

Abstract
BACKGROUND:
Cardiac contractile function requires high energy from mitochondria, and Ca2+ from the sarcoplasmic reticulum (SR). Via local Ca2+ transfer at close mitochondria-SR contacts, cardiac excitation feedforward regulates mitochondrial ATP production to match surges in demand (excitation-bioenergetics coupling, EBC). However, pathological stresses may cause mitochondrial Ca2+ overload, excessive reactive oxygen species (ROS) production and permeability transition, risking homeostatic collapse and myocyte loss. EBC involves mitochondria-SR tethers but the role of tethering in cardiac physiology/pathology is debated. Endogenous tether proteins are multifunctional; therefore non-selective targets to scrutinize inter-organelle linkage. Here, we assessed the physiological/pathological relevance of selective chronic enhancement of cardiac mitochondria-SR tethering .
METHODS:
We introduced to mice a cardiac muscle-specific engineered tether (‘linker’) transgene with a fluorescent protein core and deployed 2D/3D electron microscopy, biochemical approaches, fluorescence imaging, in vivo and ex vivo cardiac performance monitoring and stress challenges to characterize the linker phenotype.
RESULTS:
Expressed in the mature cardiomyocytes, the linker expanded and tightened individual mitochondria-junctional SR (jSR) contacts; but also evoked a marked remodeling with large dense mitochondrial clusters that excluded dyads. Yet, EBC remained well-preserved, likely due to more longitudinal mitochondria-dyad contacts and nanotunnelling between mitochondria exposed to jSR and those sealed away from jSR. Remarkably, the linker decreased female vulnerability to acute massive β-adrenergic stress. It also reduced myocyte death and mitochondrial calcium-overload-associated myocardial impairment in ex vivo ischemia/reperfusion injury.
CONCLUSIONS:
We propose that mitochondria-SR/ER contacts operate at a structural optimum. While acute changes in tethering may cause dysfunction, upon chronic enhancement of contacts from early life, adaptive remodeling of the organelles shifts the system to a new, stable structural optimum. This remodeling balances the individually enhanced mitochondrion-jSR crosstalk and EBC, by increasing the connected mitochondrial pool and, presumably, Ca2+/ROS capacity, which then improves the resilience to stresses associated with dysregulated hyperactive Ca2+ signaling.
Keywords: mitochondria, organelle contacts, mitochondria-SR tethers, adaptive remodeling
Subject Terms: Cell Biology/Structural Biology, Calcium Cycling/Excitation-Contraction Coupling, Metabolism, Heart Failure, Remodeling, Myocardial Biology, Mechanisms
Graphical Abstract
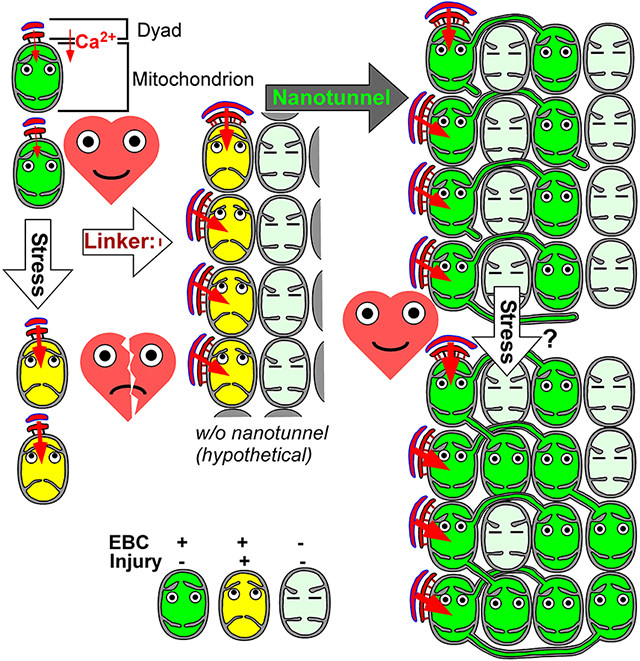
INTRODUCTION
Mitochondria and SR/ER in cardiomyocytes extensively form close contacts, secured by protein tethers, which are crucial local communication sites for molecule (e.g. lipid) exchange, Ca2+ and ROS/redox signaling1. Cytosolic [Ca2+] ([Ca2+]c) spikes that mediate cardiac excitation-contraction coupling also propagate to mitochondria and facilitate oxidative phosphorylation via stimulating Ca2+-sensitive matrix dehydrogenases of the tricarboxylic acid cycle. This regulatory loop is also known as the EBC2-4. Since endogenous mitochondria-ER/SR tether proteins have multiple functions1, their genetic targeting is not suitable for selectively studying mitochondria-SR tethering and its structural-functional roles. For instance, mitofusin-2 (Mfn2) controls mitochondrial fusion, quality control and mitophagy in cardiomyocytes5. Mfn2 and protein tyrosine phosphatase interacting protein 51 (PTPIP51) loss or gain of function have been associated with respective decrease or increase in mitochondria-SR tethering, local Ca2+ crosstalk and the EBC6, 7 Apparently, gain of function exacerbated, while loss of function protected against cardiac ischemia-reperfusion injury8-10. However, potential direct roles in mitochondrial integrity and mitochondria-associated cell death pathways (mitochondrial permeability transition pore, mPTP or Bax/Bak activation)11 could not be excluded. Here, we genetically engineered a dedicated tether (linker) composed of a monomeric red fluorescent protein (mRFP) with N-and C-terminal outer mitochondrial membrane (OMM) and ER/SR membrane-targeting domains and spacer helices, with overall tethering distance ~10-20 nm (Figure 1A and12). We introduced this linker into mouse as a cardiac muscle-specific (Myh6-driven) transgene (Tg), using a tetracycline-switchable (Tet-off) promoter system (Figure 1A). Acute adenoviral delivery of such linker enhanced the EBC and corrected the contractile deficiency in a diabetic cardiomyopathy model13. Our studies using chronic postnatal expression in the cardiac muscle revealed a profound remodeling of the mitochondria and mitochondria-SR contacts with net improved contractility, well-preserved bioenergetics, and protection in injurious β-adrenergic stress and ex vivo ischemia-reperfusion injury.
Figure 1: Mitochondria-SR/ER linker expression and basal functional phenotype in the linker-mouse hearts.

A. Schematic of the engineered tether ('linker') between the OMM and ER/SR. Helical spacers (18x, 9x repeats) keep the membrane gap distance <20 nm. B. Immunoblots of mRFP in heart lysates of control, linker- and Doxy-linker-mice. RFP is only expressed in the linker hearts. VDAC serves as loading control. C–L. Basal echocardiography parameters for male (♂) and female (♀) control (Ctr, black), linker (Lnk, red) and Doxy-linker (Lnk/Doxy+, cyan) mice. M. Body weights. Doxycycline administration is indicated under the graphs. Data are normalized to Ctr in each sex. See absolute values in Figure S2. Means±SEM. N is indicated bar wise. Statistics as shown in gray box.
METHODS
Please refer to the Expanded Materials and Methods for a detailed description of experimental methods and see the Major Resources Table in the Supplemental Materials.
Experimental Animals
All studies adhered to the NIH Guide for the Care and Use of Laboratory Animals; the protocols were approved by Thomas Jefferson and Washington Universites' Institutional Animal Care and Use Committee. Both sexes, 12-18 week-old adults and pups at ages P1-P21 were used for experiments.
Linker-Mice:
Tet-off ‘linker-mice’ (Tg/Tg(Myh6/tetO-linker) x Tg/Tg(Myh6/tTA) x Mfn2fl/fl, Figure 1A) were created in the Dorn lab using a bi-transgenic strategy14. Heterozygous breeding yielded offspring either bi-transgenic or non-transgenic (NTg) for MyH6/tetO-linker and Myh6/tTA. Hence, the NTg/NTg(Myh6/tetO-linker) x NTg/NTg(Myh6/tTA) x Mfn2fl/fl sub-colony was assigned as control. Linker-mice on regular chow constitutively express the linker in the heart. To halt expression, doxycycline-supplemented chow (‘Doxy-diet’) was provided for 3-7 weeks.
Quantification and Statistical Analysis
Statistical analysis was performed using SigmaPlot/SigmaStat (Systat/Inpixon), Statsmodels (Python) or SPSS (IBM) software. See further details within the corresponding figure legend and Expanded Methods.
Representatives.
Representative term is reserved for images that have been selected from a collection of 10+ (fluorescence) or 50+ (electron microscopy) images in a way that they reflect (fit with) the quantitative analysis. Images or graphs that are typical examples of a formation or illustrate an original recording are termed ‘exemplar image’ or ‘exemplar graph/recording’.
RESULTS
Validation of the linker-mouse.
Given a robust mitochondrial and SR remodeling phenotype in the linker-mice (see later), we focused this study to the impact of the linker itself on cardiac structure-function under normal and stressed conditions. Linker mRNA was detectable in the heart from age P7 (Figure S1C). Linker-expressing mRFP-fluorescent ventricular cardiomyocytes (VCMs) were evident from P14 (~40% of the cells) and by P21, >80% of the myocytes were red-fluorescent (Figure S1A,B), consistent with the mouse Myh6 promoter activity to follow myocyte maturation (Myh7/Myh6 switch)15, 16. Linker-mice appeared healthy, grew, bred, and generally lived/thrived normally. No linker mRNA or mRFP was detectable in the adult brain, liver or skeletal muscle of linker-mice (not shown). Mitochondria of adult linker-VCMs were decorated by mRFP (Figure S1E immunogold, Figure 3I/ii), consistent with extensive close interfaces between SR and mitochondrial networks (Figure S1F,G). Doxy-diet effectively halted linker/mRFP expression (Figures 1B, S1A). Returning to normal diet could not resume linker expression (not shown), likely owing to long-term doxycycline depot formation17. Linker- VCMs were similarly sized to control (based on fura2-fluorescent pixels/cell, not shown) or to VCMs of linker-mice on Doxy-diet (Doxy-linker-mice, Figure S1A Adult vs Adult Doxy). Since the genetic background included the C57Bl/6J strain carrying a null mutation in the nicotinamide nucleotide transhydrogenase (NNT) gene, we tested if the linker-mice carried the l mutation. Importantly, NNT plays protective role in oxidative stress. PCR genotyping showed that the linker-mice expressed the wild-type Nnt gene (Figure S1D).
Linker-mouse heart is functionally normal with increased contractility and well-maintained exercise performance.
Basal cardiac parameters were assessed in adult male and female control, linker-and Doxy-linker-mice. Linker-mice had normotrophic hearts (similar heart weight/body weight ratios to control, not shown). Transthoracic echocardiography revealed a compensated left-ventricular (LV) hypercontractility in both sexes with decreased LV end-systolic/diastolic volumes (LVESV/LVEDV Figures 1C,D; S2.A,B), preserved stroke volume (SV, Figures 1E, S2C), increased ejection fraction (EF, Figures 1F, S2D) and fractional shortening (FS, Figures 1G, S2E). Consistently, both end-systolic and end-diastolic LV anterior wall thicknesses (LVAW, s&d) were increased in the linker-hearts (Figures 1H,I; S2F,G). This hypercontractility could be reversed by removing the linker (Doxy-linker-mice, cyan-filled in Figures 1C-I; S2A-G). Heart rate (HR, Figures 1J, S2H), cardiac output (CO, Figures 1K, Figure S2I) and the cardiac index (CI, ~CO normalized to BW2/3, Figures 1L, S2J) were not significantly affected by linker expression; nor was the systolic blood pressure (Figure S3A) suggesting no significant impact on the peripheral circulation. LV wall histology of the linker-hearts was similar to the control; with no significant difference in fibrotic areas (Figure S3B,C; fibrotic/total area 2.05±0.5% vs 2.6±0.6%, means±S.D.). Collectively, linker-mouse hearts displayed increased LV contractility with otherwise normal cardiac function, without an overt hypertrophic phenotype.
Cardiac exercise performance was compared using a graded maximal exercise protocol on treadmill (Figure S3D and Expanded Methods)18. Performance was assessed by the maximum speed reached (Figure S3E) and the time spent on the treadmill belt (Figure S3F) and no significant difference was found between control and linker groups.
Mitochondria and SR remodeling in the hearts of adult linker-mice.
VCM mitochondria distribute to three regional groups: perinuclear, subsarcolemmal (SSM), and interfibrillar mitochondria (IFM). Transmission electron microscopy (TEM) of linker-expressing LV muscle revealed a vast redistribution of mitochondrial clusters. IFM that normally often form longitudinal beads-on-string clusters between myofibrils, also tended to form massive clusters, often connected with the perinuclear pool (Figure 2A). Morphometric analysis revealed an increase in mitochondrial density (Figure 2B). Consistently, western blot analysis found the matrix protein mtHsp70 abundance higher in linker-VCMs (Figure 2C). Protein levels of Pgc1α, a transcriptional coactivator for mitochondrial biogenesis, were not elevated in linker-VCMs (Figure 2C), indicating that the mitochondrial population was in a net steady state. The large mitochondrial clusters appeared well-infiltrated by network SR (nSR, Figure S1F,G). Protein abundances of the luminal jSR marker calsequestrin, nSR/ER marker calreticulin and the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA2a) in linker-VCM lysates were not significantly different from the control (Figure 2C/ii). Notably, the increased mitochondrial density in the linker-hearts was not overall reflected in the abundance of IMM proteins. MCU, the pore-forming subunit of the mitochondrial Ca2+ uniporter (mtCU) (Figure S4A) and components of the respiratory complexes I-V (Figure S4C) were not changed significantly in the linker; while the transmembrane MCU regulator EMRE (Figure S4A), and housekeeper prohibitin (Figure S4C) were elevated. For potential functional significance of the increase in EMRE, we performed a crude assessment of the Ca2+ activation of mitochondrial Ca2+ uptake in permeabilized VCMs (Figure S4B). We compared the mitochondrial Ca2+ uptake rates as the initial rate of post-peak [Ca2+] clearance of added Ca2+ pulses (from Figure S7B). No significant difference was detected in the mitochondrial Ca2+ uptake rates in Lnk vs Ctr, suggesting no major alteration in the MCU complex activity. The abundance of proteins with reported mitochondria-jSR tether function in the heart, Mfn2 and PTPIP51 was not significantly altered in the linker-heart (Figure S4D).
Figure 2: Mitochondrial and SR remodeling in the linker myocardium.

A. Representative TEM montages of a control (Ctr) and a linker-expressing (Lnk) cardiomyocyte (yellow shade) in perfusion-fixed papillary muscles. Note the large mitochondrial (cyan shade) clusters in Lnk that can reach from the perinuclear area to the intercalated disc. Nu, nucleus; m, mitochondria; LD, lipid droplet. B. Mitochondrial densities (mitochondria-occupied area of the sarcoplasm). n=14 myocytes each from N=4 Lnk and 4 Ctr hearts; 3-4 myocytes/heart. Means±SEs. P values from mixed-effects regression/Fisher’s LSD. C. Relative expression of matrix protein mtHsp70, mitochondrial biogenesis regulator Pgc1α, jSR-resident calsequestrin (Csq), nSR/ER-resident calreticulin (Crt) and SERCA2a. Pooled data from VCM lysates (N=4 hearts each). Band densities were normalized first to the total protein in the lane (from Ponceau-S gel image), then to the mean of the respective Ctr. Means±SEs. P was calculated for each protein by Mann-Whiney Rank Sum test. See all p values in Online Dataset S1.
IFM are often spaced by Z lines, where the transversal side of the mitochondria forms close contact with the dyadic jSR (Figure 3A), to support the EBC. TEM analysis found the individual mitochondria-jSR contacts in the linker-LV myocardium to be larger, with a greater segment of transversal OMM within ≤50 nm proximity to jSR (Figure 3C), and tighter, with shorter minimum gap distance (Figure 3E) and almost doubled OMM sub-segment in 0-20 nm proximity but unchanged in 20-50 nm proximity (Figure 3D). These data indicated that the linker selectively enhanced the tight (≤20 nm gap) mitochondria-jSR interfaces. Importantly, there was no significant difference between control and linker in the transversal-side OMM length and in the perimeter of contact-forming mitochondria (Figure 3F,G). However, in the linker-myocytes, the count of canonical (transversal-side) mitochondria-jSR contacts per sarcoplasmic area was decreased (Figure 3H). This was largely due to exclusion of dyads from the large dense mitochondrial clusters (Figure 3I).
Figure 3: Mitochondria-jSR contacts are individually enhanced but less frequent in the linker heart.

A-H. TEM analyses of mitochondria-jSR contacts in longitudinal sections of LV papillary muscles. A. Images of typical mitochondrion-jSR contacts occurring at Z-lines from a control (Ctr) and a linker (Lnk) myocyte. Shades: Mitochondrion-yellow, jSR-red, transversal tubule (TT)-blue, nSR-purple. L,T indicate longitudinal and transversal sides of the IFM (T-side faces jSR in the canonical orientation). Zoomed image: T-side traced for the contact length normalization. B. Mitochondria-jSR contact analysis using the MitoCare Tools ImageJ/Fiji plugin; here, instead of surface segments of the whole mitochondrion (Mito), T-side segments are binned. C,D. Gap distance distribution of mitochondrial T-side segments at <50 nm proximity of jSR. C: cumulative (10 nm) binning. D: exclusive, 0-20nm and 20-50 nm distance bins (larger T-side portion in the 0-20nm bin indicate tighter contact in the Lnk). E. Minimum OMM-jSR distances in the individual mitochondria-jSR contacts. F. Mean perimeter of the mitochondria forming jSR contact. G. Lengths of the mitochondrial T-sides involved in contact formation. H. Prevalence of mitochondria-jSR contacts per sarcoplasmic area per cell. For all, n=14 (Ctr) and 17 (Lnk) whole myocyte cross-sectional areas from N=4 Ctr and 4 Lnk hearts. 3-5 cells/heart with 8-198 (median 37) contacts/cell). Bars represent Means±SEs. P values were obtained as described in the gray text box. I. Exemplar immunofluorescence image of RyR2 (i, green, 488 nm) and its overlay with mRFP (ii, green/red respective components) in a linker-VCM. RyR2 leaves large ‘empty areas’ (e.g. the white dashed contour), which are filled with tightly packed mRFP-decorated mitochondrial clusters. 2x zoom: packed rings of linker mRFP, contouring the mitochondria and lacking green/RyR2 fluorescence.
Collectively, the engineered linker expressed from the time of cardiomyocyte maturation appeared to remodel the mitochondria and SR, leading to the enhancement of individual close mitochondrial interfaces with the SR, but with fewer mitochondria engaging in jSR contacts along the Z-discs.
Well-preserved EBC and metabolic fitness in the linker-myocytes.
Next, we examined how the linker-associated remodeling affected the EBC and respiratory fitness. EBC was assessed in freshly isolated VCMs by the changes in the respiratory chain electron donors NAD(P)H and FADH2 in response to electric field stimulation in the presence of isoproterenol. To let the myocytes ‘work’, contractile activity was not inhibited. The pre-stimulation NAD(P)H autofluorescence was not significantly altered in the linker-VCMs. Maximum fluorescence upon cyanide and Antimycin A/Rotenone addition was elevated (Figure 4A), likely due to more densely packed mitochondria per cell (more epifluorescence per pixel from the clusters). The ratio of NAD(P)H and FAD autofluorescence was taken to eliminate motion artifacts during stimulation. After correcting the fluorescence bleed-through from linker-mRFP to FAD autofluorescence, the effects of the linker and increasing stimulation frequencies were modeled using generalized estimating equations (GEE) with a nested, hierarchical covariance structure suited for repeated measurements done in several technical replicates from each animal. By the GEE model, the basal NAD(P)H/FAD ratio was significantly lower in linker than control (Figure 4C/0Hz, 11.7±3.3 vs. 16.3±2.7, p=0.010).
Figure 4. Assessment of EBC, metabolic fitness and Ca2+ signaling in the linker myocytes.

A-C. NAD(P)H and FAD autofluorescence imaging in VCMs freshly isolated from Ctr and Lnk. Isoproterenol 100 nM was added to the superfusate to provide sympathetic tone. After baseline recording, electric field stimulation (FS) at increasing frequencies (1-2-5 Hz) was applied then stopped (ns), followed by sequential additions of caffeine (Caf 10 mM), NaCN (CN 40 μM), and antimycin A+rotenone (A+R, 10 μM/0.25 μM). (A) NAD(P)H autofluorescence at baseline (basal), after respiratory complex IV (NaCN) and additional complex I&III inhibition (A+R). Means±S.E., N=8 hearts each. B. Representative timecourses of NAD(P)H/FAD ratio from individual Ctrl and Lnk myocytes (n>170 each). C. Cumulated corrected NAD(P)H/FAD ratio levels at the indicated steps of the stimulation protocol, collected from corresponding timecourse trace segments. Before forming the ratio, minimum FAD fluorescence at NaCN or A+R was subtracted to correct for mRFP crosstalk (see details in Expanded Methods). Means±SEs, n=7 independent experiments (one Ctr and Lnk heart each; 4-8 technical replicates/coverslips; 2-8 VCMs/field). D-I. [Ca2+]c (fura-2 ratio) responses of primary VCMs to sequential FS periods at 1Hz, 5Hz, 1Hz, each followed by a short (~30s) pause. After the last pause, caffeine (caff, 10 mM) is added to discharge SR Ca2+. Isoproterenol 100 nM is present throughout to provide sympathetic tone. D. Representative timecourses from a Ctr and Lnk (red) VCM (n(cells)=39 for Ctr and 53 for Lnk). Boxed segments in (i-ii) are overlaid on expanded timescale in (iii). E-I. Cumulated parameters extracted from the timecourses. For each experiment (pair of hearts), individual cells were normalized to the mean of the control (see Figure S5E-G for values without normalization to Ctr). E. Basal Fura-2 ratios. F. Diastolic [Ca2+]c at the indicated FS frequencies. G. [Ca2+]c spike amplitudes (systolic-diastolic difference) during FS. E-G: N=4 pairs of hearts; 2-6 coverslips each (2-7 VCM/field). Means+s.e. P was calculated using mixed random intercept model for E; for F-G, 2-way RM ANOVA on rank-transformed data, Holms-Sidak all-pairwise post hoc comparison. H. Exemplar timecourse of the caffeine response in a linker-VCM (n=10 cells). I. Amplitudes of caffeine responses, (see also Figure S5G). n=15(ctr) and 10(Lnk) myocytes (N=3 pairs of hearts, 3-7 VCMs each. Means ± SEs. P: mixed random intercept model).
EBC ‘pre-gauges’ the workload by the Ca2+ signals diverted from the EC coupling to the mitochondrial matrix under β-adrenergic and electric stimuli. The net effect on NAD(P)H/FADH2 levels will depend on the workload increase and related ATP usage, which, in the isolated plated VCMs can vary cell-to-cell. Accordingly, upon field-stimulation with increasing pacing frequency (1Hz→2Hz→5Hz) cells may present decrease or no change in NAD(P)H/FAD ratio, likely reflecting the EBC to undershoot or evenly balance the workload increase; or an increase, suggesting EBC overshoot (Figure 4B). Overshoot may occur in electrically paced VCMs7, owing to smaller workload (contractions meet less resistance) out of the tissue context. According to the GEE model, with the increasing pacing frequency, the control myocytes’ NAD(P)H/FAD ratio tended to decrease (p=0.077); importantly, this response was significantly different from the slight increase in the linker-myocytes (p=0.007) (Figure 4C, Table S1). These data may reflect basally somewhat more active (perhaps in connection with the hypercontractility) linker-VCMs that, under the combined stress (isoproterenol) and electric stimulation effectively raise matrix reducing equivalents to similar levels with the control.
Together, these data suggest a well-preserved net EBC in the linker-myocytes; despite having fewer mitochondria-jSR contacts.
Overall metabolic fitness was assessed based on respiration of primary VCMs in glucose/L-Glutamine-supplemented medium. Basal O2 consumption rate (OCR) and its oligomycin-sensitive component (oxidative phosphorylation) were similar between control and linker-myocytes (Figure S5A/basal, OM). These parameters carry limited relevance in non-working unstimulated VCMs. Maximum, uncoupled OCR (FCCP, Figure S5A) and maximum mitochondrial respiration (Figure S5B) that reflect the overall metabolic capacity of the mitochondrial pool were not significantly higher in the linker-VCMs, despite the increased mitochondrial density; consistently with the unchanged protein levels of respiratory complexes (Figure S4C).
To assess the proton motive force, the accumulation of the potentiometric dye tetramethylrhodamine methyl ester (TMRM) was compared in VCMs. TMRM loading caused similar %-increases of the baseline (background) fluorescence of linker-and control myocytes, indicating that ΔΨm in the linker-mitochondria was as well maintained as in the control or even better (% increase started from higher baseline because of the mRFP) (Figure S5C,D).
Linker-myocytes have enhanced Ca2+ signaling capacity.
The increased contractility and substantial reorganization of mitochondria and SR urged us to examine how the linker impacted VMC Ca2+ signaling. [Ca2+]c responses to electric field-stimulation were monitored in freshly isolated VCMs loaded with Fura2/AM using an ultra-fast epifluorescence imaging system. The superfusate contained isoproterenol 100 nM, to provide β-adrenergic tone and the following field-stimulation protocol was applied: 0Hz (rest/baseline)-1Hz-0Hz-5Hz-0Hz-1Hz-0Hz (as shown in Figure 4D). Pre-stimulation [Ca2+]c (Fura-2 ratio) was similar between control and linker (Figures 4E, S5E) and so were the diastolic [Ca2+]c levels during field-stimulation (Figure 4F,Figure S5F bottom). However, the corresponding [Ca2+]c spike amplitudes were significantly greater in the linker-myocytes at all frequencies (Figure 4G, Figure S5F top). As a crude assessment of the SR Ca2+ pool, it was fully discharged with a saturating dose (10 mM) of caffeine following the post-stimulation rest period (Figure 4H,I; Figure S5G). The corresponding [Ca2+]c spike had substantially (2.5 fold) greater magnitude in the linker-myocytes, relative to Ctr (Figure 4I).
These results indicate an increased SR Ca2+ pool, possibly due to the expanded nSR infiltrating the large mitochondrial clusters (Figure S1F,G). The pacing-associated larger Ca2+ spikes may have contributed to the increased contractility of the linker myocardium and to the well-maintained EBC.
Clustered mitochondria in linker-myocytes form non-transversal contacts with jSR and are more interconnected via nanotunnels.
While EBC efficacy in the linker-myocytes was structurally supported at the individual IFM-jSR contacts (expansion and tightening), this was countered by scarcer canonical contacts (Figure 3H), owing to the exclusion of dyads from the large dense mitochondrial clusters (Figure 3I). To understand if/how these large mitochondrial clusters contributed to the EBC, we looked for potential structural adaptations and identified two major differences from control. (1) Mitochondria on the surface of the cluster frequently contacted dyads in longitudinal orientation (Figure 5A). Longitudinal dyad contacts were >2.5 times more prevalent in the linker (Figure 5B). (2) To participate in EBC, ‘inner mitochondria’ of the dense clusters would need to connect with the ‘outer mitochondria’ that can form dyad contacts. TEM analyses (greater sampling power than tomography), revealed >3 fold more frequent nanotunnel-type mitochondrial continuities19 in the linker-myocytes (Figure 5C,D). High-resolution EM tomography (focused ion beam scanning electron microscope, FIB-SEM) of linker-LV corroborated these results by capturing elaborate nanotunnel networking between mitochondria (Figure 5E arrows, Movie S1). To further investigate if nanotunnels were connecting ‘outer mitochondria’ of the cluster (‘eligible’ for dyad contact) with the ‘inner mitochondria’, first, via TEM analysis we sorted nanotunnel-connected mitochondria by their localization relative to myofibrils into 3 groups (Figure 5F). 1) Periphery⥃Inside: either an ‘inner mitochondrion’-connected nanotunnel contacts the myofibril (Figure 5Ci&Fi) or a myofibril-contacting ‘outer mitochondrion’ forms a nanotunnel-like extension ‘directed’ inside the cluster (Figure 5C/ii&F/ii). 2) Inside⥃Inside: ‘inner nanotunnel’ is connected with an ‘inner mitochondrion’ (no myofibril contact) (Figure 5C/iii,F/iii), 3) Periphery⥃Periphery: both the nanotunnel and connected mitochondrion are in close proximity to the myofibril (Figure 5C/iv,F/iv). In the linker-myocytes, the majority (>60%) of nanotunnels belonged to the 1st group and just a minuscule (5%) portion to the 3rd group (Figure 5F bar graph). 3D analysis of the co-occurrence of jSR contacts revealed that >80% of nanotunnel-forming mitochondria in the linker-LV also had close contact with a dyad (44 out of 49, counted in 2 independent FIB-SEM volumes, Figure 5G).
Figure 5. Altered communication of mitochondria in large clusters with the jSR and each-other.

A. Exemplar TEM image from a linker-mouse LV papillary muscle showing a mitochondrion-jSR contact along the longitudinal (L) side of the mitochondrion at the periphery of a large mitochondrial cluster. B. L-side Mito-jSR contact occurrences as % of all (L+T-side) contacts in Ctr and Lnk myocardium. C. Exemplar TEM images of nanotunnels and connected mitochondria (cyan shade) with different positioning in the large clusters of the linker myocytes (i-iii) and in a row-like IFM cluster of a control myocyte (iv). D. Occurrence of nanotunnel-type intermitochondrial communication in Ctr and Lnk (from 2D TEM analysis) per myocyte (top) and per sarcoplasmic area (bottom). E. FIB-SEM tomographic visualization of nanotunnels (arrows) in a linker LV wall sample. The high 3D-resolution (5 nm voxels) gives high confidence in membrane continuities. Individual mitochondria are distinctly colored to follow continuities. Note the multiple nanotunnels connecting the green mitochondrial ‘bodies’ and the blunt-ended nanotunnel-like processes indicative of additional (dynamic) future or past continuities. See also Supplemental Movie 1. F. Schematic for grouping the nanotunnels by their relation to the cluster periphery/border (myofibrils). Periphery⥃inside (Periph⥃inside) peripheral nanotunnel(s) contact(s) inner mitochondrion (i) or peripheral/outer mitochondrion forms nanotunnel ‘inward’ (ii). Inside⥃inside: nanotunnel and connected mitochondrion are both ‘deep’ in the clusters (iii). Periph⥃Periph nanotunnel and connected mitochondrion are both adjacent to myofibril (iv). Each group is exemplified by the TEM images in C, with matching labels. Bar graph: Distribution of the three groups expressed as % of all nanotunnels in Lnk myocytes. TEM analysis: N=4 hearts/condition. n=3-5 cell/heart, Means ± SEs. P determined by mixed random intercept model (B,D) and Chi square test (F, against even distribution; with nanotunnel counts 142, 79 and 15 in the respective groups). G. Right: Exemplar volume rendering of nanotunnel-connected mitochondria (yellow) and nearby dyads from a FIB-SEM (Thermo/FEI Helios 4) tomography volume. Segmentation of the T-tubule (cyan) and SR (red) networks is partial; focused on the dyad junction areas (see also Supplemental Movie 2). Scheme and table: 3D analysis of the prevalence of nanotunnel-forming mitochondria that also contact dyad(s) (jSR). Mitochondrial cross sections from the top and bottom slices of FIB-SEM tomography stacks (from 2 linker hearts) were followed inside the whole volume. Limitation: mitochondria with a cross-sectional surface at the top/bottom slice will have a part outside the volume. Even so, almost all nanotunnel-forming mitochondria have jSR contact (Table).
Thus, the linker-induced reorganization of mitochondria to large clusters with fewer canonical mitochondria-jSR contacts drew multiple structural adaptations to maintain effective EBC. This included direct enhancement of individual contacts, frequent contact formation with longitudinally oriented dyads and nanotunnel-type continuities between peripheral and internal mitochondria of the clusters.
Improved performance of the linker-heart helps to overcome acute high-dose catecholamine-induced decompensation in females.
Mitochondria-SR/ER contact enhancement has been implicated in multiple cell/tissue injury paradigms1. Here, we investigated how the linker affected cardiac vulnerability to injurious stresses. In the first stress paradigm pathologically high-dose isoproterenol (300 mg/kg) bolus was administered intraperitoneally. We focused on the acute/subacute 24h period post-injection, to avoid potential tissue remodeling/fibrosis. Cardiac function was monitored by echocardiography before (basal) isoproterenol injection, then 30s, 30min, and 24h post-injection. Similar early time-points were examined in a rat model of acute stress-induced heart failure (takotsubo syndrome)20. The initial (30s) response was consistent with an increased sympathetic tone: male/female control and linker groups all showed an increase in HR (Figures 6E) and contractility (EF increase Figures 6C; decrease in LVESV and LVEDV, Figures 6A,B). Doxy-linker-mice responded similarly to controls (Figure S6), except that, for unclear reasons, they had slightly higher basal HR with no significant increase at 30s (Figure S6E). The post-initial (30min) phase was dominated by a drop in HR in all groups to below-basal levels (except linker-and Doxy-linker-males, Figures 6E, S6E), and a recovery from the increased contractility in linker and control (Figure 6A,B 30s vs 30min). This likely involved a compensatory circulatory reaction with a transient ‘freezing’ behavior (not shown) that had been also observed by others using similar treatment21 and considered as a vagal reflex22. To preserve basal CO, the drop in HR at 30min was compensated by an increase in SV in all males and linker-females but not/less in the control and Doxy-linker-females (Figures 6D,F,G, S6D,F,G). The increase in SV upon the HR drop prevented a drop in CO in most (7/10) of the linker-males, 4/8 linker-females, 4/9 control males but in only 2/10 control females (Figure 6G, ΔHR<1, points on or above the ‘isoCO’ line). In the rest, the HR drop @30min was followed by a CO drop; in some mice, including 5 control females (50% of those with HR drop), down below 10 ml/min (Figure 6F, below dashed line). These 5 control females also had the lowest CI (Figure 6H, bins 0-1.2 ml/min/body surface*) and three of them died within an hour (Figure 6G/crosses, 6I). Of 6 Doxy-linker-females, one died just before the 30min post-injection echocardiography, 4 presented a drop in HR, all decompensated (Figure S6G, open cyan symbols in pink-shaded area). One decompensated Doxy-linker-female died after a further plunge in HR (214/min), CO (6.1 ml/min) and CI (0.68 ml/min/body-surface*) at 60 min post-injection (time-point not shown; recorded only for a subset of animals) (Figures S6G/cyan cross, S6I).
Figure 6. Improved resistance to acute massive β-adrenergic stress in the linker-mice.

Mice were injected intraperitoneally with a single 300 mg/kg isoproterenol bolus. Echocardiography was performed before (Basal), then 30s, 30min and 24h post injection. A-F. Cumulated time-points for the indicated parameters for male (♂) and female (♀) mice. Means are shown as pseudo-timecourses (Ctr, black; Lnk, red). Individual animals are shown as vertical scatters. Means±SEs. N=13 Ctr♀, 10 Ctr♂, 11 Lnk♀, 15 Lnk♂. Gray box: statistical analysis information. G. Individual SV vs. HR fold changes from basal to the 30min point. To maintain CO (blue dashed ‘iso-CO’ line), an HR drop would be compensated by a proportional increase in SV. Mice with HR<1x under the iso-CO line (pink-shaded area) are not fully compensating (those with HR<1x & SV<1x are decompensating). Most control females are in the pink-shaded area. ✞, animal died in the first hour. H. CI distribution at 30 minute post-isoproterenol. Bins contain %portions of all Ctr or Lnk mice (actual males/female distribution is indicated by the symbols on top of the bins). Note that 5 Ctr and only one linker female have CI<1.2 ml/min/body-surface*. I. Survival traces (as labeled) at the indicated time points. Only control females died in 1 hour post-injection. See Doxy-linker mice in Figure S6.
Thus, the reaction to pathologically high-dose isoproterenol involved a critical post-initial phase with a plunge in HR. Many of the control and Doxy-linker-females became severely decompensated, with dangerously low CIs and early fatalities. Strikingly, linker-females compensated much better, without mortality, presumably due to the better-maintained contractility and energetics of the linker-hearts.
Similar isoproterenol bolus has been reported to induce myocyte injury that was reversible23 or progressed to ‘type 2’ (non-occlusive, relative hypoxia) myocardial infarction21 in C56Bl/6J mice. 2 weeks post-injection, EF was normal in23, decreased in21, while in the current study, in a subcohort of 32 mice sampled from all groups, it was rather increased (by 20±33% mean±S.D.) but so was in a small cohort of saline-injected sham animals (12±21% mean±S.D., n=6, not shown). The better long-term tolerance in our mice might be due to the mixed background, carrying the WT Nnt gene providing better resistance to oxidative stress24.
Linker-hearts are protected from ex vivo ischemia/reperfusion (I/R) injury.
Increased expression of an endogenous mitochondria-ER/SR tether component, PTPIP51 has been reported to positively correlate with the severity of cardiac I/R10. Here, we tested, using an ex vivo model, how long-term enhancement of mitochondria-ER/SR tethering by the linker affected myocardial I/R injury. Control and linker-hearts were Langendorff-perfused with Krebs-Henseleit/bicarbonate buffer for 20 min (equilibration), then subjected to 40 min of no-flow global ischemia followed by 60 min of reperfusion, while continuously monitoring the perfusion pressure and LV pressure (Figure 7A). The loss and recovery in LV contractility (+dP/dtmax) were used as functional readouts for the I/R injury (Figure 7B), and the infarct size for the extent of the injury (Figure 7C). Since I/R injury is more pronounced in males25, only male cohorts were used. dP/dtmax dropped near-zero in both groups during ischemia (Figure 7B 20-60 min). Strikingly, while in the control hearts, even after 60 minutes of reperfusion, the recovery was small (still significant increase from the ischemic levels, Figure 7B black ###), the linker-hearts showed relatively rapidly developing substantial recovery (Figure 7B 60-120 min and inset i). After 60 min of reperfusion, the recovered contractility (dP/dtmax) was >6 times greater in the linker than in the control heart (Figure 7B, inset ii, 0.68±0.2 mmHg/ms vs. 0.1±0.02 mmHg/ms). The infarct size was also substantially (by ~27%) reduced in the linker-hearts (51±6% vs 70±6%, Figure 7C red vs black). To assess the injury component involving the activation of the mPTP, the experiments were repeated in the presence of the cyclophilin D inhibitor, cyclosporine A (CSA 2 μM), upon which the recovery of control LV contractility markedly improved (Figure 7B, inset ii gray vs black), to an extent comparable to that in the untreated or CSA-treated linker-hearts (Figure 7B, inset ii gray vs red and pink ). Notably, the infarct size was not affected significantly by the CSA treatment (Figure 7C black vs gray, red vs pink), suggesting that the bulk of cell death was anoxic and mPTP (hyper)activity impacted more the functional recovery of the surviving myocardium.
Figure 7. Linker-mediated protection from ex vivo I/R injury.

A Exemplar LabChart record of LV pressure (LVP) and perfusion pressure (PP). Three phases of the experiment are indicated: equilibration/stabilization (Stabil, 20 minutes), no-flow ischemia (Isch, 40 min), reperfusion (60 min). B Timecourses of LV contractility (dP/dtmax) from control (black/gray) and linker (red/pink) hearts without (black/red) or with (gray/pink) CSA (2 μM) in the buffer. Inset i, the initial period of reperfusion on expanded timescale. Inset ii, bar graph representation of +dP/dtmax values at the end of reperfusion. C Infarct sizes (weighted average of triphenyltertazolium chloride-positive %areas of LV slices). Means±SE., N=6 animals/group/condition. Gray boxes: statistical analysis information.
Thus, linker expression seems to be protective against both, the anoxic myocyte death and the loss in function of surviving myocytes during reperfusion; and this latter portion of the protective effect likely involves a dampened activation of the mPTP. To differentiate if the protection took place via mitigating the upstream Ca2+ overload and ROS exposure or via altering the mPTP sensitivity or mitochondrial Ca2+ tolerance, the mitochondrial Ca2+ retention capacity (mCRC) of linker and control VCMs was compared. In suspensions of permeabilized myocytes the mitochondrial clearance of sequential Ca2+ boluses (CaCl2 3.75 μM) was monitored fluorometrically, simultaneously with ΔΨm. mCRC was defined as the number of boluses until mitochondrial uptake either stopped (no post-peak decrease in [Ca2+]c) or reverted to Ca2+ release (turn from decrease to increase in [Ca2+]c) before the next addition. To better isolate mitochondrial Ca2+ uptake, SERCA and the mitochondrial Na+/Ca2+ exchanger, NCLX, were respectively blocked by thapsigargin and CGP37157. To capture mPTP activity, the experiments were performed either in the absence (Figure S7A,C) or presence (Figure S7B,D) of CSA. mCRC was not significantly different between control and linker-VCMs, regardless of CSA. CSA effectively doubled mCRC in both groups. Collectively, the linker was protective in ex vivo I/R injury likely via mitigating anoxic cell death and myocyte damage linked to the activation of the mPTP. Since the mCRC of permeabilized linker-VCMs is unchanged, it is conceivable that the core site(s) of linker-mediated protection is/are other than the bulk mitochondrial matrix Ca2+ handling. Perhaps, the linker-associated mitochondria/SR remodeling and adaptive reformatting of mitochondria with individually expanded dyad-contacts and presumably enhanced Ca2+/ROS crosstalk, also mitigate the reperfusion-associated Ca2+ and ROS dysregulations.
DISCUSSION.
We aimed to assess the physiological/pathological relevance of constitutive mitochondria-SR tethering in the cardiac muscle using an engineered mitochondrion-SR tether/linker. Postnatal cardiac muscle-specific constitutive expression of the linker brought about major multi-level remodeling of the mitochondrial and SR networks in mature myocytes with mitochondrial crowding and individually enhanced but fewer mitochondria-jSR contacts. Functionally, linker-hearts had improved contractility, VCMs had stronger sarcoplasmic Ca2+ signaling, well-preserved EBC, ΔΨm and mitochondrial respiration capacity. The linker seemed to protect from early decompensation during massive adrenergic stress in the most vulnerable female cohort and ameliorated ex vivo I/R injury.
The choice of linker.
Mitochondria-SR/ER contacts are spatially-temporally defined by inter-organelle protein tethers. The linker was expressed under the cardiac muscle-specific Myh6 promoter, to avoid potential indirect cardiac effects upon expression in circulatory or autonomic nervous systems. Engineered tethers are either monomeric (linkage via membrane insertion), or bipartite (linkage via drug/light-inducible protein-protein interaction)1. We chose our monomeric linker12 to allow preferential enhancement/reinforcement of existing contacts, with less chance of snapping together brief stochastic membrane encounters1. Also, an inducible linker would require administration of rapamycin26 or rapalog that, for this purpose, has not been established in animals. Although the linker does not differentiate between ER and SR, by its length (<20 nm) it is limited to the smooth ER (ribosomes are too large for such gap), which effectively equals to the SR in muscle cells27.
The linker-induced mitochondrial and SR proliferation.
The chronic expression of the linker rearranged mitochondria into large clusters that were richly infiltrated and likely kept together by the nSR (like densely-bunched grapes by an elaborately branching grape stem). Interference with Parkin-and Mfn2-dependent metabolic maturation of cardiac mitochondria also produced mitochondrial clusters; however, mitochondria were immature, causing a severe phenotype28. The observed increase in mitochondrial density might have been prompted by a more restricted mitochondrial motility/traffic towards the peripheral IFM pool during VCM maturation. This might explain the lack of increased mitochondrial biogenesis in the adult linker-myocytes (no increase in PGC1α). Since the onset of linker protein expression seemed to overlap with VCM maturation, studying the effect of the linker in immature myocytes was not feasible in this model. Interestingly, although the housekeeping matrix HSPA protein, mtHsp7029 and IMM protein prohibitin were elevated in the linker-hearts, the abundance of respiratory complex components were not higher, nor was maximum mitochondrial respiration of linker-VCMs. It is conceivable that VCMs bear a rheostat system to keep the global mitochondrial respiratory (and related ATP/ROS-producing) capacities around a set-point, with checkpoints at transcriptional, translational or post-translational regulation of the involved proteins. Along this line, the mtCU pore-forming subunit, MCU was unchanged but the other essential channel-forming subunit EMRE protein levels were significantly elevated in the linker-heart. Although mitochondrial Ca2+ uptake activity at [Ca2+]c in the low-μM range (relevant in [Ca2+] nanodomains30) appeared unaffected, further studies will be needed to fully characterize potential shifts in the activation properties or strategic localization (at jSR contacts30) of the mtCU in the linker-hearts.
If indeed the linker-induced increase in mitochondrial mass was reactive to less effective traffic to the cell periphery (‘mitochondrial traffic jam’) during maturation, it would be plausible that some of the ‘jammed’ mitochondria in the large clusters were rendered less active. Indeed mitochondrial sizes greatly vary in the clusters with smaller-sized mitochondria having less dense cristae. Detailed characterization of this rheostatic mechanism and related cristae analysis of the massive heteromorphic mitochondrial pools of linker-myocytes is one of our future goals. ΔΨm measurements in VCMs using TMRM did not reveal significant differences between control and linker mitochondrial pools, nor detected heterogeneities amongst individual mitochondria. However, small mitochondria in clusters are not individually distinguishable via light microscopy.
Mitochondria-smooth-ER/SR tethering is critical for inter-organelle lipid exchange (similar linker to ours complemented phosphatidylethanolamine/phosphatidylcholine synthesis upon loss of the mitochondria-ER tethering ERMES complex)31. Thus, the linker may have facilitated membrane lipid synthesis, potentially supporting nSR and mitochondrial membrane expansions. Further, future studies will clarify potential linker-expression-associated changes in VCM lipid metabolism and related changes in membrane composition.
Collectively, the linker-induced mitochondrial (and potential nSR) proliferation likely involved reactive components to mechanical restrictions and anabolic components due to reinforced phospholipid synthesis.
Impact of the linker on the mitochondria-SR contacts and EBC.
The linker-induced mitochondria-jSR contact remodeling was structurally/functionally bifurcated. Individually expanded and tightened contacts were reduced in their number, owing to the exclusion of dyads from the dense mitochondrial clusters. SR and mitochondria are major sources of Ca2+ and ROS. Balanced Ca2+ transfer at the mitochondria-jSR contacts supports EBC, while excessive/prolonged Ca2+-exposure may feed a vicious cycle of ROS overshoot, ROS-sensitized RyR2-mediated Ca2+ release culminating in mitochondrial Ca2+ overload-associated mPTP activation3. Such paradigm has been implicated in acute enhancement of mitochondria-SR/ER contacts by PTPIP51-containing tethers in I/R injury10.
EBC appeared well-preserved in linker-VCMs, assessed by NAD(P)H/FAD responses to increasing electric field-stimulation at increased adrenergic tone. The latter was important since the main Ca2+ effector in EBC is pyruvate dehydrogenase for which substrate utilization becomes more significant under β-adrenergic stress, when the adult myocyte’s fuel preference for fatty acids shifts towards glucose32, 33. Since, for technical reasons, we could not establish direct [Ca2+]c/[Ca2+]m correlation, we cannot exclude that the EBC was preserved partly via the improved SR Ca2+ loading and greater systolic [Ca2+]c spikes, which also likely contributed to the observed hypercontractility.
Mitochondria-jSR encounters had to overcome multiple potential shortcomings in linker-hearts. Decreased contact count was compensated by individual expansion/tightening and more longitudinally-orientated contacts. Such remodeling, combined with the increase in SR Ca2+ release, could have put the jSR-contact-forming mitochondria at risk of Ca2+ overload via the above-mentioned vicious cycle (RyR2→Ca2+→matrixCa2+→ROS→RyR2→Ca2+)34,35. Likely as an adaptive measure, linker-myocytes had more nanotunnel-connected mitochondria. Mitochondrial nanotunnels have been envisioned as dynamic organelle stress-responses19. High occurrence of nanotunnels was reported upon Ca2+ imbalance in RyR2-mutant mice (decreased channel conductance causing SR Ca2+ overload with periodic bursts of sustained Ca2+ release)36. Consistently, our study suggest that nanotunnels were formed primarily by mitochondria in contact with jSR (indirect 2D TEM and 3D FIB-SEM data) that presumably were or would have been stressed, and at risk of Ca2+ overload in linker VCMs. We propose that the resulting continuity with mitochondria lacking jSR contact mitigates the stress while extending the EBC by distributing (“diluting”) the sequestered Ca2+ to multiple mitochondria (Figure S7E). SERCA pumps of the extensive meshwork of nSR infiltrating the dense mitochondrial clusters would then recollect the Ca2+ extruded via NCLX (Figure S7E). Thus, the composite remodeling with individually enhanced mitochondria-jSR contacts in the linker-myocytes may represent a quasi-stress-preconditioning that renders the mitochondrial network more connected, via dynamic nanotunnels, and homeostatically/energetically more durable. Further studies will be needed to establish the molecular/mechanistic details of nanotunnel dynamics.
The linker-induced remodeling is adaptive.
The organ-level functional outcome of the linker-induced remodeling was an increase in contractility; consistent with a recent report of improved contractility upon adenoviral delivery of a similar linker transgene that ameliorated experimental diabetic cardiomyopathy in mice13.
One surprising outcome of our study is the increased cardiac resilience to injurious stresses in the linker-mice. The tested stress paradigms both involved energetic deficit and oxidative stress. 1) In the in vivo acute massive adrenergic experiments, there was a critical period ~30 min after isoproterenol injection, likely in part as a vagal rebound, when in most mice the HR dropped below baseline levels. To preserve basal CO, the majority of linker and control males and linker-females compensated via proportional increases in SV; however, most control and Doxy-linker-females failed to do so. Although Doxy-linker-males also failed to compensate, their HR decrease was small and corresponding CI in the upper range (>1.8); in contrast to many decompensated control and Doxy-linker-females with very low CI at 30 min post-injection and fatalities. These occurrences of severe decompensation are reminiscent of acute stress-induced cardiomyopathy that mostly affects females and in humans, a severe (fatal unless treated) early complication can be cardiac arrest37. Proposed etiologies for such acute cardiomyopathy (takotsubo syndrome) involve transient cardiodepression owing to a change in the β-adrenergic signal transduction20 and energy demand-supply imbalance38-40. Thus, the linker provided a reversible (Doxy-linker) protection against acute stress-associated severe decompensation and fatalities affecting the female cohorts. We attribute this protective effect to the improved LV contractility at well-preserved EBC (more relevant under stress32, 33) that likely helped passing through the otherwise critical point in the females.
2) I/R-injury was assessed ex vivo to isolate the systemic stress reaction from the primary insult. Only males were included since I/R injury is known to affect males more severely than fertile females25. Potential sex-specific differences in the effect of the linker on I/R injury will be examined in future studies. Forty minutes of ischemia was sufficient to pass the rigor (ATP depletion) peak (see Figure 7A, rigor). Although the reperfusion-associated oxidative stress (ROS burst) usually peaks at the beginning of reperfusion41, the one-hour reperfusion period allowed better evaluation of the true functional recovery (beating and contractility), past the period of calcium paradox-related hypercontracture (Figure 7A). The linker showed remarkable protection both functionally and in the infarct size; seemingly conflicting earlier reports with short-term endogenous mitochondria-SR tether manipulation10. Besides non-tether roles of PTPIP511, we attribute this difference to the adaptive remodeling that occurs when the linker is chronically, constitutively expressed. One plausible mechanism for the protection in anoxic cell death might be related to a relatively hypoxic milieu that presumably built deep inside the large dense mitochondrial clusters, rendering a subpopulation of mitochondria preconditioned for ischemia. The finding that CSA in the perfusate improved the functional recovery in the control hearts comparably to the transgenic linker (while causing no significant further improvement in the linker-heart), suggested that a key target of linker-mediated protection was an mPTP-dependent component of the I/R injury. Although our exploratory tests did not reveal statistically significant further improvement in the functional recovery of the linker-hearts upon CSA-treatment, we cannot exclude that future more robust comparisons with larger cohorts would capture additional improvement by pharmacological mPTP inhibition. Indeed, it is unlikely that the linker would fully avert mPTP activation and associated cell damage. mPTP-linked damage involves a range of Ca2+ signaling dysregulations and ROS-mediated oxidative stress that eventually drive mitochondria to Ca2+ overload and to undergo PT, ultimately risking mitochondrial apoptosis and myocyte loss. Since the linker did not improve bulk mitochondrial Ca2+ tolerance (mCRC assays of permeabilized myocytes with energized mitochondria), most likely the linker-associated remodeling mitigated insults upstream or at the level of individual mitochondrial matrix Ca2+ handling. As discussed earlier, it is conceivable that the remodeling in the linker hearts to safely maintain EBC at individually enhanced mitochondria-jSR contacts via nanotunneling (Figure S7E) ‘preconditions’ the system to dysregulations in the Ca2+/ROS signaling that occur in I/R injury and impinge on the mPTP.
Taken together, our study delineates important long-term homeostatic adaptive mechanisms in cardiomyocytes and a potential new strategy to utilize these mechanisms to increase resilience to injuries. Mitochondria-SR/ER contacts as local signaling and homeostatic hubs, operate structurally-functionally by an optimum-based ‘Goldilocks principle’3, 42: the gap distance and interface area are tailored so that the local Ca2+/ROS crosstalk is effective but safe. Sudden loss or gain in tethering, unless it is compensatory13, can respectively offset the system towards energetic deficit (EBC disruption) or hyperactive signaling and associated Ca2+/ROS injury paradigms (enhancements)10. Enhanced mitochondria-SR contact formation by the engineered linker with an onset at the time of cardiomyocyte maturation brings about profound remodeling in the two organelle networks, attaining a ‘new normal’ with increased stability. The effectively irreversible doxycycline off-switch of the linker transgene (likely owing to long-term depot formation in the muscle17), prevented us to study the linker-associated remodeling with adult onset. Regardless, our study warrants further investigations to fully explore the potential therapeutic implications of chronically expressed mitochondria-SR linker transgene in the cardiac muscle as genetic preconditioning for susceptible individuals.
Supplementary Material
Novelty and Significance.
What is known?
Cardiac excitation-contraction coupling is mediated by local Ca2+ crosstalk between the transversal tubules and Ca2+ release units of the junctional sarcoplasmic reticulum (SR).
Mitochondria, which fuel the heart with ATP, form close contacts with the SR via protein tethers, providing platform for local exchanges, including Ca2+ transfer from the Ca2+ release units to the mitochondrial matrix, which then enhances respiratory chain activity and ATP production (excitation-bioenergetics coupling, EBC).
Up-or downregulation of endogenous tethering proteins alters the geometry of mitochondria-junctional SR contacts and EBC efficacy but upregulation may also lead to increased reactive oxygen species production and vulnerability to injuries involving oxidative stress.
What new information does this article contribute?
Since endogenous mitochondria-SR tethering proteins are multifunctional non-selective targets for studying the relevance of inter-organelle linkage, we studied the effects of an engineered dedicated mitochondria-SR/ER tether (‘linker’) expressed from the time of cardiomyocyte maturation under the Myh6 promoter in mice.
Such chronic linker expression brought about profound mitochondrial and SR remodeling characterized by the formation of large dense mitochondrial clusters infiltrated by the network SR but not junctional SR with individually enhanced but fewer mitochondria-junctional SR contacts.
EBC and contractile function remained well-preserved and linker-mice were more tolerant to injurious in vivo (acute massive adrenergic stress) and ex vivo (ischemia/reperfusion) insults; likely due to adaptive increase in nanotunnel-type mitochondrial networking that spread the benefits and eased the risks of individually enhanced mitochondria-junctional SR contacts.
Cardiac muscle is energized by mitochondria. Mitochondria-SR contacts locally direct Ca2+ from cardiac excitation to mitochondria to regulate mitochondrial ATP (EBC). However, overshoot in mitochondrial Ca2+ may cause oxidative stress and myocyte loss. Mitochondria-SR tethers support EBC but their role in cardiac physiology/pathology is debated. Endogenous tether proteins are multifunctional; therefore non-selective targets to address the significance of inter-organelle linkage. Here, we introduced to mice a cardiac muscle-specific dedicated engineered tether (‘linker’) transgene. Expressed from cardiomyocyte maturation, the linker enhanced individual mitochondria-junctional SR (jSR) contacts but evoked a marked remodeling with large dense mitochondrial clusters that excluded dyads. Yet, EBC remained well-preserved, likely due to more longitudinal mitochondria-jSR contacts and nanotunnelling between jSR-connected and jSR-free mitochondria. Unexpectedly, the linker improved tolerance to stressors: vulnerability to acute massive adrenergic stress, infarct size and myocardial impairment upon ex vivo ischemia/reperfusion injury were all dampened. We propose, mitochondria-SR contacts have a structural optimum. While acute increase in tethering may correct for pre-existing leftward-shift, a rightward-shift from the optimum may augment injuries. By contrast, chronic enhancement by the linker and the ensuing adaptive remodeling resets the optimum to a more stable and stress-tolerant state, opening a perspective for structural preconditioning as genetic therapeutic strategy.
ACKNOWLEDGEMENTS
We are grateful for Professor György Hajnóczky for the critical reading of the manuscript and helpful discussions; to Pal Pedersen and Rachna Parwani from Zeiss and Ken Wu from ThermoFisher Scientific for FIB-SEM demos and training; for Professor Nan Yao and John Schreiber for the scanning electron microscopy and FIB-SEM training at Princeton University’s Imaging and Analysis Center, which is partially supported through the Princeton Center for Complex Materials (PCCM), a National Science Foundation (NSF)-MRSEC program (DMR-2011750). We thank Dr. Keith Scott from TJU Biostatistics and Áron Andrési software developer at MitoCare for help with the statistical analysis.
SOURCES OF FUNDING
From NIH/NHLBI, R01HL142864 and R01 HL122124 to GC and SSS, R35 HL135736 to GWD. From NIH/NIGMS, R01 GM123771 to ELS. From the Hungarian National Research, Development and Innovation Fund, NKFI K134357 to PV. The VisualSonics ultrasound system for mouse echocardiography, housed in the Jefferson Small Animal High-Speed Ultrasound Imaging Facility, was funded by NIH grant 1 S10 OD030483-01.
Non-standard Abbreviations and Acronyms:
- BW
body weight
- CI
cardiac index
- CO
cardiac output
- Ctr
control
- Doxy
doxycycline
- EBC
excitation bioenergetics coupling
- EF
ejection fraction
- FCCP
Carbonyl cyanide p-(tri-fluromethoxy)phenyl-hydrazone
- FIB-SEM
focused ion beam scanning electron microscopy
- FS
fractional shortening
- HR
heart rate
- IFM
interfibrillar mitochondria
- IMM
inner mitochondrial membrane
- I/R
ischemia/reperfusion
- jSR
junctional SR
- Lnk
linker
- LV
left ventricle/ventricular
- LVAW
LV anterior wall
- LVEDV/LVESV
LV end diastolic/systolic volume
- MCU
mitochondrial Ca2+ uniporter
- Mfn2
mitofusin-2
- Mito-
mitochondria (connected to-)
- mRFP
monomeric red fluorescent protein
- nSR
network SR
- OCR
oxygen consumption rate
- OMM
outer mitochondrial membrane
- RyR
ryanodine receptor
- ROS
reactive oxygen species
- SR
sarcoplasmic reticulum
- SR/ER
sarco-endoplasmic reticulum
- SERCA
sarco-endoplasmic reticulum Ca2+ ATPase
- SV
stroke volume
- Tg
transgene
- TEM
transmission electron microscopy
- VCM
ventricular myocyte
Footnotes
DISCLOSURES
None.
References
- 1.Csordas G, Weaver D and Hajnoczky G. Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends in cell biology. 2018;28:523–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic calcium. Journal of molecular and cellular cardiology. 2002;34:1259–71. [DOI] [PubMed] [Google Scholar]
- 3.Brookes PS, Yoon Y, Robotham JL, Anders MW and Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American journal of physiology Cell physiology. 2004;287:C817–33. [DOI] [PubMed] [Google Scholar]
- 4.Csordas G, Thomas AP and Hajnoczky G. Calcium signal transmission between ryanodine receptors and mitochondria in cardiac muscle. Trends in cardiovascular medicine. 2001;11:269–75. [DOI] [PubMed] [Google Scholar]
- 5.Dorn GW 2nd. Mitochondrial dynamism and cardiac fate--a personal perspective. Circ J. 2013;77:1370–9. [DOI] [PubMed] [Google Scholar]
- 6.Beikoghli Kalkhoran S, Hall AR, White IJ, Cooper J, Fan Q, Ong SB, Hernandez-Resendiz S, Cabrera-Fuentes H, Chinda K, Chakraborty B, Dorn GW 2nd, Yellon DM and Hausenloy DJ. Assessing the effects of mitofusin 2 deficiency in the adult heart using 3D electron tomography. Physiol Rep. 2017;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen Y, Csordas G, Jowdy C, Schneider TG, Csordas N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S, Nerbonne JM, Dorn GW 2nd and Maack C. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circulation research. 2012;111:863–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorn GW 2nd, Song M and Walsh K. Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. Journal of molecular and cellular cardiology. 2015;78:123–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O'Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, Stanley WC and Walsh K. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiao X, Jia S, Ye J, Fang X, Zhang C, Cao Y, Xu C, Zhao L, Zhu Y, Wang L and Zheng M. PTPIP51 regulates mouse cardiac ischemia/reperfusion through mediating the mitochondria-SR junction. Sci Rep. 2017;7:45379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papanicolaou KN, Phillippo MM and Walsh K. Mitofusins and the mitochondrial permeability transition: the potential downside of mitochondrial fusion. American journal of physiology Heart and circulatory physiology. 2012;303:H243–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA and Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J Cell Biol. 2006;174:915–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dia M, Gomez L, Thibault H, Tessier N, Leon C, Chouabe C, Ducreux S, Gallo-Bona N, Tubbs E, Bendridi N, Chanon S, Leray A, Belmudes L, Coute Y, Kurdi M, Ovize M, Rieusset J and Paillard M. Reduced reticulum-mitochondria Ca(2+) transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy. Basic Res Cardiol. 2020;115:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Syed F, Odley A, Hahn HS, Brunskill EW, Lynch RA, Marreez Y, Sanbe A, Robbins J and Dorn GW 2nd. Physiological growth synergizes with pathological genes in experimental cardiomyopathy. Circulation research. 2004;95:1200–6. [DOI] [PubMed] [Google Scholar]
- 15.Guo Y and Pu WT. Cardiomyocyte Maturation: New Phase in Development. Circulation research. 2020;126:1086–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Padula SL, Velayutham N and Yutzey KE. Transcriptional Regulation of Postnatal Cardiomyocyte Maturation and Regeneration. Int J Mol Sci. 2021;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anders K, Buschow C, Charo J and Blankenstein T. Depot formation of doxycycline impairs Tet-regulated gene expression in vivo. Transgenic Res. 2012;21:1099–107. [DOI] [PubMed] [Google Scholar]
- 18.Petrosino JM, Heiss VJ, Maurya SK, Kalyanasundaram A, Periasamy M, LaFountain RA, Wilson JM, Simonetti OP and Ziouzenkova O. Graded Maximal Exercise Testing to Assess Mouse Cardio-Metabolic Phenotypes. PLoS One. 2016;11:e0148010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vincent AE, Turnbull DM, Eisner V, Hajnoczky G and Picard M. Mitochondrial Nanotunnels. Trends in cell biology. 2017;27:787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paur H, Wright PT, Sikkel MB, Tranter MH, Mansfield C, O'Gara P, Stuckey DJ, Nikolaev VO, Diakonov I, Pannell L, Gong H, Sun H, Peters NS, Petrou M, Zheng Z, Gorelik J, Lyon AR and Harding SE. High levels of circulating epinephrine trigger apical cardiodepression in a beta2-adrenergic receptor/Gi-dependent manner: a new model of Takotsubo cardiomyopathy. Circulation. 2012;126:697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forte E, Panahi M, Baxan N, Ng FS, Boyle JJ, Branca J, Bedard O, Hasham MG, Benson L, Harding SE, Rosenthal N and Sattler S. Type 2 MI induced by a single high dose of isoproterenol in C57BL/6J mice triggers a persistent adaptive immune response against the heart. J Cell Mol Med. 2021;25:229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porges SW. The polyvagal perspective. Biol Psychol. 2007;74:116–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wallner M, Duran JM, Mohsin S, Troupes CD, Vanhoutte D, Borghetti G, Vagnozzi RJ, Gross P, Yu D, Trappanese DM, Kubo H, Toib A, Sharp TE 3rd, Harper SC, Volkert MA, Starosta T, Feldsott EA, Berretta RM, Wang T, Barbe MF, Molkentin JD and Houser SR. Acute Catecholamine Exposure Causes Reversible Myocyte Injury Without Cardiac Regeneration. Circulation research. 2016;119:865–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nickel AG, von Hardenberg A, Hohl M, Loffler JR, Kohlhaas M, Becker J, Reil JC, Kazakov A, Bonnekoh J, Stadelmaier M, Puhl SL, Wagner M, Bogeski I, Cortassa S, Kappl R, Pasieka B, Lafontaine M, Lancaster CR, Blacker TS, Hall AR, Duchen MR, Kastner L, Lipp P, Zeller T, Muller C, Knopp A, Laufs U, Bohm M, Hoth M and Maack C. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015;22:472–84. [DOI] [PubMed] [Google Scholar]
- 25.Murphy E and Steenbergen C. Gender-based differences in mechanisms of protection in myocardial ischemia-reperfusion injury. Cardiovascular research. 2007;75:478–86. [DOI] [PubMed] [Google Scholar]
- 26.Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, Balla T and Hajnoczky G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Molecular cell. 2010;39:121–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berridge MJ. The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium. 2002;32:235–49. [DOI] [PubMed] [Google Scholar]
- 28.Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ and Dorn GW 2nd. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science. 2015;350:aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, Cheetham ME, Chen B and Hightower LE. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De La Fuente S, Fernandez-Sanz C, Vail C, Agra EJ, Holmstrom K, Sun J, Mishra J, Williams D, Finkel T, Murphy E, Joseph SK, Sheu SS and Csordas G. Strategic Positioning and Biased Activity of the Mitochondrial Calcium Uniporter in Cardiac Muscle. The Journal of biological chemistry. 2016;291:23343–23362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS and Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyman L, Chikando AC, Williams GS, Khairallah RJ, Kettlewell S, Ward CW, Smith GL, Kao JP and Lederer WJ. Calcium movement in cardiac mitochondria. Biophys J. 2014;107:1289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanley WC, Recchia FA and Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–129. [DOI] [PubMed] [Google Scholar]
- 34.Hamilton S, Terentyeva R, Kim TY, Bronk P, Clements RT, J OU, Csordas G, Choi BR and Terentyev D. Pharmacological Modulation of Mitochondrial Ca(2+) Content Regulates Sarcoplasmic Reticulum Ca(2+) Release via Oxidation of the Ryanodine Receptor by Mitochondria-Derived Reactive Oxygen Species. Frontiers in physiology. 2018;9:1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamilton S, Terentyeva R, Martin B, Perger F, Li J, Stepanov A, Bonilla IM, Knollmann BC, Radwanski PB, Gyorke S, Belevych AE and Terentyev D. Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic Res Cardiol. 2020;115:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lavorato M, Iyer VR, Dewight W, Cupo RR, Debattisti V, Gomez L, De la Fuente S, Zhao YT, Valdivia HH, Hajnoczky G and Franzini-Armstrong C. Increased mitochondrial nanotunneling activity, induced by calcium imbalance, affects intermitochondrial matrix exchanges. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E849–E858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh K, Carson K, Hibbert B and Le May M. Natural history of cardiac arrest in patients with takotsubo cardiomyopathy. Am J Cardiol. 2015;115:1466–72. [DOI] [PubMed] [Google Scholar]
- 38.Dawson DK, Neil CJ, Henning A, Cameron D, Jagpal B, Bruce M, Horowitz J and Frenneaux MP. Tako-Tsubo Cardiomyopathy: A Heart Stressed Out of Energy? JACC Cardiovasc Imaging. 2015;8:985–7. [DOI] [PubMed] [Google Scholar]
- 39.Godsman N, Kohlhaas M, Nickel A, Cheyne L, Mingarelli M, Schweiger L, Hepburn C, Munts C, Welch A, Delibegovic M, Van Bilsen M, Maack C and Dawson DK. Metabolic alterations in a rat model of Takotsubo syndrome. Cardiovascular research. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hirano KS K; Ikeda Y; Shirakami Y; Nagasaka H Energy Failure Hypothesis for Takotsubo Cardiomyopathy. Annals of Nuclear Cardiology. 2017;3:105–109. [Google Scholar]
- 41.Fernandez-Sanz C, Ruiz-Meana M, Castellano J, Miro-Casas E, Nunez E, Inserte J, Vazquez J and Garcia-Dorado D. Altered FoF1 ATP synthase and susceptibility to mitochondrial permeability transition pore during ischaemia and reperfusion in aging cardiomyocytes. Thromb Haemost. 2015;113:441–51. [DOI] [PubMed] [Google Scholar]
- 42.Dorn GW 2nd and Scorrano L. Two close, too close: sarcoplasmic reticulum-mitochondrial crosstalk and cardiomyocyte fate. Circulation research. 2010;107:689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bartok A, Weaver D, Golenar T, Nichtova Z, Katona M, Bansaghi S, Alzayady KJ, Thomas VK, Ando H, Mikoshiba K, Joseph SK, Yule DI, Csordas G and Hajnoczky G. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nature communications. 2019;10:3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De La Fuente S, Lambert JP, Nichtova Z, Fernandez Sanz C, Elrod JW, Sheu SS and Csordas G. Spatial Separation of Mitochondrial Calcium Uptake and Extrusion for Energy-Efficient Mitochondrial Calcium Signaling in the Heart. Cell reports. 2018;24:3099–3107 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grynkiewicz G, Poenie M and Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. The Journal of biological chemistry. 1985;260:3440–50. [PubMed] [Google Scholar]
- 46.Hamilton S, Terentyeva R, Bogdanov V, Kim TY, Perger F, Yan J, Ai X, Carnes CA, Belevych AE, George CH, Davis JP, Gyorke S, Choi BR and Terentyev D. Ero1alpha-Dependent ERp44 Dissociation From RyR2 Contributes to Cardiac Arrhythmia. Circulation research. 2022;130:711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.