amunugama R., Hajdu I., Primack B., Petalcorin M.I.R., O’Connor K.W., Konstantinopoulos P.A., Elledge S.J., Boulton S.J.et al.. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature. 2015; 518:258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kais Z., Rondinelli B., Holmes A., O’Leary C., Kozono D., D’Andrea A.D., Ceccaldi R.. FANCD2 Maintains fork stability in BRCA1/2-deficient tumors and promotes alternative end-joining DNA repair. Cell Rep. 2016; 15:2488–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Patel P.S., Abraham K.J., Guturi K.K.N., Halaby M.-J., Khan Z., Palomero L., Ho B., Duan S., St-Germain J., Algouneh A.et al.. RNF168 regulates R-loop resolution and genomic stability in BRCA1/2-deficient tumors. J. Clin. Invest. 2021; 131:e140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Patel P.S., Algouneh A., Hakem R.. Exploiting synthetic lethality to target BRCA1/2-deficient tumors: where we stand. Oncogene. 2021; 40:3001–3014. [DOI] [PubMed] [Google Scholar]
- 14. Schuster A., Erasimus H., Fritah S., Nazarov P.V., van Dyck E., Niclou S.P., Golebiewska A.. RNAi/CRISPR screens: from a pool to a valid hit. Trends Biotechnol. 2019; 37:38–55. [DOI] [PubMed] [Google Scholar]
- 15. Jeronimo C., Forget D., Bouchard A., Li Q., Chua G., Poitras C., Thérien C., Bergeron D., Bourassa S., Greenblatt J.et al.. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Mol. Cell. 2007; 27:262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xue Y., Yang Z., Chen R., Zhou Q.. A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Res. 2010; 38:360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang Y., Eichhorn C.D., Wang Y., Cascio D., Feigon J.. Structural basis of 7SK RNA 5’-γ-phosphate methylation and retention by MePCE. Nat. Chem. Biol. 2019; 15:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barboric M., Lenasi T., Chen H., Johansen E.B., Guo S., Peterlin B.M.. 7SK snRNP/P-tefb couples transcription elongation with alternative splicing and is essential for vertebrate development. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:7798–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nguyen V.T., Kiss T., Michels A.A., Bensaude O.. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature. 2001; 414:322–325. [DOI] [PubMed] [Google Scholar]
- 20. Yang Z., Zhu Q., Luo K., Zhou Q.. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature. 2001; 414:317–322. [DOI] [PubMed] [Google Scholar]
- 21. Yik J.H.N., Chen R., Nishimura R., Jennings J.L., Link A.J., Zhou Q.. Inhibition of P-tefb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell. 2003; 12:971–982. [DOI] [PubMed] [Google Scholar]
- 22. Kwak H., Lis J.T.. Control of transcriptional elongation. Annu. Rev. Genet. 2013; 47:483–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gan W., Guan Z., Liu J., Gui T., Shen K., Manley J.L., Li X.. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011; 25:2041–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen F.X., Woodfin A.R., Gardini A., Rickels R.A., Marshall S.A., Smith E.R., Shiekhattar R., Shilatifard A.. PAF1, a molecular regulator of promoter-proximal pausing by RNA polymerase II. Cell. 2015; 162:1003–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Soares F., Chen B., Lee J.B., Ahmed M., Ly D., Tin E., Kang H., Zeng Y., Akhtar N., Minden M.D.et al.. CRISPR screen identifies genes that sensitize AML cells to double-negative T-cell therapy. Blood. 2021; 137:2171–2181. [DOI] [PubMed] [Google Scholar]
- 26. Roberti A., Valdes A.F., Torrecillas R., Fraga M.F., Fernandez A.F.. Epigenetics in cancer therapy and nanomedicine. Clin. Epigenetics. 2019; 11:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Y., Bernhardy A.J., Cruz C., Krais J.J., Nacson J., Nicolas E., Peri S., van der Gulden H., van der Heijden I., O’Brien S.W.et al.. The BRCA1-Δ11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016; 76:2778–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Campisi J., Robert L.. Robert L., Fulop T.. Cell senescence: role in aging and age-related diseases. Interdisciplinary Topics in Gerontology. 2014; 39:Basel: S. KARGER AG; 45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee B.Y., Han J.A., Im J.S., Morrone A., Johung K., Goodwin E.C., Kleijer W.J., DiMaio D., Hwang E.S.. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006; 5:187–195. [DOI] [PubMed] [Google Scholar]
- 30. Hatchi E., Skourti-Stathaki K., Ventz S., Pinello L., Yen A., Kamieniarz-Gdula K., Dimitrov S., Pathania S., McKinney K.M., Eaton M.L.et al.. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell. 2015; 57:636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aguilera A., Gómez-González B.. DNA–RNA hybrids: the risks of DNA breakage during transcription. Nat. Struct. Mol. Biol. 2017; 24:439–443. [DOI] [PubMed] [Google Scholar]
- 32. Patel P.S., Krishnan R., Hakem R.. Emerging roles of DNA topoisomerases in the regulation of R-loops. Mutat. Res. 2022; 876–877:503450. [DOI] [PubMed] [Google Scholar]
- 33. Crossley M.P., Brickner J.R., Song C., Zar S.M.T., Maw S.S., Chédin F., Tsai M.-S., Cimprich K.A.. Catalytically inactive, purified RNase H1: a specific and sensitive probe for RNA-DNA hybrid imaging. J. Cell Biol. 2021; 220:e202101092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shivji M.K.K., Renaudin X., Williams Ç.H., Venkitaraman A.R.. BRCA2 Regulates transcription elongation by RNA polymerase II to prevent R-loop accumulation. Cell Rep. 2018; 22:1031–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hamperl S., Cimprich K.A.. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst). 2014; 19:84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chao S.H., Price D.H.. Flavopiridol inactivates P-tefb and blocks most RNA polymerase II transcription in vivo. J. Biol. Chem. 2001; 276:31793–31799. [DOI] [PubMed] [Google Scholar]
- 37. Albert T.K., Rigault C., Eickhoff J., Baumgart K., Antrecht C., Klebl B., Mittler G., Meisterernst M.. Characterization of molecular and cellular functions of the cyclin-dependent kinase CDK9 using a novel specific inhibitor. Br. J. Pharmacol. 2014; 171:55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang X., Chiang H.-C., Wang Y., Zhang C., Smith S., Zhao X., Nair S.J., Michalek J., Jatoi I., Lautner M.et al.. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat. Commun. 2017; 8:15908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Egloff S., Studniarek C., Kiss T.. 7SK small nuclear RNA, a multifunctional transcriptional regulatory RNA with gene-specific features. Transcription. 2018; 9:95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Aguilera A., García-Muse T.. R loops: from transcription byproducts to threats to genome stability. Mol. Cell. 2012; 46:115–124. [DOI] [PubMed] [Google Scholar]
- 41. Skourti-Stathaki K., Proudfoot N.J.. A double-edged sword: r loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014; 28:1384–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zeman M.K., Cimprich K.A.. Causes and consequences of replication stress. Nat. Cell Biol. 2014; 16:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tsegay P.S., Lai Y., Liu Y.. Replication stress and consequential instability of the genome and epigenome. Molecules. 2019; 24:3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bacon C.W., D’Orso I.. CDK9: a signaling hub for transcriptional control. Transcription. 2019; 10:57–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Abraham K.J., Khosraviani N., Chan J.N.Y., Gorthi A., Samman A., Zhao D.Y., Wang M., Bokros M., Vidya E., Ostrowski L.A.et al.. Nucleolar RNA polymerase II drives ribosome biogenesis. Nature. 2020; 585:298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Scull C.E., Schneider D.A.. Coordinated control of rRNA processing by RNA polymerase I. Trends Genet. 2019; 35:724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aymard F., Bugler B., Schmidt C.K., Guillou E., Caron P., Briois S., Iacovoni J.S., Daburon V., Miller K.M., Jackson S.P.et al.. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014; 21:366–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burger K., Mühl B., Rohrmoser M., Coordes B., Heidemann M., Kellner M., Gruber-Eber A., Heissmeyer V., Strässer K., Eick D.. Cyclin-dependent kinase 9 links RNA polymerase II transcription to processing of ribosomal RNA. J. Biol. Chem. 2013; 288:21173–21183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bensaude O. Inhibiting eukaryotic transcription. Which compound to choose? How to evaluate its activity?: which compound to choose? How to evaluate its activity?. Transcription. 2011; 2:103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wu C.-H., Yamaguchi Y., Benjamin L.R., Horvat-Gordon M., Washinsky J., Enerly E., Larsson J., Lambertsson A., Handa H., Gilmour D.. NELF and DSIF cause promoter proximal pausing on the hsp70 promoter in Drosophila. Genes Dev. 2003; 17:1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harlen K.M., Churchman L.S.. The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat. Rev. Mol. Cell Biol. 2017; 18:263–273. [DOI] [PubMed] [Google Scholar]
- 52. Shelton S.B., Shah N.M., Abell N.S., Devanathan S.K., Mercado M., Xhemalçe B.. Crosstalk between the RNA methylation and histone-binding activities of MePCE regulates P-tefb activation on chromatin. Cell Rep. 2018; 22:1374–1383. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials or have been deposited into a public database (GEO Accession: GSE173223). Additional data related to this paper may be requested from the authors.