

Abstract
INTRODUCTION:
Deciphering the molecular and genetic changes that differentiate humans from our closest primate relatives is critical for understanding our origins. Although earlier studies have prioritized how newly gained genetic sequences or variations have contributed to evolutionary innovation, the role of sequence loss has been less appreciated. Alterations in evolutionary conserved regions that are enriched for biological function could be particularly more likely to have phenotypic effects. We thus sought to identify and characterize sequences that have been conserved across evolution, but are then surprisingly lost in all humans. These human-specific deletions in conserved regions (hCONDELs) may play an important role in uniquely human traits.
RATIONALE:
Sequencing advancements have identified millions of genetic changes between chimpanzee and human genomes; however, the functional impacts of the ~1 to 5% difference between our species is largely unknown. hCONDELs are one class of these predominantly noncoding sequence changes. Although large hCONDELs (>1 kb) have been previously identified, the vast majority of all hCONDELs (95.7%) are small (<20 base pairs) and have not yet been functionally assessed. We adapted massively parallel reporter assays (MPRAs) to characterize the effects of thousands of these small hCONDELs and uncovered hundreds with functional effects. By understanding the effects of these hCONDELs, we can gain insight into the mechanistic patterns driving evolution in the human genome.
RESULTS:
We identified 10,032 hCONDELs by examining conserved regions across diverse vertebrate genomes and overlapping with confidently annotated, human-specific fixed deletions. We found that these hCONDELs are enriched to delete conserved sequences originating from stem amniotes. Overlap with transcriptional, epigenomic, and phenotypic datasets all implicate neuronal and cognitive functional impacts. We characterized these hCONDELs using MPRA in six different human cell types, including induced pluripotent stem cell–derived neural progenitor cells. We found that 800 hCONDELs displayed species-specific regulatory effect effects. Although many hCONDELs perturb transcription factor–binding sites in active enhancers, we estimate that 30% create or improve binding sites, including activators and repressors.
Some hCONDELs exhibit molecular functions that affect core neurodevelopmental genes. One hCONDEL removes a single base in an active enhancer in the neurogenesis gene HDAC5, and another deletes six bases in an alternative promoter of PPP2CA, a gene that regulates neuronal signaling. We deeply characterized an hCONDEL in a putative regulatory element of LOXL2, a gene that controls neuronal differentiation. Using genome engineering to reintroduce the conserved chimpanzee sequence into human cells, we confirmed that the human deletion alters transcriptional output of LOXL2. Single-cell RNA sequencing of these cells uncovered a cascade of myelination and synaptic function–related transcriptional changes induced by the hCONDEL.
CONCLUSION:
Our identification of hundreds of hCONDELs with functional impacts reveals new molecular changes that may have shaped our unique biological lineage. These hCONDELs display predicted functions in a variety of biological systems but are especially enriched for function in neuronal tissue. Many hCONDELs induced gains of regulatory activity, a surprising discovery given that deletions of conserved bases are commonly thought to abrogate function. Our work provides a paradigm for the characterization of nucleotide changes shaping species-specific biology across humans or other animals.
Graphical Abstract
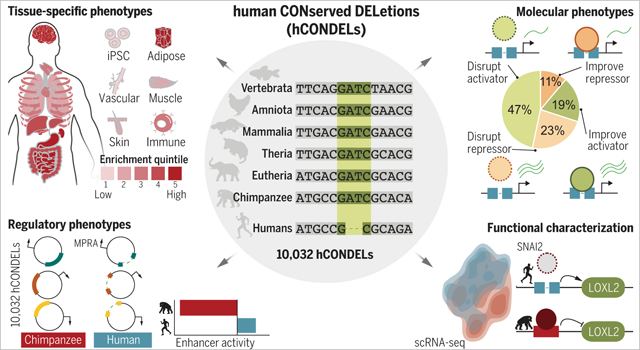
Human-specific deletions that remove nucleotides from regions highly conserved in other animals (hCONDELs). We assessed 10,032 hCONDELs across diverse, biologically relevant datasets and identified tissue-specific enrichment (top left). The regulatory impact of hCONDELs was characterized by comparing chimp and human sequences in MPRAs (bottom left). The ability of hCONDELs to either improve or perturb activating and repressing gene-regulatory elements was assessed (top right). The deleted chimpanzee sequence was reintroduced back into human cells, causing a cascade of transcriptional differences for an hCONDEL regulating LOXL2 (bottom right).
Conserved genomic sequences disrupted in humans may underlie uniquely human phenotypic traits. We identified and characterized 10,032 human-specific conserved deletions (hCONDELs). These short (average 2.56 base pairs) deletions are enriched for human brain functions across genetic, epigenomic, and transcriptomic datasets. Using massively parallel reporter assays in six cell types, we discovered 800 hCONDELs conferring significant differences in regulatory activity, half of which enhance rather than disrupt regulatory function. We highlight several hCONDELs with putative human-specific effects on brain development, including HDAC5, CPEB4, and PPP2CA. Reverting an hCONDEL to the ancestral sequence alters the expression of LOXL2 and developmental genes involved in myelination and synaptic function. Our data provide a rich resource to investigate the evolutionary mechanisms driving new traits in humans and other species.
The genetic basis of uniquely human phenotypes such as an expanded neocortex, upright morphology, and complex sociocultural abilities remains largely unknown. Characterizing these human-specific traits will improve our understanding of the evolutionary mechanisms underlying our species’ history and of the diseases associated with those traits. However, progress is hindered by difficulties in interpreting millions of sequence changes between humans and other primates in cis-regulatory elements (CREs) (1, 2).
Most evolutionary studies to date have focused on large differences between species hoping to identify substantial phenotypic impacts, potentially overlooking small changes of important effect. These previous studies include new sequences in the human genome (3), many clustered occurrences of sequence accelerations (4), or long (>1 kb) deletions in the human genome (5). However, small alterations may also be an important avenue of evolutionary change, and short deletions in conserved genomic elements are one such source. Because deep sequence conservation is an indicator of biological function (6), deletion of conserved elements in a species is surprising.
We thus set out to characterize human-specific conserved deletions (hCONDELs). We focused on identifying high-confidence small deletions, a set that comprises most hCONDELs [95.7% < 20 base pairs (bp)]. These deletions have yet to be functionally characterized in prior published studies (5, 7–9) and can be validated for complete fixation using short-read data. This approach benefits by pinpointing deletions to the precise bases that are also more experimentally tractable.
Results
Discovering hCONDELs
To discover hCONDELs at maximal resolution, we developed a rigorous computational pipeline on high-quality primate and vertebrate genomes to identify any human deletions overlapping phastCons-derived conserved elements. We first constructed a chimpanzee-anchored multiple sequence alignment across 11 vertebrate species to detect statistically significant conserved sequences (1,371,766). These elements ranged from being deeply conserved throughout vertebrates to being conserved only through primates. We then intersected our conserved elements with called deletions (2,42,706) between the human (hg38) and chimpanzee (panTro4) genomes to yield 43,588 putative hCONDELs (Fig. 1A). To ensure that putative hCONDELs were not misidentified because of polymorphisms in either species, we confirmed that conserved bases were present in several primate genomes and fully deleted in diverse human genomes (see the materials and methods).
Fig. 1. hCONDELs are dispersed in noncoding genomic regions that are enriched for developmental function.
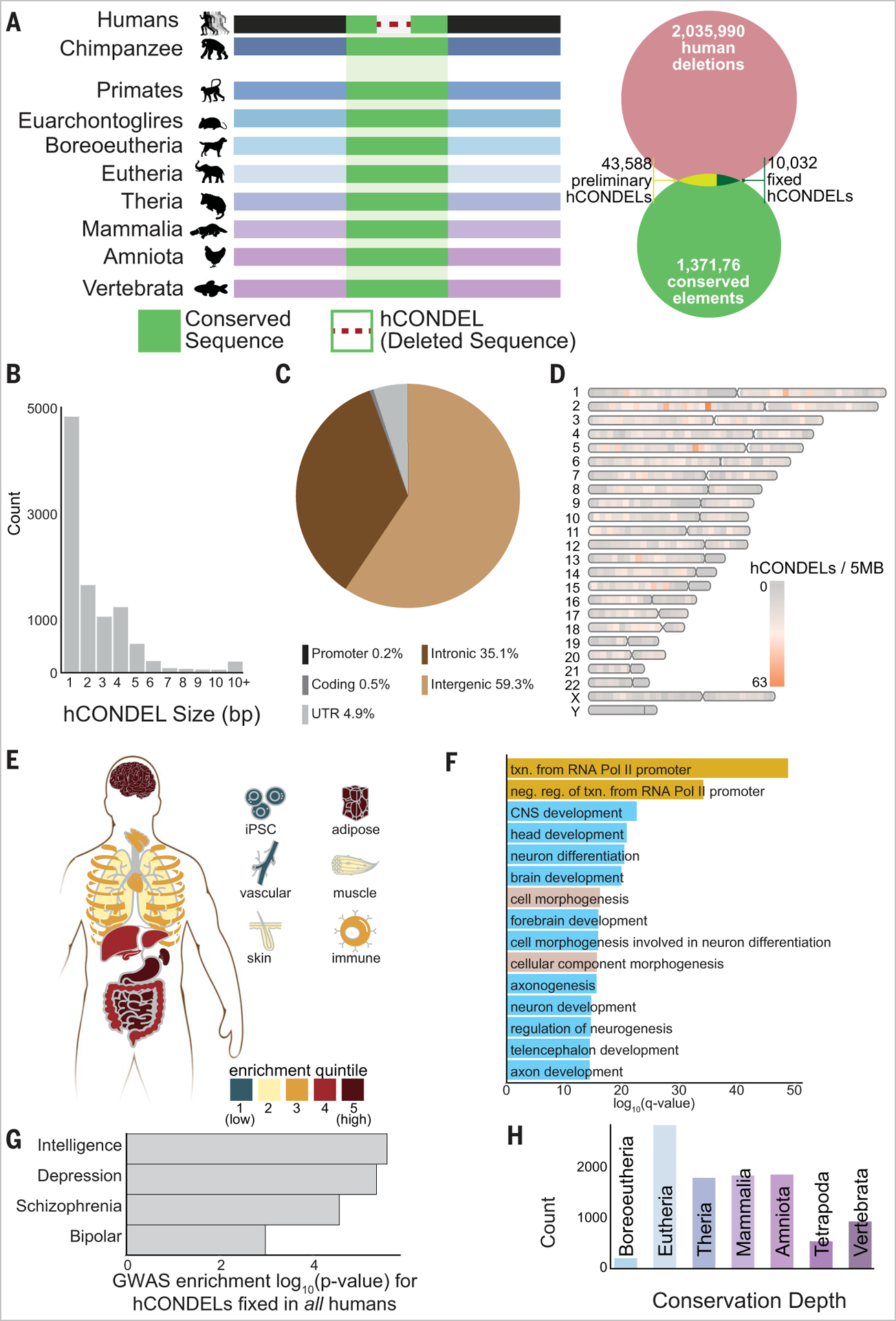
(A) hCONDEL identification strategy. (B) Distribution of hCONDEL lengths (in base pairs). (C) Overlap with genomic annotation. (D) Chromosomal distribution of hCONDELs. (E) Enrichment z score of hCONDELs in tissue-specific H3K27ac-CREs. (F) hCONDEL gene ontology enrichments include gene regulation (yellow), neurodevelopment (blue), and development (mauve). (G) Enrichment log P value of hCONDEL association with neurological GWAS (t test P < 0.01 for all bars). (H) Distribution of hCONDEL ages by most recent common ancestor.
Altogether, we identified 10,032 fixed hCONDELs (Fig. 1, A and B, materials and methods, and table S1), which are short (average 2.56 bp, range 1 to 31 bp) and mostly noncoding (intronic 35.1%, intergenic 59.3%) (Fig. 1C). Compared with permuted matched controls, hCONDELs are enriched in introns and intergenic regions (z scores = 8.32 and 2.22, respectively) (fig. S1A) and depleted from coding regions (z score = −30.5), suggesting that negative selection may deplete deletions from altering protein structures. They are also depleted from the Y chromosome (Fig. 1D). Although 11.4% of hCONDELs delete bases from repeat elements (fig. S1B), they are not enriched as a whole or in specific classes (materials and methods), suggesting that their role is distinct from repeat-based evolutionary innovations (10).
Genomic and evolutionary features of hCONDELs
We next examined the properties and potential functional impacts of coding hCONDELs. Coding hCONDELs are significantly longer compared with intergenic ones (average = 3.5 bp, two-sided t test P = 0.011;fig. S2A), a finding explained by most (42 of 47) being inframe triplet deletions. The remaining coding hCONDELs include pseudogenization of keratin (KRT87) and neuropoeitin (CTF2), whereas others create new human isoforms of PPP1CA and the neuronal plasticity gene PLPPR1. An 8-bp frame-shift hCONDEL fully abrogates human function of CTF2, which is highly expressed in mouse embryonic neuroepithelia and promotes neuronal progenitor proliferation (11).
Because most hCONDELs are noncoding, we examined their overlap with genetic and epigenetic datasets to understand the phenotypes that hCONDELs may affect. hCONDELs are strongly enriched to overlap candidate CREs (17.5%) (12) compared with genomic background (7.9%), and they show specific enrichment in multiple tissues, including multiple brain regions, as well as adipose, heart, and muscle tissues (Fig. 1E and fig. S3A). Genes near hCONDELs are enriched for neurodevelopmental, morphological, and transcriptional regulatory functions (Fig. 1F, fig. S3B, and table S2) and are uniquely differentially expressed in specific brain subregions such as the amygdala, cortex, and cerebellum [Benjamini-Hochberg (BH) adjusted P < 0.05] (fig. S3C and materials and methods). We also found that hCONDELs are enriched to overlap genes identified in cognitive genome-wide association studies (GWASs) (Fig. 1G and table S2), further suggesting their role in the brain across all humans.
We also considered hCONDEL evolutionary constraint and age. hCONDELs remove sequences that are less constrained than controls (z score =−30.7), but we found that they overlap sequences of ancient and recent phylogenetic origins (Fig. 1H). hCONDELs occur in sequences originating from stem amniotes more often than expected on the basis of matched controls (z score = 5.65) (fig. S4A), suggesting that functional elements born in this lineage are more amenable to evolutionary innovation. Most hCONDELs overlap short blocks from a single evolutionary age (fig. S4, B and C), which have been associated with more tissue-specific effects compared with multiage, complex blocks (13). hCONDEL deletion size was not correlated with age (fig. S4D), although deletion of the most ancient sequences occurred predominantly in coding regions (fig. S4E), providing evidence that the most ancient vertebrate sequences were still amenable to alteration.
Functional characterization of hCONDELs using MPRA
To functionally characterize which hCONDELs directly alter cis-regulatory potential, we deployed a massively parallel reporter assay (MPRA) across six diverse cell types: HEK293 (embryonic kidney), HepG2 (hepatocellular carcinoma), GM12878 (lymphoblastoid), K562 (leukemia), SK-N-SH (neuroblastoma), and human induced pluripotent stem cell (hiPSC)–derived neural progenitor cells (NPCs) (14). Using these cell lines, we compared the regulatory potential of human sequences bearing a deletion versus intact chimpanzee sequences (Fig. 2A). Testing human and chimpanzee regulatory sequences in the same cell lines isolates intrinsic sequence-based regulatory changes by removing trans-environment differences. The MPRA is highly reproducible (mean replicate correlation = 0.97;fig. S5A) and reflects cell type–specific regulatory states (fig. S5B). Human and chimpanzee sequences display no systematic activity differences (Wilcoxon rank-sum test P = 0.64;fig. S5C), illustrating the suitability of testing candidate CREs from the two species in our system.
Fig. 2. Identification of hCONDELs with species-specific activity perturb TF-binding motifs.
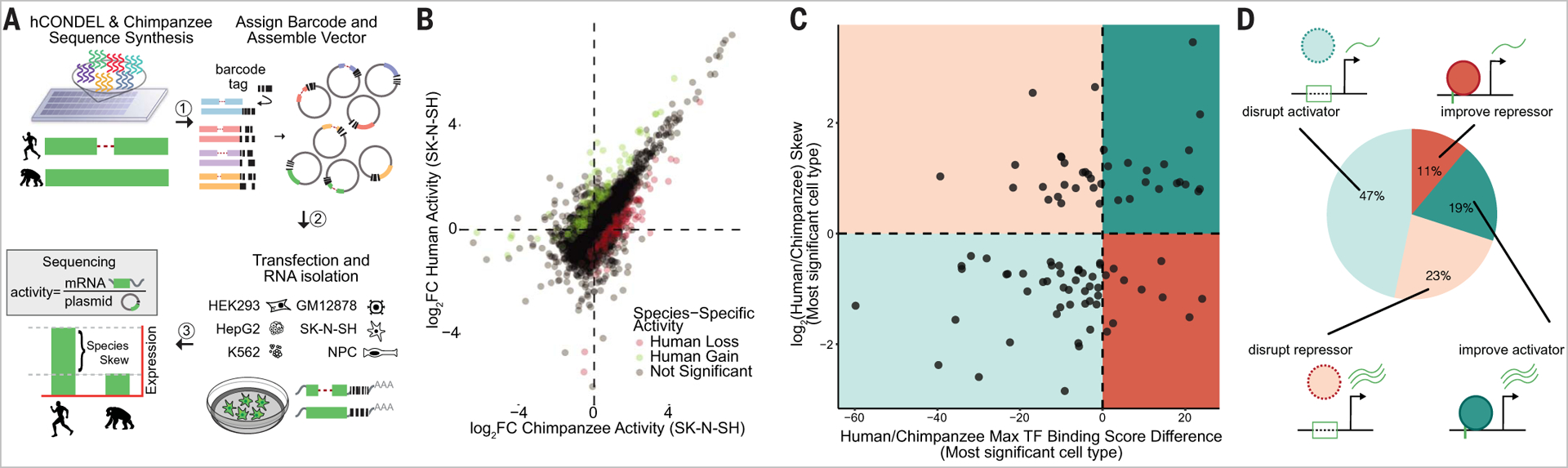
(A) MPRA characterization strategy. (B) Identification of hCONDELs with significant (BH adjusted P < 0.05) species-specific activity. Regulatory activity for chimpanzee sequence x axis versus orthologous human sequence (y axis) showing significant human loss (red) and gain (green). Illustrative SK-N-SH data are plotted. (C) Species activity correlated with predicted TF alteration score [difference in log-likelihood (base 2) in human versus chimpanzee sequence motif match]. Data from the cell type with the most significant MPRA-measured effect are shown. (D) Breakdown of regulatory activity and TF-binding differences categorized into activators (teal) and repressors (red), with either improved (solid line) or diminished (dashed line) motif prediction.
Across all tested cell types, MPRA identified 800 (7.97%) hCONDELs with significant regulatory differences between species (Fig. 2B and table S1). Of these 800, we estimate one-third to have cell type–specific effects (fig. S5, D to F, and materials and methods). As expected, hCONDELs perturbing transcription factor (TF)–binding motifs (two-sided t test P = 1.93 × 10−3) and those that had higher conservation scores over the deleted bases (two-sided t test P = 0.02) were enriched for species-specific activity (fig. S6A and materials and methods). After filtering strong repressive elements, we were able to correlate the directionality and magnitude of species-specific activity observed in the MPRA with the change in predicted TF binding between species (Pearson correlation = 0.37, P = 1.9 × 10−4) (fig. S6B). This highlights our ability to predict specific alterations to regulatory grammar that underlie species-specific activity. Subsetting TF-binding predictions on the most conserved motifs using Zoonomia 240 mammalian species phyloP scores increases concordance with species-specific activity, demonstrating the value of higher-resolution evolutionary data (fig. S6C) (15). We highlight several hCONDELs that we sequence verified in seven chimpanzee individuals; each display large regulatory changes with clearly perturbed human TF motifs (4, 5) (fig. S7, A to H).
Although deletions may be expected to abrogate function, we found that many actually increase regulatory activity, demonstrating that disruption of repressive elements or improvement of an activating site may be common (Fig. 2C). To investigate this further, for hCONDELs that altered a TF motif in a sequence background with enhancer activity, we classified the type of change by comparing the directionality of predicted TF-binding difference with the directionality of species-specific activity (see the materials and methods). Of the 42% of hCONDELs with increased human regulatory activity, 23% are predicted to disrupt TF-binding sites and 19% to improve sites (Fig. 2D). For the other 58% that decrease regulatory activity in humans, 47% and 11% disrupt or improve a TF motif, respectively. Overall, we estimate that 30% of hCONDEL TF alterations created or improved a TF-binding site. This indicates that sequence loss leading to creation or strengthening of activating motifs or disruption of repressive motifs may be a frequent event important for evolutionary change.
We clustered TF motifs by sequence similarity and identified 19 TF motifs (in 13 clusters) enriched for perturbation by hCONDELs (fig. S6D and table S2). EGR4 (z score = 3.98) and ZNF148 (z score = 5.02), two developmental neuronal TFs (16, 17), are frequently altered by hCONDELs and are the only enriched TFs in their respective clusters. FOXD3 and FOXJ3 (z scores = 3.38 and 11.7, respectively) both involved with neural differentiation (18, 19) and are both enriched TFs in the same motif cluster. These TFs may have causal motifs preferentially perturbed by our hCONDELs, and additional experimental support may refine this list (see the materials and methods).
Neurological impacts of hCONDELs
Following that hCONDELs may especially function during neuronal development, we further investigated our MPRA hits in developmentally relevant neural progenitor cells. We found 83 of the 800 hCONDELs to only have species-specific skew in NPCs, highlighting the importance of phenotype-relevant cell types. One hCONDEL overlaps a peak of H3K27ac, is predicted to regulate the neurogenesis gene HDAC5, and displays increased repression in humans (BH adjusted P = 1.6 × 10−2) (fig. S8, A and B). Another hCONDEL that deletes a single T conserved through chicken (fig. S8C) displays decreased enhancer activity in humans (BH adjusted P = 3 × 10−2) (fig. S8D) and is predicted to affect CPEB4, a gene controlling forebrain volume (20). CPEB4 is also found to be significantly down-regulated in different human neurons compared with chimpanzee [log2 fold change (log2FC) = −0.72, adjusted P = 2.72 × 10−118 in cerebellum neurons and log2FC = −0.76, adjusted P = 9.51 × 10−13 in cerebellum interneurons (21)], providing support for the hCONDEL inducing expression change. We tested the ability of two hCONDELs to drive enhancer activity in vivo. Two active hCONDELs near PPP2CA and LOXL2 both drive robust gene expression in the developing neural tube in embryonic mouse lac-Z reporter assays using site-specific insertion of transgenes at the H11 locus (22)(four of four lacZ-positive embryos for PPP2CA and nine of nine for LOXL2;fig. S9, A and B).
We further investigated one of the most conserved hCONDELs located in the promoter of an alternative isoform of PPP2CA, a crucial regulator of neuronal signaling associated with cognitive ability (Fig. 3A) (23, 24). The hCONDEL alters a motif for the TF YY2 (Fig. 3B), and the human sequence shows significantly higher activity in MPRA (species log2FC = 0.96, BH adjusted P = 9.38 × 10−5;Fig. 3C). This site also displays human-specific H3K27ac signal in the developing cortex compared with rhesus macaque (25) (P = 8.44 × 10−3;Fig.3D). Using a luciferase assay, we confirmed the hCONDEL confers human-specific increased regulatory activity to the alternative PPP2CA promoter (negative strand). These findings suggest that the hCONDEL directly increases PPP2CA transcription through an alternative promoter (Fig. 3E). We also did not observe a significant difference in regulatory activity between the human and chimpanzee testing the positive strand. Concordantly, further CRISPR-induced deletions at the human deletion caused increased expression of the alternative isoform (log2FC = 3.2, two-sided t test P = 1.9 × 10−3;Fig. 3F). Other members of this gene family also show brain functions, including PPP1CA (26), which contains an hCONDEL potentially pseudogenizing it, and PPP1R17, a gene that slows neural progenitor cell cycle progression and was found to be putatively regulated by a human accelerated region (HAR) (9).
Fig. 3. PPP2CA-associated hCONDEL induces species-specific regulatory changes.
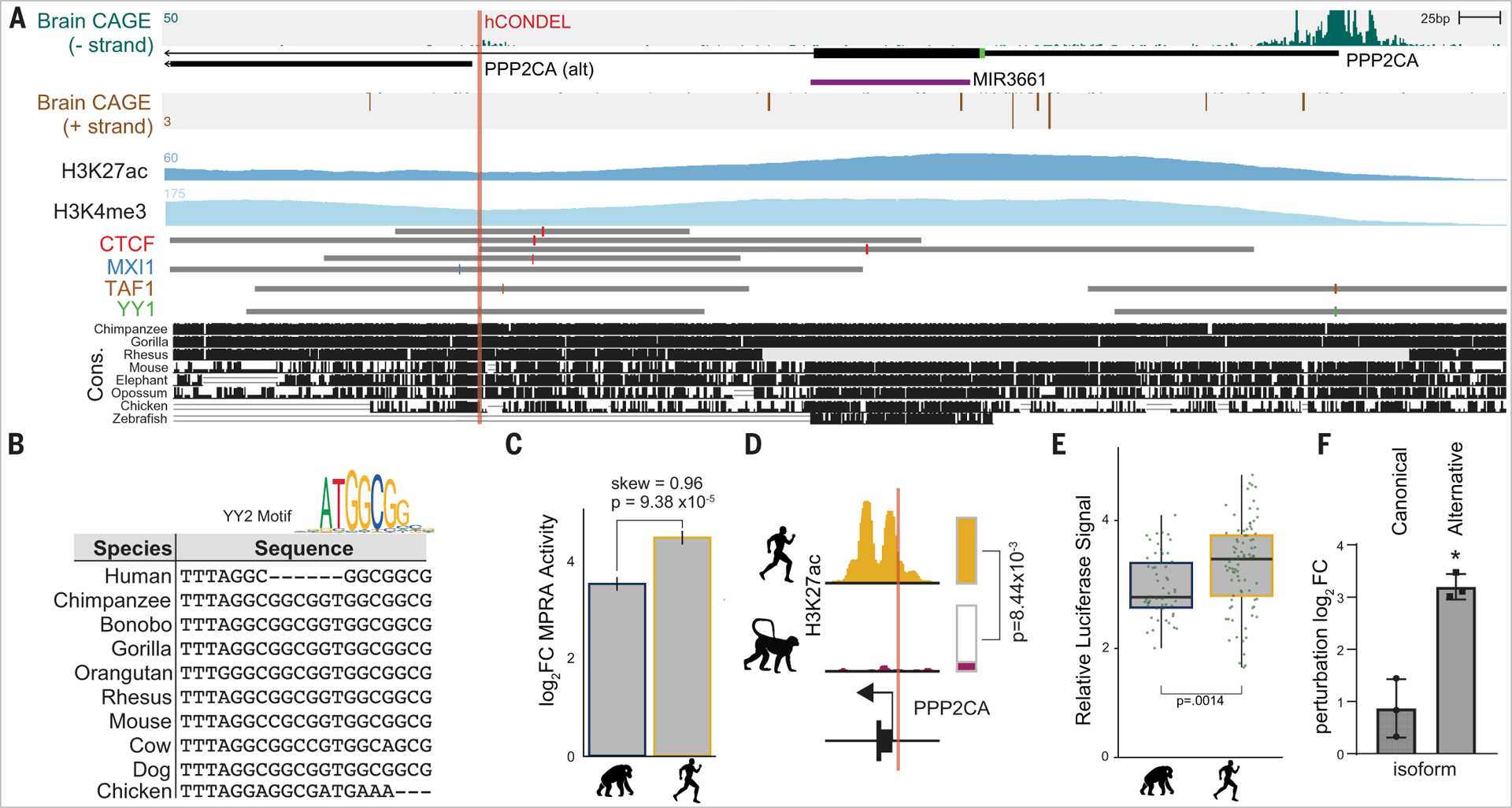
(A) Genome track of hCONDEL position. Strand-specific CAGE, H3K27ac, H3K4me3, and TF chromatin immunoprecipitation signals are depicted along with conservation. (B) Vertebrate sequences aligned to the hCONDEL position with perturbed TF motif. (C) MPRA result plotting human (blue) and chimpanzee (yellow) sequence activities. Error bars indicate SD of chimpanzee and human activity. (D) hCONDEL H3K27ac signal between human and rhesus macaque. (E) hCONDEL luciferase assay result (two-sided t test P = 0.0014). Boxes indicate the median (thick line), 25th percentile (bottom end of box), and 75th percentile (top end of box); whiskers indicate ±interquartile range. (F) qPCR results for canonical and alternative isoform of PPP2CA from CRISPR mutagenesis of human sequence surrounding hCONDEL (two-sided t test P = 1.9 × 10−3). Bar height is the mean from three biological replicates. Error bars, SD.
Endogenous characterization of a LOXL2-associated hCONDEL
We also investigated one of the strongest species-specific effects in our screen at the lysyl oxidase gene LOXL2, which maintains the extracellular matrix (27). This hCONDEL, a single base deletion, perturbs a repressive SNAI2 motif present in the chimpanzee genome (Fig. 4, A and B) (28). The hCONDEL overlaps H3K27ac and DNase accessibility CRE signatures in the human brain, and the human sequence drives regulatory activity in our MPRA in SK-N-SH cells (log2FC activity = 0.39). Comparatively, the chimpanzee version displays strong transcriptional repression (log2FC activity = −1.21), significantly lower than that of human (BH adjusted P = 5.12 × 10−7)(Fig. 4C). This is consistent with the human deletion disrupting repressor binding in the chimpanzee genome, leading to activation.
Fig. 4. hCONDEL at LOXL2 induces transcriptomic changes related to myelination and calcium signaling.
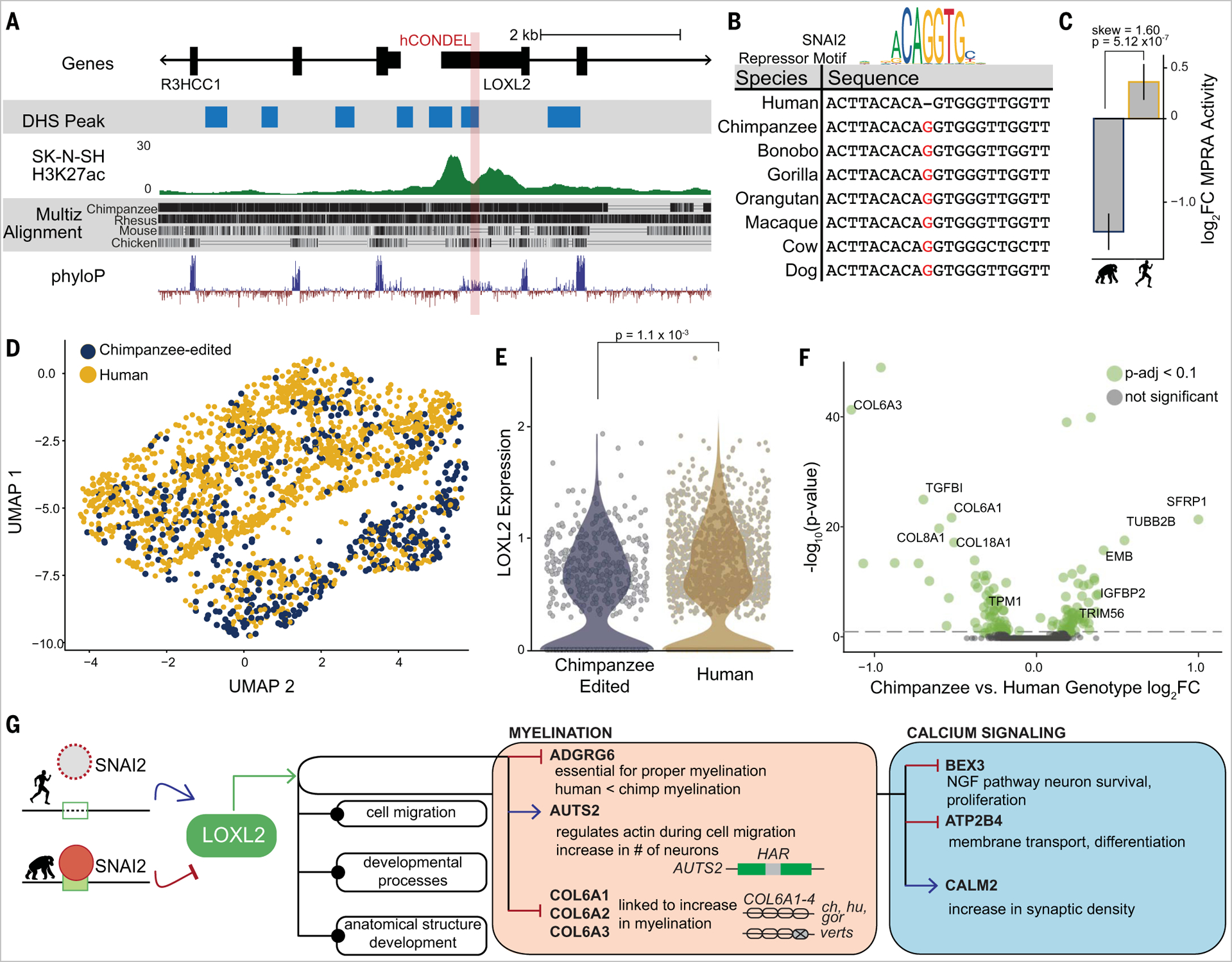
(A) Genome track of hCONDEL position in LOXL2, including H3K27ac and DNase I hypersensitive site signals from SK-N-SH and conservation scores. (B) Sequence alignment at hCONDEL with perturbed TF motif (top) and deleted conserved base (red). (C) MPRA result for LOXL2-associated hCONDEL (skew and BH adjusted P). Error bars indicate SD of chimpanzee/human activity. (D) UMAP of SK-N-SH–edited cells, with species genotype labeling for human (yellow) or chimpanzee reference (blue). (E) LOXL2 expression of SK-N-SH cells bearing the chimpanzee versus human base (Wilcoxon rank-sum test P value).(F) Volcano plot for most differentially expressed genes comparing SK-N-SH cells bearing the chimpanzee versus human sequence (genes with BH adjusted P < 0.1 highlighted in green). (G) Highlighted GO enrichments of differentially expressed genes from (F).
To investigate the direct transcriptional and downstream pathways of this hCONDEL, we genome edited human neuroblastoma SK-N-SH cells to reintroduce the conserved chimpanzee “G” base (fig. S10A). We then performed hybridization chain reaction fluorescence in situ hybridization coupled with flow cytometry (HCR-FlowFISH) to determine LOXL2 transcription levels in a pool of cells with mixed unaltered or reverted chimpanzee sequence. We recapitulate the result seen from MPRA, demonstrating the hCONDEL’s direct endogenous control of LOXL2 transcription (Fisher’s test P < 2.2 × 10−16 for two replicates;fig. S10B) (29).
We then performed single-cell genotyping and RNA sequencing on the pool of mixedspecies LOXL2 genotypes to assess broader transcriptional changes occurring caused by the introduced chimpanzee base (see the materials and methods). We found human and chimpanzee genotype cells clustering together after performing unbiased transcriptional profile clustering and overlaying the mutational profile of each cell (human versus chimpanzee base) (Fig. 4D and fig. S10C). This orthogonal analysis also confirmed the higher levels of LOXL2 expression in human versus chimpanzee-edited cells (Wilcoxon rank-sum test P = 1.1 × 10−3) (Fig. 4E).
We detected 145 genes that were differentially expressed because of the LOXL2 hCONDEL (BH adjusted P < 0.1) (Fig. 4F and table S3). These genes revealed broad enrichment in processes related to cell migration (P = 3.43 × 10−7) and development (P = 7.95 × 10−8), consistent with known LOXL2 function in neural progenitor differentiation in both mouse embryonic stem cells and during brain development in zebrafish (30, 31) (fig. S10D and Fig. 4G). One strongly down-regulated gene is ADGRG6 (FC = 0.8, P = 1.03 × 10−6), which is a crucial regulator of myelination, and more plastic myelination during development has been hypothesized to play a role in human cognitive abilities (32). Concomitantly, we observed down-regulation in multiple genes in some COL6A collagens also linked to myelination levels (33). Calcium ion transport and synaptic function may also be affected by this hCONDEL because of the differential expression of BEX3, which has been shown to cause brain morphological differences in murine models (34).
Discussion
In this study, we characterized an overlooked yet evolutionarily important set of human-specific sequences. We elucidated how thousands of conserved sequences specifically missing in humans alters TF binding, catalogued species-specific gene-regulatory activity, and identified altered gene-expression pathways. Deletion-induced human regulatory changes are enriched for brain and neuronal function, including hCONDELs regulating LOXL2 and PPP2CA, which contribute to phenotypes uniquely altered in humans, such as myelination levels, vestibular structure, and neural progenitor proliferation.
Our work provides a paradigm for characterizing the genetic basis of uniquely human traits that can also be extended to studying how sequence loss may impart unique traits across other species, such as hind limb loss in whales or echolocation in bats. Proliferation of high-quality genomes with reference-free alignments from consortiums such as Zoonomia (15) will enable the discovery of thousands more species-specific deletions and uncover new hCONDELs. The improved resolution of conservation along with MPRAs could better inform the role of evolution for interpreting sequence variation related to human biology.
These findings extend our understanding of the interplay between gene regulation and evolutionary innovation. Although sequence loss may be expected to eliminate genomic functions, we observed nearly equal gains versus loss of regulatory activity. This suggests that abrogation of repression may be as important for phenotypic change as more commonly described regulatory activity loss. In contrast to previous studies of large-scale deletions (5), we found that small evolutionary change can have large regulatory and transcriptional effects. Moreover, these effects arise, not from complete loss or invention of functional CREs (13, 35), but rather from evolutionary “tinkering” to a CRE’s regulatory potential to yield phenotypic gain.
Materials and Methods
Computational identification of hCONDELs
At the start of our project, multiple sequence alignments either did not have chimpanzee as the target genome or used older primate reference genomes. To circumvent these deficiencies, a chimpanzee (panTro4)–anchored multiple sequence alignment was created using Multiz (v. 11.2) (36). In addition to panTro4, the alignment was created with the following species (genome builds): bonobo (panPan1), macaque (rheMac8), gorilla (gorGor4), orangutan (ponAbe2), mouse (mm10), cow (bosTau8), dog (canFam3), opossum (monDom5), platypus (ornAna2), and chicken (galGal4), yielding 11 total genomes including panTro4. We followed a template multiple sequence alignment pipeline from the University of California Santa Cruz (UCSC), which produced an older chimpanzee-anchored 12-way multiple sequence alignment using the panTro3 chimpanzee genome and species of similar phylogenetic distances as our 11-taxa alignment: https://github.com/ucscGenome-Browser/kent/blob/master/src/hg/makeDb/doc/panTro3.txt). Furthermore, MultiZ requires pairwise alignments of the mentioned animal genomes with panTro4, which was performed with lastZ (37) and processed with the chain/net workflow (38).
After building the multiple sequence alignment, the phastCons program (6) was used on our Multiz-constructed alignment to obtain 1,398,973 conserved sequences. For phastCons, the following variables were used: –rho 0.3 –expected-length 45 –target-coverage 0.3 –most-conserved –score. A neutral parameter background file that contains the substitution rate matrix, a tree with branch lengths, and estimated nucleotide equilibrium frequencies was used. This background file is also provided in our code repository (see the Acknowledgments, “Materials and data availability”) and was created from running the phyloFit program on fourfold degenerate sites obtained from our Multiz alignment using the flags and parameters: –EM –precision MED –msa-format FASTA –subst-mod REV.
Nonorthologous sequences (multiple chimpanzee conserved sequences that mapped to the same human sequence) and elements with large human-specific insertions [defined as (human-mapped conserved sequence length)/(chimpanzee conserved sequence length) ≥1.05] were removed to reduce our set to 1,371,766 chimpanzee conserved sequences.
A pairwise alignment was also created with human (hg38) and chimpanzee (panTro4) and identified initial human deletions using lastZ and the chain/net workflow. From the pairwise alignment, 2,042,706 syntenic deletions were derived that do not overlie chimpanzee reference gapped regions. Then, these initial human deletions were used to extract those overlapping the 1,371,766 chimpanzee conserved segments and obtained a total of 43,855 total deletion sites. The set derived from this initial overlap are preliminary hCONDELs.
After obtaining the preliminary set of hCONDELs, it was necessary to check whether these deletions were present in other humans outside of the human reference genome and to further validate that these sites were annotated as being in the correct position. To the best of our knowledge, the accuracy of correctly annotated deletion positions is unknown from UCSC tools. Pairwise alignments in general have been known to produce spurious indel calls, and the exact indel position may be misrepresented (39). Furthermore, deletions identified in the human reference genome may be polymorphic across other individuals, which would cause our annotated site to not be a true, complete human-specific deletion. To directly address both of these issues, chimpanzeehuman (Ch-Hu) hybrid genomes were created and screened with sequences from a diverse pool of human sequences from the Simons Genome Diversity Project (SGDP) (40). Ch-Hu hybrid genomes were made by inserting each chimpanzee conserved element/deletion element combination into the corresponding human position as annotated by liftOver (41). After creating the hybrid genomes, the SGDP dataset, which contained 263 humans across a range of different populations (40), was used as sequences to screen against the preliminary set of hCONDELs. Fermikit was used to call variants on all Ch-Hu hybrid genomes (42). After obtaining the variant calls, hCONDELs were retained if the deletion position marked by FermiKit matched the same deletion position annotated by UCSC Chain and Nets. hCONDEL sequences that differed in repeat content between the variant-normalized allele and the original hCONDEL allele were also not retained because of a computational error; this removed ~1% of hCONDELs. Our filtered set produced 17,673 hCONDELs. Any hCONDELs with N’s in the 200-bp surrounding sequence were removed for both species, leaving 17,197 hCONDELs. Any sequences with an AsiSI restriction site (GCGATCGC) were filtered for cloning purposes (see the “MPRA vector assembly” section), but no sequences contained the restriction site. For every hCONDEL in this set, 200 bp of sequence (centered on the hCONDEL position) from both the human (hg38) and chimpanzee (panTro4) sequences was used. This gave a total of 17,197*2 = 34,394 sequences. A set of 1606 positive control sequences from Tewhey et al. (14) was also included. This final set of sequences (36,000 total) was synthesized by Agilent Technologies for use in our MPRA.
The hCONDEL set was then adjusted using the following filters. First, 29.1% (5000) of the 17,197 hCONDELs that were not fixed (allele frequency does not equal 1) in chimpanzees and bonobos in the Great Ape Genome Diversity Project (GAGP) (43) were removed. hg18 coordinates from the GAGP VCFs were mapped to both the hg38 and panTro4 reference genomes using liftOver and compared with both the hCONDEL hg38 deletion breakpoint (base to the left of the hCONDEL) position and the hCONDEL panTro4 conserved bases start position. Because all nonhuman primate reads were mapped to the hg18 genome by the original authors, any hCONDEL would be classified as an insertion in those VCF files. hCONDELs that matched a fixed (allele frequency of 1) GAGP chimpanzee/bonobo insertion by position and contained the same sequence as the inserted allele from the VCF file were retained.
Next, 30.3% (5,216) hCONDELs that did not have conserved bases that were present in at least one other primate group [defined as having the conserved bases fixed in at least one other primate group in the GAGP (gorillas, Sumatran orangutan, or Bornean orangutan) or present in the macaque genome (rheMac8)] were removed. This filter was to ensure that we did not retain any chimpanzee or bonobo lineage-specific insertions. This statistic overlaps largely with the previously mentioned 5000 hCONDELs that were not found to be variable in chimpanzees and bonobos (59.2% or 3,086 of the hCONDELs in this group overlaps with the 5000 hCONDELs).
Finally, 6% (1032) of hCONDELs were removed because of the hCONDEL chimpanzee position in panTro4 being not mappable to panTro5.
After applying the above filters, 10,032 hCONDELs remained. These hCONDELs are largely not in the same conserved sequence; only 189 of the 10,032 hCONDELs shared a conserved sequence background with another hCONDEL. This set also does not contain doublesided gaps (human deletions that may have additional inserted bases, compared with the chimpanzee genome, in the deleted location). hCONDELs were further mapped to panTro6 and 59 of the 10,032 hCONDELs were not mappable. These hCONDELs are likely not spurious because the deleted bases are present in all chimpanzee genomes in GAGP (potentially signifying a panTro6-specific reference genome error). Thus, we retained these 59 elements. However, a flag is provided in table S1 if hCONDELs were not mappable to panTro6.
Our set of 10,032 hCONDELs was also found to not overlap prior studies on hCONDELs (5, 7). Earlier studies of hCONDELs (5, 7) used a minimal deletion sizes of 23 and 50 bp or larger, respectively. Our hCONDELs did not overlap most prior functional studies of human accelerated regions (8, 9, 44). In Whalen et al. (44), which tested 714 HARs, 16 hCONDELs overlapped the tested regions. In Girskis et al. (9), which tested 3129 HARs, 10 hCONDELs overlappedthe tested regions. Finally, in Uebbing et al. (8), which tested 1363 HARs and 3027 humangain enhancers (enhancers with gained H3K27acactivity compared with rhesus macaque), 89 hCONDEL-tested regions overlapped their dataset. Of the 89, only one hCONDEL had functional activity that was captured by both our MPRAs. Similarly, in the second largest overlap (44), only two had functional activity that was captured by both our MPRAs.
Confirmation of hCONDEL loci in chimpanzee genomes
For the hCONDELs described in detail in this study (fig. S7, A to E, G, and H, and Figs. 3 and 4), the chimpanzee sequence was confirmed in seven individuals. Three male and three female chimpanzee iPSC lines (45) and one adult male chimpanzee were DNA sources. Polymerase chain reaction (PCR) primers bracketing the hCONDEL sequence were designed using Primer3Plus (https://www.primer3plus.com/) and synthesized with an additional adapter for Illumina sequencing. hCONDELs were amplified individually for each region in each individual’s DNA in a 50-μl PCR using the NEB Hot Start Q5 Master Mix (NEB, M0493L) with 10 μM primers and the following cycle conditions: 98°C for 2 min, 30 cycles (98°C for 10 s, 55 to 62°C for 15 s, 72°C for 45 s), 72°C for 5 min. PCR products were isolated using 1X AMPure XP beads (Beckman Coulter, A63881). A second indexing PCR was performed on the amplicons using NEB Q5 98°C for 2 min, eight cycles (98°C for 10 s, 64°C for 15 s, 72°C for 45 s), 72°C for 5 min. Libraries were purified using 1X AMPure XP beads, quantified using the Agilent 4200 TapeStation (Agilent Technologies, G2991BA) on a D1000 ScreenTape (Agilent Technologies, 5067–5583 and 5067–5582) and pooled. Sequencing was performed using 2 × 150 bp chemistry on an Illumina MiSeq and analyzed using CRISPResso (v. 2.0.30). The initial primers designed for the BBC3-associated hCONDEL did not amplify uniquely and a second design was not attempted.
MPRA
MPRA vector assembly
hCONDEL sequences centered on the deletion site from both the human and chimpanzee genomic backgrounds were synthesized by Agilent Technologies. Two hundred base pairs of sequence was derived from the chimpanzee panTro4 reference genome, and 200-X base pairs were obtained from the human hg38 reference genome, where X is the deletion size length. Fifteen base pairs of adapter sequence were also attached at both ends of the oligo for synthesis: 5′-ACTGGCCGCTTGACG [200 bp (chimpanzee) or 200-X (human) oligo] CACTGCGGCTCCTGC-3′. After synthesis, adapters and 20-bp barcodes were attached through a 48× 50-μl PCR using the NEBNext Ultra II Q5 Master Mix (NEB, M0544L) with primers MPRA_v3_F (10 μM) and MPRA_v3_F (10 μM), 3.2 ng in each reaction, and the following cycle conditions: 98°C for 20 s, 15 cycles (98°C for 10 s, 60°C for 15 s, 72°C for 45 s), 72°C for 5 min. The product was then subject to two 1X AMPure SPRIs (solid-phase reversible immobilizations) (Beckman Coulter, A63881) and eluted in 200 μl of water. pGL4:23:DxbaDluc was then digested by SFiI (NEB, R0123S) at 50°C for 1 hour. The resulting digested backbone and oligo product were then assembled through Gibson assembly reaction (NEB, E2611L) using 1 μg of digested plasmid and 1 mg oligos and incubation at 50°C for 1 hour and then purified by a 1.2X AMPure SPRI and eluted in 20 μl. Ten microliters of the assembled construct was then electroporated (2kV, 200 ohm, 25 mF) into 100 μl 10-beta Escherichia coli (NEB, C3020K). Electroporated cells were split into eight tubes and grown in 2 μl of SOC for 1 hour at 37°C. Subsequently, the eight aliquots were independently expanded in 20 μl of Luria broth (LB) supplemented with 100 μg/ml carbenicillin for 6.5 hours at 37°C. Then, bacteria were pooled and the resulting plasmid purified using the QIAGEN Plasmid Plus Maxi Kit (Qiagen, 12963). Serial dilutions estimated the combined complexity as ~1.7 × 108 colonyforming units.
Twenty micrograms of the resulting vector was then cut with 200 units of AsiSI (NEB, R0630L) and 1x CutSmart buffer in a 500-μl reaction at 37°C for 3.75 hours, followed by a 1.5X AMPure SPRI cleanup. The linearized vector and an amplicon containing a minimal promoter, green fluorescent protein (GFP) open reading frame, and partial 3′ untranslated region (3′-UTR) was then assembled together through a Gibson reaction using 10 μg of the AsiSI linearized vector and 33 μg of the GFP amplicon in a 400-μl reaction at 50°C for 1.5 hours, followed by heat inactivation for 20 min at 80°C. The entire reaction was cleaned by a 1.5X AMPure SPRI and eluted in 55 μl. The elution from the cleanup was then digested again to remove any uncut plasmids with 50 units of AsiSI, 5 units of RecBCD (NEB, M0345S), 10 μg of bovine serum albumin, 0.1 mM adenosine triphosphate (ATP), and 1X NEB Buffer 4 in a 100-μl reaction for 1 hour and 40 min at 37°C. Subsequently, 9 μl of 10 mM ATP was added to the 100-μl reaction, and the digestion was continued at 37°C for 4 hours and 20 min (6 hours total), followed by heat inactivation for 20 min at 80°C and SPRI purification.
The final vector library was generated by electroporating four batches of 100 μl of 10-beta E. coli with 10 ml of DNA (2kV, 200 ohm, 25 μF). Each batch of bacteria was split into three separate tubes, each with 2 μl of SOC, and grown for 1 hour (12 tubes in total across all four batches). After the 1 hour of recovery, all three tubes from each batch were combined into 1.5 liters of LB with 100 μg/ml carbenicillin in a single 2.8-liter flask and subsequently grown for 9 hours (four 2.8-liter flasks with 1.5 liters of LB across all four batches). The plasmid was then prepped using the Qiagen Gigaprep kit (Qiagen, 12191).
Transfection
HEK293 cells (ThermoFisher, R70007) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (ThermoFisher, 10564) containing 10% fetal bovine serum (FBS) (ThermoFisher, A3160401). Four total replicates were transfected. For each replicate, cells were plated in two 15-cm plates and grown to a density of ~80 to 90% (~20 to 40 million cells per plate). Cells were then incubated with 80 μl of Lipofectamine 2000 (ThermoFisher, 11668027) and 20 μg of DNA for 24 hours. Then, transfected cells were split 1:3 into new 15-cm plates, keeping all transfected cells. After an additional 24 hours (48 hours after transfection), cells were pelleted by centrifugation, washed once with phosphatebuffered saline (PBS), flash-frozen using liquid nitrogen, and then stored at −80°C.
HepG2s (ATCC, HB-8065) were cultured on 15-cm plates in 25 μl of minimal essential medium (MEM) Alpha (ThermoFisher, 32561037) containing 10% FBS and 1% penicillinstreptomycin (Pen-Strep). Cells were grown to 60 to 80% confluency. Four total replicates were transfected. For each replicate (grown on different days to ~60 to 80% confluency), two 15-cm plates (~20 to 40 million cells per plate) were incubated with 87.5 μl of Lipofectamine 3000 (ThermoFisher, L3000015) and 35 mg of the MPRA library. After transfection, each replicate was recovered for 48 hours in 25 ml of MEM Alpha containing 10% FBS without Pen-Strep. Cells were then trypsinized, pelleted at 300g at 4°C, washed in PBS once, flash-frozen using liquid nitrogen, and then stored at −80°C.
GM12878s (Coriell) were cultured in RPMI medium (ThermoFisher, 61870036) containing 15% FBS (ThermoFisher, 15140122) and 1% 10× Pen-Strep (Corning, 30–002-CI). Four total replicates, grown on different days to ~1 million cells/ml, were transfected. Per replicate transfection, 150 million cells were pelleted at 300g and resuspended in 1.2 ml of RPMI medium containing 150 μg of the MPRA library. Cells were electroporated using the Neon transfection system and the setting of three pulses of 1200 V for 20 ms with the 100 μl kit (ThermoFisher, MPK10096). After transfection, each replicate was recovered for 48 hours in 150 ml of RPMI medium containing 15% FBS without Pen-Strep. After the first 24 hours of recovery, cells were split 1:2 to avoid overgrowth. After 48 hours of recovery, the cells were pelleted by centrifugation, washed in PBS once, flash-frozen using liquid nitrogen, and then stored at −80°C.
K562s (ATCC, CCL-243) were cultured in RPMI medium containing 10% FBS and 1% 10× Pen-Strep. Four total replicates, grown on different days to ~1 million cells/ml, were transfected. Per replicate transfection, 150 million cells were pelleted at 300g and resuspended in 1.2 ml of RPMI medium containing 150 μg of the MPRA library. Cells were then electroporated using the Neon transfection system and the setting of three pulses of 1450 V for 10 ms with the 100 μl kit. After transfection, each replicate was recovered for 48 hours in 150 ml of RPMI medium plus 15% FBS without Pen-Strep. After the first 24 hours of recovery, cells were split 1:2 to avoid overgrowth. After 48 hours of recovery, cells were pelleted by centrifugation, washed in PBS once, flash-frozen using liquid nitrogen, and then stored at −80°C.
SK-N-SH (ATCC, HTB-11) were cultured on Nunc Triple Flasks (VWR, 89498-706) in 90 ml of Eagle’s MEM (EMEM) (ATCC, 30-2003) containing 10% FBS and 1% Pen-Strep. Four total replicates were transfected. Each replicate was grown on different days to reach 80 to 100% confluency. Cells were then trypsinized, and 40 million cells were suspended in 400 ml of Buffer R with 25 μg of the MPRA library. Subsequently, cells were electroporated using the Neon transfection system and the settings of three pulses of 950 V for 30 ms with the 100 μl kit. After transfection, each replicate was recovered for 48 hours in 45 ml of EMEM containing 10% FBS without Pen-Strep. Cells were then trypsinized, pelleted at 300g at 4°C, washed in PBS once, flash-frozen using liquid nitrogen, and then stored at −80°C.
hiPSC-derived NPCs (NSB2607, male) were used. NPC generation and cell line validation were previously described (46). NPCs were grown in 100-mm dishes coated with 0.6 to 8.6 mg Geltrex (Gibco, A1413301) in NPC medium [DMEM/F-12 GlutaMAX, ThermoFisher), 1× N2, 1× B27-RA, 1× Antibiotic-Antimycotic (ThermoFisher), and 20 ng/ml FGF2 (Stemgent)]. NPCs were maintained at a high density of up to 30 million cells per dish, dissociated twice a week with Accutase (Innovative Cell Technologies) for 5 min at room temperature, and reseeded at 9 to 11 million cells per dish (i.e., a 1:3 split) in NPC medium onto Geltrexcoated 10-cm dishes.
The MPRA library was nucleofected into NPC as follows. For each replicate, NPCs (two 100-mm plates containing ~30 × 106 cells each) were harvested with accutase, resuspended in 12 ml of NPC medium, and counted by trypan blue staining. Twenty-four simultaneous reactions of NPCs (1.6 × 106 cells in a 20-ml reaction, total 38.4 × 106 cells) were nucleofected with 0.6 μg of MPRA plasmid library (total 14.4 μg) in P3 primary cell 4D nucleofector reagents (Lonza V4XP-3032) in a Lonza 4D-nucleofector unit (Lonza AAF-1002B, AAF-1002X) with the DS-138 program following the manufacturer’s protocol. Each nucleofection reaction was immediately plated in a well of a 24-well plate with warmed (37°C) NPC medium and incubated overnight at 37°C. Cells were harvested 24 hours after nucleofection, in plate, with 200 μl of RLT plus lysis buffer (Qiagen) per well, pooled together, homogenized with a homogenizer (Omni TH-01) at one-fourth power for 30 s, and snap-frozen for processing. NPC MPRA experiments were performed in four replicates.
Across all cell types, transfection efficiency was assessed by checking GFP fluorescence from test transfections using a control vector containing GFP. A minimum of 50% of live cells fluoresced after transfection was required. HEK293, HepG2, and K562 obtained the greatest transfection efficiency (>80%), whereas GM12878 and NPCs performed near our minimum (~20 to 50%).
Sample processing
Frozen cell samples were processed following the MPRA protocol in (14). Briefly, RNA was extracted from the Qiagen Maxi RNeasy kit (Qiagen, 75162) without the on-column DNase digest. A DNase reaction was then performed to remove remaining MPRA library vectors. The GFP in the total RNA was then captured through a hybridization reaction using streptavidin beads (ThermoFisher, 65001) and a mixture of three GFP RNA-targeted biotinylated oligos (table S4). A second DNase reaction was then performed to remove any undigested library vectors. After an RNA SPRI (Beckman Coulter, A63987) cleanup, the RNA was then converted to cDNA in a Superscript III (ThermoFisher, 18080044) reaction using MPRA_v3_Amp2Sc_R (table S4). The cDNA was then cleaned using AMPure SPRI, and the relative cDNA abundance across all cell type samples and MPRA library vector was estimated through quantitative PCR (qPCR) by comparing their cycle thresholds (number of cycles required to amplify above background). In total, there were four replicates per cell type. All cell type replicates (with the exception of NPC samples, which were processed later) were normalized to approximately the same concentration and cycled for 10 cycles in a PCR using NEBNext Ultra (NEB, M0544L) to amplify the cDNA using the primers MPRA_v3_Illumina_GFP_F and TruSeq_Universal_Adapter (table S4). Five MPRA plasmid library replicates, input normalized to achieve the same PCR output abundance, were separately amplified for 10 cycles. The five plasmid replicate counts in table S1 were derived from this amplification. Because of the lower amount of GFP RNA output from our NPC samples, about three times lower RNA was used to cycle the NPC samples two cycles higher (12 cycles total). The resulting amplified products from all cell types was then subject to another round of PCR with six cycles to attach custom p7 and p5 Illumina adapters with unique sample indices (table S4).
The Agilent 2200 TapeStation with the D1000 screentape reagents (Agilent Technologies, 5067–5585) was used to acquire molar estimates of final PCR products and pooled samples for subsequent sequencing. Samples were sequenced with a S4 flowcell (2 × 150 bp) on a NovaSeq using the sequencing service from the Broad Institute. NPC samples were sequenced separately on a NextSeq using the NextSeq 500/550 High Output Kit v2.5 (20024906) (1 × 75 bp).
Quantification of species-specific activity
DESeq2 (v. 1.26.0) was used to obtain the species-specific activities (47). For DESeq2, oligo counts from all 36,000 sequences designed in our MPRA were used. Oligo counts from all replicates in all cell types except NPCs were normalized together through DESeq2 with plasmid counts. NPCs were normalized with the plasmid counts separately because it was observed that this cell type had a higher variance across replicates, especially at lower plasmid counts, because of the potential lower transfection efficiency. The dispersion values for the five cell types except for NPCs were also obtained together. The dispersion values for NPCs were obtained separately because of the higher variance. Then, for each cell type, activity values for every human or chimpanzee sequence were obtained and species-specific activity effects computed using the following model: design = ~species + type + species:type, where “type” is either the GFP RNA or the plasmid pool. Wald tests with contrasts were used to acquire human and chimpanzee functional activity (FCs of RNA over plasmid) as well as the change between human activity and chimpanzee activity (species-specific activity). To correct for multiple hypothesis testing, the BH test correction was also implemented using DESeq2. The 800 hCONDELs that were confidently marked as having species-specific activity passed the following requirements: the species-specific activity (difference in activity between human and chimpanzee) BH adjusted P value was <0.05 and the activity BH adjusted P value in the human or chimpanzee sequence was <0.1. Plasmid count filters were set for each cell line such that the proportion of skew hits in the lowest of 10% average plasmid counts (across both chimpanzee and human combined) comprised <2.5% of all reported hits in the cell type. This filter removed hCONDELs with extremely low representation in the library. Sequences with extremely low plasmid representation would have lower power to detect activity. The output from the DESeq2 analysis is reported in table S1.
hCONDEL cell-specificity analysis
Mash was used to infer species-specific effect sharing from the MPRA tested cell types (48) following a computational framework similar to (49). User-specified data-driven covariance matrices are required by mash. These matrices were made by using hCONDELs with MPRA-measured species-specific effects (BH adjusted P < 0.05, human or chimpanzee activity BH adjusted P < 0.1, and average human and average chimpanzee plasmid count ≥ 60 across all replicates). From these effects, the following data-driven covariance matrices were made: (i) the empirical covariance matrix, (ii) flash matrix factorization of the empirical covariance matrix (50), and (iii) a rank 4 SVD approximation of the empirical covariance matrix. Rank 1 covariance matrices derived from flash factors containing at least two rows with values >1/sqrt(6) were included in the data-driven covariance matrices. Extreme deconvolution (ED) was applied to the entire set of data-driven covariance matrices (51). The resulting ED output matrices were used as the final matrices for analysis. From cross-validation, it was found that the exchangeable effects model performed better than the exchangeable Z model as determined by likelihood values, and that model was used for mash. hCONDEL species effects were classified as shared across cell types A and B if the local false sign rate was <0.05 for both A and B.
hCONDEL genomic annotation, TF perturbation, and enrichment analyses
Genomic region, age, repeat, conservation, and CRE annotations
The chimpanzee 2.1.4 genomic annotations from Ensembl (Ensembl 90) were used to annotate the hCONDELs. For genomic feature annotation, if an hCONDEL fell into more than one class (i.e., is located in the 5′UTR of one gene but coding for an overlapping gene), the following mutually exclusive order was used: coding, promoter [100 bp upstream of the transcription start site (TSS)], 5′-UTR, 3′-UTR, intronic, and intergenic. The collapsing was performed to prioritize annotations with the largest potential functional impact if hCONDELs overlapped multiple annotations and affected only <2% of hCONDELs. These mutually exclusive genomic annotations were used in all analyses except for the genomic region permutation/enrichment analyses, which did not include the collapsing step. Permuted hCONDELs were separately overlapped with each genomic annotation region.
The total number of mismatches and unaligned bases in the MPRA-tested flanking sequence surrounding the hCONDEL was estimated using the “blastn” command on the human sequence and the chimpanzee sequence with the following parameters: -penalty −3 -reward 2 -gapopen 5 -gapextend 2 -dust no -word_size 10 -evalue 1 (52).
Aged syntenic blocks in human (hg19) were obtained from a previous analysis here: https://zenodo.org/record/4734606#.YWiGnC1h2AA (13). For each hCONDEL, coordinates were mapped to hg19 using liftOver, and the syntenic block(s) overlapping the deletion was identified. For each hCONDEL, the estimated evolutionary age of the most recent common ancestor of the oldest taxon was identified.
Repeat calls on the human genome (hg38) from the RepeatMasker database were used (53). hCONDELs were intersected with repeat elements to identify overlapping significant repeat calls.
hCONDEL phyloP conservation scores were derived from a chimpanzee (panTro6)–anchored multiple sequence alignment from the Zoonomia animal sequences (240 mammalian species) (15). The Zoonomia alignment was not the same animal sequence alignment that was used to construct the initial 11-species alignment (see the “Computational identification of hCONDELs” section). At the start of this project, the Zoonomia phyloP scores were not available.
ENCODE CREs were derived from SCREEN (all human cCREs, V2, https://screen.encodeproject.org/) (12).
hCONDEL gene ontology enrichment
GREAT (v. 4.04), using the default parameters (basal plus extension gene association setting), was run to derive gene ontology (GO) enrichments for the set of hCONDELs (54). The hCONDEL hg38 coordinate positions were used and the whole genome was used as the background set. Only the top 15 enriched terms from the GO biological processes collection are plotted in Fig. 1F. The set of the top 500 enrichment terms is in table S2. For fig. S3B, semantic clustering was performed on the 500 terms using REVIGO (55).
TF analyses
A total of 741 TF motifs from the JASPAR 2020 core vertebrate nonredundant collection (56) were used to compute TF alteration scores for all hCONDELs. For the analyses in Fig. 2, C and D, and fig. S6B, for every hCONDEL, a single TF alteration score was computed for each MPRA-tested cell type (six total). Thus, six TF cell type alteration scores were calculated for each hCONDEL. To calculate the scores for each cell type, only the set of TFs that were expressed in that cell type (TPM >1) was used. For fig. S6D, for each TF motif type (741 total), alteration scores for all hCONDELs were computed regardless of TF expression level.
To compute alteration scores for the analyses in Fig. 2, C and D, and fig. S6, B and D, a set of putative binding domains was first extracted for both the chimpanzee and human hCONDEL using FIMO (57). A binding domain was required to either completely overlap the deletion breakpoint (bases to both the left and right of where the deletion occurred) in the human sequence, or completely overlap the deleted bases in the chimpanzee sequence. If an hCONDEL species sequence contained multiple binding domains, the binding domain with the maximum FIMO score was retained.
Next, to calculate a single TF alteration score for each hCONDEL, a significant (P < 0.0001) binding domain in either the human or chimpanzee sequence was required. The alteration score was calculated as the difference in FIMO binding score between the human and chimpanzee sequence sequences. The alteration score can be approximated as the difference in log-likelihood (base 2) in motif match to the human compared with the chimpanzee sequence. A difference of 1 would then indicate that the motif is twice more likely to match the human compared with the chimpanzee sequence. For the analyses in Fig. 2, C and D, and fig. S6B, if multiple TF moitfs had alterations on the hCONDEL position, the alteration with the maximum magnitude was retained. For fig. S6D, for each individual TF motif type, if multiple motifs were altered, the alteration with the maximum magnitude was also retained.
For the analyses in Fig. 2, C and D, we were interested in investigating the proportion of hCONDELs altering activating and repressor motifs in enhancers.Several filters were used to ensure that the MPRA signals were overlapped with the most confident TF perturbations. The maximum phyloP score (calculated from a chimpanchored multiple sequence alignment from the Zoonomia genomes) on the human-deleted bases was required to be >1 and the phastCons score (as calculated from the 11-species animal alignment) of the conserved block containing the hCONDEL to have a log-odds score >50. Finally, the TF alteration score comparing human and macaque was used as a filter (using the macaque reference genome rheMac8, calculated in the same manner as the human and chimpanzee TF alteration score) by requiring the sign of the TF alteration score derived from the human and chimpanzee to match the sign of the TF alteration score derived from the human and macaque score. Furthermore, only hCONDELs with enhancer activity (defined as BH adjusted P < 0.1, log2FC MPRA activity > 0) in either the chimpanzee or human sequence background were used in Fig. 2, C and D. Because our MPRA design used a minimal promoter, it was less sensitive at detecting differences if both species’ sequences displayed strong repressive effects. This lack of detection may underestimate TF disruptions in purely repressive sequence backgrounds. If an hCONDEL had significant species-specific activity (defined here as BH adjusted P < 0.2 for all cell types except NPCs, which required BH adjusted P < 0.05 because of the higher effect variance) in multiple cell types, the species-specific activity with the lowest BH adjusted P value value was used for plotting. Because the hCONDELs in Fig. 2C represent the deletions with the most confident TF perturbations, the hCONDELs in that figure were used to create Fig. 2D. The estimates in Fig. 2D were produced by classifying hCONDELs in quadrant 1 as “improve activator,” quadrant 2 as “disrupt repressor,” quadrant 3 as “disrupt activator,” and quadrant 4 as “improve repressor.”
For fig. S6B, the analysis focused on investigating the correlation between motif alteration scores and MPRA species-specific activity for TF activators. For the hCONDELs plotted in fig. S6B, enhancer activity was not required in either the human or chimpanzee sequence background (all other previously mentioned filters were kept), but potential strong repressors were further removed by requiring both the human and chimpanzee species activity to be > −0.5 log2FC. The removal of sequences with strong repressors was performed because significant MPRA species-specific effects in strong repressive backgrounds would be expected to be enriched for alterations in repressive motifs. Alterations to repressive motifs would be expcted to be anticorrelated with 9 of 16 MPRA effects. For example, if a deletion weakens or destroys a repressive TF motif (leading to a negative binding score on the x axis of fig. S6B and Fig. 2C), it would induce a gain in regulatory activity (leading to a positive MPRA skew on the y axis of fig. S6B and Fig. 2C).
For both Fig. 2, C and D, and fig. S6B, a permissive, species-specific MPRA adjusted P threshold of 0.2 was used (for all cell types except NPCs as mentioned previously). A higher false-positive rate balanced against having more total true positives was acceptable for this analysis. This larger number of potential hits in estimating hCONDEL perturbation proportions derived a more robust estimate of hCONDEL regulatory function for Fig. 2D.
To create fig. S6D, for each of the 741 TF motifs, an enrichment z score was calculated by comparing the observed amount of significant motif alterations across all 10,032 hCONDELs against 1000 permuted sets (see the “Permutation set creation and analyses” section). Figure S6D shows the positively enriched motifs (BH adjusted P < 0.05) from the set of 741 motifs. Because some TF motifs may have similar sequences, the 741 TF motifs were also clustered by following the TF clustering pipeline from Vierstra et al. (58). In total, the 741 motifs were identified in one of 149 clusters. Each cluster contains a set of unique motifs distinct from every other cluster. The clusters are available in table S2. Using this clustering information, the motif enrichments are colored in fig. S6D by clusters. In fig. S6D, 19 TF motifs are found in 13 distinct motif classes, suggesting that most TFs (such as EGR4 described in the text) are enriched for perturbations uniquely within their motif clusters.
There are two limitations with our TF enrichment analysis. First, existing motifs may have differing types of experimental evidence and some TFs have no motifs because of the lack of experimental validation. Second, without chromatin immunoprecipitation sequencing (ChIP-seq) data, the exact TF motif that hCONDELs may causally perturb cannot be causally determined. However, although these limitations could produce false-negatives, they should not affect the significant enrichments reported.
Permutation set creation and analyses
Two permuted sets were created to match the attributes of the empirical hCONDELs. One permuted set was constructed from human reference genome hg19 (PermSet #1), and the other was constructed from human reference genome hg38 (PermSet #2). PermSet #1 was used as the background set for the tissue-specific CRE/age/repeat class enrichments. PermSet #2 was used as the background set for the genomic annotation, conservation, TF motif perturbation, and Genotype-Tissue Expression (GTEx) brain subregion enrichments. PermSet #1 was originally created to sample random deletion breakpoint positions solely from the human (hg19) reference genome. PermSet #2 was additionally made to create physical deletions in the human hg38 genome and requires that all these deletion positions be mappable (using liftOver) to the chimpanzee (panTro4) reference genome.
Both permuted sets consisted of 1000 batches of 10,032 permuted hCONDELs. For both sets, an iterative method was used to match each of the 10,032 hCONDELs in our set to a permuted hCONDEL. For every hCONDEL, a conserved block was first sampled from the superset of all derived conserved elements (as extracted from the 11-species multiple sequence alignment) matching based off of conserved block chromosome, total mismatch percentage between human (hg38) and chimpanzee (panTro4) (±5% from empirical hCONDEL), length (±5%), GC content (±5%), and phastCons score (±5%). To calculate the total mismatch percentage between human and chimpanzee sequences, a conserved block was extended to at least 200 bp in both human and chimpanzee if either the chimpanzee or human sequence was <200 bp. If no conserved sequences were found with the initial settings, then the total mismatch percent was increased by 1%, length by 5%, GC percentage by 3%, and log odds by 5%, and then the sequence was redrawn. This process was repeated until a conserved sequence was drawn. After sampling a conserved sequence, for PermSet #1, a base position on hg19 was selected to serve as the deletion breakpoint. For PermSet #2, a randomly drawn position was selected on the conserved block, and then a deletion size matching the deletion size of the empirical hCONDEL was used to make actual deletions on the human sequence. Additionally, for PermSet #2, the specified human sequence position to be deleted was required to be able to be mapped (using liftOver) to the chimpanzee panTro4 sequence. Deletions created in PermSet #2 were also required to not span separate conserved blocks. For PermSet #2, if a sampled deletion was not able to be mapped or spans multiple conserved blocks, then another random deletion was drawn on the human sequence. For both permuted sets, if multiple deletions were on the same conserved sequence, then they were ensured to be in the same conserved sequence in the permutation sampling. In both permutations, permuted hCONDELs were not matched with empirical hCONDELs based on genomic region annotation. Although hCONDELs are substantially deenriched to be in coding regions (z score = −30.5), the overall proportion of hCONDELs in coding regions is low in both the empirical and permuted sets (0.47% empirical compared with permuted hCONDELs being in coding ~6 to 7%).
For the genomic region, age, and repeat class annotations, enrichment statistics were calculated as follows. For each of the 1000 batches of permuted hCONDELs, the number of hCONDELs of all 10,032 hCONDELs in the batch to be in a specific category (i.e., exon, Vertebrate age, L1M repeat class) was calculated. The number of empirical hCONDELs in a specific category was also calculated. For each specific category, a permutation P value was obtained by calculating the minimum of two proportions. The first is the proportion of batches with a permuted count greater than the empirical count and second is the proportion of batches with a permuted count less than the empirical count. Enrichment z scores were calculated as: (empirical count–mean permuted count across all batches)/(SD across all batches). For each annotation set (i.e., genomic region, age and repeat class), all permutation P values from all categories were used to perform the false discovery rate (FDR) correction (using the BH method) and a significance threshold of 0.05 was used.
For the TF motif permutation enrichment analyses, computation of alteration scores for each TF for both the permuted and empirical sets was described above (see the “TF analysis” section). For this analysis, alteration scores were not computed across separate cellular contexts; only a single TF alteration score was calculated for each hCONDEL to investigate alteration in a cell type–agnostic manner. The absolute value of the TF alteration score was used as the statistic to derive permutation statistics (P values, enrichment z scores) in the same manner as previously described. FDR correction was applied across the permutation P values from all 741 TFs and a significance threshold of 0.05 was used to call enriched motifs.
GTEx brain subregions gene enrichment analyses
GTEx v8 gene expression read counts were downloaded from https://gtexportal.org/home/datasets (GTEx_Analysis_2017-06-05_v8_RNASeQCv1.1.9_gene_reads.gct.gz). The resulting counts were normalized with the trimmed mean of M values (TMM) method from the edgeR package (59) and converted to counts per million. There were a total of 13 brainspecific annotated tissues collected from GTEx. For each gene, all tissue samples from one brain subregion were compared with samples from all other brain subregions using a Wilcoxon rank-sum test to identify region-specific gene expression. The Wilcoxon rank-sum test was used over methods that use negative binomial assumptions (i.e., edgeR or DESeq2) because prior computational simulations suggested that it has lower false-positive rates on large sample sizes (n > 100 in these GTEx samples) (60). In these comparisons, the labeled GTEx subregion “Brain Frontal Cortex (BA9)” was not compared with “Brain–Cortex,” and “Brain –Cerebellum” was not compared with “Brain Cerebellar Hemisphere” because these subregions are largely, if not completely, overlapping. ABHFDR correction was applied on the resulting gene P values. Genes were marked as differentially expressed in one brain subregion if the FDR was <0.1 and the absolute log2FC was greater than X, where X can be the following: 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10. Multiple log2FC cutoffs were used because of the potential for different brain subregions to differentially express genes across distinct FC magnitudes. This process created a total of (13 brain annotated sub regions) × (11 FC cutoffs) = 132 gene sets. A gene set was then retained for subsequent analyses if the number of genes in that set was greater than nine; this filtering kept 107 gene sets.
Using the above described gene sets, enrichment analyses were performed comparing the previously described 1000 batches of 10,032 permuted hCONDELs (PermSet #2) with the 10,032 actual hCONDELs. For a particular gene set, for each permutation set, for each hCONDEL, the distance (in base pairs) to the TSS of any gene in the gene set of interest was extracted. The same closest distance metric was also extracted for the 10,032 empirical hCONDELs. The average distance to the closest gene was taken for each permutation set, and the same average was taken for the actual 10,032 hCONDELs. An enrichment P value was derived by taking the proportion of permuted hCONDEL sets with an average closest distance less than the average from the actual hCONDELs. The same process was applied to all the remaining gene sets to acquire P values for all gene sets. A BH FDR correction was applied to all the enrichment P values. A gene set was significantly associated with the observed hCONDELs if the FDR was <0.05. Because multiple log2FC cutoffs were used to create the gene sets, it was possible for a single brain subregion to have multiple significant gene sets. In fig. S3C, the z-scores from the most significant gene sets (significance measured by FDR) belonging to each brain subregion were plotted.
Neuronal-related GWAS analyses
GWASs from the following sources were used: (i) intelligence (269,867 individuals) (61); (ii) depression (173,005 individuals, with 23andMe samples excluded) (62); (iii) bipolar (413,466 individuals) (63); and (iv) schizophrenia (65,967 individuals) (64). Also used were 4178 GWASs from the UK Biobank (UKBB; http://www.nealelab.is/uk-biobank/). The UKBB database contains more GWAS for diverse traits, but has fewer case individuals compared with the previously mentioned traits in the neurological GWAS.
For the GWAS enrichment analyses, all genes that contained a TSS within 50 kb of each hCONDEL are referred to herein as “hCONDEL-associated genes.” This gene set was combined with all human protein coding genes (GRCh38.p13, Ensembl), with each of the previously mentioned GWAS data used as input into magma (v1.09a) (65) to derive enrichment scores. To ensure that our GWAS enrichments were minimally confounded by the hCONDEL conservation levels, conservation was controlled for by using additional covariates in the magma regression. For every gene, the proportion of its genomic + regulatory regions (defined as 50,000 bp upstream of the gene, 500 bp downstream of the gene) to overlap conserved elements from all conserved elements derived from our multiple sequence alignment was used as a covariate. The number of conserved regions each gene plus its regulatory region overlapped was also used as a covariate for magma. In associating GWAS single-nucleotide polymorphisms with genes, each gene’s boundary region was also extended 35,000 bp upstream and 10,000 kb downstream for input into magma following previous studies (66–68).
Permutation analysis was also performed to further ensure the validity of the observed hCONDEL enrichments with the psychiatric GWAS in Fig. 1G. magma calculates a regression coefficient associating hCONDEL-associated genes with significance scores from a GWAS of interest. A gene was considered to be hCONDEL associated if it was within 50 kb of a TSS of a gene. This process yielded close to one-third of all protein-coding genes classified as being hCONDEL associated. To ensure that our enrichments were not being biased by the large number of genes grouped as hCONDEL-associated, genes were randomly scrambled to be hCONDEL associated from all protein-coding genes, ensuring that the number of scrambled hCONDEL-associated genes matched the original observed number. magma was then run with the scrambled set and this process was repeated 1000 times to generate 1000 regression coefficients. Then, the proportion of the 1000 coefficients greater than the observed coefficient was used as a P value. In this way, significant P values were found across all four traits shown in Fig. 1G (P = 0 across all), suggesting that our analyses were robust to the number of genes classified as hCONDEL associated.
The 4178 GWAS enrichment results from the UKBB are reported in table S2; 150 of these passed FDR significance, with the most enriched GWAS with our hCONDEL set being educational achievement. Specifically, two of the top six most enriched GWAS term associated with our hCONDELs was “qualifications: college or university degree” (BH adjusted P = 1.64 × 10−5), followed by “qualifications: none of the above” (BH adjusted P = 1.82 × 10−5).These two terms represent the extremes of education from the questionnaire and may relate to our initial finding of hCONDELs enriching for genes identified in intelligence GWAS shown in Fig. 1F. Because these GWASs share genetic correlations, it is unsurprising that an enrichment for genes in one GWAS might show enrichment for a related GWAS. We believe that identifying cognitive phenotypes most strongly with hCONDELs across all UKBB phenotypes further bolsters a link between our hCONDELs and the brain. We are cognizant of the potential confounders with this finding. For example, educational achievement is influenced by numerous environmental factors, such as access to educational resources and income status, which may confound its association with measurements of intelligence, a metric already known to have putative cultural sociological biases. Furthermore, future higher-powered GWASs or GWASs that control for geographical confounding (69) may change enrichments with hCONDELs. We think that these results present further evidence of hCONDELs to have function in the brain, but caution overinterpretation of these GWAS enrichment results to highlight specific cognitive functions.
Through our UKBB analysis, other traits highly enriched for hCONDELs were uncovered (150 in total, BH adjusted P value < 0.05, although many are highly phenotypically and genetically correlated). Many adipose-related terms, such as arm/leg/trunk and overall body fat percentage, showed up as being enriched. Other terms include age at menarche, chronotype (“morning person” or “night person”), and IGF-1 and creatinine levels. These terms potentially suggest that some hCONDELs may have effects in other tissues (table S2).
MPRA species-specific activity enrichments
To test whether hCONDELs with species-specific activity were enriched for the features displayed in fig. S6A, for every hCONDEL, the minimum species-specific BH adjusted P value across all five tested cell types was used as the single species-specific adjusted P value for that hCONDEL. The hCONDEL species-specific activity status (encoded as 1 if BH adjusted P < 0.2, 0 if not) was then regressed with the feature of interest (i.e., Zoonomia phyloP score, ENCODE candidate CRE). For features that are different across tested cell types (absolute TF binding difference), the cell type–specific feature that matched the cell type with the minimum species-specific BH adjusted P value was used. The maximum log BH adjusted P value across human and chimpanzee activity (also matched with the cell type with the minimum species-specific adjusted P value) was used as an additional covariate to control for activity being a potential confounder. In this analysis, the MPRA species-specific adjusted P filter was adjusted to 0.2 (as opposed to 0.05) to increase the number of hits for enrichment overlap.
LOXL2 and PPP2CA characterization experiments and analyses
LacZ reporter assay using site-specific transgenesis (enSERT)
Tested elements were synthesized (IDT and Twist Bioscience) (hLOXL_long_temp for human LOXL2, and PPP2CA_cons_temp for human PPP2CA;table S4) and amplified in PCRs containing 30 or 100 fmol of template, 25 μl of Q5 NEBNext Master Mix (NEB, M0541), and 0.5 μM forward and reverse primers (LOXL_PCR_F and LOXL_PCR_R for LOXL2 and hPPP2CA_ PCR_F and hPPP2CA_PCR_R for PPP2CA;table S4) cycled with the following conditions: 98°C for 30 s, 20 cycles of 98°C for 10 s, 63°C for 15 s, and 72°C for 30 s, and then 72°C for 2 min. Amplified fragments were purified using 1.5× volume of AMPure XP (Beckman Coulter, A63881) and eluted with water. PCR4-Shh::lacZH11 (Addgene, 139098) was digested by NotI-HF (NEB R3189S) and rSAP (NEB M0371S) overnight at 37°C, purified using 1× volume of AMPure XP, and eluted with water. LOXL2 was assembled using 10 μl of NEBuilder HiFi DNA Assembly Master Mix (NEB, E2621S), 100 ng of linearized vector, and 10 ng of the amplicon in 20 μl total volume for 30 min at 50°C. The PPP2CA fragment was digested by NotI-HF overnight at 37°C, purified using 1.5× volume of AMPure XP, eluted with water, and ligated using 60 ng of linearized vector, 30 ng of the insert, 0.5 μl of T4 DNA ligase (NEB, M0202S) and 1 ml of NEB4 buffer in a 10-ml total volume for 15 min at room temperature.
Transgenic mice were created following the enSERT (enhancer insertion) protocol (22). A mixture of 20 ng/μl Cas9 protein (IDT 1074181), 50 ng/μl single guide RNA (table S4), 25 ng/μl donor plasmid, 10 mM Tris, pH 7.5, and 0.1 mM EDTA was injected into the pronucleus of FVB embryos. The F0 embryos were harvested at embryonic day 11.5 (E11.5) or E13.5 and fixed in PBS supplemented with 2% paraformaldehyde, 0.2% glutaraldehyde, and 0.2% NP-40 at 4°C for 1 hour. After washing with PBS, the embryos were stained in a solution containing 0.5 mg/ml X-gal (Sigma, B4252), 5 mM potassium hexacyanoferrate(II) trihydrate, 5 mM potassium hexacyanoferrate(III), 2 mM MgCl2, and 0.2% Nonidet P-40 in PBS at 37°C overnight. The images of embryos were taken using Leica M165-FC. Positive scoring of an expression pattern required signal in three or more embryos. Transverse sections were also obtained.
All animal procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committees of The Jackson Laboratory.
PPP2CA human versus macaque differential Chip-Seq signal analysis
For Fig. 3D, human and macaque H3K27ac Chip-Seq data from Reilly et al. (25) were used. For every hCONDEL, the chimpanzee panTro4 coordinates were converted to macaque rheMac8 using liftOver. Then, 200 bp of sequence surrounding the hCONDELs was used to count the number of overlapping reads from the H3K27ac samples (8.5 postconception weeks; two human samples and one macaque sample) for both the human and macaque background. DESeq2 was used to normalize and acquire the differential expression (between human and macaque) P value for the PPP2CA-associated hCONDEL.
PPP2CA luciferase experiment
Constructs for the experiment were made using the pGL4.23[luc2/minP] vector backbone and designed from GenScript (table S4). The human sequence tested ranged from the TSS of the alternative isoform of PPP2CA (ENST00000522385) to the TSS of MIR3661 (hg38 coordinates: chr5:134,225,555–134,225,756, 1-based coordinates). The chimpanzee sequence tested was the human sequence with the hCONDEL-deleted bases inserted. Because the PPP2CA-associated hCONDEL was on a potential bidirectional promoter region, both the positive and negative strand contexts were tested (table S4). SK-N-SH cells were grown in 15 ml of EMEM supplemented with 10% FBS on Nunc flasks (ThermoFisher, 156499) to 80 to 90% confluency. Then, 1 × 106 cells were harvested in triplicate by centrifugation at 300g for 5 min at 4°C, washed with 1× PBS, centrifuged again at 300g for 5 min at 4°C, and resuspended in FBS/antibioticfree EMEM on ice. Cells were then mixed with 12.5 mg of empty pGL4.23, pGL4.23 containing the cytomegalovirus (CMV) promoter, pcDNA6.2/C-EmGFP DEST (positive control plasmid containing GFP), or pGL4.23 containing the tested element and then 2.5 μg of pGL4.74. Cells were electroporated in triplicate for each construct using the Neon Transfector (Invitrogen) and Neon Transfection System 100 μl Kit (ThermoFisher, MPK10096) by three pulses of 950 V for 30 msec. Electroporated cells were transferred into a six-well plate containing 2 ml of prewarmed EMEM supplemented with 10% FBS, and grown at 37°C and 5% CO2 for 24 hours. The GFP plasmid was used as a positive electroporation control for microscopic confirmation of transfection efficiency before assay. Cells were then harvested with 200 μl of 0.05% trypsin, and eight technical replicates of 7.5 × 104 cells from each triplicate condition were transferred to 96-well white plates before assay (Greiner, 655075). The Dual-Glo Luciferase assay system (Promega, E2940) was used to measure Firefly and Renilla luciferase activity according to the manufacturer’s protocol, and their luminescence was detected using the BioTek Cytation 5 Plate Reader (AgilentBioTek Instruments) with autogain determined by the CMV-containing wells. The Firefly/Renilla ratio of luminescence normalized to the background ratio from the empty vector condition was used to determine the activity of each replicate.
PPP2CA perturbation and qPCR
PPP2CA nonhomologous end-joining (NHEJ) experiments were performed using Cpf1-editting. PPP2CA_Cpf1_Guide_RNA (Cpf1 guide RNA;table S4) was from IDT. SK-N-SH cells were transfected 24 hours after a medium change at 80% confluency. Three replicates were electroporated for both the experimental condition [electroporation of the complete ribonucleoprotein (RNP)] and the control condition (electroporation of the Cpf1 nuclease without a guide) using 3 × 105 cells for each replicate. Per replicate, 2.25 μl of PPP2CA_Cpf1_Guide_RNA (100 mM) was diluted to 75 μM using nuclease-free water. Then, 2.90 μl of Alt-R PPP2CA_Cpf1_ Guide_RNA (or 2.90 ml of nuclease-free water for the control) was combined with 2.90 μl of Alt-R A.s. Cas12a (Cpf1) Ultra (IDT, 10001273) and incubated at room temperature for 10 to 20 min to form the RNP complex. Next, 3 × 105 cells were washed with PBS and then resuspended in 24.27 μl of Neon Resuspension Buffer R and 0.9 ml of Alt-R Cpf1 Electroporation Enhancer (IDT, 1076300). Next, 4.83 ml of the RNP complex and 25.17 ml of cells in Neon Resuspension Buffer R/Electroporation Enhancer were combined. Electroporation was performed using the Neon 10 μl Transfection Kit (ThermoFisher, MPK1025). One 10-μl tip was used three times to dispense three electroporations (consisting of 1 × 105 cells each) from the same tip into one well of a six-well plate, constituting one replicate. The following electroporation conditions were used: three pulses of 950 V for 30 ms each. A total of 3 × 105 cells from each replicate for RNA or DNA extraction were flash-frozen in liquid nitrogen after a PBS wash after 2 weeks. For routine passaging, cells were split immediately upon all wells reaching confluency and uniformly seeded at 1.5 × 105 cells.
DNA and RNA was extracted using the Qiagen AllPrep DNA/RNA Mini Kit (Qiagen, 80204). Reverse-transcriptase qPCR was performed using Applied Biosystem’s Power SYBR Green RNA to-CT 1-step Kit (ThermoFisher, 4389986) with primers that span exon-exonjunctions of PPP2CA isoforms, Ensembl IDs: ENST00000481195 (canonical) and ENST00000522385 (alternate). Canonical: PPP2CA_Cannonical_qPCR_F and PPP2CA_Cannonical_qPCR_R (table S4). Alternate: PPP2CA_Alternative_qPCR_F and PPP2CA_Alternative_qPCR_R (table S4). TBP was used as a control gene (using TBP_qPCR_F and TBP_qPCR_R;table S4). Applied Biosystems’ QuantStudio5 plate reader (Applied Biosystems, A28135) was used to monitor the qPCR; 100 ng of RNA and 100 nM primers were used in a 20-μl input volume. Values for biological replicates were derived from the average of qPCR technical replicates. Delta delta CT values were generated by first normalizing to the housekeeping gene TBP and then subtracting the control from the cutting condition. For statistical analyses, the delta delta CT values for both the canonical and alternative isoform samples were compared against zero using a two-sided t test in GraphPad Prism.
The following protocol was used to amplify the PPP2CA locus to assess CRISPR editing proportions. Across each replicate, 200 ng of DNA (extracted from the Qiagen AllPrep Kit) was used to amplify the target amplicon using PCR across four separate 50-μl reactions using the NEBNext Ultra II Q5 Master Mix with 0.5 mM PPP2CA_Fwd and PPP2CA_Rev primers (table S4) and the following cycling conditions: 95°C for 20 s, 12 cycles of 95°C for 20 s, 61°C for 20 s, and 72°C for 30 s, and then 72°C for 2 min. For each target reaction, the individual post-PCRs were then pooled together, subject to a 1X AMPure SPRI purification, and eluted in 30 μl of water. Another round of PCR was then performed (same cycling conditions as above, except with eight cycles and 64°C for the annealing temperature) to attach custom p7 and p5 Illumina adapters with unique sample indices. The PCR products for all replicates were then pooled and subject to another 2X SPRI and eluted in 30 μl. Molar concentrations were assessed using Agilent 2200 TapeStation quantifications (using D1000 screentape reagents) and subsequently sequenced using 2 × 150 bp chemistry on an Illumina MiSeq. CRISPResso (v. 2.0.30) was used to derive the allele proportions from the sequencing data (70). Forty to 45% NHEJ proportions were observed for the experimental replicates and none for the control replicates.
LOXL2 genome-editing experiments
For the LOXL2 hCONDEL target, all crRNAs and ssODNs were designed and ordered with IDT (table S4). Cas9 editing was performed on the LOXL2 target, and reagents were also ordered from IDT. LOXL2_Cas9_Guide_RNA (Cas9 crRNA) and LOXL2_ssODN (ssODN) were used the LOXL2 hCONDEL target. All experiments were performed in SK-N-SH. Cells were grown in EMEM supplemented with 10% FBS for SK-N-SH. The HDR protocol used was adapted from IDT.
The following protocol was used for the LOXL2 hCONDEL target. First, 0.9 μl of 200 μM Alt-R CRISPR-Cas9 target-specific crRNA, 0.9 μl of 200 μM Alt-R CRISPR-Cas9 tracrRNA (IDT, 1072533), and 1.5 μl of Nuclease-Free Duplex Buffer (IDT, 1072570) were combined and heated at 95°C for 5 min. The crRNA: tracrRNA solution was then cooled at room temperature. Next, 3 μl of the crRNA:tracrRNA solution was then combined with 2 μl of Alt-R S.p. HiFi Cas9 Nuclease V3 (IDT, 1081059) and incubated at room temperature for 10 to 20 min to form the RNP complex. Then, 1 × 105 cells per electroporation were washed with PBS and resuspended in 7.69 μl of Neon Resuspension Buffer R. Next, 1.61 μl of the RNP complex, 7.69 μl of 100K cells in Neon Resuspension Buffer R, 0.3 μl of 100 μM ssODN, and 0.4 μl of Alt-R Cas9 Electroporation Enhancer (IDT, 1075916) were combined for one electroporation using the Neon transfection system with the 10-μl kit (ThermoFisher, MPK1025). The target underwent two electroporations using set electroporation conditions (three pulses of 950 V for 30 ms each). Both electroporations were transferred to a well containing 0.4 μl of recovery medium [regular medium supplemented with 30 μM HDR enhancer (IDT, 1081072)] in a 24-well plate and grown for 12 to 24 hours. The recovery medium was then changed to regular medium.
LOXL2 HCR-FlowFISH experiments
Two replicates of HCR-FlowFISH were performed on LOXL2-edited SK-N-SH cells (as described in the section “LOXL genome-editing experiments”) on different days following the protocol in (29). Briefly, for replicate 1, 140 million LOXL2-edited cells (70 million for replicate 2) were fixed in 4% formaldehyde in PBST (1× PBS plus 0.1% Tween 20) at room temperature for 1 hour and then washed four times with PBST. Then, cells were resuspended in 70% cold ethanol for 10 min and stored at 4°C for 10 min, resuspended in PBST, and washed with PBST twice. Cells were subsequently prepped for probe hybridization by resuspension in probe hybridization buffer [30% formamide, 5× sodium chloride sodium citrate (SSC), 9 mM citric acid (pH 6.0), 0.1% Tween 20, 50 μg/ml heparin, 1× Denhardt’s solution, 10% lowmolecular-weight dextran sulfate] with 4 nM LOXL2, TBP, and CD44 probes purchased from Molecular Instruments. TBP, a housekeeping gene, was used to control for cell size and permeability. CD44 helped to distinguish the two populations of SK-N-SH (see below) (71). The sample was then incubated overnight at 37°C. Then, the cells were resuspended in Probe Wash [30% formamide, 5× SSC, 9 mM citric acid (pH 6.0), 0.1% Tween 20, 50 μg/ml heparin] and subsequently washed with Probe Wash four times. The cells were then resuspended in 5× SSCT (5× SSC and 0.1% Tween 20), incubated at room temperature for 5 min, and then resuspended in amplification buffer (5× SSC, 0.1% Tween 20, 10% low-molecular-weight dextran sulfate) and incubated at room temperature for 30 min with rotation. Then, 15 pmol of fluorescently labeled hairpin (per initiator and per 5 million cells) was heated for 90 s at 95°C and cooled to room temperature for 15 to 30 min. The hairpins were then added to the sample to achieve a final concentration of 60 nM in amplification buffer. The sample was then incubated in the dark for 3 hours with rotation. A 5× volume of 5× SSCT was then added to the sample mixture, and the sample was pelleted and resuspended in 5× SSCT. The cells were washed with 5× SSCT for six total washes. Finally, the cells were resuspended in PBS for subsequent fluorescence-activated cell sorting (FACS).
FACS revealed two populations of SK-N-SH cells, corresponding to the S and N-type. The top and bottom 10% most expressed cells in the larger population (S-type, which expresses LOXL2) was used for subsequent comparison. A total of 400,000 cells were sorted into both the top 10% and bottom 10% expression bins for the first replicate and 750,000 cells into both bins for the second replicate. DNA was extracted by suspension in 100 μl (per 1 million cells) of 1X Chip Lysis Buffer (1% SDS, 10 mM EDTA, and 50 mM Tris-HCL, Ph 8.1) and incubated at 65°C for 3 hours, followed by the addition of 2 μl of RNase A (per 1 million cells) and incubation at 37°C. Next, 10 ml of Proteinase K (per 1 million cells) was added and incubated at 37°C for 2 hours, followed by 95°C for 20 min. The resulting sample was then subject to a 1X AMPure SPRI followed by 5× 70% ethanol washes and elution in water. If sample purity was not adequate, the AMPure SPRI was redone. For the final water elution in AMPure SPRI, elution times were extended (as long as overnight) and samples were heated at high temperature (65°C or 37°C, for maximally ~1 hour) to ensure greater elution efficiency.
After DNA extraction, for the first replicate, 550 ng (380 ng was used for the second replicate) was then directly used to amplify the target amplicon using PCR across four separate 50-μl reactions using the NEBNext Ultra II Q5 Master Mix (NEB, M0544L) with 0.5 μM LOXL2_Fwd and LOXL2_Rev primers and the following cycling conditions: 95°C for 20 s, 15 cycles of 95°C for 20 s, 65°C for 20 s, 72°C for 30 s, and then 72°C for 2 min (table S4). For each replicate, the individual post-PCRs were then pooled together, subject to a 1X AMPure SPRI (Beckman Coulter, A63881) purification, and eluted in 30 μl of water. Another round of PCR was then performed (same cycling conditions as above, except with eight cycles and 64°C for the annealing temperature) to attach custom p7 and p5 Illumina adapters with unique sample indices (table S4). The PCR products were then subject to another 2X SPRI and eluted in 30 μl. The resulting purified PCR products across all targets were then molar pooled from Agilent 2200 TapeStation quantifications (using D1000 screentape reagents) and subsequently sequenced using 2 × 150 bp chemistry on an Illumina MiSeq. CRISPResso (v. 2.0.30) was used to derive the allele proportions from the sequencing data (70). The enrichment FC was calculated as follows: (number of human reads in top 10% bin/number of human reads in low 10% bin)/(number of chimpanzee reads in top 10% bin/number of chimpanzee reads in low 10% bin). Significance was assessed by a Fisher’s t test.
LOXL2 single-cell experiment
SK-N-SH cells were first edited as described in the “LOXL genome-editing experiments” section. These cells were processed for single-cell RNA sequencing using the 10X Genomics Chromium 3′ v. 3.1 kit following the manufacturer’s instructions. For the recommended protocol, 30 μl of the cDNA was leftover; 5 μl of that cDNA was PCR amplified to enrich for the LOXL2-edited locus in a 50-μl PCR containing 25 μl of NEBNext Ultra II Q5 Master Mix, 1.0 μM (SI)-PCR primer (10x Genomics) and 10X_LOXL2_Rev (table S4) under the following conditions: 95°C for 20 s, 15 cycles of 95°C for 20 s, 62°C for 20 s, 72°C for 30 s, and then 72°Cfor2min. 0.8XSPRIselect (BeckmanCoulter, B23317) purification was then performed, and another round of PCR (as above, except with six cycles and 64°C annealing temperature) was performed using a set of0.5 μM custom Illumina p5 index primers and a 0.5 μM (SI)-PCR (table S4). Another 0.8X SPRIselect purification was performed afterward. Samples were then pooled according to molar estimates from the Agilent 2200 TapeStation (using the D1000 screentape reagents (Agilent, 5067–5585) and then sequenced on a NextSeq 550. Sequencing resulting from the LOXL2-edited locus linked LOXL2 edits to specific cell barcodes and was processed using the GoT computational pipeline (v. 2.1) (72). Seurat (v. 3.2.3) (73) was used to process the single-cell RNA dataset. Similar to our HCR-FlowFish experiment, there were two populations of SK-N-SH cells and S-type cells predominantly expressing LOXL2 were found. The cells in this group were used for subsequent single-cell analyses. DESeq2 was used to call genes differentially expressed between cells containing the human base lines and cells harboring the introduced chimpanzee base. goseq (v. 1.38.0) (74) was used to derive enriched gene ontology terms using the analysis results from DESeq2 (which were derived on only SK-N-SH (S-type) expressed genes). Genes with a BH adjusted P < 0.1 were classified as differentially expressed.
Supplementary Material
ACKNOWLEDGMENTS
We thank L. Chylek, C. Edwards, S. Gosai, and P. Pillai for thoughtful conversations and help with editing the manuscript; The Jackson Laboratory Genetic Engineering Technologies and Microscopy Core for experimental support.
Funding:
This work was supported by the ENCODE Functional Characterization Center (grant UM1 HG009435 to P.C.S., R.T., and S.K.R.); Broad SPARC (P.C.S.); Howard Hughes Medical Institute (P.C.S.); and the National Institutes of Health (grant R00HG010669 to S.K.R., grants R00HG008179 and R35HG011329 to R.T., grant RF1AG065926 to M.F.G. and K.J.B., grant R01MH125246 to M.F.G. and K.J.B., grant R56MH125237 to M.F.G. and K.J.B., grant 5T32MH014276-45 to M.F.G., and grant R01HG008742 to E.K.); the Liweibo PhD scholarship from the University of Massachusetts Chan Medical School (X.L.); and the Distinguished professor award from the Swedish Medical Research Council (K.L.T.).
Zoonomia Consortium
Gregory Andrews1, Joel C. Armstrong2, Matteo Bianchi3, Bruce W. Birren4, Kevin R. Bredemeyer5, Ana M. Breit6, Matthew J. Christmas3, Hiram Clawson2, Joana Damas7, Federica Di Palma8,9, Mark Diekhans2, Michael X. Dong3, Eduardo Eizirik10, Kaili Fan1, Cornelia Fanter11, Nicole M. Foley5, Karin Forsberg-Nilsson12,13, Carlos J. Garcia14, John Gatesy15, Steven Gazal16, Diane P. Genereux4, Linda Goodman17, Jenna Grimshaw14, Michaela K. Halsey14, Andrew J. Harris5, Glenn Hickey18, Michael Hiller19,20,21, Allyson G. Hindle11, Robert M. Hubley22, Graham M. Hughes23, Jeremy Johnson4, David Juan24, Irene M. Kaplow25,26, Elinor K. Karlsson1,4,27, Kathleen C. Keough17,28,29, Bogdan Kirilenko19,20,21, Klaus-Peter Koepfli30,31,32, Jennifer M. Korstian14, Amanda Kowalczyk25,26, Sergey V. Kozyrev3, Alyssa J. Lawler4,26,33, Colleen Lawless23, Thomas Lehmann34, Danielle L. Levesque6, Harris A. Lewin7,35,36, Xue Li1,4,37, Abigail Lind28,29, Kerstin Lindblad-Toh3,4,Ava Mackay-Smith38, Voichita D. Marinescu3, Tomas Marques-Bonet39,40,41,42, Victor C. Mason43, Jennifer R. S. Meadows3, Wynn K. Meyer44, Jill E. Moore1, Lucas R. Moreira1,4, Diana D. Moreno-Santillan14, Kathleen M. Morrill1,4,37, Gerard Muntané24, William J. Murphy5, Arcadi Navarro39,41,45,46, Martin Nweeia47,48,49,50, Sylvia Ortmann51, Austin Osmanski14, Benedict Paten2, Nicole S. Paulat14, Andreas R. Pfenning25,26, BaDoi N. Phan25,26,52, Katherine S.Pollard28,29,53, Henry E. Pratt1, David A. Ray14, Steven K. Reilly38, Jeb R. Rosen22, Irina Ruf54, Louise Ryan23, Oliver A. Ryder55,56, Pardis C.Sabeti4,57,58, Daniel E. Schäffer25, Aitor Serres24, Beth Shapiro59,60, Arian F. A. Smit22, Mark Springer61, Chaitanya Srinivasan25, Cynthia Steiner55, Jessica M. Storer22, Kevin A. M. Sullivan14, Patrick F. Sullivan62,63, Elisabeth Sundström3, Megan A. Supple59, Ross Swofford4, Joy-El Talbot64, Emma Teeling23, Jason Turner-Maier4, Alejandro Valenzuela24, Franziska Wagner65, Ola Wallerman3, Chao Wang3, Juehan Wang16, Zhiping Weng1, Aryn P. Wilder55, Morgan E. Wirthlin25,26,66, James R. Xue4,57, Xiaomeng Zhang4,25,26
1Program in Bioinformatics and Integrative Biology, UMass Chan Medical School, Worcester, MA 01605, USA. 2Genomics Institute, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 3Department of Medical Biochemistry and Microbiology, Science for Life Laboratory, Uppsala University, Uppsala 751 32, Sweden.4Broad Institute of MIT and Harvard, Cambridge, MA 02139, USA.5Veterinary Integrative Biosciences, Texas A&M University, College Station, TX 77843, USA. 6School of Biology and Ecology, University of Maine, Orono, ME 04469, USA. 7The Genome Center, University of California Davis, Davis, CA 95616, USA. 8Genome British Columbia, Vancouver, BC, Canada. 9School of Biological Sciences, University of East Anglia, Norwich, UK. 10School of Health and Life Sciences, Pontifical Catholic University of Rio Grande do Sul, Porto Alegre 90619–900, Brazil. 11School of Life Sciences, University of Nevada Las Vegas, Las Vegas, NV 89154, USA. 12Biodiscovery Institute, University of Nottingham, Nottingham, UK. 13Department of Immunology, Genetics and Pathology, Science for Life Laboratory, Uppsala University, Uppsala 751 85, Sweden. 14Department of Biological Sciences, Texas Tech University, Lubbock, TX 79409, USA. 15Division of Vertebrate Zoology, American Museum of Natural History, New York, NY 10024, USA. 16Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA. 17Fauna Bio Incorporated, Emeryville, CA 94608, USA.18Baskin School of Engineering, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 19Faculty of Biosciences, Goethe-University, 60438 Frankfurt, Germany. 20LOEWE Centre for Translational Biodiversity Genomics, 60325 Frankfurt, Germany.21Senckenberg Research Institute, 60325 Frankfurt, Germany.22Institute for Systems Biology, Seattle, WA 98109, USA. 23School of Biology and Environmental Science, University College Dublin, Belfield, Dublin 4, Ireland. 24Department of Experimental and Health Sciences, Institute of Evolutionary Biology (UPF-CSIC), Universitat Pompeu Fabra, Barcelona 08003, Spain. 25Department of Computational Biology, School of Computer Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 26Neuroscience Institute, Carnegie Mellon University, Pittsburgh, PA 15213, USA.27Program in Molecular Medicine, UMass Chan Medical School, Worcester, MA 01605, USA. 28Department of Epidemiology & Biostatistics, University of California San Francisco, San Francisco, CA 94158, USA. 29Gladstone Institutes, San Francisco, CA 94158, USA. 30Center for Species Survival, Smithsonian’s National Zoo and Conservation Biology Institute, Washington, DC 20008, USA.31Computer Technologies Laboratory, ITMO University, St. Petersburg 197101, Russia. 32Smithsonian-Mason School of Conservation, George Mason University, Front Royal, VA 22630, USA. 33Department of Biological Sciences, Mellon College of Science, Carnegie Mellon University, Pittsburgh, PA 15213, USA. 34Senckenberg Research Institute and Natural History Museum Frankfurt, 60325 Frankfurt am Main, Germany. 35Department of Evolution and Ecology, University of California Davis, Davis, CA 95616, USA.36John Muir Institute for the Environment, University of California Davis, Davis, CA 95616, USA. 37Morningside Graduate School of Biomedical Sciences, UMass Chan Medical School, Worcester, MA 01605, USA. 38Department of Genetics, Yale School of Medicine, New Haven, CT 06510, USA. 39Catalan Institution of Research and Advanced Studies (ICREA), Barcelona 08010, Spain. 40CNAG-CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona 08036, Spain. 41Department of Medicine and Life Sciences, Institute of Evolutionary Biology (UPF-CSIC), Universitat Pompeu Fabra, Barcelona 08003, Spain.42Institut Català de Paleontologia Miquel Crusafont, Universitat Autònoma de Barcelona, 08193 Cerdanyola del Vallès, Barcelona, Spain. 43Institute of Cell Biology, University of Bern, 3012 Bern, Switzerland. 44Department of Biological Sciences, Lehigh University, Bethlehem, PA 18015, USA. 45BarcelonaBeta Brain Research Center, Pasqual Maragall Foundation, Barcelona 08005, Spain.46CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), Barcelona 08003, Spain.47Department of Comprehensive Care, School of Dental Medicine, Case Western Reserve University, Cleveland, OH 44106, USA.48Department of Vertebrate Zoology, Canadian Museum of Nature, Ottawa, ON K2P 2R1, Canada. 49Department of Vertebrate Zoology,Smithsonian Institution, Washington, DC 20002, USA. 50Narwhal Genome Initiative, Department of Restorative Dentistry and Biomaterials Sciences, Harvard School of Dental Medicine, Boston, MA 02115, USA. 51Department of Evolutionary Ecology, Leibniz Institute for Zoo and Wildlife Research, 10315 Berlin, Germany.52Medical Scientist Training Program, University of Pittsburgh School of Medicine, Pittsburgh, PA 15261, USA. 53Chan Zuckerberg Biohub, San Francisco, CA 94158, USA. 54Division of Messel Research and Mammalogy, Senckenberg Research Institute and Natural History Museum Frankfurt, 60325 Frankfurt am Main, Germany. 55Conservation Genetics, San Diego Zoo Wildlife Alliance, Escondido, CA 92027, USA. 56Department of Evolution, Behavior and Ecology, School of Biological Sciences, University of California San Diego, La Jolla, CA 92039, USA. 57Department of Organismic andEvolutionary Biology, Harvard University, Cambridge, MA 02138, USA.58Howard Hughes Medical Institute, Harvard University, Cambridge, MA 02138, USA. 59Department of Ecology and Evolutionary Biology, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 60Howard Hughes Medical Institute, University of California Santa Cruz, Santa Cruz, CA 95064, USA. 61Department of Evolution, Ecology and Organismal Biology, University of California Riverside, Riverside, CA 92521, USA. 62Department of Genetics, University of North Carolina Medical School, Chapel Hill, NC 27599, USA. 63Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden. 64Iris Data Solutions, LLC, Orono, ME 04473, USA. 65Museum of Zoology, Senckenberg Natural History Collections Dresden, 01109 Dresden, Germany. 66Allen Institute for Brain Science, Seattle, WA 98109, USA.
Footnotes
Competing interests: P.C.S. is a cofounder of and consultant to Sherlock Biosciences and Delve Bio. She is also a board member of Danaher Corporation. She is a shareholder in all three companies. The remaining authors declare no competing interests.
SUPPLEMENTARY MATERIALS
science.org/doi/10.1126/science.abn2253 Figs. S1 to S10
REFERENCES AND NOTES
- 1.Chimpanzee Sequencing and Analysis Consortium, Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437, 69–87 (2005). doi: 10.1038/nature04072; [DOI] [PubMed] [Google Scholar]
- 2.ENCODE Project Consortium, An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). doi: 10.1038/nature11247; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dennis MY, Eichler EE, Human adaptation and evolution by segmental duplication. Curr. Opin. Genet. Dev 41, 44–52 (2016). doi: 10.1016/j.gde.2016.08.001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prabhakar S et al. , Human-specific gain of function in a developmental enhancer. Science 321, 1346–1350 (2008). doi: 10.1126/science.1159974; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLean CY et al. , Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 471, 216–219 (2011). doi: 10.1038/nature09774; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siepel A et al. , Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15, 1034–1050 (2005). doi: 10.1101/gr.3715005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kronenberg ZN et al. , High-resolution comparative analysis of great ape genomes. Science 360, eaar6343 (2018).doi: 10.1126/science.aar6343; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uebbing S et al. , Massively parallel discovery of human-specific substitutions that alter enhancer activity Proc. Natl. Acad. Sci. U.S.A. 118, e2007049118 (2021). doi: 10.1073/pnas.2007049118; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Girskis KM et al. , Rewiring of human neurodevelopmental gene regulatory programs by human accelerated regions. Neuron 109, 3239–3251.e7 (2021). doi: 10.1016/j.neuron.2021.08.005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lynch VJ et al. , Ancient transposable elements transformed the uterine regulatory landscape and transcriptome during the evolution of mammalian pregnancy. Cell Rep 10, 551–561 (2015). doi: 10.1016/j.celrep.2014.12.052; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Derouet D et al. , Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor Proc. Natl. Acad. Sci. U.S.A. 101, 4827–4832 (2004). doi: 10.1073/pnas.0306178101; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore JE et al. , Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). doi: 10.1038/s41586-020-2493-4; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fong SL, Capra JA, Modeling the evolutionary architectures of transcribed human enhancer sequences reveals distinct origins, functions, and associations with human trait variation. Mol. Biol. Evol 38, 3681–3696 (2021). doi: 10.1093/molbev/msab138; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tewhey R et al. , Direct identification of hundreds of expression-modulating variants using a multiplexed reporter assay. Cell 172, 1132–1134 (2018). doi: 10.1016/j.cell.2018.02.021; [DOI] [PubMed] [Google Scholar]
- 15.Sullivan PF et al. , Leveraging base pair mammalian constraint to understand genetic variation and human disease. Science 380, eabn2937 (2023). doi: 10.1123/science.abn2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stevens SJC et al. , Truncating de novo mutations in the Krüppel-type zinc-finger gene ZNF148 in patients with corpus callosum defects, developmental delay, short stature, and dysmorphisms. Genome Med 8, 131 (2016). doi: 10.1186/s13073-016-0386-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu JQ et al. , Transcriptome sequencing revealed significant alteration of cortical promoter usage and splicing in schizophrenia. PLOS ONE 7, e36351 (2012). doi: 10.1371/journal.pone.0036351; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu H et al. , Retinoic acid-induced upregulation of miR-219 promotes the differentiation of embryonic stem cells into neural cells. Cell Death Dis 8, e2953 (2017). doi: 10.1038/cddis.2017.336; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dottori M, Gross MK, Labosky P, Goulding M, The winged-helix transcription factor Foxd3 suppresses interneuron differentiation and promotes neural crest cell fate. Development 128, 4127–4138 (2001). doi: 10.1242/dev.128.21.4127; [DOI] [PubMed] [Google Scholar]
- 20.Parras A et al. , Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Nature 560, 441–446 (2018). doi: 10.1038/s41586-018-0423-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khrameeva E et al. , Single-cell-resolution transcriptome map of human, chimpanzee, bonobo, and macaque brains. Genome Res 30, 776–789 (2020). doi: 10.1101/gr.256958.119; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kvon EZ et al. , Comprehensive in vivo interrogation reveals phenotypic impact of human enhancer variants. Cell 180, 1262–1271.e15 (2020). doi: 10.1016/j.cell.2020.02.031; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reynhout S et al. , De novo mutations affecting the catalytic Ca subunit of PP2A, PPP2CA, cause syndromic intellectual disability resembling other PP2A-related neurodevelopmental disorders. Am. J. Hum. Genet 104, 139–156 (2019).doi: 10.1016/j.ajhg.2018.12.002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reijnders MRF et al. , Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat. Commun 8, 1052 (2017). doi: 10.1038/s41467-017-00933-6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reilly SK et al. , Evolutionary genomics. Evolutionary changes in promoter and enhancer activity during human corticogenesis. Science 347, 1155–1159 (2015). doi: 10.1126/science.1260943; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Banzhaf-Strathmann J et al. , MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J 33, 1667–1680 (2014). doi: 10.15252/embj.201387576; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puente A et al. , LOXL2-A new target in antifibrogenic therapy? Int. J. Mol. Sci 20, 1634 (2019). doi: 10.3390/ijms20071634; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolós V et al. , The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci 116, 499–511 (2003). doi: 10.1242/jcs.00224; [DOI] [PubMed] [Google Scholar]
- 29.Reilly SK et al. , Direct characterization of cis-regulatory elements and functional dissection of complex genetic associations using HCR-FlowFISH. Nat. Genet 53, 1166–1176 (2021). doi: 10.1038/s41588-021-00900-4; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iturbide A et al. , LOXL2 oxidizes methylated TAF10 and controls TFIID-dependent genes during neural progenitor differentiation. Mol. Cell 58, 755–766 (2015). doi: 10.1016/j.molcel.2015.04.012; [DOI] [PubMed] [Google Scholar]
- 31.Hollosi P, Yakushiji JK, Fong KSK, Csiszar K, Fong SFT, Lysyl oxidase-like 2 promotes migration in noninvasive breast cancer cells but not in normal breast epithelial cells. Int. J. Cancer 125, 318–327 (2009). doi: 10.1002/ijc.24308; [DOI] [PubMed] [Google Scholar]
- 32.Glasser MF, Goyal MS, Preuss TM, Raichle ME, Van Essen DC, Trends and properties of human cerebral cortex: Correlations with cortical myelin content. Neuroimage 93, 165–175 (2014). doi: 10.1016/j.neuroimage.2013.03.060; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen P, Cescon M, Megighian A, Bonaldo P, Collagen VI regulates peripheral nerve myelination and function. FASEB J 28, 1145–1156 (2014). doi: 10.1096/fj.13-239533; [DOI] [PubMed] [Google Scholar]
- 34.Navas-Pérez E et al. , Characterization of an eutherian gene cluster generated after transposon domestication identifies Bex3 as relevant for advanced neurological functions. Genome Biol 21, 267 (2020). doi: 10.1186/s13059-020-02172-3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lynch VJ, Leclerc RD, May G, Wagner GP, Transposonmediated rewiring of gene regulatory networks contributed to the evolution of pregnancy in mammals. Nat. Genet 43, 1154–1159 (2011). doi: 10.1038/ng.917; [DOI] [PubMed] [Google Scholar]
- 36.Blanchette M et al. , Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14, 708–715 (2004). doi: 10.1101/gr.1933104; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harris RS, thesis, The Pennsylvania State University, Ann Arbor, MI (2007). [Google Scholar]
- 38.Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D, Evolution’s cauldron: Duplication, deletion, and rearrangement in the mouse and human genomes. Proc. Natl. Acad. Sci. U.S.A 100, 11484–11489 (2003). doi: 10.1073/pnas.1932072100; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Landan G, Graur D, Characterization of pairwise and multiple sequence alignment errors. Gene 441, 141–147 (2009).doi: 10.1016/j.gene.2008.05.016; [DOI] [PubMed] [Google Scholar]
- 40.Mallick S et al. , The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 538, 201–206 (2016). doi: 10.1038/nature18964; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hinrichs AS et al. , The UCSC Genome Browser Database: Update 2006. Nucleic Acids Res 34, D590–D598 (2006). doi: 10.1093/nar/gkj144; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li H, FermiKit: Assembly-based variant calling for Illumina resequencing data. Bioinformatics 31, 3694–3696 (2015). doi: 10.1093/bioinformatics/btv440; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prado-Martinez J et al. , Great ape genetic diversity and population history. Nature 499, 471–475 (2013). doi: 10.1038/nature12228; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whalen S et al. , Machine-learning dissection of Human Accelerated Regions in primate neurodevelopment. bioRxiv (2022), p. 256313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gallego Romero I et al. , A panel of induced pluripotent stem cells from chimpanzees: A resource for comparative functional genomics. eLife 4, e07103 (2015). doi: 10.7554/eLife.07103; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoffman GE et al. , Transcriptional signatures of schizophrenia in hiPSC-derived NPCs and neurons are concordant with post-mortem adult brains. Nat. Commun 8, 2225 (2017). doi: 10.1038/s41467-017-02330-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). doi: 10.1186/s13059-014-0550-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Urbut SM, Wang G, Carbonetto P, Stephens M, Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nat. Genet 51, 187–195 (2019). doi: 10.1038/s41588-018-0268-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Griesemer D et al. , Genome-wide functional screen of 3’UTR variants uncovers causal variants for human disease and evolution. Cell 184, 5247–5260.e19 (2021). doi: 10.1016/j.cell.2021.08.025; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang W, Stephens M, Empirical Bayes matrix factorization. arXiv:1802.06931 [stat.ME] (2018). [PMC free article] [PubMed] [Google Scholar]
- 51.Bovy J, Hogg DW, Roweis ST, Extreme deconvolution: Inferring complete distribution functions from noisy, heterogeneous and incomplete observations. arXiv:0905.2979 [stat.ME] (2009). [Google Scholar]
- 52.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ, Basic local alignment search tool. J. Mol. Biol 215, 403–410 (1990). doi: 10.1016/S0022-2836(05)80360-2; [DOI] [PubMed] [Google Scholar]
- 53.Jurka J, Repbase update: A database and an electronic journal of repetitive elements. Trends Genet 16, 418–420 (2000). doi: 10.1016/S0168-9525(00)02093-X; [DOI] [PubMed] [Google Scholar]
- 54.McLean CY et al. , GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501 (2010). doi: 10.1038/nbt.1630; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Supek F, Bošnjak M, Škunca N, Šmuc T, REVIGO summarizes and visualizes long lists of gene ontology terms. PLOS ONE 6, e21800 (2011). doi: 10.1371/journal.pone.0021800; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fornes O et al. , JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48 (D1), D87–D92 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grant CE, Bailey TL, Noble WS, FIMO: Scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011). doi: 10.1093/bioinformatics/btr064; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vierstra J et al. , Global reference mapping of human transcription factor footprints. Nature 583, 729–736 (2020). doi: 10.1038/s41586-020-2528-x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robinson MD, McCarthy DJ, Smyth GK, edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). doi: 10.1093/bioinformatics/btp616; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y, Ge X, Peng F, Li W, Li JJ, Exaggerated false positives by popular differential expression methods when analyzing human population samples. Genome Biol 23, 79 (2022).doi: 10.1186/s13059-022-02648-4; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Savage JE et al. , Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet 50, 912–919 (2018).doi: 10.1038/s41588-018-0152-6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wray NR et al. , Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet 50, 668–681 (2018). doi: 10.1038/s41588-018-0090-3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mullins N et al. , Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet 53, 817–829 (2021).doi: 10.1038/s41588-021-00857-4; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ruderfer DM et al. , Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell 173, 1705–1715.e16 (2018). doi: 10.1016/j.cell.2018.05.046; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Leeuw CA, Mooij JM, Heskes T, Posthuma D, MAGMA: Generalized gene-set analysis of GWAS data. PLOS Comput. Biol 11, e1004219 (2015). doi: 10.1371/journal.pcbi.1004219; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sey NYA et al. , A computational tool (H-MAGMA) for improved prediction of brain-disorder risk genes by incorporating brain chromatin interaction profiles. Nat. Neurosci 23, 583–593 (2020). doi: 10.1038/s41593-020-0603-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium, Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci 18, 199–209 (2015). doi: 10.1038/nn.3922; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pardiñas AF et al. , Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet 50, 381–389 (2018). doi: 10.1038/s41588-018-0059-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Abdellaoui A, Dolan CV, Verweij KJH, Nivard MG, Gene-environment correlations across geographic regions affect genome-wide association studies. Nat. Genet 54, 1345–1354 (2022). doi: 10.1038/s41588-022-01158-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clement K et al. , CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol 37, 224–226 (2019). doi: 10.1038/s41587-019-0032-3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walton JD et al. , Characteristics of stem cells from human neuroblastoma cell lines and in tumors. Neoplasia 6, 838–845 (2004). doi: 10.1593/neo.04310; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nam AS et al. , Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature 571, 355–360 (2019). doi: 10.1038/s41586-019-1367-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stuart T et al. , Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21 (2019). doi: 10.1016/j.cell.2019.05.031; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Young MD, Wakefield MJ, Smyth GK, Oshlack A, Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol 11, R14 (2010). doi: 10.1186/gb-2010-11-2-r14; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xue JR et al. , Associated data and scripts for: The functional and evolutionary impacts of human-specific deletions in conserved elements, Zenodo (2023). doi: 10.5281/zenodo.7829717 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.