

Abstract
Vaccination has become an increasingly attractive strategy for protecting against antibiotic-resistant infections. Nanovaccines based on the outer membrane from Gram-negative bacteria are appealing due to their multiantigenic nature and inherent immunogenicity. Here, we develop cellular nanodiscs made of bacterial outer membrane (OM-NDs), as a platform for antibacterial vaccination. Using Pseudomonas aeruginosa as a model pathogen, the resulting OM-NDs can effectively interact with antigen-presenting cells, exhibiting accelerated uptake and an improved capacity for immune stimulation. With their small size, the OM-NDs are also capable of efficiently transporting to the lymph nodes after in vivo administration. As a result, the nanovaccine is effective at eliciting potent humoral and cellular immune responses against P. aeruginosa. In a murine model of pneumonia, immunization with OM-NDs confers strong protection against subsequent lung infection, resulting in improved survival, reduced bacterial loads, and alleviation of immune overactivation. Overall, this report illustrates the advantages of OM-NDs, which can be readily generalized to other pathogens and may be applied towards other biomedical applications.
Keywords: biomimetic nanotechnology, nanodisc, outer membrane vesicle, antibacterial vaccine, Gram-negative infection, Pseudomonas aeruginosa
Graphical Abstract
The increasing incidence of drug-resistant pathogens can be attributed mainly to the widespread overuse of antibiotics.1–2 In order to reduce reliance on antimicrobial drugs, an increased emphasis has been placed on the development of vaccines that augment the immune system’s innate ability to resist infection.3–4 Various strategies for enhancing vaccination efficacy have been investigated.5–7 Among them, the use of nanomaterial-based systems has been of particular interest due to their beneficial properties, including improved uptake by antigen-presenting cells and the ability to co-deliver antigens with immunostimulatory adjuvants.8–10 More recently, cell membrane-based nanoparticles have become increasingly popular due to their ability to mimic many of the properties associated with living cells.11–12 These platforms are fabricated using natural cell membrane, which can be derived from a wide range of cell types such as red blood cells, platelets, white blood cells, and cancer cells, among others.13–16 The resulting biomimetic nanoparticles are inherently multiantigenic, and they exhibit unique properties based on the source of the membrane material. Biomimetic nanoparticles fabricated using the outer membrane from Gram-negative bacteria have been explored as vaccine formulations.7, 17–18 These nanoparticles mimic the antigenic profile of the original bacteria and contain pathogen-associated molecular patterns that provide self-adjuvanting properties for effective immune stimulation.19
In order to achieve vaccination efficacy, the immune system must be properly mobilized. The localization of antigens and adjuvants to the lymph nodes, which are secondary lymphoid organs that play a major role in fighting infection, is an important consideration in vaccine development.20–21 Lymph node targeting is a task for which nanovaccines are well-suited, as their small size enables effective lymphatic drainage after administration.22 Nanodiscs, which oftentimes have diameters in the sub-20 nm range, have proven to be a particularly attractive platform for this type of application.23–24 These unique nanoparticles consist of a discoidal membrane bilayer that is encircled by a scaffold protein or polymer. Traditionally, nanodiscs have been employed for studying the biochemistry and biophysics of cell membrane,25 although more recently a greater emphasis has been placed on their biomedical utility.26 Nanodiscs readily transport via the lymphatic system following subcutaneous administration, enabling the codelivery antigen and adjuvant payloads to immune cells that reside within draining lymph nodes. As such, nanodiscs have proven their effectiveness as vaccine candidates, potently stimulating adaptive immunity in an antigen-specific manner.23
Herein, to the best of our knowledge, we report on the first example of cellular nanodiscs fabricated directly from natural bacterial outer membrane (denoted ‘OM-NDs’) instead of synthetic lipid bilayers for antibacterial vaccination (Figure 1a). The platform builds on the advantages of previous generation nanovaccines by presenting naturally immunostimulatory and multiantigenic material in a format that is readily processed by the immune system. Pseudomonas aeruginosa, a common Gram-negative bacterium responsible for multidrug-resistant infections in hospitals, was selected as a model pathogen.27 OM-NDs were fabricated by incubating P. aeruginosa outer membrane vesicles (OMVs) with a styrene-maleic acid (SMA) copolymer, an amphiphilic membrane scaffold co-polymer.28 With their small size, it was observed that OM-NDs more effectively localized to the draining lymph nodes after subcutaneous administration compared to traditional OMVs. Moreover, OM-NDs induced dendritic cell maturation and elicited pathogen-specific antibody production. This ultimately prolonged the survival of vaccinated mice when challenged intratracheally with live P. aeruginosa, and this protection was also correlated with reduced local inflammation in the lungs. Overall, this work demonstrates that nanodiscs fabricated from bacterial outer membrane can be employed as vaccines to protect against bacterial infections. In the future, it is envisioned that this approach can be easily generalized to other pathogens.
Figure 1.
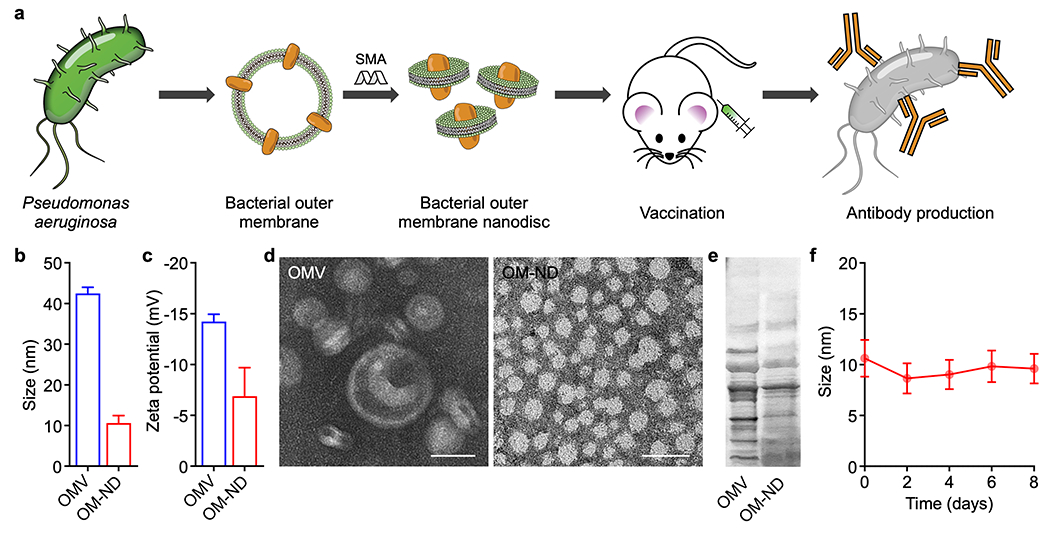
Preparation and characterization of OM-NDs. (a) Bacterial OMVs are collected from Pseudomonas aeruginosa and incubated with styrene-maleic acid (SMA). The resulting OM-ND formulation can be used as a nanovaccine to protect against bacterial infection. (b,c) Size (b) and zeta potential (c) of free OMVs and OM-NDs (n = 3, mean + SD). (d) Transmission electron microscopy images of free OMVs (left) and OM-NDs (right) after negative staining (scale bars: 20 nm). (e) Protein profile of OMVs and OM-NDs after gel electrophoresis. (f) Size of OM-NDs in PBS over time (n = 3, mean ± SD).
RESULTS AND DISCUSSION
To fabricate OM-NDs, bacterial OMVs were first collected from the P. aeruginosa strain PAO1 following a previously established protocol.29 The OMVs were then vortexed with SMA to form OM-NDs, and any large non-solubilized membrane material was removed by ultracentrifugation. It has been suggested that SMA copolymers facilitate nanodisc formation via the intercalation of their phenyl groups between the acyl chains of membrane lipids and the interaction of their carboxyl groups with membrane lipid heads.30 Finally, centrifugal ultrafiltration devices were used to remove the excess SMA and condense the OM-NDs. Free OMVs in their native form, which contain a variety of immunomodulatory pathogen-associated molecular patterns,31 were employed as a control group throughout this work. After the fabrication process, approximately 50% of the inputted outer membrane protein was incorporated into the OM-ND formulation. With an average diameter of 10 nm, OM-NDs were significantly smaller than free OMVs, which averaged just over 40 nm when measured by dynamic light scattering (Figure 1b). With the addition of SMA, the zeta potential of OM-NDs was slightly less negative compared with free OMVs (Figure 1c). The morphology of OM-NDs was characterized by transmission electron microscopy, revealing a relatively homogeneous population of spherical particles consistent with previous reports on SMA-stabilized nanodiscs32 (Figure 1d). In comparison, visualization of free OMVs showed a more heterogeneous population of vesicular structures. Following gel electrophoresis, it was confirmed that OM-NDs had a similar protein profile compared to the original OMVs, with reductions in some lower molecular weight proteins likely resulting from the purification steps during synthesis (Figure 1e). This suggested that many P. aeruginosa antigens that could serve as potential vaccine targets were retained through the fabrication procedure. The retention of immunostimulatory molecules, including potent Toll-like receptor agonists, was also confirmed (Figure S1). In terms of stability, OM-NDs maintained their size in phosphate-buffered saline (PBS) for at least 8 days (Figure 1f).
After confirming the successful fabrication of OM-NDs, we evaluated their interaction with antigen-presenting cells in vitro. First, OM-NDs were labeled with a far-red fluorophore in order to track uptake by DC2.4 dendritic cells over time using flow cytometry (Figure 2a). Compared with dye-labeled free OMVs, OM-NDs were taken up by the cells at a considerably faster rate. The results were corroborated using fluorescence microscopy, where significantly more uptake was observed for DC2.4 cells incubated with OM-NDs for 3 h (Figure 2b). Whereas the uptake of OMVs was mediated predominantly by various endocytic pathways, macropinocytosis also played a role in the uptake of OM-NDs (Figure S2). Subsequently, we evaluated the immunostimulatory properties of OM-NDs. For these experiments, nanodiscs fabricated using red blood cell (RBC) membrane (denoted ‘RBC-NDs’) were employed as a negative control group (Figure S3). First, it was confirmed in a preliminary study that OM-NDs could activate DC2.4 dendritic cells, as determined by cytokine secretion, in a dose-dependent manner (Figure S4). Interleukin 6 (IL-6) levels became significantly elevated and saturated when incubating with 0.5 μg/mL of OM-NDs for 2 days. At this dosage, which was used for further study, no cytotoxic effects were observed on the DC2.4 cells (Figure 2c). Subsequently, we assessed the in vitro production of different proinflammatory cytokines, including IL-1β, IL-6, IL-12p40, and tumor necrosis factor α (TNFα), by DC2.4 (Figure 2d–g). There was significant elevation in the levels for all the cytokines after OM-ND treatment.
Figure 2.
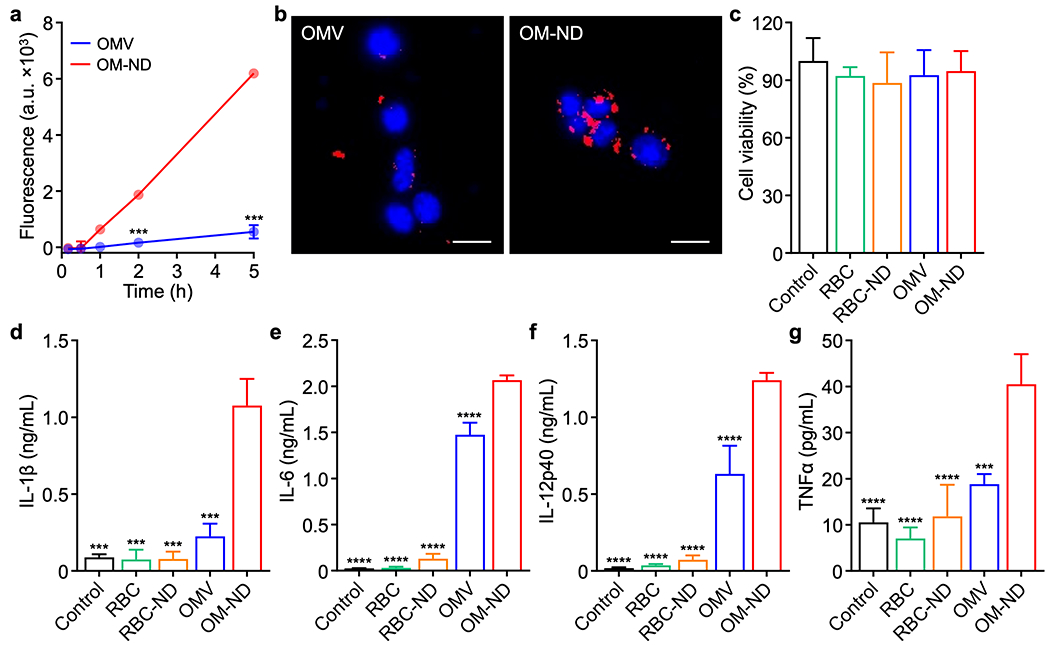
In vitro uptake and immune stimulation. (a) Uptake of dye-labeled free OMVs and OM-NDs by DC2.4 cells over time (n = 3, mean ± SD). (b) Fluorescence visualization of free OMV (left) and OM-ND (right) uptake after 3 h of incubation with DC2.4 cells (scale bars: 20 μm; red: bacterial membrane, blue: nuclei). (c) Viability of DC2.4 cells after 2 days of incubation with OM-NDs or various control samples (n = 3, mean + SD). (d-g) Concentration of IL-1β (d), IL-6 (e), IL-12p40 (f), and TNFα (g) secreted by DC2.4 after 2 days of incubation with OM-NDs or control samples (n = 3, mean + SD). ***p < 0.001 and ****p < 0.0001 (compared to OM-ND); Student’s t-test (a) or one-way ANOVA (d-g).
Notably, OM-NDs were significantly better than free OMVs at stimulating the dendritic cells. RBC vesicles and RBC-NDs were lowly immunostimulatory as expected. We concurrently investigated the surface expression of dendritic cell maturation markers (Figure S5). Consistent with the cytokine results, the expression of both CD40 and CD86 was significantly higher after treatment with OM-NDs versus free OMVs or the RBC-derived formulations. Overall, the data demonstrated that the transformation of bacterial membrane into nanodiscs resulted in better engagement with dendritic cells, suggesting that OM-NDs could be used as an effective nanovaccine.
We next proceeded to assess the immune-priming capabilities of OM-NDs in vivo. First, dye-labeled OMVs or OM-NDs were administered into mice via hock injection in order to study lymph node transport (Figure 3a,b). At 6 h, the draining popliteal lymph node was collected for analysis. Ex vivo imaging revealed that there was considerably more accumulation of OM-NDs compared with free OMVs. Quantitative analysis confirmed these observations, as there was an approximately 5-fold increase in fluorescent signal. Improved delivery was also observed on a cellular level, with antigen-presenting cells such as macrophages and dendritic cells exhibiting a higher degree of uptake (Figure S6). The enhanced lymph node accumulation of OM-NDs highlighted the benefit of their small size, enabling more efficient lymphatic drainage after administration. To evaluate the safety of OM-NDs, blood samples were collected one day after administration, and all serum chemistry parameters and cell counts were consistent with those of control mice (Figure S7). Next, mice were immunized via the hock, and the draining popliteal lymph nodes were extracted after 48 h to evaluate in vivo dendritic cell maturation. There was increased expression of CD40 and CD86 among lymph node cells derived from mice vaccinated with OM-NDs compared to those vaccinated with free OMVs (Figure 3c,d). Overall, the results here showing the higher immunostimulatory activity of OM-NDs after in vivo administration correlated well with the trends that were observed in vitro.
Figure 3.
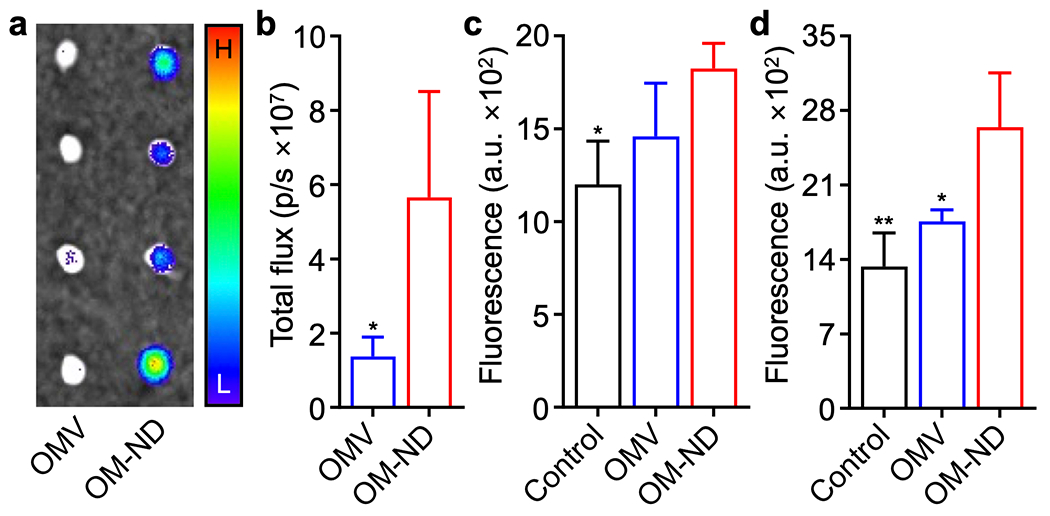
In vivo transport and immune stimulation. (a) Ex vivo fluorescent imaging of the draining popliteal lymph nodes at 6 h after the administration of dye-labeled free OMVs and OM-NDs (H: high signal, L: low signal). (b) Quantification of fluorescence in the popliteal lymph nodes at 6 h after the administration of dye-labeled free OMVs and OM-NDs (n = 3, mean + SD). (c,d) Expression of the maturation markers CD40 (c) and CD86 (d) by CD11c+F4/80− dendritic cells in the popliteal lymph nodes of mice 48 h after vaccination with free OMVs or OM-NDs (n = 4, mean + SD). *p < 0.05 and **p < 0.01 (compared to OM-ND); Student’s t-test (b) or one-way ANOVA (c,d).
To characterize adaptive immune response, we first evaluated the ability of OM-NDs to elicit antibodies specific to P. aeruginosa outer membrane. In a preliminary study to assess the appropriate vaccination schedule, mice were immunized with the nanoformulation via hock injection on day 0 followed by booster doses on days 7 and 14, and anti-outer membrane immunoglobulin G (IgG) titers in the serum were tracked over time (Figure S8). Antibody levels in mice vaccinated with OM-NDs were significantly higher compared with OMV-vaccinated mice; anti-outer membrane titers began to elevate on day 7 and saturated by day 14. These results were corroborated in a second study in which only a single booster was given on day 7, and detailed characterization of IgG production was performed on day 14 (Figure 4a–d). For all subtypes that were analyzed, including IgG1, IgG2b, and IgG3, the OM-ND formulation significantly outperformed free OMVs. The elevated production of IgG2 and IgG3 suggest a potential bias towards T helper 1 (Th1) immunity,33 which may be important in the clearance of bacterial infections.34 On day 21 after the last vaccination, it was observed that a higher proportion of B220+GL7hi germinal center B cells were present in the draining lymph node of mice vaccinated with OM-NDs (Figure 4e), which correlated well with the production of pathogen-specific IgGs. Finally, T cell responses were characterized at the same timepoint by restimulating splenocytes ex vivo with P. aeruginosa outer membrane (Figure 4f,g). The secretion of interferon γ (IFNγ) and IL-17a, which are associated respectively with Th1 and Th17 immunity, was significantly increased for the OM-ND group. Overall, the immune characterization data confirmed that OM-NDs were effective at promoting adaptive cellular immune responses important for fighting infections.34
Figure 4.
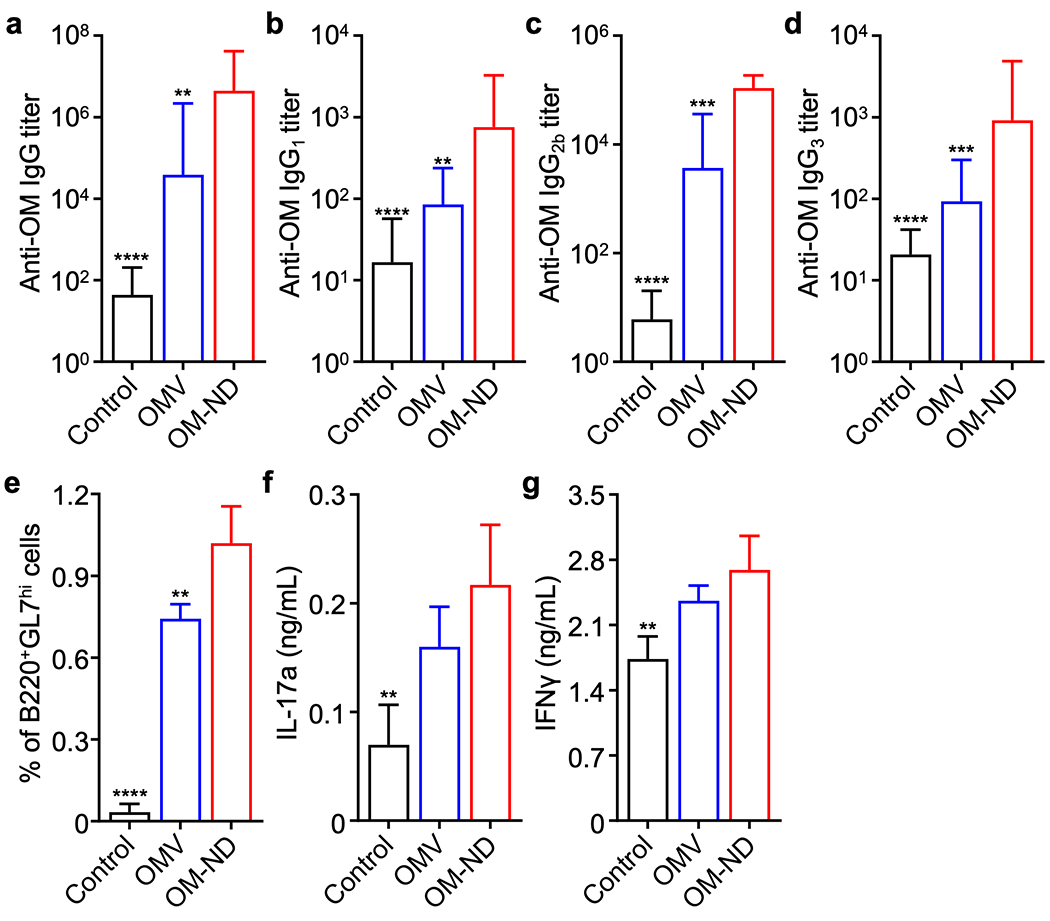
In vivo adaptive immune responses. (a-d) Total anti-outer membrane (anti-OM) IgG titers (a), anti-OM IgG1 titers (b), anti-OM IgG2b titers (c), and anti-OM IgG3 titers (d) in the serum of mice on day 14 following vaccination on days 0 and 7 with free OMVs or OM-NDs (n = 8, mean + SD). (e) Proportion of B220+GL7hi B cells in the popliteal lymph nodes on day 28 after immunization with free OMVs or OM-NDs on days 0 and 7 (n = 4, mean + SD). (f,g) Secretion of IL-17a (f) and IFNγ (g) by splenocytes collected from mice on day 28 after immunization with free OMVs or OM-NDs on days 0 and 7, followed by restimulation with outer membrane (n = 4, mean + SD). **p < 0.01, ***p < 0.001, and ****p < 0.0001 (compared to OM-ND); one-way ANOVA.
Given that P. aeruginosa is known to commonly infect the airways and lungs,35 we elected to evaluate prophylactic efficacy in a murine model of pneumonia. Mice were subcutaneously immunized with OM-NDs on days 0 and 7, followed by intratracheal challenge with P. aeruginosa on day 14. Compared with unvaccinated mice and free OMV-vaccinated mice, those receiving OM-NDs had improved long-term survival at two different bacterial challenge doses (Table S1). Bacterial burden in the lungs was also quantified 24 h after infection, and it was observed that the P. aeruginosa load was significantly reduced in mice vaccinated with OM-NDs compared to those receiving free OMVs (Figure 5a). The reduced bacterial counts and improved survival were associated with elevated levels of anti-outer membrane IgGs in bronchoalveolar lavage fluid samples collected 24 h after intratracheal bacterial challenge (Figure 5b). Previous works have demonstrated that nanovaccine-mediated immune protection correlates with reduced immune infiltration after intratracheal P. aeruginosa challenge.36 Vaccination with OM-NDs also reduced the level of proinflammatory cytokines, including IL-6, IL-12, IL-17a, and TNFα, in the lungs after infection (Figure 5c-f), demonstrating that the OM-ND nanovaccine could offer improved protection while also minimizing excessive immune activation.
Figure 5.
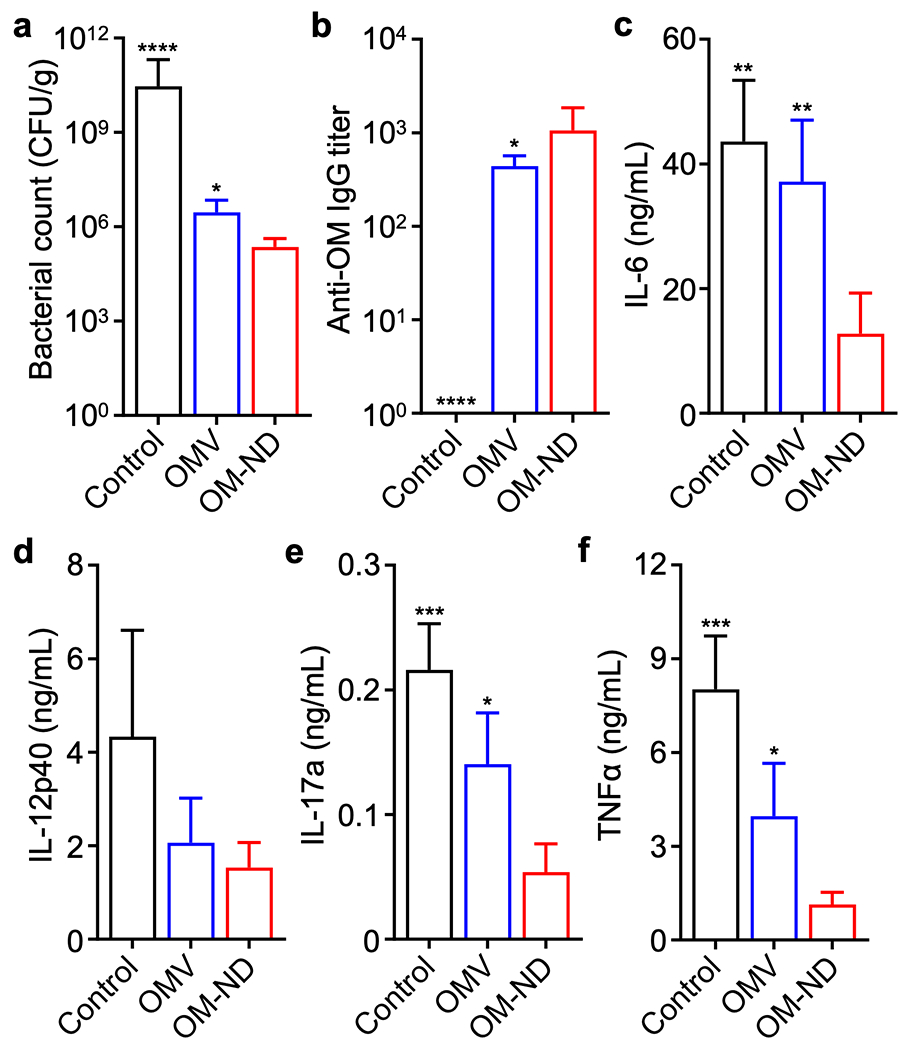
In vivo protection against P. aeruginosa pneumonia. In a-f, mice were vaccinated on days 0 and 7 with free OMVs or OM-NDs, challenged intratracheally with P. aeruginosa on day 14, and euthanized after another 24 h for analysis. (a) Bacterial load in the lungs (n = 4, mean + SD). (b) Anti-outer membrane (anti-OM) IgG titers in the bronchoalveolar lavage fluid (n = 4, mean + SD). (c-f) Concentration of IL-6 (c), IL-12p40 (d), IL-17a (e), and TNFα (f) in the bronchoalveolar lavage fluid (n = 4, mean + SD). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 (compared to OM-ND); one-way ANOVA.
CONCLUSION
In summary, we successfully fabricated a cellular nanodisc formulation using bacterial outer membrane as an effective nanovaccine to protect against bacterial infection. The resulting OM-NDs had an antigenic profile similar to that of free OMVs but were significantly smaller in size at approximately 10 nm. It should be noted that, even with significant processing using techniques such as extrusion or sonication, it would be near impossible to produce a vesicular bacterial outer membrane formulation in the same size range. With its ability to efficiently transport to the lymph nodes after in vivo administration and interact with resident antigen-presenting cells, the new nanovaccine effectively elicited antibacterial antibody production. Additionally, it was demonstrated that OM-NDs were capable of promoting Th1- and Th17-biased cellular immunity. Overall, OM-NDs were able to outperform a free OMV control at the exact same antigen dose in a murine model of pneumonia, helping to reduce bacterial burden, improve survival, and alleviate excessive inflammation. The results achieved in this work highlight the advantages of employing a nanodisc format when developing cell membrane-based nanoformulations, particularly for vaccine applications that require efficient cellular uptake and lymphatic transport. In the future, it will be important to study which antigens correlate strongly with protection against infection. We envision that this approach can be readily used to generate nanovaccines for protecting against other pathogenic diseases. Cellular nanodiscs may ultimately find use across a wide range of applications and serve as an important tool for those working in the field of biomimetic nanomedicine.
METHODS
Bacterial outer membrane derivation.
OMV collection was performed based on a modified version of a previously published protocol.29 P. aeruginosa strain PAO1 was obtained from the American Type Culture Collection. The bacteria were inoculated into Luria–Bertani (LB, Becton Dickinson) broth and cultured in a rotary shaker at 37 °C overnight. Afterwards, the culture medium was diluted 1:30 with fresh LB broth, and the bacteria were cultured for another day. P. aeruginosa were pelleted by centrifugation at 4,500 g for 40 min, and the supernatant was filtered using 0.45-μm polyethersulfone (PES) vacuum filters (Nalgene). Then, the filtered sample was concentrated using a KrosFlo KR2i tangential flow filtration system equipped with a 30 kDa molecular weight cut-off (MWCO) hollow fiber modified PES membrane column (Spectrum). The OMVs were pelleted by ultracentrifugation at 150,000 g for 2 h and resuspended with Ultrapure DNase/RNase-free distilled water (Invitrogen). Protein content was quantified using a bicinchoninic acid (BCA) protein assay kit (Pierce) following the manufacturer’s instructions, and the OMVs were stored at −80 °C for further use. RBC membrane was derived from mouse whole blood (Bioreclamation) based on a previously published protocol.13
Nanodisc preparation and characterization.
Bacterial outer membrane or RBC membrane at 1 mg/mL was mixed with SMA (Cube Biotech) at 5 mg/mL and vortexed at room temperature overnight. Afterwards, non-solubilized proteins and lipids were pelleted by ultracentrifugation at 150,000 g for 30 min. The supernatant was condensed and the free SMA was removed using 10 kDa MWCO Amicon Ultra centrifugal filters (Millipore). The protein content in the resulting RBC-ND and OM-ND samples was quantified using a BCA protein assay. The yield of OM-ND fabrication was calculated by the following equation: NDmass/OMVmass × 100%, where OMVmass was defined as the initial OMV input in terms of protein weight. Size and zeta potential were measured using a Malvern Zetasizer Lab Red Label. To evaluate stability, OM-NDs were kept in PBS at 4 °C, and size was measured every two days for 8 days total. The morphology of the nanoparticles negatively stained with 1% uranyl acetate (Electron Microscopy Sciences) was visualized using a JEOL 1200 EX II transmission electron microscope. For protein characterization, OMVs and OM-NDs were prepared in NuPAGE LDS sample buffer (Invitrogen). The samples were then run on a NuPAGE 4-12% Bis-Tris 15-well minigel (Invitrogen) in NuPAGE MOPS SDS running buffer (Invitrogen) using an Invitrogen XCell SureLock Mini-Cell electrophoresis system at 165 V for 45 min, and the gel was stained with InstantBlue Coomassie protein stain (Abcam) overnight for visualization. Endotoxin levels were quantified using a chromogenic endotoxin quantitation kit (Pierce) according to the manufacturer’s instructions.
For dye labeling, Alexa Fluor 647-NHS (excitation/emission: 651/672 nm, Invitrogen) dissolved in dimethyl sulfoxide was mixed with OMVs in distilled water, followed by incubation at room temperature for 1 h. Free dye was removed by washing the samples using 3 kDa MWCO Amicon Ultra centrifugal filters (Millipore). The dye-conjugated OMVs were stored at −80 °C for further use. Dye-labeled OM-NDs were prepared following the same procedure outlined above for unlabeled OM-NDs.
In vitro dendritic cell uptake.
DC2.4, an immortalized murine dendritic cell line, was a gift from the Dong-Er Zhang laboratory and was cultured in DMEM (Corning) with 10% bovine calf serum (Hyclone) and 1% penicillin-streptomycin (Gibco). The cells were seeded overnight into a 48-well tissue culture plate at 5 × 104 per well, followed by incubation with dye-labeled free OMVs or OM-NDs at a protein concentration of 5 μg/mL. At predetermined time points (10 min, 30 min, 1 h, 2 h, 5 h), the cells were collected into PBS containing 2 mM ethylenediaminetetraacetic acid (EDTA; Corning) and 0.5% bovine serum albumin (BSA; Sigma-Aldrich), and data was collected using a Becton Dickinson LSR II flow cytometer. Analysis was performed using FlowJo software. To visualize uptake, DC2.4 cells were seeded overnight into a glass-bottom dish. Then, dye-labeled free OMVs or OM-NDs were incubated with the cells for 3 h at 5 μg/mL. Cell nuclei were stained with Hoechst 33342 (Invitrogen) for 10 min, and imaging was performed on a Keyence BZ-X710 fluorescence microscope.
To study the uptake mechanism, 5 × 104 DC2.4 cells were pretreated for 30 min with the following inhibitors: chlorpromazine (Sigma-Aldrich) at 15 μg/mL for clathrin-mediated endocytosis, dynasore (Sigma-Aldrich) at 80 μM for clathrin-mediated and caveolae-mediated endocytosis, filipin III (Sigma-Aldrich) at 5 μg/mL for caveolae-mediated and lipid raft-mediated endocytosis, and 5-(N-ethyl-N-isopropyl)-amiloride (Sigma-Aldrich) at 100 μM for macropinocytosis.37–40 Afterwards, the cells were washed with 1% BSA in PBS and then incubated with 0.5 μg/mL of dye-labeled OMV or OM-ND samples for 1 h at 37 °C. A set of untreated cells was also incubated at 4 °C to determine if the uptake was energy-dependent. Finally, the cells were washed twice with 1% BSA in PBS, and data was collected using a Becton Dickinson LSR II flow cytometer. Analysis was performed using FlowJo software.
In vitro dendritic cell viability.
DC2.4 cells were seeded overnight into a 96-well tissue culture plate at 7 × 103 per well, followed by incubation with RBC vesicles, RBC-NDs, free OMVs, or OM-NDs at 0.5 μg/mL. After 48 h, CellTiter-Blue cell viability assay (Promega) solution was added, and the fluorescence intensity (excitation/emission: 560/590 nm) was measured using a TECAN Spark 20M microplate reader.
In vitro dendritic cell stimulation.
For the dose-response study, DC2.4 cells were seeded overnight into 48-well tissue culture plates at 4 × 104 per well, followed by incubation with various concentrations of OM-NDs. After 48 h, the supernatant was collected from each well, and IL-6 levels were analyzed using an enzyme-linked immunosorbent assay (ELISA) kit (BioLegend) following the manufacturer’s instructions. For the multiple cytokine study, DC2.4 cells were seeded overnight into 12-well tissue culture plates at 3 × 105 per well, followed by incubation with RBC vesicles, RBC-NDs, free OMVs, or OM-NDs at 0.5 μg/mL. After 48 h, the supernatant was collected from each well, and cytokine (IL-1β, IL-6, IL-12p40, and TNFα) levels were analyzed using the appropriate ELISA kits (BioLegend) following the manufacturer’s instructions. Adherent cells were also collected using a cell scraper and pelleted by centrifugation at 700 g for 5 min. The cells were blocked with TruStain FcX PLUS anti-mouse CD16/32 (BioLegend) at 4 °C for 10 min, followed by staining with a LIVE/DEAD fixable aqua dead cell stain kit (Life Technologies) and a cocktail containing FITC anti-mouse CD40 (3/23, BioLegend) and Alexa Fluor 647 anti-mouse CD86 (GL-1, BioLegend) for 30 min. Then, the cells were washed and resuspended in PBS containing EDTA and BSA. Data was collected on a Becton Dickinson LSRFortessa flow cytometer, and analysis was performed using FlowJo software.
Animal care.
Male CD-1 mice (6 to 8 weeks) were purchased from Charles River Laboratories. Mice were housed in an animal facility at the University of California San Diego (UCSD) under federal, state, local, and National Institutes of Health (NIH) guidelines. All animal experiments were performed in accordance with NIH guidelines and approved by the Institutional Animal Care and Use Committee of UCSD under protocol number S09388.
In vivo lymph node targeting.
Dye-labeled free OMVs or OM-NDs were administered into mice via the hock at a protein dose of 20 μg. After 6 h, the mice were euthanized, and the draining popliteal lymph nodes were collected. The fluorescence in each lymph node was visualized and quantified using an IVIS Lumina in vivo imaging system. To analyze the cellular-level distribution, each draining popliteal lymph node was mechanically sheared and extruded through a 40-μm cell strainer (Bel-Art) to form a single-cell suspension. Then, 5 × 105 cells were blocked with TruStain FcX PLUS anti-mouse CD16/32 at 4 °C for 10 min, followed by staining with LIVE/DEAD fixable aqua, PE/Cy7 anti-mouse CD11c (N418, BioLegend), FITC anti-mouse CD3 (17A2, BioLegend), Pacific Blue anti-mouse CD19 (6D5, BioLegend), APC/Cy7 anti-mouse CD11b (M1/70, BioLegend), PE anti-mouse F4/80 (BM8, BioLegend), and PerCP anti-mouse Ly-6G/Ly-6C (Gr-1) (RB6-8C5, BioLegend) for 30 min. Finally, the cells were washed and resuspended in PBS containing EDTA and BSA. Data was collected on a Becton Dickinson LSRFortessa flow cytometer, and analysis was performed using FlowJo software.
In vivo safety study.
Free OMVs or OM-NDs were administered into mice via the hock at a protein dose of 0.5 μg. After 1 day, blood was collected via submandibular puncture into Microvette 100 potassium-EDTA blood collection tubes (Sarstedt) for cell counting. For serum biochemistry analysis, blood was collected without an anticoagulant and allowed to coagulate for at least 30 min before centrifuging at 3000 g for 10 min. All tests were performed by the Animal Care Program Diagnostic Services Laboratory at UCSD.
In vivo dendritic cell maturation.
Free OMVs or OM-NDs were administered into mice via the hock at a protein dose of 0.5 μg. After 48 h, the mice were euthanized, and the draining popliteal lymph nodes were collected. Each lymph node was mechanically sheared and extruded through a 40-μm cell strainer to form a single-cell suspension. Then, 5 × 105 cells were blocked with TruStain FcX PLUS anti-mouse CD16/32 at 4 °C for 10 min, followed by staining with LIVE/DEAD fixable aqua, FITC anti-mouse CD40, Alexa 647 Fluor anti-mouse CD86, APC/Cy7 anti-mouse F4/80 (BM8, BioLegend), and PE/Cy7 anti-mouse CD11c for 30 min. Then, the cells were washed and resuspended in PBS containing EDTA and BSA. Data was collected on a Becton Dickinson LSRFortessa flow cytometer, and analysis was performed using FlowJo software.
Anti-P. aeruginosa titers.
Free OMVs or OM-NDs were administered to mice via the hock on days 0, 7, and 14 at a protein dose of 0.5 μg, and the serum from each mouse was sampled on days 0, 7, 14, and 21. Anti-P. aeruginosa titer levels were determined by a direct ELISA. OMVs dissolved in ELISA coating buffer (BioLegend) were coated overnight at 4 °C onto 96-well assay plates at 0.5 μg per well. The plates were then blocked at room temperature for 1 h with 5% skim milk (Apex Bioresearch Products) dissolved in PBS containing 0.05% Tween-20 (National Scientific) (PBST), incubated with serially diluted serum samples for 2 h, and probed with a horseradish peroxidase (HRP)-conjugated anti-mouse IgG (BioLegend) for another 2 h. To analyze the various IgG isotypes, HRP-conjugated anti-mouse IgG1, IgG2b, or IgG3 (SouthernBiotech) was used as the probe for the last step. The plates were developed with 3,3′,5,5′-tetramethylbenzidine substrate (BioLegend), and the reaction was stopped with 1 N HCl (Fisher Scientific). The absorbance was read at 450 nm with 570 nm as the reference using a TECAN Spark 20M microplate reader. All plates were washed at least 4 times with PBST between each step, and incubation steps were performed at room temperature on a rotary shaker.
Adaptive immune response characterization.
Free OMVs or OM-NDs were administered to mice via the hock on days 0 and 7 at a protein dose of 0.5 μg. For germinal center B cell analysis, the popliteal lymph nodes were collected on day 28, followed by mechanical shearing and then extrusion through a 40-μm cell strainer to obtain a single-cell suspension. Then, 5 × 105 cells were blocked with TruStain FcX PLUS anti-mouse CD16/32 at 4 °C for 10 min, followed by staining with LIVE/DEAD fixable aqua, Alexa Fluor 488 anti-mouse/human GL7 antigen (GL7, BioLegend), and Pacific Blue anti-mouse/human CD45R/B220 (RA3-6B2, BioLegend) for 30 min. Finally, the cells were washed and resuspended in PBS containing EDTA and BSA. Data was collected on a Becton Dickinson LSRFortessa flow cytometer, and analysis was performed using FlowJo software. For the ex vivo splenocyte restimulation assay, the spleens were collected and physically sheared, followed by RBC lysis using a commercial buffer (BioLegend) and extrusion through a 70-μm mesh cell strainer (Fisher Scientific) to obtain a single-cell suspension. Then, 2 × 106 splenocytes were seeded into a 12-well suspension culture plate in RPMI 1640 media (Gibco) containing 10% fetal bovine serum (Omega Scientific), 55 mM 2-mercaptoethanol (Gibco), and 1% penicillin-streptomycin. OMVs were added to the cells at 0.1 μg per well. After 3 days of incubation, IL-17a and IFNγ levels in the supernatant were quantified using the appropriate ELISA kits (BioLegend) according to the manufacturer’s instructions.
Prophylactic efficacy against P. aeruginosa pneumonia.
Free OMVs or OM-NDs were administered to mice via the hock on days 0 and 7 at a protein dose of 0.5 μg. On day 14, the mice were anesthetized using a cocktail of 150 mg/kg ketamine (Zoetis) and 10 mg/kg xylazine (Lloyd Laboratories), followed by intratracheal challenge with 40 μL containing 5 × 107 or 1 × 108 CFU of P. aeruginosa for survival monitoring and 5 × 107 CFU of P. aeruginosa for all other studies. For bacterial enumeration, mice were euthanized 1 day after the bacterial challenge to collect the lungs, which were then weighed and homogenized with a BioSpec Mini-Beadbeater-16 in 1 mL of PBS using 2-mm zirconia beads (BioSpec). Lung homogenates were serially diluted in PBS and plated onto LB agar plates. After 16 h of incubation at 37 °C, the number of colonies were counted. For bronchoalveolar lavage fluid analysis, mice were euthanized 1 day after bacterial challenge, and then 1 mL of the fluid was collected with cold PBS containing EDTA and BSA. After pelleting any cells that were present by centrifugation, outer membrane-specific IgG antibody titers were analyzed as described above, and cytokine levels were quantified using the appropriate ELISA kits (BioLegend) according to the manufacturer’s instructions. For survival studies, mice were monitored daily for 10 days.
Data Analysis.
All data were analyzed with GraphPad Prism 9 and presented as the mean ± standard deviation (SD). A minimum sample size of 3 was used for all studies in which statistical analysis was performed. Comparisons between two groups were done with an unpaired two-tailed Student’s t-test. For studies with three or more groups, one-way analysis of variance (ANOVA) with Turkey’s posthoc analysis was used to determine significance.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by the Defense Threat Reduction Agency Joint Science and Technology Office for Chemical and Biological Defense under award number HDTRA1-21-1-0010 and the National Institutes of Health under Award Number R21AI159492.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.nano-lett.XXXXXXX.
Preservation of immunostimulatory molecules; uptake mechanism; fabrication and characterization of RBC-NDs; dose-dependent immunostimulatory activity of OM-NDs in vitro; in vitro dendritic cell maturation; cellular distribution in the lymph nodes; in vivo safety; anti-outer membrane IgG titer monitoring over time; long-term survival in a P. aeruginosa pneumonia model (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Blair JM; Webber MA; Baylay AJ; Ogbolu DO; Piddock LJ, Molecular Mechanisms of Antibiotic Resistance. Nat. Rev. Microbiol 2015, 13, 42–51. [DOI] [PubMed] [Google Scholar]
- (2).Ventola CL, The Antibiotic Resistance Crisis: Part 1: Causes and Threats. P T 2015, 40, 277–283. [PMC free article] [PubMed] [Google Scholar]
- (3).Jansen KU; Knirsch C; Anderson AS, The Role of Vaccines in Preventing Bacterial Antimicrobial Resistance. Nat. Med 2018, 24, 10–19. [DOI] [PubMed] [Google Scholar]
- (4).Klugman KP; Black S, Impact of Existing Vaccines in Reducing Antibiotic Resistance: Primary and Secondary Effects. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 12896–12901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Kaufmann SH, The Contribution of Immunology to the Rational Design of Novel Antibacterial Vaccines. Nat. Rev. Microbiol 2007, 5, 491–504. [DOI] [PubMed] [Google Scholar]
- (6).Pati R; Shevtsov M; Sonawane A, Nanoparticle Vaccines against Infectious Diseases. Front. Immunol 2018, 9, 2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhou J; Kroll AV; Holay M; Fang RH; Zhang L, Biomimetic Nanotechnology toward Personalized Vaccines. Adv. Mater 2020, 32, 1901255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhao L; Seth A; Wibowo N; Zhao CX; Mitter N; Yu C; Middelberg AP, Nanoparticle Vaccines. Vaccine 2014, 32, 327–337. [DOI] [PubMed] [Google Scholar]
- (9).Sahdev P; Ochyl LJ; Moon JJ, Biomaterials for Nanoparticle Vaccine Delivery Systems. Pharm. Res 2014, 31, 2563–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Fang RH; Luk BT; Hu CM; Zhang L, Engineered Nanoparticles Mimicking Cell Membranes for Toxin Neutralization. Adv. Drug Deliv. Rev 2015, 90, 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Fang RH; Jiang Y; Fang JC; Zhang L, Cell Membrane-Derived Nanomaterials for Biomedical Applications. Biomaterials 2017, 128, 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Fang RH; Kroll AV; Gao W; Zhang L, Cell Membrane Coating Nanotechnology. Adv. Mater 2018, 30, 1706759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hu CM; Zhang L; Aryal S; Cheung C; Fang RH; Zhang L, Erythrocyte Membrane-Camouflaged Polymeric Nanoparticles as a Biomimetic Delivery Platform. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 10980–10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hu CM; Fang RH; Wang KC; Luk BT; Thamphiwatana S; Dehaini D; Nguyen P; Angsantikul P; Wen CH; Kroll AV; Carpenter C; Ramesh M; Qu V; Patel SH; Zhu J; Shi W; Hofman FM; Chen TC; Gao W; Zhang K, et al. , Nanoparticle Biointerfacing by Platelet Membrane Cloaking. Nature 2015, 526, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Fang RH; Hu CM; Luk BT; Gao W; Copp JA; Tai Y; O’Connor DE; Zhang L, Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery. Nano Lett. 2014, 14, 2181–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Thamphiwatana S; Angsantikul P; Escajadillo T; Zhang Q; Olson J; Luk BT; Zhang S; Fang RH; Gao W; Nizet V; Zhang L, Macrophage-Like Nanoparticles Concurrently Absorbing Endotoxins and Proinflammatory Cytokines for Sepsis Management. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 11488–11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Gnopo YMD; Watkins HC; Stevenson TC; DeLisa MP; Putnam D, Designer Outer Membrane Vesicles as Immunomodulatory Systems – Reprogramming Bacteria for Vaccine Delivery. Adv. Drug Deliv. Rev 2017, 114, 132–142. [DOI] [PubMed] [Google Scholar]
- (18).Gao W; Fang RH; Thamphiwatana S; Luk BT; Li J; Angsantikul P; Zhang Q; Hu CM; Zhang L, Modulating Antibacterial Immunity via Bacterial Membrane-Coated Nanoparticles. Nano Lett. 2015, 15, 1403–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Schwechheimer C; Kuehn MJ, Outer-Membrane Vesicles from Gram-Negative Bacteria: Biogenesis and Functions. Nat. Rev. Microbiol 2015, 13, 605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Moyer TJ; Zmolek AC; Irvine DJ, Beyond Antigens and Adjuvants: Formulating Future Vaccines. J. Clin. Invest 2016, 126, 799–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fang RH; Kroll AV; Zhang L, Nanoparticle-Based Manipulation of Antigen-Presenting Cells for Cancer Immunotherapy. Small 2015, 11, 5483–5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Reddy ST; van der Vlies AJ; Simeoni E; Angeli V; Randolph GJ; O’Neil CP; Lee LK; Swartz MA; Hubbell JA, Exploiting Lymphatic Transport and Complement Activation in Nanoparticle Vaccines. Nat. Biotechnol 2007, 25, 1159–1164. [DOI] [PubMed] [Google Scholar]
- (23).Kuai R; Ochyl LJ; Bahjat KS; Schwendeman A; Moon JJ, Designer Vaccine Nanodiscs for Personalized Cancer Immunotherapy. Nat. Mater 2017, 16, 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kuai R; Sun X; Yuan W; Xu Y; Schwendeman A; Moon JJ, Subcutaneous Nanodisc Vaccination with Neoantigens for Combination Cancer Immunotherapy. Bioconjug. Chem. 2018, 29, 771–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Denisov IG; Sligar SG, Nanodiscs in Membrane Biochemistry and Biophysics. Chem. Rev 2017, 117, 4669–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Bariwal J; Ma H; Altenberg GA; Liang H, Nanodiscs: A Versatile Nanocarrier Platform for Cancer Diagnosis and Treatment. Chem. Soc. Rev 2022, 51, 1702–1728. [DOI] [PubMed] [Google Scholar]
- (27).Horcajada JP; Montero M; Oliver A; Sorli L; Luque S; Gomez-Zorrilla S; Benito N; Grau S, Epidemiology and Treatment of Multidrug-Resistant and Extensively Drug-Resistant Pseudomonas aeruginosa Infections. Clin. Microbiol. Rev 2019, 32, e00031–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Knowles TJ; Finka R; Smith C; Lin YP; Dafforn T; Overduin M, Membrane Proteins Solubilized Intact in Lipid Containing Nanoparticles Bounded by Styrene Maleic Acid Copolymer. J. Am. Chem. Soc 2009, 131, 7484–7485. [DOI] [PubMed] [Google Scholar]
- (29).Zhou J; Karshalev E; Mundaca-Uribe R; Esteban-Fernandez de Avila B; Krishnan N; Xiao C; Ventura CJ; Gong H; Zhang Q; Gao W; Fang RH; Wang J; Zhang L, Physical Disruption of Solid Tumors by Immunostimulatory Microrobots Enhances Antitumor Immunity. Adv. Mater 2021, 33, 2103505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dorr JM; Scheidelaar S; Koorengevel MC; Dominguez JJ; Schafer M; van Walree CA; Killian JA, The Styrene-Maleic Acid Copolymer: A Versatile Tool in Membrane Research. Eur. Biophys. J 2016, 45, 3–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kaparakis-Liaskos M; Ferrero RL, Immune Modulation by Bacterial Outer Membrane Vesicles. Nat. Rev. Immunol 2015, 15, 375–387. [DOI] [PubMed] [Google Scholar]
- (32).Esmaili M; Brown CJ; Shaykhutdinov R; Acevedo-Morantes C; Wang YL; Wille H; Gandour RD; Turner SR; Overduin M, Homogeneous Nanodiscs of Native Membranes Formed by Stilbene-Maleic-Acid Copolymers. Nanoscale 2020, 12, 16705–16709. [DOI] [PubMed] [Google Scholar]
- (33).Germann T; Bongartz M; Dlugonska H; Hess H; Schmitt E; Kolbe L; Kolsch E; Podlaski FJ; Gately MK; Rude E, Interleukin-12 Profoundly Up-Regulates the Synthesis of Antigen-Specific Complement-Fixing IgG2a, IgG2b and IgG3 Antibody Subclasses In Vivo. Eur. J. Immunol 1995, 25, 823–829. [DOI] [PubMed] [Google Scholar]
- (34).Holley MM; Kielian T, Th1 and Th17 Cells Regulate Innate Immune Responses and Bacterial Clearance During Central Nervous System Infection. J. Immunol 2012, 188, 1360–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sandri A; Lleo MM; Signoretto C; Boaretti M; Boschi F, Protease Inhibitors Elicit Anti-Inflammatory Effects in CF Mice with Pseudomonas aeruginosa Acute Lung Infection. Clin. Exp. Immunol 2021, 203, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wei X; Ran D; Campeau A; Xiao C; Zhou J; Dehaini D; Jiang Y; Kroll AV; Zhang Q; Gao W; Gonzalez DJ; Fang RH; Zhang L, Multiantigenic Nanotoxoids for Antivirulence Vaccination against Antibiotic-Resistant Gram-Negative Bacteria. Nano Lett. 2019, 19, 4760–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kuhn DA; Vanhecke D; Michen B; Blank F; Gehr P; Petri-Fink A; Rothen-Rutishauser B, Different Endocytotic Uptake Mechanisms for Nanoparticles in Epithelial Cells and Macrophages. Beilstein J. Nanotechnol 2014, 5, 1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).O’Donoghue EJ; Krachler AM, Mechanisms of Outer Membrane Vesicle Entry into Host Cells. Cell. Microbiol 2016, 18, 1508–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Macia E; Ehrlich M; Massol R; Boucrot E; Brunner C; Kirchhausen T, Dynasore, a Cell-Permeable Inhibitor of Dynamin. Dev. Cell 2006, 10, 839–850. [DOI] [PubMed] [Google Scholar]
- (40).Noh I; Kim M; Kim J; Lee D; Oh D; Kim J; Kim C; Jon S; Kim YC, Structure-Inherent Near-Infrared Bilayer Nanovesicles for Use as Photoacoustic Image-Guided Chemo-Thermotherapy. J. Control. Release 2020, 320, 283–292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.