

Abstract
Purpose of Review
The objective of this review is to provide up-to-date and comprehensive discussion of tissue-specific fructose metabolism in the context of diabetes, dyslipidemia, and nonalcoholic fatty liver disease (NAFLD).
Recent Findings
Increased intake of dietary fructose is a risk factor for a myriad of metabolic complications. Tissue-specific fructose metabolism has not been well delineated in terms of its contribution to detrimental health effects associated with fructose intake. Since inhibitors targeting fructose metabolism are being developed for the management of NAFLD and diabetes, it is essential to recognize how inability of one tissue to metabolize fructose may affect metabolism in the other tissues. The primary sites of fructose metabolism are the liver, intestine, and kidney. Skeletal muscle and adipose tissue can also metabolize a large portion of fructose load, especially in the setting of ketohexokinase deficiency, the rate-limiting enzyme of fructose metabolism. Fructose can also be sensed by the pancreas and the brain, where it can influence essential functions involved in energy homeostasis. Lastly, fructose is metabolized by the testes, red blood cells, and lens of the eye where it may contribute to infertility, advanced glycation end products, and cataracts, respectively.
Summary
An increase in sugar intake, particularly fructose, has been associated with the development of obesity and its complications. Inhibition of fructose utilization in tissues primary responsible for its metabolism alters consumption in other tissues, which have not been traditionally regarded as important depots of fructose metabolism.
Keywords: Sugar, Fructose, Insulin resistance, Diabetes, NAFLD, Dyslipidemia
Introduction
There is a worldwide epidemic of obesity, insulin resistance, and type 2 diabetes mellitus (T2DM). A major risk factor for development of obesity and its complications was thought to be increased intake of calorie-dense high-fat diet. Emphasis on detrimental effects of dietary fat over the last 30 years has resulted in decreased consumption of saturated fat [1]. Unfortunately, this success did not translate into a significant decrease in the prevalence of obesity.
A recent paradigm shift has focused more attention on harmful effects of sugar as a driver of the obesity epidemic [2]. Dietary sugars are predominantly composed of fructose and glucose monosaccharides. Fructose intake is more strongly associated with obesity and particularly with development of its comorbidities, such as dyslipidemia, insulin resistance, and fatty liver disease. These effects of fructose are mediated via a complex interplay of several metabolic pathways, at least some of which appear to be independent of excess caloric intake [3, 4]. Increased awareness about negative health effects of dietary sugar prompted the FDA to propose new food labels, which emphasize disclosing the amount of added sugar in our food [5]. Furthermore, several cities in the USA [6, 7] and countries around the globe have implemented a tax on sugar-sweetened beverages as a way to reduce their intake and combat the obesity epidemic [8–10]. The general public also participates in this paradigm shift as low-sugar, high-fat diets, such as Atkins’ New Diet Revolution and South Beach Diet, are becoming increasingly more popular [11].
While the evidence linking sugar intake with obesity and its complications is strong, the mechanism how these effects are mediated is not clearly defined. This has led to unprecedented interest in understanding fructose metabolism, in an attempt to identify therapeutic targets to decrease harmful health effects associated with fructose intake.
In this review, we discuss how tissue-specific fructose metabolism contributes to the development of obesity and its complications, focusing primarily on diabetes, dyslipidemia, and nonalcoholic fatty liver disease (NAFLD). First, we review human studies linking dietary fructose intake with the development of insulin resistance and T2DM. Next, we describe organ-specific fructose metabolism in the liver, intestine, and kidney, the primary sites of fructose metabolism. Additionally, we discuss fructose metabolism in the muscle, adipose tissue, the pancreas, and the brain, which have been traditionally overlooked as important sites of fructose metabolism.
Clinical Studies Linking Fructose Consumption to Obesity and Diabetes
Increased intake of dietary sugar and fructose in particular has been associated with a myriad of metabolic complications including insulin resistance and T2DM [3]. We recently reviewed studies where hypercaloric fructose supplementation leads to the development of hepatic and systemic insulin resistance [12]. In short-term studies providing up to 25% of additional energy needs from fructose-sweetened drinks, fructose intake is associated with the development of hepatic insulin resistance, primarily manifesting as increased hepatic glucose production [13–16]. Much lower amounts of fructose providing 10–15% of additional calories, but over a longer time span, likewise result in the development of both hepatic and whole-body insulin resistance [17–19]. Studies utilizing hypercaloric fructose feeding have been criticized as increased energy intake alone can induce insulin resistance.
Interestingly, even energy neutral intake of fructose-sweetened drinks can lead to the development of insulin resistance and diabetes [20–23], suggesting that fructose intake may be an independent risk factor for the development of hepatic insulin resistance. Indeed, a meta-analysis of eight studies reporting outcomes in 310,819 participants found that individuals in the highest quintile of sugar-sweetened beverage (SSB) intake (1–2 servings/day) had a 26% greater risk of developing T2DM than those in the lowest quintile of SSB intake (none or < 1 serving/month) [24]. This increase in T2DM risk remains even after adjusting for adiposity associated with SSB intake. Another meta-analysis of 17 cohorts including 38,253 cases found that consuming one SSB serving per day was associated with 18% greater incidence of T2DM before adjustment for adiposity, whereas the risk remained 13% higher, after adjusting for adiposity [4].
Association with increased fructose intake and insulin resistance is not only observed in studies utilizing sugar- or fructose-sweetened drinks, but also with consumption of solid diets high in sugar. For example, in a cross-over trial, 24 adult men and women consumed 5%, 18%, or 33% of total daily calories as sucrose or the same amount of wheat starch for 6 weeks. Mean glucose levels, and insulin responses to a sucrose load, were higher for men and women consuming 18% and 33% sucrose diets versus the 5% sucrose diet [22]. Similarly, replacing 30% of calories from wheat with sucrose for 6 weeks in 19 glucose-intolerant adult subjects increased fasting serum insulin, glucose, and insulin response to a sucrose load [23]. In another cross-over study, 12 men with hyperinsulinemia and 12 healthy age- and BMI-matched men were provided standardized equicaloric meals that contained either 0, 7.5, or 15% of the energy requirement as fructose in solid food for 5 weeks. The subjects consuming 15% fructose diet had increased glucose and insulin responses to a 3-h oral sucrose tolerance test when compared with the 0% fructose diet group [21]. Moreover, consumption of an isocaloric diet containing high fructose (25% energy requirement) for 9 days increased hepatic glucose production during a euglycemic-hyperinsulinemic clamp in eight healthy men when compared to their baseline on low-fructose diet (starch provided in place of fructose) [20].
For the sake of completeness, it should be noted that not all studies found a positive association between increased intake of SSB with increased weight gain and T2DM [25]. However, a comprehensive analysis of systematic reviews found that most studies (83%) reporting that scientific evidence is insufficient to support a positive association with SSB consumption and weight gain had some financial conflict of interest with the food industry [26]. Thus, the preponderance of evidence suggests that increased intake of sugar, particularly fructose, is associated with increased weight gain and risk of T2DM.
Fructose Transport and Metabolism
Fructose metabolism in many ways overlaps with that of glucose; however, there are important differences (Fig. 1). Fructose is transported into tissues via facilitated diffusion, mainly through glucose transporters 2 and 5 (GLUT2 and GLUT5) [27]. GLUT5 is the sole transporter specific for fructose with limited ability to transport glucose or galactose. GLUT2, on the other hand, is an efficient carrier for glucose and glucosamine. Other transporters such as GLUT7, GLUT8, GLUT9a/b, GLUT11, and GLUT12, as well as the sodium- and glucose-linked transporters SGLT4 and SGLT5, possess varying degrees of fructose selectivity and capacity [28]. Tissue-specific distribution of these transporters determines their availability to transport fructose. The first step of intracellular fructose metabolism is mediated by ketohexokinase (KHK), also known as fructokinase, which converts fructose to fructose 1-phosphate [29]. KHK is the rate-limiting enzyme in fructose metabolism. There are two isoforms of KHK produced by alternative splicing of exon 3. KHK-C is the main isoform responsible for fructose metabolism and is highly expressed in the intestine, liver, and kidney [30]. Tissue-specific distribution of KHK-C is thought to be the reason why these organs are considered major sites of fructose metabolism. Conversely, KHK-A isoform is ubiquitously expressed, although it has much lower affinity for fructose (Km 7 mM) than KHK-C (Km 0.8 mM) [31, 32]. The second enzyme of fructose metabolism, fructose-bisphosphate aldolase, or simply aldolase (Aldo), converts fructose 1-phosphate to dihydroxyacetone phosphate (DHAP) and glyceraldehyde (GA). There are three aldolase isoforms A, B, and C, which have unique tissue distribution. AldoA is expressed in muscle and erythrocytes, AldoB is mainly expressed in the liver, whereas AldoC is expressed in the brain [33]. Aldolase is not specific for fructose metabolism, as it also catalyzes a step in glycolysis to convert fructose-1,6 bisphosphate into DHAP and glyceraldehyde 3-phosphate (GA3P). Whereas DHAP is a common metabolite between fructolysis and glycolysis, fructose-derived GA is a more specific product of fructolysis and is the center of fructose metabolic crossroads [34]. Triokinase/FMN cyclase (TKFC) converts GA to GA3P and appears to be specifically regulated by dietary fructose. Its activity increases with fructose (but not glucose) refeeding, decreases with fasting, and recovers to baseline following 20 h of refeeding [36]. GA can also be catabolized by aldehyde dehydrogenase (ALDH) to eventually form pyruvate, the end product of glycolysis. The third metabolic fate of fructose-derived GA is its conversion to glycerol by alcohol dehydrogenase (ADH), which is then phosphorylated by glycerol kinase (GK) to form glycerol 3-phosphate, the backbone of triglyceride synthesis. The specificity of an increase in ALDH and ADH following fructose versus glucose intake is uncertain; however, several reports indicate that dietary fructose, but not glucose, increases alcohol metabolism [37–39].
Fig. 1.
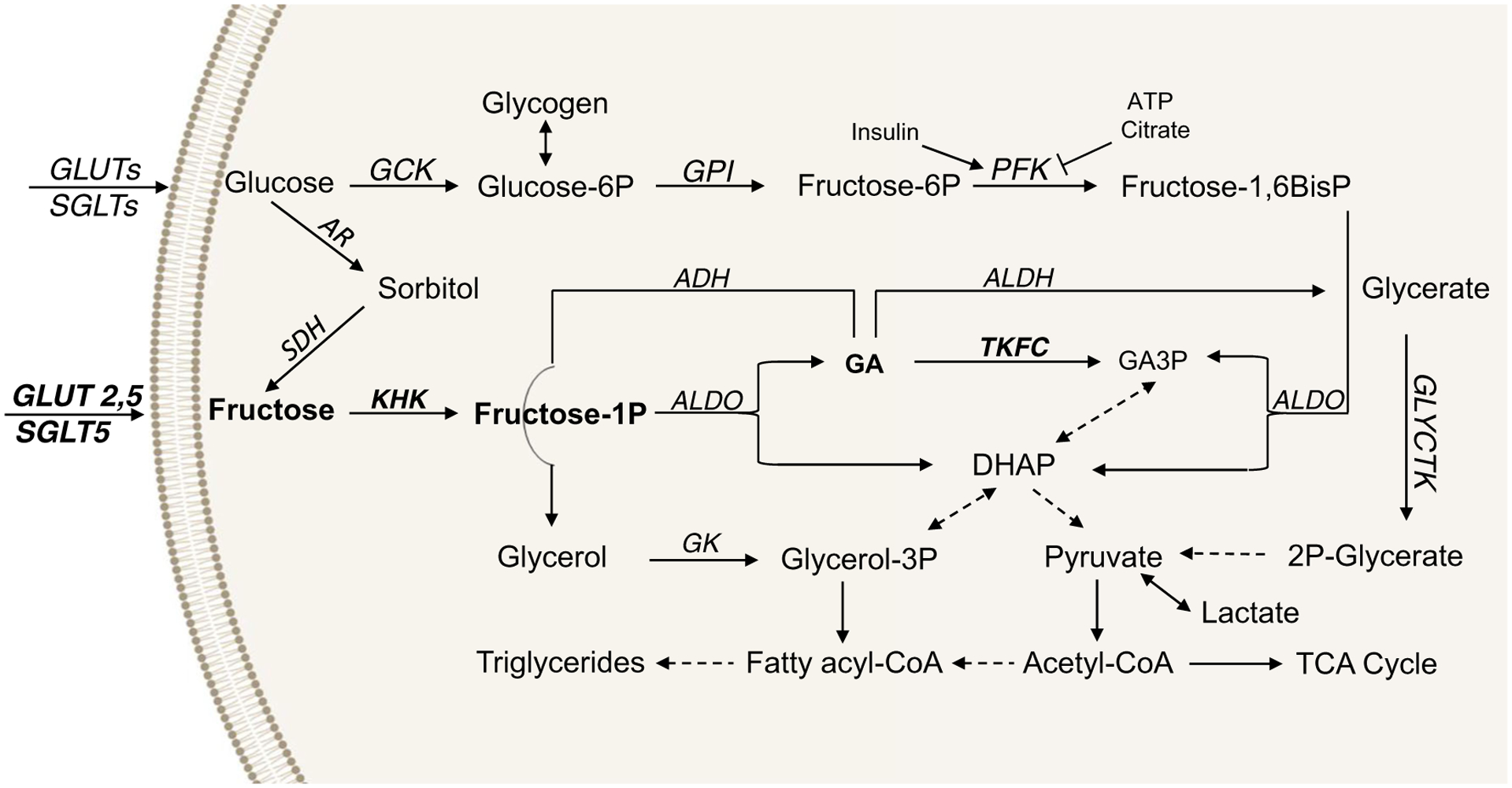
Pathways of fructose and glucose metabolism. Fructose translocation across the cellular membrane is primarily mediated by glucose transporters (GLUT2, GLUT5), as well as sodium and glucose cotransporters, such as SGLT5. Intracellular fructose metabolism is catalyzed by ketohexokinase (KHK), which converts fructose to fructose 1-phosphate. This reaction is considered to be the rate-limiting step in fructose catabolism. The aldolase (aldo) family of enzymes uses fructose 1-phosphate to produce glyceraldehyde (GA) and DHAP, a common intermediate with glycolysis, which is primarily converted into pyruvate. Pyruvate can then be metabolized to lactate or acetyl-CoA, which is a substrate for fatty acid and sterol synthesis or for incorporation into the TCA cycle. The GA produced from fructose 1-phosphate is more specifically associated with fructose as compared to glucose catabolism and has three fates: (1) it is converted to glycerol via alcohol dehydrogenase (ADH) to serve as a backbone for triglyceride synthesis; (2) it produces glycerate via aldehyde dehydrogenase (ALDH), which can contribute to the production of pyruvate via glycerate kinase (GLYCTK); or (3) it produces GA3P via triose kinase (TKFC). Induction of TKFC is another specific step of fructose metabolism. By converging onto common intermediates, fructose can be converted to glucose and thus support glycogen synthesis. On the other hand, glucose can be endogenously converted to fructose via the polyol pathway. First, glucose is reduced by aldose reductase (AR) to sorbitol, which is subsequently oxidized by sorbitol dehydrogenase (SDH) to fructose. Although the metabolism of glucose and that of fructose converge onto common metabolites, the flux through the glycolytic and fructolytic pathways is starkly different. Glycolysis is highly regulated at the level of phosphofructokinase (PFK) by insulin, ATP, and citrate. Conversely, fructose metabolism is 10 times faster than that of glucose and is not regulated by its end products or by insulin.
In addition to fructose and glucose metabolism converging onto common intermediates, glucose can endogenously be converted to fructose via the polyol pathway [40]. First, glucose is reduced by aldose reductase (AR) to form sorbitol, which is metabolized by sorbitol dehydrogenase (SDH) to form fructose. In spite of their interconversion and convergence onto the same metabolic end products, more fructose carbons, as compared to glucose, are incorporated into lactate and triglycerides, suggesting that carbon flux through fructolysis and glycolysis pathways is different. Indeed, glycolysis is regulated by insulin, citrate, and ATP via their action on phosphofructokinase, whereas fructose metabolism is not regulated by insulin or end products of carbohydrate metabolism (Fig. 1). Furthermore, phosphorylation of fructose by KHK in the liver is 10 times faster than phosphorylation of glucose by glucokinase (GCK) [32]. Due to rapid conversion of fructose and ATP to fructose 1-phosphate and ADP by KHK, fructose metabolism activates AMP deaminase leading to purine degradation and the production of uric acid. Unlike fructolysis, glycolysis is slower and regulated by feedback inhibition; therefore, glucose metabolism does not lead to ATP depletion or uric acid production. In summary, although fructose and glucose metabolism share many common intermediaries, the unique early enzymes of fructose metabolism (e.g., KHK and TKFC), and rapid and unregulated flux through the pathway may account for starkly different effects of fructose and glucose on whole-body metabolism.
Intestinal Metabolism of Fructose
The intestine is the first organ exposed to dietary fructose. Fructose is absorbed across the intestinal epithelium by the facilitative glucose transporters. GLUT2, 5, 7, and 9 are expressed on the small intestinal apical surface [41, 42]. GLUT5 is thought to be the main fructose transporter and is highly specific for fructose transport [38]. Global deletion of GLUT5 markedly decreases blood fructose levels in mice fed a high-fructose diet [43, 44]. GLUT2, localized on the basolateral side of the enterocyte, is responsible for fructose trafficking from the cytoplasm to the blood. Although GLUT7 [45] and GLUT9 [46] are expressed in the small intestine and are able to transport fructose, their role in intestinal fructose transport seems to be modest [47, 48], and GLUT9 appears to be a more specific urate transporter [49]. Thus, the main fructose transporters in the small intestine are GLUT5 and GLUT2.
Intestinal fructose metabolism is upregulated by a feed-forward loop as fructose-gavaged mice exhibit an increased expression of fructolytic genes such as GLUT5, KHK, and AldoB [50]. Fructose treatment in vitro increased Glut5 mRNA in enterocytes, Paneth and goblet cells, but not in intestinal stem cells [51]. As a consequence, perfusing the intestine with a high-fructose solution increases fructose uptake in the rat [52]. Intestinal fructose metabolism is required for GLUT5 upregulation as the non-metabolizable fructose analog 3-O-methylfructose only modestly increases GLUT5 expression [53]. Additionally, abolishing intestinal fructose metabolism by knockout of KHK prevents fructose-induced GLUT5 upregulation [50, 54]. On the contrary, Glut2 expression is not increased by high-fructose content in the intestinal lumen [55]. Thioredoxin-interacting protein (TXNIP), a member of the arrestin-like protein family regulating oxidation/reduction balance [56], regulates fructose transport by directly binding to GLUT5 and GLUT2 [57]; co-expression of TXNIP with GLUT2 and GLUT5 increases fructose uptake in a human epithelial colorectal adenocarcinoma cell line [57]. Conversely, deletion of TXNIP in mice reduces plasma levels of fructose and its uptake by peripheral tissue such as the liver, heart, and kidney after fructose gavage [57]. On a transcriptional level, mRNA expression of genes regulating fructose transport (Glut5, Txnip) and metabolism (Khk, AldoB, Tkfc) are regulated by carbohydrate response element-binding protein (ChREBP) transcription factor [58]. Thus, ChREBP knockdown leads to intestinal fructose malabsorption and symptoms resembling irritable bowel syndrome, diarrhea predominant subtype [59–61].
Traditionally, the liver has been considered the main organ that metabolizes fructose. Recently, it has been shown that the small intestine plays an important role in metabolizing a small amount of dietary fructose. Using stable isotope tracers, Jang et al. showed that the small intestine, and especially the jejunum, has the highest level of labeled fructose 1-phosphate [62•]. They concluded that 3–5 g of dietary fructose load is metabolized by the small intestine (around 90%), while only a small amount of fructose reaches the liver at this dose [62•]. Another recent study confirmed that fructose is metabolized in the intestine, but that the intestinal microbiome converts fructose to acetate, which is used as a substrate for de novo lipogenesis (DNL) in the liver [63]. Dietary fructose that reaches the liver serves primarily to turn on the lipogenic machinery. Utilizing liver- and intestine-specific KHK knockout mice, it has been concluded that intestinal fructose metabolism mediates sugar intake, whereas hepatic fructose metabolism drives metabolic dysfunction [64•]. In aggregate, these studies suggest that intestinal fructose metabolism protects the liver from excess lipid accumulation and insulin resistance. Similarly, increased intestinal fructose metabolism, achieved by knockdown of hypoxia-inducible factor 1 (HIF1), protects mice from development of NAFLD and insulin resistance [65, 66].
Intestinal Fructose Metabolism in Metabolic Diseases
Fructose metabolism by the intestine contributes to the association of fructose consumption with the development of T2DM. GLUT5 and GLUT2 are significantly enhanced in brush-border membrane vesicles from streptozotocin-diabetic rats [67, 68]. Similarly, duodenal biopsies from humans with T2DM showed a three- to fourfold increase in GLUT5, GLUT2, and SGLT1 expression levels, as compared with those from non-diabetic controls, suggestive of increased intestinal monosaccharide absorption in patients with T2DM [69]. These studies paved the way for the discovery of human GLUT5 inhibitors as potential therapeutic targets to treat T2DM [70]. In addition to intestinal monosaccharide transport, fructose intake has been linked with aberrant incretin responses. Kuhre et al. found that fructose stimulates glucagon-like peptide-1 (GLP-1), but not glucose-dependent insulinotropic polypeptide (GIP), secretion in mice, rats, and humans [71]. Similarly, acute fructose loading induced greater GLP-1 responses in obese adolescents, as compared with their lean peers [72]. In contrast, GLP-1 response was higher in the lean group after glucose ingestion and GIP response was similar in both groups. The authors concluded that fructose consumption by obese subjects might contribute to hyperinsulinemia through a GLP-1-mediated mechanism.
The intestine plays an important role in whole-body lipid metabolism, as it absorbs dietary fats and packages them into chylomicrons for systemic distribution. Increased intestinal chylomicron and VLDL production is observed in patients with metabolic syndrome, compared with lean age-matched controls [73]. Fructose feeding for 3 weeks in hamsters increased secretion of intestinally derived apolipoprotein B48 (apoB48)–containing lipoproteins in close parallel with increased intestinal de novo lipogenesis. Inhibition of fatty acid synthesis by cerulenin, a fatty acid synthase inhibitor, resulted in a dose-dependent decrease in intestinal apoB48 secretion [74]. Using mouse intestinal organoids, Al-Jawadi et al. showed that the increased fructose-induced lipogenic gene expression is dependent on KHK [75]. Furthermore, fructose feeding leads to insulin resistance in the intestine. Sixty minutes after insulin administration, circulating levels of apoB48-containing lipoproteins were decreased in chow-fed, but not in fructose-fed, animals [76]. This effect was normalized by restoring insulin sensitivity upon treatment with rosiglitazone, an insulin sensitizer [77]. Effects of fructose to increase intestinal lipoprotein production are also observed in humans. Intraduodenal infusion of Intralipid plus fructose, under pancreatic clamp conditions, increased both apoB48 and apoB100 production in healthy male subjects, when compared with Intralipid alone [78]. Together, these studies indicate that intestinal lipoprotein production contributes to fructose-induced dyslipidemia.
Intestinal fructose metabolism also contributes to the development of NAFLD via its effect on gut dysbiosis and intestinal permeability. Studies in mice fed high-fructose diet revealed that fructose increases intestinal permeability [79]. This was associated with a decrease in expression of genes involved in the regulation of the tight junction barrier in the small intestine, including Ocln and Tjp1 [79]. Moreover, Volynets et al. observed decreased Muc2 expression associated with decreased mucus thickness in the colon of mice when fructose was added to a western diet [80]. As a consequence of increased intestinal permeability, circulating levels of endotoxin were higher in the portal vein [80]. Numerous studies associate endotoxemia with progression of liver injury in patients with NAFLD ([81, 82]). Interestingly, when mice fed fructose were treated with non-absorbable antibiotics, hepatic lipid accumulation was significantly reduced ([80, 83]). High-sugar diets are known to decrease microbial diversity within 1 week of feeding ([84, 85]). These studies suggest that the intestinal microbiome, in part, mediates liver injury induced by fructose.
Fructose Metabolism in the Liver
Once absorbed by the small intestine, dietary fructose enters portal circulation and is transported into the liver to undergo rapid first-pass metabolism. Since GLUT5 is not expressed in the liver [86], GLUT2 and GLUT8 are the most abundant fructose transporters in hepatocytes ([33, 87]). However, GLUT2 has much lower affinity for fructose (Km = 11 mM) than GLUT5 (Km = 6 mM) and GLUT8 (Km = 2 mM) [88], and the portal fructose concentration after a fructose-containing meal is estimated to be only 5 mM. This opens the possibility that other transporters play an important role in fructose transport into the liver. Fructose can also be transported by GLUT7, GLUT9, GLUT11, and GLUT12, some of which have Km for fructose less than 1 mM [89]. Indeed, GLUT8, 9, and 10 are expressed in the liver [86], and at least GLUT8 has been shown to be important for fructose transport into the hepatocytes [87]. In spite of whole-body and liver-specific GLUT8 knockout mice transporting less fructose and being protected from fructose-induced NAFLD, GLUT8 is thought to account only for 25% of fructose transport into hepatocytes [90].
Once inside hepatocytes, fructose is rapidly phosphorylated by ketohexokinase-C, which is abundantly expressed in the liver. Hepatic KHK is increased in mice fed fructose [91], sucrose [92], or HFD [93]. Hypoxia increases KHK expression in the liver, but it is unknown whether this is HIF1 dependent [94•]. Hepatic KHK is also upregulated by uric acid [95], a byproduct of AMP degradation generated by metabolism of fructose. Similar to KHK regulation in the intestine, ChREBP mediates KHK upregulation in the liver [95]. The crucial role of ChREBP in fructose metabolism is demonstrated in liver-specific ChREBP knockout mice, which develop marked transaminitis and hepatomegaly when fed diets containing fructose. The ChREBP null mice are unable to upregulate KHK and display elevated glycogen accumulation when challenged with a high-fructose diet [96]. The second enzyme of fructose metabolism, AldoB, is very important for fructose metabolism in the liver. Patients with a generic deficiency of AldoB develop hereditary fructose intolerance, which is manifested by abdominal pain, vomiting, diarrhea, hypoglycemia, hyperuricemia, and in extreme cases liver failure and death, when exposed to dietary fructose [97]. In AldoB-deficient mice, these symptoms can be ameliorated by knockdown of KHK-C, which prevents hepatic fructose metabolism [98].
Hepatic Fructose Metabolism in Metabolic Diseases
On a molecular level, several properties of fructose metabolism prime the liver for development of hepatic insulin resistance. These include the strong propensity of fructose to increase hepatic de novo lipogenesis, decrease mitochondrial fatty acid oxidation, induce endoplasmic reticulum stress, and trigger hepatic inflammatory responses [12]. Furthermore, fructose metabolism may have more direct effects on insulin signaling by decreasing levels of insulin receptor and insulin receptor substrate 2 expression, and by upregulating protein tyrosine phosphatase non-receptor type 1 (PTP1b), a negative regulator of insulin signal transduction [12]. Hyperinsulinemia, as a result of insulin resistance, further stimulates hepatic lipogenesis [99] and decreases fatty acid oxidation leading to a vicious cycle of metabolic dysregulation.
Fructose strongly stimulates hepatic DNL [100] and increases lipid secretion, contributing to dyslipidemia. Indeed, in healthy subjects, 7 days of high-fructose diet increased ectopic hepatic lipid deposition and serum VLDL-triacylglycerol levels [15]. This effect was exaggerated in healthy offspring of patients with T2DM. The effects of fructose to increase serum cholesterol may be even more dramatic. In a cross-over study, fourteen healthy subjects consumed a high-fructose diet (20% of energy from fructose) for 28 days or an isocaloric high-starch diet containing < 3% fructose. A high-fructose diet resulted in significantly higher fasting serum total and LDL cholesterol when compared with a high-starch diet [101]. A meta-analysis of 37 clinical trials confirmed that higher intake of dietary sugars significantly raises total cholesterol, LDL-C, and HDL-C, an effect that was independent of changes in body weight [102]. Mechanistic studies in animals found that fructose stimulates VLDL and ApoB overproduction by doubling levels of microsomal triglyceride transfer protein, a key enzyme involved in VLDL assembly [103]. The mechanism of how fructose induces hypercholesterolemia is largely unknown and is the subject of ongoing research in our lab. We find that dietary fructose increases hepatic cholesterol levels, while knockdown of KHK specifically in the liver decreases hepatic and serum cholesterol. These data suggest that hepatic fructose metabolism is sufficient to induce hypercholesterolemia. Whereas dietary fructose induces dyslipidemia, fructose restriction improves it. Ten days of isocaloric fructose restriction in patients with metabolic syndrome reduced LDL-C, apoB, ApoC-III, and ApoE, paralleling changes in insulin sensitivity ([104, 105]). Thus, fructose induces dyslipidemia by stimulating hepatic lipid synthesis, an effect that is dependent on hepatic KHK.
Fructose was traditionally thought to be primarily metabolized by the liver in rats [106] and humans [107]. Furthermore, expression and enzymatic activity of all proteins involved in fructose metabolism are sufficiently high in human liver, pointing to the important physiologic role of this organ in fructose metabolism [108]. Indeed, as fructose intake has increased in our diet, fructose has emerged as one of the primary risk factors for development of NAFLD [109]. In the liver, fructose stimulates DNL [93•] and decreases mitochondrial fatty acid oxidation [110]. Effects of fructose to decrease fatty acid oxidation via its effects on post-translational modification of mitochondrial proteins are of particular interest to our group [110–112]. Furthermore, increased fructose intake has been found to correlate with severity of liver fibrosis [113] and KHK is elevated in humans with biopsy-proven NAFLD [109]. Fructose potentiates its metabolism, so patients with NAFLD are more sensitive to the adverse metabolic effects of dietary fructose [114]. In animal studies, knockdown of KHK protects mice from the development of NAFLD and insulin resistance [91–93]. A large body of literature associates dietary fructose intake with development of NAFLD, and this topic has been covered by other reviews [115–118]. Lastly, due to a strong association of fructose with the development and progression of NAFLD, KHK inhibitors are being developed for the treatment of NAFLD [119].
Fructose Metabolism in the Kidney
Once fructose is absorbed by the intestine and portal fructose is extracted by first-pass metabolism through the liver, maximal systemic fructose concentration is low, in the range of 1 mM [120, 121]. The kidney is the next most relevant site of fructose metabolism. About 20% of systemic fructose is taken up by the kidney [122], compared to over 50% of intravenous fructose load extracted by the liver [107]. GLUT5 is expressed in the kidney, and is increased three- to fourfold with diets high in fructose or sucrose, but not glucose [123]. Furthermore, other fructose transporters such as GLUT2 [124], GLUT9 [125], SGLT1 [126], and SGLT4 [127] are expressed in the kidney, but the contribution of these transporters to renal fructose transport is uncertain. SGLT5 is exclusively expressed in the kidney and was found to have high capacity to transport fructose and mannose [128]. SGLT5 expression increases three- to fourfold after fructose consumption and its knockout increases urinary fructose excretion [129]. Subsequent studies have confirmed this finding and proposed that SGLT5 is one of the main transporters for fructose reabsorption in the proximal tubule of the kidney [130]. In addition to fructose transporters, all fructose-metabolizing genes are present in the human kidney cortex and medulla [131]. KHK-C is also robustly expressed in the kidney of rodents, enabling robust fructose metabolism in the kidney [30].
Kidney Fructose Metabolism in Metabolic Diseases
Increased fructose concentrations have been observed in blood and urine of patients with diabetes [132]. In diabetic patients, higher fructose intake is linked with greater albuminuria, a marker of chronic kidney disease (CKD) [133]. Albuminuria was also associated with intake of two or more sugar-containing beverages in the NHANES (1999–2004) study of US adults without diabetes [134]. In contrast, lowering fructose intake from 59 to 12 g per day for 6 weeks lowered fasting insulin in patients with CKD [135]. On a molecular level, fructose metabolism leads to generation of uric acid [136, 137] and higher uric acid is linked with vascular complications and diabetic kidney disease [138]. Hyperuricemia causes oxidative stress leading to kidney dysfunction [139]. Conversely, blockade of fructose metabolism, in a mouse model of diabetes, alleviates renal injury by reducing uric acid production in tubular cells [140]. Fructose metabolism via KHK has been shown to induce inflammation in human proximal tubular cells via a uric acid–dependent mechanism [141]. Increased inflammatory response may account for progression of CKD and accelerated tubular atrophy, tubulointerstitial inflammation, and collagen deposition in rodents fed fructose [142–144]. On the other hand, KHK knockout mice (total KHK-A/C KO) have improved renal function and attenuated kidney injury as compared with diabetic wild-type mice [91]. Interestingly, isolated knockout of KHK-A led to enhanced kidney injury in streptozotocin-induced diabetic mice [145], secondary to increased fructose metabolism via KHK-C. KHK-A may also have protective properties, as it phosphorylates and activates phosphoribosyl pyrophosphate synthetase 1 (PRPS1) to promote de novo nucleic acid synthesis [146].
In addition to worsening diabetic kidney disease, recurrent dehydration-induced kidney injury is mediated via a KHK-dependent mechanism. Dehydration triggers the generation of endogenous fructose produced through the polyol pathway [147] and plays a critical role in the pathogenesis of acute kidney injury due to the accumulation of endogenous fructose and sorbitol [140, 148]. Aging-associated kidney disease is also KHK dependent, as demonstrated in KHK knockout mice [149]. Lastly, luteolin, a KHK inhibitor, ameliorated kidney damage in wild-type mice undergoing ischemic kidney injury [150].
Whereas the contribution of fructose to diabetic kidney disease is well described, kidney-specific fructose metabolism may also contribute to the development of other metabolic diseases such as NAFLD and hypertension. Interestingly, SGLT5-deficient mice exhibit increased urinary fructose loss, but they paradoxically developed increased hepatic steatosis [129]. Furthermore, dietary fructose intake has been linked with the development of hypertension [151]. In addition to inducing CKD and hyperuricemia, both of which can lead to hypertension, fructose increases intestinal salt and water absorption. This is mediated by increased expression of the apical chloride/base exchanger Slc26a6 (PAT1) and the sodium hydrogen exchanger 3 (NHE3) in the jejunum [152]. This process also occurs in the kidney, as fructose increases sodium transport via NHE3 in the renal proximal tubule [153]. This is dependent on KHK, as fructose-induced phosphorylation of NHE3 is abrogated in KHK knockout mice [154]. Overall, a significant portion of fructose is metabolized in the kidney, and this leads to the development of diabetic kidney disease and hypertension.
Fructose Metabolism in Adipose Tissue
Adipose tissue has not been traditionally considered a major site of fructose metabolism, but some reports suggest that adipose tissue metabolizes fructose to the same extent as the intestine or kidney [155]. Adipose tissue also robustly expresses fructose transporter GLUT5 ([27, 156]). Consistent with this notion, GLUT5 mediates about 80% of fructose uptake in rat adipocytes [157]. GLUT5 expression can also be influenced by several factors, including hypoxia [158], obesity ([27, 157]), LXR agonists [159], triiodothyronine ([160, 161]), and glucocorticoids [162]. However, GLUT5 expression and membrane translocation are not affected by insulin. Shepherd et al. reported that ex vivo insulin treatment of plasma membranes isolated from adipocytes of five non-obese and four obese individuals did not result in recruitment of GLUT5 to the plasma membrane in either group, in spite of a 54% insulin-stimulated increase in GLUT4 in non-obese subjects [163].
KHK-A versus KHK-C is the major isoform in adipocytes [30]; however, the overall expression of both isoforms is quite low [91]. Instead, fructose metabolism in adipose tissue is mediated by hexokinase, as evident by impaired fructose metabolism in the presence of the hexokinase inhibitor 3,5-dinitrobenzoylglucosamine [155]. Km of hexokinase for glucose (10−4 to 10−5 M) is much lower than for fructose (10−3 to 10−2 M). However, glucose transport into adipocytes is dependent on insulin, so free glucose concentration in adipocytes is low under fasting conditions. The absence of free glucose enables fructose metabolism in adipose tissue, at least in the setting of low insulin or perhaps in an insulin-resistant state [155]. To further understand metabolic fate of fructose in human adipocytes, Varma et al. utilized [U-13C6]-labeled fructose to demonstrate that fructose is robustly metabolized in pre-adipocytes and fully differentiated adipocytes [164]. Most of the labeled fructose entered the TCA cycle through the pyruvate dehydrogenase pathway and robustly stimulated anabolic processes, including glutamate formation and de novo fatty acid synthesis [164]. Moreover, fructose supplementation induces adipogenesis in the pre-adipocyte 3T3-L1 cell line [165]. GLUT5 overexpression in 3T3-L1 cells can induce adipocyte differentiation, while GLUT5 knockdown impairs adipogenesis [165]. GLUT5-deficient mice display reduced fat mass and impaired adipogenesis ex vivo [165]. Taken together, these data suggest that fructose is readily taken up in adipocytes via GLUT5 and metabolized via hexokinase to support adipose tissue development and hypertrophy.
Indeed, fructose supplementation promotes visceral obesity, adipose tissue inflammation, and insulin resistance [166]. Visceral adiposity is a known risk factor for cardiovascular disease and T2DM. In adolescents, excess fructose consumption is associated with visceral adiposity, plasma C-reactive protein, and increased HOMA-IR [167]. This is consistent with data demonstrating that high-fructose feeding in rhesus monkeys produces insulin resistance and aspects of metabolic syndrome, including central obesity and inflammation [168]. Adipose tissue development and function has marked effects on NAFLD [169], as most liver triglycerides are derived from adipose tissue lipolysis. The contribution of adipose tissue–specific fructose metabolism in the development of NAFLD warrants further exploration.
Fructose Metabolism in Skeletal Muscle
Skeletal muscle is another tissue that has not been considered a primary site of fructose metabolism. However, fructose metabolism can occur in the skeletal muscle. Individuals with KHK deficiency have a benign condition called essential fructosuria and are unable to metabolize fructose in the intestine, liver, and kidney; thus, their plasma fructose concentration is elevated [170]. Interestingly, they only excrete about 10–20% of fructose load in the urine [170], so that majority of fructose is still metabolized within the body, mainly in adipose tissue and skeletal muscle [171]. Indeed, when fructose is infused into exercising healthy subjects to maintain a concentration of 5.5 mmol/L, there is considerably more fructose oxidation by exercising and resting muscles [172].
Similar to adipose tissue, GLUTs 1, 4, and 5 are expressed in skeletal muscle. Unlike GLUT4, which translocates from intracellular vesicles to the plasma membrane [173] in response to insulin, GLUT5 is localized primarily in the sarcolemma of skeletal muscle [174]. Moreover, GLUT5 is not responsive to acute insulin treatment ([157, 163]); however, chronic insulin treatment (~ 24 h) can stimulate GLUT5 expression, promoter activity, and fructose uptake in rat L6 myotubes in vitro [175]. Furthermore, increased GLUT5 expression in muscle from T2DM patients compared with healthy patients is reversed by pioglitazone [176], suggesting an insulin-dependent effect. The predominant KHK isoform in muscle is low-affinity KHK-A isoform [91]. Despite lower affinity for fructose, KHK-A can metabolize fructose at physiological concentrations [91]. Mice with whole-body deficiency of both KHK isoforms display 30-fold increased levels of fructose in serum, and markedly increased levels of fructose and fructose-6-phosphate in muscle relative to wild-type (WT) animals. This suggests that fructose is phosphorylated by hexokinase in the absence of both KHK isoforms in skeletal muscle [177].
Fructose can impair glucose metabolism in skeletal muscle by inducing advanced glycation end products (AGEs) [178] and reducing plasma membrane GLUT4 levels [179]. Furthermore, fructose supplementation induces the expression of the proinflammatory cytokine TNFα in the soleus and the extensor digitorum longus in rats [180], potentially contributing to impaired insulin-mediated glucose uptake ([181, 182]). Interestingly, fructose feeding impairs insulin-stimulated glucose transport in the skeletal muscle of male rats, but not in female or ovariectomized female rats [183]. Taken together, these data demonstrate that fructose metabolism occurs in the skeletal muscle, at least in the setting of high serum fructose levels and affects skeletal muscle glucose handling.
Fructose Metabolism in the Pancreas
The primary fructose transporter GLUT5 shows little to no expression in the pancreas [184]. GLUT2, however, is expressed on the plasma membrane of pancreatic β cells, but exhibits low affinity to fructose (Km ~ 76 mM) relative to glucose (Km ~ 17 mM) [185]. Despite poor uptake in the pancreas, fructose can activate sweet taste receptors on β cells to amplify glucose-stimulated insulin release in mouse and human islets [186]. Compared to glucose, however, fructose alone is not considered an insulin secretagogue [187].
Previous studies have measured low KHK expression in the pancreas of several species and in pancreatic insulinoma cell line MIN6 [30]. In agreement with minimal KHK expression in the pancreas ([188, 189]), fructose metabolism in the pancreas occurs by a KHK-independent mechanism via hexokinase [190]. This was based on a few observations: (1) pancreatic fructose phosphorylation is K+ independent unlike KHK metabolism which requires K+; (2) glucose and G6P inhibited fructose phosphorylation in the pancreas but not in the liver; and (3) fructose phosphorylation kinetics in islet homogenates coincided with purified hexokinase activity [190]. Furthermore, Malaisse et al. used heat to discern between KHK and hexokinase activity, since KHK, unlike other hexokinases, is stable at elevated temperatures ~ 70 °C. They demonstrated that phosphorylation of fructose in heated islet homogenates is 90% lower than in non-heated homogenates and is not affected by glucose or G6P. Fructose metabolism in heated islets is completely dependent on K+, indicating KHK-dependent metabolism [191]. Consistently, other groups have shown that the activity of KHK is lower in the islets than in the liver, ileum, or whole pancreas [192] with no observable differences in KHK activity between β and non-β cells [192]. Collectively, phosphorylation of fructose in the pancreas is catalyzed by hexokinase, which coincides with minimal KHK expression in this tissue.
Fructose Effects on the Brain
Glucose is the principal energy source for the brain, but fructose can cross the blood-brain barrier and be metabolized by the brain ([193, 194]). GLUT5 is expressed on microglia [195], blood-brain barrier [196], and cerebellum [197]. The expression of GLUT5 is increased in the brain of fructose-fed rats [198], during hypoxia/ischemia and other factors that induce microglial stress [199]. Additionally, GLUT1 which typically transports glucose also has a low affinity for fructose [200]. However, the function of fructose transporters in the brain remains uncertain since radiolabeled fructose injected into the arterial circulation in rats results in minimal accumulation of labeled fructose in the brain [201]. Alternatively, fructose can be endogenously produced in the human brain from glucose, in the setting of hyperglycemia [202]. Thus, the concentration of fructose in the brain often correlates with serum glucose levels [203]. Endogenous fructose production in the brain could explain the 20-fold higher levels of fructose in cerebrospinal fluid compared to serum [202]. In the brain, fructose is metabolized by both KHK and hexokinase since fructose metabolism is synergistically inhibited by both glucose and a KHK-specific inhibitor [204]. Interestingly, KHK enzymatic activity was 5–10 times higher and rates of fructose oxidation were 15–150 times higher in the brain compared to liver [204]. This is in agreement with higher glycolytic capacity of the brain relative to the liver [205].
Fructose utilization in the brain exerts profound effects on central control of appetite [206]. In healthy subjects, consumption of fructose versus glucose drinks led to greater hunger and desire for food, as assessed by functional MRI [207]. Intriguingly, fructose is naturally sweeter than glucose and has high affinity for sweet taste receptors [208]. However, fructose intake does not reduce cerebral blood flow in brain regions involved with appetite and reward pathways, as does glucose [209]. On a molecular level, fructose metabolism leads to rapid depletion of ATP, lowers malonyl-CoA levels in the hypothalamus, and as a result increases food intake [210]. In addition to its central effects, hormonal response to fructose by peripheral tissue may also play a role in regulation of appetite. First, fructose does not induce robust insulin secretion, which signals in the brain to reduce food intake [211]. Next, fructose is less potent than glucose to increase leptin secretion [212]. Lastly, rats consuming fructose diet had a twofold higher fasting serum ghrelin, as compared with glucose-fed rats [213]. Taken together, fructose is metabolized in the brain and effects food intake.
Fructose Metabolism in Other Tissues
Fructose is found in human semen and is used as an energy source for spermatozoa. GLUT5 is found in human testis and spermatozoa [214], but fructose is also endogenously produced in the seminal vesicles. In the testes, fructose is metalized by KHK to fructose 1-phosphate. Increased fructose intake has been associated with decreased testicular weight in rats [215]. Fructose metabolism is documented in human red blood cells, which strongly express GLUT5 [216]. Inside erythrocytes, fructose is converted to fructose 3-phosphate by 3-phosphokinase [217]. Fructose 3-phosphate leads to rapid formation of advanced glycation end products [218]. Lastly, fructose is endogenously produced in the lens of the eye, in the setting of hyperglycemia [219]. Similar to RBCs, fructose is metabolized to fructose 3-phosphate in the lens [220]. Endogenous fructose production in the lens is thought to contribute to cataract formation [221].
Summary and Future Perspective
Dietary fructose is metabolized by a variety of tissues, albeit at different levels (Table 1). The primary sites of fructose metabolism are the liver, kidney, and intestine. Fructose metabolism by these tissues is highly associated with development of obesity and its complications, such as diabetes, dyslipidemia, and NAFLD. Fructose metabolism can also occur in adipose tissue and the muscle, but likely only in the setting of high circulating fructose, induced by genetic absence of KHK or by high dietary fructose intake. Fructose can also be sensed and metabolized by the pancreas and brain, where it modifies their essential functions, including insulin secretion and regulation of appetite, respectively. Lastly, small amounts of fructose are metabolized in the testes, red blood cells, and lens of the eye. Understanding tissue-specific fructose metabolism is paramount, as systemic and liver-specific KHK inhibitors are being developed for management of fatty liver disease and diabetes. Future studies are needed to understand what effects inhibition of KHK has on fructose metabolism in tissues that do not rely on KHK for fructose metabolism.
Table 1.
An overview of tissue-specific fructose metabolism
| Importance | Organ | Amount of fructose metabolized | Primary Fructose Transporters | Secondary Fructose Transporters | First Enzyme of Metabolism | Second Enzyme of Metabolism | Third Enzyme of Metabolism | Metabolism Regulated by |
|---|---|---|---|---|---|---|---|---|
| Primary sites of metabolism | Intestine | 20% | GLUT 5 GLUT 2* |
GLUT 7 GLUT 9 |
KHK-C | ALDOB | TKFC | HIF1a TXNIP ChREBP |
| Liver | 50–70% | GLUT 2 GLUT8* |
GLUT 5 GLUT 9 |
KHK-C | ALDOB | TKFC | Hypoxia Uric acid ChREBP |
|
| Kidney | 20% | GLUT 5 SGLT 5 |
GLUT 2 SGLT 4 |
KHK-C | ALDOB | TKFC | HIF1a Uric acid Dehydration |
|
| Inducible metabolism | Adipose | 0–20% | GLUT 5 | - | HK2 KHK-A |
ALDOC | - | Hypoxia LXR agonists Glucocorticoid |
| Muscle | Inducible | GLUT 5 | - | HK2 KHK-A |
ALDOA | - | High Fructose Chronic Insulin Tx |
|
| Affects organ function | Pancreas | Minimal | GLUT 2 | T1R2# | HK1 KHK-A |
ALDOB * | - | - |
| Brain | Endogenous production | GLUT 5 | GLUT 1 | HK1 KHK-A |
ALDOC | - | Ischemia Microglia stress |
|
| Metabolism by other tissues | Testes | Endogenous production | GLUT 5 | - | KHK-A | - | TKFC | - |
| RBC | Minimal | GLUT 5 | - | FN3K | ALDOA | - | - | |
| Lense | Endogenous production | - | - | FN3K | - | - | - |
denotes less important contribution;
indicates the data was not found in the literature;
stands for sweet taste receptor, not a transporter;
GLUT glucose transporter; SGLT5 sodium glucose cotransporter; KHK ketohexokinase; HK hexokinase; FN3K fructosamine 3 kinase; T1R2 taste receptor type 1 member 2; Aldo aldolase; TKFC triokinase; RBC red blood cells; HIF1a hypoxia-inducible factor 1a; TXNIP thioredoxin-interacting protein; ChREBP carbohydrate response element-binding protein; LXR liver X receptor
Footnotes
Conflict of Interest The authors declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Chanmugam P, Guthrie JF, Cecilio S, Morton JF, Basiotis PP, Anand R. Did fat intake in the United States really decline between 1989–1991 and 1994–1996? J Am Diet Assoc. 2003;103: 867–72. [DOI] [PubMed] [Google Scholar]
- 2.Bray GA, Nielsen SJ, Popkin BM. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am J Clin Nutr. 2004;79:537–43. [DOI] [PubMed] [Google Scholar]
- 3.Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ. Fructose, weight gain, and the insulin resistance syndrome. Am J Clin Nutr. 2002;76:911–22. [DOI] [PubMed] [Google Scholar]
- 4.Imamura F, O’Connor L, Ye Z, Mursu J, Hayashino Y, Bhupathiraju SN, et al. Consumption of sugar sweetened beverages, artificially sweetened beverages, and fruit juice and incidence of type 2 diabetes: systematic review, meta-analysis, and estimation of population attributable fraction. BMJ. 2015;351: h3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collier R FDA proposes new food labels. CMAJ. 2014;186:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cawley J, Frisvold D, Hill A, Jones D. The impact of the Philadelphia beverage tax on purchases and consumption by adults and children. J Health Econ. 2019;67:102225. [DOI] [PubMed] [Google Scholar]
- 7.Falbe J, Thompson HR, Becker CM, Rojas N, McCulloch CE, Madsen KA. Impact of the Berkeley excise tax on sugar-sweetened beverage consumption. Am J Public Health. 2016;106:1865–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bridge G, Lomazzi M, Bedi R. Implementation of a sugar-sweetened beverage tax in low- and middle-income countries: recommendations for policymakers. J Public Health Policy. 2020;41:84–97. [DOI] [PubMed] [Google Scholar]
- 9.Alviso-Orellana C, Estrada-Tejada D, Carrillo-Larco RM, Bernabe-Ortiz A. Sweetened beverages, snacks and overweight: findings from the Young Lives cohort study in Peru. Public Health Nutr. 2018;21:1627–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caro JC, Corvalan C, Reyes M, Silva A, Popkin B, Taillie LS. Chile’s 2014 sugar-sweetened beverage tax and changes in prices and purchases of sugar-sweetened beverages: an observational study in an urban environment. PLoS Med. 2018;15:e1002597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makris A, Foster GD. Dietary approaches to the treatment of obesity. Psychiatr Clin North Am. 2011;34:813–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Softic S, Stanhope KL, Boucher J, Divanovic S, Lanaspa MA, Johnson RJ, et al. Fructose and hepatic insulin resistance. Crit Rev Clin Lab Sci. 2020:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beck-Nielsen H, Pedersen O, Lindskov HO. Impaired cellular insulin binding and insulin sensitivity induced by high-fructose feeding in normal subjects. Am J Clin Nutr. 1980;33:273–8. [DOI] [PubMed] [Google Scholar]
- 14.Faeh D, Minehira K, Schwarz JM, Periasamy R, Park S, Tappy L. Effect of fructose overfeeding and fish oil administration on hepatic de novo lipogenesis and insulin sensitivity in healthy men. Diabetes. 2005;54:1907–13. [DOI] [PubMed] [Google Scholar]
- 15.Le KA, Ith M, Kreis R, Faeh D, Bortolotti M, Tran C, et al. Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of type 2 diabetes. Am J Clin Nutr. 2009;89:1760–5. [DOI] [PubMed] [Google Scholar]
- 16.Lecoultre V, Egli L, Carrel G, Theytaz F, Kreis R, Schneiter P, et al. Effects of fructose and glucose overfeeding on hepatic insulin sensitivity and intrahepatic lipids in healthy humans. Obesity (Silver Spring). 2013;21:782–5. [DOI] [PubMed] [Google Scholar]
- 17.Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119:1322–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taskinen MR, Soderlund S, Bogl LH, Hakkarainen A, Matikainen N, Pietilainen KH, et al. Adverse effects of fructose on cardiometabolic risk factors and hepatic lipid metabolism in subjects with abdominal obesity. J Intern Med. 2017;282:187–201. [DOI] [PubMed] [Google Scholar]
- 19.Aeberli I, Hochuli M, Gerber PA, Sze L, Murer SB, Tappy L, et al. Moderate amounts of fructose consumption impair insulin sensitivity in healthy young men: a randomized controlled trial. Diabetes Care. 2013;36:150–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwarz JM, Noworolski SM, Wen MJ, Dyachenko A, Prior JL, Weinberg ME, et al. Effect of a high-fructose weight-maintaining diet on lipogenesis and liver fat. J Clin Endocrinol Metab. 2015;100:2434–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hallfrisch J, Ellwood KC, Oet M, Reiser S, Odorisio TM, Prather ES. Effects of dietary fructose on plasma glucose and hormone responses in normal and hyperinsulinemic men. J Nutr. 1983;113: 1819–26. [DOI] [PubMed] [Google Scholar]
- 22.Reiser S, Bohn E, Hallfrisch J, OEt M, Keeney M, Prather ES. Serum insulin and glucose in hyperinsulinemic subjects fed three different levels of sucrose. Am J Clin Nutr. 1981;34:2348–58. [DOI] [PubMed] [Google Scholar]
- 23.Reiser S, Handler HB, Gardner LB, Hallfrisch JG, OEt M, Prather ES. Isocaloric exchange of dietary starch and sucrose in humans. II. Effect on fasting blood insulin, glucose, and glucagon and on insulin and glucose response to a sucrose load. Am J Clin Nutr. 1979;32:2206–16. [DOI] [PubMed] [Google Scholar]
- 24.Malik VS, Popkin BM, Bray GA, Despres JP, Willett WC, Hu FB. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: a meta-analysis. Diabetes Care. 2010;33:2477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mattes RD, Shikany JM, Kaiser KA, Allison DB. Nutritively sweetened beverage consumption and body weight: a systematic review and meta-analysis of randomized experiments. Obes Rev. 2011;12:346–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bes-Rastrollo M, Schulze MB, Ruiz-Canela M, Martinez-Gonzalez MA. Financial conflicts of interest and reporting bias regarding the association between sugar-sweetened beverages and weight gain: a systematic review of systematic reviews. PLoS Med. 2013;10:e1001578 dicsussion e1001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Douard V, Ferraris RP. Regulation of the fructose transporter GLUT5 in health and disease. Am J Physiol Endocrinol Metab. 2008;295:E227–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thorens B, Mueckler M. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab. 2010;298(2):E141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bonthron DT, Brady N, Donaldson IA, Steinmann B. Molecular basis of essential fructosuria: molecular cloning and mutational analysis of human ketohexokinase (fructokinase). Hum Mol Genet. 1994;3:1627–31. [DOI] [PubMed] [Google Scholar]
- 30.Diggle CP, Shires M, Leitch D, Brooke D, Carr IM, Markham AF, et al. Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. J Histochem Cytochem. 2009;57: 763–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diggle CP, Shires M, McRae C, Crellin D, Fisher J, Carr IM, et al. Both isoforms of ketohexokinase are dispensable for normal growth and development. Physiol Genomics. 2010;42A:235–43. [DOI] [PubMed] [Google Scholar]
- 32.Asipu A, Hayward BE, O’Reilly J, Bonthron DT. Properties of normal and mutant recombinant human ketohexokinases and implications for the pathogenesis of essential fructosuria. Diabetes. 2003;52:2426–32. [DOI] [PubMed] [Google Scholar]
- 33.Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. J Clin Invest. 2018;128:545–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sillero MA, Sillero A, Sols A. Enzymes involved in fructose metabolism in lir and the glyceraldehyde metabolic crossroads. Eur J Biochem. 1969;10:345–50. [DOI] [PubMed] [Google Scholar]
- 35.Liu L, Li T, Liao Y, Wang Y, Gao Y, Hu H, Huang H, Wu F, Chen Y-G, Xu S, Fu S. Triose kinase controls the lipogenic potential of fructose and dietary tolerance. Cell Metab. 20200;S1550–4131(20):30413–7. [DOI] [PubMed] [Google Scholar]
- 36.Adelman RC, Spolter PD, Weinhouse S. Dietary and hormonal regulation of enzymes of fructose metabolism in rat liver. J Biol Chem. 1966;241:5467–72. [PubMed] [Google Scholar]
- 37.Mascord D, Smith J, Starmer GA, Whitfield JB. The effect of fructose on alcohol metabolism and on the [lactate]/[pyruvate] ratio in man. Alcohol Alcohol. 1991;26:53–9. [PubMed] [Google Scholar]
- 38.Thorne M, Carpenter RCL. The effects of fructose on the metabolism of ethyl alcohol in man. J Pharmacol Exp Ther. 1937;60: 286–95. [Google Scholar]
- 39.Thieden HIDLF. The influence of fructose and its metabolites on ethanol metabolism in vitro. Biochem J. 1967;102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lorenzi M The polyol pathway as a mechanism for diabetic retinopathy: attractive, elusive, and resilient. Exp Diabetes Res. 2007;2007:61038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romero A, Gomez O, Terrado J, Mesonero JE. Expression of GLUT8 in mouse intestine: identification of alternative spliced variants. J Cell Biochem. 2009;106:1068–78. [DOI] [PubMed] [Google Scholar]
- 42.Mueckler M Facilitative glucose transporters. Eur J Biochem. 1994;219:713–25. [DOI] [PubMed] [Google Scholar]
- 43.Barone S, Fussell SL, Singh AK, Lucas F, Xu J, Kim C, et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J Biol Chem. 2009;284:5056–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel C, Douard V, Yu S, Gao N, Ferraris RP. Transport, metabolism, and endosomal trafficking-dependent regulation of intestinal fructose absorption. FASEB J. 2015;29:4046–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Q, Manolescu A, Ritzel M, Yao S, Slugoski M, Young JD, et al. Cloning and functional characterization of the human GLUT7 isoform SLC2A7 from the small intestine. Am J Physiol Gastrointest Liver Physiol. 2004;287:G236–42. [DOI] [PubMed] [Google Scholar]
- 46.Kim HR, Park SW, Cho HJ, Chae KA, Sung JM, Kim JS, et al. Comparative gene expression profiles of intestinal transporters in mice, rats and humans. Pharmacol Res. 2007;56:224–36. [DOI] [PubMed] [Google Scholar]
- 47.Ferraris RP, Choe JY, Patel CR. Intestinal absorption of fructose. Annu Rev Nutr. 2018;38:41–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ebert K, Ludwig M, Geillinger KE, Schoberth GC, Essenwanger J, Stolz J, et al. Reassessment of GLUT7 and GLUT9 as putative fructose and glucose transporters. J Membr Biol. 2017;250:171–82. [DOI] [PubMed] [Google Scholar]
- 49.DeBosch BJ, Kluth O, Fujiwara H, Schurmann A, Moley K. Early-onset metabolic syndrome in mice lacking the intestinal uric acid transporter SLC2A9. Nat Commun. 2014;5:4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patel C, Douard V, Yu S, Tharabenjasin P, Gao N, Ferraris RP. Fructose-induced increases in expression of intestinal fructolytic and gluconeogenic genes are regulated by GLUT5 and KHK. Am J Physiol Regul Integr Comp Physiol. 2015;309:R499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kishida K, Pearce SC, Yu S, Gao N, Ferraris RP. Nutrient sensing by absorptive and secretory progenies of small intestinal stem cells. Am J Physiol Gastrointest Liver Physiol. 2017;312:G592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang L, David ES, Espina N, Ferraris RP. GLUT-5 expression in neonatal rats: crypt-villus location and age-dependent regulation. Am J Physiol Gastrointest Liver Physiol. 2001;281:G666–74. [DOI] [PubMed] [Google Scholar]
- 53.Jiang L, Ferraris RP. Developmental reprogramming of rat GLUT-5 requires de novo mRNA and protein synthesis. Am J Physiol Gastrointest Liver Physiol. 2001;280:G113–20. [DOI] [PubMed] [Google Scholar]
- 54.Merino B, Fernandez-Diaz CM, Cozar-Castellano I, Perdomo G. Intestinal fructose and glucose metabolism in health and disease. Nutrients. 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cui XL, Soteropoulos P, Tolias P, Ferraris RP. Fructose-responsive genes in the small intestine of neonatal rats. Physiol Genomics. 2004;18:206–17. [DOI] [PubMed] [Google Scholar]
- 56.Yodoi J, Masutani H, Nakamura H. Redox regulation by the human thioredoxin system. Biofactors. 2001;15:107–11. [DOI] [PubMed] [Google Scholar]
- 57.Dotimas JR, Lee AW, Schmider AB, Carroll SH, Shah A, Bilen J, et al. Diabetes regulates fructose absorption through thioredoxin-interacting protein. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee HJ, Cha JY. Recent insights into the role of ChREBP in intestinal fructose absorption and metabolism. BMB Rep. 2018;51:429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oh AR, Sohn S, Lee J, Park JM, Nam KT, Hahm KB, et al. ChREBP deficiency leads to diarrhea-predominant irritable bowel syndrome. Metabolism. 2018;85:286–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim M, Astapova II, Flier SN, Hannou SA, Doridot L, Sargsyan A, et al. Intestinal, but not hepatic, ChREBP is required for fructose tolerance. JCI Insight. 2017;2:e96703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kato T, Iizuka K, Takao K, Horikawa Y, Kitamura T, Takeda J. ChREBP-knockout mice show sucrose intolerance and fructose malabsorption. Nutrients. 2018;10:340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61•.Jang C, Hui S, Lu W, Cowan AJ, Morscher RJ, Lee G, et al. The small intestine converts dietary fructose into glucose and organic acids. Cell Metab. 2018;27:351–361 e353 [DOI] [PMC free article] [PubMed] [Google Scholar]; This original research article demonstrates that low doses of dietary fructose are primarily metabolized by the intestine; however, high doses of dietary fructose reach both the liver and colonic microbiota for further metabolism.
- 63.Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature. 2020;579:586–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63•.Andres-Hernando A, Orlicky DJ, Kuwabara M, Ishimoto T, Nakagawa T, Johnson RJ, et al. Deletion of fructokinase in the liver or in the intestine reveals differential effects on sugar-induced metabolic dysfunction. Cell Metab. 2020;32:117–127 e113 [DOI] [PMC free article] [PubMed] [Google Scholar]; This original research article demonstrates that global deletion of KHK reduces sugar intake and prevents fructose-induced metabolic syndrome in mice. Meanwhile, sugar intake is regulated by intestinal KHK activity while metabolic syndrome is controlled by hepatic fructose metabolism.
- 65.Laitakari A, Tapio J, Makela KA, Herzig KH, Dengler F, Gylling H, et al. HIF-P4H-2 inhibition enhances intestinal fructose metabolism and induces thermogenesis protecting against NAFLD. J Mol Med (Berl). 2020;98:719–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Helsley RN, Softic S. Fructose metabolism by the guts cuts liver fat. J Mol Med (Berl). 2020;98:733–4. [DOI] [PubMed] [Google Scholar]
- 67.Corpe CP, Basaleh MM, Affleck J, Gould G, Jess TJ, Kellett GL. The regulation of GLUT5 and GLUT2 activity in the adaptation of intestinal brush-border fructose transport in diabetes. Pflugers Arch. 1996;432:192–201. [DOI] [PubMed] [Google Scholar]
- 68.Burant CF, Flink S, DePaoli AM, Chen J, Lee WS, Hediger MA, et al. Small intestine hexose transport in experimental diabetes. Increased transporter mRNA and protein expression in enterocytes. J Clin Invest. 1994;93:578–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dyer J, Wood IS, Palejwala A, Ellis A, Shirazi-Beechey SP. Expression of monosaccharide transporters in intestine of diabetic humans. Am J Physiol Gastrointest Liver Physiol. 2002;282:G241–8. [DOI] [PubMed] [Google Scholar]
- 70.George Thompson AM, Ursu O, Babkin P, Iancu CV, Whang A, Oprea TI, et al. Discovery of a specific inhibitor of human GLUT5 by virtual screening and in vitro transport evaluation. Sci Rep. 2016;6:24240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kuhre RE, Gribble FM, Hartmann B, Reimann F, Windelov JA, Rehfeld JF, et al. Fructose stimulates GLP-1 but not GIP secretion in mice, rats, and humans. Am J Physiol Gastrointest Liver Physiol. 2014;306:G622–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Galderisi A, Giannini C, Van Name M, Caprio S. Fructose consumption contributes to hyperinsulinemia in adolescents with obesity through a GLP-1-mediated mechanism. J Clin Endocrinol Metab. 2019;104:3481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shojaee-Moradie F, Ma Y, Lou S, Hovorka R, Umpleby AM. Prandial hypertriglyceridemia in metabolic syndrome is due to an overproduction of both chylomicron and VLDL triacylglycerol. Diabetes. 2013;62:4063–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Haidari M, Leung N, Mahbub F, Uffelman KD, Kohen-Avramoglu R, Lewis GF, et al. Fasting and postprandial overproduction of intestinally derived lipoproteins in an animal model of insulin resistance. Evidence that chronic fructose feeding in the hamster is accompanied by enhanced intestinal de novo lipogenesis and ApoB48-containing lipoprotein overproduction. J Biol Chem. 2002;277:31646–55. [DOI] [PubMed] [Google Scholar]
- 75.Al-Jawadi A, Patel CR, Shiarella RJ, Romelus E, Auvinen M, Guardia J, et al. Cell-type-specific, ketohexokinase-dependent induction by fructose of lipogenic gene expression in mouse small intestine. J Nutr. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Federico LM, Naples M, Taylor D, Adeli K. Intestinal insulin resistance and aberrant production of apolipoprotein B48 lipoproteins in an animal model of insulin resistance and metabolic dyslipidemia: evidence for activation of protein tyrosine phosphatase-1B, extracellular signal-related kinase, and sterol regulatory element-binding protein-1c in the fructose-fed hamster intestine. Diabetes. 2006;55:1316–26. [DOI] [PubMed] [Google Scholar]
- 77.Lewis GF, Uffelman K, Naples M, Szeto L, Haidari M, Adeli K. Intestinal lipoprotein overproduction, a newly recognized component of insulin resistance, is ameliorated by the insulin sensitizer rosiglitazone: studies in the fructose-fed Syrian golden hamster. Endocrinology. 2005;146:247–55. [DOI] [PubMed] [Google Scholar]
- 78.Xiao C, Dash S, Morgantini C, Lewis GF. Novel role of enteral monosaccharides in intestinal lipoprotein production in healthy humans. Arterioscler Thromb Vasc Biol. 2013;33:1056–62. [DOI] [PubMed] [Google Scholar]
- 79.Do MH, Lee E, Oh MJ, Kim Y, Park HY. High-glucose or - fructose diet cause changes of the gut microbiota and metabolic disorders in mice without body weight change. Nutrients. 2018;10:639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Volynets V, Louis S, Pretz D, Lang L, Ostaff MJ, Wehkamp J, et al. Intestinal barrier function and the gut microbiome are differentially affected in mice fed a Western-style diet or drinking water supplemented with fructose. J Nutr. 2017;147:770–80. [DOI] [PubMed] [Google Scholar]
- 81.Pang J, Xu W, Zhang X, Wong GL, Chan AW, Chan HY, et al. Significant positive association of endotoxemia with histological severity in 237 patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2017;46:175–82. [DOI] [PubMed] [Google Scholar]
- 82.Cho YE, Kim DK, Seo W, Gao B, Yoo SH, Song BJ. Fructose promotes leaky gut, endotoxemia, and liver fibrosis through ethanol-inducible cytochrome P450–2E1-mediated oxidative and nitrative stress. Hepatology. 2019. 10.1002/hep.30652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bergheim I, Weber S, Vos M, Kramer S, Volynets V, Kaserouni S, et al. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: role of endotoxin. J Hepatol. 2008;48:983–92. [DOI] [PubMed] [Google Scholar]
- 84.Sen T, Cawthon CR, Ihde BT, Hajnal A, DiLorenzo PM, de La Serre CB, et al. Diet-driven microbiota dysbiosis is associated with vagal remodeling and obesity. Physiol Behav. 2017;173:305–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ferrere G, Leroux A, Wrzosek L, Puchois V, Gaudin F, Ciocan D, et al. Activation of Kupffer cells is associated with a specific dysbiosis induced by fructose or high fat diet in mice. PLoS One. 2016;11:e0146177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wood IS, Trayhurn P. Glucose transporters (GLUT and SGLT): expanded families of sugar transport proteins. Br J Nutr. 2003;89:3–9. [DOI] [PubMed] [Google Scholar]
- 87.Debosch BJ, Chen Z, Saben JL, Finck BN, Moley KH. Glucose transporter 8 (GLUT8) mediates fructose-induced de novo lipogenesis and macrosteatosis. J Biol Chem. 2014;289:10989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schmidt S, Joost HG, Schurmann A. GLUT8, the enigmatic intracellular hexose transporter. Am J Physiol Endocrinol Metab. 2009;296:E614–8. [DOI] [PubMed] [Google Scholar]
- 89.Manolescu AR, Witkowska K, Kinnaird A, Cessford T, Cheeseman C. Facilitated hexose transporters: new perspectives on form and function. Physiology (Bethesda). 2007;22:234–40. [DOI] [PubMed] [Google Scholar]
- 90.Zhang Y, Shaikh N, Ferey JL, Wankhade UD, Chintapalli SV, Higgins CB, et al. Lactotrehalose, an analog of trehalose, increases energy metabolism without promoting Clostridioides difficile infection in mice. Gastroenterology 2020;158:1402–1416 e1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, Maclean PS, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc Natl Acad Sci U S A. 2012;109:4320–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ishimoto T, Lanaspa MA, Rivard CJ, Roncal-Jimenez CA, Orlicky DJ, Cicerchi C, et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology. 2013;58:1632–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92•.Softic S, Gupta MK, Wang GX, Fujisaka S, O’Neill BT, Rao TN, et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J Clin Invest. 2017;127:4059–74 [DOI] [PMC free article] [PubMed] [Google Scholar]; This original research article demonstrates that fructose supplementation on a high-fat diet worsens metabolic complications as compared with a glucose-supplemented diet, despite similar caloric intake. Dietary fructose feeding led to increased SREBP1c-regulated gene expression and impaired hepatic insulin signaling, as compared to mice fed glucose.
- 94.Park TJ, Reznick J, Peterson BL, Blass G, Omerbasic D, Bennett NC, et al. Fructose-driven glycolysis supports anoxia resistance in the naked mole-rat. Science. 2017;356:307–11. [DOI] [PubMed] [Google Scholar]
- 95.Lanaspa MA, Sanchez-Lozada LG, Cicerchi C, Li N, Roncal-Jimenez CA, Ishimoto T, et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS One. 2012;7:e47948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shi JH, Lu JY, Chen HY, Wei CC, Xu X, Li H, et al. Liver ChREBP protects against fructose-induced glycogenic hepatotoxicity by regulating L-type pyruvate kinase. Diabetes. 2020;69:591–602. [DOI] [PubMed] [Google Scholar]
- 97.Ali M, Rellos P, Cox TM. Hereditary fructose intolerance. J Med Genet. 1998;35:353–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lanaspa MA, Andres-Hernando A, Orlicky DJ, Cicerchi C, Jang C, Li N, et al. Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J Clin Invest. 2018;128:2226–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Softic S, Kirby M, Berger NG, Shroyer NF, Woods SC, Kohli R. Insulin concentration modulates hepatic lipid accumulation in mice in part via transcriptional regulation of fatty acid transport proteins. PLoS One. 2012;7:e38952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Softic S, Cohen DE, Kahn CR. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig Dis Sci. 2016;61:1282–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Swanson JE, Laine DC, Thomas W, Bantle JP. Metabolic effects of dietary fructose in healthy subjects. Am J Clin Nutr. 1992;55:851–6. [DOI] [PubMed] [Google Scholar]
- 102.Te Morenga LA, Howatson AJ, Jones RM, Mann J. Dietary sugars and cardiometabolic risk: systematic review and meta-analyses of randomized controlled trials of the effects on blood pressure and lipids. Am J Clin Nutr. 2014;100:65–79. [DOI] [PubMed] [Google Scholar]
- 103.Taghibiglou C, Carpentier A, Van Iderstine SC, Chen B, Rudy D, Aiton A, et al. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J Biol Chem. 2000;275:8416–25. [DOI] [PubMed] [Google Scholar]
- 104.Lustig RH, Mulligan K, Noworolski SM, Tai VW, Wen MJ, Erkin-Cakmak A, et al. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity (Silver Spring). 2016;24:453–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gugliucci A, Lustig RH, Caccavello R, Erkin-Cakmak A, Noworolski SM, Tai VW, et al. Short-term isocaloric fructose restriction lowers apoC-III levels and yields less atherogenic lipoprotein profiles in children with obesity and metabolic syndrome. Atherosclerosis. 2016;253:171–7. [DOI] [PubMed] [Google Scholar]
- 106.Topping DL, Mayes PA. The concentration of fructose, glucose and lactate in the splanchnic blood vessels of rats absorbing fructose. Nutr Metab. 1971;13:331–8. [DOI] [PubMed] [Google Scholar]
- 107.Mendeloff AI, Weichselbaum TE. Role of the human liver in the assimilation of intravenously administered fructose. Metabolism. 1953;2:450–8. [PubMed] [Google Scholar]
- 108.Heinz F, Lamprecht W, Kirsch J. Enzymes of fructose metabolism in human liver. J Clin Invest. 1968;47:1826–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ouyang X, Cirillo P, Sautin Y, McCall S, Bruchette JL, Diehl AM, et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol. 2008;48:993–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109••.Softic S, Meyer JG, Wang GX, Gupta MK, Batista TM, Lauritzen H, et al. Dietary sugars alter hepatic fatty acid oxidation via transcriptional and post-translational modifications of mitochondrial proteins. Cell Metab. 2019;30:735–753 e734 [DOI] [PMC free article] [PubMed] [Google Scholar]; This original research article demonstrates that high-fat diet supplemented with fructose impairs mitochondrial size, function, and protein acetylation resulting in decreased fatty acid oxidation.
- 111.Meyer JG, Softic S, Basisty N, Rardin MJ, Verdin E, Gibson BW, et al. Temporal dynamics of liver mitochondrial protein acetylation and succinylation and metabolites due to high fat diet and/or excess glucose or fructose. PLoS One. 2018;13:e0208973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang G, Meyer JG, Cai W, Softic S, Li ME, Verdin E, et al. Regulation of UCP1 and mitochondrial metabolism in brown adipose tissue by reversible succinylation. Mol Cell. 2019;74:844–57 e847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Abdelmalek MF, Suzuki A, Guy C, Unalp-Arida A, Colvin R, Johnson RJ, et al. Nonalcoholic steatohepatitis clinical research N: increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jin R, Le NA, Liu S, Farkas Epperson M, Ziegler TR, Welsh JA, et al. Children with NAFLD are more sensitive to the adverse metabolic effects of fructose beverages than children without NAFLD. J Clin Endocrinol Metab. 2012;97:E1088–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jensen T, Abdelmalek MF, Sullivan S, Nadeau KJ, Green M, Roncal C, et al. Fructose and sugar: a major mediator of non-alcoholic fatty liver disease. J Hepatol. 2018;68:1063–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251–64. [DOI] [PubMed] [Google Scholar]
- 117.Yilmaz Y Review article: fructose in non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2012;35:1135–44. [DOI] [PubMed] [Google Scholar]
- 118.Softic S, Kahn CR. Fatty liver disease: is it nonalcoholic fatty liver disease or obesity-associated fatty liver disease? Eur J Gastroenterol Hepatol. 2019;31:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Attia SL, Softic S, Mouzaki M. Evolving role for pharmacotherapy in NAFLD/NASH. Clin Transl Sci. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Macdonald I, Turner LJ. Serum-fructose levels after sucrose or its constituent monosaccharides. Lancet. 1968;1:841–3. [DOI] [PubMed] [Google Scholar]
- 121.Holdsworth CD, Dawson AM. Absorption of fructose in man. Proc Soc Exp Biol Med. 1965;118:142–5. [DOI] [PubMed] [Google Scholar]
- 122.Bjorkman O, Felig P. Role of the kidney in the metabolism of fructose in 60-hour fasted humans. Diabetes. 1982;31:516–20. [DOI] [PubMed] [Google Scholar]
- 123.Burant CF, Saxena M. Rapid reversible substrate regulation of fructose transporter expression in rat small intestine and kidney. Am J Phys. 1994;267:G71–9. [DOI] [PubMed] [Google Scholar]
- 124.Leturque A, Brot-Laroche E, Le Gall M. GLUT2 mutations, translocation, and receptor function in diet sugar managing. Am J Physiol Endocrinol Metab. 2009;296:E985–92. [DOI] [PubMed] [Google Scholar]
- 125.Keembiyehetty C, Augustin R, Carayannopoulos MO, Steer S, Manolescu A, Cheeseman CI, et al. Mouse glucose transporter 9 splice variants are expressed in adult liver and kidney and are upregulated in diabetes. Mol Endocrinol. 2006;20:686–97. [DOI] [PubMed] [Google Scholar]
- 126.Horiba N, Masuda S, Ohnishi C, Takeuchi D, Okuda M, Inui K. Na(+)-dependent fructose transport via rNaGLT1 in rat kidney. FEBS Lett. 2003;546:276–80. [DOI] [PubMed] [Google Scholar]
- 127.Tazawa S, Yamato T, Fujikura H, Hiratochi M, Itoh F, Tomae M, et al. SLC5A9/SGLT4, a new Na+-dependent glucose transporter, is an essential transporter for mannose, 1,5-anhydro-D-glucitol, and fructose. Life Sci. 2005;76:1039–50. [DOI] [PubMed] [Google Scholar]
- 128.Grempler R, Augustin R, Froehner S, Hildebrandt T, Simon E, Mark M, et al. Functional characterisation of human SGLT-5 as a novel kidney-specific sodium-dependent sugar transporter. FEBS Lett. 2012;586:248–53. [DOI] [PubMed] [Google Scholar]
- 129.Fukuzawa T, Fukazawa M, Ueda O, Shimada H, Kito A, Kakefuda M, et al. SGLT5 reabsorbs fructose in the kidney but its deficiency paradoxically exacerbates hepatic steatosis induced by fructose. PLoS One. 2013;8:e56681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gonzalez-Vicente A, Cabral PD, Hong NJ, Asirwatham J, Saez F, Garvin JL. Fructose reabsorption by rat proximal tubules: role of Na(+)-linked cotransporters and the effect of dietary fructose. American Journal of Physiology Renal Physiology. 2019;316: F473–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Heinz F, Schlegel F, Krause PH. Enzymes of fructose metabolism in human kidney. Enzyme. 1975;19:85–92. [DOI] [PubMed] [Google Scholar]
- 132.Kawasaki T, Akanuma H, Yamanouchi T. Increased fructose concentrations in blood and urine in patients with diabetes. Diabetes Care. 2002;25:353–7. [DOI] [PubMed] [Google Scholar]
- 133.Gomez-Samano MA, Almeda-Valdes P, Cuevas-Ramos D, Navarro-Flores MF, Espinosa-Salazar HD, Martinez-Saavedra M, et al. A higher fructose intake is associated with greater albuminuria in subjects with type 2 diabetes mellitus. International Journal of Nephrology. 2018;2018:5459439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shoham DA, Durazo-Arvizu R, Kramer H, Luke A, Vupputuri S, Kshirsagar A, et al. Sugary soda consumption and albuminuria: results from the National Health and Nutrition Examination Survey, 1999–2004. PLoS One. 2008;3:e3431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Brymora A, Flisinski M, Johnson RJ, Goszka G, Stefanska A, Manitius J. Low-fructose diet lowers blood pressure and inflammation in patients with chronic kidney disease. Nephrol Dial Transplant. 2012;27:608–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cirillo P, Sato W, Reungjui S, Heinig M, Gersch M, Sautin Y, et al. Uric acid, the metabolic syndrome and renal disease. Journal of the American Society of Nephrology. 2006;17:S165–8. [DOI] [PubMed] [Google Scholar]
- 137.Johnson RJ, Nakagawa T, Sanchez-Lozada LG, Shafiu M, Sundaram S, Le M, et al. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes. 2013;62:3307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Maahs DM, Caramori L, Cherney DZ, Galecki AT, Gao C, Jalal D, et al. Uric acid lowering to prevent kidney function loss in diabetes: the Preventing Early Renal Function Loss (PERL) allopurinol study. Current Diabetes Reports. 2013;13:550–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sanchez-Lozada LG, Soto V, Tapia E, Avila-Casado C, Sautin YY, Nakagawa T, et al. Role of oxidative stress in the renal abnormalities induced by experimental hyperuricemia. American Journal of Physiology Renal Physiology. 2008;295:F1134–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lanaspa MA, Ishimoto T, Cicerchi C, Tamura Y, Roncal-Jimenez CA, Chen W, et al. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. Journal of the American Society of Nephrology. 2014;25: 2526–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cirillo P, Gersch MS, Mu W, Scherer PM, Kim KM, Gesualdo L, et al. Ketohexokinase-dependent metabolism of fructose induces proinflammatory mediators in proximal tubular cells. Journal of the American Society of Nephrology. 2009;20:545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gersch MS, Mu W, Cirillo P, Reungjui S, Zhang L, Roncal C, et al. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. American Journal of Physiology Renal Physiology. 2007;293:F1256–61. [DOI] [PubMed] [Google Scholar]
- 143.Nakayama T, Kosugi T, Gersch M, Connor T, Sanchez-Lozada LG, Lanaspa MA, et al. Dietary fructose causes tubulointerstitial injury in the normal rat kidney. American Journal of Physiology Renal Physiology. 2010;298:F712–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Burch HB, Choi S, Dence CN, Alvey TR, Cole BR, Lowry OH. Metabolic effects of large fructose loads in different parts of the rat nephron. J Biol Chem. 1980;255:8239–44. [PubMed] [Google Scholar]
- 145.Doke T, Ishimoto T, Hayasaki T, Ikeda S, Hasebe M, Hirayama A, et al. Lacking ketohexokinase-A exacerbates renal injury in streptozotocin-induced diabetic mice. Metab Clin Exp. 2018;85: 161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Li X, Qian X, Peng LX, Jiang Y, Hawke DH, Zheng Y, et al. A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat Cell Biol. 2016;18:561–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Roncal Jimenez CA, Ishimoto T, Lanaspa MA, Rivard CJ, Nakagawa T, Ejaz AA, et al. Fructokinase activity mediates dehydration-induced renal injury. Kidney Int. 2014;86:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dvornik E, Simard-Duquesne N, Krami M, Sestanj K, Gabbay KH, Kinoshita JH, et al. Polyol accumulation in galactosemic and diabetic rats: control by an aldose reductase inhibitor. Science. 1973;182:1146–8. [DOI] [PubMed] [Google Scholar]
- 149.Roncal-Jimenez CA, Ishimoto T, Lanaspa MA, Milagres T, Hernando AA, Jensen T, et al. Aging-associated renal disease in mice is fructokinase dependent. American Journal of Physiology Renal Physiology. 2016;311:F722–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Andres-Hernando A, Li N, Cicerchi C, Inaba S, Chen W, Roncal-Jimenez C, et al. Protective role of fructokinase blockade in the pathogenesis of acute kidney injury in mice. Nat Commun. 2017;8:14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Madero M, Perez-Pozo SE, Jalal D, Johnson RJ, Sanchez-Lozada LG. Dietary fructose and hypertension. Curr Hypertens Rep. 2011;13:29–35. [DOI] [PubMed] [Google Scholar]
- 152.Singh AK, Amlal H, Haas PJ, Dringenberg U, Fussell S, Barone SL, et al. Fructose-induced hypertension: essential role of chloride and fructose absorbing transporters PAT1 and Glut5. Kidney Int. 2008;74:438–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Queiroz-Leite GD, Crajoinas RO, Neri EA, Bezerra CN, Girardi AC, Reboucas NA, et al. Fructose acutely stimulates NHE3 activity in kidney proximal tubule. Kidney & blood pressure research. 2012;36:320–34. [DOI] [PubMed] [Google Scholar]
- 154.Hayasaki T, Ishimoto T, Doke T, Hirayama A, Soga T, Furuhashi K, et al. Fructose increases the activity of sodium hydrogen exchanger in renal proximal tubules that is dependent on ketohexokinase. J Nutr Biochem. 2019;71:54–62. [DOI] [PubMed] [Google Scholar]
- 155.Froesch ER, Ginsberg JL. Fructose metabolism of adipose tissue. I. Comparison of fructose and glucose metabolism in epididymal adipose tissue of normal rats. J Biol Chem. 1962;237:3317–24. [PubMed] [Google Scholar]
- 156.Kayano T, Burant CF, Fukumoto H, Gould GW, Fan YS, Eddy RL, et al. Human facilitative glucose transporters. Isolation, functional characterization, and gene localization of cDNAs encoding an isoform (GLUT5) expressed in small intestine, kidney, muscle, and adipose tissue and an unusual glucose transporter pseudogene-like sequence (GLUT6). J Biol Chem. 1990;265:13276–82. [PubMed] [Google Scholar]
- 157.Hajduch E, Darakhshan F, Hundal HS. Fructose uptake in rat adipocytes: GLUT5 expression and the effects of streptozotocin-induced diabetes. Diabetologia. 1998;41:821–8. [DOI] [PubMed] [Google Scholar]
- 158.Wood IS, Wang B, Lorente-Cebrian S, Trayhurn P. Hypoxia increases expression of selective facilitative glucose transporters (GLUT) and 2-deoxy-D-glucose uptake in human adipocytes. Biochem Biophys Res Commun. 2007;361:468–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zwarts I, van Zutphen T, Kruit JK, Liu W, Oosterveer MH, Verkade HJ, et al. Identification of the fructose transporter GLUT5 (SLC2A5) as a novel target of nuclear receptor LXR. Sci Rep. 2019;9:9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Matosin-Matekalo M, Mesonero JE, Laroche TJ, Lacasa M, Brot-Laroche E. Glucose and thyroid hormone co-regulate the expression of the intestinal fructose transporter GLUT5. Biochem J. 1999;339(Pt 2):233–9. [PMC free article] [PubMed] [Google Scholar]
- 161.Mochizuki K, Yagi E, Sakaguchi N, Mochizuki H, Takabe S, Kuranuki S, et al. The critical period for thyroid hormone responsiveness through thyroid hormone receptor isoform alpha in the postnatal small intestine. Biochim Biophys Acta. 2007;1770:609–16. [DOI] [PubMed] [Google Scholar]
- 162.Douard V, Choi HI, Elshenawy S, Lagunoff D, Ferraris RP. Developmental reprogramming of rat GLUT5 requires glucocorticoid receptor translocation to the nucleus. J Physiol. 2008;586: 3657–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shepherd PR, Gibbs EM, Wesslau C, Gould GW, Kahn BB. Human small intestine facilitative fructose/glucose transporter (GLUT5) is also present in insulin-responsive tissues and brain. Investigation of biochemical characteristics and translocation. Diabetes. 1992;41:1360–5. [DOI] [PubMed] [Google Scholar]
- 164.Varma V, Boros LG, Nolen GT, Chang CW, Wabitsch M, Beger RD, et al. Metabolic fate of fructose in human adipocytes: a targeted (13)C tracer fate association study. Metabolomics. 2015;11:529–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Du L, Heaney AP. Regulation of adipose differentiation by fructose and GluT5. Mol Endocrinol. 2012;26:1773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Marek G, Pannu V, Shanmugham P, Pancione B, Mascia D, Crosson S, et al. Adiponectin resistance and proinflammatory changes in the visceral adipose tissue induced by fructose consumption via ketohexokinase-dependent pathway. Diabetes. 2015;64:508–18. [DOI] [PubMed] [Google Scholar]
- 167.Pollock NK, Bundy V, Kanto W, Davis CL, Bernard PJ, Zhu H, et al. Greater fructose consumption is associated with cardiometabolic risk markers and visceral adiposity in adolescents. J Nutr. 2012;142:251–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bremer AA, Stanhope KL, Graham JL, Cummings BP, Wang W, Saville BR, et al. Fructose-fed rhesus monkeys: a nonhuman primate model of insulin resistance, metabolic syndrome and type 2 diabetes. Clin Transl Sci. 2011;4:243–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Softic S, Boucher J, Solheim MH, Fujisaka S, Haering MF, Homan EP, et al. Lipodystrophy due to adipose tissue-specific insulin receptor knockout results in progressive NAFLD. Diabetes. 2016;65:2187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Laron Z Essential benign fructosuria. Arch Dis Child. 1961;36: 273–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tran C Inborn errors of fructose metabolism. What can we learn from them? Nutrients. 2017;9:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ahlborg G, Bjorkman O. Splanchnic and muscle fructose metabolism during and after exercise. J Appl Physiol (1985). 1990;69: 1244–51. [DOI] [PubMed] [Google Scholar]
- 173.Thurmond DC, Pessin JE. Molecular machinery involved in the insulin-regulated fusion of GLUT4-containing vesicles with the plasma membrane (review). Mol Membr Biol. 2001;18:237–45. [DOI] [PubMed] [Google Scholar]
- 174.Darakhshan F, Hajduch E, Kristiansen S, Richter EA, Hundal HS. Biochemical and functional characterization of the GLUT5 fructose transporter in rat skeletal muscle. Biochem J. 1998;336(Pt 2): 361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hajduch E, Litherland GJ, Turban S, Brot-Laroche E, Hundal HS. Insulin regulates the expression of the GLUT5 transporter in L6 skeletal muscle cells. FEBS Lett. 2003;549:77–82. [DOI] [PubMed] [Google Scholar]
- 176.Stuart CA, Howell ME, Yin D. Overexpression of GLUT5 in diabetic muscle is reversed by pioglitazone. Diabetes Care. 2007;30:925–31. [DOI] [PubMed] [Google Scholar]
- 177.Miller CO, Yang X, Lu K, Cao J, Herath K, Rosahl TW, et al. Ketohexokinase knockout mice, a model for essential fructosuria, exhibit altered fructose metabolism and are protected from diet-induced metabolic defects. Am J Physiol Endocrinol Metab. 2018;315:E386–93. [DOI] [PubMed] [Google Scholar]
- 178.Rai AK, Jaiswal N, Maurya CK, Sharma A, Ahmad I, Ahmad S, et al. Fructose-induced AGEs-RAGE signaling in skeletal muscle contributes to impairment of glucose homeostasis. J Nutr Biochem. 2019;71:35–44. [DOI] [PubMed] [Google Scholar]
- 179.Baena M, Sanguesa G, Davalos A, Latasa MJ, Sala-Vila A, Sanchez RM, et al. Fructose, but not glucose, impairs insulin signaling in the three major insulin-sensitive tissues. Sci Rep. 2016;6:26149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Togashi N, Ura N, Higashiura K, Murakami H, Shimamoto K. The contribution of skeletal muscle tumor necrosis factor-alpha to insulin resistance and hypertension in fructose-fed rats. J Hypertens. 2000;18:1605–10. [DOI] [PubMed] [Google Scholar]
- 181.Austin RL, Rune A, Bouzakri K, Zierath JR, Krook A. siRNA-mediated reduction of inhibitor of nuclear factor-kappaB kinase prevents tumor necrosis factor-alpha-induced insulin resistance in human skeletal muscle. Diabetes. 2008;57:2066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.del Aguila LF, Claffey KP, Kirwan JP. TNF-alpha impairs insulin signaling and insulin stimulation of glucose uptake in C2C12 muscle cells. Am J Phys. 1999;276:E849–55. [DOI] [PubMed] [Google Scholar]
- 183.Rattanavichit Y, Chukijrungroat N, Saengsirisuwan V. Sex differences in the metabolic dysfunction and insulin resistance of skeletal muscle glucose transport following high fructose ingestion. Am J Physiol Regul Integr Comp Physiol. 2016;311:R1200–12. [DOI] [PubMed] [Google Scholar]
- 184.Douard V, Ferraris RP. The role of fructose transporters in diseases linked to excessive fructose intake. J Physiol. 2013;591: 401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Asp Med. 2013;34:121–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kyriazis GA, Soundarapandian MM, Tyrberg B. Sweet taste receptor signaling in beta cells mediates fructose-induced potentiation of glucose-stimulated insulin secretion. Proc Natl Acad Sci U S A. 2012;109:E524–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Grant AM, Christie MR, Ashcroft SJ. Insulin release from human pancreatic islets in vitro. Diabetologia. 1980;19:114–7. [DOI] [PubMed] [Google Scholar]
- 188.Hayward BE, Bonthron DT. Structure and alternative splicing of the ketohexokinase gene. Eur J Biochem. 1998;257:85–91. [DOI] [PubMed] [Google Scholar]
- 189.Springer N, Lindbloom-Hawley S, Schermerhorn T. Tissue expression of ketohexokinase in cats. Res Vet Sci. 2009;87:115–7. [DOI] [PubMed] [Google Scholar]
- 190.Sener A, Giroix MH, Malaisse WJ. Hexose metabolism in pancreatic islets. The phosphorylation of fructose. Eur J Biochem. 1984;144:223–6. [DOI] [PubMed] [Google Scholar]
- 191.Malaisse WJ, Malaisse-Lagae F, Davies DR, Van Schaftingen E. Presence of fructokinase in pancreatic islets. FEBS Lett. 1989;255:175–8. [DOI] [PubMed] [Google Scholar]
- 192.Giroix MH, Jijakli H, Courtois P, Zhang Y, Sener A, Malaisse WJ. Fructokinase activity in rat liver, ileum, parotid gland, pancreas, pancreatic islet, B and non-B islet cell homogenates. Int J Mol Med. 2006;17:517–22. [PubMed] [Google Scholar]
- 193.Thurston JH, Levy CA, Warren SK, Jones EM. Permeability of the blood-brain barrier to fructose and the anaerobic use of fructose in the brains of young mice. J Neurochem. 1972;19:1685–96. [DOI] [PubMed] [Google Scholar]
- 194.Wolfgang MJ, Cha SH, Sidhaye A, Chohnan S, Cline G, Shulman GI, et al. Regulation of hypothalamic malonyl-CoA by central glucose and leptin. Proc Natl Acad Sci U S A. 2007;104:19285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Payne J, Maher F, Simpson I, Mattice L, Davies P. Glucose transporter Glut 5 expression in microglial cells. Glia. 1997;21:327–31. [DOI] [PubMed] [Google Scholar]
- 196.Mantych GJ, James DE, Devaskar SU. Jejunal/kidney glucose transporter isoform (Glut-5) is expressed in the human blood-brain barrier. Endocrinology. 1993;132:35–40. [DOI] [PubMed] [Google Scholar]
- 197.Nualart F, Godoy A, Reinicke K. Expression of the hexose transporters GLUT1 and GLUT2 during the early development of the human brain. Brain Res. 1999;824:97–104. [DOI] [PubMed] [Google Scholar]
- 198.Shu HJ, Isenberg K, Cormier RJ, Benz A, Zorumski CF. Expression of fructose sensitive glucose transporter in the brains of fructose-fed rats. Neuroscience. 2006;140:889–95. [DOI] [PubMed] [Google Scholar]
- 199.Vannucci SJ, Maher F, Simpson IA. Glucose transporter proteins in brain: delivery of glucose to neurons and glia. Glia. 1997;21:2–21. [DOI] [PubMed] [Google Scholar]
- 200.Cunningham P, Afzal-Ahmed I, Naftalin RJ. Docking studies show that D-glucose and quercetin slide through the transporter GLUT1. J Biol Chem. 2006;281:5797–803. [DOI] [PubMed] [Google Scholar]
- 201.Oldendorf WH. Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am J Phys. 1971;221:1629–39. [DOI] [PubMed] [Google Scholar]
- 202.Hwang JJ, Johnson A, Cline G, Belfort-DeAguiar R, Snegovskikh D, Khokhar B, et al. Fructose levels are markedly elevated in cerebrospinal fluid compared to plasma in pregnant women. PLoS One. 2015;10:e0128582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Hwang JJ, Jiang L, Hamza M, Dai F, Belfort-DeAguiar R, Cline G, et al. The human brain produces fructose from glucose. JCI Insight. 2017;2:e90508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Oppelt SA, Zhang W, Tolan DR. Specific regions of the brain are capable of fructose metabolism. Brain Res. 2017;1657:312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gaitonde MK, Richter D. Changes with age in the utilization of glucose carbon in liver and brain. J Neurochem. 1966;13:1309–16. [DOI] [PubMed] [Google Scholar]
- 206.Lowette K, Roosen L, Tack J, Vanden Berghe P. Effects of high-fructose diets on central appetite signaling and cognitive function. Front Nutr. 2015;2(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Luo S, Monterosso JR, Sarpelleh K, Page KA. Differential effects of fructose versus glucose on brain and appetitive responses to food cues and decisions for food rewards. Proc Natl Acad Sci U S A. 2015;112:6509–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bantle JP. Is fructose the optimal low glycemic index sweetener? Nestle Nutr Workshop Ser Clin Perform Programme. 2006;11:83–95. [DOI] [PubMed] [Google Scholar]
- 209.Page KA, Chan O, Arora J, Belfort-Deaguiar R, Dzuira J, Roehmholdt B, et al. Effects of fructose vs glucose on regional cerebral blood flow in brain regions involved with appetite and reward pathways. JAMA. 2013;309:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Cha SH, Wolfgang M, Tokutake Y, Chohnan S, Lane MD. Differential effects of central fructose and glucose on hypothalamic malonyl-CoA and food intake. Proc Natl Acad Sci U S A. 2008;105:16871–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Woods SC, Lotter EC, McKay LD, Porte D Jr. Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature. 1979;282:503–5. [DOI] [PubMed] [Google Scholar]
- 212.Suga A, Hirano T, Kageyama H, Osaka T, Namba Y, Tsuji M, et al. Effects of fructose and glucose on plasma leptin, insulin, and insulin resistance in lean and VMH-lesioned obese rats. Am J Physiol Endocrinol Metab. 2000;278:E677–83. [DOI] [PubMed] [Google Scholar]
- 213.Lindqvist A, Baelemans A, Erlanson-Albertsson C. Effects of sucrose, glucose and fructose on peripheral and central appetite signals. Regul Pept. 2008;150:26–32. [DOI] [PubMed] [Google Scholar]
- 214.Burant CF, Takeda J, Brot-Laroche E, Bell GI, Davidson NO. Fructose transporter in human spermatozoa and small intestine is GLUT5. J Biol Chem. 1992;267:14523–6. [PubMed] [Google Scholar]
- 215.Shibata K, Fukuwatari T. High d(+)-fructose diet adversely affects testicular weight gain in weaning rats horizontal line protection by moderate d(+)-glucose diet. Nutr Metab Insights. 2013;6:29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Concha II, Velasquez FV, Martinez JM, Angulo C, Droppelmann A, Reyes AM, et al. Human erythrocytes express GLUT5 and transport fructose. Blood. 1997;89:4190–5. [PubMed] [Google Scholar]
- 217.Petersen A, Kappler F, Szwergold BS, Brown TR. Fructose metabolism in the human erythrocyte. Phosphorylation to fructose 3-phosphate. Biochem J. 1992;284(Pt 2):363–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hamada Y, Araki N, Koh N, Nakamura J, Horiuchi S, Hotta N. Rapid formation of advanced glycation end products by intermediate metabolites of glycolytic pathway and polyol pathway. Biochem Biophys Res Commun. 1996;228:539–43. [DOI] [PubMed] [Google Scholar]
- 219.Kuck JF Jr. Glucose metabolism and fructose synthesis in the diabetic rat lens. Investig Ophthalmol. 1962;1:390–5. [PubMed] [Google Scholar]
- 220.Lal S, Szwergold BS, Taylor AH, Randall WC, Kappler F, Wells-Knecht K, et al. Metabolism of fructose-3-phosphate in the diabetic rat lens. Arch Biochem Biophys. 1995;318:191–9. [DOI] [PubMed] [Google Scholar]
- 221.Lee AY, Chung SK, Chung SS. Demonstration that polyol accumulation is responsible for diabetic cataract by the use of transgenic mice expressing the aldose reductase gene in the lens. Proc Natl Acad Sci U S A. 1995;92:2780–4. [DOI] [PMC free article] [PubMed] [Google Scholar]