

Abstract
Genetic liability to substance use disorders can be parsed into loci that confer general or substance-specific addiction risk. We report a multivariate genome-wide association meta-analysis that disaggregates general and substance-specific loci for published summary statistics of problematic alcohol use, problematic tobacco use, cannabis use disorder, and opioid use disorder in a sample of 1,025,550 individuals of European descent and 92,630 individuals of African descent. Nineteen independent SNPs were genome-wide significant (P < 5e-8) for the general addiction risk factor (addiction-rf), which showed high polygenicity. Across ancestries, PDE4B was significant (among other genes), suggesting dopamine regulation as a cross-substance vulnerability. An addiction-rf polygenic risk score was associated with substance use disorders, psychopathologies, somatic conditions, and environments associated with the onset of addictions. Substance-specific loci (9 for alcohol, 32 for tobacco, 5 for cannabis, 1 for opioids) included metabolic and receptor genes. These findings provide insight into genetic risk loci for substance use disorders that could be leveraged as treatment targets.
The lives lost, impacts on individuals and families, and socioeconomic costs attributable to substance use reflect a growing public health crisis1. For example, in the United States, 13.5% of deaths among young adults2 are attributable to alcohol, smoking is the leading risk factor for mortality in males3, and the odds of dying by opioid overdose are greater than those of dying in a motor vehicle crash4. Despite the large impact of substance use and disorders5, there is limited knowledge of the molecular genetic underpinnings of addiction broadly.
Individual SUDs are heritable (h2 ~ 50–60%) and highly polygenic6,7. Recent large-scale genome-wide association studies (GWASs) have identified loci associated with problematic drinking8,9, alcohol use disorder (AUD)10,11, cigarettes smoked per day12, nicotine dependence13,14, cannabis use disorder (CUD)15 and opioid use disorder (OUD)16. Echoing evidence from twin and family studies17, these GWASs show that the genetic architecture of SUDs is characterized by a high degree of commonality18, i.e., a general addiction genetic factor likely conveys vulnerability to multiple substance use disorders. Even after accounting for genetic correlations with non-problematic substance use and with other psychiatrically-relevant traits and disorders, there is considerable variance that is unique to this general risk for addiction, indicating that a liability to addiction reflects more than just the combined genetic liability to substance use and psychopathology18–21.
We conducted a multivariate GWAS of the largest available discovery GWASs of substance use disorders, including problematic alcohol use (PAU: N=435,563; continuous)8, problematic tobacco use (PTU: N=270,120; continuous)12,13,18, cannabis use disorder (CUD: N=384,032, N cases = 14,080)15 and opioid use disorder (OUD: N=79,729, N cases = 10,544 cases)16. First, we partitioned SNP effects into 5 sources of variation: (1) a general addiction risk factor (referred to as addiction-rf), and risks specific to (2) alcohol, (3) nicotine, (4) cannabis and (5) opioids. Second, we identified biological pathways underlying risk for these 5 SUD phenotypes using gene, eQTL, and pathway enrichment analyses. Third, we examined whether currently available medications could potentially be repurposed to treat SUDs22. Fourth, we assessed the association of a polygenic risk score (PRS) derived from addiction-rf with general and specific SUD phenotypes in an independent case/control sample. Fifth, we examined the extent to which genetic liability to addiction-rf is shared with other phenotypes (e.g., physical and mental health outcomes). Sixth, we tested whether the addiction-rf PRS was associated with: (a) medical diagnoses derived from electronic medical records and (b) behavioral phenotypes in largely substance-naïve 9–10-year-old children.
Results
European ancestry GWAS: Addiction risk factor.
As in our prior study18, we estimated a single factor model, scaled the variance of addiction-rf to 1, and allowed loadings to be estimated freely. The single factor model that loaded on Opioid Use Disorder (OUD; Neffective = 30,443), Problematic Alcohol Use (PAU; Neffective=300,789), Problematic Tobacco Use (PTU; Neffective = 270,120), and Cannabis Use Disorder (CUD; Neffective = 46,351) fit the data well (χ2(1) = .017, p = 0.895, CFI = 1, SRMR = 0.002). The latent factor loaded significantly on all indicators (standardized loadings on OUD = 0.83, PAU= 0.58, PTU = 0.36, CUD = 0.93 see Supplemental Figure 1 for full model). The addiction-rf was associated with 19 independent (r2< 0.1) genome-wide significant (GWS) SNPs that mapped to 17 genomic risk loci (Figure 1; Table 1; Supplemental Table 1 for lead SNPs and Supplemental Table 2 for genomic risk loci). The most significant SNP (rs6589386, p=2.9e-12) was intergenic, but closest to DRD2, which was GWS in gene-based analyses (p=7.9e-12; Supplemental Table 3). Further, rs6589386 was an expression quantitative trait locus (eQTL) for DRD2 in the cerebellum, and Hi-C analyses (in FUMA)23 revealed that the variant made chromatin contact with the promoter of the gene (Supplemental Figure 2).
Figure 1. Manhattan Plot of Addiction-rf Genome-Wide Significant Results.
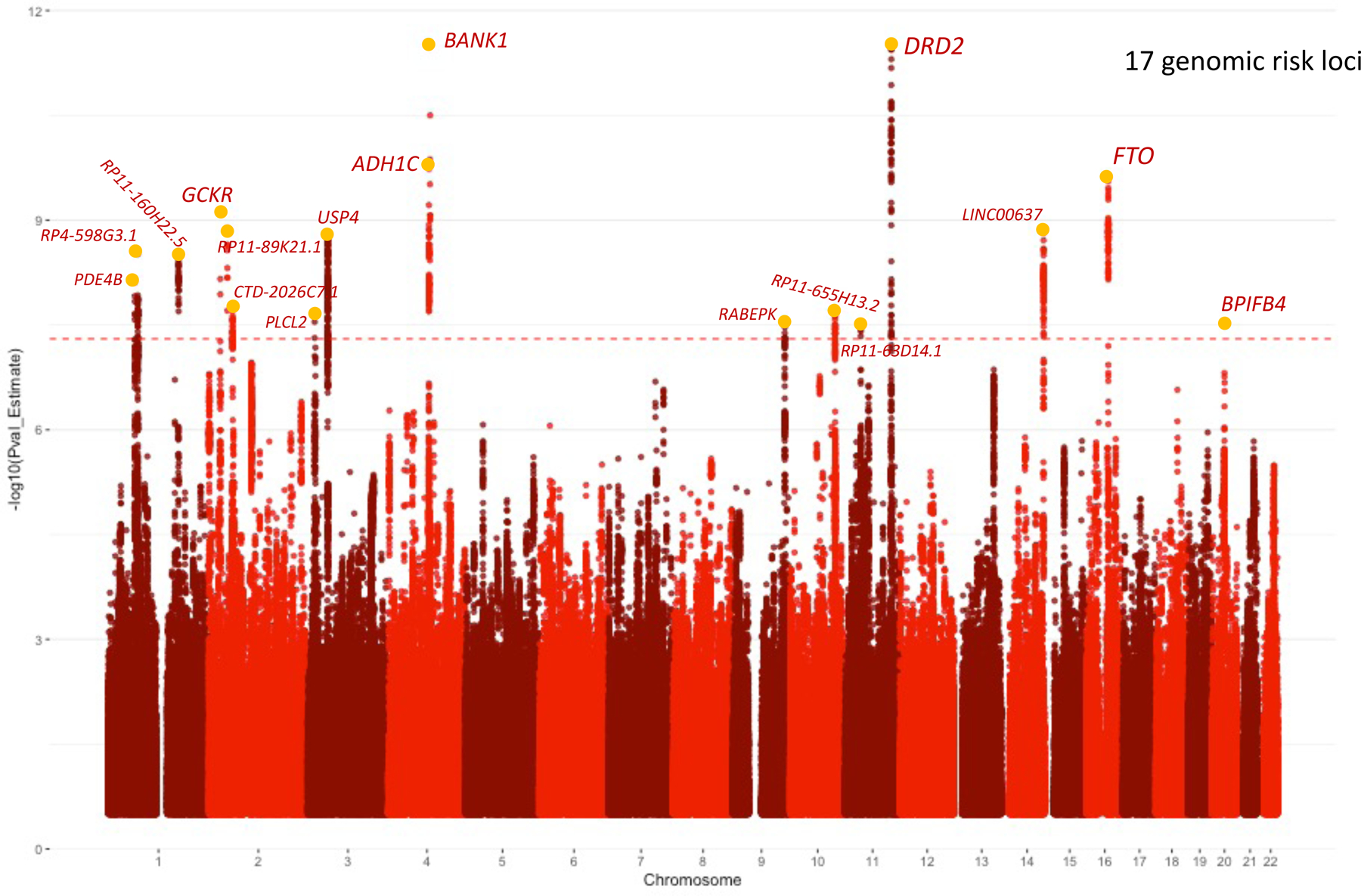
The dotted line represents genome-wide significance at 5e-8. Each SNP peak is annotated with the closest mapped gene from FUMA (Table 1). We have not included all SNPs in the credible set in Table 1, but they are shown in Supplementary Table 4. Significance is set at genome-wide significance Bonferroni correction in a two-sided test (P < 5e-8).
Table 1.
Lead GWAS significant variants
| European Ancestry | African Ancestry | Cross-ancestry | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| rsID | chr | A1 | position | Genomic SEM Beta | GenomicSEM P | SusieR SNPs in Credible set | FUMA GWAS SNPs | ASSET Beta | ASSET p | METASOFT Beta | METASOFT p |
| rs1937455 | 1 | A | 66416939 | 0.013 | 7.74E-09 | 2 | 43 | 0.024 | 3.00E-03 | 0.013 | 0.100 |
| rs1475064 | 1 | A | 73882478 | 0.014 | 3.06E-09 | 3 | 161 | 0.09 | 6.25E-03 | 0.011 | 0.051 |
| rs2860846 | 1 | T | 174075924 | 0.014 | 3.37E-09 | 34 | 36 | NA | NA | NA | NA |
| rs1260326 | 2 | T | 27730940 | −0.015 | 7.60E-10 | 17 | 10 | NA | NA | NA | NA |
| rs570436 | 2 | C | 45142673 | −0.015 | 1.31E-09 | 15 | 8 | NA | NA | NA | NA |
| rs2717054 | 2 | G | 58046683 | −0.014 | 1.97E-08 | 8 | 44 | NA | NA | NA | NA |
| rs55855024 | 3 | C | 16850764 | −0.014 | 2.37E-08 | 8 | 35 | NA | NA | NA | NA |
| rs6795772 | 3 | C | 49365269 | 0.014 | 1.58E-09 | 2 | 265 | NA | NA | NA | NA |
| rs3114045 | 4 | T | 100252560 | −0.023 | 1.85E-10 | 3 | 134 | −0.019 | 0.554 | −0.012 | 2.60E-15 |
| rs1662031 | 4 | A | 100256793 | −0.015 | 1.17e-09 | 3 | 134 | NA | NA | NA | NA |
| rs1813006 | 4 | G | 103001649 | 0.037 | 3.18E-12 | 9 | 11 | NA | NA | NA | NA |
| rs864882 | 9 | C | 127968109 | 0.015 | 3.41E-08 | 2 | 26 | NA | NA | NA | NA |
| rs7073987 | 10 | C | 110565868 | 0.015 | 2.04E-08 | 3 | 61 | NA | NA | NA | NA |
| rs2861190 | 11 | C | 38517941 | −0.014 | 3.66E-08 | 5 | 117 | NA | NA | NA | NA |
| rs17602038 | 11 | T | 113364691 | 0.017 | 6.64e-12 | 2 | 117 | NA | NA | NA | NA |
| rs6589386 | 11 | C | 113443753 | 0.017 | 2.92E-12 | 2 | 65 | NA | NA | NA | NA |
| rs10083370 | 14 | G | 104314182 | 0.014 | 1.53E-09 | 3 | 89 | NA | NA | NA | NA |
| rs28567725 | 16 | T | 53826028 | 0.016 | 2.50E-10 | 5 | 83 | 0.012 | .457 | 0.007 | 6.49E-12 |
| rs2424952 | 20 | T | 31685873 | 0.030 | 3.21E-08 | 2 | 4 | NA | NA | NA | NA |
Lead GWAS variants from European ancestry addiction-rf meta-analysis (in Genomic SEM), African ancestry addiction-rf meta-analysis (based on ASSET), and trans-ancestry meta-analysis (in METASOFT) of the common SNPs underlying PAU, PTU, CUD, and OUD. We generated 4 cross-substance meta-analyses. First, we used a model that leverages genetic overlap across different SUDs via genomic SEM (Figure 1). The GWAS of European ancestry individuals, run with GSEM, forms the primary analysis for most downstream analyses (i.e., TWAS, genetic correlation, PheWAS, genetic causality). The GSEM results for the addiction-rf are shown first (Genomic SEM Beta, Genomic SEM p); the number of credible SNPs in each set (SusieR; r2= .6) with GWAS lead SNPs (from FUMA) are also shown. Next, results for the cross-substance meta-analysis in African ancestry individuals, using ASSET, is shown (ASSET Beta, ASSET p). ASSET splits groups of SNPs into pleiotropic versus non-pleiotropic SNPs, which produces a sparser set of GWAS results (as all SNPs must be pleiotropic to estimate a Beta and P-value), hence, NAs. Finally, to conduct a cross-ancestry meta-analysis, we applied ASSET to the European ancestry sample and then meta-analyzed the European and African ancestry summary data using METASOFT (METASOFT beta, METASOFT p; see Table 2). Direction of all betas corresponds to the effect allele (A1). rsID=rs number, chr=Chromosome, A1=effect allele, position=bp genomic position. Significance is set at Bonferroni corrected genome-wide significance in a two-sided test (P < 5e-8).
Gene-based analyses identified 42 significantly associated genes (Supplemental Table 3); the most significant signals were FTO (p=1.86E-13), DRD2 (p=7.9e-12), and PDE4B (p=9.63E-11). Fine-mapping identified 123 GWS SNPs (of 660 non-independent GWS SNPs) in credible sets as potential causal SNPs based on the posterior probability of inclusion (Supplemental Table 4). Mapping the lead independent SNPs in the credible sets to their nearest gene based on posterior probability of 1, the following SNPs showed the strongest causal potential: rs1937455 (PDE4B), rs3739095 (GTF3C2), rs6718128 (ZNF512), rs4143308 (RP11–89K21.1), rs4953152 (SIX3), rs41335055 (CTD-2026C7.1), rs2678900 (VRK2), rs7620024 (TCTA), rs283412 (ADH1C), rs901406 (BANK1), rs359590 (RABEPK), rs10083370 (LINC00637), rs1477196 (FTO), rs291699 (CDK5RAP1) (Supplemental Table 4 and Figure 1). Pathway analysis of gene-based results revealed several significant GO terms including double-stranded DNA binding (pbonferroni=0.005), sequence-specific double-stranded DNA binding (pbonferroni=0.01), regulation of nervous system development (2 terms: pbonferroni=0.011 – 0.037), and positive regulation of transcription by RNA polymerase (pbonferroni=0.038) (Supplemental Table 6).
European ancestry GWAS: Substance-specific risk.
To identify loci associated with only a single substance (i.e. not pleiotropic), we used ASSET24 (1-sided p < 5e-8). SNPs that were associated at GWS with only a single individual substance (PAU, PTU, CUD, or OUD) were considered substance-specific (e.g., CHRNA5 SNPs were only associated with PTU; Supplemental Figure 3B–E).
Problematic Alcohol Use.
ASSET analyses revealed 9 independent SNPs in 6 loci associated specifically with PAU (Supplemental Figure 3B; Supplemental Table 7 and 8). As expected8, the top signal was rs1229984 in ADH1B (p-value=4.11E-68). Gene-based enrichment analyses also implicated the alcohol dehydrogenase activity zinc dependent pathway (pbonferroni=0.035; Supplemental Table 9).
Problematic Tobacco Use.
PTU was specifically associated with 32 independent SNPs in 12 loci (Supplemental Figure 3C; Supplemental Table 10 and 11). The top SNP was rs10519203 (p=5.12e-267) in HYKK and a robust eQTL for CHRNA5; the signal is likely driven by the CHRNA5 missense variant, rs16969968 (p=2.79e-175), which has previously been linked to tobacco use (r2= .87)12. Several other SNPs were closest to genes encoding nicotinic acetylcholine receptors, including CHRNA4, CHRNB4, CHRNB3, and CHRNB2 (Supplemental Table 10). Gene-based enrichment implicated multiple pathways and gene sets related to nicotinic acetylcholine receptors (Supplemental Table 12). Specific dopamine-related associations were also noted (e.g., PDE1C: rs215600; p=2.35e-18; DBH: rs1108581; p=1.00e-14).
Cannabis Use Disorder.
ASSET identified 5 substance-specific loci for CUD (Supplemental Table 13 and 14), with lead signals at rs11913634 (FAM19A5; p=1.20e-15), rs8104317 (CACNA1A; p=1.17e-13), rs72818514 (ATP10B; p=1.57e-09), and rs11715758 (GNAI2/HYAL3; p=4.84e-08; Supplemental Figure 3D) and rs11778040 (p=1.77e-09; annotated to the GULOP pseudogene). rs11778040 mapped to the previously discovered signal for CUD near CHRNA2 and EPHX215 and is an eQTL for CHRNA2, EPHX2, and CCDC25. CUD-specific signals showed no significant gene-based enrichment.
Opioid Use Disorder.
The only significant substance-specific signal for OUD was the well-characterized16 mu opioid receptor (OPRM1) SNP, rs1799971 (p=1.63e-08; Figure 2E). Gene-based analyses produced no significant findings.
Figure 2. Manhattan Plot of TWAS Results for Addiction-rf.
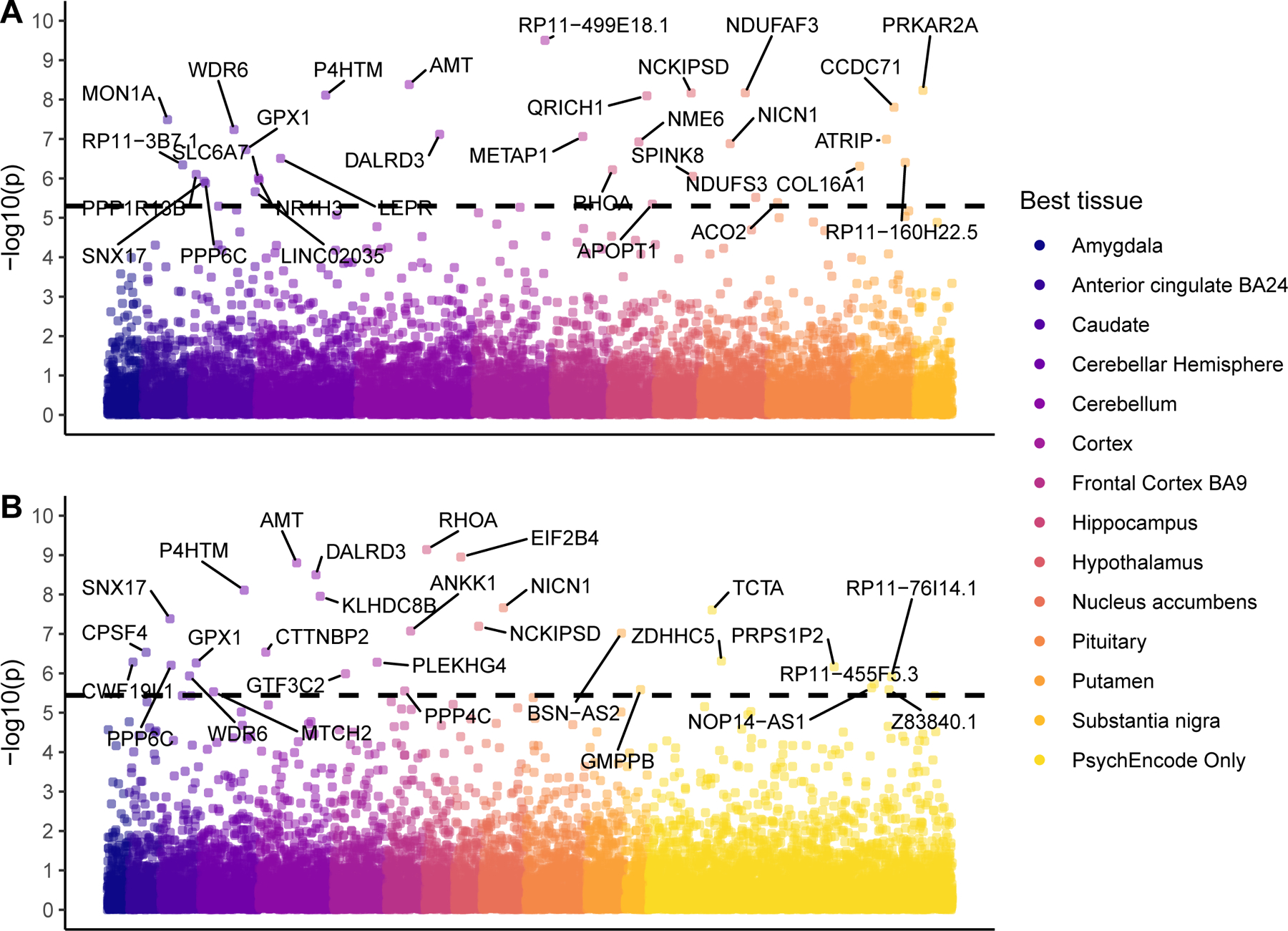
A Transcriptome-Wide Association Study (TWAS) of the addiction-rf, plotted as a Manhattan plot. In Panel A, Analyses were conducted in S-MultiXcan with GTeX v8 data. In Panel B, the analysis was run using S-PredixCan with weights trained from PsychEncode. The y-axis is presented as −log10(p), the color of the data point represents the tissue in which correlation between gene expression and outcome was the highest. The dotted black line represents Bonferroni corrected TWAS significance of a two-sided test (Plot A has 9,944 genes, Bonferroni = 5×10^-6 and the line is at 5.3, Plot B has 13,850 genes, Bonferroni = 3.6×10^-6, line is at 5.4)
African ancestry GWAS: cross-substance risk.
The ASSET-based meta-analysis of GWAS data for AUD (N=82,705)11, tobacco dependence (based on the Fagerstrom Test for Nicotine Dependence, N=9,925)13, CUD (N=9,745)15, and OUD (N=32,088)16 in individuals of African ancestry yielded only 1 GWS pleiotropic SNP, rs77193269 (p=4.92e-8); this SNP was GWS for AUD and tobacco dependence when considering ASSET loci pleiotropic for 2 substances (Supplemental Figure 4B). For substance-specific signals, only one SNP was GWS significant: rs2066702, an ADH1B variant that was alcohol-specific (Supplemental Figure 4A).
Cross-Ancestry GWAS: cross-substance risk.
We found 68 GWS SNPs (Supplemental Figure 5), which are challenging to map to nearby regions or candidate genes due to ancestral differences in LD structure. Table 2 lists the SNP with the lowest GWAS p-value on each chromosome. The most significant association was noted near the FUT2 gene (rs507766, p = 3.47e-19). Many GWS signals were consistent with genes found in the European GWAS, including FTO (rs9928094, p = 6.50e-32) and PDE4B (rs1937439, p = 8.56e-12). We also identified two SNPs in genes which have previously been implicated in SUDs including CADM2 (rs62250713, p=1.00E-18) and FOXP2 (rs4727799, p=3.90E-15), both of which were within r2= 0.6 of lead signals from the European GWAS.
Table 2.
Top results from the Cross-Ancestry meta-analysis in METASOFT
| Chr | SNPID | Gene | A1 | Pheno_Eur | Pheno_AA | Cross_Beta | Cross_P | AA_Beta | AA_P | Eur_Beta | Eur_P |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 19 | rs507766 | FUT2 | T | CUD,PAU | CUD,AUD | −0.009 | 3.47E-19 | 0.007 | 0.914 | −0.004 | 2.66E-04 |
| 16 | rs9928094 | FTO | A | PTU,OUD,PAU | OUD,AUD | 0.006 | 6.50E-32 | 0.011 | 0.321 | 0.006 | 1.63E-11 |
| 2 | rs472140 | NA | C | PTU,PAU | CUD,PTU | −0.007 | 3.79E-43 | −0.02 | 0.722 | −0.007 | 7.66E-13 |
| 4 | rs6846266 | NA | A | PTU,PAU | OUD,AUD | −0.005 | 6.05E-23 | −0.011 | 0.317 | −0.005 | 3.61E-06 |
| 3 | rs62250713 | CADM2 | G | CUD,PAU | CUD,AUD | 0.009 | 1.00E-18 | 0.018 | 0.268 | 0.009 | 6.97E-11 |
| 1 | rs784601 | NA | G | CUD,PTU,PAU | CUD,PTU | −0.004 | 3.57E-15 | −0.014 | 0.519 | −0.004 | 2.47E-05 |
| 7 | rs4727799 | FOXP2 | C | CUD,PTU | PTU,OUD | −0.004 | 3.90E-15 | −0.024 | 0.752 | −0.004 | 2.74E-06 |
| 22 | rs6002381 | NA | T | CUD,PTU,PAU | PTU,AUD | −0.007 | 5.23E-12 | −0.022 | 0.551 | −0.007 | 1.13E-06 |
| 12 | rs1497253 | IRAG2 | A | CUD,PTU,OUD,PAU | CUD,PTU,AUD | −0.004 | 4.01E-15 | 0.007 | 0.914 | −0.004 | 2.66E-04 |
| 14 | rs7147171 | PP1R13B | G | PTU,PAU | PTU,OUD,AUD | −0.007 | 3.71E-12 | −0.01 | 0.37 | −0.007 | 3.09E-10 |
| 15 | rs35175834 | SEMA6D | G | CUD,PTU,PAU | CUD,PTU,OUD,AUD | −0.007 | 5.84E-12 | 0.007 | 0.959 | −0.007 | 3.51E-07 |
| 20 | rs293553 | C20orf112 | A | PTU,OUD | PTU,OUD | −0.009 | 2.03E-09 | −0.018 | 0.24 | −0.009 | 1.43E-07 |
| 9 | rs7033815 | SCAI | G | PTU,OUD | CUD,OUD,AUD | 0.006 | 2.71E-09 | 0.018 | 0.171 | 0.006 | 2.83E-06 |
| 17 | rs587880 | FBXL20 | G | OUD,PAU | OUD,AUD | −0.009 | 1.07E-09 | −0.014 | 0.119 | −0.009 | 1.36E-05 |
| 18 | rs4996482 | RP11–397A1 6.1 | C | CUD,PTU,OUD | CUD,OUT | 0.006 | 3.54E-09 | 0.028 | 0.006 | 0.261 | 4.18E-07 |
| 5 | rs7708715 | TMEM161B-AS1,CTC-498M16.2 | C | CUD,PTU | CUD,OUD,AUD | 0.003 | 3.59E-09 | 0.013 | 0.34 | 0.003 | 6.16E-05 |
| 8 | rs2321459 | LEKR1 | T | PTU,PAU | PTU,OUD | −0.006 | 3.68E-09 | −0.009 | 0.974 | −0.006 | 4.51E-05 |
| 6 | rs62394558 | NA | G | CUD,PTU | CUD,OUD | −0.003 | 3.89E-09 | −0.02 | 0.962 | −0.003 | 8.00E-03 |
| 1* | rs1937439 | PDE4B | A | CUD,PTU,PAU | CUD,AUD | 0.007 | 5.18E-12 | 0.025 | 0.173 | 0.007 | 4.54e-10 |
| 4* | rs10031172 | BANK1 | T | CUD,OUD,PAU | Tob,AUD | −0.006 | 1.93E-09 | −0.014 | 0.090 | −0.006 | 8.03e-05 |
Note. Chr = Chromosome, SNPID = snp rsID, Gene is closest gene, A1 is effect allele. Pheno_AA lists the phenotypes associated with the SNP in the African Ancestry Sample GWAS. Pheno_Eur lists the phenotypes associated with the SNP in the European sample, Cross_Beta is the cross-ancestry Beta from the METASOFT random effects meta-analysis. Cross_P is the P from the P-value of the cross ancestry meta-analysis. AA_Beta is the transformation of the OR from ASSET when run in the African American Sample. AA_P is the pvalue from ASSET. Eur_Beta is the transformation of the Odds Ratio (to a Beta) from ASSET when run in the European sample. Eur_P is the P-value from ASSET. Significance is set at Bonferroni corrected genome-wide significance in a two-sided test (P < 5e-8).
Polygenic architecture and power.
We used a likelihood estimation-based approach to calculate the probability distribution of effect sizes for the addiction-rf and each of the constituent input GWASs (i.e., PAU, PTU, CUD, OUD) to examine relative differences in polygenicity (see Methods). The addiction-rf showed a narrow distribution of small effect sizes with almost all values falling close to 0. On the other hand, the original substance specific GWASs were characterized by larger average effects (see Supplemental Figure 6 for shape of probability density distribution). For example, only 26% of genes associated with Problematic Tobacco Use showed effect sizes as close to the mean threshold of the probability distribution as addiction-rf did. These findings suggest that the addiction-rf is characterized by greater polygenicity than specific substances.
Transcriptome-wide Association and Drug Repurposing.
A transcriptome-wide association analysis25 of the addiction-rf using multiple tissues simultaneously from GTEx in MetaXcan (See Methods) identified 35 genes in 13 brain regions (Figure 2; Supplemental Table 15). Gene-set analysis using FUMA23 revealed that these genes were enriched for gene sets and pathways related to neural cells and T-cell processes (Supplemental Figure 7; Supplemental Table 16). Transcriptome-wide analyses with PsychEncode data found 29 significantly associated genes and 11 genes that overlapped with those identified in the GTEx analysis (AMT, DALRD3, GPX1, KLHDC8B, NCKIPSD, NICN1, P4HTM, PPP6C, RHOA, SNX17, WDR6) (Figure 2). Linking transcriptome-wide patterns from our GTEx MetaXcan analysis to perturbagens that cross the blood-brain barrier from the Library of Integrated Network-Based Cellular Signatures (LINCS)26 database, identified 104 medications approved by the U. S. Food and Drug Administration that reverse the addiction-rf transcriptional profile (Supplemental Table 17). Medications currently used to treat SUDs (e.g., varenicline, smoking cessation), other psychiatric conditions (e.g., reboxetine, depression) as well as those used for other purposes (e.g., mifepristone, pregnancy termination; currently under clinical investigation for treating AUD; riluzole, amyotrophic lateral sclerosis) were identified.
LD Score Regression and Genetic Correlations.
After Bonferroni correction (p < .05/1,547 =3.20e-5), the addiction-rf was genetically correlated with 251 phenotypes (Figure 3; Supplemental Table 18). Notably, 38 of these (15%) included somatic diseases linked to specific substances (e.g., lung cancer with tobacco, and pain-related conditions with opioids). As expected, we found significant genetic correlations between the addiction-rf and serious, trans-diagnostic psychopathological behaviors, including suicide attempt (rg=0.62, p=2.89e-33) and self-medication (e.g., using non-prescribed drugs or alcohol for anxiety, rg=0.64, p=3.18e-6). The addiction-rf was correlated with, but remained separable based on 95% confidence intervals (rg=0.63 ±.037, p=2.33e-231), from an externalizing factor27 that included similar indices of problematic substance use and behavioral measures.
Figure 3. PheWAS of Genetic Correlations using MASSIVE.
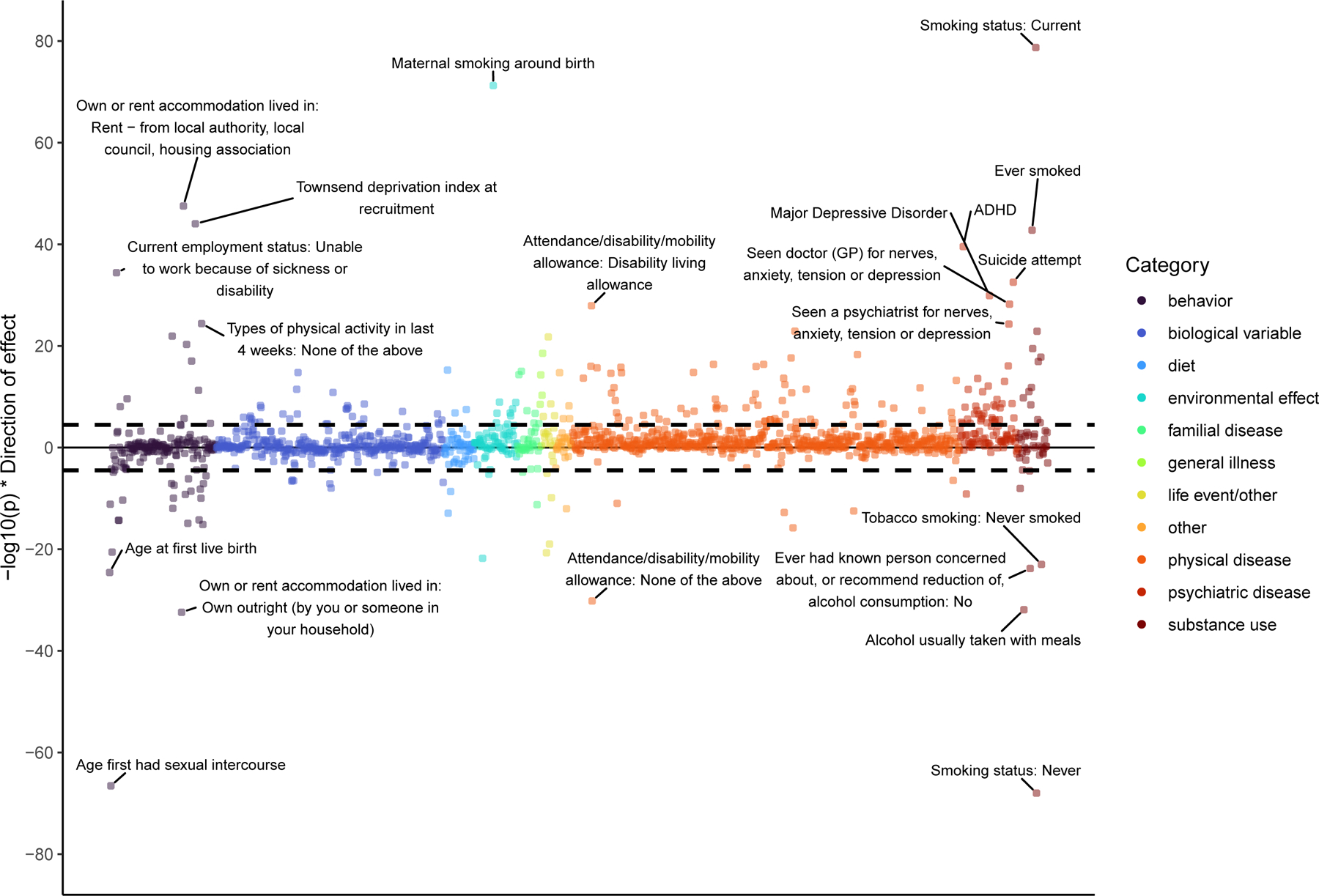
Genetic correlations between 1,547 traits and the addiction-rf, calculated in MASSIVE, mapped by their statistical significance (-log10(p) on the y-axis), and broad category. The top 20 correlations are annotated. The black dashed line represents Bonferroni significance for association of a two-sided test (pbon= .05/1,574 = 3.232e-05).
Latent Causal Variable Analysis.
We used MASSIVE to conduct Latent Causal Variable (LCV)28 analyses on the same 251 phenotypes significant in our genetic correlation analyses (Supplemental Table 19). Post multiple corrections (p = .05/250 = 1.98e-4), the only significant causal processes were medication codes. Specifically, addiction-rf was estimated as a potential risk factor for “Medication for cholesterol, blood pressure or diabetes: Cholesterol lowering medication” (gcp = −.739(.078), p =4.51e21), “treatment/medication code: atorvastatin” (gcp = −.373(.050), p = 7.93e-14) and “Medication for cholesterol, blood pressure, diabetes, or take exogenous hormones: Cholesterol lowering medication” (gcp= −.315(.071), p = 8.3128e-06). The negative gcp estimates suggest a causal role of addiction on physical disease (addiction-rf is trait 2 in all instances).
Polygenic risk score (PRS) analyses.
PRS analyses with measures addiction and SUDs.
In the independent Yale-Penn 3 sample16 (EUR n=1,986), the addiction-rf PRS was significantly associated with a phenotypic factor loading on several SUDs (p<.001), polysubstance use disorder (2 or more substance use disorders; p<2e-16), and each individual SUD (DSM-IV29: AUD, CUD, OUD, tobacco dependence or TD, and cocaine use disorder [CoUD] (all ps < 7.71E-06; Figure 4; Supplemental Table 20). Nagelkerke’s R2 values ranged from 2.4% for CUD to 5.9% for TD, and 6.6% for a phenotype similar to addiction-rf that represents phenotypic commonality across AUD, CUD, OUD, TD and CoUD. Odds ratios varied from 1.41 for CUD to 1.73 for OUD.
Figure 4. Polygenic Risk Score Prediction in Yale-Penn.
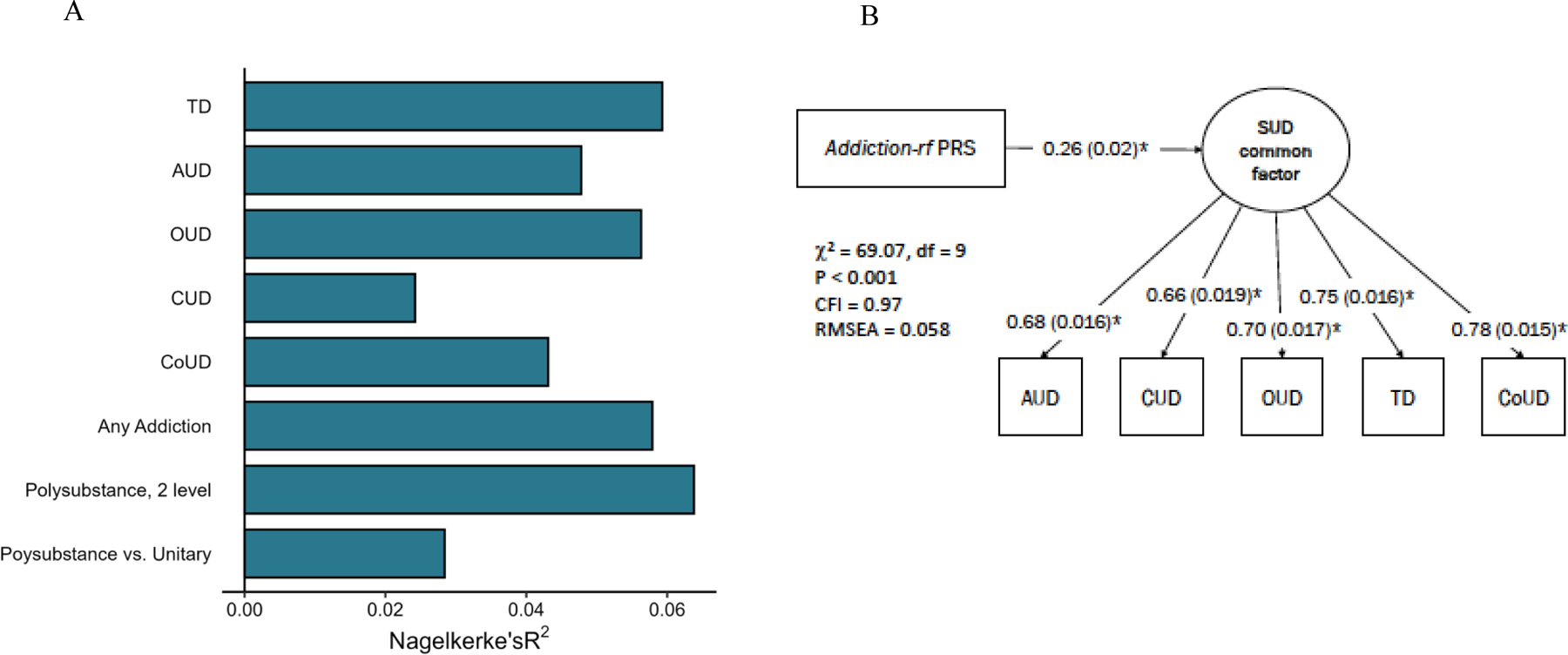
(A) Polygenic risk score (PRS) of the addiction-rf predicts lifetime alcohol (AUD), cannabis (CUD), opioid (OUD), tobacco (TD), and cocaine (CoUD) use disorders, and variables representing more than one lifetime substance use disorder diagnosis vs no SUDs diagnosis (Polysubstance Use Disorder, 2 level), more than one lifetime diagnosis vs. one lifetime diagnosis (polysubstance vs. unitary), as well as any substance use disorder diagnosis (Any Addiction) in an independent sample (Yale Penn 3; N=1,986 individuals of European genetic ancestry). (B) The addiction-rf PRS was associated with a comparable phenotypic substance use disorders (SUD) common factor in the Yale-Penn sample. Controlling for age, sex and 10 genetic principal components of ancestry, all path estimates are fully standardized. Estimates were significant at p < .001 of a two-sided test (LAVAAN does not report P-values lower than .001).
PheWAS in Electronic Health Record data.
In the BioVU sample (EUR N=66,914)30, the addiction-rf PRS was associated with SUDs (p=3.31e-29; Supplemental Figure 8), various types of substance involvement [e.g., Tobacco Use Disorder p=9.79e-24, alcoholism (so named in EHR, we note the term “alcohol use disorder” is more appropriate), p=1.12e-21), chronic airway obstruction (p=4.99e-10)], and several psychiatric disorders, with the strongest being bipolar disorder (p=2.44e-11). Controlling for any SUD diagnosis to account for causal effects found similar associations with alcoholism, mood disorders, respiratory disease, and heart disease (Supplemental Figure 9A). Controlling for tobacco dependence diagnosis did not significantly modify associations (Supplemental Figure 9B).
Behavioral Phenotypes in Substance-Naïve Children.
Among 4,491 substance-naïve children aged 9–10 years who completed the baseline session of the ABCD Study®31, the addiction-rf PRS was positively correlated (after Bonferroni correction) with BAS fun-seeking (an aspect of externalizing behavior; p=2.09e-05), family history of drug addiction (p=7.04e-07), family history of hospitalization due to mental health concerns (including suicidal behavior; p=4.64e-06), childhood externalizing behaviors (e.g., antisocial; p=1.62e-05), childhood thought problems (p=3.51e-06), sleep duration (p=1.52e-07), parental externalizing and substance use behaviors (e.g., prenatal tobacco exposure; p=2.87e-11), maternal pregnancy characteristics (e.g., urinary tract infection during pregnancy, p=2.70e-7), socio-economic disadvantage (e.g., child’s neighborhood deprivation; p= 9.84e-07), and child’s likeliness to play sports (p=2.80e-06) (Supplemental Figure 10; Supplemental Table 21 for results from all phenotypes and Supplementary Table 23 for measure inclusion criteria).
Discussion
We found 17 genomic loci significantly associated with addiction-rf, and 47 substance-specific loci. Post hoc fine-mapping, annotation, and exploratory drug repurposing analyses highlight the potential therapeutic relevance of the discovered loci. The addiction-rf PRS was associated with many medical conditions characterized by high morbidity and mortality rates, including psychiatric illnesses, self-harming behaviors, and somatic diseases that could be consequences of chronic substance use (e.g., chronic airway obstruction) or precursors to heavy substance use (e.g., chronic pain). Finally, in a sample of drug-naïve children, the addiction-rf PRS was correlated with parental substance use problems and externalizing behavior.
Our analyses suggest that the regulation/modulation of dopaminergic genes, rather than variation in dopaminergic genes themselves, is central to general addiction liability. DRD2 was the top gene signal, which was mapped via chromatin refolding, suggesting a regulatory mechanism. The role of striatal dopamine in positive drug reinforcement is well established32. DRD2 plays a role in reward sensitivity and may also be central to executive functioning33 – the interplay of reward and cognition is likely relevant throughout the course of addiction. These complementary observations reinforce the role of dopamine signaling in addiction32.
Other regulatory effects on dopaminergic pathways were supported by the signal at PDE4B, which has been implicated in prior GWASs of disinhibition traits27. The phosphodiesterase (PDE) system has been proposed as a dopaminergic regulation mechanism34. Further, animal studies suggest that the PDE system is associated with down-regulation of drug-seeking behaviors across opioids, alcohol, and psychostimulants35. Notably, The PDE4B antagonist, ibudilast, has been shown to reduce heavy drinking among patients with AUD36,37 and also shown to reduce inflammation in methamphetamine use disorder38, and was significant in our drug repurposing analysis.
The addiction-rf PRS was associated with general and specific SUD liabilities in an independent sample. The addiction-rf PRS predicted ~6% of OUD variance, which is nearly half the total SNP-heritability of OUD16. The addiction-rf PRS also predicted variance in cocaine use disorder (CoUD); as CoUD was not included in the development of the addiction-rf (due to lack of a well-powered CoUD GWAS), these findings highlight the generalizability of the addiction-rf beyond alcohol, tobacco, cannabis and opioids.
Substance-specific genetic signals fell primarily into three broad categories: drug-specific metabolism (e.g., ADH1B for PAU), drug receptors (e.g., CHRNA5 for PTU, OPRM1 for OUD), and general neurotransmitter mechanisms (e.g., CACNA1A for CUD). Surprisingly, even after accounting for the addiction-rf, dopaminergic genes (DBH and PDE1C in particular) were implicated in substance-specific effects for tobacco (PTU). On the other hand, CUD-specific genes did not include well-studied receptor targets (e.g., CNR1) or metabolic mechanisms (e.g., Cytochrome P450 genes).
The current addiction-rf is distinct from recent genetic factors21,27,39 that were based upon analyses of SUDs with other substance use, psychiatric, and behavioral traits. We focus on SUDs rather than measures of substance use or other externalizing traits, which prior data indicate have differing etiologies and relationships with psychiatric health9,40,41. Our study also parses substance-general (i.e., addiction-rf) and substance-specific loci. This approach distinguishes the addiction-rf from other genetic factors that have include substance use measures. For example, despite genetic overlap between the addiction-rf and a recent index of externalizing behaviors (rG=0.63)27, a significant portion of the variance in addiction-rf was distinct.
Our analyses highlight the robust genetic association of addiction-rf with serious mental and somatic illness. The addiction-rf PRS was more strongly associated with using drugs to cope with internalizing disorder symptoms (anxiety, depression; rg=0.60–0.62) than with the individual psychiatric traits and disorders themselves (rg=0.3), suggesting that genetic correlations between SUDs and mood disorders may partially be attributable to a predisposition to use substances to alleviate negative mood states (“self-medication”)42.
The PheWAS also provided insight into potentially complex mechanisms of genetic liability to environmental pathways of risk. In addition to indices of socio-economic status (SES), the addiction-rf was correlated with maternal tobacco smoking during pregnancy and ADHD, in line with evidence that effects ascribed to the prenatal environment may also be mediated by the inheritance of risk loci43,44. The addiction-rf PRS was associated with a family history of serious mental illness, which likely represents an amalgam of genetic and environmental vulnerability45. Finally, disability and SES were also associated with polygenic risk, further supporting the association between environmental risk factors and common genetic effects on SUD liability9,41,46
This study has limitations. First, our GWAS in individuals of African-ancestry had few discoveries, underscoring the need for systematic data collection on SUDs in globally representative populations. Still, we chose to analyze and present these data as their exclusion only furthers disparities in genetic discoveries. Second, although we discovered many loci, they accounted for only a small proportion of the total variance. More samples, particularly those in diverse populations, and the integration of rarer variants are needed to discover the biological pathways that fall below genome-wide significance or are missed in GWAS. Finally, despite interesting associations between our PRS and SUDs, our findings do not apply to prognostication of future disease risk.
Conclusion
A common and highly polygenic genetic architecture underlies multiple SUDs, a finding that merits integration into medical knowledge on addictions.
METHODS
Summary statistics from each SUD-related GWAS
Summary statistics from the largest available discovery GWAS were used to represent genetic risk for each construct. These include four measures of problematic substance use or substance use disorder (SUD): 1) Problematic Alcohol Use8, 2) Problematic Tobacco Use12,13,18, 3) Cannabis Use Disorder15, 4) Opioid Use Disorder16 (described below). All GWAS summary statistics were filtered to retain variants with minor allele frequencies > 0.01 and INFO score > 0.90 for GSCAN12 and PGC15 and INFO score > 0.70 for the MVP8,16.
For the current cross-trait GWAS, we maintained the same QC metrics and only analyzed SNPs that were present in all four input GWAS, i.e., variants that passed QC thresholds at all levels, resulting in 3,513,381 SNPs in samples of European ancestry and 5,303,643 SNPs in samples of African American ancestry. The LD scores used for the genomic structural equation modeling (GenomicSEM)47 were estimated in the European ancestry samples only using the 1000 Genomes European data48. We restricted analyses to HapMap3 SNPs49 as these tend to be well imputed and produce accurate estimates of heritability. We used the effective N, which was estimated for each GWAS50. For traits with a binary distribution, the effective sample size for an equivalently powered case-control study under a 50–50 case control balance was estimated using the equation: Neffective = 4/((1/Ncase) + (1/Ncontrol))51. Continuous and quasi-continuous traits used the given N or if from MTAG, the equation Neffective = ((Z/β)2)/(2*MAF*(1-MAF))8 to approximate an equivalently powered GWAS of a single trait. Effective Ns ranged from 46,351 (CUD) to 300,789 (PAU) and are described for each substance-specific GWAS in the Results below. Individual GWAS details can be found in the Online Methods.
Genome-wide Analyses in European Ancestry
We conducted a GWAS of a unidimensional addiction risk factor (addiction-rf) underlying the genetic covariance among PAU, PTU, CUD and OUD by applying GenomicSEM47 to these European ancestry summary statistics. GenomicSEM conducts genome-wide association analyses in two stages. First, a multivariate version of LD score regression is used to estimate the genetic covariance matrix among all GWAS phenotypes, which is then combined with each individual SNP to calculate SNP-specific genetic covariance matrices. Second, these matrices are then used to estimate the SEM using the lavaan package in R52. Variable and unknown extents of sample overlap across contributing GWASs are automatically accounted for in the estimation procedure. The unifactor model fit the data well53 [X2(1) = .017, p = 0.895, CFI = 1, SRMR = 0.002; residual r = 0.51, p = 0.016; Supplemental Figure 1; see also our prior work18 and Online Methods).
As the sample size of summary data derived from African Americans (n range = 9,835–56,648) was not sufficient for LD Score54 analyses, we used ASSET24 to conduct the addiction-rf GWAS, as opposed to GenomicSEM, as described in the subsequent ASSET section below.
ASSET: Trans-ancestry Analyses
Association analysis based on Subsets (ASSET)24 was used to identify pleiotropic (i.e., SNPs that show associations with more than one SUD) and substance-specific (i.e., SNPs only associated with a single SUD) SNPs within the European and African American ancestry samples in addition to GenomicSEM in Europeans). ASSET was used in our African American ancestry addiction-rf GWAS as the sample size was not sufficient for the Genomic SEM approach used in the European addiction-rf GWAS. As a result, there are important differences in the primary addiction-rf GWAS and GWAS run in ASSET. First, the ASSET-based addiction-rf GWAS contain SNPs that may influence 2, 3, or all 4 individual SUDs, while the GenomicSEM-based addiction-rf GWAS in European ancestry includes SNPs associated with a common factor across included SUDs. ASSET identified pleiotropic SNPs in the European ancestry sample to facilitate method-consistent cross-ancestry meta-analysis GWAS (see subsequent Cross-Ancestry Meta-analysis section below) and cross validate primary GenomicSEM results.
ASSET does not leverage the genetic correlation to identify variants of interest (as GenomicSEM does); instead, subset searches scaffold effects into pleiotropic and non-pleiotropic variants based on effect size and standard error derivations that estimate the degree to which the SNP-trait association is due to pooled effects across the phenotypes, vs. a single phenotype driving variant association. Loci were designated as substance-specific when they were only significantly associated with only 1 SUD. As ASSET does not automatically account for sample overlap; we used LDSC-estimated genetic correlations to adjust for overlap within the European ancestry ASSET covariance term.
Cross-Ancestry Meta-analysis
We conducted a cross-ancestry meta-analysis of ASSET-derived (to maintain analytic consistency) European and African ancestry addiction-rf summary statistics. First, SNPs with evidence of SUD pleiotropy (i.e., effects on 2 SUDs, 3 SUDs, or all 4 SUDs, including different sets of SUDs in each ancestry) in both ancestral groups were extracted. SNPs with evidence of cross-ancestral heterogeneity (i.e., Cochran’s Q statistic <5e-8) were removed, leaving 317,447 SNPs. A meta-analysis in METASOFT55 using a random-effects meta-analysis with ancestry group as a random effect was used to identify cross-ancestral effects. We report the random effects BETA and p-value as cross-ancestry effects.
Specific Genetics in European Ancestry Individuals
To validate substance-specific SNPs, we used ASSET for discovery of these variants and, in the European ancestry GWAS, also examined Q-SNP results derived from GenomicSEM. Q-SNP14 indexes violation of the null hypothesis that a SNP acts on a trait entirely through a common factor (e.g., addiction-rf). For example, if a SNP has a particular effect on one SUD trait (such as SNPs in CHRNA5 influencing PTU), then it should have significant Q-SNP statistics because it violates the assumption that its effect on PTU is via the addiction-rf. We identified Q-SNPs by estimating the association between each SNP and the addiction-rf. Then, we fit a model where the SNP predicted the indicators underlying the addiction-rf, i.e., PAU, PTU, CUD, OUD. We compared the Chi-square difference statistic between the two models; those with significant decrement of fit (X2 for Δdf = 4) in the model where the SNP predicted the addiction-rf alone relative to the SNP predicting the indicators themselves was considered a significant Q-SNP above genome-wide significance (i.e. Q p < 5e-8). SNPs with significant Q-SNP statistics were removed from the addiction-rf summary statistics for all post-hoc analyses, including fine-mapping, gene-based tests, transcriptome-wide association analyses, LD score genetic correlations and polygenic risk score analyses.
Q-SNP analysis also identified several SNPs that appeared to be specific to a single substance. However, as Q-SNP cannot be used for precise identification of substance-specific (trait-specific) SNPs, we relied on ASSET analyses (with a 1-sided p-value), to identify the subset of SNPs with effects (at genome-wide significance, p<5e-8) limited to only 1 SUD-related trait (e.g., PAU-specific). It is worth noting that the ASSET analysis used to determine specific SNPSs is the same analysis that went in the cross-ancestry results, except pleotropic loci were retained for the cross-ancestry analysis and specific SNPs for the analysis described here.
Post-hoc analyses of European ancestry GWAS results
Estimation of expected SNP effect sizes
We estimated the distribution of genetic effect-sizes of addiction-rf (Genomic SEM) and the 4 input GWAS (PAU, PTU, CUD, OUD) using Genetic effect-size distribution inference from summary-level data (GENESIS). GENESIS is a likelihood-based approach56. In this approach, GWAS summary statistics and an external panel of linkage disequilibrium (in our case, the 1000 Genomes Phase 3 reference panel) are used to estimate a projected distribution of SNP effect sizes. A flexible normal mixture model based on the number of tagged SNPs and LD scores is estimated. A 3-component model is fit, where SNP effect sizes are estimated to belong to one of three components based on bins of effect sizes (large, medium, small). If the distribution of SNPs is multivariate normal, the estimation of the SNPs with large and medium effect sizes can be done via their independent effect sizes. The third component represents SNPs with null and small effect sizes, and these should follow a similar distribution. Therefore, this model reweights SNPs and generates a projected distribution of effect sizes, and from this projection, we can draw conclusions about the distribution of effect sizes54.
Biological Characterization
FUMA23 was used for post-hoc bioinformatic analyses of our five GWAS (i.e., the addiction-rf (from Genomic SEM), PAU-specific, PTU-specific, CUD-specific, OUD-specific (from ASSET) loci) in European ancestry samples and to determine lead and independent variants. Within FUMA, gene-based tests and gene-set enrichment were conducted via MAGMA57; gene annotation, and identification of SNP-to-gene associations via expression quantitative trait loci (eQTLs) and/or chromatin interactions (via Hi-C data) in PsychEncode58 and Roadmap Epigenomics tissues for Prefrontal cortex, hippocampus, ventricles, and neural progenitor cells59,60. For each specific SUD GWAS run using ASSET, the distribution of p-values included all non-pleiotropic SNPs (i.e., SNPs only associated with a single SUD, n SNP CUD-Specific = 312,661, n SNP PTU-Specific = 560,983, n SNP PAU-Specific = 193,647, n SNP OUD-Specific = 425,665). Additional details on the FUMA analyses are available in Online Methods.
Fine-mapping with SusieR.
We fine-mapped the association statistics of the four phenotypes (addiction-rf, PAU-specific, PTU-specific, CUD-specific; OUD-specific only had one significant loci, and that loci has known mechanism of effect) that had more than 1 genome-wide significant SNP in a 1 MB region around the lead SNP to determine the 95% credible set using susieR61 with at most 10 causal variants (this analysis reduces the total number of SNPs at a lead genome-wide signal to those that can credibly be considered as causal SNPs). The credible set reports include the likelihood of being a causal variant; the marginal posterior inclusion probability (PIP) ranges from 0 to 1, with values closer to 1 being most likely causal.
Transcriptome-Wide Association Analysis.
We conducted two transcriptome-wide analyses. First, we used MetaXcan/S-MultiXcan38 to conduct a cross-tissue analysis of all brain tissues in the GTEx v8 data37. S-MultiXcan returns a broad Z-score across all tissues in the model, along with the top and lowest scores at each tissue. S-MultiXcan combines information across individual tissues, which improves the power for discovery by reducing the multiple correction burden. It also produces z-score and p-values for top-associated tissues. Second, we also used S-Predixcan62 to predict transcription using the weights trained from on psychiatric cases vs controls transcriptional differences from the frontal and temporal cortex using the PsychEncode63 dataset. As these data were very densely sampled for psychiatrically relevant traits, it serves to complement the relatively healthy GTEx sample.
Drug Repurposing
Our signature matching technique used data from the Library of Integrated Network-based Cellular Signatures (LINCs) L1000 database64. The LINCs L1000 database catalogues in vitro gene expression profiles (signatures) from thousands of compounds in over 80 human cell lines (level 5 data from phase I: GSE92742 and phase II: GSE70138)26. We selected compounds that were currently FDA approved or in clinical trials (via https://clue.io/repurposing#download-data; updated 3/24/20). Our analyses included signatures of 829 chemical compounds (590 FDA approved, 239 in clinical trials) in five neuronal cell-lines (NEU, NPC, MNEU.E, NPC.CAS9 and NPC.TAK), a total of 3,897 signatures were present as not all compounds were tested in all cell lines in the LINCs dataset.
In vitro medication signatures were matched with addiction-rf signatures from the transcriptome-wide association analyses (conducted using S-MultiXcan)25,62 via multi-level meta-regression. We computed weighted (by its proportion of heritability explained (h2MULTI-XCAN)) Pearson correlations between transcriptome-wide brain associations and in vitro L1000 compound signatures using the metafor package in R65. We treated each L1000 compound as a fixed effect incorporating the effect size (rweighted) and sampling variability (se2r_weighted) from all signatures of a compound (e.g., across all time points, cell lines, doses). Analyses included brain region as a random effect to estimate tissue specific heterogeneity. Only genes that were Bonferroni significant in the S-PrediXcan(transcriptome-wide correction = .05/14,389 = 3.48e-06) were entered into the model. We only report those perturbagens that were associated after Bonferroni correction (perturbagen correction = .05/3,897 = 1.28e-05).
Polygenic Risk Score Analyses in Yale-Penn
Yale-Penn.
The Yale-Penn16,66 sample includes 11,332 genotyped and phenotyped individuals recruited across three phases (i.e., Yale-Penn 1, Yale-Penn 2, and Yale-Penn 3) based on the time of recruitment and genotyping array used. All cohorts were ascertained via recruitment at substance use treatment centers or targeted advertisements for genetic studies of cocaine, opioid, and alcohol dependence, resulting in a sample highly enriched for problematic substance use, as well as control subjects and relatives. All participants were assessed using the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA)67. Analyses based on Yale-Penn 1 and 2 have been published previously66, and were used in the discovery sample of the present study. Here, we used data from Yale-Penn 316 for replication analyses and as a target sample for polygenic risk score analyses; the Yale-Penn 3 sample is independent from our discovery GWASs. Yale-Penn 3 comprises 3,026 genotyped and phenotyped Americans of European (EUR; N=1,986) and African (AFR; N=1,040) ancestry passing standard quality control. Genotyping was performed at the Gelernter lab at Yale University using the Illumina Multi-ethnic Global Array containing 1,779,819 markers, followed by genotype imputation using Minimac368 and the Haplotype Reference Consortium reference panel69 as implemented on the Michigan imputation server (https://imputationserver.sph.umich.edu).
For the present analysis, only Yale-Penn 3 EUR subjects (N=1,986) were included. DSM-IV29 substance abuse and dependence diagnoses (combined as abuse or dependence to represent use disorder) based on SSADDA assessments were used to determine case and control status for alcohol use disorder (AUD), cannabis use disorder (CUD), cocaine use disorder (CoUD), tobacco dependence (TD), and opioid use disorder (OUD). Of the 1,986 EUR subjects, 42.4% met criteria for AUD (N=843), 25.9% met criteria for CUD (N=515), 25.3% met criteria for CoUD (N=503), 31% met criteria for TD (N=615), and 22.6% met criteria for OUD (N=448). The mean age of Yale-Penn 3 EUR subjects is 41.5 (SE=15.1) and 51.5% are female (N=1,023).
We calculated the addiction-rf PRS using the PRS-cs auto approach70. This method assumes a general distribution of effect sizes across the genome, and then reweights SNPs based on this assumption, their effect size in the original GWAS, and their linkage disequilibrium (LD); weights for every SNP were then summed to create a final score. PRS were associated with phenotypes (OUD, TD, CUD, AUD, CoUD) in Yale-Penn 3 via a logistic regression controlling for first 10 PCs, age, sex and age by sex. PRS were scaled to unit variance. These logistic regression analyses were also examined for the following contrasts: 1) Those with any SUD (n=985) vs those with no SUD (n=1,001), to represent Any SUD; 2) Those with ≥2 SUDs (n=729) vs those with <2 (including 0) SUDs (n=1,257) to represent Polysubstance Use Disorder (PSUD); and 3) Those with ≥2 SUDs (n=729) vs those with 1 SUD (n=256) to represent PSUD within those with SUD. The association between the addiction-rf PRS and the SUD Common Factor was estimated with lavaan52 where a common factor loaded on the 5 disorders.
Genetic Correlations and Latent Causal Variable modeling
To examine phenotypes that were genetically correlated with the addiction-rf, we calculated genetic correlations using LD score regression54,71 through the MASSIVE pipeline72, which conducts LD score regression13,46 and Latent Causal Variable Analysis28 on 1,547 summary statistics for various phenotypic traits, including a mixture of ICD codes and self-reported traits from the UK Biobank and publicly available meta-analyses from GWAS consortiums.
Phenome-wide Association Studies (PheWAS)
PheWAS in adult samples.
As MASSIVE includes a fairly sparse set of diagnoses (not all ICD codes are available) for genetic correlation analyses, we conducted additional and theoretically relevant PheWASs using the addiction-rf PRS. We used electronic health records (EHR) data for 66,914 genotyped individuals of European-ancestry from the Vanderbilt University Medical Center biobank (BioVU)30. BioVU is a repository of leftover blood samples (~240,000 samples) from clinical testing, that are sequenced, de-identified, and linked to clinical and demographic data. Genotyping and quality control of this sample have been described elsewhere30. The addiction-rf PRS was used to predict 1,335 diseases in a logistic regression model, controlling for median age on record, reported gender, and first 10 genetic ancestry PCs. For an individual to be considered a case, they were required to have two separate ICD codes for the index phenotype, and each phenotype needed at least 100 cases to be included in the analysis. A Bonferroni-corrected phenome-wide significance threshold of 0.05/1335=3.7E-05 was used73.
ABCD PheWAS of phenotypes collected in childhood.
To identify phenotypes that were associated with the addiction-rf before the onset of regular substance use, we used data from the Adolescent Brain and Cognitive Development (ABCD®) Study release 2.0 for genome data and 3.0 for phenotypes to conduct a phenome-wide association analysis of behavioral, social, and environmental phenotypes in adolescence. The ABCD Study is an ongoing multi-site longitudinal study of child health and development (see Online Methods for details)31,74. Children (N=11,875; including twins and siblings) ages 8.9–11 were recruited from 22 sites across the United States to complete the ABCD Study baseline assessment. We restricted our sample to participants of genomically-confirmed European ancestry (based on principal components) who were not missing any covariate measures (N=4,490).
PRS were generated using the PRS-cs software package70 consistent with our other (i.e., Yale-Penn 4, BioVU) PRS analyses described above. Associations between addiction-rf PRS and phenotypes were estimated using mixed-effects models in the lme475 package in R. PRS were scaled to unit variance. Family ID and site were included as random effects to account for non-independence of measurement associated with relatedness and scanner/site. We controlled for the first 10 ancestry principal components, age, sex, age by sex. We used a Bonferroni-corrected phenome-wide significance threshold of 0.05/1480= 3.38e-05; all results are presented in the Supplemental Table 21.
Supplementary Material
Acknowledgements
The Authors would like to thank Cold Harbor Labroatory for posting a preprint of this work on MedArchive (https://www.medrxiv.org/). The authors thank Million Veteran Program (MVP) staff, researchers, and volunteers, who have contributed to MVP, and especially participants who previously served their country in the military and now generously agreed to enroll in the study.
Funding:
K01 AA030083 (ASH), T32 DA007261 (ASH), DA54869 (AA, JG, HE), R01 DA54750 (AA, RB), K02 DA32573 (AA), R21 AA027827 (RB), U01 DA055367 (RB), K01 DA51759 (ECJ), K23 MH121792 (NK), DP1 DA54394 (SS-R), T32 MH014276 (GAP), R01 AA027522 (AE), F31 AA029934 (SEP), R01 MH120219 (EMTD, ADG), RF1 AG073593 (EMTD, ADG), P30 AG066614 (EMTD), P2CHD042849 (EMTD), R33 DA047527 (RP, GAP), T32 AA028259 (JDD)
Footnotes
Disclosure: Dr. Kranzler is a member of advisory boards for Dicerna Pharmaceuticals and Sophrosyne Pharmaceuticals, and Enthion Pharmaceuticals; a consultant to Sobrera Pharmaceuticals; and a member of the American Society of Clinical Psychopharmacology’s Alcohol Clinical Trials Initiative, which was supported in the last three years by Alkermes, Dicerna, Ethypharm, Lundbeck, Mitsubishi, and Otsuka. Drs. Kranzler and Gelernter hold U..S. Patent 10900,082: “Genotype-guided dosing of opioid agonists,” issued 26 January 2021. The remaining authors declare not conflicts of interest.
Data Availability
The MVP summary statistics were obtained via an approved dbGaP application (phs001672.v4.p1). For details on the MVP, see https://www.research.va.gov/mvp/ and Gaziano, J.M. et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J Clin Epidemiol 70, 214–23 (2016)). This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration, and was supported by the Veterans Administration (VA) Cooperative Studies Program (CSP) award #G002.
Publicly available data were also downloaded from the psychiatric genomics consortium: https://www.med.unc.edu/pgc/, and the GSCAN consortium: https://conservancy.umn.edu/handle/11299/201564
The dataset(s) used for the BioVU analyses described were obtained from Vanderbilt University Medical Center’s biorepository which is supported by numerous sources: institutional funding, private agencies, and federal grants. These include the NIH funded Shared Instrumentation Grant S10RR025141; and CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711; and additional funding sources listed at https://victr.vumc.org/biovu-funding/
Data from Yale-Penn 1 are available through dbGAP accession no phs000425.v1.p1 including 1889 African-American (AA) subjects and 1020 European-American (EA) subjects. Yale-Penn 1 data are also available through dbGAP accession no phs000952.v1.p1 including 1,531 AA subjects and 1,339 self-reported EA subjects. Summary statistics for all Yale-Penn data are available on request by contacting joel.gelernter@yale.edu
References
- 1.Degenhardt L et al. The impact of cohort substance use upon likelihood of transitioning through stages of alcohol and cannabis use and use disorder: Findings from the Australian National Survey on Mental Health and Wellbeing. Drug Alcohol Rev 37, 546–556 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peacock A et al. Global statistics on alcohol, tobacco and illicit drug use: 2017 status report. Addiction 113, 1905–1926 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Reitsma MB et al. Spatial, temporal, and demographic patterns in prevalence of smoking tobacco use and initiation among young people in 204 countries and territories, 1990–2019. Lancet Public Health 6, e472–e481 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Council NS Odds of Dying. in Injury Facts ((accessed dec 3 2021), https://injuryfacts.nsc.org/all-injuries/preventable-death-overview/odds-of-dying/, 2019). [Google Scholar]
- 5.Vanyukov MM An Eternal Epidemic: 1. Why substance use problems persist. https://psyarxiv.com/tkm5u/ (2021). [Google Scholar]
- 6.Deak JD & Johnson EC Genetics of substance use disorders: a review. Psychol Med 51, 2189–2200 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gelernter J & Polimanti R Genetics of substance use disorders in the era of big data. Nat Rev Genet (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou H et al. Genome-wide meta-analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat Neurosci 23, 809–818 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mallard TT et al. Item-Level Genome-Wide Association Study of the Alcohol Use Disorders Identification Test in Three Population-Based Cohorts. Am J Psychiatry, appiajp202020091390 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walters RK et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat Neurosci 21, 1656–1669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kranzler HR et al. Genome-wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat Commun 10, 1499 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu M et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 51, 237–244 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hancock DB et al. Genome-wide association study across European and African American ancestries identifies a SNP in DNMT3B contributing to nicotine dependence. Mol Psychiatry 23, 1911–1919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quach BC et al. Expanding the genetic architecture of nicotine dependence and its shared genetics with multiple traits. Nat Commun 11, 5562 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson EC et al. A large-scale genome-wide association study meta-analysis of cannabis use disorder. Lancet Psychiatry 7, 1032–1045 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou H et al. Association of OPRM1 Functional Coding Variant With Opioid Use Disorder: A Genome-Wide Association Study. JAMA Psychiatry 77, 1072–1080 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kendler KS et al. Recent advances in the genetic epidemiology and molecular genetics of substance use disorders. Nat Neurosci 15, 181–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatoum AS et al. The addiction risk factor: A unitary genetic vulnerability characterizes substance use disorders and their associations with common correlates. Neuropsychopharmacology (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdellaoui A, Smit DJA, van den Brink W, Denys D & Verweij KJH Genomic relationships across psychiatric disorders including substance use disorders. Drug Alcohol Depend 220, 108535 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Waldman ID, Poore HE, Luningham JM & Yang J Testing structural models of psychopathology at the genomic level. World Psychiatry 19, 350–359 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grotzinger AD et al. Genetic architecture of 11 major psychiatric disorders at biobehavioral, functional genomic and molecular genetic levels of analysis. Nat Genet 54, 548–559 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duan Q et al. LINCS Canvas Browser: interactive web app to query, browse and interrogate LINCS L1000 gene expression signatures. Nucleic Acids Res 42, W449–60 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat Commun 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhattacharjee S et al. A subset-based approach improves power and interpretation for the combined analysis of genetic association studies of heterogeneous traits. Am J Hum Genet 90, 821–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barbeira AN et al. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet 15, e1007889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian A et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 171, 1437–1452 e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karlsson Linner R et al. Multivariate analysis of 1.5 million people identifies genetic associations with traits related to self-regulation and addiction. Nat Neurosci (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Connor LJ & Price AL Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat Genet 50, 1728–1734 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Association AP Diagnostic and Statistical Manual (DSM-IV), (American Psychiatric Press Inc, Washington D.C., 1994). [Google Scholar]
- 30.Roden DM et al. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin Pharmacol Ther 84, 362–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Volkow ND et al. The conception of the ABCD study: From substance use to a broad NIH collaboration. Dev Cogn Neurosci 32, 4–7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wise RA & Robble MA Dopamine and Addiction. Annu Rev Psychol 71, 79–106 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Hatoum AS et al. Genome-Wide Association Study Shows that Executive Functioning Is Influenced by GABAergic Processes and Is a Neurocognitive Genetic Correlate of Psychiatric Disorders. Biological Psychiatry Pre-proof(2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Snyder GL & Vanover KE PDE Inhibitors for the Treatment of Schizophrenia. Adv Neurobiol 17, 385–409 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Olsen CM & Liu QS Phosphodiesterase 4 inhibitors and drugs of abuse: current knowledge and therapeutic opportunities. Front Biol (Beijing) 11, 376–386 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burnette EM, Ray LA, Irwin MR & Grodin EN Ibudilast attenuates alcohol cue-elicited frontostriatal functional connectivity in alcohol use disorder. Alcohol Clin Exp Res 45, 2017–2028 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grodin EN et al. Ibudilast, a neuroimmune modulator, reduces heavy drinking and alcohol cue-elicited neural activation: a randomized trial. Transl Psychiatry 11, 355 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li MJ, Briones MS, Heinzerling KG, Kalmin MM & Shoptaw SJ Ibudilast attenuates peripheral inflammatory effects of methamphetamine in patients with methamphetamine use disorder. Drug Alcohol Depend 206, 107776 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schoeler T et al. Novel biological insights into the common heritable liability to substance involvement: a multivariate genome-wide association study. Biological Psychiatry Pre-proof(2022). [Google Scholar]
- 40.Sanchez-Roige S, Palmer AA & Clarke TK Recent Efforts to Dissect the Genetic Basis of Alcohol Use and Abuse. Biol Psychiatry 87, 609–618 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marees AT et al. Genetic correlates of socio-economic status influence the pattern of shared heritability across mental health traits. Nat Hum Behav 5, 1065–1073 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khantzian EJ Addiction as a self-regulation disorder and the role of self-medication. Addiction 108, 668–9 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Thapar A & Rice F Family-Based Designs that Disentangle Inherited Factors from Pre- and Postnatal Environmental Exposures: In Vitro Fertilization, Discordant Sibling Pairs, Maternal versus Paternal Comparisons, and Adoption Designs. Cold Spring Harb Perspect Med 11(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haan E et al. Prenatal smoking, alcohol and caffeine exposure and maternal reported ADHD symptoms in childhood: triangulation of evidence using negative control and polygenic risk score analyses. Addiction (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cornelis MC, Zaitlen N, Hu FB, Kraft P & Price AL Genetic and environmental components of family history in type 2 diabetes. Hum Genet 134, 259–67 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wendt FR et al. Multivariate genome-wide analysis of education, socioeconomic status and brain phenome. Nat Hum Behav 5, 482–496 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grotzinger AD et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat Hum Behav 3, 513–525 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Genomes Project C et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.International HapMap C et al. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52–8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grotzinger AD, de la Fuente J, Nivard MG & Tucker-Drob EM Pervasive downward bias in estimates of liability scale heritability in GWAS meta-analysis: A simple solution. 10.1101/2021.09.22.21263909 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Turley P et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat Genet 50, 229–237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosseel Y Lavaan: An R Package for structural equation modeling. J. Stat Software 48, 1–36 (2012). [Google Scholar]
- 53.Hu L & Bentler PM Cutoff criteria for fit indexes in covariance strcuture analysis: Conventional criteria versus new alternatives. Structural Equation Modeling 6, 1 (1999). [Google Scholar]
- 54.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 47, 291–5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han B & Eskin E Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am J Hum Genet 88, 586–98 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang Y, Qi G, Park JH & Chatterjee N Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat Genet 50, 1318–1326 (2018). [DOI] [PubMed] [Google Scholar]
- 57.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Psych EC et al. The PsychENCODE project. Nat Neurosci 18, 1707–12 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roadmap Epigenomics C et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–30 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ernst J & Kellis M ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9, 215–6 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang G, Sarkar A, Carbonetto P & Stephens M A simple new approach to variable selection in regression, with application to genetic fine mapping. Royal Statistical Society, Series B 10.1111/rssb.12388(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barbeira AN et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat Commun 9, 1825 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gandal MJ et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu C et al. Compound signature detection on LINCS L1000 big data. Mol Biosyst 11, 714–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Viechtbauer W Accounting for heterogeneity via random-effects models and moderator analyses in meta-analysis. Zeitschrift für Psychologie / Journal of Psychology 215, 104–121 (2007). [Google Scholar]
- 66.Sherva R et al. Genome-wide Association Study of Cannabis Dependence Severity, Novel Risk Variants, and Shared Genetic Risks. JAMA Psychiatry 73, 472–80 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pierucci-Lagha A et al. Diagnostic reliability of the Semi-structured Assessment for Drug Dependence and Alcoholism (SSADDA). Drug Alcohol Depend 80, 303–12 (2005). [DOI] [PubMed] [Google Scholar]
- 68.Das S et al. Next-generation genotype imputation service and methods. Nat Genet 48, 1284–1287 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McCarthy S et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48, 1279–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ge T, Chen CY, Ni Y, Feng YA & Smoller JW Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun 10, 1776 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 47, 1236–41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cuellar-Partida G et al. Complex-Traits Genetics Virtual Lab: A community-driven web platform for post-GWAS analyses. 10.1101/518027 (2021). [DOI] [Google Scholar]
- 73.Carroll RJ, Bastarache L & Denny JC R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics 30, 2375–6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lisdahl KM et al. Substance use patterns in 9–10 year olds: Baseline findings from the adolescent brain cognitive development (ABCD) study. Drug Alcohol Depend 227, 108946 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bates D, Machler M, Bolker BM & Walker SC Fitting linear mixed-effects models using lme4. J. Stat Software 67, 1–48 (2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MVP summary statistics were obtained via an approved dbGaP application (phs001672.v4.p1). For details on the MVP, see https://www.research.va.gov/mvp/ and Gaziano, J.M. et al. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. J Clin Epidemiol 70, 214–23 (2016)). This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration, and was supported by the Veterans Administration (VA) Cooperative Studies Program (CSP) award #G002.
Publicly available data were also downloaded from the psychiatric genomics consortium: https://www.med.unc.edu/pgc/, and the GSCAN consortium: https://conservancy.umn.edu/handle/11299/201564
The dataset(s) used for the BioVU analyses described were obtained from Vanderbilt University Medical Center’s biorepository which is supported by numerous sources: institutional funding, private agencies, and federal grants. These include the NIH funded Shared Instrumentation Grant S10RR025141; and CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, R01HD074711; and additional funding sources listed at https://victr.vumc.org/biovu-funding/
Data from Yale-Penn 1 are available through dbGAP accession no phs000425.v1.p1 including 1889 African-American (AA) subjects and 1020 European-American (EA) subjects. Yale-Penn 1 data are also available through dbGAP accession no phs000952.v1.p1 including 1,531 AA subjects and 1,339 self-reported EA subjects. Summary statistics for all Yale-Penn data are available on request by contacting joel.gelernter@yale.edu