

Abstract
Adeno-associated virus (AAV)-mediated systemic micro-dystrophin (μDys) therapy is currently in clinical trials. The hope is to permanently improve the life quality of Duchenne muscular dystrophy (DMD) patients. Numerous preclinical studies have been conducted to support these trials. However, none examined whether a single therapy at a young age can lead to lifelong disease amelioration. To address this critical question, we injected 1 × 1013 vg particles/mouse of an AAV serotype-9 μDys vector to 3-month-old mdx mice through the tail vein. Therapeutic outcomes were evaluated at the age of 11 months (adulthood, 8 months postinjection) and 21 months (terminal age, 18 months postinjection). Immunostaining and Western blot showed saturated supraphysiological levels of μDys expression in skeletal muscle and heart till the end of the study. Treatment significantly improved grip force and treadmill running, and significantly reduced the serum creatine kinase level at both time points. Since cardiac death is a major threat in late-stage patients, we evaluated cardiac electrophysiology and hemodynamics by ECG and the closed-chest cardiac catheter assay, respectively. Significant improvements were observed in these assays. Importantly, many ECG and hemodynamic parameters (heart rate, PR interval, QRS duration, QTc interval, end-diastolic/systolic volume, dP/dt max and min, max pressure, and ejection fraction) were completely normalized at 21 months of age. Our results have provided direct evidence that a single systemic AAV μDys therapy has the potential to provide lifelong benefits in the murine DMD model.
Keywords: DMD, micro-dystrophin, AAV, mdx, heart
INTRODUCTION
Adeno-associated virus (AAV)-mediated micro-dystrophin (μDys) gene therapy is a promising approach to treat Duchenne muscular dystrophy (DMD), a lethal muscle-wasting disease in boys.1 DMD is caused by the loss of a large cytoskeletal protein called dystrophin.2 Dystrophin is expressed from the DMD gene, one of the largest genes in the genome.3 Mutations in the DMD gene abolish dystrophin expression. As a monogenic disease, DMD is a perfect candidate for gene replacement therapy.
AAV has emerged as the most appealing vector for systemic gene therapy of neuromuscular diseases.4,5 One AAV drug has been approved by the Food and Drug Administration USA to treat spinal muscular atrophy.6 However, AAV is one of the smallest viruses.7 The maximum genome that can be efficiently packaged in the 25 nm AAV capsid is ∼5 kb.8–10 The full-length DMD gene is ∼2.4 mb. The full-length dystrophin coding sequence is ∼11.4 kb. They greatly exceed the carrying capacity of the AAV vector. Although the engineered triple-AAV system can deliver a full-length dystrophin expression cassette, the efficiency is too low for therapy.11 An alternative approach is to truncate the size of the dystrophin coding sequence.12,13 For the past two decades, numerous highly abbreviated microgenes were engineered.14–18 These minimized μDys genes are <4 kb and can be efficiently delivered with the AAV vector.19
Preclinical studies suggest that local and systemic AAV μDys gene therapy can effectively reduce muscle pathology and improve skeletal muscle and heart function (reviewed in Ref.19). Although these results have set a strong premise for ongoing human trials, lifelong follow-up studies are missing. DMD is a chronic disease and as such, an ideal therapy for DMD would be a single systemic delivery that can protect dystrophin-deficient muscles throughout life.
Long-term AAV persistence has been reported in normal human muscle.20 However, whether this is the case in the dystrophic muscle remains unclear. To address this important question, we injected an AAV serotype-9 (AAV9) μDys vector into 3-month-old mdx mice through the tail vein at the dose of 1 × 1013 vector genome (vg) particles/mouse. The μDys-treated mice were followed until they reached the terminal age of 21 months.21,22 We examined μDys expression, serum creatine kinase (CK), grip force, treadmill running, ECG, and hemodynamic function of the heart. Immunostaining and Western blot revealed robust μDys expression in multiple limb muscles and heart. Serum CK level was significantly reduced. Skeletal muscle fibrosis was significantly decreased. Grip force and treadmill running distance were significantly improved. Multiple ECG and cardiac hemodynamic parameters were normalized. Our results suggest that a single systemic AAV μDys therapy may provide long-lasting benefits in mdx mice.
MATERIALS AND METHODS
Experimental animals
All animal experiments were approved by the institutional animal care and use committee and were in accordance with NIH guidelines. C57/BL10 (wild-type [WT] control) and dystrophin-deficient mdx mice were generated in a barrier facility using breeders purchased from The Jackson Laboratory (Bar Harbor, ME) (mdx, C57BL/10ScSn-Dmdmdx/J, stock number 001801; BL10, C57BL/10ScSnJ, stock number 000476). Female mice were used in the study because aged female mdx mice are the only model that faithfully (both genetically and phenotypically) reproduced dilated cardiomyopathy in human patients.23,24 Since the median survival of female mdx mice is 21.25 months,22 we terminated treated mice at 21 months of age (range, 20.27–21.93 months; median, 21.77 months; mean ± standard, 21.49 ± 0.58 months). All mice were maintained in a specific-pathogen-free animal care facility on a 12-h light (25 lux):12-h dark cycle with access to food and water ad libitum.
AAV production and delivery
The cis μDys packaging plasmid (also called SJ46 and ΔR2-15/ΔR18-19/ΔR20-23/ΔC μDys) was published before.25 It contained the N-terminal domain, hinge 1 (H1), spectrin-like repeat 1 (R1), R16, R17, H3, R24, H4, the cysteine-rich domain, and the Dys-2 epitope at the end of the dystrophin C-terminal domain. Transgene expression was regulated by the cytomegalovirus promoter and the simian virus 40 polyadenylation signal. The μDys construct was packaged in AAV9. Recombinant AAV vector was produced, purified, and titrated according to our published protocol.26 A total of 1 × 1013 vg particles/mouse of the AAV9 μDys vector were injected through the tail vein to conscious 3-month-old mdx mice.
Morphological studies
Tissues were harvested at the end of the study. Harvested tissues were snap-frozen in the Tissue-Plus® optimal cutting temperature compound (Scigen Scientific, Gardena, CA, USA) in a liquid nitrogen-cooled 2-methylbutane bath. General histology was examined by hematoxylin and eosin (HE) staining. Fibrosis was examined by Masson trichrome (MT) staining. Dystrophin expression was evaluated by immunostaining using a monoclonal antibody that recognizes dystrophin H1-R1-R2 (NCL-Dys B, 1:80; Leica Biosystems, Product code DYSB, clone 34C5). Slides were viewed at the identical exposure setting using a Nikon E800 fluorescence microscope. Photomicrographs were taken with a Leica DFC7000 camera.
Fibrotic areas in the heart and skeletal muscle were quantified using the lasso tool in the Photoshop software on MT-stained whole tissue cross-section images. In brief, the micrometer scale was defined with the set measurement scale option in the software. The fibrotic area was marked using the quick selection tool. The sum of all fibrotic areas was then represented as a percentage of the whole cross-sectional area.
Western blot
To prepare for whole heart and muscle lysates, the tissues were snap-frozen in liquid nitrogen. The frozen tissue samples were ground to fine powder in liquid nitrogen followed by homogenization in a buffer containing 10% sodium dodecyl sulfate, 5 mM ethylenediaminetetraacetic acid, 62.5 mM Tris-HCl at pH6.8, and the protease inhibitor cocktail (Roche, Indianapolis, IN, USA). The crude lysate was chilled on ice for 2 min, and then centrifuged at 16,000 g for 2 min. The supernatant was collected as the whole muscle lysate. Protein concentration was measured using the DC protein assay kit (Bio-Rad, Hercules, CA, USA). The whole muscle lysate was heated at 95°C for 3 min to denature before loading in the gel.
Dystrophin was detected with a monoclonal antibody that recognizes the Dys-2 epitope at the C-terminal end of dystrophin (NCL-Dys2, 1:100; Leica Biosystems). Western blot was viewed using Li-Cor Odessey Fc imaging system and quantification was performed using the LI-COR Image Studio Version 5.0.21 software. The intensity of the respective protein band was normalized to the corresponding loading control in the same blot. The relative band intensity was further normalized to the WT control. Sarcomeric alpha-actinin was used as a loading control and was detected with a rabbit polyclonal antibody (1:2,000; Abcam, Cambridge, MA, USA).
Treadmill running
Treadmill endurance assay was performed as we described before with modifications.27 In brief, mice were subjected to 5-day treadmill acclimation at a 7° uphill treadmill (Columbus Instruments, Columbus, OH, USA). The acclimation protocol begins with placing the animal on an unmoving flat treadmill for 2 min followed by 5 min in 7° uphill inclined treadmill for each day. All running acclimations were done at 7° inclined treadmill only. On the first day, the mouse ran at 5 m/min for 15 min followed by 10 m/min for 5 min. On day 2, the mouse ran at 5 m/min for 5 min, 10 m/min for 15 min, and 12 m/min for 5 min in that order. On day 3, the mouse ran at 5 m/min for 5 min, 10 m/min for 15 min, and 12 m/min for 10 min.
On days 4 and 5, the mouse ran for 5 m/min for 5 min, 10 m/min for 20 min, 12 m/min for 5 min, and 15 m/min for 5 min. The running distance was measured on day 6. On the day of distance measurement, the mouse was placed on an unmoving treadmill for 2 min and then ran at 5 m/min for 5 min. The treadmill speed was then increased by 1 m/min every 5 min. The total running distance was calculated after the mouse became exhausted. Exhaustion is diagnosed when the animal gives up running and ends up in contact with the shocker (at the minimal setting) for typically 1–3 s without attempting to re-enter the treadmill. Animals that did not run were excluded from the analysis.
Serum CK activity assay
Fresh serum was collected by tail vein bleeding. The CK activity was determined using the CK Liqui-UV test kit from Stanbio Laboratory (Boerne, TX, USA) according to the manufacturer's guidelines.
Forelimb grip strength measurement
Forelimb grip strength was measured with a computerized grip strength meter (Columbus Instruments) as we described previously.28 The grip strength meter has a pulling bar attached to a force transducer and a digital display. The mouse was first acclimated to the apparatus for ∼5 min. The mouse was then allowed to grab the pulling bar while being held from the tip of the tail. The mouse was gently pulled away from the grip bar. When the mouse can no longer grasp the bar, the reading was recorded. The protocol was repeated five times with at least 30 s rest between trials. The highest three values were averaged to obtain the absolute grip strength. Normalized grip strength was obtained by dividing the absolute grip strength by the body weight.
ECG and hemodynamic assay
Cardiac functions were evaluated using our published protocols as described in the standard operating protocol in the Cardiac Protocols for Duchenne Animal Models.29,30,31 Specifically, a 12-lead ECG assay was performed using a commercial system from AD Instruments (Colorado Springs, CO, USA). The Q wave amplitude was determined using the lead I tracing. Other ECG parameters were analyzed using the lead II tracing.
The QTc interval was determined by correcting the QT interval with the heart rate. The cardiomyopathy index was calculated by dividing the QT interval by the PQ segment. Left ventricular hemodynamics was evaluated using a closed-chest approach as we previously described.29 The resulting PV loops were analyzed with the PVAN software (Millar Instruments, Houston, TX, USA). The cardiac relaxation time constant Tau was calculated according to Weiss et al.32 The body surface area was calculated as described by Cheung et al.33
Statistics
Data are presented as mean ± standard error of the mean. For all the physiological assays, data are presented using scatterplots. One-way analysis of variance (ANOVA) with Tukey's multiple comparison analysis was performed using GraphPad PRISM software version 7.0 for Mac OSX (GraphPad Software, La Jolla, CA, USA). A p < 0.05 was considered statistically significant.
RESULTS
Systemic therapy in young adult mice resulted in robust μDys expression in skeletal muscle and heart throughout life
To study the longitudinal effects of systemic AAV μDys gene therapy, we used a μDys construct that has been validated in murine and canine DMD models by local and systemic delivery.25,34–36 We packaged the μDys expression cassette in AAV9, a serotype that is currently being investigated in multiple clinical trials in DMD patients (NCT03362502, NCT04281485, and NCT05429372 sponsored by Pfizer; NCT03368742 sponsored by Solid Biosciences) (Fig. 1A).37–39 The μDys vector was injected into the tail vein of 3-month-old mdx mice at the dose of 1 × 1013 viral genome copies per animal (vg/mouse). Mice were euthanized at 21 months of age (Fig. 1A).
Figure 1.

Single intravenous delivery of an AAV9 micro-dystrophin vector to 3-month-old mdx mice resulted in robust protein expression up to 18 months postinjection. (A) Top panel is a cartoon illustration of R2-15/ΔR18-19/ΔR20-23/ΔC micro-dystrophin vector. Bottom panel shows the study plan. AAV [1 × 1013 viral genome particles (vg)/mouse] was injected at 3 months of age through the tail vein. Noninvasive assays were performed when animals were 11-month old (8 months postinjection). Terminal studies were performed when the animals reach 21 months of age (18 months postinjection). (B) Representative dystrophin immunofluorescence staining photomicrographs from different muscles, including the TA, quadriceps, gastrocnemius, and upper arm muscle. (C) Representative skeletal muscle Western blot. Quad, quadriceps; TA, tibialis anterior; Gas, gastrocnemius; Arm, upper arm muscle. Black arrowhead, full-length dystrophin; gray arrowhead, micro-dystrophin; white arrowhead, sarcomeric alpha-actinin (loading control). Densitometry quantification of the dystrophin level is shown in the scatterplot. (D) Representative dystrophin immunofluorescence staining photomicrographs of the heart. Dys B is the primary antibody used in the staining. Dys B recognizes H1-R1 region of the dystrophin protein. (E) Representative heart Western blot. Black arrowhead, full-length dystrophin; gray arrowhead, micro-dystrophin; white arrowhead, sarcomeric alpha-actinin (loading control). Densitometry quantification of the dystrophin level is shown in the scatterplot. AAV, adeno-associated virus; AAV9, AAV serotype-9; CMV, cytomegalovirus promoter; H1, hinge 1; R1, repeat 1; TA, tibialis anterior.
Immunostaining revealed saturated μDys expression at the sarcolemma in multiple limb muscles and the heart (Fig. 1B, D). Importantly, widespread μDys expression was achieved in muscles that are critical for respiration, including the diaphragm, intercostal muscle, and abdominal muscle (Supplementary Fig. S1). On Western blots, the μDys level exceeded the level of full-length dystrophin in WT mice (Fig. 1C, E, Supplementary Fig. S2). On quantification, micro-dystrophin expression in the treated skeletal muscle and heart was ∼9- and ∼2.5-fold higher than that of full-length dystrophin in normal skeletal muscle and heart, respectively (right panels in Fig. 1C, E).
AAV9 μDys therapy reduced fibrosis in skeletal muscle and heart
A hallmark of DMD is the replacement of muscle by noncontractile fibrotic tissue. We examined fibrosis by MT staining. On histochemical staining, the fibrotic replacement was clearly reduced in both heart and skeletal muscle (Fig. 2A, B; Supplementary Fig. S3). On quantification, ∼0.90% of the heart area was fibrotic in μDys-treated mice (Fig. 2C). This was comparable with that of the normal heart. Quantification confirmed the reduction of fibrosis in the tibialis anterior muscle, but the difference did not reach statistical significance (Fig. 2D). We also examined the general histology by HE staining. Infiltrating mononucleated cells appeared greatly reduced in skeletal muscle and heart of μDys-treated mice (Fig. 2A, B; Supplementary Fig. S3A, C, E, and F). However, the percentage of myofibers with centrally located myonuclei did not change in μDys-treated mice (Fig. 2E).
Figure 2.

Long-term systemic AAV9 micro-dystrophin therapy significantly reduced fibrosis in heart and skeletal muscle. (A) Representative high-power photomicrographs from MT staining and HE staining of the heart of WT, mdx, and AAV9.micro-dystrophin (AAV-μDys)-treated mdx mice. Fibrotic tissue is stained in blue, and muscle is stained in dark red in MT staining. (B) Representative high-power photomicrographs from MT staining and HE staining of the TA muscle of WT, mdx, and AAV9.micro-dystrophin (AAV-μDys)-treated mdx mice. The yellow asterisk marks the same myofiber in serial sections. (C) Quantification of the fibrotic area in the heart (WT n = 5, mdx n = 6, AAV n = 4). (D) Quantification of the fibrotic area in the TA muscle (WT n = 4, mdx n = 4, AAV n = 3). (E) Quantification of the percentage of myofiber with centrally located nuclei in the TA muscle (WT n = 3, mdx n = 3, AAV n = 3). *Significantly different from the indicated group(s), p < 0.05. HE, hematoxylin and eosin; MT, Masson trichrome; WT, wild type.
AAV9 μDys therapy reduced serum CK, improved grip strength, and enhanced treadmill running
To quantify muscle damage, we evaluated serum CK activity (Fig. 3 top panels). The CK level was significantly increased in untreated mdx mice. Systemic μDys therapy significantly reduced the serum CK level at 8 and 18 months post-AAV delivery. Noninvasive grip force and treadmill running were used to quantify gross muscle physiology at the middle and end of the study (Fig. 3 middle and bottom panels). Grip force was significantly enhanced and treadmill running distance was significantly increased when treated mice were examined at 11 months of age (8 months post-therapy) (Fig. 3A middle and bottom panels). Improvements in grip force and treadmill running lasted till 21 months of age (18 months post-therapy, the end of the study) (Fig. 3B middle and bottom panels).
Figure 3.
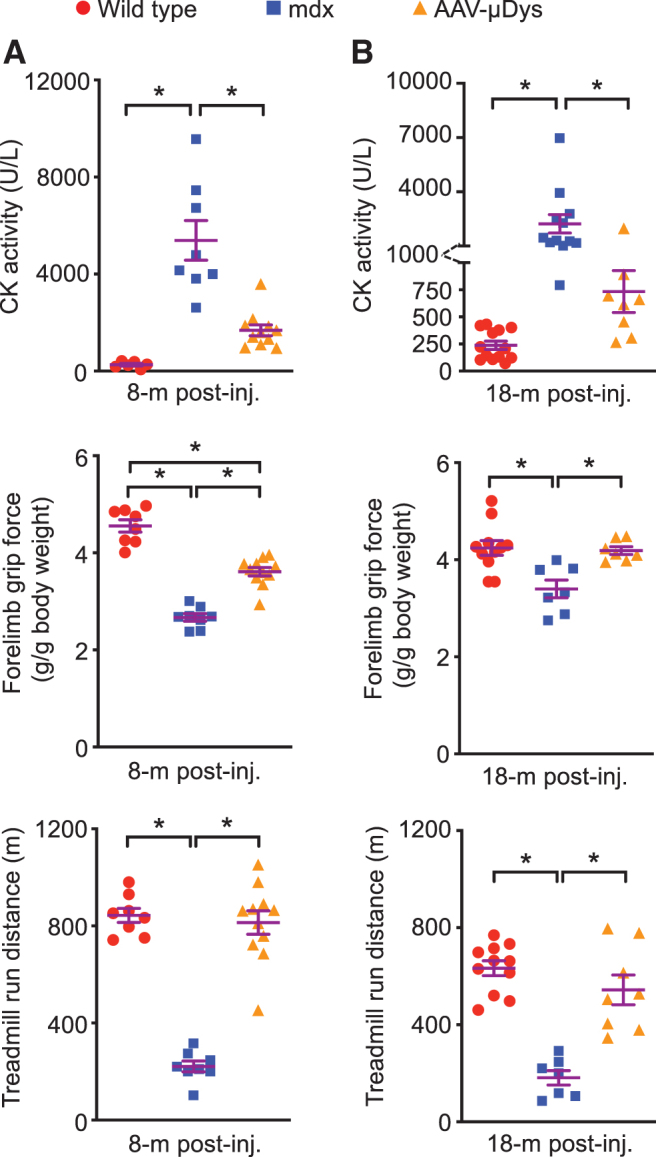
Long-term AAV9.micro-dystrophin therapy significantly reduced the serum CK level, and improved forelimb grip force and treadmill running distance. (A) Serum CK (WT n = 6, mdx n = 8, AAV n = 11), grip force (WT n = 8, mdx n = 8, AAV n = 11), and treadmill running distance (WT n = 8, mdx n = 8, AAV n = 11) at 8 months post-AAV injection. (B) Serum CK (WT n = 13, mdx n = 12, AAV n = 8), grip force (WT n = 11, mdx n = 7, AAV n = 7), and treadmill running distance (WT n = 11, mdx n = 7, AAV n = 8) at 18 months post-AAV injection. *Significantly different from the indicated group, p < 0.05. CK, creatine kinase.
AAV9 μDys therapy effectively prevented dilated cardiomyopathy in aged mdx mice
Heart function was examined by ECG and left-ventricle catheterization (Fig. 4; Supplementary Fig. S4). At 11 months of age, ECG examination showed significant improvements in PR interval, QRS duration, and cardiomyopathy index (Supplementary Fig. S4). Interestingly, nearly all ECG parameters (except for the Q amplitude) were normalized at 21 months of age in μDys-treated mdx mice (Fig. 4A). The hemodynamic function of the heart was examined right before the mice were euthanized. Several systolic (end-systolic volume, dP/dt max, and maximum pressure) and diastolic (end-diastolic volume, dP/dt min, and heart relaxation constant Tau) parameters were fully normalized (Fig. 4B top two panels).
Figure 4.

Long-term systemic AAV9.micro-dystrophin therapy resulted in persistent heart function improvement till 21 months of age. (A) Quantitative ECG parameters from WT, untreated mdx and AAV9.micro-dystrophin-treated mdx mice. (B) Quantitative cardiac pump function parameters from left-ventricular catheterization assay showing systolic (top panel), diastolic (middle panel) and overall heart pump function (bottom panel) indices from WT, untreated mdx and AAV9.micro-dystrophin-treated mdx mice. *Significantly different from the groups indicated, p < 0.05.
The ejection fraction of treated mice was significantly higher than that of untreated mdx mice (Fig. 4B bottom left panel). However, the stroke volume and cardiac output were not changed (Fig. 4B bottom middle and right panels). We also observed significant improvements in multiple other hemodynamics parameters, including the end-systolic pressure, stroke work, pressure at the max value of dV/dt, pressure and volume at the maximum value of dP/dt, volume at the minimum value of dP/dt, and maximum power (Supplementary Table S1). Anatomical parameters of the treated heart (weights and weight ratios) showed a trend of improvement (Supplementary Table S2).
DISCUSSION
In this study, we evaluated lifelong systemic AAV μDys gene therapy in a mouse DMD model. We showed that a single treatment in young adult mdx mice resulted in persistent and robust μDys expression in skeletal muscle and heart till the mice reached the terminal age (Fig. 1; Supplementary Figs. S1 and S2). Gene therapy reduced fibrosis and inflammation in muscle and heart, lowered serum CK, and increased grip force and treadmill running (Figs. 2 and 3; Supplementary Fig. S3). Importantly, it significantly improved the electric and mechanical function of the heart and prevented dilated cardiomyopathy (Fig. 4; Supplementary Fig. S4 and Supplementary Table S1).
The size of the full-length dystrophin protein is 427 kD.2 It contains four major domains, including the N-terminal domain, rod domain, cysteine-rich domain, and the C-terminal domain. The rod domain contains 24 spectrin-like repeats and four hinges. Patient studies suggest that highly truncated dystrophins can significantly reduce dystrophic phenotype.40,41 Based on clinical observations, abbreviated μDys genes were developed to fit into the AAV vector, the only viral vector that can efficiently deliver a transgene to all muscles in the body.4 A variety of μDys genes have been tested in rodent and canine DMD models. In most studies, the μDys AAV vector was delivered to young animals and the therapeutic effect was examined when the treated animals reached adulthood. In a subset of studies, the AAV μDys vector was administered to old animals and examined at their terminal age.42–44 However, lifelong therapy has not been examined.
There are two fundamental questions in DMD gene replacement therapy. First, whether permanent dystrophin expression is required for lifelong benefits. Second, whether it is possible to achieve permanent dystrophin expression from a single therapy. An RNA interference study found that persistent knockdown of dystrophin for 1 year did not cause muscle disease in adult mice.45 However, when a floxed dystrophin gene was removed by Cre recombinase in adult mice, we observed significant dystrophic phenotype and function reduction in both skeletal muscle and heart.46 Our data suggest that an effective DMD gene therapy depends on enduring dystrophin expression.
Several studies have shown that AAV vectors can persist in normal tissues, including muscle, for years.20,47 However, this may not be the case in a dystrophic muscle. Cordier et al. demonstrated immune-mediated AAV loss in dystrophic but not normal muscle, suggesting a dystrophic microenvironment may potentiate the cytotoxic T cell response against the AAV vector.48 Le Hir et al. found that leaky sarcolemma and muscle degeneration play critical roles in AAV loss from dystrophic muscle.49 This is especially true when a nontherapeutic AAV vector was delivered or when therapeutic dystrophin restoration was suboptimal. Dupont et al. further showed that mRNA produced from the AAV vector was selectively damaged by oxidative stress in dystrophic muscle.50 Several different strategies have been used to prevent AAV loss in dystrophic muscle.
The use of the muscle-specific promoter greatly reduced immunological AAV loss.48 Pretreatment with antisense oligonucleotide mediated exon-skipping also effectively prevented AAV loss, likely due to transient stabilization of the sarcolemma by exon-skipping rescued dystrophin.51,52 A third approach is to prevent muscle degeneration and turnover.49,53 In our study, we achieved supraphysiological level saturated μDys expression. We believe this has effectively stopped AAV loss from degeneration-induced myofiber death. We would like to emphasize that our findings may not directly translate to human patients, because the level of μDys expression we saw in this study is hard to achieve in a clinical trial. Furthermore, the highly minimized μDys cannot fully protect muscle from damage induced by contraction, especially eccentric contraction.
A reduction of centrally nucleated myofibers is often used to evaluate μDys therapy.14,16,54 However, we did not see a statistically significant decrease in centronucleation, although a trend of reduction was noted (Fig. 2E). We believe this is likely due to the age of AAV injection rather than the ineffectiveness of the therapy. We delivered the AAV.μDys vector to 3-month-old mdx mice. At this age, mdx muscle disease has become stable.55 Migration of the centrally located myonuclei to the peripheral is a lengthy process and may take up to 2 years.56
Improved respiratory care (such as assisted ventilation) in the past several decades has unmasked cardiomyopathy as a leading cause of death in Duchenne patients. With this backdrop, we have paid special attention to cardiac rescue in our study. AAV μDys therapy has been evaluated in the heart of dystrophin-deficient rodents by many groups.42,43,57–64 These studies revealed correct sarcolemmal localization of μDys in cardiomyocytes, restoration of dystrophin-associated glycoprotein complex, prevention of Evans blue dye uptake, suppression of myocardial fibrosis, and improvements in electrocardiography and hemodynamic function. Interestingly, only H2 Dys (also called ΔR4-23/ΔC μDys and M1 μDys) and its derivatives have been studied in the heart.
H2 μDys contains the N-terminal domain, H1, R1, R2, R3, H2, R24, H4, and the cysteine-rich domain. A recent proteomic study suggests that H2 μDys has an altered association with syntrophin and cavin in the heart.65 Modified H2 μDys variants were investigated in two newly published studies. In one study, the authors examined H3 μDys. This μDys is identical to H2 μDys except for the replacement of H2 by H3. Similar to H2 μDys, H3 μDys effectively prevented cardiac disease in a new DMD cardiomyopathy model.64 In another study, the authors added different lengths of the C-terminal domain to H2 μDys.63 The C-terminal domain has been implicated in heart protection in a genotype–phenotype correlation analysis.66 However, no added cardiac benefits were observed in the new study.63
Three different versions of μDys are currently in human trials (NCT03362502, NCT04281485, and NCT05429372 sponsored by Pfizer; NCT03368742 sponsored by Solid Biosciences; NCT03375164, NCT03769116, NCT04626674, and NCT05096221 sponsored by Sarepta; and EudraCT Number 2020-002093-27 sponsored by Genethon).19 They all contain the N-terminal domain and cysteine-rich domain. The major differences are (1) the number of spectrin-like repeats (four repeats in trials sponsored by Sarepta and Genethon, five repeats in trials sponsored by Pfizer and Solid Biosciences), and (2) with or without a central hinge (with a central hinge in trials sponsored by Pfizer, Sarepta, and Genethon; no central hinge in trials sponsored by Solid Biosciences).
Several trials used H2 μDys (NCT03375164, NCT03769116, NCT04626674, and NCT05096221 sponsored by Sarepta; EudraCT Number 2020-002093-27 sponsored by Genethon).67,68 However, other trials used μDys construct that does not carry hinge 2 (NCT03362502, NCT04281485, and NCT05429372 sponsored by Pfizer; NCT03368742 sponsored by Solid Biosciences).37–39 It is currently unclear whether μDys can protect the heart in the absence of a central hinge. To address this issue, we used a μDys gene similar to the one used in Solid Bioscience trial (NCT03368742).38,39
Specifically, we replaced R2 and R3 of the R1-R2-R3 membrane-binding domain with R16 and R17,69 the two repeats essential for anchoring neuronal nitric oxide synthase to the sarcolemma in skeletal muscle.34,70,71 Given the robust cardiac rescue observed in our study, we conclude that a central hinge is not required to protect the heart by μDys. In support, we recently found that a novel R16-19 containing μDys isoform completely normalized arrhythmia-inducing Na current abnormality in heart Purkinje fibers.17
We have previously studied cardiac protection of ΔH2-R19 mini-dystrophin in heart-specific transgenic mice.28 ΔH2-R19 mini-dystrophin is two times larger than μDys used in this study. Interestingly, we detected better heart function rescue with μDys. We believe the improved protection is likely due to simultaneous treatment of both heart and skeletal muscle with systemic μDys therapy. Generation and analysis of ΔH2-R19 mini-dystrophin heart/skeletal muscle double transgenic mice will help to delineate whether larger dystrophin can better protect the heart.
Our study has several limitations. First, the study was not designed to evaluate the effect of systemic μDys therapy on the lifespan. Second, we only examined the long-term benefits in female mice. Third, we did not quantify the contractile properties of the skeletal muscle. Future studies are warranted to determine whether μDys therapy can extend the lifespan, whether similar benefits can be achieved in male mdx mice, and whether lifelong μDys therapy improves the contractile properties of the limb muscle and diaphragm.28,72 Most importantly, there is an urgent need to investigator long-term μDys therapy in large animal models (such as dystrophin-deficient canines) because the canine model better recapitulates the clinical phenotype and is more suitable to investigate the immune response to AAV μDys therapy.73
In summary, we demonstrated the lifelong benefits of a single systemic AAV μDys gene therapy in young mdx mice. Our findings provide additional support to continue μDys therapy in DMD patients.
Supplementary Material
AUTHORs' CONTRIBUTIONS
N.B.W. and D.D. conceived the idea and designed the study. N.B.W., Y.Y., B.H., and J.H.S., conducted experiments. N.B.W., A.S., G.Y., and D.D. analyzed the data. N.B.W., G.Y., and D.D. wrote the paper. A.S., G.Y., and D.D. edited the paper. A.S. and D.D. secured the funding. All authors approved the submission.
AUTHOR DISCLOSURE
D.D. is a member of the scientific advisory board for Solid Biosciences and equity holders of Solid Biosciences. D.D. is a member of the scientific advisory board for Sardocor Corp. D.D. is an inventor on several issued and filed patents on micro-dystrophin gene therapy and recombinant AAV vectors. The Duan lab received research support unrelated to this project from Solid Biosciences in the past 3 years. The Duan lab has received research support unrelated to this project from Edgewise Therapeutics in the past 3 years. A.S. is a cofounder of, and has equity in, Lacerta Therapeutics. He is also a consultant for Passage Bio and AgeX Therapeutics and an inventor on several issued and filed patents on recombinant AAV vectors that have been or are being licensed to various AAV gene therapy companies.
FUNDING INFORMATION
This study was supported by grants from the National Institutes of Health (AR-70517 to D.D.; AR-81018 to A.S. and D.D.), Jesse Davidson Foundation-Defeat Duchenne Canada (to D.D.), Jett Foundation (to D.D.), and Jackson Freel DMD Research Fund (to D.D.). B.H. was supported by the University of Missouri Life Science Fellowship.
SUPPLEMENTARY MATERIAL
REFERENCES
- 1. Duan D. Micro-dystrophin gene therapy goes systemic in Duchenne muscular dystrophy patients. Hum Gene Ther 2018;29:733–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Duan D, Goemans N, Takeda S, et al. . Duchenne muscular dystrophy. Nat Rev Dis Primers 2021;7:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kunkel LM. 2004 William Allan award address. cloning of the DMD gene. Am J Hum Genet 2005;76:205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Duan D. Systemic delivery of adeno-associated viral vectors. Curr Opin Virol 2016;21:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Verhaart IEC, Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol 2019;15:373–386. [DOI] [PubMed] [Google Scholar]
- 6. Mendell JR, Al-Zaidy S, Shell R, et al. . Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med 2017;377:1713–1722. [DOI] [PubMed] [Google Scholar]
- 7. Carter BJ. Adeno-associated virus and the development of adeno-associated virus vectors: a historical perspective. Mol Ther 2004;10:981–989. [DOI] [PubMed] [Google Scholar]
- 8. Wang D, Tai PWL, Gao GP. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov 2019;18:358–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li C, Samulski RJ. Engineering adeno-associated virus vectors for gene therapy. Nat Rev Genet 2020;21:255–272. [DOI] [PubMed] [Google Scholar]
- 10. Lai Y, Yue Y, Duan D. Evidence for the failure of adeno-associated virus serotype 5 to package a viral genome > or = 8.2 kb. Mol Ther 2010;18:75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lostal W, Kodippili K, Yue Y, et al. . Full-length dystrophin reconstitution with adeno-associated viral vectors. Hum Gene Ther 2014;25:552–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scott J, Li S, Harper S, et al. . Viral vectors for gene transfer of micro-, mini-, or full-length dystrophin. Neuromuscul Disord 2002;12 Suppl:S23. [DOI] [PubMed] [Google Scholar]
- 13. Duan D. From the smallest virus to the biggest gene: marching towards gene therapy for Duchenne muscular dystrophy. Discov Med 2006;6:103–108. [PMC free article] [PubMed] [Google Scholar]
- 14. Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci U S A 2000;97:13714–13719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harper SQ, Hauser MA, DelloRusso C, et al. . Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med 2002;8:253–261. [DOI] [PubMed] [Google Scholar]
- 16. Ramos JN, Hollinger K, Bengtsson NE, et al. . Development of novel micro-dystrophins with enhanced functionality. Mol Ther 2019;27:623–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ebner J, Pan X, Yue Y, et al. . Microdystrophin therapy rescues impaired Na currents in cardiac Purkinje fibers from dystrophin-deficient Mdx mice. Circ Arrhythm Electrophysiol 2022;15:e011161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wasala LP, Watkins T, Wasala N, et al. . The implication of hinge 1 and hinge 4 in micro-dystrophin gene therapy for Duchenne muscular dystrophy. Hum Gene Ther 2022. [Epub ahead of print]; DOI: 10.1089/hum.2022.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duan D. Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol Ther 2018;26:2337–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Buchlis G, Podsakoff GM, Radu A, et al. . Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood 2012;119:3038–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chamberlain JS, Metzger J, Reyes M, et al. . Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. Faseb J 2007;21:2195–2204. [DOI] [PubMed] [Google Scholar]
- 22. Li D, Long C, Yue Y, et al. . Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum Mol Genet 2009;18:1209–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bostick B, Yue Y, Long C, et al. . Prevention of dystrophin-deficient cardiomyopathy in twenty-one-month-old carrier mice by mosaic dystrophin expression or complementary dystrophin/utrophin expression. Circ Res 2008;102:121–130. [DOI] [PubMed] [Google Scholar]
- 24. Bostick B, Yue Y, Duan D. Gender influences cardiac function in the mdx model of Duchenne cardiomyopathy. Muscle Nerve 2010;42:600–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shin J-H, Pan X, Hakim CH, et al. . Microdystrophin ameliorates muscular dystrophy in the canine model of Duchenne muscular dystrophy. Mol Ther 2013;21:750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shin J-H, Yue Y, Duan D. Recombinant adeno-associated viral vector production and purification. Methods Mol Biol 2012;798:267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bostick B, Yue Y, Long C, et al. . Cardiac expression of a mini-dystrophin that normalizes skeletal muscle force only partially restores heart function in aged mdx mice. Mol Ther 2009;17:253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hakim CH, Li D, Duan D. Monitoring murine skeletal muscle function for muscle gene therapy. Methods Mol Biol 2011;709:75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bostick B, Yue Y, Duan D. Phenotyping cardiac gene therapy in mice. Methods Mol Biol 2011;709:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duan D, Rafael-Fortney JA, Blain A, et al. . Standard operating procedures (SOPs) for evaluating the heart in preclinical studies of Duchenne muscular dystrophy. J Cardiovasc Transl Res 2016;9:85–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parent project muscular dystrophy. Standard operating procedures (sops) for Duchenne animal models. Washington, DC; 2023. Available from https://www.parentprojectmd.org/research/for-researchers-industry/resources/standard-operating-procedures-for-duchenne-animal-models/ [Last accessed: April 17, 2023].
- 32. Weiss JL, Frederiksen JW, Weisfeldt ML. Hemodynamic determinants of the time-course of fall in canine left ventricular pressure. J Clin Invest 1976;58:751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheung MC, Spalding PB, Gutierrez JC, et al. . Body surface area prediction in normal, hypermuscular, and obese mice. J Surg Res 2009;153:326–331. [DOI] [PubMed] [Google Scholar]
- 34. Lai Y, Thomas GD, Yue Y, et al. . Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest 2009;119:624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li D, Yue Y, Lai Y, et al. . Nitrosative stress elicited by nNOSmu delocalization inhibits muscle force in dystrophin-null mice. J Pathol 2011;223:88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yue Y, Pan X, Hakim CH, et al. . Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum Mol Genet 2015;24:5880–5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moorehead T, Yong F, Neelakantan S, et al. . Safety and tolerability of PF-06939926 in ambulatory boys with Duchenne muscular dystrophy: a phase 1b Multicenter, Open-Label, Dose Ascending Study. Mol Ther 2020;28:S273–S274. [Google Scholar]
- 38. Gonzalez JP, Brown KJ, Lawlor MW, et al. . SGT-001 microdystrophin gene therapy for Duchenne muscular dystrophy. Mol Ther 2020;28:S220. [Google Scholar]
- 39. Morris CA, Dreghici RD, Redican S, et al. . IGNITE DMD study of SGT-001 microdystrophin gene therapy for Duchenne muscular dystrophy: long-term outcomes and biomarker update. Mol Ther 2022;30:S553–S554. [Google Scholar]
- 40. England SB, Nicholson LV, Johnson MA, et al. . Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 1990;343:180–182. [DOI] [PubMed] [Google Scholar]
- 41. Beggs AH, Hoffman EP, Snyder JR, et al. . Exploring the molecular basis for variability among patients with Becker muscular dystrophy: dystrophin gene and protein studies. Am J Hum Genet 1991;49:54–67. [PMC free article] [PubMed] [Google Scholar]
- 42. Bostick B, Shin J-H, Yue Y, et al. . AAV-microdystrophin therapy improves cardiac performance in aged female mdx mice. Mol Ther 2011;19:1826–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bostick B, Shin J-H, Yue Y, et al. . AAV micro-dystrophin gene therapy alleviates stress-induced cardiac death but not myocardial fibrosis in >21-m-old mdx mice, an end-stage model of Duchenne muscular dystrophy cardiomyopathy. J Mol Cell Cardiol 2012;53:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gregorevic P, Blankinship MJ, Allen JM, et al. . Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol Ther 2008;16:657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ghahramani Seno MM, Graham IR, Athanasopoulos T, et al. . RNAi-mediated knockdown of dystrophin expression in adult mice does not lead to overt muscular dystrophy pathology. Hum Mol Genet 2008;17:2622–2632. [DOI] [PubMed] [Google Scholar]
- 46. Wasala NB, Lai Y, Shin J-H, et al. . Genomic removal of a therapeutic mini-dystrophin gene from adult mice elicits a Duchenne muscular dystrophy-like phenotype. Hum Mol Genet 2016;25:2633–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stieger K, Schroeder J, Provost N, et al. . Detection of intact rAAV particles up to 6 years after successful gene transfer in the retina of dogs and primates. Mol Ther 2009;17:516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cordier L, Gao GP, Hack AA, et al. . Muscle-specific promoters may be necessary for adeno-associated virus-mediated gene transfer in the treatment of muscular dystrophies. Hum Gene Ther 2001;12:205–215. [DOI] [PubMed] [Google Scholar]
- 49. Le Hir M, Goyenvalle A, Peccate C, et al. . AAV genome loss from dystrophic mouse muscles during AAV-U7 snRNA-mediated exon-skipping therapy. Mol Ther 2013;21:1551–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dupont JB, Tournaire B, Georger C, et al. . Short-lived recombinant adeno-associated virus transgene expression in dystrophic muscle is associated with oxidative damage to transgene mRNA. Mol Ther Methods Clin Dev 2015;2:15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peccate C, Mollard A, Le Hir M, et al. . Antisense pre-treatment increases gene therapy efficacy in dystrophic muscles. Hum Mol Genet 2016;25:3555–3563. [DOI] [PubMed] [Google Scholar]
- 52. Forand A, Muchir A, Mougenot N, et al. . Combined treatment with peptide-conjugated phosphorodiamidate morpholino oligomer-PPMO and AAV-U7 rescues the severe DMD phenotype in mice. Mol Ther Methods Clin Dev 2020;17:695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pacak CA, Conlon T, Mah CS, et al. . Relative persistence of AAV serotype 1 vector genomes in dystrophic muscle. Genet Vaccines Ther 2008;6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu M, Yue Y, Harper SQ, et al. . Adeno-associated virus-mediated microdystrophin expression protects young mdx muscle from contraction-induced injury. Mol Ther 2005;11:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McGreevy JW, Hakim CH, McIntosh MA, et al. . Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis Model Mech 2015;8:195–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Meyer GA. Evidence of induced muscle regeneration persists for years in the mouse. Muscle Nerve 2018;58:858–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yue Y, Li Z, Harper SQ, et al. . Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation 2003;108:1626–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bostick B, Yue Y, Lai Y, et al. . Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum Gene Ther 2008;19:851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gregorevic P, Allen JM, Minami E, et al. . rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med 2006;12:787–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Townsend D, Blankinship MJ, Allen JM, et al. . Systemic administration of micro-dystrophin restores cardiac geometry and prevents dobutamine-induced cardiac pump failure. Mol Ther 2007;15:1086–1092. [DOI] [PubMed] [Google Scholar]
- 61. Shin J-H, Nitahara-Kasahara Y, Hayashita-Kinoh H, et al. . Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Ther 2011;18:910–919. [DOI] [PubMed] [Google Scholar]
- 62. Malerba A, Sidoli C, Lu-Nguyen N, et al. . Dose-dependent microdystrophin expression enhancement in cardiac muscle by a cardiac-specific regulatory element. Hum Gene Ther 2021;32:1138–1146. [DOI] [PubMed] [Google Scholar]
- 63. Bourdon A, Francois V, Zhang L, et al. . Evaluation of the dystrophin carboxy-terminal domain for micro-dystrophin gene therapy in cardiac and skeletal muscles in the DMD(mdx) rat model. Gene Ther 2022;29:520–535. [DOI] [PubMed] [Google Scholar]
- 64. Howard ZM, Dorn LE, Lowe J, et al. . Micro-dystrophin gene therapy prevents heart failure in an improved Duchenne muscular dystrophy cardiomyopathy mouse model. JCI Insight 2021;6:e146511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wang H, Marrosu E, Brayson D, et al. . Proteomic analysis identifies key differences in the cardiac interactomes of dystrophin and micro-dystrophin. Hum Mol Genet 2021;30:1321–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tandon A, Jefferies JL, Villa CR, et al. . Dystrophin genotype-cardiac phenotype correlations in Duchenne and Becker muscular dystrophies using cardiac magnetic resonance imaging. Am J Cardiol 2015;115:967–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mendell JR, Sahenk Z, Lehman K, et al. . Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol 2020;77:1122–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Genethon. Genethon announces first patient dosed in clinical trial of investigational gene therapy GNT 0004 for Duchenne muscular dystrophy. https://www.genethon.fr/wp-content/uploads/2021/04/PR_GENETHON_DMD-1.pdf (last accessed April 20, 2021).
- 69. Zhao J, Kodippili K, Yue Y, et al. . Dystrophin contains multiple independent membrane-binding domains. Hum Mol Genet 2016;25:3647–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lai Y, Zhao J, Yue Y, et al. . alpha2 and alpha3 helices of dystrophin R16 and R17 frame a microdomain in the alpha1 helix of dystrophin R17 for neuronal NOS binding. Proc Natl Acad Sci U S A 2013;110:525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li D, Bareja A, Judge L, et al. . Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J Cell Sci 2010;123:2008–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hakim CH, Lessa TB, Jenkins GJ, et al. . An improved method for studying mouse diaphragm function. Sci Rep 2019;9:19453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Duan D. Duchenne muscular dystrophy gene therapy in the canine model. Hum Gene Ther Clin Dev 2015;26:57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.