

Summary
Although chronic kidney disease (CKD) is a major health problem worldwide, its underlining mechanism is incompletely understood. We previously identified adipolin as an adipokine which provides benefits for cardiometabolic diseases. Here, we investigated the role of adipolin in the development of CKD. Adipolin-deficiency exacerbated urinary albumin excretion, tubulointerstitial fibrosis and oxidative stress of remnant kidneys in mice after subtotal nephrectomy through inflammasome activation. Adipolin positively regulated the production of ketone body, β-hydroxybutyrate (BHB) and expression of a catalytic enzyme producing BHB, HMGCS2 in the remnant kidney. Treatment of proximal tubular cells with adipolin attenuated inflammasome activation through the PPARα/HMGCS2-dependent pathway. Furthermore, systemic administration of adipolin to wild-type mice with subtotal nephrectomy ameliorated renal injury, and these protective effects of adipolin were diminished in PPARα-deficient mice. Thus, adipolin protects against renal injury by reducing renal inflammasome activation through its ability to induce HMGCS2-dependent ketone body production via PPARα activation.
Subject areas: Molecular biology, Immunology, Cell biology
Graphical abstract
Highlights
-
•
Adipolin deficiency enhances renal injury through activation of renal inflammasome
-
•
Adipolin prevents renal damage through PPARα-dependent inflammasome inhibition
-
•
Adipolin reduces inflammasome activity in tubular cells via PPARα/HMGCS2 pathway
Molecular biology; Immunology; Cell biology
Introduction
Chronic kidney disease (CKD) has become a major public health issue in recent years, with the number of cases worldwide increasing to 700 million in 2017.1,2 CKD contributes to an increased risk of cardiovascular mortalities. Obesity is an important risk factor for CKD development and participates in the pathogenesis of CKD, at least in part by increasing the risk of type 2 diabetes, atherosclerosis, and hypertension.3 Furthermore, obesity appears to promote CKD progression through its direct effects on the kidneys.3,4 However, the precise mechanism by which obesity causes CKD development has not yet been fully elucidated.
Adipose-derived hormonal factors, also known as adipokines, participate in the pathogenesis of various obesity disorders.5,6,7 Tumor necrosis factor (TNF) α and interleukin (IL) 6 are pro-inflammatory adipokines, and their circulating levels are positively correlated with CKD prevalence.8 Leptin is upregulated by obesity and promotes glomerulosclerosis and renal fibrosis with pro-inflammatory properties.9,10 In contrast, adiponectin is an anti-inflammatory adipokine with reno-protective functions.11,12 Therefore, obesity may cause an imbalance of pro- and anti-inflammatory adipokines, leading to the development of CKD.
We previously identified adipolin, also referred to as C1q/Tnf-related protein 12, as an insulin-sensitizing adipokine downregulated by obesity.13 Adipolin improves insulin resistance in obese mice by attenuating adipose tissue inflammation. We also found that adipolin improves the pathological remodeling of vascular walls by reducing smooth muscle cell proliferation and macrophage inflammatory responses.14 Recently, we found that adipolin reduces adverse myocardial remodeling in response to myocardial infarction by suppressing inflammation.15 Thus, it is conceivable that adipolin acts as an anti-inflammatory adipokine that exerts beneficial actions on obesity-related metabolic and cardiovascular diseases. However, little is known about the role of adipolin in renal diseases. Here, we investigated whether adipolin could modulate CKD development using a mouse subtotal nephrectomy model.
Results
Disrupt of adipolin exacerbates renal damage following subtotal nephrectomy
To examine the role of adipolin in renal injury, adipolin-knockout (APL-KO) and wild-type (WT) mice were subjected to 5/6 nephrectomy. Eight weeks after subtotal nephrectomy or sham operation, APL-KO and WT mice were sacrificed for analyses after the collection of urine and blood samples. No differences were observed in body weight, systolic blood pressure, or heart rate between WT and APL-KO mice after subtotal nephrectomy or sham operation (Table 1). Subtotal renal ablation significantly increased urinary albumin excretion and circulating levels of urea nitrogen (UN) and creatinine (Cr) in WT mice (Figure 1A). Urinary albumin excretion and plasma UN levels were further increased in APL-KO mice after subtotal nephrectomy compared to those in WT mice (Figure 1A). In contrast, plasma Cr levels after the renal injury did not significantly differ between APL-KO and WT mice.
Table 1.
Characteristics of WT and APL-KO mice at 8 weeks after subtotal nephrectomy
| Sham |
Nephrectomy |
|||
|---|---|---|---|---|
| WT | APL-KO | WT | APL-KO | |
| Body weight (g) | 26.0 ± 0.4 | 25.3 ± 0.6 | 24.1 ± 0.4 | 24.2 ± 0.3 |
| SBP (mmHg) | 100.5 ± 3.4 | 101.4 ± 2.4 | 103.0 ± 2.1 | 101.1 ± 4.3 |
| Heart rate (bpm) | 624.7 ± 17.6 | 619.5 ± 5.4 | 627.8 ± 22.1 | 629.0 ± 15.9 |
Data are presented as mean ± S.E.M.
WT; wild-type mice, APL-KO; adipolin-knockout mice, SBP; systolic blood pressure, bpm: beat per minute.
Figure 1.

Adipolin-deficiency leads to exacerbation of albuminuria and tubulointerstitial fibrosis after renal ablation
(A) Urine and plasma parameters of renal function in wild-type (WT) and adipolin knockout (APL-KO) mice after subtotal nephrectomy or sham operation. Left panel shows urinary albumin excretion normalized to urinary Cr. Center and right panels show plasma concentration of UN and Cr. Sham WT group; N = 6, sham APL-KO group; N = 6, nephrectomy WT group; N = 7, nephrectomy APL-KO group; N = 7.
(B) Histological evaluation of interstitial fibrosis. Left panels show representative photos of the kidneys from WT and APL-KO mice after subtotal nephrectomy or sham operation as determined by Masson Trichrome staining. Right panel shows quantitative analysis of fibrosis area as measured by ImageJ. N = 6 in each group. Scale bars show 100 μm.
(C) mRNA levels of tumor necrosis factor (TNF) α, interleukin (IL) 6, monocyte chemoattractant protein (MCP) 1 and F4/80 in the kidney in WT and APL-KO mice after subtotal nephrectomy or sham operation. N = 6 in each group.
(D) mRNA levels of inflammasome-related genes (NLRP3, Caspase 1, IL1β and IL18) in the kidney in WT and APL-KO mice after subtotal nephrectomy or sham operation. N = 6 in each group.
(E) mRNA levels of oxidative stress markers (gp91phox, p47phox, p67phox and p22phox) in the kidney in WT and APL-KO mice after subtotal nephrectomy or sham operation. N = 6 in each group.One-way ANOVA with Tukey’s multiple comparisons test ((A) (urine albumin and plasma UN), (C) (TNFα, IL6 and F4/80), (D) (NLRP3, Caspase 1 and IL18) and Steel-Dwass test ((A) (plasma Cr), (B, C) (MCP1), (D) (IL1β) (E) (p67phox)) were used to produce the p values. N.S, not significant.
To evaluate interstitial fibrosis after renal injury, the kidneys of WT and APL-KO mice after subtotal nephrectomy or sham operation were stained with Masson’s trichrome. APL-KO mice exhibited an increased fibrotic area in injured kidneys after subtotal renal ablation compared to WT mice (Figure 1B). APL-KO mice also showed increased expression levels of fibrosis markers, including collagen I, collagen III, and transforming growth factor (TGF) β1, in the remnant kidney after partial nephrectomy compared to WT mice (Figure S1A).
To assess glomerular hypertrophy, the kidneys of WT and APL-KO mice after subtotal nephrectomy or sham operation were stained with Hematoxylin and Eosin (H-E). APL-KO mice exhibited an increased glomerular cross-sectional area after subtotal nephrectomy compared with WT mice (Figure S1B).
To evaluate renal tubular cell apoptosis, the kidneys of WT and APL-KO mice after subtotal nephrectomy or sham operation were stained with a terminal deoxynucleotidyl transferase-mediated dUTP-nick end labeling (TUNEL). APL-KO mice exhibited increased numbers of TUNEL-positive apoptotic cells in the remnant kidneys after subtotal nephrectomy compared with WT mice (Figure S1C). In contrast, no significant differences were observed in renal fibrosis area, glomerular cross-sectional area and renal apoptosis after sham operation between WT and APL-KO mice.
Inflammation, inflammasome activation, and oxidative stress contribute to CKD progression.16,17,18,19 Thus, to investigate the participation of inflammation, inflammasome activation, and oxidative stress in exacerbated renal injury in APL-KO mice, mRNA levels of inflammatory mediators, inflammasome-associated factors, and components of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in the kidneys of WT and APL-KO mice were measured using quantitative real-time PCR. APL-KO mice exhibited higher mRNA levels of pro-inflammatory mediators, including TNFα, IL6, monocyte chemoattractant protein (MCP) 1 and F4/80 in the injured kidneys compared to WT mice (Figure 1C). In contrast, no differences were observed in mRNA levels of toll-like receptor (TLR) 2 and TLR4 in the injured kidneys between WT and APL-KO mice (Figure S1D). APL-KO mice also exhibited increased mRNA levels of inflammasome-related genes, including the nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing (NLRP) 3, caspase 1, IL1β, and IL18 in the remnant kidneys after subtotal nephrectomy compared with WT mice (Figure 1D). Furthermore, APL-KO mice showed increased protein levels of NLRP3, caspase 1, and IL1β in the remnant kidneys after subtotal nephrectomy compared to WT mice (Figure S2). APL-KO mice also showed higher expression levels of gp91phox, p47phox, p67phox, and p22phox, which are components of NADPH oxidase, in the remnant kidneys after subtotal nephrectomy than WT mice (Figure 1E). In contrast, there were no significant differences in the expression levels of pro-inflammatory inflammasome-associated genes and components of NADPH oxidase in the kidneys after sham operation between WT and APL-KO mice (Figure 1E). Therefore, these data indicate that adipolin deficiency enhances renal damage following subtotal nephrectomy, with accompanying increases in inflammation, inflammasome activation, and oxidative stress in the kidney.
Systemic administration of adipolin ameliorates renal injury after subtotal nephrectomy
To assess the therapeutic effects of adipolin on renal injury, an adenoviral vector expressing adipolin (Ad-APL) or Ad-β-gal as control was intravenously injected into WT mice 4 weeks after subtotal nephrectomy. Ad-APL-treated WT mice showed a 2.2 ± 0.3-fold increase in plasma adipolin levels 7 days after Ad-APL injection. Ad-APL administration significantly reduced urinary albumin excretion and plasma UN levels in WT mice 4 weeks after Ad-APL injection (i.e., 8 weeks after renal ablation) (Figure 2A). In contrast, Ad-APL treatment did not affect the plasma Cr levels in WT mice after subtotal nephrectomy. Ad-APL treatment reduced the fibrotic area in the remnant kidney of WT mice after subtotal nephrectomy compared with Ad-β-gal treatment (Figure 2B). Similarly, Ad-APL treatment significantly reduced the mRNA levels of fibrosis-related genes, including collagen I, collagen III, and TGFβ1 in the remnant kidneys of WT mice (Figure S3A). Ad-APL treatment also significantly reduced glomerular cross-sectional area in the injured kidneys in WT mice compared with Ad-β-gal treatment (Figure S3B). Moreover, Ad-APL treatment significantly reduced the numbers of TUNEL-positive apoptotic cells in the remnant kidneys of WT mice (Figure S3C). In addition, Ad-APL-treated WT mice showed reduced expression levels of TNFα, IL6, MCP1, F4/80, inflammasome-associated factors, including NLRP3, caspase 1, IL1β, and IL18, and oxidative stress markers, including gp91phox, p47phox, p67phox, and p22phox, in the injured kidneys compared with Ad-β-gal-treated WT mice (Figures 2C and 2D).
Figure 2.

Systemic administration of adipolin ameliorates renal injury in WT mice after subtotal nephrectomy
(A) Urine and plasma parameters of renal function in WT mice treated with Ad-β-gal or Ad-APL at 8 weeks after subtotal nephrectomy. Left panel shows quantitative analysis of urinary albumin excretion normalized to urinary Cr. Center and right panels show plasma concentration of UN and Cr. N = 8 in each group.
(B) Evaluation of interstitial fibrosis in the injured kidney. Left panels show representative photos of the remnant kidney from Ad-β-gal-treated or Ad-APL-treated WT mice as determined by Masson Trichrome staining. Right panel shows quantitative analysis of fibrosis area as measured by ImageJ. N = 6 in each group. Scale bars show 100 μm.
(C) mRNA levels of pro-inflammatory mediators (TNFα, IL6, MCP1 and F4/80) in WT mice treated with Ad-β-gal or Ad-APL.
(D) mRNA levels of inflammasome-related genes (NLRP3, Caspase 1, IL1β and IL18) and oxidative stress markers (gp91phox, p47phox, p67phox and p22phox) in WT mice treated with Ad-β-gal or Ad-APL. N = 6 in each group. Student’s t-test ((A) (plasma UN), (B, C and D)) and Wilcoxon test ((A) (urine albumin and plasma Cr)) were used to produce the p values. N.S, not significant.
Inhibition of inflammasomes attenuates exacerbated renal injury in APL-KO mice after subtotal nephrectomy
To investigate the contribution of inflammasome activation to enhanced renal damage in APL-KO mice, continuous administration of MCC950, an inhibitor of the NLRP3 inflammasome, was performed for 6 weeks (from 2 weeks to 8 weeks following subtotal nephrectomy). Compared with WT mice, APL-KO mice showed increased urinary albumin excretion and plasma UN levels, which were diminished by treatment with MCC950 (Figure 3A). In contrast, the treatment of WT mice with MCC950 did not significantly affect urinary albumin excretion or plasma UN levels. In addition, treatment of APL-KO mice with MCC950 significantly reduced tubulointerstitial fibrosis, glomerular cross-sectional area and the numbers of TUNEL-positive apoptotic cells in the remnant kidneys after subtotal nephrectomy (Figures 3B, S4A, and S4B). Treatment of APL-KO mice with MCC950 significantly attenuated the expression of inflammasome-associated genes and oxidative stress markers in injured kidneys (Figures 3C and 3D). These data indicate that adipolin deficiency contributes to enhanced inflammasome activation in response to renal ablation, thereby exacerbating the renal injury.
Figure 3.

Inhibition of inflammasome activation ameliorates the exacerbated renal damage of APL-KO mice after subtotal nephrectomy
(A) Urine and plasma parameters of renal function in WT and APL-KO mice treated with vehicle (Veh) or MCC950 (MCC) at 8 weeks after subtotal nephrectomy. Left panel shows urinary albumin excretion normalized to urinary Cr. Right panel shows plasma concentration of UN. N = 7 in each group.
(B) Evaluation of interstitial fibrosis in the injured kidneys. Left panels show representative photos of the remnant kidney from WT and APL-KO mice treated with vehicle (Veh) or MCC950 (MCC) as stained with Masson Trichrome. Right panel shows quantitative analysis of fibrosis area as measured by ImageJ. N = 6 in each group. Scale bars show 100 μm.
(C and D) mRNA levels of inflammasome-related genes (NLRP3, Caspase 1, IL1β and IL18) (D) and oxidative stress markers (gp91phox, p47phox, p67phox and p22phox) (E) in the remnant kidney in WT and APL-KO mice treated with vehicle (Veh) or MCC950 (MCC). N = 6 in each group. One-way ANOVA with Tukey’s multiple comparisons test (A, B, C (IL18) and D) and Steel-Dwass test (C (NLRP3, Caspase1, IL1 β)) were used to produce the p values. N.S, not significant.
Adipolin suppresses activation of the inflammasome and oxidative stress in renal tubular cells and podocytes
To dissect the precise mechanism of adipolin-mediated attenuation of inflammasome activation, human renal proximal tubular epithelial cell line, HK-2 cells were pretreated with recombinant adipolin protein or vehicle, followed by stimulation with angiotensin (Ang) II. Pretreatment of HK-2 cells with adipolin significantly decreased Ang II-stimulated mRNA expression of inflammasome-related genes, including NLRP3, Caspase 1, IL1β, and IL18 (Figure 4A). Consistently, pretreatment of HK-2 cells with adipolin significantly decreased the protein levels of NLRP3, Caspase 1, and IL1β after stimulation with Ang II (Figure S5). In addition, pretreatment of HK-2 cells with adipolin attenuated Ang II-induced expression of oxidative stress markers, including gp91phox, p47phox, and p22phox (Figure 4B).
Figure 4.

Adipolin attenuates activation of inflammasome and oxidative stress in renal proximal tubular cells
(A and B) Effect of adipolin (APL) on angiotensin (Ang) II-stimulated expression of inflammasome-related genes (NLRP3, Caspase 1, IL1β and IL18) (A) and oxidative stress markers (gp91phox, p47phox and p22phox) (B) in HK-2 cells. Human proximal tubular cell lines, HK-2 cells were pretreated with APL (300 ng/mL) or vehicle for 1 h followed by incubation in the presence or absence of Ang II (1 μmol/L). N = 5 in each group. One-way ANOVA with Tukey’s multiple comparisons test were used to produce the p values. N.S, not significant.
To investigate whether adipolin modulates inflammasome activation and oxidative stress in podocytes, cultured podocytes, MPC-5 cells were pretreated with adipolin protein or vehicle, followed by stimulation with Ang II. Pretreatment of MPC-5 cells with adipolin significantly decreased Ang II-stimulated mRNA expression of inflammasome-related genes, including NLRP3, Caspase 1 and IL18, and oxidative stress markers, including gp91phox and p47phox (Figures S6A and S6B). Thus, it is conceivable that adipolin reduces inflammasome activation in renal tubular cells and podocytes, resulting in renal protection.
Adipolin increased the production of ketone bodies and β-hydroxybutyrate in the remnant kidney after subtotal nephrectomy
Recent reports have shown that ketone bodies exert beneficial effects on kidney disease.20,21 Thus, we assessed the levels of ketone body β-hydroxybutyrate (BHB) in the remnant kidney after subtotal nephrectomy. APL-KO mice showed reduced levels of renal BHB after subtotal nephrectomy compared with WT mice (Figure 5A). Systemic delivery of Ad-APL increased the renal BHB levels in WT mice in response to subtotal nephrectomy (Figure 5B). We also evaluated the expression of 3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS) 2, a key enzyme producing BHB, in the remnant kidney. APL-KO mice showed reduced expression levels of HMGCS2 in injured kidneys following subtotal nephrectomy compared to WT mice (Figure 5C). Systemic administration of Ad-APL to WT mice increased HMGCS2 expression in the remnant kidney after subtotal nephrectomy (Figure 5D). Similarly, treatment with adipolin protein increased mRNA levels of HMGCS2 in HK-2 cells (Figure 5E).
Figure 5.

Adipolin positively regulates β-hydroxybutyrate production and HMGCS2 expression in the remnant kidney after subtotal nephrectomy
(A) β-hydroxybutyrate (BHB) levels in the remnant kidney from WT and APL-KO mice at 8 weeks after subtotal nephrectomy. N = 6 in each group.
(B) BHB levels in the remnant kidney of WT mice treated with Ad-β-gal or Ad-APL at 8 weeks after subtotal nephrectomy. N = 6 in each group.
(C) mRNA levels of HMGCS2, which is a key productive enzyme of BHB, in the remnant kidney of WT and APL-KO mice at 8 weeks after subtotal nephrectomy. N = 6 in each group.
(D) mRNA levels of HMGCS2 in the remnant kidney of WT mice treated with Ad-β-gal or Ad-APL at 8 weeks after subtotal nephrectomy. N = 6 in each group.
(E) mRNA levels of HMGCS2 in HK-2 cells treated with adipolin (APL) protein (300 ng/mL) or vehicle for 6 h. N = 6 in each group.
(F) Contribution of HMGCS2 to the inhibitory effects of APL on Ang II-stimulated expression of inflammasome-related genes, including NLRP3, Caspase 1 and IL1β in HK-2 cells. HK-2 cells were treated with siRNA targeting HMGCS2 (10 nmol/L) or control unrelated siRNA for 8 h. After 16 h of serum starvation, HK-2 cells were treated with APL protein (300 ng/mL) or vehicle for 1 h followed by stimulation with Ang II (1 μmol/L) or vehicle for 24 h. N = 5 in each group. Student’s t-test (B, D and E), Wilcoxon test (A and C) and One-way ANOVA with Tukey’s multiple comparisons test (F) were used to produce the p values.
To examine whether HMGCS2 was involved in the inhibitory effect of adipolin on inflammasome activation in vitro, HK-2 cells were transfected with siRNA targeting HMGCS2 or unrelated siRNA. Transduction of HK-2 cells with siRNA against HMGCS2 decreased mRNA levels of HMGCS2 by 80.2 ± 2.0% (Figure S7). Ablation of HMGCS2 blocked the inhibitory effect of adipolin on Ang II-induced expression of NLRP3, Caspase 1, and IL1β in HK-2 cells (Figure 5F). Thus, it is likely that adipolin could positively regulate ketone body production in the injured kidney through the induction of HMGCS2 expression, thereby contributing to the suppression of inflammasome activation.
Adipolin attenuates inflammasome activation in renal proximal tubular cells through the PPARα/HMGCS2-dependent pathway
Because endogenous expression of HMGCS2 is induced by peroxisome proliferator-activated receptor (PPAR) α,22 we assessed the effect of adipolin on PPARα binding to peroxisome proliferator-activated receptor response element (PPRE) in HK-2 cells. Treatment of HK-2 cells with adipolin protein increased PPARα binding to PPRE (Figure 6A). Similarly, adipolin treatment significantly increased the PPRE luciferase activity in HK-2 cells (Figure 6B). Adipolin treatment also increased the mRNA levels of lipoprotein lipase (LPL), uncoupling protein (UCP), and acyl CoA oxidase (ACOX), which are downstream target genes of PPARα in HK-2 cells and MPC-5 podocytes (Figures 6C and S8). In addition, APL-KO mice showed reduced mRNA levels of LPL, UCP2, and ACOX1 in the remnant kidney after subtotal nephrectomy compared with WT mice (Figure 6D). Furthermore, Ad-APL administration to WT mice significantly increased the mRNA levels of LPL, UCP2, and ACOX1 in the remnant kidney after subtotal nephrectomy compared to Ad-β-gal treatment (Figure 6E). These findings indicate that adipolin promotes PPARα-dependent signaling pathways in the injured kidneys.
Figure 6.

Adipolin reduces inflammasome activation in proximal tubular cells through the PPARα/HMGCS2 pathway
(A) PPARα activity in HK-2 cells stimulated with adipolin (APL) protein (300 ng/mL) or vehicle for 6 h. N = 6 in each group.
(B) PPRE luciferase activity in HK-2 cells stimulated with APL protein (300 ng/mL) or vehicle for 24 h. N = 6 in each group.
(C) mRNA expression of lipoprotein lipase (LPL), uncoupling protein (UCP) 2 and acyl CoA oxidase (ACOX)-1, which are downstream molecules of PPARα, in HK-2 cells after stimulation with APL protein (300 ng/mL) or vehicle for 6 h. N = 6 in each group.
(D) mRNA levels of LPL, UCP2 and ACOX-1 in the remnant kidney of WT and APL-KO mice at 8 weeks after subtotal nephrectomy. N = 6 in each group.
(E) mRNA levels of LPL, UCP2 and ACOX-1 in the remnant kidney of WT mice treated with Ad-β-gal or Ad-APL at 8 weeks after subtotal nephrectomy. N = 6 in each group.
(F) Contribution of PPARα, TGFβ/Smad and PI3-kinase/Akt pathways to the suppressive effects of adipolin on angiotensin (Ang) II-induced expression of NLRP3, Caspase 1 and IL1β in HK-2 cells. HK-2 cells were pretreated with GW6471 (GW; PPARα antagonist, 50 μmol/L), SB431542 (SB; TGFβ/Smad inhibitor, 1 μmol/L) or LY294002 (LY; PI3-kinase/Akt inhibitor, 20 μmol/L) or vehicle for 30 min and treated with APL protein (300 ng/mL) or vehicle for 1 h followed by stimulation with Ang II (1 μmol/L) or vehicle for 24 h. N = 5 in each group.
(G) Contribution of PPARα to the stimulatory effect of adipolin on HMGCS2 expression in HK-2 cells. HK-2 cells were pretreated with siRNA targeting PPARα (10 nmol/L) or control unrelated siRNA for 24 h followed by treatment with APL protein (300 ng/mL) or vehicle for 6 h. N = 4 in each group.
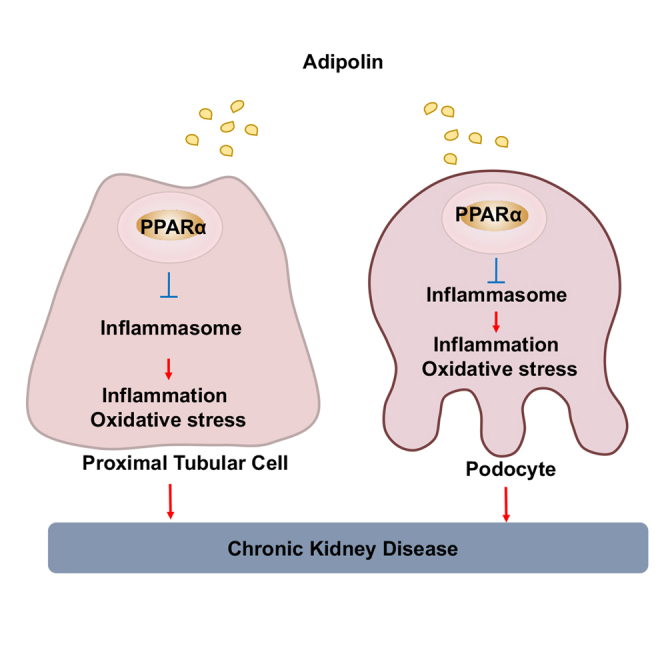
(H) Proposed scheme of the mechanism of renal protection by adipolin. Adipolin attenuates inflammation and oxidative stress by reducing inflammasome activation in proximal tubular cells through induction of PPARα/HMGCS2/β-hydroxybutyrate-dependent pathway, thereby leading to protection against chronic kidney disease. One-way ANOVA with Tukey’s multiple comparisons test (F and G), Student’s t-test (A–E) were used to produce the p values.
To investigate the contribution of PPARα to the adipolin-mediated inhibition of inflammasome activation in response to Ang II, HK-2 cells were treated with the PPARα antagonist GW6471. Pretreatment of HK-2 cells with GW6471 reversed the suppressive effects of adipolin on the Ang II-stimulated expression of NLRP3, caspase 1, and IL1β (Figure 6F). We previously reported that adipolin suppresses the inflammatory response of macrophages through TGFβ/Smad signaling.14 We also showed that adipolin reduced cardiomyocyte apoptosis after myocardial infarction through the PI3-kinase/Akt-dependent pathway.15 Thus, to examine the possible involvement of TGFβ/Smad signaling or PI3-kinase/Akt signaling in the suppressive actions of adipolin on Ang II-stimulated inflammasome activation, HK-2 cells were pretreated with an inhibitor of TGFβ/Smad (SB431542) or an inhibitor of PI3-kinase/Akt (LY294002). Pretreatment of HK-2 cells with SB431542 or LY294002 did not affect the inhibitory effects of adipolin on the Ang II-stimulated increase in the expression of NLRP3, Caspase 1, and IL1β (Figure 6F). Transduction of HK-2 cells with siRNA against PPARα reduced the mRNA levels of PPARα by 84.1 ± 2.8% compared with transduction with unrelated siRNA as a control (Figure S9A). Knockdown of PPARα by siRNA targeting PPARα abrogated the adipolin-stimulated expression of HMGCS2 in HK-2 cells (Figure 6G). Moreover, ablation of PPARα reversed the suppressive effects of adipolin on the Ang II-stimulated expression of NLRP3 and IL1β in HK-2 cells (Figure S9B). These findings indicate that adipolin reduces inflammasome activation in proximal tubular cells through induction of the PPARα/HMGCS2-dependent pathway (Figure 6H).
To dissect the upstream molecule of adipolin-induced PPARα activation, HK-2 cells were transduced with siRNA targeting AdipoR1 or AdipoR2, or unrelated siRNA. Transduction of HK-2 cells with siRNA against AdipoR1 and AdipoR2 reduced the mRNA levels of AdipoR1 and AdipoR2 by 82.6 ± 1.1% and 78.9 ± 1.8% compared with transduction with unrelated siRNA (Figure S10A). Ablation of AdipoR2, but not knockdown of AdipoR1, abrogated the adipolin-stimulated expression of LPL and HMGCS2 in HK-2 cells. (Figure S10B). These results indicate that adipolin stimulates PPARα activation in proximal tubular cells through an AdipoR2-dependent pathway.
Adipolin ameliorates renal injury after subtotal nephrectomy in a PPARα-dependent manner
Finally, to assess the contribution of PPARα to the renal protective effects of adipolin in vivo, Ad-APL or Adβ-gal as control was intravenously injected into WT or PPARα-knockout (KO) mice 4 weeks after subtotal nephrectomy. Although Ad-APL treatment significantly reduced urinary albumin excretion and plasma UN levels in WT mice, it did not affect urinary albumin excretion or plasma UN levels in PPARα-KO mice (Figure 7A). In contrast to WT mice, Ad-APL treatment had no significant effect on tubulointerstitial fibrosis after subtotal nephrectomy in PPARα-KO mice (Figure 7B). Similarly, Ad-APL treatment did not affect the expression levels of pro-inflammatory mediators, including TNFα and IL6, and inflammasome-associated factors, including NLRP3 and IL18 in the remnant kidney of PPARα-KO mice after subtotal nephrectomy (Figure 7C). Collectively, adipolin exerts a renoprotective function by inhibiting inflammasome activation via a PPARα-dependent pathway.
Figure 7.

Systemic administration of adipolin ameliorates renal injury after subtotal nephrectomy through the PPARα dependent mechanism in vivo
(A) Urine and plasma parameters of renal function in WT and PPARα knockout (KO) mice treated with Ad-β-gal or Ad-APL at 8 weeks after subtotal nephrectomy. Left panel shows quantitative analysis of urinary albumin excretion normalized to urinary Cr. Right panel shows plasma concentration of UN. N = 5 in each group.
(B) Evaluation of interstitial fibrosis as assessed by Masson Trichrome staining. Quantitative analysis of fibrosis area in the injured kidneys from WT and PPARα-KO mice treated with Ad-β-gal or Ad-APL as measured by ImageJ. N = 5 in each group.
(C) mRNA levels of pro-inflammatory mediators (TNFα and IL6) and inflammasome-related genes (NLRP3 and IL18) in the inured kidneys in WT mice and PPARα-KO mice treated with Ad-β-gal or Ad-APL. N = 5 in each group. One-way ANOVA with Tukey’s multiple comparisons test were used to produce the p values.
Discussion
The present study provides the first evidence that adipolin exerts renoprotective effects in a mouse model of subtotal nephrectomy. Disruption of adipolin led to the enhancement of urinary albumin secretion, tubulointerstitial fibrosis, and renal inflammasome activation in mice following subtotal nephrectomy. Inflammasome inhibition by MCC950 blocked the enhancement of damaged kidney fibrosis under conditions of adipolin deficiency. Systemic administration of adipolin to mice with subtotal nephrectomy improved fibrosis and inflammasome activation in injured kidneys. Pretreatment of renal proximal tubular cells with adipolin attenuates inflammasome activation. These findings indicate that adipolin protects against renal dysfunction and injury by reducing inflammasome activation, suggesting that adipolin plays a crucial role in preventing CKD development.
NLRP3 inflammasome is involved in the pathogenesis of various non-microbial diseases, including CKD.23,24 NLRP3 deficient mice exhibit reduced tubular injury and fibrosis after unilateral urinary occlusion (UUO).18 Lack of apoptosis-associated speck-like protein containing CARD, an integral component of the inflammasome, results in attenuated renal injury and fibrosis after UUO.25 Ang II has been reported to activate NLRP3-inflammasome in proximal tubular cells by increasing endoplasmic reticulum (ER) stress.26 In addition, albumin dose-dependently increases expression of angiotensin converting enzyme (ACE) 1 in proximal tubular cells, leading to an increased production of Ang II.27 Thus, it is likely that excessive exposure to albumin or Ang II could cause inflammasome activation in proximal tubular cells during CKD progression, thereby contributing to exacerbation of renal injury. Our present findings demonstrate that adipolin attenuates renal dysfunction and damage in a mouse model of subtotal nephrectomy by reducing inflammasome activation.
Ketone bodies, including BHB, are produced mainly in the liver and play an important role as an alternative energy source for the brain, heart, and skeletal muscle during nutrient starvation.28,29 Recently, sodium-glucose cotransporter 2 inhibitors were reported to protect against diabetic kidney disease by elevating BHB levels.21 Furthermore, it has been reported that BHB inhibits NLRP3 inflammasome activation in mouse models of NLRP3-mediated inflammation.30 Our data indicated that adipolin positively regulates BHB production and HMGCS2 expression in injured kidneys. Our gene knockdown experiments also demonstrated that ablation of HMGCS2 canceled the adipolin-mediated reduction of Ang II-induced expression of inflammasome-related genes, including NLRP3, in renal proximal tubular cells. These results suggested that the ability of adipolin to protect against renal inflammasome activation in vitro and in vivo is dependent, at least in part, on its ability to increase ketone body production.
PPARα is a member of the nuclear hormone receptor superfamily of ligand-activated transcription factors.31 PPARα is abundantly expressed in the liver, skeletal muscle, heart, and kidney, and regulates lipid metabolism and the inflammatory response.32,33 Fenofibrate, a PPARα agonist, reduces lipotoxicity-induced renal injury.34 Fenofibrate also has a beneficial effect on fatty acid-induced apoptosis in renal tubular cells.35 Transgenic mice expressing PPARα in the proximal tubule show improved renal function during acute kidney injury.36 These findings indicate that PPARα activation contributes to protection against kidney disease. In the present study, we found that adipolin enhanced PPARα activity and expression of PPARα target genes in proximal tubular cells and that adipolin positively regulated the expression of PPARα-dependent genes in the injured kidney of mice. Thus, it is likely that adipolin has a PPARα ligand activity. HMGCS2 has a peroxisome proliferator response element in the promoter region and is transcriptionally regulated by PPARα.22 In agreement with this finding, our experiments using siRNA showed that ablation of PPARα blocked adipolin-induced expression of HMGCS2 in renal proximal tubular cells. Our in vitro experiments also showed that the suppressive effects of adipolin on Ang II-stimulated inflammasome activation in renal proximal tubular cells were abrogated by ablation of PPARα or HMGCS2. Importantly, the protective effects of adipolin on injured kidneys in vivo were diminished in the background of PPARα deficiency. Taken together, these data suggest that adipolin can attenuate renal inflammasome activation in response to subtotal renal ablation, at least in part, through its ability to increase HMGCS2 expression via PPARα activation, thereby leading to renal protection (Figure 6H).
In conclusion, we demonstrated for the first time that adipolin, a novel adipokine, functions as a renoprotective factor that can mitigate renal inflammasome activity through a PPARα/HMGCS2-dependent mechanism. We, and others, have shown that adipolin improves metabolic dysfunction, pathological remodeling of the vascular wall, and adverse cardiac remodeling after myocardial infarction.13,14,15,37 It has been reported that reduced levels of serum adipolin are associated with renal dysfunction in patients with type 2 diabetes.38 Furthermore, patients with type 2 diabetes and hemodialysis exhibit significantly lower blood adipolin levels compared with patients with type 2 diabetes.39 These clinical data indicate the inverse association between circulating adipolin levels and renal function. Thus, adipolin could represent a novel therapeutic target for the prevention or treatment of kidney and cardiometabolic diseases.
Limitations of the study
In the present study, we used mice subjected to subtotal (5/6) nephrectomy as CKD models. Although the mouse 5/6 nephrectomy model is widely used, this model shows mild renal dysfunction compared with other models of CKD such as lupus nephritis and diabetic kidney disease. In addition, we used 5/6 nephrectomized WT mice fed normal diets. It would be better to employ obese mice to clarify the role of adipolin in obesity-associated CKD. Thus, future studies will be needed to dissect the role of adipolin in severe renal dysfunction using different CKD models.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-NLRP3 antibody | Adipogen | AG-20B-0014 |
| Anti-NLRP3 antibody | Novus | NBP2-12446SS |
| Anti-Caspase 1 antibody | Adipogen | AG-20B-0042 |
| Anti-Caspase 1 antibody | Proteintech | 22915-1-AP |
| Anti-IL1β antibodies | R&D Systems | AF-401-SP |
| Bacterial and virus strains | ||
| Adenovirus producing β-galactosidase | This paper | N/A |
| Adenovirus producing adipolin | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| MCC950 | Adipogen | AG-CR1-3615 |
| GW6471 | Cayman | 11697 |
| SB431542 | Cayman | 13031 |
| LY294002 | Cell Signaling Technology | #9901 |
| Recombinant angiotensin II protein | Sigma-Aldrich | A9525 |
| Critical commercial assays | ||
| Masson’s trichrome assay kit | Sigma-Aldrich | HT15 |
| β-hydroxybutyrate and a ketone Body assay kit | Cayman | 700190 |
| TUNEL Assay kit-HRP-DAB | Abcam | AB20638 |
| Plasma UN and Cr measurement | SRL | commercial laboratory |
| Urine Cr measurement | SRL | commercial laboratory |
| murine albumin enzyme-linked immunosorbent assay kit | Exocell | 1011 |
| PPARα transcription factor assay kit | Cayman | 10006915 |
| dual-luciferase reporter assay system | Promega | E1910 |
| Experimental models: Cell lines | ||
| Human HK-2 cell | ATCC | CRL2190 |
| Mouse MPC-5 podocyte clonal cell | University of Bristol | N/A |
| Experimental models: Organisms/strains | ||
| Mouse C57BL/6J | Jackson Laboratories | Strain No. 000664 |
| Adipolin knockout mice | This paper | N/A |
| PPARα knockout mice (B6; 129S4-Pparatm1Gonz/J) | Jackson Laboratories | Strain No. 008154 |
| Oligonucleotides | ||
| siRNA targeting nontargeting pool | Horizon Discovery | D-001810-10 |
| siRNA targeting HMGCS2 (ON-TARGETplus, SMARTpool) | Horizon Discovery | L-010179-00 |
| siRNA targeting PPARα (ON-TARGETplus, SMARTpool) | Horizon Discovery | L-003434-00 |
| siRNA targeting AdipoR1 (ON-TARGETplus, SMARTpool) | Horizon Discovery | L-007800-01 |
| siRNA targeting AdipoR2 (ON-TARGETplus, SMARTpool) | Horizon Discovery | L-007801-01 |
| Software and algorithms | ||
| image J analysis system | Schneider et al.40 | Nat Methods 9, 671–675. 10.1038/nmeth.2089 |
| JMP Pro 15 software | SAS | N/A |
Resource availability
Lead contact
Further information and requests of resources and reagents should be directed to the lead contact, Koji Ohashi (ohashik@med.naogya-u.ac.jp).
Materials availability
Antibodies were commercial sources described in the STAR Methods key resources table. All unique materials generated In this study will be available from the lead contact after completing a Material Transfer Agreement.
Experimental model and subject details
Animal and surgical procedure
Adipolin knockout (APL-KO) mice were generated as previously described (Accession No. CDB1102K website: http://www2.clst.riken.jp/arg/mutant%20mice%20list.html).14 Homozygous APL-KO and wild-type (WT) mice with a C57BL/6J background at 8 weeks of age were used in this study. The genotyping primers for the adipolin (Fam 132a/CTRP12) WT allele were as follows:5′-GCCTGAATCCCCCACTAACT-3′ (P1) and 5′-TCTGGTAGCCCTGAGAATCG-3′ (P2). Primers for the adipolin (Fam 132α/CTRP12) null allele were as follows:5′-GGAAGTGCCCAATGAGTCC-3′ (P3) and 5′-GTGGATGTGGAAATGTGTGC-3′ (P4). PPARα knockout (PPARα-KO) mice were purchased from the Jackson Laboratory at Osaka University and were provided by Yuya Fujishima, Norikazu Maeda, and Iichiro Shimomura at Osaka University.41 Male APL-KO and WT mice at the age of 8 weeks were assigned to two groups with or without subtotal renal ablation. Subtotal (5/6 of) nephrectomies were performed using surgical excision.11,42 Briefly, the upper and lower poles of the left kidney (two-thirds of the left kidney) were resected. After one week, the remaining right kidney was removed through a right paramedian incision after ligation of the right renal artery, vein, and ureter. Eight weeks after ablation, APL-KO and WT mice were sacrificed for analysis. In some experiments, Alzet mini-osmotic pumps (Model 2006, Durect Corp.) were used to deliver 5 mg/kg/day of MCC950 (Adipogen) two weeks after ablation for a period of 42 days. Pumps were implanted subcutaneously in the murine mid-scapular region through a small incision in the back of the neck, which was closed with clips. Before the surgical procedure, anesthesia (medetomidine, midazolam, and butorphanol at doses of 0.15, 2.0, and 2.5 mg/kg, respectively) was administered intraperitoneally. Adequacy of anesthesia was confirmed by the lack of a toe-pinch withdrawal response during the surgical procedure. In some experiments, Ad-APL, or Ad-β-gal (2×109 plaque-forming units/mouse) was intravenously injected into WT or PPARα-KO mice 4 weeks after renal ablation. The study protocol was approved by the Institutional Animal Care and Use Committee of Nagoya University. All animal procedures were performed in accordance with the National Institutes of Health guidelines (Guide for the Care and Use of Laboratory Animals).
Cell culture
HK-2 cells were purchased from AmericanType Culture Collection (Rockville, MD, USA). The cells were cultured in a medium consisting of DMEM/F12 (Gibco) supplemented with 10% FBS at intervals of 3-4 days to continuously passaged. HK-2 cells were cultured in DMEM/F12 medium for 16 h and cultured in the presence or absence of adipolin (300 ng/ml) for 1 h, followed by stimulation with Ang (1 μmol/L) or vehicle for 24 h. Knockdown of PPARα, HMGCS2, AdipoR1, or AdipoR2 was achieved by siRNA transduction at 10 nM with Lipofectamine RNAiMAX (Invitrogen) 24 h before experiments. Lipofectamine RNAiMAX and siRNAs were dissolved in Opti-MEM (Gibco). The ON-TARGETplus siRNA SMART pools targeting each gene were purchased from Horizon Discovery. Control cultures were transfected with an unrelated scrambled siRNA (ON-TARGET plus Control Non-Targeting Pool, Horizon Discovery).
Conditionally immortalized murine MPC-5 podocyte clonal cells (University of Bristol) were maintained in RPMI1640 medium containing 10% fetal bovine serum, 10 IU/ml recombinant murine interferon-γ (Wako) at 33°C for proliferating cells. To induce differentiation, MPC-5 cells were incubated at 5% CO2 at 37°C without interferon-γ for 14 days.
Method details
Materials
A polyclonal antibody against mouse adipolin was generated, as previously described.43 Anti-NLRP3 antibody was purchased from Adipogen and Novus. Anti-caspase 1 antibodies were purchased from Adipogen and Proteintech. Anti-IL1β antibodies were purchased from R&D Systems. MCC950 was purchased from Adipogen. GW6471 and SB431542 were purchased from Cayman Chemicals. LY294002 was purchased from Cell Signaling Technology. Recombinant angiotensin II protein was purchased from Sigma-Aldrich. β-hydroxybutyrate and a ketone Body assay kit were purchased from Cayman. Adenoviral vectors expressing the genes encoding β-galactosidase (Ad-β-gal) and FLAG-tagged adipolin (Ad-APL) were constructed under the control of the CMV promoter.13,14
Histological analyses
Tissue samples were fixed with 4% paraformaldehyde and embedded in paraffin. Serial tissue sections (5 μm) of the kidneys were stained with Masson’s trichrome (Sigma-Aldrich) and hematoxylin and eosin (H-E). Renal apoptosis was assessed by a terminal deoxynucleotidyl transferase-mediated dUTP-nick end labeling (TUNEL) staining using TUNEL Assay kit-HRP-DAB (Abcam). The fibrotic area, glomerular size and apoptotic cell number were measured by using an image J analysis system.11,40
Laboratory methods
Eight weeks after the operation, the mice were sacrificed, and blood and urine samples were collected for analysis. Plasma concentrations of urea nitrogen (UN) and creatinine (Cr) and urine concentrations of Cr were measured in a commercial laboratory (SRL). The urinary albumin concentration was measured using a murine albumin enzyme-linked immunosorbent assay kit (Exocell). Urinary albumin excretion was evaluated as albumin per gram of urinary Cr. The specific DNA-binding activity of PPARα was evaluated using a PPARα transcription factor assay kit (Cayman).
Western blot analysis
Tissue samples and cell samples were prepared in lysis buffer containing 20 mM Tris-HCl (pH 8.0), 1% NP-40, 150 mM NaCl, 0.5% deoxycholic acid, 1 mM sodium orthovanadate, and protease inhibitor cocktail (Sigma Chemical Co.). The protein concentration was determined by a BCA protein assay kit (Thermo Scientific). The equal amounts of proteins were separated by denaturing SDS-PAGE. Proteins were transferred onto PVDF membrane (GE Healthcare) and probed with the primary antibody followed by incubation with the HRP-conjugated secondary antibody. ECL prime system (GE Healthcare) were used for detection of the protein signal. The expression levels were determined by measurement of the corresponding band intensities by using image J software (National Institute of Health),40 and the relative values were expressed relative to α-tubulin signal.
Luciferase assay
HK-2 cells in 24 well dishes were transfected with 500 ng of ptkLUC vector (Promega) containing a fragment of human PPREs along with 500 ng of pCMV6-AC-GFP (PS100010) expression vector (Origene) containing human PPARα in the presence of 50 ng of pRL-SV40 (Promega) using Lipofectamine 2000, as previously described.44 After 24 h, cells were lysed in Passive Lysis Buffer (Promega), and the lysates were assayed for firefly and Renilla luciferase activities using a dual-luciferase reporter assay system (Promega). Promoter activity was determined as the ratio of firefly luciferase activity to Renilla luciferase activity.
Determination of mRNA levels
Gene expression levels were analyzed using quantitative real-time PCR. RNA from HK-2 cells and kidney tissue was extracted using the RNeasy Mini Kit (Qiagen), and cDNA was synthesized using ReverTra Ace (TOYOBO). Real-time PCR was performed with a Bio-Rad real-time PCR detection system using the THUNDERBIRD SYBR qPCR mix (TOYOBO) as a double-standard DNA-specific dye. The primers used are listed in Tables S1 and S2. The expression levels of the examined transcripts were compared with those of 36B4 and normalized to the mean value of the controls.
Quantification and statistical analysis
Data are presented as mean ± S.E.M. The differences in variables with normal distributions between the two groups were evaluated using an unpaired Student’s t-test. Differences between three or more groups were evaluated using one-way analysis of variance (ANOVA) with post-hoc Tukey’s test. Differences between groups for variables with non-normal distribution were analyzed using the Wilcoxon signed-rank test (for two groups) or the Steel-Dwass test (for three or more groups). Data distributions were evaluated using the Shapiro-Wilk test. P < 0.05 denoted the presence of a statistically significant difference. All statistical analyses were performed using the JMP Pro 15 software (SAS).
Acknowledgments
We gratefully thank for the technical assistance of Yoko Inoue. We would like to thank Editage (www.editage.com) for English language editing. This work was supported by Grant-in-Aid for Scientific Research A, Grant-in-Aid for Challenging Exploratory Research and grants from Takeda Science Foundation to N Ouchi. K. Ohashi was supported by Grant-in-Aid for Scientific Research C.
Author contributions
L.F. designed the research study, conducted the experiments, acquired the data, analyzed the data and wrote the manuscript. K.O. designed the research study, conducted the experiments, acquired the data, analyzed the data, provided expertise related to the experiments and wrote the manuscript. S.H., H.O., N. Otaka, H.K., T.T., Y.O., K.T., and M. Tatsumi conducted the experiments, acquired the data, analyzed the data. M. Takefuji, Y.S., Y.B.K., and T.M. designed the research study, provided expertise related to the experiments. Y.F., N.M., and I.S. provided PPARα deficient mice and expertise related to the experiments. N. Ouchi designed the research study, analyzed the data, provided expertise related to the experiments and wrote the manuscript. All authors participated in interpreting the results and revising the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: April 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106591.
Contributor Information
Koji Ohashi, Email: ohashik@med.nagoya-u.ac.jp.
Noriyuki Ouchi, Email: nouchi@med.nagoya-u.ac.jp.
Supplemental information
Data and code availability
-
•
All data for evaluating the contributions in the paper are presented in the paper and/or the supplemental information.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Jha V., Garcia-Garcia G., Iseki K., Li Z., Naicker S., Plattner B., Saran R., Wang A.Y.M., Yang C.W. Chronic kidney disease: global dimension and perspectives. Lancet. 2013;382:260–272. doi: 10.1016/S0140-6736(13)60687-X. [DOI] [PubMed] [Google Scholar]
- 2.GBD Chronic Kidney Disease Collaboration Global, regional, and national burden of chronic kidney disease, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2020;395:709–733. doi: 10.1016/S0140-6736(20)30045-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stenvinkel P., Zoccali C., Ikizler T.A. Obesity in CKD--what should nephrologists know? J. Am. Soc. Nephrol. 2013;24:1727–1736. doi: 10.1681/ASN.2013040330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ejerblad E., Fored C.M., Lindblad P., Fryzek J., McLaughlin J.K., Nyrén O. Obesity and risk for chronic renal failure. J. Am. Soc. Nephrol. 2006;17:1695–1702. doi: 10.1681/ASN.2005060638. [DOI] [PubMed] [Google Scholar]
- 5.Lakkis J.I., Weir M.R. Obesity and kidney disease. Prog. Cardiovasc. Dis. 2018;61:157–167. doi: 10.1016/j.pcad.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Zhu Q., Scherer P.E. Immunologic and endocrine functions of adipose tissue: implications for kidney disease. Nat. Rev. Nephrol. 2018;14:105–120. doi: 10.1038/nrneph.2017.157. [DOI] [PubMed] [Google Scholar]
- 7.Ouchi N., Parker J.L., Lugus J.J., Walsh K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011;11:85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee B.T., Ahmed F.A., Hamm L.L., Teran F.J., Chen C.S., Liu Y., Shah K., Rifai N., Batuman V., Simon E.E., et al. Association of C-reactive protein, tumor necrosis factor-alpha, and interleukin-6 with chronic kidney disease. BMC Nephrol. 2015;16:77. doi: 10.1186/s12882-015-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rhee C.M., Ahmadi S.F., Kalantar-Zadeh K. The dual roles of obesity in chronic kidney disease: a review of the current literature. Curr. Opin. Nephrol. Hypertens. 2016;25:208–216. doi: 10.1097/MNH.0000000000000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kümpers P., Gueler F., Rong S., Mengel M., Tossidou I., Peters I., Haller H., Schiffer M. Leptin is a coactivator of TGF-beta in unilateral ureteral obstructive kidney disease. Am. J. Physiol. Renal Physiol. 2007;293:F1355–F1362. doi: 10.1152/ajprenal.00003.2007. [DOI] [PubMed] [Google Scholar]
- 11.Ohashi K., Iwatani H., Kihara S., Nakagawa Y., Komura N., Fujita K., Maeda N., Nishida M., Katsube F., Shimomura I., et al. Exacerbation of albuminuria and renal fibrosis in subtotal renal ablation model of adiponectin-knockout mice. Arterioscler. Thromb. Vasc. Biol. 2007;27:1910–1917. doi: 10.1161/ATVBAHA.107.147645. [DOI] [PubMed] [Google Scholar]
- 12.Sharma K., Ramachandrarao S., Qiu G., Usui H.K., Zhu Y., Dunn S.R., Ouedraogo R., Hough K., McCue P., Chan L., et al. Adiponectin regulates albuminuria and podocyte function in mice. J. Clin. Invest. 2008;118:1645–1656. doi: 10.1172/JCI32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enomoto T., Ohashi K., Shibata R., Higuchi A., Maruyama S., Izumiya Y., Walsh K., Murohara T., Ouchi N. Adipolin/C1qdc2/CTRP12 protein functions as an adipokine that improves glucose metabolism. J. Biol. Chem. 2011;286:34552–34558. doi: 10.1074/jbc.M111.277319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ogawa H., Ohashi K., Ito M., Shibata R., Kanemura N., Yuasa D., Kambara T., Matsuo K., Hayakawa S., Hiramatsu-Ito M., et al. Adipolin/CTRP12 protects against pathological vascular remodelling through suppression of smooth muscle cell growth and macrophage inflammatory response. Cardiovasc. Res. 2020;116:237–249. doi: 10.1093/cvr/cvz074. [DOI] [PubMed] [Google Scholar]
- 15.Takikawa T., Ohashi K., Ogawa H., Otaka N., Kawanishi H., Fang L., Ozaki Y., Eguchi S., Tatsumi M., Takefuji M., et al. Adipolin/C1q/Tnf-related protein 12 prevents adverse cardiac remodeling after myocardial infarction. PLoS One. 2020;15:e0243483. doi: 10.1371/journal.pone.0243483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foresto-Neto O., Ávila V.F., Arias S.C.A., Zambom F.F.F., Rempel L.C.T., Faustino V.D., Machado F.G., Malheiros D.M.A.C., Abensur H., Camara N.O.S., et al. NLRP3 inflammasome inhibition ameliorates tubulointerstitial injury in the remnant kidney model. Lab. Invest. 2018;98:773–782. doi: 10.1038/s41374-018-0029-4. [DOI] [PubMed] [Google Scholar]
- 17.Komada T., Chung H., Lau A., Platnich J.M., Beck P.L., Benediktsson H., Duff H.J., Jenne C.N., Muruve D.A. Macrophage uptake of necrotic cell DNA activates the AIM2 inflammasome to regulate a proinflammatory phenotype in CKD. J. Am. Soc. Nephrol. 2018;29:1165–1181. doi: 10.1681/ASN.2017080863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vilaysane A., Chun J., Seamone M.E., Wang W., Chin R., Hirota S., Li Y., Clark S.A., Tschopp J., Trpkov K., et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 2010;21:1732–1744. doi: 10.1681/ASN.2010020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shimizu M.H.M., Coimbra T.M., de Araujo M., Menezes L.F., Seguro A.C. N-acetylcysteine attenuates the progression of chronic renal failure. Kidney Int. 2005;68:2208–2217. doi: 10.1111/j.1523-1755.2005.00677.x. [DOI] [PubMed] [Google Scholar]
- 20.Hattori Y. Beneficial effects on kidney during treatment with sodium-glucose cotransporter 2 inhibitors: proposed role of ketone utilization. Heart Fail. Rev. 2021;26:947–952. doi: 10.1007/s10741-020-10065-7. [DOI] [PubMed] [Google Scholar]
- 21.Tomita I., Kume S., Sugahara S., Osawa N., Yamahara K., Yasuda-Yamahara M., Takeda N., Chin-Kanasaki M., Kaneko T., Mayoux E., et al. SGLT2 inhibition mediates protection from diabetic kidney disease by promoting ketone body-induced mTORC1 inhibition. Cell Metab. 2020;32:404–419.e6. doi: 10.1016/j.cmet.2020.06.020. [DOI] [PubMed] [Google Scholar]
- 22.Rodríguez J.C., Gil-Gómez G., Hegardt F.G., Haro D. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase gene by fatty acids. J. Biol. Chem. 1994;269:18767–18772. [PubMed] [Google Scholar]
- 23.Martinon F., Pétrilli V., Mayor A., Tardivel A., Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 24.Bani-Hani A.H., Leslie J.A., Asanuma H., Dinarello C.A., Campbell M.T., Meldrum D.R., Zhang H., Hile K., Meldrum K.K. IL-18 neutralization ameliorates obstruction-induced epithelial-mesenchymal transition and renal fibrosis. Kidney Int. 2009;76:500–511. doi: 10.1038/ki.2009.216. [DOI] [PubMed] [Google Scholar]
- 25.Komada T., Usui F., Shirasuna K., Kawashima A., Kimura H., Karasawa T., Nishimura S., Sagara J., Noda T., Taniguchi S., et al. ASC in renal collecting duct epithelial cells contributes to inflammation and injury after unilateral ureteral obstruction. Am. J. Pathol. 2014;184:1287–1298. doi: 10.1016/j.ajpath.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 26.Wang J., Wen Y., Lv L.L., Liu H., Tang R.N., Ma K.L., Liu B.C. Involvement of endoplasmic reticulum stress in angiotensin II-induced NLRP3 inflammasome activation in human renal proximal tubular cells in vitro. Acta Pharmacol. Sin. 2015;36:821–830. doi: 10.1038/aps.2015.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu B.C., Gao J., Li Q., Xu L.M. Albumin caused the increasing production of angiotensin II due to the dysregulation of ACE/ACE2 expression in HK2 cells. Clin. Chim. Acta. 2009;403:23–30. doi: 10.1016/j.cca.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 28.Newman J.C., Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014;25:42–52. doi: 10.1016/j.tem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cotter D.G., Schugar R.C., Crawford P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013;304:H1060–H1076. doi: 10.1152/ajpheart.00646.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Youm Y.H., Nguyen K.Y., Grant R.W., Goldberg E.L., Bodogai M., Kim D., D'Agostino D., Planavsky N., Lupfer C., Kanneganti T.D., et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015;21:263–269. doi: 10.1038/nm.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dreyer C., Krey G., Keller H., Givel F., Helftenbein G., Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell. 1992;68:879–887. doi: 10.1016/0092-8674(92)90031-7. [DOI] [PubMed] [Google Scholar]
- 32.Delerive P., Fruchart J.C., Staels B. Peroxisome proliferator-activated receptors in inflammation control. J. Endocrinol. 2001;169:453–459. doi: 10.1677/joe.0.1690453. [DOI] [PubMed] [Google Scholar]
- 33.Barbier O., Torra I.P., Duguay Y., Blanquart C., Fruchart J.C., Glineur C., Staels B. Pleiotropic actions of peroxisome proliferator-activated receptors in lipid metabolism and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2002;22:717–726. doi: 10.1161/01.atv.0000015598.86369.04. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka Y., Kume S., Araki S.i., Isshiki K., Chin-Kanasaki M., Sakaguchi M., Sugimoto T., Koya D., Haneda M., Kashiwagi A., et al. Fenofibrate, a PPARalpha agonist, has renoprotective effects in mice by enhancing renal lipolysis. Kidney Int. 2011;79:871–882. doi: 10.1038/ki.2010.530. [DOI] [PubMed] [Google Scholar]
- 35.Zuo N., Zheng X., Liu H., Ma X. Fenofibrate, a PPARalpha agonist, protect proximal tubular cells from albumin-bound fatty acids induced apoptosis via the activation of NF-kB. Int. J. Clin. Exp. Pathol. 2015;8:10653–10661. [PMC free article] [PubMed] [Google Scholar]
- 36.Li S., Nagothu K.K., Desai V., Lee T., Branham W., Moland C., Megyesi J.K., Crew M.D., Portilla D. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-alpha in mice confers protection during acute kidney injury. Kidney Int. 2009;76:1049–1062. doi: 10.1038/ki.2009.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei Z., Peterson J.M., Lei X., Cebotaru L., Wolfgang M.J., Baldeviano G.C., Wong G.W. C1q/TNF-related protein-12 (CTRP12), a novel adipokine that improves insulin sensitivity and glycemic control in mouse models of obesity and diabetes. J. Biol. Chem. 2012;287:10301–10315. doi: 10.1074/jbc.M111.303651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du J., Xu J., Wang X., Liu Y., Zhao X., Zhang H. Reduced serum CTRP12 levels in type 2 diabetes are associated with renal dysfunction. Int. Urol. Nephrol. 2020;52:2321–2327. doi: 10.1007/s11255-020-02591-y. [DOI] [PubMed] [Google Scholar]
- 39.Alipoor E., Salmani M., Yaseri M., Kolahdouz-Mohammadi R., Esteghamati A., Hosseinzadeh-Attar M.J. Role of type 2 diabetes and hemodialysis in serum adipolin concentrations: a preliminary study. Hemodial. Int. 2019;23:472–478. doi: 10.1111/hdi.12787. [DOI] [PubMed] [Google Scholar]
- 40.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fujita K., Maeda N., Sonoda M., Ohashi K., Hibuse T., Nishizawa H., Nishida M., Hiuge A., Kurata A., Kihara S., et al. Adiponectin protects against angiotensin II-induced cardiac fibrosis through activation of PPAR-alpha. Arterioscler. Thromb. Vasc. Biol. 2008;28:863–870. doi: 10.1161/ATVBAHA.107.156687. [DOI] [PubMed] [Google Scholar]
- 42.Hayakawa S., Ohashi K., Shibata R., Kataoka Y., Miyabe M., Enomoto T., Joki Y., Shimizu Y., Kambara T., Uemura Y., et al. Cardiac myocyte-derived follistatin-like 1 prevents renal injury in a subtotal nephrectomy model. J. Am. Soc. Nephrol. 2015;26:636–646. doi: 10.1681/ASN.2014020210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Enomoto T., Shibata R., Ohashi K., Kambara T., Kataoka Y., Uemura Y., Yuasa D., Murohara T., Ouchi N. Regulation of adipolin/CTRP12 cleavage by obesity. Biochem. Biophys. Res. Commun. 2012;428:155–159. doi: 10.1016/j.bbrc.2012.10.031. [DOI] [PubMed] [Google Scholar]
- 44.Enomoto T., Ohashi K., Shibata R., Kambara T., Uemura Y., Yuasa D., Kataoka Y., Miyabe M., Matsuo K., Joki Y., et al. Transcriptional regulation of an insulin-sensitizing adipokine adipolin/CTRP12 in adipocytes by Kruppel-like factor 15. PLoS One. 2013;8:e83183. doi: 10.1371/journal.pone.0083183. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data for evaluating the contributions in the paper are presented in the paper and/or the supplemental information.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.