

Significance
Clinical studies have long suggested a link between type 2 diabetes (T2D) and Alzheimer’s disease (AD): T2D patients have higher rates of AD and exhibit more rapid disease progression, whereas AD patients often have impaired glucose metabolism and altered insulin resistance. We sought to understand how dysregulated insulin signaling in astrocytes contributes to AD onset and progression. Using in vitro and in vivo models, we demonstrate a role of astrocyte insulin signaling in mediating the comorbidity of T2D and AD. This work may help identify potential targets to treat patients with T2D and AD.
Keywords: diabetes, insulin resistance, neurons, astrocytes, Alzheimer’s disease
Abstract
Brain insulin signaling controls peripheral energy metabolism and plays a key role in the regulation of mood and cognition. Epidemiological studies have indicated a strong connection between type 2 diabetes (T2D) and neurodegenerative disorders, especially Alzheimer’s disease (AD), linked via dysregulation of insulin signaling, i.e., insulin resistance. While most studies have focused on neurons, here, we aim to understand the role of insulin signaling in astrocytes, a glial cell type highly implicated in AD pathology and AD progression. To this end, we created a mouse model by crossing 5xFAD transgenic mice, a well-recognized AD mouse model that expresses five familial AD mutations, with mice carrying a selective, inducible insulin receptor (IR) knockout in astrocytes (iGIRKO). We show that by age 6 mo, iGIRKO/5xFAD mice exhibited greater alterations in nesting, Y-maze performance, and fear response than those of mice with the 5xFAD transgenes alone. This was associated with increased Tau (T231) phosphorylation, increased Aβ plaque size, and increased association of astrocytes with plaques in the cerebral cortex as assessed using tissue CLARITY of the brain in the iGIRKO/5xFAD mice. Mechanistically, in vitro knockout of IR in primary astrocytes resulted in loss of insulin signaling, reduced ATP production and glycolic capacity, and impaired Aβ uptake both in the basal and insulin-stimulated states. Thus, insulin signaling in astrocytes plays an important role in the control of Aβ uptake, thereby contributing to AD pathology, and highlighting the potential importance of targeting insulin signaling in astrocytes as a site for therapeutics for patients with T2D and AD.
Thirty-four million people in the United States have diabetes (9.4% of the adult population), and this number continues to increase at epidemic proportions (1). Over 95% of these individuals have type 2 diabetes (T2D), a chronic condition that results from insulin resistance and a limited ability of β-cells to compensate for this resistance. Insulin resistance is also a driving force in many other common disorders, including obesity, metabolic syndrome, nonalcoholic fatty liver, and atherosclerosis. T2D, obesity, and metabolic syndrome are associated with increased risk of neurodegenerative disorders, especially Alzheimer’s disease (AD) (2, 3). Conversely, patients with AD are at increased risk of T2D and related metabolic disorders (4–6). Together, these drive increased health-care costs and much patient suffering.
Accumulating evidence has shown that insulin signaling is altered in brains from AD patients and that insulin resistance, including alterations in the phosphorylation of IRS-1 and IRS-2, the two major substrates of the insulin receptor, in the brain is an early feature of AD pathology (7–10). Alterations in IRS phosphorylation have also been observed in some mouse models of AD (11, 12). However, exactly what cell types in the brain might be insulin resistant is unclear. In previous studies, we have shown that NIRKO mice with brain-specific insulin receptor knockout created using nestin-Cre, which deletes IR in both neurons and glial cells, exhibit neurobehavioral dysfunction in cognition and emotion (13), and these phenotypes increase with aging (14), reinforcing the clinical observations that brain insulin resistance is associated with increased risks of age-related cognitive decline (15). We also showed that NIRKO mice have increased levels of tau phosphorylation, a change also observed in the pathogenesis of human AD (14).
While most attention has focused on the possibility that it is the loss of insulin action in neurons that contributes to neurodegeneration, numerous studies have suggested that astrocytes regulate neuronal functions and also contribute to AD pathogenesis. We have previously shown that astrocytes express active insulin receptors and that astrocytic insulin signaling regulates ATP release from astrocytes, which then acts to modulate the activity of dopaminergic neurons in the midbrain (16), as well as systemic glucose metabolism (17). Astrocytes are also a major site of cholesterol synthesis in the brain, and this process is regulated by insulin (16, 17). Targeted deletion of astrocytic cholesterol synthesis in mice showed that astrocytes are a main source of brain cholesterol (18, 19). Wang et al. have shown that this cholesterol is carried by ApoE, a cholesterol transport protein genetically linked to AD, and can regulate β-amyloid production in neurons and interact with β- and γ-secretases to enhance β-amyloid production (20), which may contribute to the loss of cognition in patients with dementia.
Although these studies imply a role of astrocyte function in mediating the comorbidity of T2D and AD, a direct link between astrocytic insulin signaling and AD pathogenesis is absent. To this end, in the present study, we crossed mice with an inducible astrocyte-specific knockout of IR (iGIRKO) (16) with 5xFAD mice, which carry transgenes for five familial mutations for early onset of AD (21), to determine how astrocyte insulin resistance might contribute to AD pathogenesis. We find that insulin signaling in astrocytes is not only important in the regulation of astrocytic energy and mitochondrial metabolism, but that dysregulation of this signaling impairs Aβ uptake and accelerates the pathogenesis of AD. Thus, modifying astrocyte insulin signaling may provide targets for therapeutics in patients with T2D and AD.
Results
Knockout of IR in Astrocytes Impairs Mitochondrial Respiration and Glycolysis.
To begin to determine the role of insulin action in astrocytes, we isolated and purified astrocytes from male adult IR floxed (IRf/f) mice and infected them with an adenovirus encoding a Cre:GFP fusion protein to induce Insr gene deletion (SI Appendix, Fig. S1A). Compared with infection by adenovirus-encoding GFP-only controls, astrocytes expressing Cre exhibited a 75% decrease in the mRNA levels of insulin receptor (Insr), demonstrating efficient Cre-dependent Insr deletion (SI Appendix, Fig. S1B). This was associated with nonsignificant trends to increase the expression of two astrocyte marker genes, Gfap and Apoe, indicating that loss of IR in astrocytes does not alter cell identify or reactivity (SI Appendix, Fig. S1B).
Mitochondrial function in astrocytes has been suggested to play an important role in brain function (22), and mitochondrial dysfunction is evident in neurodegenerative diseases including AD (23, 24). Loss of IR in mature astrocytes significantly reduced the content of mitochondrial DNA (mtDNA)-encoded genes, including mtND1, mtCytB, and mtAPT6 (Fig. 1A), and a 50% reduction in mitochondrial content as assessed by Mitotracker staining (Fig. 1B). This resulted in an ~20% reduction of mitochondrial ATP production and maximal mitochondrial respiratory capacity in IR knockout (IRKO) astrocytes as assessed by Seahorse analysis of oxygen consumption rate (OCR) and extracellular flux acidification rate (ECAR), while basal respiration remained unaltered (Fig. 1 C–E). In addition, IRKO astrocytes had a ~20% reduction in glycolysis rate and 33% reduction in maximal glycolytic capacity (Fig. 1 F–H). Thus, loss of insulin receptor signaling in astrocytes significantly impaired mitochondrial respiration and glycolysis.
Fig. 1.
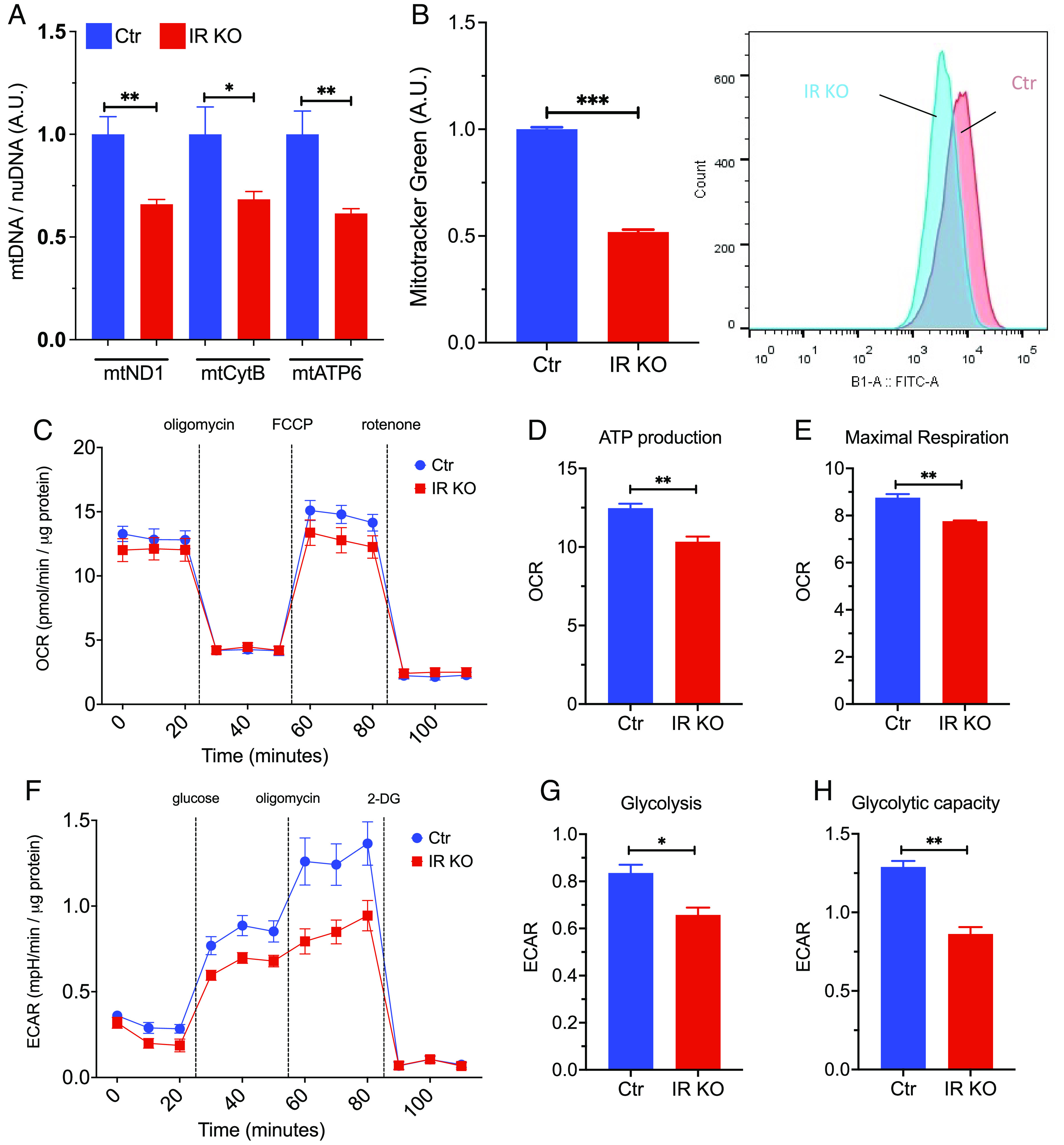
Loss of insulin signaling impairs mitochondrial respiration and glycolytic capacity in astrocytes. (A) qPCR analysis showing copy number of the mitochondrial DNA versus genomic DNA (mtND1, mtCytB, mtATP6) in control and IRKO astrocytes. N = 6. (B) Mean fluorescent intensity (MFI) in control and IRKO astrocytes loaded with mitotracker green and quantified by flow cytometry. N = 5. (C) OCR of control and IRKO astrocytes sequentially treated with oligomycin (2 µM), FCCP (2 µM), and rotenone (2.5 µM) was measured using a Seahorse X24 Bioanalyzer. All data were normalized to the protein content of the corresponding wells (N = 9 to 10). (D and E) Mitochondrial ATP production and maximal respiration of control and IRKO astrocytes were calculated from the Seahorse data (25) (N = 9 to 10). (F) ECAR of control and IRKO astrocytes sequentially treated with glucose (10 mM), oligomycin (2 µM), and 2-deoxyglucose (50 µM) was measured using a Seahorse X24 Bioanalyzer. All the data were normalized to the protein content of the corresponding wells (N = 10). (G and H) Glucose-induced glycolysis and maximal glycolic capacity of control and IRKO astrocytes (N = 10), *P < 0.05, **P < 0.01, ***P < 0.001, in an unpaired t test. Data are presented as mean ± SEM.
Effect of IR Knockout in Astrocytes on Body Weight Gain and Metabolism.
Ablation of insulin receptors in astrocytes has been shown to reduce glucose-induced activation of hypothalamic proopiomelanocortin neurons and impair physiological responses to changes in glucose availability (17). Growing evidence has shown that metabolic dysfunction in astrocytes can also play a role in the development of AD and affect amyloid generation and/or clearance (26). Since Gfap can be expressed in neurons and neuronal precursors early in development (27), for in vivo physiological studies, we crossed the IRf/f mice with mice carrying a tamoxifen-inducible GFAP-CreERT2 (16) to produce inducible astrocyte-specific IRKO or iGIRKO mice. These mice were then crossed with 5xFAD mice, which carry transgenes for five familial AD gene mutations designed to increase β-amyloid deposition (21), to study the interaction between astrocyte insulin action and AD pathogenesis (Fig. 2A). To induce IR deletion in astrocytes, tamoxifen (100 mg/kg) was administered to mice at age 6 wk (Materials and Methods). Tamoxifen was also administered to IRf/f mice crossed with 5xFAD mice but not carrying the GFAP-CreERT2 transgene (hereafter referred as “5xFAD”) as controls. At 6 mo of age, when compared to the controls, both male and female iGIRKO/5xFAD mice showed a mild (12 to 13%), but significant, reduction in body weight (Fig. 2B and SI Appendix, Fig. S2A), with intermediate reductions in body weight in both the iGIRKO and 5xFAD alone. This was accompanied by a trend to lower fasting glucose and serum insulin levels, but neither of these reached statistical significance (Fig. 2 C and D). qPCR analysis of cortex samples from 8-mo-old male mice showed increases in Gfap and Apoe mRNA in the two 5xFAD groups (with and without iGIRKO), while GLT-1 glutamate transporter mRNA remained unchanged (Fig. 2E and SI Appendix, Fig. S2B). In addition, while qPCR analysis of the whole cortex revealed only a trend to decreased levels of IR mRNA, there was a significant ~30% decrease in IR protein in iGIRKO mice, but no significant difference between the iGIRKO/5xFAD mice and 5xFAD mice (Fig. 2F and SI Appendix, Fig. S2 C and D). Insulin-stimulated phosphorylation of IR/IGF1R in the cortex following peripheral insulin injection also tended to be decreased in the iGIRKO mice compared to control, but this did not reach statistical significance in the iGIRKO/5xFAD group in this bulk tissue analysis (SI Appendix, Fig. S2 E and F).
Fig. 2.
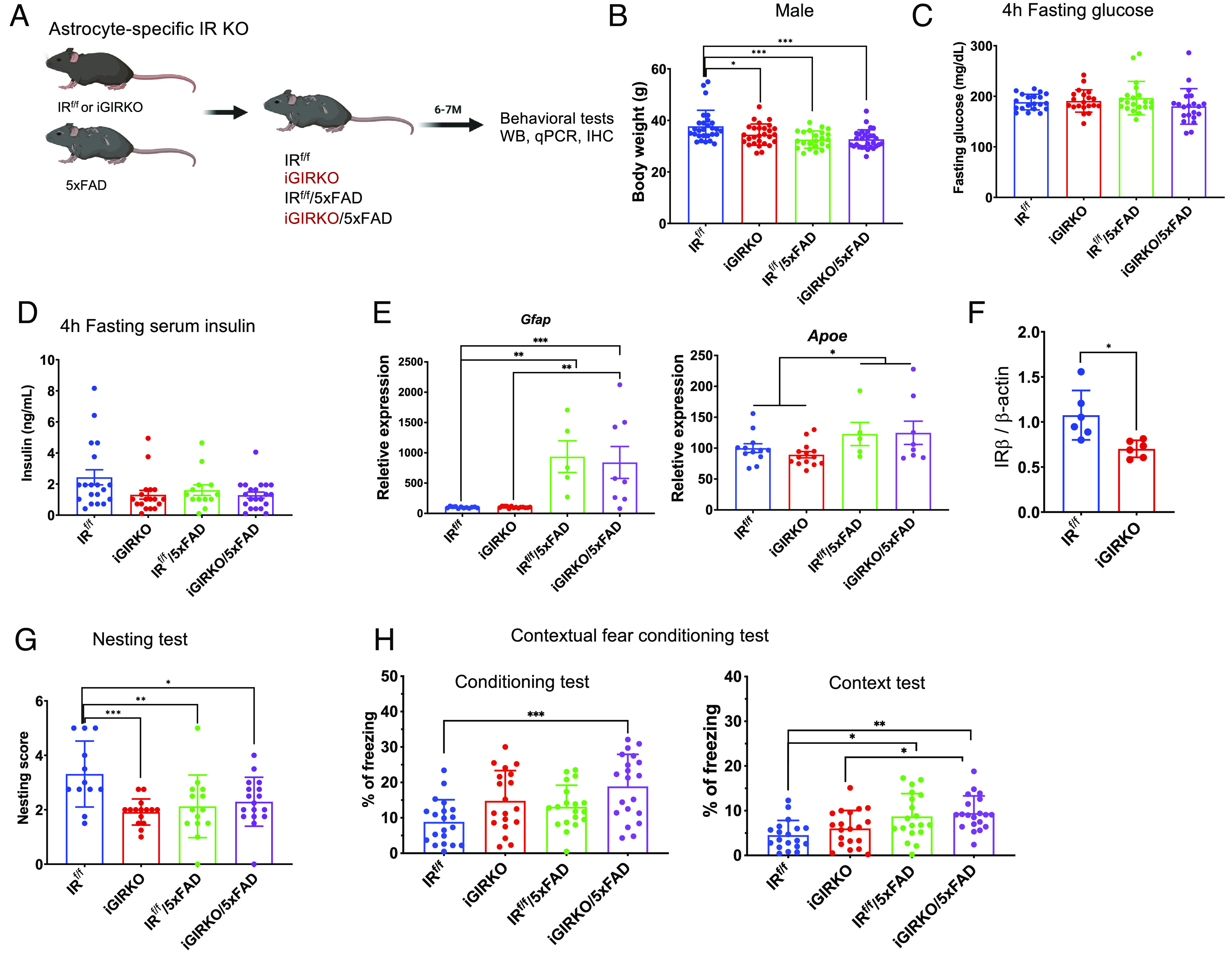
Establishment and characterization of 5xFAD mouse strains with inducible IR KO in astrocytes (iGIRKO/5FAD). (A) Schematic of creation of the iGIRKO/5xFAD mouse. (B) Measurement of body weight (g) at 6-mo-old in male mice. N = 23 to 30 mice per condition. (C and D) 4-h fasting glucose (C) and serum insulin levels (D) in 6-mo-old male mice. N = 20 to 21 mice per condition in C and N = 13 to 21 per condition in D. (E) qPCR analysis of astrocyte marker genes Gfap and Apoe in the cortex of 8-mo-old male mice. N = 5 to 14 mice per condition. (F) Quantification of insulin receptor β-subunit (IRβ) in protein extracted from the cortex of 8-mo-old male mice. N = 4 to 6 per condition. Related to SI Appendix, Fig. S2 C and D. (G) Analysis of scores in nest building test in 6-mo-old male mice (N = 12 to 17 mice per condition). (H) Percent of time with freezing behavior in contextual fear conditioning test in 6-mo-old male mice (N = 19 to 20 mice per condition). *P < 0.05, **P < 0.01, ***P < 0.001, by two-way ANOVA analysis followed by Tukey’s multiple comparisons test. Data are presented as mean ± SEM. Schematic was created with BioRender.
Conditional IRKO in Astrocytes Impairs Behaviors in 5xFAD Mice.
To assess the behavioral effects of astrocyte IRKO and 5xFAD overexpression, we performed a battery of behavioral tests on male mice at the age of 6 mo, an age when behavioral deficits are known to start to develop in 5xFAD mice (21). We found that, compared to controls, both iGIRKO and 5xFAD mice showed decreased nesting building performance, and this persisted in the iGIRKO/5xFAD mice, but without additivity (Fig. 2G). In the Y-maze test using the new-arm exploration paradigm, which assesses the willingness of rodents to explore new environments, there was no alteration in the performance of either iGIRKO alone or 5xFAD alone, but iGIRKO/5xFAD mice carrying both defects showed a significantly lower percentage of new-arm entries than either, suggesting that loss of astrocytic insulin signaling can interact with the 5xFAD lesion to exacerbate memory loss in mice (SI Appendix, Fig. S2G). Likewise, in the contextual fear conditioning test, during both the training/conditioning sessions, iGIRKO/5xFAD mice showed increased freezing response when electronic foot shocks were delivered, whereas mice with either single defect, i.e., only 5xFAD or iGIRKO mice, showed intermediate responses (Fig. 2H), suggesting an additive effect of loss of insulin signaling and 5xFAD lesion on the level of fear response to foot shock. Likewise, when mice were returned to the chambers on the next day but without foot shock (context test), again both iGIRKO and 5xFAD showed a trend toward increased percentage of freezing time compared to the IRf/f control mice, and this increase became significant in the iGIRKO/5xFAD mice, indicating additivity between the 5xFAD effect and loss of astrocytic IR signaling (IRf/f vs iGIRKO/5xFAD, P < 0.001, Fig. 2H). Finally, when the mice were placed in the chambers where the visual and olfactory cues were altered, i.e., altered context phase, both groups carrying the 5xFAD transgenes showed increased freezing time, but this was not altered by knockout of the IR in astrocytes (SI Appendix, Fig. S2H). In summary, these tests show that both loss of astrocytic IR signaling and the presence of the 5xFAD transgenes can lead to altered behavioral responses, and, at least for some behaviors, these defects are additive.
IRKO in Astrocytes Enhances Tau Phosphorylation, Autophagy, and Mitophagy.
To better understand the molecular mechanisms that underlie behavioral impairments in iGIRKO/5xFAD mice, we performed qPCR and western blot analyses on homogenates from the cortex of 6-mo-old mice for markers of neurotransmission. Expression of synaptic marker genes Stx1a (syntaxin 1A) were decreased in both 5xFAD and iGIRKO/5xFAD groups, and Dlg4 tended to be decreased (Fig. 3A), as was the level of postsynaptic density 95 protein (PSD-95), the protein encoded by Dlg4 (SI Appendix, Fig. S3 A and B). mRNA and protein expression of Syp (synaptophysin), as estimated by qPCR and immunostaining, showed somewhat variable results, but was significantly reduced at the mRNA level in the 5xFAD group (Fig. 3A) and at the protein level in the iGIRKO mice (SI Appendix, Fig. S3 C and D), with trends to lower levels in the other groups. Importantly, phosphorylation of Tau protein at Thr231, that contributes to the development of neurofibrillary tangles in AD pathology and is the major site of GKS3β phosphorylation (28), was increased in the cortex homogenates of both iGIRKO and 5xFAD mice and was further increased in the iGIRKO/5xFAD group, with no change in the level of total Tau protein (Fig. 3B). There was also no change in the phosphorylation of Tau at T212/S214 and S396 (SI Appendix, Fig. S3E).
Fig. 3.
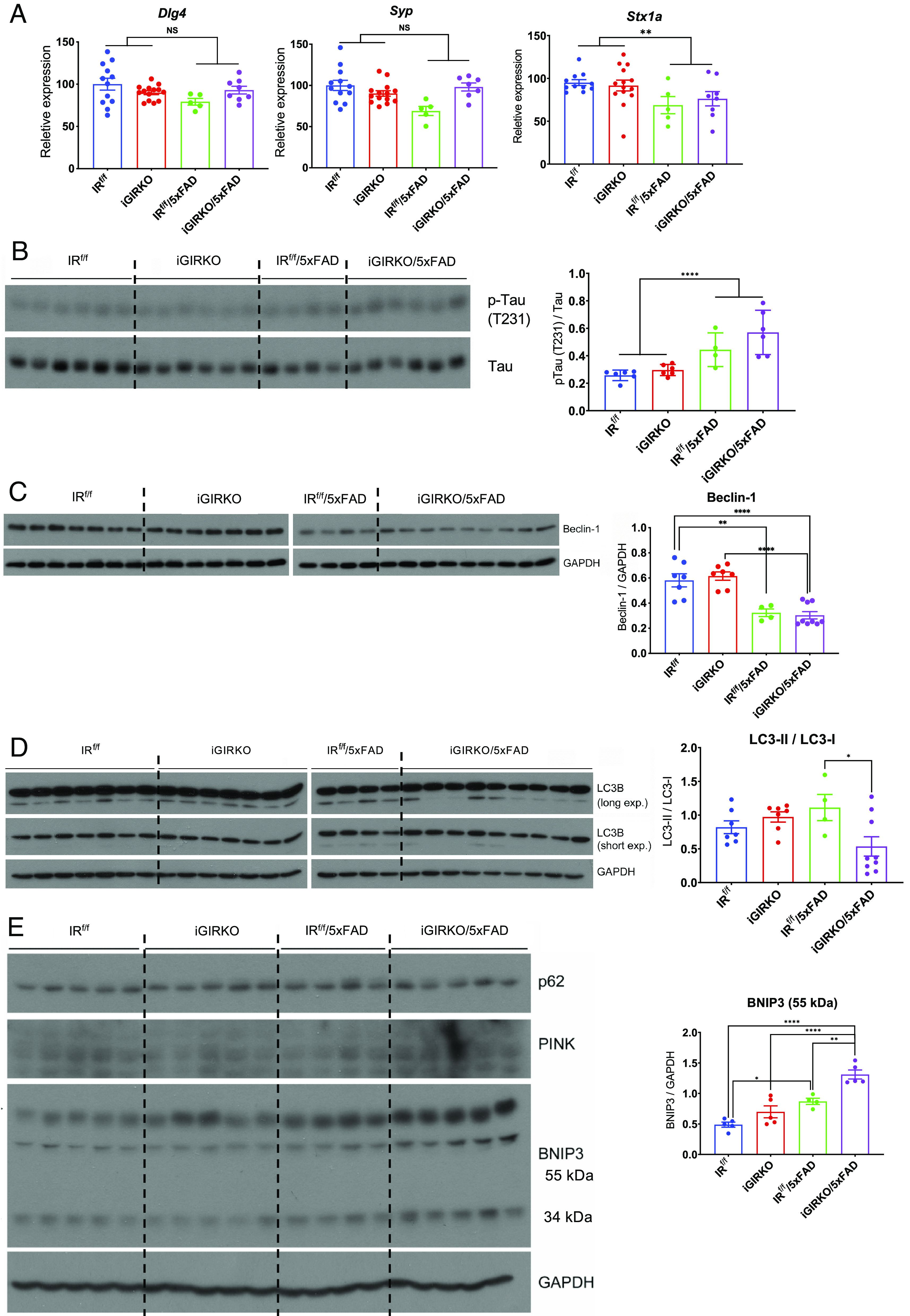
Loss of insulin signaling in astrocytes enhances Tau phosphorylation, mitophagy, and autophagy. (A) qPCR analysis of synaptic marker genes (Dlg4, Syp, Stx1a) in 8-mo-old male mouse cortex. N = 5 to 14 per condition. (B) Representative western blots and quantification of phosphorylated Tau proteins in protein extracted from the cortex of 6-mo-old male mice. N = 4 to 6 per condition. (C) Representative western blots and quantification of Beclin-1 in proteins extracted from the cortex of 6-mo-old male mice. N = 4 to 9 per condition. (D) Representative western blots and quantification of mitophagy pathway-related proteins LC3-I and LC3-II and ratio of LC3-II to LC3-I in protein extracted from the cortex of 6-mo-old male mice. N = 4 to 9 per condition. (E) Representative western blots and quantification of autophagy marker proteins BNIP3 in protein extracted from the cortex of 6-mo-old male mice. N = 4 to 5 per condition. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by two-way ANOVA analysis followed by Tukey’s multiple comparisons test. Quantitative data are presented as mean ± SEM.
Both altered insulin signaling (5, 29) and mitochondrial dysfunction (24, 30) have been suggested to contribute to the pathogenesis of AD. As noted above, there was a significant reduction in mitochondrial content associated with knockout of the IR in astrocytes, raising the possibility that altered insulin signaling may contribute to increased mitophagy and/or autophagy in the 5xFAD mice. Immunoblotting of cortical homogenates from brains of 6-mo-old 5xFAD and iGIRKO/5xFAD mice revealed a significant reduction in Beclin-1 protein (Fig. 3C). Beclin1 has been previously indicated to function in mitophagy by initiating autophagosome formation at damaged mitochondria (31). Levels of the autophagy marker LC3-I (microtubule-associated protein 1 light chain 3) in the cortex homogenates of 6-mo-old mice showed a trend to increase in iGIRKO and 5xFAD mice and were dramatically increased in the iGRIKO/5xFAD group (Fig. 3D and SI Appendix, Fig. S3F), while levels of LC3-II (membrane-bound) showed no significant changes (Fig. 3D and SI Appendix, Fig. S3G), resulting in a reduction in the ratio of LC3-II/LC3-I (Fig. 3D), also suggesting decreased autophagy. On the contrary, PTEN-induced kinase 1 (PINK1) and Parkin, two regulators of mitophagy that are altered in some patients with early-onset familial Parkinson’s disease (32), and Bnip3, a member of the Bcl2 family of proteins which can also participate in mitochondrial dysfunction and mitophagy (33), were significantly increased in iGIRKO/5xFAD mouse cortex compared to both the iGIRKO and 5xFAD mice and the IRf/f control (Fig. 3E and SI Appendix, Fig. S3H). Likewise, p62 (sequestosome1 or SQSTM1), a protein that connects parkin in pink1/parkin mitophagy pathway which has been associated with neurodegenerative diseases (34), was also increased in the two 5xFAD groups compared to the two non-5xFAD groups (SI Appendix, Fig. S3I). Taken together, these findings suggest that loss of insulin signaling in astrocytes results in a decrease in autophagic flux and altered mitophagy, which could contribute to the progression of AD.
Loss of IR Signaling in Astrocytes Leads to Accumulation of Aβ Plaque.
To determine whether loss of IR signaling in astrocytes could lead to altered formation or clearance of Aβ, we performed both conventional immunostaining and SHIELD-based intact tissue immunofluorescent imaging (35) using cleared brain hemispheres. Conventional immunostaining of hippocampus of brains from all the four mouse genotypes revealed markedly increased β-amyloid staining in both 5xFAD mice and iGIRKO/5xFAD mice, with a trend to increase plaque size in the latter (SI Appendix, Fig. S4 A–D). To obtain better resolution within the three-dimensional context of the whole brain, brain hemispheres from 6-mo-old 5xFAD and iGIRKO/5xFAD mice were subjected to tissue clearing, immunolabeled with antibodies against GFAP and β-amyloid, and imaged using a SmartSPIM light sheet microscope. For each brain hemisphere, 1,650 sagittal images were generated (SI Appendix, Fig. S5A and Movies S1 and S2). A SmartAnalytics program (LifeCanvas Inc.) was then used to generate heatmaps of β-amyloid plaque deposit in different brain areas aligned to the Allen Brain Atlas (SI Appendix, Fig. S5B). Focusing on the cerebral cortex, we found that compared with 5xFAD mice, iGIRKO/5xFAD mouse brains showed a significant 12% increase in β-amyloid plaque area, a 17% increase in the average size of plaques, and a 10% increase in the percentage of total plaque area, but no increase in the total number of β-amyloid plaques (Fig. 4A). Analysis of the whole hemispheres revealed similar increases in the average size of the amyloid plaques and percent area of β-amyloid (SI Appendix, Fig. S5C).
Fig. 4.

Loss of astrocyte insulin signaling in mice leads to accumulation of Aβ plaque in the cortex. (A) Representative images and quantification of Aβ plaque in the cerebral cortex of male 5xFAD and iGIRKO/5xFAD mice. N = 160 images from four mice were analyzed. (Scale bar: 1 mm.) (B) Representative images of astrocyte processes surround Aβ plaque in the cerebral cortex and quantification of this colocalization. N = 8 to 23 ROI images from two mice were analyzed per condition. (Scale bar: 1 mm.) (C) Schematic (up) showing the workflow of in situ Aβ uptake analysis in brain slice preparation. Representative images (down) and quantification of percent area of Aβ plaque on brain slices treated with control medium, isolated WT astrocytes, or isolated IR KO astrocytes. *P < 0.05, **P < 0.01, ****P < 0.0001, by unpaired t test. Quantitative data are presented as mean ± SEM. Schematic was created with BioRender.
Using these cleared images, we could also examine the colocalization of astrocytes to amyloid plaques by measuring overlapping of GFAP+ and Aβ+ signals in brain regions that showed substantial Aβ deposition (36). The results showed a significant increase of colocalization of astrocytic processes to amyloid plaques in the cortex in iGIRKO/5xFAD compared to the 5xFAD mice (Fig. 4B). These results suggest that a failure in astrocytic phagocytosis might contribute to the larger size of plaques in the cortex in the iGIRKO/5xFAD mice. To test this hypothesis, we used an ex vivo system consisting of purified astrocytes from male adult IRf/f mice in which we induced IR deletion using adenovirus-encoding Cre-GFP as described above, after which we incubated these astrocytes with 100 µm slices cut from 9-mo-old male 5xFAD mouse brain. As expected, 5xFAD mouse brain slices incubated in astrocyte culture media only as a control contained large amount of Aβ plaques (Fig. 4 C, Left). When similar brain slices were incubated for 16 h with astrocytes from IRf/f control mice, the area of Aβ plaques was reduced by ~65% (Fig. 4 C, Middle). By contrast, when the brain slices were incubated with astrocytes isolated from IRKO mice, the areas of Aβ plaques remained the same as in the brain slices incubated with cell-free astrocyte culture media (Fig. 4 C, Right). Thus, normal astrocytes are able to uptake and/or degrade Aβ protein in the plaques present in the brain slices in vitro, and loss of IR in the astrocytes significantly impairs this process.
Microglia activation has been reported as an early pathological feature following Aβ deposition, and this can trigger inflammatory processes leading to damage and loss of brain function, including cognition (37, 38). Hippocampal staining for lBA1, a marker of microglial activation, revealed a trend to higher levels in both the 5xFAD and slight reductions in both the iGIRKO and iGIRKO/5xFAD mice, but these did not reach statistical significance (SI Appendix, Fig. S6 A and B). On the contrary, immunostaining for C1q protein (a marker of the microglial complement pathway) showed significantly higher levels of staining in the 5xFAD mice and deletion of the insulin receptor in astrocytes (i.e., addition of the iGIRKO lesion) tended to reduce the level of staining in both control and iGIRKO/5xFAD mice (SI Appendix, Fig. S6 C–E), suggesting that loss of astrocytic insulin signaling may blunt the process of microglial activation.
Loss of IR Signaling in Astrocytes Impairs Uptake of Aβ Peptide.
The increased size and accumulation of amyloid plaques in the iGIRKO/5xFAD brain, as well as the in vitro data in which astrocytes have been added to brain slices and amyloid clearance measured, suggest that reducing insulin action in astrocytes by IR knockout might impair astrocyte clearance of β-amyloid. To further explore this process, we isolated astrocytes from brains of neonatal mice carrying floxed IR alleles and infected them with either an adenovirus encoding the Cre recombinase to create IR knockout astrocytes or adenovirus-carrying luciferase as a control (SI Appendix, Fig. S7A). Following 30 min stimulation with either media alone or media supplemented with 100 nM insulin, both control and IRKO astrocytes were incubated with HiLyte Fluor647-conjugated Aβ1-42 protein for 1 h, and Aβ1-42 uptake was assessed by flow cytometry (Fig. 5A). Consistent with the previous findings, knockout of IR in astrocytes reduced basal Aβ1-42 uptake by ~25% when compared with astrocytes with intact insulin signaling (P < 0.0001). Following insulin stimulation, both control and IRKO astrocytes demonstrated an ~35% increase in the uptake of Aβ1-42, but the uptake of Aβ1-42 uptake in the IRKO astrocytes remained reduced when compared with insulin-stimulated control astrocytes. A similar result was seen when the duration of Aβ1-42 uptake was extended to 24 h (SI Appendix, Fig. S7B). Likewise, similar effects of insulin to increase and IR knockout to decrease Fluor647-conjugated Aβ1-42 were observed when astrocytes were derived from young adult (2-mo-old) IRf/f mice and used to create an induced IR deletion (Fig. 5B), as well as when Aβ1-40 uptake was assessed (Fig. 5C). Taken together, these results indicate that insulin can regulate Aβ uptake in astrocytes, and loss of insulin signaling in astrocytes impairs the uptake of Aβ peptides.
Fig. 5.

Effect of Aβ on insulin signaling and insulin signaling on uptake of Aβ peptides by astrocytes. (A) MFI analysis of Aβ1-42 HiLyte Fluor 647 labeled uptake by sorted neonatal astrocytes (IRf/f) following a 1-h incubation. N = 5 to 6 per condition. (B and C) FACS analysis of Aβ1-42 HiLyte Fluor 647 labeled peptide (B) and Aβ1-40 HiLyte Fluor 647 labeled uptake by isolated adult astrocytes following a 2-h incubation (C). (D) Representative western blots showing the effects of Aβ1-42 (2 µM) on insulin (10 nM)-stimulated signaling pathway. (E) Quantification of insulin signaling molecules and their phosphorylation. N = 3 per condition. *P < 0.05, **P < 0.01, ***P < 0.001 by unpaired t test. Data are presented as mean ± SEM.
Given the highly homologous nature of insulin and IGF-1 receptors (IGF1R), the ability of each ligand to bind to the receptor of the other ligand, and the residual, albeit reduced, effect of insulin in the IRKO cells, we investigated to what extent the insulin’s effects on Aβ1-42 uptake may be via action through the IGF1R. To this end, we created primary cultured astrocytes with an IGF1R-KO using a similar approach as for the IRKO astrocytes (Materials and Methods and SI Appendix, Fig. S7A). While loss of IGF1R led to only a modest reduction in Aβ1-42 uptake under basal conditions, there was significantly impaired insulin-induced Aβ1-42 uptake in these cells, indicating that insulin stimulation of an Aβ1-42 uptake by astrocytes is mediated by IGF1R, as well as the insulin receptor (SI Appendix, Fig. S7 C and D).
Aβ Modifies Insulin Signaling in Astrocytes.
While altered insulin signaling can lead to altered Aβ uptake, it is also possible that the amyloid proteins themselves may modify insulin signaling. To test this hypothesis, we incubated primary cultured astrocytes with and without 2 mM Aβ42 peptide overnight and then assessed insulin signaling. We found that in control primary astrocytes in vitro, 10 nM insulin elicited robust stimulation of phosphorylation of IR, IRS-1, Akt, and ERK1/2 (Fig. 5D). Overnight pretreatment of astrocytes with 2 mM Aβ42, on the contrary, significantly blunted insulin-stimulated phosphorylation of both IRS-1 and ERK1/2 (Fig. 5 D and E) but did not affect insulin stimulation of phosphorylation of Akt at S473 (Fig. 5 D and E). Thus, altered insulin signaling in astrocytes may impair Aβ uptake and clearance, while high levels of Aβ may induce insulin resistance, at least at the level of IRS-1 and the MAP kinase cascade.
Discussion
Insulin resistance and T2D have been linked to increased incidence and accelerated AD progression (2, 5, 29). In the present study, we tested the hypothesis that insulin resistance in astrocytes is, at least in part, responsible for this exacerbated AD pathology observed in insulin-resistant states. We show that loss of insulin signaling in astrocytes in vitro leads to a reduction in mitochondrial mass, decreased mitochondrial respiration, and impaired ATP production and glycolytic capacity. More importantly, loss of insulin action in astrocytes by genetic knockout of the insulin receptors significantly impairs the ability of astrocytes to take up and degrade Aβ both in vitro and in vivo. In addition, when mice with an inducible astrocyte IRKO (iGIRKO) are crossed with an AD mouse model (5xFAD), there are increased Tau phosphorylation, altered mitophagy/autophagy, increased size of Aβ plaques in the brain, and exacerbation of some behavioral abnormalities. For some, but not all, behavioral measures, the loss of insulin action in astrocytes also exacerbates the defect in the 5xFAD mouse. Together, these data indicate that astrocytic insulin signaling plays an important role in maintaining normal astrocyte function, regulating the uptake of β-amyloid, and maintaining brain function in the presence of increased β-amyloid load.
Astrocytes are a major site for regulation of brain function, not only providing neurons with energy support, but also regulating and maintaining neuronal function and ultimately modulating behavior. The glycolytic nature of astrocytes in energy metabolism has been suggested in regulating synaptic plasticity and formation (39, 40), as well as learning and memory, whereas its dysregulation has been suggested in neuropathogenesis of AD (41, 42). Here, we show that loss of insulin signaling in astrocytes significantly impairs mitochondrial respiration and glycolysis, and in vivo, both the 5xFAD transgenes and selective astrocytic IRKO resulted in a decrease in body weight, consistent with previous studies of 5xFAD mice (43, 44). Whether this reflects an interaction of cognitive decline and metabolic changes in AD pathogenesis or simply a subtle effect on food intake or energy expenditure will require further study. More importantly from a neurobehavioral point of view, loss of insulin receptors in astrocytes is associated with decreased nesting behavior and, when combined with transgenes which promote an AD-like phenotype, altered behavior in a Y-maze and altered response in fear conditioning tests. Thus, loss of insulin signaling in astrocytes in vivo can contribute to metabolic and behavioral abnormalities and, for some responses, enhance the abnormalities produced by overexpression of genes promoting AD. The manner in which these responses are altered may also vary depending on other factors within the experimental setting. For example, while some studies have observed blunted responses in the fear conditioning test in murine models of AD (45–47), in our experimental setting, both the presence of the five FAD transgenes and the loss of insulin receptors in astrocytes result in enhanced responses to fear conditioning.
Mechanistically, it is known that astrocytes release gliotransmitters, including glutamate, D-serine, and ATP, which can bind to their respective receptors on neurons and modulate neuronal functions. In previous work, we showed that loss of astrocyte insulin receptors reduces exocytosis of ATP from astrocytes, and this in turn reduces purinergic signaling on dopaminergic neurons, resulting in depressive-like behaviors in mice (16). In the present study, when mice with astrocyte IRKO are crossed with the AD mouse model, we observe aggravated AD pathology and behavioral performances, and that this is, at least in part, due to increased Aβ peptide deposition. Astrocytes are also a major site of cholesterol synthesis in the brain (48), and cholesterol is an important component of synaptic vesicles, as well as myelin sheaths (49, 50). We previously demonstrated that the cholesterol synthesis in astrocytes is also regulated by insulin signaling via its action on SREBP2 (19), and astrocyte-specific KO of SREBP2 leads to decreased brain cholesterol synthesis associated with a profound disruption in neuronal functions, brain development, and learning and memory (19). Aβ accumulation in neurons may also be regulated by cholesterol synthesis and ApoE transport from astrocytes (20). Indeed, ApoEε4 is one of the strongest known genetic markers for sporadic AD (51). Increased cholesterol in neurons increases APP association with β- and γ-secretases in lipid clusters, and this association appears to regulate Aβ accumulation and Aβ levels in insoluble plaque (20). Thus, suppressing cholesterol production by genetic deletion of SREBP2 in mouse models of AD has been shown to attenuate the production of Aβ plaques (20). This process appears to be responsive to changes in circulating insulin; thus, mice subjected to a euglycemic–hyperinsulinemic clamp, in which peripheral insulin levels are raised without changing glucose levels, exhibit a marked upregulation of genes involved in cholesterol biosynthesis in the brain (52). To what extent loss of insulin signaling in astrocytes and the associated reduction in astrocyte cholesterol synthesis contributes to Aβ generation and/or clearance in AD-prone animals needs further study.
In addition to the deposition of amyloid plaques and altered cholesterol metabolism, AD brains are characterized by neurofibrillary tangles, an important component of which is hyperphosphorylated Tau protein (28). Multiple different serine/threonine sites of tau phosphorylation have been reported, and how these are modified in humans and murine models of AD varies considerably depending on the model and the site of phosphorylation being studied. For example, previous studies of 5xFAD mice using immunostaining found significantly increased phosphorylation of Ser396, but in increases in Tau phosphorylation at Ser202 or Thr205 (21, 53). In our cohorts of mice, we observed significantly higher levels of tau phosphorylation at Thr231 in both the 5xFAD and iGIRKO/5xFAD mice, with the latter trending to be even higher, but no change in the phosphorylation of Tau at Ser202, Thr205, or Ser396. Whether this relates to differences in the animal colony or in the antibodies used for determination is unclear. Hyperphosphorylation of Tau reduces its ability to promote tubulin assembly and formation of bundles, and Thr231 is a major site of Tau phosphorylation by GSK3β (54). Normally, insulin increases GSK3β via stimulation of the PI3K/Akt pathway; however, this phosphorylation is a negative regulator of GSK3β activity (55). Thus, loss of insulin action on GSK3β will increase its activity and thus increase Tau phosphorylation and its potential for formation of neurofibrillary tangles. In vitro we find that Aβ treatment reduces Erk but not activity upon stimulation. Since these pathways can be activated by other stimuli including cytokines and inflammation, understanding these differential alterations and how they relate to astrocytic insulin signaling might provide a target for therapy of AD.
One of the most striking findings of our study was the observation that insulin stimulation of primary astrocytes in vitro, especially more prolonged stimulation, can increase astrocyte Aβ uptake. Targeting Aβ generation and degradation to delay cognitive impairment in AD patients has been regarded as one important direction in AD drug development. Recent Phase 2 clinical trials using donanemab, a humanized Aβ antibody, have shown some promising results in reducing plaque and Tau protein load; however, cognitive scores were not improved (56), indicating the complexity of AD pathogenesis and the likelihood that inhibiting plaque formation may only affect AD development to a limited extent. In addition, cerebral microhemorrhages have been observed during Aβ-lowering treatment (57), suggesting secondary effects of this treatment on blood–brain barrier (BBB) permeability. Insulin receptors on endothelial cells have also been suggested to play a role in the BBB, but insulin may also gain access to the brain through regions of the vasculature where the barrier is reduced or through other mechanisms, such as tanycytes (58). Astrocytes express both insulin and IGF-1 receptors (59), and for the effects on Aβ uptake, both receptors appear to have potential regulatory roles. Thus, knockout of either the IR or IGF1R in astrocytes partially reduces basal and insulin-stimulated Aβ uptake in astrocytes, but neither alone obliterated this effect. While the specific roles of IGF1R action in astrocytes are unclear, in the iGIRKO mouse, IGF1R remains intact. IGF1R signaling has been implicated in brain development, function, and repair following injury (60, 61), and pharmacologic inhibition of the IGF1R has been shown to alter AD-related pathological features including Aβ levels and gliosis (62). While the unique contributions of IR and IGF1R to signaling and metabolism have been well characterized in adipocytes (59), muscle (63), and other cell types (64), determining the unique contributions of these two complementary receptors in astrocytes may help identify postreceptor sites for therapeutic intervention in patients with diabetes and AD. The need to engage both receptors may also explain why clinical investigations using intranasal insulin for treatment of AD have produced only modest effects in cognitive improvement (reviewed in ref. 5).
As shown in this study and our previous work (13–16), insulin resistance in the brain and in astrocytes specifically leads to decreased mitochondrial content and/or oxidative function, which may also contribute to the impairment in Aβ uptake and neuronal dysfunction. The brain has high energy demands for maintaining normal brain activities, including myelination, synaptic remodeling, and memory formation, all of these processes depend to some extent on mitochondrial energy production and cellular metabolism (5, 19). We and others have shown that insulin signaling plays a critical role in the regulation of mitochondria biogenesis and physiology in the brain (65–67). Brain-wide knockout of insulin receptor in mice (NIRKO) leads to mitochondrial dysfunction characterized by a decrease in mitochondrial mass and size and increased levels of reactive oxygen species (ROS) in the brain (14). It has been demonstrated that insulin action prevents accumulation of ROS in the hippocampus, a brain area involved in high-order cognition (68). ROS can also contribute to the insulin resistance (69, 70), creating a vicious cycle contributing to the development of AD. Whether the loss of insulin signaling and altered mitochondrial function in astrocytes leads to an increase of ROS production in the brain remains to be determined, but it has been shown that administration of intranasal insulin treatment to normal mice can increase mitochondrial ATP production in the brain (71). Rosiglitazone, a PPARγ agonist that improves insulin resistance, can also reduce brain mitochondrial ROS production (68), suggesting that targeting insulin action on brain mitochondria could be a potential therapeutic approach to treat dementia or AD.
The observation that loss of astrocytic insulin signaling increases mitophagy while decreasing autophagic flux further provides another factor in AD pathogenesis. Mitophagy is a process that selectively removes damaged mitochondria, providing a consistent quality control on these important organelles. We find a lower ratio of LC3-II to LC3-I in 5xFAD brains with loss of astrocytic insulin signaling, suggesting impaired macroautophagy (72), and loss of macroautophagy has been shown to accelerate disease progression in an AD mouse model (73). We also found significant increases in the levels of PINK1 and BINP3 mRNA in brains of iGIRKO/5xFAD mice. PINK1 regulates PARKIN translocation in mitochondria and drives their removal via the process known as mitophagy (74). BNIP3 is a proapoptotic protein that may also increase mitophagy (33). Taken together, these alterations in autophagy and mitophagy, along with the impairment in Aβ uptake and altered cholesterol synthesis, might create synergistic deleterious effects on AD pathology.
In summary, insulin signaling in astrocytes plays an important role in the regulation of mitochondrial function and Aβ uptake by astrocytes and dysfunction of this signaling system can result in an impairment in Aβ clearance and exacerbate AD-like phenotypes in a 5xFAD mouse model. Understanding the mechanisms of this interaction may provide insights into potential sites for therapeutic development in T2D patients with cognitive loss and AD patients with metabolic syndrome.
Materials and Methods
Animals.
All experimental procedures and animal welfare care were approved by the Institutional Animal Care and Use Committee of the Joslin Diabetes Center and/or Brandeis University. Both animal facilities provide veterinary services on a 24-h basis. Mice were housed in a temperature- and humidity-controlled animal facility (23 °C and 80% humidity) with a 12 h/12 h light/dark photoperiod (lights on at 6:30 AM). The animals were provided with standard chow diet (Mouse diet 9F 5020; PharmaServ) and water ad libitum unless otherwise specified. IR floxed mice (IRf/f) were created as previously described (13) and maintained in house. GFAPCreERT2 mice (Stock # 012849) and 5xFAD mice (Stock # 008730) were purchased from Jackson Laboratory (Bar Harbor, Maine). For generation of experimental animals and tamoxifen induction in these animals, detailed Materials and Methods are available in SI Appendix.
Brain Homogenate Preparation.
Freshly isolated brain tissue was kept on ice and cut in a sagittal manner. The right hemisphere was immediately frozen in liquid nitrogen and stored at −80 °C, while the left hemisphere was stored in precold 4% PFA for immunohistochemistry (IHC) analysis. For brain lysate preparation and subsequent analyses including western blotting, RT-qPCR, and IHC staining, detailed Materials and Methods are available in SI Appendix.
Mouse Brain Clearing, Staining, and Imaging.
Whole-brain volumetric immunofluorescent imaging was performed using a previously reported SHIELD protocol (35). Postfixed brains were processed and imaged, and 3D image datasets from light sheet microscope were analyzed (LifeCanvas Technologies, MA, USA). Detailed methods are available in SI Appendix.
Mouse Behavioral Assays.
Nesting tests, Y maze, and contextual fear conditioning tests were conducted to assess behavioral performances in mice. Detailed methods are available in SI Appendix.
Statistics.
All the data were presented as mean ± SEM. Two-group comparison was performed using unpaired two-tailed Student’s t test. For experiments involving analysis of the multiple groups with two factors (i.e., IR KO or 5xFAD), a two-way ANOVA analysis was performed to detect the interactions between the two main factors, followed by a Tukey’s post hoc multiple comparison test when appropriate. A P-value < 0.05 was considered significant.
Supplementary Material
Appendix 01 (PDF)
2D visualization of sagittal images (related to Figure 4) Movie showing accelerated playing of sagittal images across a brain hemisphere. Movie was created using Image J.
3D visualization of brain hemispheres (related to Figure 4). 3D movie showing a brain hemisphere cleared by LifeCanvas’ SHIELD technology, with red fluorescent signals represent β-amyloid staining. Movie was created using Imaris.
Acknowledgments
This work was supported by NIH grants R01 DK031036 (to C.R.K.) and R01 MH125903 (to W. Cai.). The Joslin Diabetes Center DRC Advanced Microscopy Core, Flow Cytometry Core, and Animal Physiology Core (P30 DK036836) provided important help. We thank LifeCanvas Inc. for providing important help on whole-brain clearing, imaging, and analysis. We also thank the support of the Harvard Medical School Neurobiology Imaging Facility (supported by National Institute of Neurological Disorders and Stroke (NINDS) Core Center Grant P30NS072030). We thank all the Kahn lab members for their helpful discussion.
Author contributions
W. Chen and C.R.K. designed research; W. Chen, Q.H., E.K.L., A.G., M.W., T.G.R., D.F., and W. Cai performed research; W. Chen, Q.H., E.K.L., A.G., E.S., T.G.R., D.F., and W. Cai analyzed data; and W. Chen and C.R.K. wrote the paper.
Competing interests
The lead author, C.R.K., and one reviewer, D.A., are co-authors of a recent publication (DOI: 10.1038/s41467-022-33008-2) in which they each provided different reagents to a third group whose publication this represents. In this co-authored publication, there was no collaboration between C.R.K. and D.A. themselves.
Footnotes
Reviewers: D.A., Columbia University; and S.C., Wake Forest University.
Contributor Information
Dan Frenkel, Email: dfrenkel@tauex.tau.ac.il.
Weikang Cai, Email: wcai04@nyit.edu.
C. Ronald Kahn, Email: c.ronald.kahn@joslin.harvard.edu.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Sun H., et al. , IDF diabetes atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 183, 109119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barbagallo M., Dominguez L. J., Type 2 diabetes mellitus and Alzheimer’s disease. World J. Diabetes 5, 889–893 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kahn C. R., Suzuki R., “Insulin action in the brain and the pathogenesis of alzheimer’s disease” in Diabetes, Insulin and Alzheimer’s Disease, Craft S., Christen Y., Eds. (Springer, Berlin Heidelberg, Berlin, Heidelberg, 2010), pp. 1–20, 10.1007/978-3-642-04300-0_1. [DOI] [Google Scholar]
- 4.Kleinridders A., Ferris H. A., Cai W., Kahn C. R., Insulin action in brain regulates systemic metabolism and brain function. Diabetes 63, 2232–2243 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen W., Cai W., Hoover B., Kahn C. R., Insulin action in the brain: Cell types, circuits, and diseases. Trends Neurosci. 45, 384–400 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bedse G., Di Domenico F., Serviddio G., Cassano T., Aberrant insulin signaling in Alzheimer’s disease: Current knowledge. Front. Neurosci. 9, 204 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frolich L., et al. , Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J. Neural. Transm. (Vienna) 105, 423–438 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Talbot K., et al. , Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 122, 1316–1338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bomfim T. R., et al. , An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J. Clin. Invest. 122, 1339–1353 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mullins R. J., Mustapic M., Goetzl E. J., Kapogiannis D., Exosomal biomarkers of brain insulin resistance associated with regional atrophy in Alzheimer’s disease. Hum. Brain Mapp. 38, 1933–1940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ochiai T., et al. , Differential involvement of insulin receptor substrate (IRS)-1 and IRS-2 in brain insulin signaling is associated with the effects on amyloid pathology in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 159, 105510 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Nakamura N., et al. , Apomorphine Therapy for neuronal insulin resistance in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 58, 1151–1161 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Bruning J. C., et al. , Role of brain insulin receptor in control of body weight and reproduction. Science 289, 2122–2125 (2000). [DOI] [PubMed] [Google Scholar]
- 14.Kleinridders A., et al. , Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. U.S.A. 112, 3463–3468 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biessels G. J., Deary I. J., Ryan C. M., Cognition and diabetes: A lifespan perspective. Lancet Neurol. 7, 184–190 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Cai W., et al. , Insulin regulates astrocyte gliotransmission and modulates behavior. J. Clin. Invest. 128, 2914–2926 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garcia-Caceres C., et al. , Astrocytic insulin signaling couples brain glucose uptake with nutrient availability. Cell 166, 867–880 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu C. C., Liu C. C., Kanekiyo T., Xu H., Bu G., Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferris H. A., et al. , Loss of astrocyte cholesterol synthesis disrupts neuronal function and alters whole-body metabolism. Proc. Natl. Acad. Sci. U.S.A. 114, 1189–1194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H., et al. , Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol. Proc. Natl. Acad. Sci. U.S.A. 118, e2102191118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oakley H., et al. , Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gollihue J. L., Norris C. M., Astrocyte mitochondria: Central players and potential therapeutic targets for neurodegenerative diseases and injury. Ageing Res. Rev. 59, 101039 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bell S. M., et al. , Mitochondrial dysfunction in Alzheimer’s disease: A biomarker of the future? Biomedicines 9, 63 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang W., Zhao F., Ma X., Perry G., Zhu X., Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener 15, 30 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kleinridders A., et al. , Leptin regulation of Hsp60 impacts hypothalamic insulin signaling. J. Clin. Invest. 123, 4667–4680 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zulfiqar S., Garg P., Nieweg K., Contribution of astrocytes to metabolic dysfunction in the Alzheimer’s disease brain. Biol. Chem. 400, 1113–1127 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Casper K. B., McCarthy K. D., GFAP-positive progenitor cells produce neurons and oligodendrocytes throughout the CNS. Mol. Cell Neurosci. 31, 676–684 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Lee V. M., Balin B. J., Otvos L. Jr., Trojanowski J. Q., A68: A major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251, 675–678 (1991). [DOI] [PubMed] [Google Scholar]
- 29.De Felice F. G., Goncalves R. A., Ferreira S. T., Impaired insulin signalling and allostatic load in Alzheimer disease. Nat. Rev. Neurosci. 23, 215–230 (2022). [DOI] [PubMed] [Google Scholar]
- 30.Wang X., et al. , Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 1842, 1240–1247 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quiles J. M., et al. , Deciphering functional roles and interplay between Beclin1 and Beclin2 in autophagosome formation and mitophagy. Sci. Signal 16, eabo4457 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amadoro G., et al. , Morphological and bioenergetic demands underlying the mitophagy in post-mitotic neurons: The pink-parkin pathway. Front. Aging Neurosci. 6, 18 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao A., Jiang J., Xie F., Chen L., Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta 506, 72–83 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Liu H., et al. , From autophagy to mitophagy: The roles of P62 in neurodegenerative diseases. J. Bioenerg. Biomembr. 49, 413–422 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Park Y. G., et al. , Protection of tissue physicochemical properties using polyfunctional crosslinkers. Nat. Biotechnol., 10.1038/nbt.4281 (2018). [DOI] [PMC free article] [PubMed]
- 36.Gail Canter R., et al. , 3D mapping reveals network-specific amyloid progression and subcortical susceptibility in mice. Commun. Biol. 2, 360 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forner S., et al. , Systematic phenotyping and characterization of the 5xFAD mouse model of Alzheimer’s disease. Sci. Data 8, 270 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jawhar S., Trawicka A., Jenneckens C., Bayer T. A., Wirths O., Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal abeta aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 33, e29-40 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Vezzoli E., et al. , Ultrastructural evidence for a role of astrocytes and glycogen-derived lactate in learning-dependent synaptic stabilization. Cereb. Cortex 30, 2114–2127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zehnder T., et al. , Mitochondrial biogenesis in developing astrocytes regulates astrocyte maturation and synapse formation. Cell Rep. 35, 108952 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Mulica P., Grunewald A., Pereira S. L., Astrocyte-neuron metabolic crosstalk in neurodegeneration: A mitochondrial perspective. Front. Endocrinol. (Lausanne) 12, 668517 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Newington J. T., Harris R. A., Cumming R. C., Reevaluating metabolism in Alzheimer’s disease from the perspective of the astrocyte-neuron lactate shuttle model. J. Neurodegener. Dis. 2013, 234572 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kameno K., et al. , Loss of body weight in old 5xFAD mice and the alteration of gut microbiota composition. Exp. Gerontol. 166, 111885 (2022). [DOI] [PubMed] [Google Scholar]
- 44.Gendron W. H., et al. , Age related weight loss in female 5xFAD mice from 3 to 12 months of age. Behav. Brain Res. 406, 113214 (2021). [DOI] [PubMed] [Google Scholar]
- 45.Kimura R., Ohno M., Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol. Dis. 33, 229–235 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Devi L., Ohno M., Genetic reductions of beta-site amyloid precursor protein-cleaving enzyme 1 and amyloid-beta ameliorate impairment of conditioned taste aversion memory in 5XFAD Alzheimer’s disease model mice. Eur. J. Neurosci. 31, 110–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohno M., Failures to reconsolidate memory in a mouse model of Alzheimer’s disease. Neurobiol. Learn. Mem. 92, 455–459 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suzuki R., et al. , Diabetes and insulin in regulation of brain cholesterol metabolism. Cell Metab. 12, 567–579 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takamori S., et al. , Molecular anatomy of a trafficking organelle. Cell 127, 831–846 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Rosa P., Fratangeli A., Cholesterol and synaptic vesicle exocytosis. Commun. Integr. Biol. 3, 352–353 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saunders A. M., et al. , Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43, 1467–1472 (1993). [DOI] [PubMed] [Google Scholar]
- 52.Cai W., et al. , Peripheral insulin regulates a broad network of gene expression in hypothalamus, hippocampus, and nucleus accumbens. Diabetes 70, 1857–1873 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kanno T., Tsuchiya A., Nishizaki T., Hyperphosphorylation of Tau at Ser396 occurs in the much earlier stage than appearance of learning and memory disorders in 5XFAD mice. Behav. Brain Res. 274, 302–306 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Lin Y. T., et al. , The binding and phosphorylation of Thr231 is critical for Tau’s hyperphosphorylation and functional regulation by glycogen synthase kinase 3beta. J. Neurochem. 103, 802–813 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Yang L., Wang H., Liu L., Xie A., The Role of Insulin/IGF-1/PI3K/Akt/GSK3beta signaling in parkinson’s disease dementia. Front. Neurosci. 12, 73 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mintun M. A., et al. , Donanemab in early Alzheimer’s disease. N. Engl. J. Med. 384, 1691–1704 (2021). [DOI] [PubMed] [Google Scholar]
- 57.Sperling R. A., et al. , Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s association research roundtable workgroup. Alzheimers Dement 7, 367–385 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Porniece Kumar M., et al. , Insulin signalling in tanycytes gates hypothalamic insulin uptake and regulation of AgRP neuron activity. Nat. Metab. 3, 1662–1679 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cai W., et al. , Domain-dependent effects of insulin and IGF-1 receptors on signalling and gene expression. Nat. Commun. 8, 14892 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Joseph D’Ercole A., Ye P., Expanding the mind: Insulin-like growth factor I and brain development. Endocrinology 149, 5958–5962 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cardoso S., Lopez I. P., Pineiro-Hermida S., Pichel J. G., Moreira P. I., IGF1R deficiency modulates brain signaling pathways and disturbs mitochondria and redox homeostasis. Biomedicines 9, 158 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sohrabi M., Floden A. M., Manocha G. D., Klug M. G., Combs C. K., IGF-1R inhibitor ameliorates neuroinflammation in an Alzheimer’s disease transgenic mouse model. Front. Cell Neurosci. 14, 200 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Neill B. T., et al. , Differential role of insulin/IGF-1 receptor signaling in muscle growth and glucose homeostasis. Cell Rep. 11, 1220–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nagao H., et al. , Distinct signaling by insulin and IGF-1 receptors and their extra- and intracellular domains. Proc. Natl. Acad. Sci. U.S.A. 118, e2019474118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cheng Z., Tseng Y., White M. F., Insulin signaling meets mitochondria in metabolism. Trends Endocrinol. Metab. 21, 589–598 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wardelmann K., et al. , Insulin action in the brain regulates mitochondrial stress responses and reduces diet-induced weight gain. Mol. Metab. 21, 68–81 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Montgomery M. K., Turner N., Mitochondrial dysfunction and insulin resistance: An update. Endocr. Connect. 4, R1–R15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pipatpiboon N., Pratchayasakul W., Chattipakorn N., Chattipakorn S. C., PPARgamma agonist improves neuronal insulin receptor function in hippocampus and brain mitochondria function in rats with insulin resistance induced by long term high-fat diets. Endocrinology 153, 329–338 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Houstis N., Rosen E. D., Lander E. S., Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440, 944–948 (2006). [DOI] [PubMed] [Google Scholar]
- 70.Furukawa S., et al. , Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 114, 1752–1761 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ruegsegger G. N., et al. , Insulin deficiency and intranasal insulin alter brain mitochondrial function: A potential factor for dementia in diabetes. FASEB J. 33, 4458–4472 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kadowaki M., Karim M. R., Cytosolic LC3 ratio as a quantitative index of macroautophagy. Methods Enzymol. 452, 199–213 (2009). [DOI] [PubMed] [Google Scholar]
- 73.Bourdenx M., et al. , Chaperone-mediated autophagy prevents collapse of the neuronal metastable proteome. Cell 184, 2696–2714.e2625 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Quinn P. M. J., Moreira P. I., Ambrosio A. F., Alves C. H., PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 8, 189 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
2D visualization of sagittal images (related to Figure 4) Movie showing accelerated playing of sagittal images across a brain hemisphere. Movie was created using Image J.
3D visualization of brain hemispheres (related to Figure 4). 3D movie showing a brain hemisphere cleared by LifeCanvas’ SHIELD technology, with red fluorescent signals represent β-amyloid staining. Movie was created using Imaris.
Data Availability Statement
All study data are included in the article and/or supporting information.