

Abstract
The novel coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has spread worldwide. The main protease (Mpro) of SARS-CoV-2 plays a central role in viral replication and transcription and represents an attractive drug target for fighting COVID-19. Many SARS-CoV-2 Mpro inhibitors have been reported, including covalent and noncovalent inhibitors. The SARS-CoV-2 Mpro inhibitor PF-07321332 (Nirmatrelvir) designed by Pfizer has been put on the market. This paper briefly introduces the structural characteristics of SARS-CoV-2 Mpro and summarizes the research progress of SARS-CoV-2 Mpro inhibitors from the aspects of drug repurposing and drug design. These information will provide a basis for the drug development of treating the infection of SARS-CoV-2 and even other coronaviruses in the future.
Keywords: COVID-19, SARS-CoV-2, Mpro inhibitors, Drug repurposing, Drug design
Graphical abstract
1. Introduction
At the end of 2019, COVID-19 caused by SARS-CoV-2 brought about an unprecedented public health emergency around the globe. The severity of infection varies from asymptomatic to severe fatal disease [1,2]. SARS-CoV-2 mainly spreads through small droplets discharged by infected individuals when they breathe or cough [3,4]. As of February 18, 2023, 756 million confirmed cases and 6.8 million deaths have been reported globally (https://www.who.int/emergencies/diseases/novel-coronavirus-2019).
Coronaviruses (CoVs) are positive single-stranded RNA viruses being diversely prevalent in humans and wildlife [5]. According to the International Committee on Taxonomy of Viruses, Coronaviruses belong to the subfamily Coronavirinae in the family Coronaviridae of the order Nidovirales. The Coronavirinae are divided into four genera, including α-, β-, γ- and δ-coronaviruses (Fig. 1 ). There are seven coronaviruses that can infect humans. HCoV-229E and HCoV-NL63 belong to α-coronavirus, and HCoV-OC43, HCoV-HKU1, SARS-CoV-1, Middle East respiratory syndrome coronavirus (MERS-CoV) and SARS-CoV-2 belong to type β-coronavirus [[6], [7], [8]]. Coronavirus has posed a persistent threat to human. In 2002, SARS-CoV-1 infected 8096 people, of whom 774 died. MERS-CoV, which appeared in 2012, affected a total of 1841 individuals, 652 of whom died [9,10]. Therefore, it is an urgent task to develop more efficient antiviral drugs [11].
Fig. 1.

Schematic representation of the taxonomy of Coronaviruses.
Recently, a large number of drug targets of SARS-CoV-2 have been identified through relevant studies on COVID-19. The main protease (Mpro) is vital for the replication of SARS-CoV-2, so SARS-CoV-2 Mpro is considered as a prominent drug target of COVID-19 therapy. This review briefly introduces the structural characteristics of SARS-CoV-2 Mpro and focuses on the research progress of SARS-CoV-2 Mpro inhibitors in recent years from all sources, including drug repurposing and drug design. These information will provide a basis for the drug development of treating the infection of SARS-CoV-2 and even other coronaviruses in the future.
2. Structure and function of SARS-CoV-2 Mpro
SARS-CoV-2 is composed of genomic RNA complexes and membrane, which consists of at least three viral proteins: (a) Spinous protein (S); (b) Transmembrane membrane protein (M); and (c) Envelope protein (E) (Fig. 2 A) [[12], [13], [14]]. The spike protein (S) of SARS-CoV-2 is responsible for binding to its host cell surface receptor, angiotensin converting enzyme 2 (ACE2) [15]. After entering the host cells, viral RNA encodes two overlapping open-reading frames (ORF1a and ORF1b), which are translated into two large polyproteins, pp1a and pp1ab. These polyproteins are further processed by the main protease (Mpro) and papain-like protease (PLpro) to generate 16 nonstructural proteins (NSPs) (Fig. 2B) [16,17]. The replicase genes encoding 16 NSPs occupy the most of the genome, approximately two thirds [18]. These NSP4 -NSP16 released by cleavage with Mpro are responsible for viral genome replication and transcription [19,20]. Therefore, effectively blocking Mpro can stop the viral RNA replication and transcription, thereby reducing the virus proliferation.
Fig. 2.

Structure and function of SARS-CoV-2 Mpro. (A) The structure of coronavirus. (B) Coronavirus (SARS-CoV-2) genome. (C) Overall scheme of natural amide substrate hydrolysis by Cys145 and His41 at the active site of Mpro: a. The first step in the process is the deprotonation of the Cys145-thiol; b. In the second step, the anionic sulfur next attacks the substrate carbonyl carbon; c. Then, the product with an amino terminus is released, while the His41 is restored its deprotonated form; d. Next, the generated thioester is hydrolyzed to release the carboxylic acid. e. In the last step, the Cys-His catalytic binary is formed again for the next proteolytic cycle.
Mpro, also known as 3-chymotrypsin like protease (3CLpro), is a cysteine protease composed of about 300 amino acids and contains three domains [19,21]. Mpro, with a nonclassical Cys-His catalytic binary (Cys145 and His41) in the gap between domains I and II, cleaves the polyproteins at 11 positions with stringent substrate specificity and shows self-proteolytic activity [13,20,22]. In the Cys-His catalytic binary, Cys145 acts as a nucleophile and His41 plays a general acid-base role in the proteolytic process(Fig. 2C) [23]. Mpro is embedded in the polyproteins as the nsp5 domain, thus Mpro must use its own proteolytic activity to separate itself from the polyproteins in order to release the mature protease [[24], [25], [26], [27]]. The active site of Mpro is composed of four sites (S1′, S1, S2, and S4), which often accommodate four fragments (P1′, P1, P2, and P3, respectively) of peptidomimetic inhibitors. Notably, the S1′ is formed by His41, Gly143, Ser144, and Cys145 [28,29]. Mpro exclusively cleaves polypeptides after a glutamine (Gln) residue, and no known human protease displays the same cleavage specificity as Mpro [28,30], so Mpro inhibitors have extremely low toxicity to host cells. Mpro has become one of the ideal specific drug targets in the development of antiviral drugs.
3. The development of SARS-CoV-2 Mpro inhibitors
Because of the crucial role of Mpro in viral replication and transcription, it has been considered as a promising target for fighting COVID-19 drug development. Based on drug repurposing and drug design, scientists and researchers have developed a number of SARS-CoV-2 Mpro inhibitors. This section summarizes the research progress of SARS-CoV-2 Mpro inhibitors.
3.1. Approved SARS-CoV-2 Mpro inhibitors
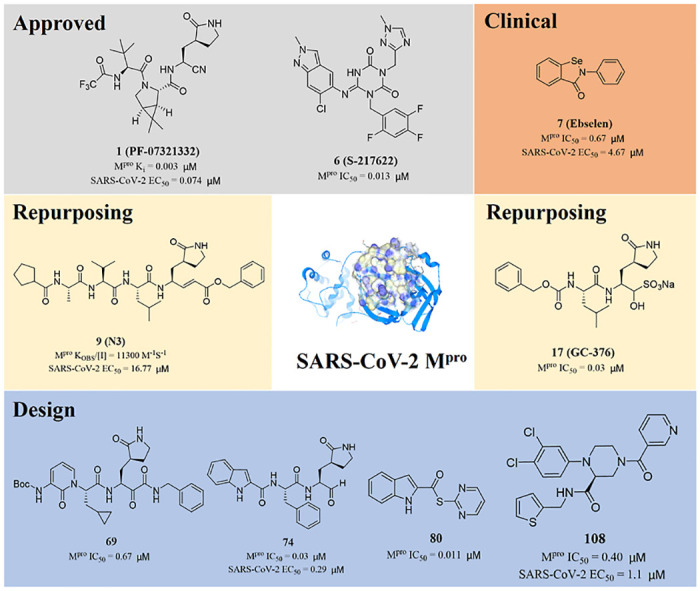
After the outbreak of SARS in 2003, PF-00835231 (2) was identified as an effective inhibitor of SARS-CoV-1 Mpro [31]. In 2021, in order to improve the low passive absorptive permeability and poor oral absorption of PF-00835231 (2) in animals [32], Owen and his colleagues reported an oral SARS-CoV-2 Mpro inhibitor (PF-07321332 (1), Ki = 0.003 μM) (Fig. 3 ). At the same time, PF-07321332 (1) had strong Vero E6 antiviral activity with the half maximum effective concentration (EC50) values of 0.074 μM and exhibited excellent selectivity and safety in vivo [33]. Furthermore, in order to block the rapid metabolism of PF-07321332 (1) by CYP3A, the HIV protease inhibitor Ritonavir was added to form a new drug, Paxlovid. It is used to treat adults with mild and moderate COVID-19 symptoms authorized by the United States Food and Drug Administration on December 22, 2021. The data showed an 89% reduction in the risk of COVID-19 related death or hospitalization in adults treated with Paxlovid, compared to placebo, within three days of symptom onset.
Fig. 3.

Approved and clinical drugs of SARS-CoV-2.
In addition to Paxlovid, Japan approved Ensitrelvir (S-217622) developed by Hokkaido University and Shionogi & Co, Ltd on November 22, 2022. S-217622 (6) was discovered via virtual screening followed by biological assay, and optimization of the hit compound 4 using a structure-based drug design strategy(Fig. 3) [34,35]. The specific steps are as following: first, they optimized the P1' ligand on the basis of compound 4 to obtain compound 5. The P1′ ligand of compound 5 is 6-chloro-2-methyl-2H-indayzole, and its enzyme inhibitory activity is increased by 90 times. Next, the methylamide moiety of P1 was substituted by a series of heterocyclic compounds to obtain S-217622 (6). S-217622 (6) exhibited significant inhibitory activity against SARS-CoV-2 Mpro with IC50 value of 0.013 μM, and effective antiviral activity with EC50 value of 0.37 μM. In addition, S-217622 (6) showed antiviral activity against a series of SARS-CoV-2 variants and coronavirus family in vitro, favorable drug metabolism and pharmacokinetic (DMPK) profiles for the oral dosing, such as high metabolic stability (96% and 88% in human and rat liver microsomes, respectively), high oral absorption (97%), and low clearance (1.70 mL/min/kg) in rats [34].
The National Medical Products Administration (NMPA) approved the SARS-CoV-2 Mpro inhibitor SIM0417, which is used to treat adults with mild and moderate COVID-19 symptoms on January 28, 2023. The structure of SIM0417 has not yet been disclosed. SIM0417 and PF-07321332 (1) need to be combined with low-dose Ritonavir, which helps to delay their metabolism in vivo and improve the antiviral effect.
PF-07321332 (1) is a covalent peptidomimetic inhibitor with a nitrile warhead. Conversely, S-217622 (6) is a noncovalent, nonpeptidic inhibitor. The nonpeptidic character provides metabolic stability, and the lack of a covalent warhead reduces potential off-target toxicity issues. Most of the antiviral protease (HIV and HCV) inhibitors in clinic are peptidic, with short plasma half-life and easy to be filtered and cleared by the kidney. The method adopted by Unoh et al. eliminates the bias towards molecules containing amide bond, thus avoiding potential metabolic degradation.
3.2. SARS-CoV-2 Mpro inhibitors in clinical trials
Following the outbreak of SARS in 2003, Boras and his colleagues found a potential SARS-CoV-1 Mpro inhibitor PF-00835231 (2) based on structure-based drug design, and its phosphate prodrug is PF-07304814 (3) (Fig. 3). Then, PF-00835231 (2), as a single agent, showed potent antiviral activity against SARS-CoV-2 in vitro [31,36]. Currently, NCT04627532, NCT04535167 and NCT05050682 are in clinical trials (Table 1 ).
Table 1.
SARS-CoV-2 Mpro inhibitors in clinical trials (www.clinicaltrials.gov).
| Inhibitor | Sponsor | Phase | Status | Identifier |
|---|---|---|---|---|
| PF-07304814 | Pfizer | 1 | Completed | NCT04627532 |
| PF-07304814 | Pfizer | 1 | Completed | NCT04535167 |
| PF-07304814 | Pfizer | 1 | Completed | NCT05050682 |
| Ebselen | Sound Pharmaceuticals, Incorporated | 2 | Enrolling by invitation | NCT04484025 |
| Ebselen | Sound Pharmaceuticals, Incorporated | 2 | Enrolling by invitation | NCT04483973 |
| Masitinib | AB Science | 2 | Recruiting | NCT04622865 |
| Masitinib | AB Science | 2 | Recruiting | NCT05047783 |
Jin and his colleagues screened a large number of compounds (about 10000) by using fluorescence resonance energy transfer (FRET) analysis, including natural products, approved drugs and candidate drugs for clinical trials. Among them, Ebselen (7) had the strongest inhibitory effect on Mpro activity, with an IC50 value of 0.67 μM [37] (Fig. 3). In addition, Ebselen (7) has extremely low cytotoxicity (the median lethal dose in rats is > 4600 mg/kg), and its safety in humans has been evaluated in many clinical trials [[38], [39], [40]]. Considering the therapeutic potential of Ebselen (7) in COVID-19, two clinical trials have been registered to evaluate the safety and efficacy of this drug in patients with moderate and severe COVID-19 (Table 1) [41].
In 2021, Drayman and his colleagues screened a library of 1900 clinically used drugs that were either approved for human use or had extensive safety data in humans (phase 2 or 3 clinical trials). Eight of these drugs inhibited the activity of SARS-CoV-2 Mpro, of which Masitinib (8) is the most effective (Fig. 3). After receiving Masitinib (8) treatment, the virus titer in the lungs and nose of mice infected with SARS-CoV-2 decreased by more than 200 times, and the lung inflammation was also alleviated. The two phase 2 clinical trials have been registered with clinicaltrials.gov (identifier: NCT04622865 and NCT05047783) to test the effect of the combination of Masitinib (8) and isoquercetin on COVID-19 hospitalized patients [42] and evaluate the anti-viral efficacy of three different dosages of masitinibin patients with mild symptom (Table 1).
Ebselen (7) is a glutathione peroxidase mimic that can permeate the blood-brain barrier and has anti-inflammatory, anti-tumor and antiviral activities, making it a novel anti-inflammatory agent approved by FDA. Masetinib (8), an oral tyrosine kinase inhibitor, was approved by FDA in 2009 and 2015, respectively, for the treatment of pancreatic cancer and amyotrophic lateral sclerosis (ALS). The security of Ebselen (7) and Masitinib (8) have been proved repeatedly, thus the treatment of COVID-19 may become their new mission.
3.3. SARS-CoV-2 Mpro inhibitors obtained by drug repurposing
The high infectivity and pathogenicity of SARS-CoV-2 have made it a global pandemic, so it is urgent to develop effective drugs to treat COVID-19. Drug repurposing is considered to be one of the most practical and rapid methods to find such therapeutic drugs. Drug repurposing has the advantages of lower cost and higher safety, especially for the drugs that have been studied their clinical safety, which is attracting people's attention.
In 2020, Jin and his colleagues used molecular docking to observe whether N3 (9) could target SARS-CoV-2 Mpro (Table 2 ). A docking result indicates that N3 (9) could fit inside the substrate-binding pocket [37]. To evaluate the efficacy of N3 (9) against SARS-CoV-2 Mpro, they performed kinetic analysis. The results showed that N3 (9) was a time-dependent irreversible inhibitor and showed a very potent inhibitory effect on SARS-CoV-2 Mpro. They determined the value of kobs/[I] of N3 (9) for SARS-CoV-2 Mpro as 11,300 ± 880 M−1s−1, and the kobs/[I] was used as an approximation of the pseudo second-order rate constant (k3/ki) to evaluate the inhibitory effect.
Table 2.
Structures and biological activity data of compounds 9–22.
| Drug | Structure | Class | IC50a (μM) |
|---|---|---|---|
| N3 (9) | 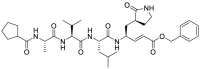 |
Mpro inhibitor | – |
| Disulfiram (10) | 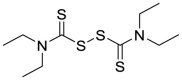 |
Alcohol deterrent | 9.35 |
| Carmofur (11) | 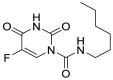 |
Antineoplastic agent | 1.82 |
| Shikonin (12) | 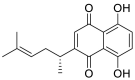 |
Pyruvate kinase M2 inhibitor | 15.75 |
| Tideglusib (13) | 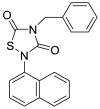 |
Anti-inflammatory and neuroprotective agents | 1.55 |
| PX-12 (14) | 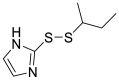 |
Thioredoxin-1 inhibitor | 21.39 |
| TDZD-8 (15) | 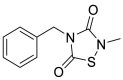 |
GSK-3β inhibitor | 2.15 |
| Boceprevir (16) | 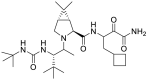 |
Hepatitis C virus NS3/4A protease inhibitor | 4.13 |
| GC-376 (17) | 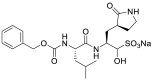 |
Mpro inhibitor | 0.03 |
| MG-132 (18) | 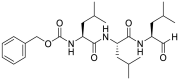 |
Proteasome inhibitor | 3.90 |
| 19 | 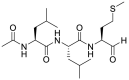 |
Calpain inhibitors II | 0.97 |
| 20 | 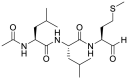 |
Calpain inhibitors XII | 0.45 |
| YH-53 (21) | 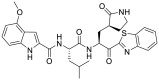 |
SARS-CoV-1 Mpro inhibitor |
– |
| Teicoplanin (22) |  |
Glycopeptide antibiotic | 1.50 |
SARS-CoV-2 Mpro IC50.
In addition, Jin and his colleagues found seven hits by screening with FRET analysis, including approved drugs (Disulfiram (10) and Carmofur (11)) and preclinical or clinical trial candidate drugs (Ebselen (7), Shikonin (12), Tideglusib (13), PX-12 (14) and TDZD-8 (15)) (Table 2). The IC50 values of these seven compounds were determined as 0.67–21.4 μM. Besides, the tandem mass spectrometry data showed that Ebselen (7), PX-12 (14) and Carmofur (11) could covalently bind to Cys145 of SARS-CoV-2 Mpro catalytic binary. Ebselen (7) and N3 (9) at the concentration of 10 μM showed the strongest antiviral effect on Vero cells infected with SARS-CoV-2. They further evaluated the efficacy of these two compounds in protecting cells. Ebselen (7) and N3 (9) showed inhibitory effect on SARS-CoV-2, with EC50 values of 4.67 and 16.77 μM, respectively [37,43].
In 2020, Ma and his colleagues found several potent SARS-CoV-2 Mpro inhibitors with effective cellular antiviral activity (Table 2). Boceprevir (16), an FDA approved HCV drug, inhibited SARS-COV-2 Mpro with an IC50 of 4.13 μM. In the cellular viral CPE assay, the EC50 against SARS-CoV-2 is 1.90 μM. Because Boceprevir (16) is a drug approved by FDA, the dose, toxicity, dosage form and pharmacokinetic properties have been clearly defined, which will greatly accelerate the design of follow-up studies [44,45]. GC-376 (17) is a research veterinary drug that is being developed the treatment of feline infectious peritonitis (FIP). GC-376 (17) showed promising antiviral activity against SARS-CoV-2 virus (EC50 = 3.37 μM). The enzyme inhibition of Mpro was the strongest, with IC50 value of 0.03 μM. Besides, it has promising in vivo therapeutic effect on cats infected with FIP and favorable in vivo pharmacokinetic properties. Therefore, GC-376 (17) can be detected in relevant animal models of SARS-CoV-2 at any time [[44], [45], [46], [47]]. Three calpain/cathepsin inhibitors, MG-132 (18), calpain inhibitor II (19) and XII (20), are effective inhibitors of Mpro, with single digit to submicromolar efficacy in enzyme detection. Calpain inhibitors II (19) and XII (20) inhibited SARS-CoV-2 in CPE assay, with EC50 values of 2.07 and 0.49 μM, respectively [44]. This result indicates that calpain/cathepsin inhibitors are rich sources of drug candidates for SARS-CoV-2, and also suggests that it may be feasible to design dual inhibitors against viral Mpro and host calpains/cathepsins. Further development based on these drugs may generate clinically useful COVID-19 antiviral drugs.
Konno et al. repositioned and investigated the potential of their SARS-CoV-1 inhibitors as anti-SARS-CoV-2 drugs [48]. In particular, YH-53 (21) contains an indole group at the P3- position and benzothiazolyl ketone as the reactive warhead, which demonstrated strong inhibitory activity against SARS-CoV-2 Mpro (Ki = 0.034 μM) (Table 2). X-ray structure analysis showed that YH-53 (21) formed multiple hydrogen bond interactions with the main chain amino acids and a covalent bond with the active site of Mpro.
Tripathi and his colleagues tested the different classes of drugs-nucleoside analogues, antiretroviral drugs, HIV protease inhibitors and neuraminidase inhibitors to research their potential antiviral effects. They found that Teicoplanin (22) was an effective drug against SARS-CoV-2 Mpro with an IC50 value of 1.5 μM [49] (Table 2). Teicoplanin is an effective glycopeptide antibiotic and has been reported to have anti-MERS-CoV activity [50,51]. Zhou et al. pointed out that Teicoplanin (22) acts at the early stage of the life cycle of coronavirus virus, and its mechanism of action is to inhibit the low-pH cleavage of the viral spike protein by late endosomal cathepsin L, thereby preventing the release of genomic viral RNA and viral replication [52]. In addition, Zhang and his colleagues showed that Teicoplanin blocked virus entry by specifically inhibiting the activity of cathepsin L of SARS-CoV-2 virus [53].
Zhu et al. performed a quantitative high-throughput screening (qHTS) of 10755 compounds using a self-quenching fluorescent peptide substrates, including approved drugs, candidate drugs for clinical studies and bioactive compounds. They identified twenty-three SARS-CoV-2 Mpro inhibitors with IC50 values ranging from 0.26 to 28.85 μM. Among these inhibitors, Walrycin B (24) showed the best enzyme inhibitory activity with IC50 value of 0.26 μM (Table 3 ). Hydroxycobalamin (23) (IC50 = 3.29 μM), Z-DEVD-FMK (25) (IC50 = 6.81 μM), LLL-12 (26) (IC50 = 9.84 μM), Suramin sodium (27) (IC50 = 6.5 μM) and Z-FA-FMK (28) (IC50 = 11.39 μM) are also potent inhibitors of SARS-CoV-2 Mpro [54].
Table 3.
Structures and biological activity data of compounds 23–39.
| Drug | Structure | Class | IC50a (μM) |
|---|---|---|---|
| Hydroxycobalamin (23) | 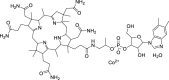 |
Vitamin B12a | 3.29 |
| Walrycin B (24) | 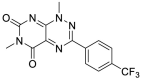 |
Antibacterial agent | 0.26 |
| Z-DEVD-FMK (25) |
 |
Caspase-3 inhibitor | 6.81 |
| LLL-12 (26) | 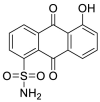 |
Signal transducer and activator of transcriptionl 3 inhibitor | 9.48 |
| Suramin sodium (27) |  |
Antiprotozoal drug | 6.5 |
| Z-FA-FMK (28) | 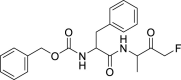 |
Cysteine protease inhibitor | 11.39 |
| Ethacrynic acid (29) | 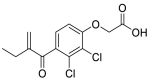 |
Diuretic | 1.11 |
| Naproxen (30) | 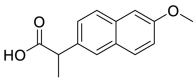 |
Anti-inflammatory drug | 3.45 |
| Allopurinol (31) | 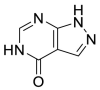 |
Xanthine oxidase inhibitor | 3.77 |
| Butenafine hydrochloride (32) | 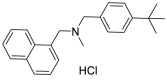 |
Antifungal | 5.40 |
| Raloxifene hydrochloride (33) | 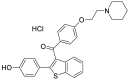 |
Estrogen receptor modulator | 5.61 |
| Tranylcypromine hydrochloride (34) | 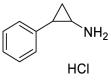 |
Antidepressant drug | 8.64 |
| Saquinavir mesylate (35) | 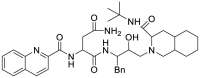 |
HIV protease inhibitor | 9.92 |
| JCP400 (36) | 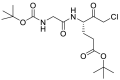 |
cysteine proteases inhibitor | 1.74 |
| JCP403 (37) | 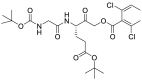 |
cysteine proteases inhibitor | 2.99 |
| JCP474 (38) | 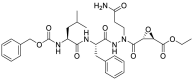 |
cysteine proteases inhibitor | 0.96 |
| JCP543 (39) | 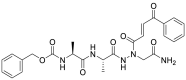 |
cysteine proteases inhibitor | 0.49 |
SARS-CoV-2 Mpro IC50.
The utilization of FDA approved drug library is an efficient tool for drug reuse in antiviral research [[55], [56], [57]]. In 2021, Zhao and his colleagues screened a library containing 774 FDA approved drugs to find potential SARS-CoV-2 Mpro inhibitors. Among them, seven drugs have superior inhibitory activity against SARS-CoV-2 Mpro (Table 3), namely, Ethacrynic acid (29), Naproxen (30), Allopurinol (31), Butenafine hydrochloride (32), Raloxifene hydrochloride (33), Tranylcypromine hydrochloride (34) and Saquinavir mesylate (35) [58].
Steuten and his colleagues screened a library of approximately 650 diverse covalent inhibitor scaffolds against the two major SARS-CoV-2 cysteine proteases, Mpro and PLpro. They identified seven inhibitors containing various electrophiles, of which six exhibited time-dependent inhibition of recombinant SARS-CoV-2 Mpro. Notably, they did not find any viable PLpro inhibitors. In the cellular infectivity assays of A549 epithelial lung cells, only chloromethyl ketone JCP400 (36) and acyloxymethyl ketone JCP403 (37) were active (Table 3). The two compounds demonstrated relatively weak potency with greater than 75% inhibition only when applied at concentrations above 20 μM. The two most effective inhibitors against SARS-CoV-2 Mpro in vitro, JCP474 (38) and JCP543 (39), were not active in the cellular infectivity assays [59].
Coelho et al. performed biochemical high-throughput screening (HTS) on the recombinantly expressed SARS-CoV-2 Mpro. A fluorescent assay was used to identify inhibitors from a compound library containing known drugs, bioactive molecules and natural products [60]. The screening led to the identification of 13 compounds with IC50 values ranging from 0.2 to 23 μM, and several known SARS-CoV-1 Mpro inhibitors were identified as inhibitors of SARS-CoV-2 Mpro, such as the organomercury compounds Thiomersal (40) and Phenylmercury acetate (41) (Table 4 ). Benzophenone derivative (42) was identified as the most effective screening hits. In addition, Evans blue (43), a sulfonic acid-containing dye, was also identified as a SARS-CoV-2 Mpro inhibitor [60].
Table 4.
Structures and biological activity data of compounds 40–54.
| Drug | Structure | Class | IC50a (μM) |
|---|---|---|---|
| Thiomersal (40) | 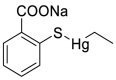 |
antiseptic and antifungal agent | 0.60 |
| Phenylmercury acetate (41) | 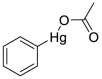 |
fungicide and slimicide | 0.40 |
| 42 | 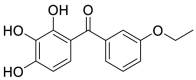 |
10.60 | |
| Evans blue (43) | l-glutamate uptake inhibitor | 0.20 | |
| 44 | 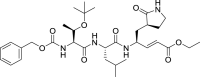 |
SARS-CoV-1 Mpro inhibitor |
0.15 |
| MAC-5576 (45) | 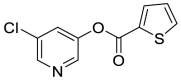 |
Mpro inhibitor | 0.08 |
| Baicalein (46) | 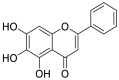 |
Xanthine oxidase inhibitor | 0.39 |
| Scutellarein (47) | 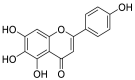 |
Anti-inflammatory | 5.80 |
| Dihydromyricetin (48) | 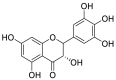 |
Dihydropyrimidinase inhibitor | 1.20 |
| Quercetagetin (49) | 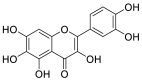 |
Pim-1 kinase inhibitor | 1.24 |
| Myricetin (50) | 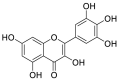 |
Antioxidant | 2.86 |
| Acriflavine (51) | 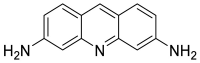 |
Antibacterial agent | 5.60 |
| Proflavine Hemisulfate (52) | 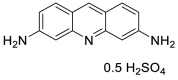 |
Antibacterial agent | 2.07 |
| 53 | 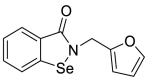 |
Metallo-β-lactamase inhibitor | 0.07 |
| 54 | 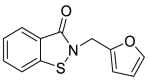 |
Metallo-β-lactamase inhibitor | 0.11 |
SARS-CoV-2 Mpro IC50.
Iketani and his colleagues also identified that compound 44, GC376 (17) and MAC-5576 (45) were inhibitors of SARS-CoV-2 Mpro (Table 4). In the cell-based assay, compound 44 and GC376 (17) also inhibited the SARS-CoV-2 virus, while MAC-5576 (45) did not [47].
Liu and his colleagues reported that Baicalein (46) (Table 4)and the four active Baicalein analogues compounds 47–50 could effectively inhibit the SARS-CoV-2 Mpro in vitro, and Baicalein (46) had the strongest inhibitory activity, with an IC50 value of 0.39 μM [61,62]. These compounds can be used as lead compounds to develop more effective drugs for COVID-19 disease through further optimization and modification.
In 2022, Chen research group established an enzymatic assay that used a fluorogenic substrate to screen the Mpro inhibitors [63]. The results showed that Acriflavine (51) and Proflavine Hemisulfate (52) had good inhibitory activity against Mpro with IC50 values of 5.60 and 2.07 μM, respectively (Table 4). They also exhibited nanomolar activities against SARS-CoV-2, which were superior to GC376 (17) for anti-HCoV-43. Acriflavine (51) has previously been reported as a PLpro inhibitor [64], therefore these two compounds might be dual-targeted inhibitors against coronaviruses.
In 2021, Yang and his colleagues used FRET experiments to identify potential inhibitors of Mpro. A total of thirty-six compounds were screened, with IC50 values ranging from 0.074 to 0.91 μM. Among them, compounds 53 and 54 had the strongest inhibitory effect on Mpro, with IC50 values of 0.074 and 0.11 μM, and Ki values of 0.031 and 0.078 μM, respectively (Table 4). The preincubation, jump dilution assays and fluorescent labeling experiments showed that the two compounds were covalently and irreversibly bound to Mpro, and molecular docking suggested that compound 54 formed an S–S bond with Cys145 at the active site of the enzyme [65]. The research of Yang and his colleagues provides two very effective scaffolds Ebsulfur and Ebselen (7) for the development of Mpro covalent inhibitors to combat COVID-19.
Most of the above compounds with potential SARS-CoV-2 Mpro inhibitory activity were obtained by scientists and researchers screening approved, clinical, and preclinical trials drugs. These compounds usually have different biological significance, such as anticancer, anti-inflammatory, antivirus, alcohol abstinence activities, etc. Baicalin (46) shows stronger antiviral effects and higher clinical efficacy than Ribavirin in the treatment of hand-foot-mouth disease [66]. N3 (9) and GC376 (17) have effect on SARS-CoV and MERS-CoV, two zoonotic coronaviruses infecting human beings [67]. GC376 (17) could also inhibit the main protease of ferret and mink coronavirus [68]. Multitarget antiviral inhibitors can more effectively overcome the drug resistance and usually show more potent therapeutical effect. Scientists and researchers are trying to find broadspectrum antiviral drugs, which will help fight SARS-CoV-2 and future coronaviruses.
3.4. SARS-CoV-2 Mpro inhibitors obtained by drug design
To date, drug repurposing efforts have not produced safe and effective Mpro inhibitors for approved clinical use in humans. Therefore, scientists and researchers have optimized and modified the above structures, and designed and synthesized many new SARS-CoV-2 Mpro inhibitors. SARS-CoV-2 Mpro inhibitors can be divided into covalent and non-covalent inhibitors according to their inhibitory mechanisms.
3.4.1. Covalent SARS-CoV-2 Mpro inhibitors
Covalent SARS-CoV-2 Mpro inhibitors, including peptidomimetic inhibitors and nonpeptidic small molecule inhibitors, bear different warhead groups and act through a two-step mechanism. First, these inhibitors bind to the active site and form non-covalent complexes with the target protease. Then, the warhead forms a covalent bond with nucleophilic residues, especially the catalytic Cys145 or other key cysteines (such as Cys300 and Cys44) [69,70], whose covalent bond modification may further cause the inactivation of SARS-CoV-2 Mpro.
Vankadara and his colleagues synthesized Nirmatrelvir and its 10 analogues with different electrophilic warheads, and determined its inhibitory activity against SARS-CoV-2 and HCoV-229E Mpro by using FRET based biochemical detection method [71]. Compound 55 partially substituted nitrile of Nirmatrelvir with a hydroxymethylketone moiety, and the IC50 values for SARS-CoV-2 and HCoV-229E Mpro were 0.008 and 0.013 μM, respectively (Fig. 4 ). Notably, these IC50 values were 4–11 fold higher than that of Nirmatrelvir (IC50 of 0.031 and 0.145 μM, respectively), indicating that the hydroxymethylketone warhead is more active than nitrile. Compound 56 replaced the nitrile of Nirmatrelvir with a ketobenzothiazole warhead, and the IC50 values for SARS-CoV-2 and HCoV-229E Mpro were 0.027 and 0.239 μM, respectively, similar to Nirmatrelvir. A subsequent HCoV-229E cell inhibition (EC50) assay showed that compounds 55 and 56 had the same potency as Nirmatrelvir, indicating that both inhibitors may be candidates for further drug development [71].
Fig. 4.

Structures and biological activity data of derivatives 53–58 of PF-07321332 and Ebselen.
Huff et al. designed a series of 2-phenyl-1,2-benzoselenzole-3-one derivatives targeting SARS-CoV-2 Mpro. Their substitutions are mainly concentrated in the N-phenyl moiety, including halogen (F, Cl and Br) and hydrophobic groups (CH3, CF3, SCH3, OCH3 and CH2CH3) as well as several lipophilic substituted phenyl moiety [72]. These compounds were determined to inhibit the replication of SARS-CoV-2 in infected Vero E6 and Calu-3 cells, and their potency was comparable to that of the clinical therapeutic drug Remdesivir. The most potent compound 57 demonstrated a nanomolar antiviral activity with EC50 value of 0.84 μM [72] (Fig. 4). This study provides a structural framework and mechanism of SARS-CoV-2 Mpro inhibitor, which will help to develop drugs for treating COVID-19.
Qiao and his colleagues designed and synthesized a series of Ebselen (7) derivatives, which replaced the critical groups of Ebselen (7) through non-covalent binding with SARS-CoV-2 Mpro, thus reducing the steric hindrance and ultimately improving the antiviral activity. Nine Ebselen (7) derivatives (EBs) had more potent inhibitory effects on SARS-CoV-2 Mpro, with IC50 values of 0.07–0.38 μM. Further evaluation of these derivatives showed that compound 58 exhibited the strongest inhibitory effect of SARS-CoV-2 virus replication, with an IC50 value of 4.08 μM in HPAepiC cells, as compared to the Ebselen (7) at 24.61 μM [73] (Fig. 4).
Dampalla and his colleagues synthesized a series of deuterated variants of an Mpro inhibitor GC376 (17) and proved that the deuterated GC376 (17) showed potent inhibitory activity against SARS-CoV-2 Mpro. In addition, the fatally infected mice were treated with deuterated derivatives of GC376 (17). After 24h of infection, K18-hACE2 mice treated with compound 59 had an increased survival rate compared with vehicle-treated mice [74] (Fig. 5 ). The crystal structures of compound 59 and SARS-CoV-2 Mpro revealed that heavy deuteration did not change the interaction between GC376 (17) and Mpro [44,75].
Fig. 5.

Structures and biological activity data of derivatives 59–66 of GC-376.
Sacco and his colleagues explored two series of SARS-CoV-2 inhibitors, one is the dual inhibitors targeting both Mpro and cathepsin L, such as calpain inhibitors II (19) and XII (20), and the other is the Mpro specific inhibitor, such as GC-376 (17) analogues UAWJ246 (60), UAWJ247 (61), and UAWJ248 (62) [76] (Fig. 5). In the plaque experiment, the EC50 values of GC-376 (17), UAWJ246 (60), UAWJ247 (61), and UAWJ248 (62) for inhibiting virus replication were 0.48, 4.61, 2.06, and 11.1 μM, respectively. Overall, these three GC-376 (17) analogues had confirmed the antiviral activity in cell culture. Comparing the Mpro binding and antiviral efficacy of GC-376 (17) analogues, it was found that the aldehyde warhead might be more suitable for cell activity than the α-ketoamide.
In 2021, Vederas and his colleagues designed and synthesized some new dipeptide derivatives with IC50 enzyme inhibition and EC50 antiviral value based on the antiviral peptide aldehyde GC373 and its bisulfite prodrug GC-376 (17) [77]. Compared with GC-376 (17), inhibitors 63 and 64 emerged as key compounds for Mpro inhibition with better IC50 and cellular EC50 values (Fig. 5). In addition, the newly designed and synthesized bisulfite adduct displayed a higher binding affinity for Mpro, which is 2.5–5.0 times higher, compared to GC-376 (17). The Na+ cation was replaced with K+ cation (65) or choline (66), which showed very similar IC50 values to that of the parent Na + form, and demonstrated retained effectiveness combined with increased aqueous solubility [77]. The results of this study provide new insights for drug development based on cysteine protease inhibitors, whose significance is not limited to SARS-CoV-2.
Zhang et al. developed the lead compound 67 as a potent inhibitor of SARS-CoV-2 Mpro through structural optimization and transformation. To improve the half-life of compound 67 in plasma, they hid the amide bond in the pyridine ring. In addition, in order to increase the solubility of the compound in plasma, they replaced the hydrophobic cinnamoyl moiety with a less hydrophobic Boc group to obtain compound 68. To enhance its antiviral activity on SARS-CoV-2, they replaced the cyclohexyl group in compound 68 with the smaller cyclopropyl group in compound 69 (Fig. 6 ). Compound 69 can effectively inhibit SARS-CoV-2 Mpro with IC50 value of 0.67 μM. In human Calu-3 cells infected with SARS-CoV-2, an EC50 of 4–5 μM was observed, whereas compound 70 lacking the Boc group was almost inactive [78]. This suggests that hydrophobic and bulky Boc groups are necessary for crossing the cell membrane, and here even more hydrophobic moieties may be advantageous.
Fig. 6.

Structures and biological activity data of compounds 67–70.
Ojida et al. designed and synthesized a series of chlorofluoroacetamide (CFA) derivatives as potential SARS-CoV-2 Mpro covalent inhibitors by introducing a CFA unit into an azapeptide scaffold. The data showed that compound 71 strongly blocked SARS-CoV-2 replication in infected cells, and its effect is equivalent to that of Nimatralvir (Fig. 7 ). X-ray structural analysis revealed that compound 71 formed a covalent bond with Cys145 at the catalytic center of Mpro [79].
Fig. 7.

Structures and biological activity data of derivatives 3, 21 and 71–77 of PF-00835231.
Dai et al. designed and synthesized two lead compounds 72 and 73 targeting SARS-CoV-2 Mpro based on common structural fragments of SARS-CoV-1 Mpro inhibitors and an aldehyde as a new warhead (Fig. 7). Compounds 72 and 73 exhibited high SARS-CoV-2 Mpro inhibitory activity with IC50 values of 0.053 and 0.040 μM, respectively. Besides, compounds 72 and 73 showed good anti-SARS-CoV-2 activity in cell culture, with EC50 values of 0.53 and 0.72 μM by plaque assay, respectively. Besides, they evaluated the pharmacokinetic properties of these two compounds. Compound 72 given to mice intraperitoneally (5 mg/kg) and intravenously (5 mg/kg) displayed a half-life (T1/2) of 4.27 h and 4.41 h, respectively. They were also observed a high maximal concentration (Cmax = 2394 ng/mL) and a good bioavailability of 87.8% when compound 72 was given intraperitoneally. The metabolic stability of compound 72 in mice was also good (clearance = 17.4 mL/min/kg). Compared with compound 72 administered intravenously to CD-1 mice, compound 73 displayed a shorter T1/2 time (1.65 h) and a faster clearance rate (clearance = 20.6 mL/min/kg). The above pharmacokinetic results indicate that both compounds are promising candidate drugs [80].
In addition, Dai and his colleagues designed and synthesized a series of novel peptidomimetic aldehydes against the Mpro of enterovirus 71 (EV71). Among them, compound 74 demonstrated broad-spectrum antiviral activity and could effectively inhibit the activity of SARS-CoV-2 with EC50 value of 0.29 μM. Besides, it also exhibited good inhibitory activity against SARS-CoV-2 Mpro (IC50 = 0.034 μM) [81] (Fig. 7). Compound 74 may be a good starting point for the optimization of SARS-CoV-2 Mpro inhibitors.
Shang's research group has previously developed a series of covalent and noncovalent inhibitors of Mpro. Among those inhibitors, peptidomimetic aldehyde 75 showed certain inhibitory activity against SARS-CoV-2 Mpro with an IC50 value of 3.889 μM [[82], [83], [84], [85]]. In 2022, Shang and his colleagues designed a series of peptide mimetic inhibitors based on compound 75. Among them, compound 76 had an excellent inhibitory effect on SARS-CoV-2 Mpro with an IC50 value of 0.148 μM. Besides, compound 77 had significant antiviral activity against SARS-CoV-2 with EC50 value of 1.06 μM [86] (Fig. 7). These peptide mimetic inhibitors may be used as encouraging candidate drugs for further development of antiviral drugs.
In 2022, Pillaiya and his colleagues conducted a virtual screening of the Tübingen Kinase Inhibitor Collection (TüKIC). Then they docked the two screening hits, IPA-3 (78) and LN5535 (79), at the Mpro active site (PDB ID: 7RC0) and observed that they showed a similar orientation to the indole chloropyridyl ester derivatives. Finally, they designed a novel class of small molecule thioesters as SARS-CoV-2 Mpro inhibitors through inserting variable cyclic systems from IPA-3 (78) and the (hetero)aryl thiols from LN5535 (79) (Fig. 8 ). Compound 80 showed excellent SARS-CoV-2 Mpro inhibition, and its kinac/Ki was 58700 M−1s−1 (Ki = 0.0141 μM). In Calu-3 and Vero E6 cells, several compounds exhibited antiviral activity in the nanomolar range, but were non-toxic to host cells [87]. The potent SARS-CoV-2 Mpro inhibitors also inhibited the Mpro of other β-coronaviruses, including SARS-CoV-1 and MERS-CoV, suggesting that they may contribute to the treatment of a wider range of coronavirus infections.
Fig. 8.

The structures of compounds 78–80 and the crystal structure docking diagram with SARS-CoV-2 Mpro (PDB ID: 7RC0).
Liu's research group designed and synthesized 30 covalent inhibitors with a piperazine scaffold containing different warheads [88]. Among them, compound 81 showed the most potent inhibitory effect on SARS-CoV-2 Mpro with IC50 value of 0.18 μM, and displayed excellent antiviral potency against SARS-CoV-2 (EC50 = 2.64 μM) (Fig. 9 ). In addition, compound 81 presented favorable target selectivity for SARS-CoV-2 Mpro versus human cysteine proteases.
Fig. 9.

Structures and biological activity data of compounds 81–98.
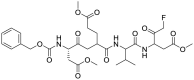
Qiao et al. designed and synthesized 32 new SARS-CoV-2 Mpro inhibitors containing bicyclic proline. First, an aldehyde was used in P1′ as a fighting part to form a covalent bond with the catalytic site Cys145, which is essential for antiviral activity [80]. Second, P2 fragment is from Boceprevir or Telaprevir, both of which are approved antiviral drugs. Finally, they decided to use a medium-sized P3 hydrophobic subgroups to enhance the potency and pharmacokinetic (PK) properties of the resulting inhibitors. Therefore, 32 compounds with different P3 fragments were designed and synthesized. Among them, MI-23 (82) had the strongest inhibitory activity with IC50 of 0.007 μM. Both MI-09 (83) and MI-30 (84) showed excellent antiviral activity in cell-based assays (Fig. 9). In the transgenic mouse model of SARS-CoV-2 infection, oral or intraperitoneal injection of MI-09 (83) or MI-30 (84) significantly reduced the lung viral load and lung injury. They also showed good pharmacokinetic properties and safety in rats. After i.p. administration, MI-09 (83) displayed a half-life (T1/2) of 4.53 h, a bioavailability of 78.0%, and a clearance rate (CL) of 22.67 mL/min/kg. The corresponding values for MI-30 (84) were T1/2 = 3.88 h, bioavailability = 76.2%, and CL = 17.10 mL/min/kg [89].
In 2022, Jiao and his colleagues reported 2-(furan-2-ylmethylene)hydrazine-1- carbothioamide derivatives as novel inhibitors of SARS-CoV-2 Mpro. Through library screening and similarity search, they identified compound 85 as an inhibitor of SARS-CoV-2 Mpro with the enzymatic IC50 value of 10.76 μM. The 4-nitrophenyl group in compound 85 was located at the S1 site and formed a hydrogen bond with His163, and the furan ring was deeply buried in the S2 site and had a π–π stacking with the imidazole of His41 [90]. At the same time, the thiourea linker of compound 85 formed two hydrogen bonds with the main chain carbonyl of Cys44, therefore, the removal or replacement of the thiourea linker led to the loss of inhibitory ability. In addition, the right part of compound 85 was located at the solvent exposed region and forms a hydrogen bond with Ser46. Then, Jiao and his colleagues discovered compounds 86 and 87, which are non-peptidomimetic inhibitors of Mpro with IC50 values of 1.57 and 1.55 μM, respectively, based on three rounds of optimization of drug structure design and synthetic modification (Fig. 9). Meanwhile, enzyme kinetics and mass spectrometry studies showed that compound 86 was a reversible covalent inhibitor of Mpro. Moreover, compound 86 had no obvious cytotoxicity in Vero and MDCK cells with CC50 values over 100 μM [90]. The SAR of the newly identified scaffolds was also discussed, which provides useful guidance for the further development of SARS-CoV-2 Mpro inhibitors.
Ghosh and his colleagues reported 5-chloropyridyl indole carboxylate derivatives as a class of potent SARS-CoV-2 Mpro inhibitors based on previous research. A number of compounds exhibited nanomolar Mpro inhibitory activity. The inhibition mode includes catalyzing cys145 nucleophilic attack on the ester carbonyl group of the inhibitor and forming a covalent bond between Cys145 and the carbonyl group of the active ester. Compound 88 inhibited SARS-CoV-2 Mpro with an IC50 of 0.25 μM and an antiviral EC50 of 2.8 μM in Vero E6 cells. Compound 89 with an N-allyl derivative demonstrated the most potent SARS-CoV-2 Mpro with IC50 value of 0.07 μM [91] (Fig. 9).
The 3,7-diazabicyclo[3.3.1]nonan (bispidine) framework belongs to the “privileged structures” in medicinal chemistry. For the first time, Shcherbakov et al. proposed the derivatives of 3,7-diazabicyclo- [3.3.1]nonane (bispidine) as potential SARS-CoV-2 Mpro inhibitors. The results of the experiments performed with bispidine compounds showed that 14 compounds exhibited activity in the concentration range 1–10 μM, and 3 samples exhibited submicromolar activity [92]. Compound 90 exhibited the strongest inhibitory activity against SARS-CoV-2 Mpro with an IC50 value of 0.75 μM (Fig. 9). The SAR studies exhibited that the molecule containing carbonyl group at the C-9 position of the bicycle had the greatest activity [92].
In order to identify Cys145 reactive electrophilic molecules with potent SARS-CoV-2 Mpro inhibition and high target selectivity, Ma and his colleagues systematically explored a series of new electrophilic reagents substituted by P1′ furan in compound 101, which led to the discovery of several new cysteine reactive warheads. The most promising lead compounds 91 with the dichloroacetamide warhead and 92 with the tribromoacetamide inhibited SARS-CoV-2 Mpro with IC50 values of 0.43 and 0.08 μM, respectively [93] (Fig. 9). These two compounds showed potent inhibitory effects on SARS-CoV-2 in both Vero E6 and Caco2-hACE2 cells, with EC50 values ranging from micromolar to submicromolar. It is worth noting that both compounds 91 and 92 had high target specificity for Mpro and did not inhibit host proteases including calpain I, cathepsin B, cathepsin K, cathepsin L, caspase-3 and trypsin [93]. In contrast, GC-376 (17) is not selective and inhibits calpain I, cathepsin B, cathepsin K, and cathepsin L with potency comparable to Mpro. The X-ray crystal structures of SARS-CoV-2 Mpro with compounds 91 and 92 showed that Cys145 forms covalent adducts with the reaction warhead.
In 2021, Malla et al. designed and synthesized penicillin derivatives, which were potent SARS-CoV-2 Mpro inhibitors. These derivatives form a stable acyl-enzyme complexes by reaction with the nucleophilic cysteine. Malla and his colleagues discovered the most potent compounds, which were the C6 dibromo-penicillin sulfones 93, 94 and 95, with IC50 values of 0.7, 0.6 and 0.5 μM, respectively [94] (Fig. 9). β-Lactams showed considerable potential as SARS-CoV-2 Mpro inhibitors.
In 2021, Moitessier research group transformed the non-covalent inhibitor 96 acting on SARS-CoV-1 Mpro into covalent inhibitors acting on SARS-CoV-2 Mpro [95]. The study of the crystal structure shows that compound 96 might be modified by adding a covalent warhead near the catalytic cysteine residue. As shown in Fig. 9, the sulfur atom of Cys145 is located at 3.2 Å from the imidazole part, which is the same position as the covalent warhead of PF-00835231 (2). Therefore, they developed covalent inhibitors of SARS-CoV-2 Mpro by replacing imidazole with covalent warheads. They first used the docking program FITTED, which was specially modified to accommodate covalent inhibitors, and screened a group of covalent warheads. The docking poses confirmed that the reactive group substituting the imidazole ring should lead to effective covalent inhibition. It is satisfactory that the inhibitory efficacy of the lead compounds 97 and 98 developed by them is an order of magnitude higher than that of compound 96 [95].
The warhead groups of covalent SARS-CoV-2 Mpro inhibitors mainly include ketones, aldehydes and different types of Michael receptors and form a covalent bond with Cys145 residues in the Mpro S1′ pocket. Comparing the Mpro binding and antiviral efficacy of covalent inhibitors, it is found that the aldehyde warhead might be more effective to inhibit SARS-CoV-2 Mpro. The aldehyde compounds 72, 73, 83 and 84, the peptidomimetic inhibitors designed and synthesized for SARS-CoV-2 Mpro by Dai and Qiao et al., exhibited good pharmacokinetic properties in vivo.
Covalent SARS-CoV-2 Mpro inhibitors generally include peptide drugs and small molecule drugs [96]. The main component of Pfizer's antiviral drug Paxlovid, PF-07321332(1), is a peptidomimetic and covalent SARS-CoV-2 Mpro inhibitor [97]. Several significant peptidomimetic inhibitors of SARS-CoV-2 Mpro have been reported so far, including compounds N3 (9), 69, 72 and GC-376 (17), which exhibited high SARS-CoV-2 Mpro inhibitory activity and SARS-CoV-2 inhibition at micromolar to submicromolar levels [37,44,78,80]. However, their peptide scaffolds are easily vulnerable to cell metabolism, resulting in low oral bioavailability. Therefore, PF-07321332 (1) is taken orally in combination with the CYP3A inhibitor Ritonavir to avoid extensive metabolism. Nevertheless, the addition of Ritonavir increases the risk of drug interactions and side effects. Considering the shortcomings of the current covalent Mpro inhibitors, there is an urgent need for candidate drugs with better performance.
3.4.2. Non-covalent SARS-CoV-2 Mpro inhibitors
Covalent inhibitors possibly have off target problems, which may lead to unpredictable toxic and side effects in the clinic [98]. In addition, non-covalent inhibitors may have a higher selectivity for SARS-CoV-2 Mpro. Therefore, scientists and researchers have designed and synthesized some novel potent non-covalent SARS-CoV-2 Mpro inhibitors.
Previously, Liu et al. reported a series of N-substituted 5-carboxamide-isatin compounds as inhibitors of SARS-CoV-1 Mpro. The optimal compound has a submicromolar IC50 for SARS-CoV-1 Mpro [99]. In order to verify the inhibitory effect of isatin compounds on SARS-CoV-2 Mpro, they screened a series of isatin compounds from the internally synthesized compound library. Liu and his colleagues tested their inhibitory effects against SARS-CoV-2 Mpro, and the results showed that 29 N-substituted isatin derivatives showed inhibitory effect on SARS-CoV-2 Mpro [100]. Compound 99 was the most potent SARS-CoV-2 Mpro inhibitor with an IC50 value of 0.045 μM (Fig. 10 ). At the same time, these inhibitors have high cytotoxicity, which also hinders the quantitative determination of their anti-SARS-CoV-2 activity.
Fig. 10.

Structures and biological activity data of compounds 99–108.
The sequence and structure of SARS-CoV-1 and SARS-CoV-2 Mpro are similar, and the binding modes of ML188 (100) and Calpain inhibitor XII (20) are similar. Therefore, based on the noncovalent SARS-CoV-1 Mpro inhibitor ML188 (100), Kitamura and his colleagues found the noncovalent inhibitor 101 of SARS-CoV-2 Mpro [101,102] (Fig. 10). It was found that compound 101 was an active diastereomer with an IC50 value of 0.20 μM. The antiviral activity of compound 101 was tested against SARS-CoV-2 in Vero E6 cells by the immunofluorescence assay. It was found that the EC50 value of compound 101 was 1.27 μM. The second antiviral assay was carried out in human lung epithelial Calu-3 cell line, with an EC50 value of 3.03 μM. The X-ray crystal structure of SARS-CoV-2 Mpro with compound 101 complex revealed that there was a ligand-induced binding pocket between the S2 and S4 sites [102]. This study showed a promising noncovalent Mpro inhibitor 101, which has potent cellular antiviral activity for further development.
Dengue virus (DENV) and SARS-CoV-2 pose a serious threat to global human health, and their proteases (NS2B/NS3 and Mpro) are considered as promising targets for drug development [103,104]. In 2022, KüHL et al. designed and synthesized several compounds with benzoxaborole motif and tested them against the DENV-2 protease and SARS-CoV-2 Mpro. Compound 102 has obvious inhibitory activity against SARS-CoV-2 Mpro with IC50 value of 6.1 μM. Besides, the EC50 values against DENV-2 protease was 2.4 μM in the cell-based DENVproHeLa assay [105] (Fig. 10). The majority of benzoxaboroles did not show relevant cytotoxicity or obvious off-target inhibition. These compounds provide an opportunity to develop drugs with anti-SARS-CoV-2 and DENV protease activities.
In 2021, Gao's research group reported 9,10-dihydrophenanthrene derivatives as non-peptidomimetic and non-covalent inhibitors of SARS-CoV-2 Mpro (Fig. 10). Among all tested 9,10-dihydrophenanthrene derivatives, compounds 103 and 104 had the strongest SARS-CoV-2 Mpro inhibition activity, with IC50 values of 1.55 and 1.81 μM, respectively. Besides, compound 103 exhibited excellent metabolic stability in the gastrointestinal tract, human plasma, and human liver microsomes [96]. This study can provide more structural references for the development of SARS-CoV-2 Mpro inhibitors.
In order to improve the affinity of compound 105 and explore the chemical determinants for ligand binding to S1 and S2, Kneller and his colleagues designed a series of derivatives of compound 105 (HL-3 series). Isothermal titration calorimetry was used to evaluate the thermodynamic binding properties of compound 105 and the two most potent inhibitors HL-3-68 (106) and Mcule-CSR-494190-S1 (107) [106] (Fig. 10). HL-3-68 (106) showed submicromolar affinity for SARS-CoV-2 Mpro, and its binding ability to the enzyme is about 2 times higher than the other two compounds. Notably, none of the compounds showed antiviral activity against SARS-CoV-2 in cell-based assays.
Liu's research group designed and synthesized potent non-covalent non-peptide SARS-CoV-2 Mpro inhibitors with 1,2,4-triasubstituted piperazine scaffold by modifying the piperazine nitrogen atoms and the carboxyl side chain of compound 105, respectively. Compound 108 exhibited potent inhibitory effect on Mpro with IC50 values of 0.40 μM, and displayed excellent antiviral activity (EC50 = 1.1 μM) (Fig. 10). It is noteworthy that compound 108 showed low cytotoxicity (CC50 > 100 μM) and good target selectivity for SARS-CoV-2 Mpro (IC50 > 50 μM for cathepsins B, F, K, L, and caspase 3) [107].
Japan approved a non-covalent, non-peptidomimetic SARS-CoV-2 Mpro inhibitor S-217622 (6) on November 22, 2022. S-217622 (6) had significant inhibitory activity with IC50 value of 0.13 μM and also showed good drug metabolism and pharmacokinetic characteristics [34]. Compounds 101 and 103 are non-covalent inhibitors of SARS-CoV-2 Mpro, with IC50 values of 0.20 and 1.55 μM, respectively. Compound 103 displayed excellent metabolic stability in the gastrointestinal tract, human plasma and human liver microsomes [96,102]. Non-covalent drugs not only have potent biological activity, but also exhibit excellent pharmacokinetic characteristics and good cell permeability. So far, scientists and researchers have found relatively few non-covalent Mpro inhibitors of SARS-CoV-2, and further efforts are still needed.
4. Conclusion and future perspective
Coronavirus have caused three pandemics in the past 20 years, including SARS, MERS and COVID-19. With the continuous epidemic of COVID-19, SARS-CoV-2 vaccines have been approved and used worldwide, but the emergence of virus mutation compromised the effectiveness of the vaccine [108]. In addition, the percentage of vaccinated people has remained at a moderate to low level in many countries. Therefore, the development of antiviral drugs targeting SARS-CoV-2 is still needed to reduce the incidence and symptom severity and fatality rates. Mpro plays an important role in the process of viral replication and transcription, and it is considered as a promising target for the development of antiviral drugs. Through drug repurposing and drug design, many SARS-CoV-2 Mpro inhibitors have been developed.
At present, the drug repurposing methods of SARS-CoV-2 Mpro inhibitors mainly include: (1) Reuse of homologous virus Mpro inhibitors: studying the potential of SARS-CoV-1 Mpro inhibitors as anti-SARS-CoV-2 drugs; (2) High throughput screening based on activity: using fluorescence technology to screen the activity of compounds from natural products, approved drugs, and clinical trial drugs libraries; (3) Virtual screening: using the reported Mpro crystal structure to reasonably narrow the range of active screening compounds. Through drug repurposing, a variety of scaffold structures can be provided for the design of SARS-CoV-2 Mpro inhibitors, and broad-spectrum antiviral agents that overcome drug resistance can also be obtained. In conclusion, drug repurposing can provide a reliable and efficient method to discover lead compounds (Fig. 11 ).
Fig. 11.

Discovery of SARS-CoV-2 Mpro inhibitors assisted by drug repurposing.
In the drug design section, relevant researchers designed and synthesized some covalent and non-covalent SARS-CoV-2 Mpro inhibitors by modifying and optimizing the structure of cysteine protease inhibitors. Through the analysis of SARS-CoV-2 Mpro peptidomimetic covalent inhibitors, we can summarize the most suitable molecular fragments (P1', P1, P2, P3) for interacting with Mpro active sites (S1', S1, S2, S3): (1) The P1' is the warhead group mainly consisting of ketones, aldehydes and different types of Michael receptors; (2) The most common fragment in the P1 is the γ-lactamic ring; (3) The P2 is usually aromatic or alkane groups, such as cyclopropyl, cyclohexyl, etc; (4) The P3 may be an aromatic group, such as indole and substituted phenyl (especially with halogens) (Fig. 12 ). The warhead groups form a covalent bond with the Cys145 residues in the Mpro S1' pocket, which determines the inhibitory activity of the Mpro inhibitors. The γ-lactamic ring in the P1 has the ability to mimic the natural amino acid glutamine and can penetrate into the S1 pocket to form a stablilizing interaction with key residues. Therefore, it can be maintained in the design of new Mpro inhibitors. The main component of Pfizer's antiviral drug Paxlovid, PF-07321332(1), is a peptidomimetic covalent SARS-CoV-2 Mpro inhibitor. On December 22, 2021, the United States Food and Drug Administration approved Paxlovid. Therefore, peptidomimetic covalent inhibitors can still be considered as the most promising antiviral drugs for the development of Mpro inhibitors. Notably, the covalent binding mode with Mpro may lead to the reduction of oral bioavailability and metabolic stability, which usually requires coadministration with pharmacokinetic enhancers such as Ritonavir. Non-peptidomimetic covalent inhibitor Ebselen (7) may be a potential lead compound as Mpro inhibitors.
Fig. 12.

The most common fragment in P1', P1, P2, P3.
Non-covalent drugs not only have potent biological activity, but also exhibit excellent pharmacokinetic characteristics and good cell permeability. In addition, S-217622 (6) has been proved to be effective for most SARS-CoV-2 variants and to possess significant selectivity for Mpro versus host protease [34], indicating that non-covalent non-peptide inhibitors have great potential for the therapy of COVID-19. The mutation and recombination rate of SARS-CoV-2 is relatively high, thus multitarget antiviral inhibitors can more effectively overcome the drug resistance. Scientists and researchers are trying to find broadspectrum antiviral drugs for fighting SARS-CoV-2 and future coronaviruses. In addition, SARS-CoV-2 Mpro inhibitors with diverse chemical scaffolds and improved pharmaceutical profile need to be further explored and developed.
SARS-CoV-2 Mpro is currently targeted by small molecular inhibitors, which may become a potential target of proteolysis targeting chimeras (PROTACs). PROTACs are a newly developed technology that degrades target protein through ubiquitin-proteasome pathway [109]. After the PROTAC molecule enters the cell, it first binds to the target protein and recruits E3 ligase for ubiquitination labeling, and then degrades the target protein through the proteasome in vivo [110]. The feasibility of antiviral PROTACs was first established with the degradation of the hepatitis C virus (HCV) NS3/4A protease. We firmly believe that the PROTAC technique will be used in the research of COVID-19 drugs.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors thank the National Natural Science Foundation of China (NSFC) (No. 82141216), Chunhui Program-Cooperative Research Project of the Ministry of Education, Liaoning Province Natural Science Foundation (No. 2022-MS-241), and Shenyang Young and Middle-aged Innovative Talents Support Program (No. RC210446) for financial supports. We acknowledged the support from Joint National Local Engineering Research Center of Fujian and Taiwan Chinese Medicine Molecular Biotechnology, Fujian Key Laboratory of Chinese Materia Medica, Fujian University Key Laboratory for Research and Development of TCM Resources, at Fujian University of Traditional Chinese Medicine.
Data availability
Data will be made available on request.
References
- 1.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie X., Hu L., Xue H., Xiong Y., Panayi A.C., Lin Z., Chen L., Yan C., Zhou W., Mi B., Liu G. Prognosis and treatment of complications associated with COVID-19: a systematic review and meta-analysis. Acta Materia Medica. 2022;1:124–137. [Google Scholar]
- 3.Wu J.T., Leung K., Leung G.M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. Lancet. 2020;395:689–697. doi: 10.1016/S0140-6736(20)30260-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan J.F.W., Yuan S., Kok K.H., To K.K.W., Chu H., Yang J., Xing F., Liu J., Yip C.C.Y., Poon R.W.S., Tsoi H.W., Lo S.K.F., Chan K.H., Poon V.K.M., Chan W.M., Ip J.D., Cai J.P., Cheng V.C.C., Chen H., Hui C.K.M., Yuen K.Y. A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: a study of a family cluster. Lancet. 2020;395:514–523. doi: 10.1016/S0140-6736(20)30154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amin S.A., Jha T. Fight against novel coronavirus: a perspective of medicinal chemists. Eur. J. Med. Chem. 2020;201 doi: 10.1016/j.ejmech.2020.112559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pillaiyar T., Meenakshisundaram S., Manickam M. Recent discovery and development of inhibitors targeting coronaviruses. Drug Discov. Today. 2020;25:668–688. doi: 10.1016/j.drudis.2020.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nakagawa K., Lokugamage K.G., Makino S. Viral and cellular mRNA translation in coronavirus-infected cells. Adv. Virus Res. 2016;96:165–192. doi: 10.1016/bs.aivir.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Groot Raoul J., Baker Susan C., Baric Ralph S., Brown Caroline S., Drosten C., Enjuanes L., Fouchier Ron A.M., Galiano M., Gorbalenya Alexander E., Memish Ziad A., Perlman S., Poon Leo L.M., Snijder Eric J., Stephens Gwen M., Woo Patrick C.Y., Zaki Ali M., Zambon M., Ziebuhr J. Commentary: Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the coronavirus study group. J. Virol. 2013;87:7790–7792. doi: 10.1128/JVI.01244-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woo Patrick C.Y., Lau Susanna K.P., Chu C.m., Chan K.h., Tsoi H.w., Huang Y., Wong Beatrice H.L., Poon Rosana W.S., Cai James J., Luk W.k., Poon Leo L.M., Wong Samson S.Y., Guan Y., Peiris J.S.M., Yuen K.y. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 2005;79:884–895. doi: 10.1128/JVI.79.2.884-895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.World Health O. World Health Organization; 2019. Clinical Management of Severe Acute Respiratory Infection when Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Infection Is Suspected: Interim Guidance. [Google Scholar]
- 11.Li H., Wei W., Xu H. Drug discovery is an eternal challenge for the biomedical sciences. Acta Materia Medica. 2022;1:1–3. [Google Scholar]
- 12.Xu X., Chen P., Wang J., Feng J., Zhou H., Li X., Zhong W., Hao P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020;63:457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ren Z., Yan L., Zhang N., Guo Y., Yang C., Lou Z., Rao Z. The newly emerged SARS-Like coronavirus HCoV-EMC also has an “Achilles' heel”: current effective inhibitor targeting a 3C-like protease. Protein & Cell. 2013;4:248–250. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramajayam R., Tan K.P., Liang P.H. Recent development of 3C and 3CL protease inhibitors for anti-coronavirus and anti-picornavirus drug discovery. Biochem. Soc. Trans. 2011;39:1371–1375. doi: 10.1042/BST0391371. [DOI] [PubMed] [Google Scholar]
- 15.Dong N., Yang X., Ye L., Chen K., Chan E.W.C., Yang M., Chen S. bioRxiv; China: 2020. Genomic and Protein Structure Modelling Analysis Depicts the Origin and Infectivity of 2019-nCoV, a New Coronavirus Which Caused a Pneumonia Outbreak in Wuhan. [Google Scholar]
- 16.Forni D., Cagliani R., Clerici M., Sironi M. Molecular evolution of human coronavirus genomes. Trends Microbiol. 2017;25:35–48. doi: 10.1016/j.tim.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marra M.A., Jones S.J.M., Astell C.R., Holt R.A., Brooks-Wilson A., Butterfield Y.S.N., Khattra J., Asano J.K., Barber S.A., Chan S.Y., Cloutier A., Coughlin S.M., Freeman D., Girn N., Griffith O.L., Leach S.R., Mayo M., McDonald H., Montgomery S.B., Pandoh P.K., Petrescu A.S., Robertson A.G., Schein J.E., Siddiqui A., Smailus D.E., Stott J.M., Yang G.S., Plummer F., Andonov A., Artsob H., Bastien N., Bernard K., Booth T.F., Bowness D., Czub M., Drebot M., Fernando L., Flick R., Garbutt M., Gray M., Grolla A., Jones S., Feldmann H., Meyers A., Kabani A., Li Y., Normand S., Stroher U., Tipples G.A., Tyler S., Vogrig R., Ward D., Watson B., Brunham R.C., Krajden M., Petric M., Skowronski D.M., Upton C., Roper R.L. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y., Grunewald M., Perlman S. Coronaviruses: an updated overview of their replication and pathogenesis. Methods Mol. Biol. 2020;2203:1–29. doi: 10.1007/978-1-0716-0900-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 20.Yang H., Bartlam M., Rao Z. Drug design targeting the main protease, the achilles heel of coronaviruses. Curr. Pharmaceut. Des. 2006;12:4573–4590. doi: 10.2174/138161206779010369. [DOI] [PubMed] [Google Scholar]
- 21.Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S.A., Freire E. Identification of novel inhibitors of the SARS coronavirus main protease 3CLpro. Biochemistry. 2004;43:4906–4912. doi: 10.1021/bi0361766. [DOI] [PubMed] [Google Scholar]
- 22.Fan K., Ma L., Han X., Liang H., Wei P., Liu Y., Lai L. The substrate specificity of SARS coronavirus 3C-like proteinase. Biochem. Biophys. Res. Commun. 2005;329:934–940. doi: 10.1016/j.bbrc.2005.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 24.Chen S., Jonas F., Shen C., Higenfeld R. Liberation of SARS-CoV main protease from the viral polyprotein: N-terminal autocleavage does not depend on the mature dimerization mode. Protein & Cell. 2010;1:59–74. doi: 10.1007/s13238-010-0011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomar S., Johnston M.L., John S.E. St, Osswald H.L., Nyalapatla P.R., Paul L.N., Ghosh A.K., Denison M.R., Mesecar A.D. Ligand-induced dimerization of Middle East respiratory syndrome (MERS) coronavirus nsp5 protease (3CLpro): IMPLICATIONS for nsp5 regulation and the development of antivirals. J. Biol. Chem. 2015;290:19403–19422. doi: 10.1074/jbc.M115.651463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu M.F., Kuo C.J., Chang K.T., Chang H.C., Chou C.C., Ko T.P., Shr H.L., Chang G.G., Wang A.H.J., Liang P.H. Mechanism of the maturation process of SARS-CoV 3CL protease. J. Biol. Chem. 2005;280:31257–31266. doi: 10.1074/jbc.M502577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li C., Qi Y., Teng X., Yang Z., Wei P., Zhang C., Tan L., Zhou L., Liu Y., Lai L. Maturation mechanism of severe acute respiratory syndrome (SARS) coronavirus 3C-like proteinase. J. Biol. Chem. 2010;285:28134–28140. doi: 10.1074/jbc.M109.095851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.H. An overview of severe acute respiratory syndrome–coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300:1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J., von Brunn A., Leyssen P., Lanko K., Neyts J., de Wilde A., Snijder E.J., Liu H., Hilgenfeld R. α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020;63:4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- 31.Hoffman R.L., Kania R.S., Brothers M.A., Davies J.F., Ferre R.A., Gajiwala K.S., He M., Hogan R.J., Kozminski K., Li L.Y., Lockner J.W., Lou J., Marra M.T., Mitchell L.J., Murray B.W., Nieman J.A., Noell S., Planken S.P., Rowe T., Ryan K., Smith G.J., Solowiej J.E., Steppan C.M., Taggart B. Discovery of ketone-based covalent inhibitors of coronavirus 3CL proteases for the potential therapeutic treatment of COVID-19. J. Med. Chem. 2020;63:12725–12747. doi: 10.1021/acs.jmedchem.0c01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veber D.F., Johnson S.R., Cheng H.Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 33.Owen D.R., Allerton C.M.N., Anderson A.S., Aschenbrenner L., Avery M., Berritt S., Boras B., Cardin R.D., Carlo A., Coffman K.J., Dantonio A., Di L., Eng H., Ferre R., Gajiwala K.S., Gibson S.A., Greasley S.E., Hurst B.L., Kadar E.P., Kalgutkar A.S., Lee J.C., Lee J., Liu W., Mason S.W., Noell S., Novak J.J., Obach R.S., Ogilvie K., Patel N.C., Pettersson M., Rai D.K., Reese M.R., Sammons M.F., Sathish J.G., Singh R.S.P., Steppan C.M., Stewart A.E., Tuttle J.B., Updyke L., Verhoest P.R., Wei L., Yang Q., Zhu Y. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 34.Unoh Y., Uehara S., Nakahara K., Nobori H., Yamatsu Y., Yamamoto S., Maruyama Y., Taoda Y., Kasamatsu K., Suto T., Kouki K., Nakahashi A., Kawashima S., Sanaki T., Toba S., Uemura K., Mizutare T., Ando S., Sasaki M., Orba Y., Sawa H., Sato A., Sato T., Kato T., Tachibana Y. Discovery of S-217622, a noncovalent oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 2022;65:6499–6512. doi: 10.1021/acs.jmedchem.2c00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tyndall J.D.A. S-217622, a 3CL protease inhibitor and clinical candidate for SARS-CoV-2. J. Med. Chem. 2022;65:6496–6498. doi: 10.1021/acs.jmedchem.2c00624. [DOI] [PubMed] [Google Scholar]
- 36.Boras B., Jones R.M., Anson B.J., Arenson D., Aschenbrenner L., Bakowski M.A., Beutler N., Binder J., Chen E., Eng H., Hammond H., Hammond J., Haupt R.E., Hoffman R., Kadar E.P., Kania R., Kimoto E., Kirkpatrick M.G., Lanyon L., Lendy E.K., Lillis J.R., Logue J., Luthra S.A., Ma C., Mason S.W., McGrath M.E., Noell S., Obach R.S., O'Brien M.N., O'Connor R., Ogilvie K., Owen D., Pettersson M., Reese M.R., Rogers T.F., Rossulek M.I., Sathish J.G., Shirai N., Steppan C., Ticehurst M., Updyke L.W., Weston S., Zhu Y., Wang J., Chatterjee A.K., Mesecar A.D., Frieman M.B., Anderson A.S., Allerton C. bioRxiv; 2021. Discovery of a Novel Inhibitor of Coronavirus 3CL Protease for the Potential Treatment of COVID-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 38.Lynch E.D., Kil J. Development of ebselen, a glutathione peroxidase mimic, for the prevention and treatment of noise-induced hearing loss. Semin. Hear. 2009;30:47–55. [Google Scholar]
- 39.Kil J., Lobarinas E., Spankovich C., Griffiths S.K., Antonelli P.J., Lynch E.D., Le Prell C.G. Safety and efficacy of ebselen for the prevention of noise-induced hearing loss: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2017;390:969–979. doi: 10.1016/S0140-6736(17)31791-9. [DOI] [PubMed] [Google Scholar]
- 40.Masaki C., Sharpley A.L., Cooper C.M., Godlewska B.R., Singh N., Vasudevan S.R., Harmer C.J., Churchill G.C., Sharp T., Rogers R.D., Cowen P.J. Effects of the potential lithium-mimetic, ebselen, on impulsivity and emotional processing. Psychopharmacology. 2016;233:2655–2661. doi: 10.1007/s00213-016-4319-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haritha C.V., Sharun K., Jose B. Ebselen, a new candidate therapeutic against SARS-CoV-2. Int. J. Surg. 2020;84:53–56. doi: 10.1016/j.ijsu.2020.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drayman N., DeMarco J.K., Jones K.A., Azizi S.A., Froggatt H.M., Tan K., Maltseva N.I., Chen S., Nicolaescu V., Dvorkin S., Furlong K., Kathayat R.S., Firpo M.R., Mastrodomenico V., Bruce E.A., Schmidt M.M., Jedrzejczak R., Muñoz-Alía M.Á., Schuster B., Nair V., Han K.y., O'Brien A., Tomatsidou A., Meyer B., Vignuzzi M., Missiakas D., Botten J.W., Brooke C.B., Lee H., Baker S.C., Mounce B.C., Heaton N.S., Severson W.E., Palmer K.E., Dickinson B.C., Joachimiak A., Randall G., Tay S. Masitinib is a broad coronavirus 3CL inhibitor that blocks replication of SARS-CoV-2. Science. 2021;373:931–936. doi: 10.1126/science.abg5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Menéndez C.A., Byléhn F., Perez-Lemus G.R., Alvarado W., de Pablo J.J. Molecular characterization of ebselen binding activity to SARS-CoV-2 main protease. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abd0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma C., Sacco M.D., Hurst B., Townsend J.A., Hu Y., Szeto T., Zhang X., Tarbet B., Marty M.T., Chen Y., Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020;30:678–692. doi: 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu L., Ye F., Feng Y., Yu F., Wang Q., Wu Y., Zhao C., Sun H., Huang B., Niu P., Song H., Shi Y., Li X., Tan W., Qi J., Gao G.F. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020;11:4417. doi: 10.1038/s41467-020-18233-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hung H.C., Ke Y.Y., Huang S.Y., Huang P.N., Kung Y.A., Chang T.Y., Yen K.J., Peng T.T., Chang S.E., Huang C.T., Tsai Y.R., Wu S.H., Lee S.J., Lin J.H., Liu B.S., Sung W.C., Shih S.R., Chen C.T., Hsu J.T. Discovery of M Protease inhibitors encoded by SARS-CoV-2. Antimicrob. Agents Chemother. 2020;64 doi: 10.1128/AAC.00872-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iketani S., Forouhar F., Liu H., Hong S.J., Lin F.Y., Nair M.S., Zask A., Huang Y., Xing L., Stockwell B.R., Chavez A., Ho D.D. Lead compounds for the development of SARS-CoV-2 3CL protease inhibitors. Nat. Commun. 2021;12 doi: 10.1038/s41467-021-22362-2. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Konno S., Kobayashi K., Senda M., Funai Y., Seki Y., Tamai I., Schakel L., Sakata K., Pillaiyar T., Taguchi A., Taniguchi A., Gutschow M., Muller C.E., Takeuchi K., Hirohama M., Kawaguchi A., Kojima M., Senda T., Shirasaka Y., Kamitani W., Hayashi Y. 3CL protease inhibitors with an electrophilic arylketone moiety as anti-SARS-CoV-2 agents. J. Med. Chem. 2022;65:2926–2939. doi: 10.1021/acs.jmedchem.1c00665. [DOI] [PubMed] [Google Scholar]
- 49.Tripathi P.K., Upadhyay S., Singh M., Raghavendhar S., Bhardwaj M., Sharma P., Patel A.K. Screening and evaluation of approved drugs as inhibitors of main protease of SARS-CoV-2. Int. J. Biol. Macromol. 2020;164:2622–2631. doi: 10.1016/j.ijbiomac.2020.08.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramos-Martín V., Johnson A., McEntee L., Farrington N., Padmore K., Cojutti P., Pea F., Neely M.N., Hope W.W. Pharmacodynamics of teicoplanin against MRSA. J. Antimicrob. Chemother. 2017;72:3382–3389. doi: 10.1093/jac/dkx289. [DOI] [PubMed] [Google Scholar]
- 51.Baron S.A., Devaux C., Colson P., Raoult D., Rolain J.-M. Teicoplanin: an alternative drug for the treatment of COVID-19? Int. J. Antimicrob. Agents. 2020;55 doi: 10.1016/j.ijantimicag.2020.105944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou N., Pan T., Zhang J., Li Q., Zhang X., Bai C., Huang F., Peng T., Zhang J., Liu C., Tao L., Zhang H. Glycopeptide antibiotics potently inhibit cathepsin L in the late endosome/lysosome and block the entry of ebola virus, Middle East respiratory syndrome coronavirus (MERS-CoV), and severe acute respiratory syndrome coronavirus (SARS-CoV) J. Biol. Chem. 2016;291:9218–9232. doi: 10.1074/jbc.M116.716100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu F., Pan T., Huang F., Ying R., Liu J., Fan H., Zhang J., Liu W., Lin Y., Yuan Y., Yang T., Li R., Zhang X., Lv X., Chen Q., Liang A., Zou F., Liu B., Hu F., Tang X., Li L., Deng K., He X., Zhang H., Zhang Y., Ma X. 2021. Glycopeptide Antibiotic Teicoplanin Inhibits Cell Entry of SARS-CoV-2 by Suppressing the Proteolytic Activity of Cathepsin L, bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu W., Xu M., Chen C.Z., Guo H., Shen M., Hu X., Shinn P., Klumpp-Thomas C., Michael S.G., Zheng W. Identification of SARS-CoV-2 3CL protease inhibitors by a quantitative high-throughput screening. ACS Pharmacol Transl Sci. 2020;3:1008–1016. doi: 10.1021/acsptsci.0c00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang P., Liu Y., Zhang G., Wang S., Guo J., Cao J., Jia X., Zhang L., Xiao G., Wang W. Screening and identification of lassa virus entry inhibitors from an FDA-approved drug library. J. Virol. 2018;92 doi: 10.1128/JVI.00954-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 57.Afshar S., Bahmani A., Saidijam M. Molecular docking and fragment-based QSAR modeling for in silico screening of approved drugs and candidate compounds against COVID-19. Avicenna Journal of Medical Biochemistry. 2020;8:83–88. [Google Scholar]
- 58.Chiou W.C., Hsu M.S., Chen Y.T., Yang J.M., Tsay Y.G., Huang H.C., Huang C. Repurposing existing drugs: identification of SARS-CoV-2 3C-like protease inhibitors. J. Enzym. Inhib. Med. Chem. 2021;36:147–153. doi: 10.1080/14756366.2020.1850710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Steuten K., Kim H., Widen J.C., Babin B.M., Onguka O., Lovell S., Bolgi O., Cerikan B., Neufeldt C.J., Cortese M., Muir R.K., Bennett J.M., Geiss-Friedlander R., Peters C., Bartenschlager R., Bogyo M. Challenges for targeting SARS-CoV-2 proteases as a therapeutic strategy for COVID-19. ACS Infect. Dis. 2021;7:1457–1468. doi: 10.1021/acsinfecdis.0c00815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coelho C., Gallo G., Campos C.B., Hardy L., Wurtele M. Biochemical screening for SARS-CoV-2 main protease inhibitors. PLoS One. 2020;15 doi: 10.1371/journal.pone.0240079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Su H.X., Yao S., Zhao W.F., Li M.J., Liu J., Shang W.J., Xie H., Ke C.Q., Hu H.C., Gao M.N., Yu K.Q., Liu H., Shen J.S., Tang W., Zhang L.K., Xiao G.F., Ni L., Wang D.W., Zuo J.P., Jiang H.L., Bai F., Wu Y., Ye Y., Xu Y.C. Anti-SARS-CoV-2 activities in vitro of Shuanghuanglian preparations and bioactive ingredients. Acta Pharmacol. Sin. 2020;41:1167–1177. doi: 10.1038/s41401-020-0483-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu H., Ye F., Sun Q., Liang H., Li C., Li S., Lu R., Huang B., Tan W., Lai L. Scutellaria baicalensis extract and baicalein inhibit replication of SARS-CoV-2 and its 3C-like protease in vitro. J. Enzym. Inhib. Med. Chem. 2021;36:497–503. doi: 10.1080/14756366.2021.1873977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liang J., Zheng M., Xu W., Chen Y., Tang P., Wu G., Zou P., Li H., Chen L. Acriflavine and proflavine hemisulfate as potential antivirals by targeting Mpro. Bioorg. Chem. 2022;129 doi: 10.1016/j.bioorg.2022.106185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Napolitano V., Dabrowska A., Schorpp K., Mourao A., Barreto-Duran E., Benedyk M., Botwina P., Brandner S., Bostock M., Chykunova Y., Czarna A., Dubin G., Frohlich T., Holscher M., Jedrysik M., Matsuda A., Owczarek K., Pachota M., Plettenburg O., Potempa J., Rothenaigner I., Schlauderer F., Slysz K., Szczepanski A., Greve-Isdahl Mohn K., Blomberg B., Sattler M., Hadian K., Popowicz G.M., Pyrc K. Acriflavine, a clinically approved drug, inhibits SARS-CoV-2 and other betacoronaviruses. Cell Chem Biol. 2022;29:774–784 e778. doi: 10.1016/j.chembiol.2021.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun L.Y., Chen C., Su J., Li J.Q., Jiang Z., Gao H., Chigan J.Z., Ding H.H., Zhai L., Yang K.W. Ebsulfur and Ebselen as highly potent scaffolds for the development of potential SARS-CoV-2 antivirals. Bioorg. Chem. 2021;112 doi: 10.1016/j.bioorg.2021.104889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin H., Zhou J., Lin K., Wang H., Liang Z., Ren X., Huang L., Xia C. Efficacy of scutellaria baicalensis for the treatment of hand, foot, and mouth disease associated with encephalitis in patients infected with EV71: a multicenter, retrospective analysis. BioMed Res. Int. 2016 doi: 10.1155/2016/5697571. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rathnayake A.D., Zheng J., Kim Y., Perera K.D., Mackin S., Meyerholz D.K., Kashipathy M.M., Battaile K.P., Lovell S., Perlman S., Groutas W.C., Chang K.O. 3C-like protease inhibitors block coronavirus replication in vitro and improve survival in MERS-CoV-infected mice. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.abc5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perera K.D., Galasiti Kankanamalage A.C., Rathnayake A.D., Honeyfield A., Groutas W., Chang K.O., Kim Y. Protease inhibitors broadly effective against feline, ferret and mink coronaviruses. Antivir. Res. 2018;160:79–86. doi: 10.1016/j.antiviral.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xiong Y., Zhu G.H., Zhang Y.N., Hu Q., Wang H.N., Yu H.N., Qin X.Y., Guan X.Q., Xiang Y.W., Tang H., Ge G.B. Flavonoids in Ampelopsis grossedentata as covalent inhibitors of SARS-CoV-2 3CLpro: inhibition potentials, covalent binding sites and inhibitory mechanisms. Int. J. Biol. Macromol. 2021;187:976–987. doi: 10.1016/j.ijbiomac.2021.07.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Verma N., Henderson J.A., Shen J. Proton-coupled conformational activation of SARS coronavirus main proteases and opportunity for designing small-molecule broad-spectrum targeted covalent inhibitors. J. Am. Chem. Soc. 2020;142:21883–21890. doi: 10.1021/jacs.0c10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vankadara S., Dawson M.D., Fong J.Y., Oh Q.Y., Ang Q.A., Liu B., Chang H.Y., Koh J., Koh X., Tan Q.W., Joy J., Chia C.S.B. A warhead substitution study on the coronavirus main protease inhibitor Nirmatrelvir. ACS Med. Chem. Lett. 2022;13:1345–1350. doi: 10.1021/acsmedchemlett.2c00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huff S., Kummetha I.R., Tiwari S.K., Huante M.B., Clark A.E., Wang S., Bray W., Smith D., Carlin A.F., Endsley M., Rana T.M. Discovery and mechanism of SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2022;65:2866–2879. doi: 10.1021/acs.jmedchem.1c00566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qiao Z., Wei N., Jin L., Zhang H., Luo J., Zhang Y., Wang K. The Mpro structure-based modifications of ebselen derivatives for improved antiviral activity against SARS-CoV-2 virus. Bioorg. Chem. 2021;117 doi: 10.1016/j.bioorg.2021.105455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dampalla C.S., Zheng J., Perera K.D., Wong L.R., Meyerholz D.K., Nguyen H.N., Kashipathy M.M., Battaile K.P., Lovell S., Kim Y., Perlman S., Groutas W.C., Chang K.O. Postinfection treatment with a protease inhibitor increases survival of mice with a fatal SARS-CoV-2 infection. Proc. Natl. Acad. Sci. U.S.A. 2021;118 doi: 10.1073/pnas.2101555118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vuong W., Khan M.B., Fischer C., Arutyunova E., Lamer T., Shields J., Saffran H.A., McKay R.T., van Belkum M.J., Joyce M.A., Young H.S., Tyrrell D.L., Vederas J.C., Lemieux M.J. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020;11:4282. doi: 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sacco M.D., Ma C., Lagarias P., Gao A., Townsend J.A., Meng X., Dube P., Zhang X., Hu Y., Kitamura N., Hurst B., Tarbet B., Marty M.T., Kolocouris A., Xiang Y., Chen Y., Wang J. Structure and inhibition of the SARS-CoV-2 main protease reveal strategy for developing dual inhibitors against Mpro and cathepsin L. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vuong W., Fischer C., Khan M.B., van Belkum M.J., Lamer T., Willoughby K.D., Lu J., Arutyunova E., Joyce M.A., Saffran H.A., Shields J.A., Young H.S., Nieman J.A., Tyrrell D.L., Lemieux M.J., Vederas J.C. Improved SARS-CoV-2 M(pro) inhibitors based on feline antiviral drug GC376: structural enhancements, increased solubility, and micellar studies. Eur. J. Med. Chem. 2021;222 doi: 10.1016/j.ejmech.2021.113584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hirose Y., Shindo N., Mori M., Onitsuka S., Isogai H., Hamada R., Hiramoto T., Ochi J., Takahashi D., Ueda T., Caaveiro J.M.M., Yoshida Y., Ohdo S., Matsunaga N., Toba S., Sasaki M., Orba Y., Sawa H., Sato A., Kawanishi E., Ojida A. Discovery of chlorofluoroacetamide-based covalent inhibitors for severe acute respiratory syndrome coronavirus 2 3CL protease. J. Med. Chem. 2022;65:13852–13865. doi: 10.1021/acs.jmedchem.2c01081. [DOI] [PubMed] [Google Scholar]
- 80.Dai W., Zhang B., Jiang X.M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F., Li C., Li Y., Bai F., Wang H., Cheng X., Cen X., Hu S., Yang X., Wang J., Liu X., Xiao G., Jiang H., Rao Z., Zhang L.K., Xu Y., Yang H., Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dai W., Jochmans D., Xie H., Yang H., Li J., Su H., Chang D., Wang J., Peng J., Zhu L., Nian Y., Hilgenfeld R., Jiang H., Chen K., Zhang L., Xu Y., Neyts J., Liu H. Design, synthesis, and biological evaluation of peptidomimetic aldehydes as broad-spectrum inhibitors against enterovirus and SARS-CoV-2. J. Med. Chem. 2022;65:2794–2808. doi: 10.1021/acs.jmedchem.0c02258. [DOI] [PubMed] [Google Scholar]
- 82.Ma Y., Shang C., Yang P., Li L., Zhai Y., Yin Z., Wang B., Shang L. 4-Iminooxazolidin-2-one as a bioisostere of the cyanohydrin moiety: inhibitors of enterovirus 71 3C protease. J. Med. Chem. 2018;61:10333–10339. doi: 10.1021/acs.jmedchem.8b01335. [DOI] [PubMed] [Google Scholar]
- 83.Zhai Y., Zhao X., Cui Z., Wang M., Wang Y., Li L., Sun Q., Yang X., Zeng D., Liu Y., Sun Y., Lou Z., Shang L., Yin Z. Cyanohydrin as an anchoring group for potent and selective inhibitors of enterovirus 71 3C protease. J. Med. Chem. 2015;58:9414–9420. doi: 10.1021/acs.jmedchem.5b01013. [DOI] [PubMed] [Google Scholar]
- 84.Ma Y., Li L., He S., Shang C., Sun Y., Liu N., Meek T.D., Wang Y., Shang L. Application of dually activated Michael acceptor to the rational design of reversible covalent inhibitor for enterovirus 71 3C protease. J. Med. Chem. 2019;62:6146–6162. doi: 10.1021/acs.jmedchem.9b00387. [DOI] [PubMed] [Google Scholar]
- 85.Zhai Y., Ma Y., Ma F., Nie Q., Ren X., Wang Y., Shang L., Yin Z. Structure–activity relationship study of peptidomimetic aldehydes as enterovirus 71 3C protease inhibitors. Eur. J. Med. Chem. 2016;124:559–573. doi: 10.1016/j.ejmech.2016.08.064. [DOI] [PubMed] [Google Scholar]
- 86.Wang H., Pei R., Li X., Deng W., Xing S., Zhang Y., Zhang C., He S., Sun H., Xiao S., Xiong J., Zhang Y., Chen X., Wang Y., Guo Y., Zhang B., Shang L. The structure-based design of peptidomimetic inhibitors against SARS-CoV-2 3C like protease as Potent anti-viral drug candidate. Eur. J. Med. Chem. 2022;238 doi: 10.1016/j.ejmech.2022.114458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pillaiyar T., Flury P., Kruger N., Su H., Schakel L., Barbosa Da Silva E., Eppler O., Kronenberger T., Nie T., Luedtke S., Rocha C., Sylvester K., Petry M.R.I., McKerrow J.H., Poso A., Pohlmann S., Gutschow M., O'Donoghue A.J., Xu Y., Muller C.E., Laufer S.A. Small-molecule thioesters as SARS-CoV-2 main protease inhibitors: enzyme inhibition, structure-activity relationships, antiviral activity, and X-ray structure determination. J. Med. Chem. 2022;65:9376–9395. doi: 10.1021/acs.jmedchem.2c00636. [DOI] [PubMed] [Google Scholar]
- 88.Gao S., Song L., Claff T., Woodson M., Sylvester K., Jing L., Weisse R.H., Cheng Y., Strater N., Schakel L., Gutschow M., Ye B., Yang M., Zhang T., Kang D., Toth K., Tavis J., Tollefson A.E., Muller C.E., Zhan P., Liu X. Discovery and crystallographic studies of nonpeptidic piperazine derivatives as covalent SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2022;65:16902–16917. doi: 10.1021/acs.jmedchem.2c01716. [DOI] [PubMed] [Google Scholar]
- 89.Qiao J., Li Y.S., Zeng R., Liu F.L., Luo R.H., Huang C., Wang Y.F., Zhang J., Quan B., Shen C., Mao X., Liu X., Sun W., Yang W., Ni X., Wang K., Xu L., Duan Z.L., Zou Q.C., Zhang H.L., Qu W., Long Y.H.P., Li M.H., Yang R.C., Liu X., You J., Zhou Y., Yao R., Li W.P., Liu J.M., Chen P., Liu Y., Lin G.F., Yang X., Zou J., Li L., Hu Y., Lu G.W., Li W.M., Wei Y.Q., Zheng Y.T., Lei J., Yang S. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371:1374–1378. doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dou X., Sun Q., Xu G., Liu Y., Zhang C., Wang B., Lu Y., Guo Z., Su L., Huo T., Zhao X., Wang C., Yu Z., Song S., Zhang L., Liu Z., Lai L., Jiao N. Discovery of 2-(furan-2-ylmethylene)hydrazine-1-carbothioamide derivatives as novel inhibitors of SARS-CoV-2 main protease. Eur. J. Med. Chem. 2022;238 doi: 10.1016/j.ejmech.2022.114508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ghosh A.K., Raghavaiah J., Shahabi D., Yadav M., Anson B.J., Lendy E.K., Hattori S.I., Higashi-Kuwata N., Mitsuya H., Mesecar A.D. Indole chloropyridinyl ester-derived SARS-CoV-2 3CLpro inhibitors: enzyme inhibition, antiviral efficacy, structure-activity relationship, and X-ray structural studies. J. Med. Chem. 2021;64:14702–14714. doi: 10.1021/acs.jmedchem.1c01214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shcherbakov D., Baev D., Kalinin M., Dalinger A., Chirkova V., Belenkaya S., Khvostov A., Krut'ko D., Medved'ko A., Volosnikova E., Sharlaeva E., Shanshin D., Tolstikova T., Yarovaya O., Maksyutov R., Salakhutdinov N., Vatsadze S. Design and evaluation of bispidine-based SARS-CoV-2 main protease inhibitors. ACS Med. Chem. Lett. 2022;13:140–147. doi: 10.1021/acsmedchemlett.1c00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ma C., Xia Z., Sacco M.D., Hu Y., Townsend J.A., Meng X., Choza J., Tan H., Jang J., Gongora M.V., Zhang X., Zhang F., Xiang Y., Marty M.T., Chen Y., Wang J. Discovery of di- and trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. 2021;143:20697–20709. doi: 10.1021/jacs.1c08060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Malla T.R., Brewitz L., Muntean D.G., Aslam H., Owen C.D., Salah E., Tumber A., Lukacik P., Strain-Damerell C., Mikolajek H., Walsh M.A., Schofield C.J. Penicillin derivatives inhibit the SARS-CoV-2 main protease by reaction with its nucleophilic cysteine. J. Med. Chem. 2022;65:7682–7696. doi: 10.1021/acs.jmedchem.1c02214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stille J.K., Tjutrins J., Wang G., Venegas F.A., Hennecker C., Rueda A.M., Sharon I., Blaine N., Miron C.E., Pinus S., Labarre A., Plescia J., Burai Patrascu M., Zhang X., Wahba A.S., Vlaho D., Huot M.J., Schmeing T.M., Mittermaier A.K., Moitessier N. Design, synthesis and in vitro evaluation of novel SARS-CoV-2 3CL(pro) covalent inhibitors. Eur. J. Med. Chem. 2022;229 doi: 10.1016/j.ejmech.2021.114046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang J.W., Xiong Y., Wang F., Zhang F.M., Yang X., Lin G.Q., Tian P., Ge G., Gao D. Discovery of 9,10-dihydrophenanthrene derivatives as SARS-CoV-2 3CL(pro) inhibitors for treating COVID-19. Eur. J. Med. Chem. 2022;228 doi: 10.1016/j.ejmech.2021.114030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Owen D.R., Allerton C.M.N., Anderson A.S., Aschenbrenner L., Avery M., Berritt S., Boras B., Cardin R.D., Carlo A., Coffman K.J., Dantonio A., Di L., Eng H., Ferre R., Gajiwala K.S., Gibson S.A., Greasley S.E., Hurst B.L., Kadar E.P., Kalgutkar A.S., Lee J.C., Lee J., Liu W., Mason S.W., Noell S., Novak J.J., Obach R.S., Ogilvie K., Patel N.C., Pettersson M., Rai D.K., Reese M.R., Sammons M.F., Sathish J.G., Singh R.S.P., Steppan C.M., Stewart A.E., Tuttle J.B., Updyke L., Verhoest P.R., Wei L., Yang Q., Zhu Y. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 98.Erak M., Bellmann-Sickert K., Els-Heindl S., Beck-Sickinger A.G. Peptide chemistry toolbox – transforming natural peptides into peptide therapeutics. Bioorg. Med. Chem. 2018;26:2759–2765. doi: 10.1016/j.bmc.2018.01.012. [DOI] [PubMed] [Google Scholar]
- 99.Zhou L., Liu Y., Zhang W., Wei P., Huang C., Pei J., Yuan Y., Lai L. Isatin compounds as noncovalent SARS coronavirus 3C-like protease inhibitors. J. Med. Chem. 2006;49:3440–3443. doi: 10.1021/jm0602357. [DOI] [PubMed] [Google Scholar]
- 100.Liu P., Liu H., Sun Q., Liang H., Li C., Deng X., Liu Y., Lai L. Potent inhibitors of SARS-CoV-2 3C-like protease derived from N-substituted isatin compounds. Eur. J. Med. Chem. 2020;206 doi: 10.1016/j.ejmech.2020.112702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Saldanha S.A., Chase P., Eggler A., Dawson E.S., Baez-Santos Y.M., Tomar S., Mielech A.M., Baker S.C., Lindsley C.W., Hodder P., Mesecar A., Stauffer S.R. Discovery, synthesis, and structure-based optimization of a series of N-(tert-Butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamides (ML188) as potent noncovalent small molecule inhibitors of the severe acute respiratory syndrome coronavirus (SARS-CoV) 3CL protease. J. Med. Chem. 2013;56:534–546. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kitamura N., Sacco M.D., Ma C., Hu Y., Townsend J.A., Meng X., Zhang F., Zhang X., Ba M., Szeto T., Kukuljac A., Marty M.T., Schultz D., Cherry S., Xiang Y., Chen Y., Wang J. Expedited approach toward the rational design of noncovalent SARS-CoV-2 main protease inhibitors. J. Med. Chem. 2022;65:2848–2865. doi: 10.1021/acs.jmedchem.1c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bhatt S., Gething P.W., Brady O.J., Messina J.P., Farlow A.W., Moyes C.L., Drake J.M., Brownstein J.S., Hoen A.G., Sankoh O., Myers M.F., George D.B., Jaenisch T., Wint G.R.W., Simmons C.P., Scott T.W., Farrar J.J., Hay S.I. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nitsche C., Holloway S., Schirmeister T., Klein C.D. Biochemistry and medicinal chemistry of the dengue virus protease. Chem. Rev. 2014;114:11348–11381. doi: 10.1021/cr500233q. [DOI] [PubMed] [Google Scholar]
- 105.Kuhl N., Lang J., Leuthold M.M., Klein C.D. Discovery of potent benzoxaborole inhibitors against SARS-CoV-2 main and dengue virus proteases. Eur. J. Med. Chem. 2022;240 doi: 10.1016/j.ejmech.2022.114585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kneller D.W., Li H., Galanie S., Phillips G., Labbe A., Weiss K.L., Zhang Q., Arnould M.A., Clyde A., Ma H., Ramanathan A., Jonsson C.B., Head M.S., Coates L., Louis J.M., Bonnesen P.V., Kovalevsky A. Structural, electronic, and electrostatic determinants for inhibitor binding to subsites S1 and S2 in SARS-CoV-2 main protease. J. Med. Chem. 2021;64:17366–17383. doi: 10.1021/acs.jmedchem.1c01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Gao S., Sylvester K., Song L., Claff T., Jing L., Woodson M., Weisse R.H., Cheng Y., Schakel L., Petry M., Gutschow M., Schiedel A.C., Strater N., Kang D., Xu S., Toth K., Tavis J., Tollefson A.E., Muller C.E., Liu X., Zhan P. Discovery and crystallographic studies of trisubstituted piperazine derivatives as non-covalent SARS-CoV-2 main protease inhibitors with high target specificity and low toxicity. J. Med. Chem. 2022;65:13343–13364. doi: 10.1021/acs.jmedchem.2c01146. [DOI] [PubMed] [Google Scholar]
- 108.Keehner J., Horton L.E., Binkin N.J., Laurent L.C., Pride D., Longhurst C.A., Abeles S.R., Torriani F.J. Resurgence of SARS-CoV-2 infection in a highly vaccinated health system workforce. N. Engl. J. Med. 2021;385:1330–1332. doi: 10.1056/NEJMc2112981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Churcher I. Protac-induced protein degradation in drug discovery: breaking the rules or just making new ones? J. Med. Chem. 2018;61:444–452. doi: 10.1021/acs.jmedchem.7b01272. [DOI] [PubMed] [Google Scholar]
- 110.Bondeson D.P., Mares A., Smith I.E.D., Ko E., Campos S., Miah A.H., Mulholland K.E., Routly N., Buckley D.L., Gustafson J.L., Zinn N., Grandi P., Shimamura S., Bergamini G., Faelth-Savitski M., Bantscheff M., Cox C., Gordon D.A., Willard R.R., Flanagan J.J., Casillas L.N., Votta B.J., den Besten W., Famm K., Kruidenier L., Carter P.S., Harling J.D., Churcher I., Crews C.M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015;11:611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.