

Abstract
Proteolysis targeting chimera (PROTAC) technology has become a powerful strategy in drug discovery, especially for undruggable targets/proteins. A typical PROTAC degrader consists of three components: a small molecule that binds to a target protein, an E3 ligase ligand (consisting of an E3 ligase and its small molecule recruiter), and a chemical linker that hooks first two components together. In the past 20 years, we have witnessed advancement of multiple PROTAC degraders into the clinical trials for anticancer therapies. However, one of the major challenges of PROTAC technology is that only very limited number of E3 ligase recruiters are currently available as E3 ligand for targeted protein degradation (TPD), although human genome encodes more than 600 E3 ligases. Thus, there is an urgent need to identify additional effective E3 ligase recruiters for TPD applications. In this review, we summarized the existing RING‐type E3 ubiquitin ligase and their small molecule recruiters that act as effective E3 ligands of PROTAC degraders and their application in anticancer drug discovery. We believe that this review could serve as a reference in future development of efficient E3 ligands of PROTAC technology for cancer drug discovery and development.
Keywords: E3 ligase ligands, PROTAC degraders, targeted therapy
We summarized the existing RING‐type E3 ubiquitin ligase and their small molecule recruiters to act as effective E3 ligands of PROTAC degraders. This review could serve as a reference for future development of efficient E3 ligands for PROTAC technology.
1. INTRODUCTION
Discovery of effective small molecule drugs for targeted therapy face several major challenges due to developed drug resistance, undruggable targets and poor selectivity among target family members. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 Small‐molecule substitutes have emerged and gained clinical significance. 9 , 10 , 11 The most representative alternative approach is the development of targeted protein degradation (TPD), with proteolysis targeting chimeras (PROTACs) as a typical example. 12 , 13 , 14 , 15 , 16 , 17 , 18 TPD has entered its third decade with both opportunities and challenges. 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 TPD techniques rely on the ubiquitin–proteasome system (UPS), which couples ubiquitylation, catalyzed by E1 activating enzyme, E2 conjugate enzyme and E3 ligase, and proteasome for degradation of target substrates. 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 , 40 , 41
The concept of PROTAC was first proposed in 2001, and the PROTAC technology has developed rapidly in the past 20 years. 5 , 8 , 19 , 42 , 43 , 44 , 45 At the present, more than 10 PROTAC degraders have entered clinical Phase I‐II trials with ARV‐471 and ARV‐110 from Arvinas, Inc as the most advanced ones for the treatment of recurrent breast cancer and prostate cancer, respectively. 46 , 47 , 48 , 49 However, PROTAC still faces many challenges. 50 , 51 , 52 , 53 , 54 , 55 , 56 , 57 , 58 , 59 , 60 , 61 , 62 , 63 , 64 , 65 A typical PROTAC degrader consists of three components: the target‐binding small molecule ligand (protein of interest, POI), the E3 ligase ligand and the appropriate linker that connects the two components (Figure 1). 50 , 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 E3 ligases and the corresponding ligands are important as the driving force of protein degradation. 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 , 83 , 84 , 85 , 86 , 87 , 88 , 89 , 90 , 91 Good drug‐like small‐molecule ligands for a E3 ligase system are still limited. 92 , 93 , 94 , 95 , 96 , 97 Although more than 600 E3 ligases are encoded by human genome, only less than 10 E3 ligases were developed as ligands for the development of PROTAC degraders, including von Hippel‐Lindau (VHL), cereblon (CRBN), mouse double minute 2 (MDM2), cellular IAP1 (cIAP1), Kelch‐like ECH‐associated protein‐1 (KEAP1), DDB1‐cullin 4‐associated factor (DCAF), RING finger protein (RNF), aryl hydrocarbon receptor (AHR), and others (Figure 1). 66 , 74 , 98 , 99 , 100 , 101 , 102 , 103 , 104 Therefore, it is urgent to develop new and efficient E3 ligands to expand the role of PROTAC technology in drug discovery and development. 105 , 106 , 107 , 108 , 109 , 110 , 111 , 112 In this review, we systematically summarize all reported E3 ligases and their corresponding small molecule ligands for the successful development of PROTAC degraders.
FIGURE 1.
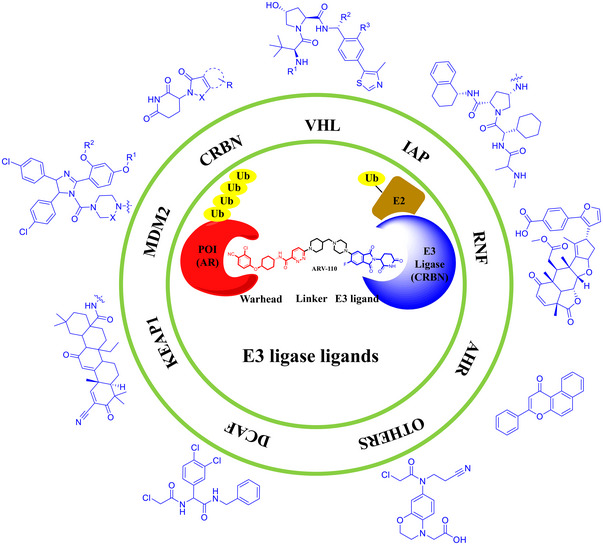
A summary of current ligands for few E3 ubiquitin ligases used in PROTACs (PDB ID: 7B5S). Structures or general structural formulas of these E3 ligands and a working model of PROTAC with ARV‐110 as an example.
2. VHL LIGANDS AND THEIR UTILIZATIONS IN PROTACS FOR CANCER DRUG DISCOVERY
2.1. Common VHL ligands
VHL protein, together with elongin B and C, Cullin 2, and RBX‐1, is part of a multiprotein complex with E3 ubiquitin ligase activity. 113 , 114 , 115 , 116 , 117 , 118 In the complex, VHL is responsible for binding to specific substrates, most notably hypoxia‐inducible factor (HIF‐1 α), for its ubiquitination and proteasome degradation. 119 , 120 In 2012, Crews group disclosed the first small‐molecule ligands for VHL E3 ligase based on skeleton of hydroxyproline. 121 Since then, a series of novel and highly effective VHL E3 ligase ligands have been discovered and reported, typified by compounds VHL‐1–VHL‐8 (Figure 2) with improved lipophilicity. 119 , 120 , 122 The studies on the eutectic structure of the VHL ligand with the protein helps to locate the solvent‐exposed region, leading to revelation of four possible linking sites without negatively affecting the interaction between the protein and the corresponding ligand (Figure 2; PDB ID: 4W9H). 123 , 124 , 125 , 126 , 127 , 128 , 129 , 130 , 131 , 132 , 133 , 134 These sites are (a) terminal amino; (b) sulfhydryl; (c) benzyl; and (d) phenolic hydroxyl group on the benzene ring. 113 , 135 , 136 , 137 , 138 , 139 , 140 , 141 At present, VHL E3 ligand has been widely and successfully applied in the design and synthesis of PROTAC as one of the most commonly used E3 ligands (Table 1). 13 , 17 , 18 , 142 , 143 , 144 , 145 , 146 , 147 , 148 , 149 , 150 , 151 , 152 , 153 , 154
FIGURE 2.
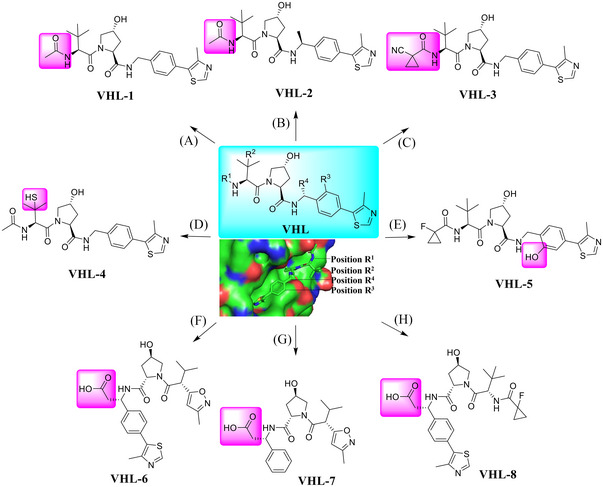
A variety of small molecules that serve as the ligands for VHL E3 ligase. (A–C) The VHL ligands retain the R1 tethering position based upon a cocrystal structure of a VHL‐1 ligand in complex with pVHL:EloB:EloC (PDB ID: 4W9H). (D) The VHL ligand retains the R2 tethering position. (E) The VHL ligand retains the R3 tethering position. (F‐H) The VHL ligands retain the R4 tethering position.
TABLE 1.
Chemical structures and biological activities of representative PROTAC degraders.
| No. | Name | Structure | DC50 (μM) | POI | E3 ligand |
|---|---|---|---|---|---|
| 1 | ARCC‐4 |
|
0.005 (VCaP) | AR | VHL‐1/VHL‐3 |
| 2 | PROTAC‐1 |
|
NA a | AR | VHL‐2 |
| 3 | ARD‐61 |
|
0.001 (VCaP) | AR | VHL‐6 |
| 4 | ARD‐69 |
|
0.00074 (VCaP) | AR | VHL‐8 |
| 5 | ARD‐266 |
|
0.001 (VCaP) | AR | VHL‐7 |
| 6 | SJF‐0628 |
|
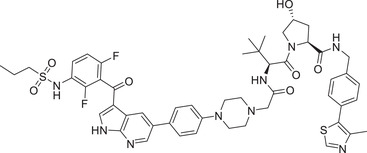
0.0068 (SK‐MEL‐28) | BRAF | VHL‐1/VHL‐3 |
| 7 | MS‐39 |
|
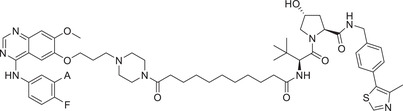
0.0033 (H3255) | EGFR | VHL‐1/VHL‐3 |
| 8 | 14o |
|
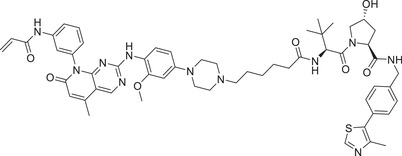
0.0059 (H1975) | EGFR | VHL‐1/VHL‐3 |
| 9 | PROTAC‐2 |
|
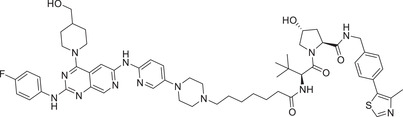
0.0348 (HCC‐827) | EGFR | VHL‐1/VHL‐3 |
| 10 | LC‐2 |
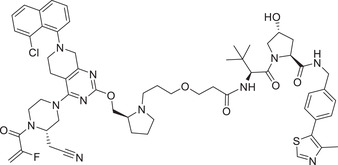
|
0.59 (NCI‐H2030) | KRAS | VHL‐1/VHL‐3 |
| 11 | YF‐135 |
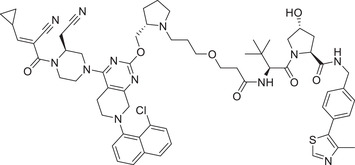
|
3.61 (NCI‐H358) | KRAS | VHL‐1/VHL‐3 |
| 12 | GMB‐805 |
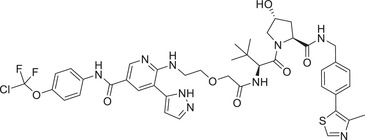
|
0.03 (K562) | BCR–ABL | VHL‐1/VHL‐3 |
| 13 | SIAIS178 |
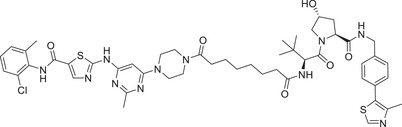
|
0.0085 (K562) | BCR–ABL | VHL‐1/VHL‐3 |
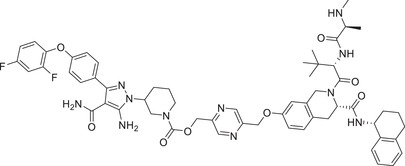
| 14 | JNJ‐1013 |
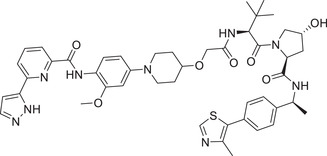
|
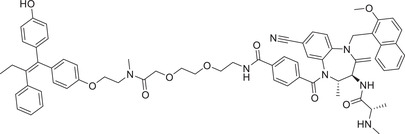
0.003 (HBL‐1) | IRAK1 | VHL‐2 |
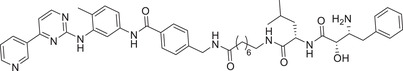
| 15 | PROTAC‐3 |
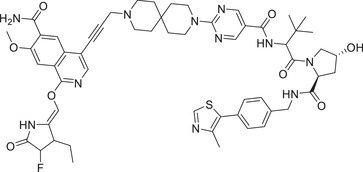
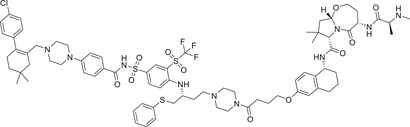
|
0.15 (PBMC) | IRAK4 | VHL‐1/VHL‐3 |
| 16 | ERD‐308 |
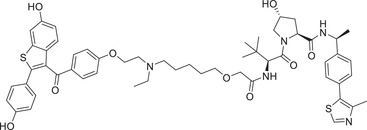
|
0.00017 (MCF‐7) | ER | VHL‐2 |
| 17 | AM‐A3 |
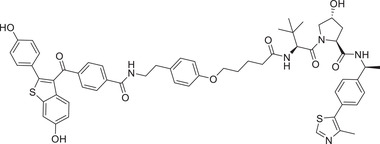
|
0.0011 | ER | VHL‐2 |
| 18 | PROTAC‐4 |
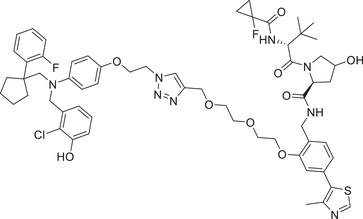
|
0.037 | ER | VHL‐5 |
| 19 | SHP2‐D26 |
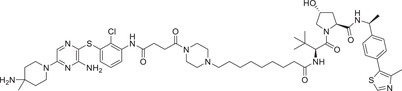
|
0.0026 (MV4; 11) | SHP2 | VHL‐2 |
| 20 | AT‐1 |
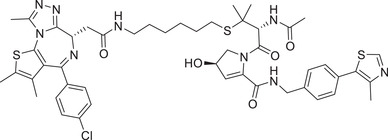
|
0.1 (Hela) | BRD4 | VHL‐4 |
| 21 | AGB1 |
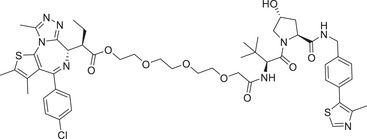
|
0.015 | BRD4 | VHL‐1/VHL‐3 |
| 22 | 3j |
|
0.0071 (MM.1S) | HDAC | VHL‐1/VHL‐3 |
| 23 | XH‐07‐189 |
|
NA | HDAC | VHL‐5 |
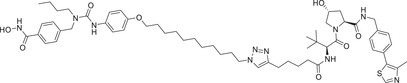
| 24 b | ARV‐110 b |
|
0.0016 (VCaP) | AR | CRBN‐1/CRBN‐2 |
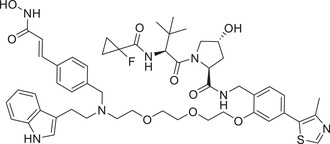
| 25 | PROTAC‐5 |
|
NA | AR | CRBN‐12 |
| 26 | ARD‐2128 |
|
0.00028 (VCaP) | AR | CRBN‐1/CRBN‐2 |
| 27 | ARD‐2585 |
|
0.00004 (VCaP) | AR | CRBN‐1/CRBN‐2 |
| 28 | MS910 |
|
0.094/MEK1; 0.038/MEK2 (SK‐MEL‐28) | MEK | CRBN‐1/CRBN‐2 |
| 29 | CPS2 |
|
0.002 (MV4; 11) | CDK2 | CRBN‐1/CRBN‐2 |
| 30 | TMX‐2172 |
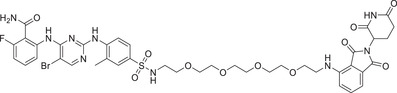
|
NA | CDK2/5 | CRBN‐1/CRBN‐2 |
| 31 | PROTAC‐6 |
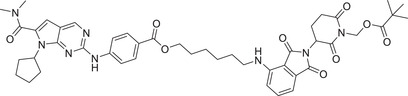
|
NA | CDK2/4/6 | CRBN‐5 |
| 32 | B03 |
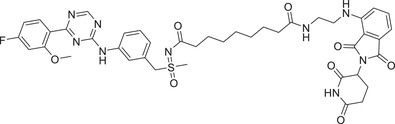
|
0.0076 (MV4;11) | CDK9 | CRBN‐1/CRBN‐2 |
| 33 | SIAIS001 |
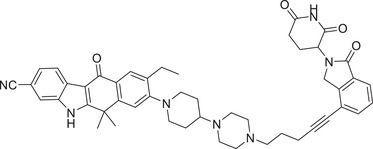
|
0.0039 (SR) | ALK | CRBN‐3 |
| 34 | PROTAC‐7 |
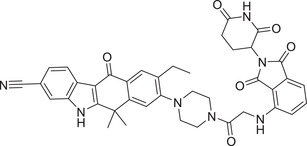
|
0.027 (H3122) | ALK | CRBN‐1/CRBN‐2 |
| 35 | MD‐224 |
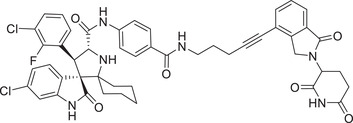
|
0.001 (VCaP) | MDM2 | CRBN‐3 |
| 36 | WB214 |
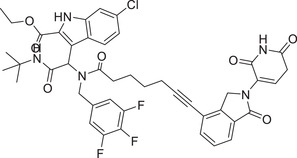
|
0.0041 (RS4;11) | MDM2 | CRBN‐3 |
| 37 | INY‐03‐041 |
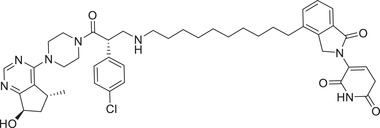
|
NA | AKT | CRBN‐3 |
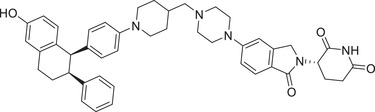
| 38 | MS170 |
|
0.032 (BT474) | AKT | CRBN‐1/CRBN‐2 |
| 39 | SD‐36 |
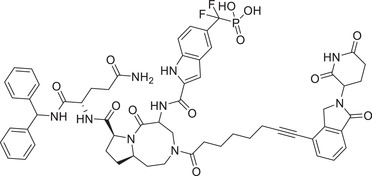
|
0.06 (Molm‐16) | STAT3 | CRBN‐3 |
| 40 | AK‐2292 |
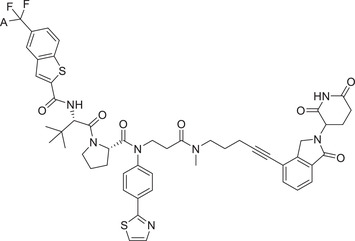
|
0.1 (SKNO1) | STAT5 | CRBN‐3 |
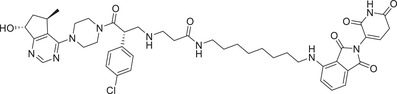
| 41 c | ARV‐471 c |
|
0.002 (MCF‐7) | ER | CRBN‐4 |
| 42 | PROTAC‐8 |
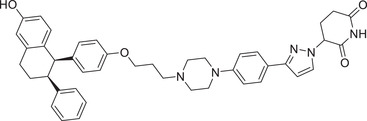
|
NA | ER | CRBN‐11 |
| 43 | SIAIS125 |
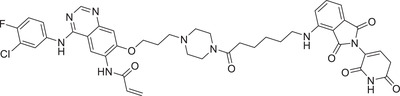
|
0.1 (PC9) | EGFR | CRBN‐1/CRBN‐2 |
| 44 | PROTAC‐9 |
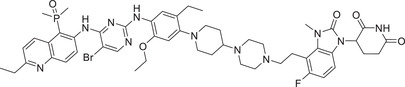
|
NA | EGFR | CRBN‐6 |
| 45 | QCA570 |
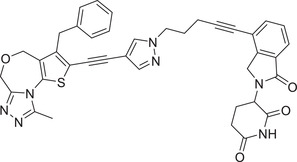
|
<0.00001 (RS4;11) | BET | CRBN‐3 |
| 46 | PROTAC‐10 |
|
0.00032 (22Rv1) | BET | CRBN‐7 |
| 47 | PROTAC‐11 |
|
NA | BET | CRBN‐10 |
| 48 | SJ10542 |
|
0.014 (JAK2) 0.011 (JAK3) (PDX cells SJBALL020589) |
JAK2/3 | CRBN‐9 |
| 49 | DGY‐08‐097 |
|
0.05 (HCV‐NS3) | HCV | CRBN‐8 |
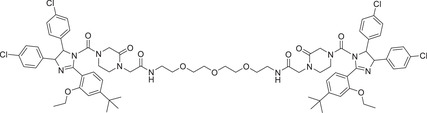
| 50 | PROTAC‐12 |
|
10 (Hela) | AR | MDM2‐1 |
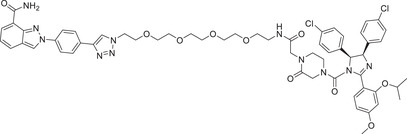
| 51 | A1874 |
|
0.023 (HCT116) | BRD4 | MDM2‐2 |
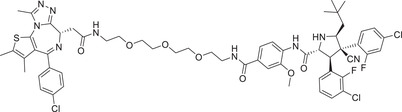
| 52 | PROTAC‐13 |
|
5 (MDA‐MB‐231) | PARP1 | MDM2‐3 |
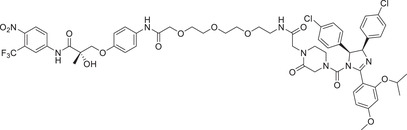
| 53 | PROTAC‐14 |
|
NA | MDM2 | MDM2‐1/MDM‐4 |
| 54 | PROTAC‐15 |
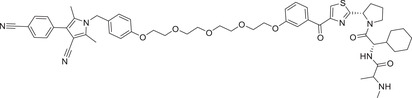
|
2 (22Rv1) | AR | IAP‐1 |
| 55 | PROTAC‐16 |
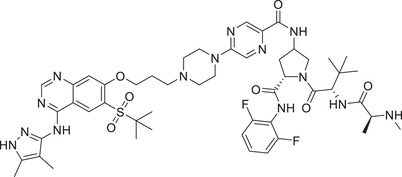
|
7.9 (THP‐1) | RIPK2 | IAP‐2 |
| 56 | PROTAC‐17 |
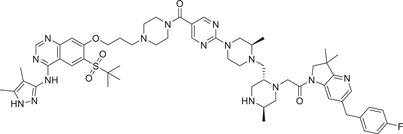
|
1.6 (THP‐1) | RIPK2 | IAP‐3 |
| 57 | PROTAC‐18 |
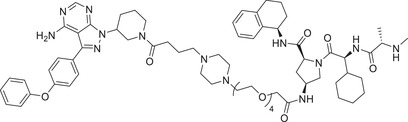
|
0.2 (THP‐1) | BTK | IAP‐4 |
| 58 | BCPyr |
|
0.8 (THP‐1) | BTK | IAP‐6 |
| 59 | PROTAC‐19 |
|
0.68 (MCF‐7) | ER | IAP‐5 |
| 60 | SNIPER‐2 |
|
30 (K562) | BCR–ABL | IAP‐7 |
| 61 | PROTAC‐20 |
|
0.1 (MyLa 1929) | BCL‐XL | IAP‐8 |
| 62 | CDDO‐JQ1 |
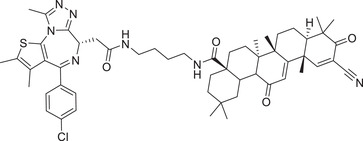
|
0.05 (231MFP) | BRD4 | KEAP1‐1 |
| 63 | MS‐83 |
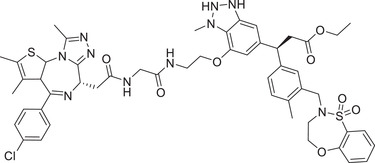
|
0.25 (MDA‐MB‐231) | BRD2/3/4 | KEAP1‐3 |
| 64 | PROTAC‐21 |
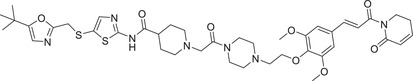
|
0.009 (MOLT4) | CDK9 | KEAP1‐2 |
| 65 | KB02‐SLF |
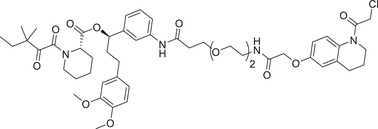
|
0.5 (HEK293T) | FKBP12 | DCAF16 |
| 66 | 21‐SLF |
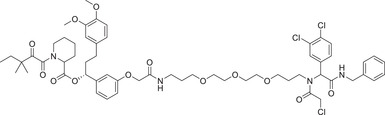
|
NA | FKBP12 | DCAF11 |
| 67 | YT117R |
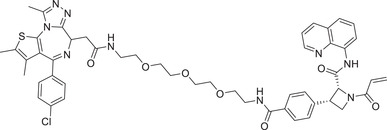
|
NA | BRD4 | DCAF1 |
| 68 | DP‐1 |
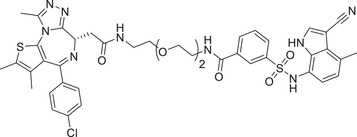
|
10.8 (SU‐DHL‐4) | BRD2/3/4 | DCAF15 |
| 69 | XH‐2 |
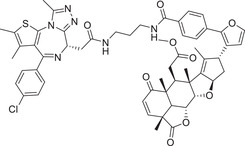
|
0.5 (231MFP) | BRD4 | RNF114 |
| 70 | ML 2‐14 |
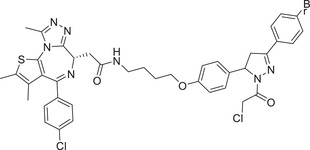
|
0.014 (231MFP) | BRD4 |
RNF114 (EN219) |
| 71 | CCW 28‐3 |
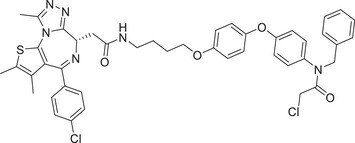
|
0.1 (231MFP) | BRD4 | RNF4 |
| 72 | NJH‐1‐106 |
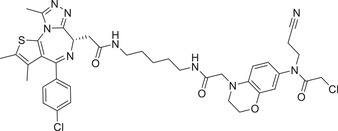
|
0.25 (HEK293T) | BRD4 | FEM1B |
| 73 | KL‐7 |
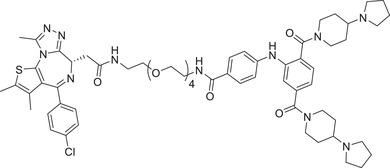
|
5 (HCT116) | BRD2 | L3MBTL3 |
| 74 | KL‐4 |
|
1 (HEK293_HFNES) | FKBP12F36V | L3MBTL3 |
| 75 | BT‐1 |
|
1 (K562) | BCR–ABL | RNF114 |
| 76 | β‐NF‐ATRA |
|
NA | CRABPs | β‐NF |
Not Applicable.
Phase II.
Phase III, NCT05654623.
Abbreviations: AKT, protein kinase B; ALK, anaplastic lymphoma kinase; AR, androgen receptor; BCL‐XL, recombinant human B‐cell leukemia/lymphoma XL; BCR–ABL, breakpoint cluster region‐Abelson; BET, the bromo and extral terminal domain family; BRAF, v‐raf murine sarcoma viral oncogene homolog B1; BRD4, bromodomain‐containing protein 4; BTK, Bruton's tyrosine kinase; CDK, cyclin‐dependent kinase; CRABPs, cellular retinoic acid binding proteins; CRBN, cereblon; DC50, half‐maximal degradation concentrations; DCAF, DDB1‐cullin 4‐associated factor; EGFR, epidermal growth factor receptor gene; ER, estrogen receptor; FKBP12, FK506 binding protein 12 kDa; HCV, hepatitis C virus; HDAC, histone deacetylase; IAP, inhibitor of apoptosis protein; IRAK, interleukin‐1 receptor‐activated kinase; JAK, Janus kinase; KEAP1, the Kelch‐like ECH‐associated protein‐1; KRAS, Kirsten rat sarcoma viral oncogene; MDM2, murine double minute 2; MEK, mitogen activated protein kinase; PARP1, poly(ADP‐ribose) polymerase; POI, protein of interest; RIPK2, receptor Interaction serine/threonine kinase 2; RNF, RING finger protein.; SHP2, Src homology region 2‐containing protein tyrosine phosphatase 2; STAT, signal transducer and activator of transcription; VHL, Von Hippel‐Lindau.
2.2. The utilizations in PROTACs
A variety of PROTAC degraders have been designed and discovered based on the four potential binding sites of VHL ligands.
2.2.1. AR
As an important member of the nuclear hormone receptor superfamily, androgen receptor (AR) plays a crucial role in the maintenance and development of male prostatic secondary sex characteristics. 155 The dysfunction of AR is a major cause for the development of human prostate cancer. At the present, metastatic castration‐resistant prostate cancer (mCRPC) remains incurable and fatal. AR antagonists are effective for the treatment of mCRPC, significantly improves the survival of prostate cancer patients with well tolerance. 156 , 157 A plenty of AR antagonists have been developed for the treatment of advanced prostate cancer, exemplified by enzalutamide. In prostate cancer patients, resistance to enzalutamide usually appears within 18 months. 158 , 159 , 160 The AR signaling pathway remained functional in the majority of patients with enzalutamide resistance. Targeting AR protein degradation is a very promising therapeutic strategy for the treatment of mCRPC, particularly enzalutamide‐resistant mCRPC with potential being more effective than AR antagonists.
In 2018, a PROTAC AR degrader named ARCC‐4 (Table 1) was reported by Crews group, using VHL‐1/VHL‐3 (Figure 2). 161 ARCC‐4 showed good in vitro potency including low‐nanomolar DC50 (half‐maximal degradation concentrations) value, moderate inhibition on proliferation of prostate cancer cells with better antiproliferative effects on enzalutamide‐resistant AR mutant cells. In 2019, Arvinas’ patent disclosed a series of potent PROTACs targeting AR and Kargbo, showing that the chiral methyl group in VHL ligand (Figure 1; VHL‐2) improved the degradation capacity, as exemplified by PROTAC‐1 (Table 1). 162 Recently, the Wang group discovered a series of new VHL ligands (Figure 1; VHL‐6–8) in the process of designing AR degraders ARD‐61, ARD‐69, and ARD‐266 (Table 1). 163 , 164 , 165 , 166 The (S)‐methyl group in VHL‐2 is exposed to the solvent zone, which can be used as a possible tethering point in the design of PROTAC AR degraders, based on the cocrystal structure of VHL‐1 and VHL protein (Figure 2). Compounds ARD‐61 and ARD‐69 effectively induced the degradation of AR with DC50 values as low as 0.74–1 nM in VCaP cells. These two compounds also showed excellent prostate cancer cell growth inhibition in vitro and tumor inhibition in vivo.
2.2.2. BRAF
The RAF kinase family mainly regulates cell proliferation, growth, differentiation, survival, and other physiological activities via the RAS–RAF–MEK–ERK signaling pathway. 167 Mutational activation of BRAF, in a form of BRAFV600E, occurs in many human cancers, which is associated with the occurrence and development of human cancers. 168 Drug resistance severely limits the clinical effectiveness of BRAF V600E inhibitors. In addition, BRAF V600E small molecule inhibitors mainly act by binding with the catalytic bag of RAF with incomplete inhibition, because they failed to inhibit the BRAF dimer, another key mechanism of RAF activation. 169 , 170 New drug discovery techniques, such as PROTAC, are urgently needed to overcome the limitations of existing RAF inhibitors and provide the basis for new targeted alternative therapy strategies for the treatment of BRAF V600E ‐causing cancer.
In 2021, Crews group disclosed a series of VHL‐based degraders by coupling BRAF inhibitor vemurafenib to VHL ligands (Figure 1; VHL‐1/VHL‐3), as exemplified by compound SJF‐0628 via a rigid piperazine linker (Table 1). 171 BRAF degrader SJF‐0628 could effectively induce degradation of BRAF V600E , but not wild type BRAF protein in multiple cell lines. The DC50 of SJF‐0628 for BRAF V600E was impressive 6.8 nM in SK‐MEL‐28 cell line, SJF‐0628 also has good inhibitory effect on other tumor cells. 171
2.2.3. EGFR
Epidermal growth factor receptor (EGFR) is a glycoprotein of tyrosine kinase‐type receptors, which consists of three regions: the intracellular kinase region, the transmembrane region and the extracellular ligand‐binding region. 172 EGFR is involved in a variety of physiological activities, overexpression, and abnormal activation in a variety of solid tumors, and is closely related to angiogenesis, cell proliferation, tumorigenesis, migration, invasion, and tumor metastasis. 173 , 174 Three generations of small molecule EGFR inhibitors have been approved by the United States Food and Drug Administration (US FDA) to treat non‐small cell lung cancer (NSCLC) and other human cancers, severe resistance in clinical patients due to persistent isomerism mutations (EGFR C797S ) remains an insurmountable problem for these inhibitors. PROTAC is a revolutionary technology to overcome the resistance of EGFR inhibitors for effective cancer therapy.
In 2019, Jin group developed a novel class of EGFR degraders based VHL E3 ligase ligands (Figure 1; VHL‐1/VHL‐3), as exemplified by compound MS‐39 (Table 1). 175 Compound MS‐39 could effectively induce the degradation of EGFR mutants with good selectivity but not EGFR WT with DC50 value of 3.3 nM in H3255 (EGFR L858R ) cells. This compound also inhibited the proliferation of other lung cancer cells. In addition, MS‐39 is a potential candidate compound for in vivo pharmacodynamic study due to its good PK properties. In 2020, Ding group reported a class of new PROTACs based on a selective EGFR L858R/T790M inhibitor (XTF‐262) that can efficiently target EGFR L858R/T790M degradation. 176 They compared the abilities of four E3 ligases (VHL, CRBN, cIAP1, and MDM2) in the design of novel EGFR degraders, and found that degradation of EGFR L858R/T790M was effectively induced by compound 14o (Table 1) with VHL ligand (Figure 1; VHL‐1/VHL‐3) and DC50 value of 5.9 nM in H1915 cell line. In 2020, Zhang group reported a series of EGFR degraders based on VHL E3 ligands (Figure 1; VHL‐1/VHL‐3). 177 , 178 The most potent degrader PROTAC‐2 (Table 1) induced degradation of EGFR with a DC50 value of 34.8 nM in HCC‐827 (EGFR e19d ) cell line.
2.2.4. KRAS
Rat sarcoma virus oncogene homologues (RAS) and KRAS are among the most common RAS genes associated with human cancer. 179 The mutational activation of KRAS is one of most frequently events occurring in human cancers and causally related to tumorigenesis, thus KRAS is an effective antitumor drug target. 180 The discovery of KRAS drugs has been challenged in past 30 years because it has a flat structure without approachable pocket for small molecules to bind, thus being an undruggable target. 181 , 182 , 183 , 184 Few small molecules targeting KRAS‐G12C, known as the switch II Pocket (G12C) via covalent binding, was recently approved in May 2021 by Amgen (AMG510‐sotorasib) and Mirati (MRTX849‐adagrasib) for clinical trials. 185 However, it has not been possible to rapidly develop inhibitors of other KRAS onco‐alleles, such as G12D, which is dominant in pancreatic cancer and new drug development strategies still need to be explored.
In 2020, Crews group reported endogenous KRAS G12C degradation by connecting the covalent KRAS inhibitor MRTX849 to VHL ligands (Figure 1; VHL‐1/VHL‐3). 186 After screening, compound LC‐2 (Table 1) was identified as the most potent KRAS G12C degrader with a D max value of 80% and a DC50 value of 0.59 μM in inducing degradation of endogenous KRAS G12C in NCI‐H2030 cell line. At the same time, Lu group reported a new series of KRAS G12C degraders based on the structure of LC‐2. In their study, the promising compound YF‐135 (Table 1) could induce the degradation of KRAS G12C protein with a moderate DC50 value of 3.61 μM in the H358 cells. 187 Compound YF‐135 is the first reversible covalent PROTAC degrader that can induce KRAS G12C degradation through recruitment of VHL‐mediated proteasome.
2.2.5. BCR–ABL
BCR/ABL fusion gene is an antiapoptotic gene with high tyrosine kinase activity, which leads to overproliferation of cells and disorder of cell regulation. 188 Constitutively active BCR–ABL activates the downstream proliferative signaling pathways to cause chronic myelogenous leukemia (CML). 189 Currently, three generations of BCR–ABL inhibitors have been approved to treat CML, mainly including Imatinib (Gleevec), Nilotinib, and Ponatinib. However, drug resistance develops after initial success and various side effects also limit its clinical application. 190 , 191 , 192 Therefore, the discovery of BCR–ABL degraders seems to overcome these problems.
In 2016, Crews group disclosed the first Dasatinib‐based BCR–ABL PROTAC degrader, while this compound achieved only micromolar (>60% at 1 μM) degradation of BCR–ABL, and unable to overcome generic resistant mutants, especially for the T315I mutant. 193 Subsequently, the same group successively designed and synthesized a new VHL‐based (Figure 1; VHL‐1/VHL‐3) BCR–ABL PROTAC degrader GMB‐805 (Table 1), 194 which had more than ten‐times increase in inducing BCL–ABL degradation with improved pharmacokinetic properties and in vivo activity. In 2019, Jiang group combined the same BCR–ABL inhibitor Dasatinib and VHL E3 ligand (Figure 1; VHL‐1/VHL‐3) through extensive optimization of the linker to discover a new class of BCR–ABL degraders. 195 The compound SIAIS178 (Table 1) was identified as the most promising BCR–ABL degrader to induce an effective degradation of wild‐type BCR–ABL in K562 cells as well as several clinically relevant drug‐resistant mutations with a DC50 value of 8.5 nM.
2.2.6. IRAK
Interleukin‐1 receptor‐activated kinase (IRAK) plays a critical role in signal transduction of innate immune responses. 196 The IRAK kinase family consists of four members, IRAK1, IRAK2, IRAK3, and IRAK4, which are potential targets for therapeutic interventions in autoinflammatory diseases. 197 , 198 , 199 IRAK1 is a kind of serine‐threonine protein kinase that involves in many downstream signal transduction of TLRs and IL‐1R, and is an effective target for drug development for the treatment of chronic inflammatory diseases. Despite some progress in the development of IRAK1 inhibitors, IRAK1 remains a very challenging target due to the lack of a domain primarily responsible for its scaffold function. 200 Furthermore, the role of IRAK4 in tumor growth and progression and the mode of action for IRAK4 inhibitors are still unclear. 201
In 2021, the Dai group reported that an IRAK1 degrader JNJ‐1013 can effectively degrade cellular IRAK1 protein with a DC50 value of 3 nM in HBL‐1 cell line (Table 1 and Figure 2; VHL‐2). 202 In addition, JNJ‐1013 could effectively inhibit the downstream signaling pathway of IRAK1 and showed strong antiproliferation effect in activated B‐cell‐like (ABC) DLBCL cell line mutated with MyD88. 202 This study shows that IRAK1 degrader have the potential to treat cancers dependent on the function of IRAK1 stents compared to IRAK1 inhibitors. In 2019, Anderson group developing a series of PROTACs by binding the IRAK4 inhibitor PF‐06650833 to different E3 ligases. 203 The most effective degrader of IRAK4 PROTAC‐3 (Table 1) in PBMC cells was induced by VHL‐based E3 ligands (Figure 1; VHL‐1/VHL‐3) with a DC50 value of 151 nM.
2.2.7. ER
Estrogen receptors (ER) are located in the nucleus and consist of two types of classical nuclear receptors, ERα and ERβ, which mediate the effects of estrogen. 204 , 205 Studies have shown that ER is closely related to the occurrence and development of breast cancer, in which the overexpression of ER α (ER‐positive) accounts for 70% of breast cancer, 206 thus being a well‐established target for the treatment of breast cancer. 207 Currently approved endocrine treatments for breast cancer include aromatase inhibitors (AIs), like letrozole, selective ER modulators (SERMs), like tamoxifen, and selective ER degraders (SERDs), like fulvestrant. 208 However, drug resistance caused by long‐term clinical use limited their therapeutic efficacy. Furthermore, the oral bioavailability of existing breast cancer drugs, such as selective ER degrader fulvestrant, can only be administered by intramuscular injection. 209 , 210 The emergence of PROTACs provides a new approach for the discovery and development of ERα targeted drugs.
In 2019, Wang group developed a new class of highly potent ER degraders, as exemplified by compound ERD‐308 (Table 1 and Figure 2; VHL‐2). 211 ERD‐308 effectively reduced the degradation of ER to achieve DC50 values of 0.17 and 0.43 nM in MCF‐7 and T47D ER+ breast cancer cells, respectively. More importantly, ERD‐308 induced more complete ER degradation than fulvestrant, the only SERD approved by the US FDA, and achieved better cell growth inhibition than fulvestrant in MCF‐7 cell line. In 2020, Tang group designed and developed a series of ER degraders based on PROTACs strategy using the same ER antagonist and VHL ligand in compound ERD‐308. 212 The promising compound AM‐A3 (Table 1) showed a good degradation activity on ER with a DC50 value of 1.1 nM and a D max value of 98% in MCF‐7 cell line. In addition, AM‐A3 can also effectively induce potent antiproliferation with an IC50 value of 13.2 nM in MCF‐7 cells. In 2021, X‐Chem Inc. disclosed a new class of ER PROTAC degraders based on ER antagonist and VHL E3 ligand (Figure 2; VHL‐5) with a new linking position on the phenolic hydroxyl group. 213 With the DNA‐encoded chemical library screening, they found a promising compound PROTAC‐4 (Table 1) which could induce the degradation of ERα with a DC50 value of 37 nM and inhibit the growth of MCF‐7 cells efficiently.
2.2.8. SHP2
Protein tyrosine phosphatase Src homology region 2‐containing protein tyrosine phosphatase 2 (SHP2) mutations in the Src homologous domain of the protein tyrosine phosphatase family exist in most tumor cells and are closely associated with cancer development and poor prognosis of cancer patients 214 , 215 by activating several proliferative signaling pathways for cancer occurrence. 216 Thus, SHP2 was proposed as an attractive target for cancer treatment.
In 2020, Wang group reported a novel class of SHP2 degraders by conjugating with the SHP2 inhibitor SHP099 and the VHL ligand VHL‐2 (Figure 2). 217 The most potent compound SHP2‐D26 (Table 1) effectively induced the degradation of SHP2 protein with a DC50 value of 2.6 nM in MV4;11 cells and with D max to reduce SHP2 protein levels in cancer cells by more than 95%. In addition, compound SHP2‐D26 showed 30‐fold more potent than the SHP2 inhibitor SHP099 in MV4;11 cells.
2.2.9. BRD
The bromo and extral terminal domain family (BET) proteins, including BRD2, BRD3, BRD4, and BRDT, are a class of bromodomain‐containing proteins that have been extensively studied. 218 Over the years, a large number of inhibitors targeting BET proteins have been developed, which have good anticancer efficacy, and some compounds have successfully entered clinical studies. 219 , 220 However, for highly homologous BET proteins, it is difficult to achieve highly selective targeting of BET subtypes. PROTAC technology can improve the selectivity of target protein degradation through the binding of triprimary complex and the recognition of polyubiquitination. Thus, it is a new strategy for the discovery and development of selective BET targeting drugs.
In 2017, Ciulli group reported a novel BRD4 degrader AT‐1 (Table 1) based on VHL ligand by linking on the sulfydryl of VHL‐4 (Figure 2). 221 Recently, they also developed a novel class of BRD4 degraders based on VHL E3 ligands (Figure 2; VHL‐1/VHL‐3), as exemplified by compound AGB1 (Table 1). The degrader AGB1 could completely induce the degradation of BromoTagged target proteins with a low DC50 value of 15 nM and is highly selective to native wild type BET protein. 222
2.2.10. HDAC
Histone deacetylases (HDACs) removes acetyl‐group mainly from histones, thus acting as “epigenetic erases” to play a crucial role in modification of chromosome structure and regulation of gene expression. 223 Frequent alterations of HDACs in human cancers make them attractive cancer targets for the discovery and development of small molecule inhibitors. 224 , 225 Indeed, HDAC inhibitors were found to induce cell cycle arrest, cell death, and differentiation, to blocked angiogenesis as well as to modulate immune response, and few inhibitors have been approved for the treatment of some T‐cell lymphoma and multiple myeloma. 226
Currently, the major challenge of achieving complex selectivity with HDAC inhibitors is considered to be a major source of off‐target toxicity and adverse reactions associated with HDAC inhibitors. 227 , 228 Using PROTAC to improve selectivity may be an effective strategy to solve this problem. 229 , 230
In 2020, Tang group reported the first class of selective HDAC6 degraders employing VHL E3 ubiquitin ligase. 231 The representative compound 3j (Table 1 and Figure 2; VHL‐1/VHL‐3) could effectively induce HDAC6 degradation with high selectivity, not have activity on any known neo‐substrates. The DC50 and D max values of the most potent compound 3j are 7.1 nM and 90%, respectively, in human MM.1S cells. VHL‐5 (Figure 2) was also used in the discovery of HDAC degrader XH‐07‐189 through linking the phenolic hydroxyl by Fischer group in 2021 (Table 1). 232
Although VHL E3 ligand was among the first in the development of PROTAC degraders, the difficulty in reaching the oral availability limited its clinical usage. 233 , 234
3. CRBN LIGANDS AND THEIR UTILIZATIONS IN PROTACS
3.1. Common CRBN ligands
CRBN is a protein composed of 442 amino acids that is substrate‐recognizing subunit, complexed with scaffold protein Cullin‐4 and adaptor protein: damaged DNA binding protein 1 (DDB1) to form the Cullin‐4‐RING ligase (CRL4). 235 , 236 , 237 , 238 , 239 , 240 , 241 CRBN, a substrate receptor of CRL4, has been widely studied for its regulation of a variety of biological processes. 242 , 243 , 244 , 245 , 246 The discovery of CRBN E3 ligase ligands can be traced back to the developed drug thalidomide (Figure 3) as sedative used to treat insomnia in the 1950s. 247 , 248 , 249 , 250 , 251 , 252 , 253 However, it was quickly discontinued in clinic due to its teratogenic effects. 254 , 255 Although it has since been found to treat a number of other diseases, its molecular mechanism remains unclear. 256 , 257 It was not until 2010 that thalidomide was found to directly bind to CRBN, acting as an immunomodulatory drug (IMiD), for its mechanism of action. 4 , 5 , 50 , 258 , 259 , 260 , 261 , 262 , 263 , 264 , 265 In the subsequent decade, the research based on thalidomide and its analogues (e.g., pomalidomide and lenalidomide) (Figure 3) has grown rapidly, mainly in the area of PROTAC and molecule glue. 67 , 266 , 267 , 268 , 269 , 270 , 271 , 272 , 273 , 274 , 275 , 276 , 277 , 278 , 279 , 280 The eutectic structure of lenalidomide and CRBN revealed that the exposure of lenalidomide benzene ring to the solvent region is a potential good binding site (Figure 3; PDB ID: 4CI2). 281 , 282 , 283
FIGURE 3.
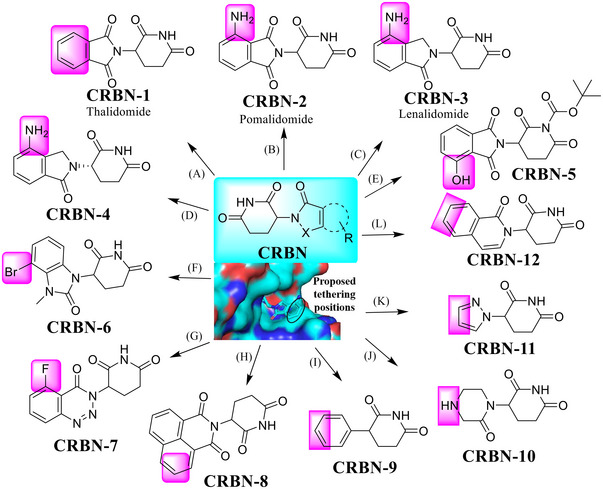
A variety of small molecules that serve as the ligands for CRBN E3 ligase. (A and B) Various small molecules acting as the ligands for CRBN E3 ligase and proposed tethering position in the CRBN ligand based upon a cocrystal structure of lenalidomide in complex with DDB1–CRBN–CUL4 E3 ubiquitin ligase (PDB ID: 4CI2). (C and D) The lenalidomide and its S‐isomer. (E) CRBN E3 ligand for prodrug strategy. (F) Imidazole‐CRBN E3 ligand. (G) Triazin‐CRBN E3 ligand. (H) Isoquinoline‐CRBN E3 ligand. (I) Phenyl glutarimide (PG)‐CRBN E3 ligand. (J) Oxopiperazin glutarimide‐CRBN E3 ligand. (K) Pyrazol glutarimide‐CRBN E3 ligand. (L) Oxoisoquinolin glutarimide‐CRBN E3 ligand.
3.2. The utilizations in PROTACs
A variety of PROTAC degraders have been discovered and developed based on the four potential binding sites of CRBN E3 ligands. 284 , 285
3.2.1. AR
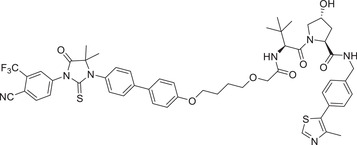
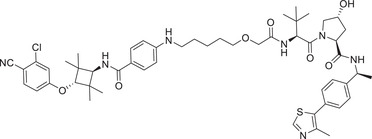
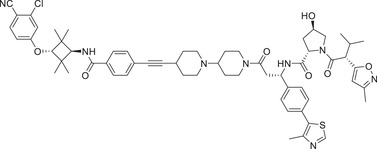
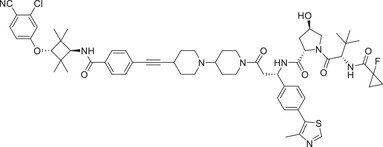
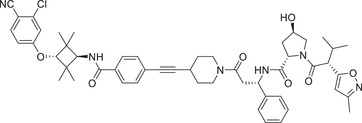
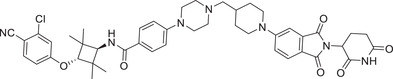
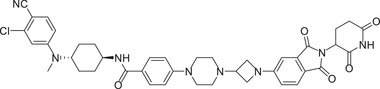
Thalidomide, one of the most classic CRBN E3 ligase ligands, has been successfully applied in the design and discovery of a variety of highly efficient PROTAC degraders, including the first clinical AR degrader ARV‐110 (Table 1 and Figure 3; CRBN‐1/CRBN‐2), the first two published oral AR degraders ARD‐2128 and ARD‐2585 (Table 1 and Figure 3; CRBN‐1/CRBN‐2). 47 , 48 , 49 , 286 , 287 Compound ARV‐110 is an AR degrader drug discovered by Crews group and Arvinas, Inc. to overcome the developed drug resistance of prostate cancer. After further optimized dose escalation exposure with different AR antagonists, ARV‐110 completely degraded AR protein levels in all tested cell lines (LNCaP, VCaP, MCF7, etc.) with DC50 values <1 nM in vitro. In addition, in vivo trials as well as in prostate cancer resistant to enzalutamide showed good efficacy. ARV‐110 is currently in the clinical phase II in patients with mCRPC. In 2018, Arvinas, Inc. disclosed a patent to show a new type of AR degrader PROTAC‐5 (Table 1) using oxoisoquinolin glutarimide ligand CRBN‐12 (Figure 3) as the CRBN recruiter. 242 In 2021, Wang group developed two class of potent AR degraders ARD‐2128 and ARD‐2585 with good oral bioavailability (Table 1 and Figure 3; CRBN‐1/CRBN‐2). The oral bioavailability of compounds ARD‐2128 and ARD‐2585 in mice reached 67 and 51%, respectively, which effectively reduced the level of AR protein and significantly inhibit tumor growth in mice without any observable toxicity. Furthermore, ARD‐2585 showed an excellent degradation ability in MDA‐PCa‐2b (L701H and T877A) cell lines, which laid a solid foundation for overcoming drug resistance.
3.2.2. MEK
MEK1 and MEK2 (mitogen activated protein kinase) are two family members of MAP kinase, which are important signaling molecules in the Ras–RAF–MEK–ERK pathway. 288 MEK1 and MEK2 activate downstream ERK to promote growth and survival of cancer cells, thus playing an important role in tumor development. 289 , 290
The Jin group has been engaged in the discovery of a variety of MEK degraders based on VHL and CRBN E3 ligands. Specifically, in 2020 the group reported several novel and potent VHL recruited MEK1/2 degraders, and the first CRBN recruited MEK1/2 degrader MS910 (Table 1). These compounds effectively and selectively degrade MEK1/2, thus inhibiting downstream signaling pathways and growth of cancer cells. 291
3.2.3. CDK
Currently, 20 proteins have been classified as members of the CDK family, a large part of which are widely recognized as key regulatory factors of cell cycle, transcription, metabolism and/or cell differentiation, and are closely related to the occurrence and development of tumors, with CDK2/4/6 as the best examples of cancer targets. 292 , 293 The inactivation of cyclin‐dependent kinase 2 (CDK2) effectively overcomes the blocking of cell differentiation in acute myeloid leukemia (AML) and is therefore a promising and potential approach for the treatment of AML. 294
In 2021, the Rao group disclosed a highly potent CDK2 depressant CPS2 (Table 1) via the conjugated CRBN ligands (Figure 3; CRBN‐1/CRBN‐2) and the nonselective CDK2 inhibitor JNJ‐7706621. 295 The compound CPS2 effectively induced rapid and efficient degradation of CDK2 with a DC50 value of 2 nM in MV4;11 cell line. In 2020, the Gray group developed a selective CDK2/5 degraders over the other CDKs, as exemplified by compound TMX‐2172 (Table 1 and Figure 3; CRBN‐1/CRBN‐2). 296 Compound TMX‐2172 selectively induced the degradation of CDK2/5 in a time and dose‐dependent manner. Biologically, they demonstrated that degradation of CDK2 protein has antiproliferative activity in ovarian cancer cells (OVCAR8). In 2021, the Yang group developed a novel PROTAC CDK2/4/6 degrader PROTAC‐6 (Table 1 and Figure 3; CRBN‐5), based on the structure of CDK inhibitor Ribociclib's derivatives. Compound PROTAC‐6 was designed through prodrug strategy by introducing a leaving group. 297 In 2021, the Bian group developed a series of selective CDK9 degraders by conjugating a selective CDK9 ligand BAY‐1143572 and CRBN ligand pomalidomide (Figure 3; CRBN‐1/CRBN‐2), as exemplified by compound B03 (Table 1). Compound B03 could also effectively induce the degradation of CDK9 with a DC50 value of 7.6 nM in MV4;11 cells.
3.2.4. ALK
As a tyrosine kinase in the insulin receptor kinase subfamily, anaplastic lymphoma kinase (ALK) is a potent and promising oncogenic factor in lung cancer. 298 Therefore, targeting ALK fusion proteins is an emerging and effective target for the treatment of cancer, especially NSCLC. 299 However, the durability of clinical efficacy of ALK tyrosine kinase inhibitors in NSCLC is often limited by drug resistance within 1−2 years, partly due to acquired ALK resistance mutations. 300 , 301 Therefore, new effective treatment strategies are needed to overcome the defects of clinical resistance to ALK inhibitors.
In 2020, the Jiang group developed a series of ALK degraders based on ALK inhibitor Alectinib and lenalidomide (Figure 3; CRBN‐3). 302 The promising compound SIAIS001 (Table 1) showed good ALK degradation activity with a DC50 value of 3.9 nM and a D max of 70.3% in SR cell line. In 2021, the Xu group developed, based on the same ALK inhibitor Alectinib, a class of ALK degraders employing CRBN ligase. 303 The promising compound PROTAC‐7 (Table 1 and Figure 3; CRBN‐1/CRBN‐2) could effectively induce the degradation of ALK with a DC50 value of 27 nM in H3122 NSCLC cell line.
3.2.5. MDM2
MDM2 is an onco‐protein that acts as an E3 ubiquitin ligase to promote ubiquitylation and degradation of p53, a classical tumor suppressor, which induces growth arrest or apoptosis of cancer cells to suppress tumor formation. 304 , 305 Therefore, targeting MDM2 protein by chemical means would reactivate p53 as a promising strategy for the discovery and development of antitumor drugs. 306 , 307 Over the years, a number of small molecule inhibitors that disrupt MDM2‐p53 interaction have been reported and some were developed in clinic, 308 but none of them has yet been approved by the US FDA for anticancer therapy. The cumulative toxicity of MDM2, as a result of p53 activation to transactivate MDM2 expression, is a major problem affecting the development of these MDM2‐p53 binding inhibitors. 309 Thus, the discovery and development of PROTAC for targeted degradation of MDM2 is a promising strategy to overcome this side‐effect of small molecule inhibitors.
In 2019, the Wang group reported a PROTAC MDM2 degrader. 310 The most active compound MD‐224 (Table 1 and Figure 3; CRBN‐3) effectively induced rapid degradation of MDM2 at the concentrations <1 nM in human leukemia cell lines. The compound showed good anticancer activity in both in vitro and in vivo. Specifically, compound MD224 achieved complete and lasting tumor regression in vivo with a well‐tolerated dose schedule in RS4;11 xenograft tumor models. 310 In 2021, the Tang group disclosed a series of MDM2 degraders based on MDM2 inhibitors and lenalidomide (Figure 3; CRBN‐3). 311 After extensive optimization, the most potent WB214 (Table 1) was shown to have an impressive MDM2‐degradation activity with DC50 value of 4.1 nM against leukemia cells.
3.2.6. AKT
As a downstream component of the phosphoinositol 3‐kinase (PI3K) signaling cascade, serine/threonine kinase AKT positively regulates several key processes in cell proliferation, survival, and metabolism. 312 AKT is overactivated by acquired functional mutation, amplification of upstream oncogenes (receptor tyrosine kinases and PI3K) or inactivation of tumor suppressor genes (PTEN, INPP4B, and PHLPP), which contributes to the malignant phenotypes associated with tumorigenesis. 313 Therefore, targeting AKT is an attractive therapeutic strategy for cancer treatment.
In 2020, the Toker group developed a class of pan‐AKT degraders consisting of a recruiter of the ATP competitive AKT ligand GDC‐0068 coupled with CRBN ligand lenalidomide, CRBN‐3 (Figure 3). 314 Compared with GDC‐0068, the most promising compound INY‐03‐041 effectively induced degradation of all three AKT isoforms and showed enhanced antiproliferative effects. Using the same GDC‐0068, the Jin group also developed a potent AKT degrader MS170 (Table 1), employing CRBN by pomalidomide ligands (Figure 3; CRBN‐1/CRBN‐2). 315 The degrader MS170 showed AKT‐degradation activities with a DC50 value of 32 nM in BT474 breast cancer cells. Compound MS170 also showed good growth inhibition activity against different cancer cell lines, including PC‐3 prostate cancer cells, BT474 and MDA‐MB‐468 breast cancer cells. 314
3.2.7. STAT
Signal transducer and activator of transcription (STAT) is a unique family of proteins that can bind to DNA. 316 The STAT family consists of STAT1, STAT2, STAT3, STAT4, STAT5, and STAT6. 317 , 318 STAT proteins, particularly STAT3 and 5 transduce signals from cytokines and growth factors through their receptors to the nucleus to regulate expression of a variety of genes, leading to cell proliferation, apoptosis inhibition, and chemo‐resistance 319 , 320 (PMID: 36596870). As promising classical targets for the treatment of human cancers, several small molecule inhibitors targeting STAT3 and STAT5 have entered clinical trials, but none of them have been in clinical use. 321 , 322 , 323 PROTAC degraders targeting STAT3/5 has been recently developed.
In 2019, The Wang group first developed a novel STAT3 degrader SD‐36 (Table 1) by employing ligands for CRBN/cullin 4A E3 ligase (Figure 3; CRBN‐3) and STAT3 inhibitors. 324 , 325 , 326 SD‐36 highly selectively induces rapid degradation of STAT3 at low nanomolar concentration in cancer cells. Significantly, complete and lasting tumor regression was achieved by compound SD‐36 in a Molm‐16 xenograft tumor model with a well‐tolerated dose schedule. Very recently, the same group reported a new class of highly effective STAT5 degraders, typically by compound AK‐2292 (Table 1 and Figure 3; CRBN‐3). 327 , 328 The most active compound AK‐2292 effectively induced degradation of STAT5A, STAT5B, and phosphorylated STAT5 proteins in AML cells in a concentration‐ and time‐dependent manner, and exhibited excellent degradation selectivity for STAT5 over all other STAT members. The in vivo tumor model study also showed that AK‐2292 effectively reduced STAT5 protein levels with strong antitumor activity under a tolerated dose schedule. 327 , 328
3.2.8. ER
The compound ARV‐471 (Table 1), a potent oral ER degrader, was discovered by using the S‐isomer of lenalidomide CRBN‐4 (Figure 3). 46 , 49 In 2019, the ER‐targeted PROTAC ARV‐471 developed by Crews group and Arvinas, Inc. was approved by the US FDA as an investigational new drug for the treatment of locally advanced or metastatic ER‐positive/HER2‐negative breast cancer. 46 At present, ARV‐471 has successfully passed the phase II clinical study and started the Phase III clinical study at the beginning of 2023, which is the furthest clinical progress of a degrader in the field of PROTAC technology. The same group also developed a new series of potent ER degrader PROTAC‐8 (Table 1) utilizing a pyrazol glutarimide ligand CRBN‐11 (Figure 3) as the CRBN recruiter described in their patent application. 242
3.2.9. EGFR
In 2021, Jiang group reported a novel class of highly selective and functional PROTAC degraders targeting EGFR, as exemplified by compound SIAIS125 (Table 1) based on Canertinib and CRBN E3 ligand (Figure 3; CRBN‐1/CRBN‐2). 329 Compound SIAIS125 effectively induced the degradation of EGFR protein with a DC50 value of 100 nM in PC9 cell line. The Lei group disclosed a series of novel bifunctional compounds as EGFR degraders using CRBN‐6 (Figure 3) in 2022, as exemplified by compound PROTAC‐9 (Table 1). 330
3.2.10. BET
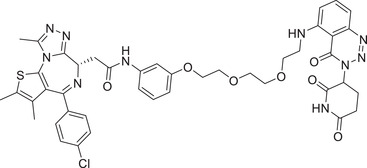
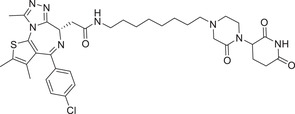
In 2018, the Wang group reported a new BET inhibitor in the class of [1,4]oxazepines, and then synthesized a new class of highly efficient small molecule BET degraders. Through systematic modification and activity screening, the optimal compound QCA570 (Table 1 and Figure 3; CRBN‐3) can effectively induce the degradation of BET protein and inhibit the growth of human acute leukemia cell line at low picmolal concentration. The in vivo study showed that QCA570 achieved complete and sustained tumor regression in a mouse leukemia xenograft model and was well tolerated. In 2019, the Hwang group disclosed a series of bifunctional compounds by conjugation of BET inhibitor JQ‐1 with a novel CRBN modulator (Figure 3; CRBN‐7), as exemplified by compound PROTAC‐10 (Table 1). 331 Compound PROTAC‐10 could effectively induce the degradation of BET proteins with a good DC50 value of 0.32 nM in 22Rv1 cell line. In 2017, the C4 Therapeutics disclosed a patent to show various derivatives of glutarimide as novel CRBN ligands and their applications in discovering BET degrader, as exemplified by compounds oxopiperazin glutarimide‐CRBN‐10 (Figure 3) as CRBN recruiter and PROTAC‐11 as BET degrader (Table 1). 242
3.2.11. JAK2/3
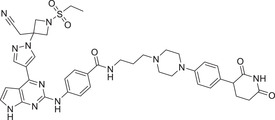
Common Janus kinases (JAKs), JAK2 and JAK3, are involved in a variety of cell signal transduction related to T‐ and B‐cell‐mediated diseases and are closely associated with the pathogenesis of common lymphogenic diseases and leukemia. 332 , 333 Therefore, the development of targeted inhibitors or degraders by intervening in JAK2/3 is a valuable research strategy for reducing the risk of these diseases. In 2022, the Rankovic group developed a novel and potent PROTAC degrader targeting JAK2/3 kinases utilizing a phenyl glutarimide (PG) inhibitor as the CRBN recruiter. 243 The most potent compound SJ10542 (Table 1 and Figure 3; CRBN‐9) could effectively induce the degradation of JAK2/3 with DC50 values of 14 nM and 11 nM in PDX cells. Compound SJ10542 can also inhibit the PDX cells (JAK2‐fusion ALL) with IC50 value of 24 nM.
3.2.12. HCV
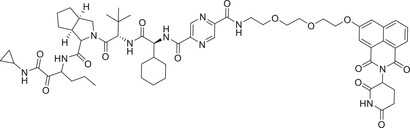
PROTAC techniques are also increasingly being used to degrade viral proteins for potential therapeutic virus‐related indications, like Hemagglutinin 1 Neuraminidase 1 virus (HIN1) and hepatitis C virus (HCV). Recently, the Yang group discovered a novel class of small molecule antivirals that induce proteasomal degradation of HCV proteins. 334 The most promising degrader DGY‐08‐097 (Table 1 and Figure 3; CRBN‐8) could effectively induce the degradation of HCV proteins and contributes to the inhibition of HCV replication in cells.
These reports laid a solid foundation for future discovery of CRBN‐based degraders to treat a variety of human cancers. So far, CRBN‐based E3 ligands confer the best oral availability in all known PROTAC degraders, thus having high potential for clinical application. 46 , 47 , 286 , 287
4. MDM2 LIGANDS AND THEIR UTILIZATIONS IN PROTACS
4.1. Common MDM2 ligands
MDM2 is an E3 ubiquitin ligase that promotes tumor development by binding to the tumor suppressor p53 for degradation. 335 , 336 , 337 , 338 , 339 , 340 , 341 , 342 Over the past 20 years, a number of MDM2 inhibitors were used in the discovery of PROTAC degraders, like RG7388 and Nutlin‐3 series (Figure 4). 19 , 343 , 344 , 345 , 346 From the cocrystal structure of the MDM2 inhibitor nutlin‐3 with MDM2 (Figure 4; PDB ID: 4J3E), 347 the pink moiety is exposed to the solvent and is a potentially ideal linking position in the design of PROTAC degraders.
FIGURE 4.
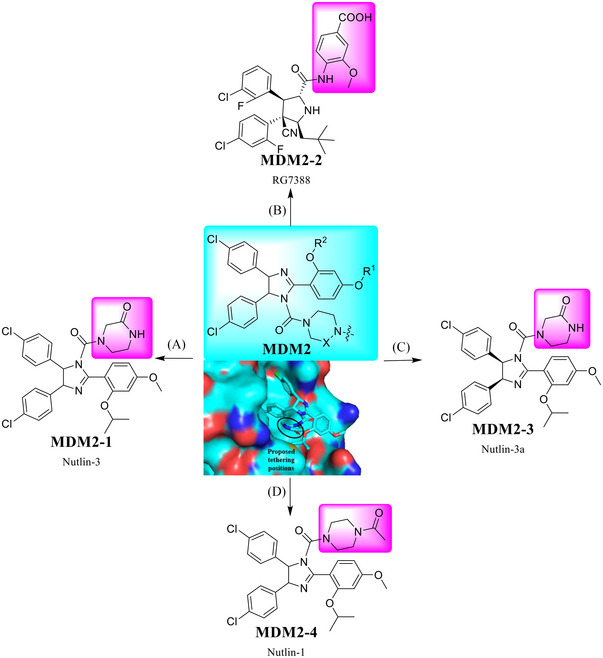
A variety of small molecules that serve as the ligands for MDM2 E3 ligase. (A‐D) Various small molecules (MDM2‐1–4) acting as MDM2 E3 ligase ligands and proposed tethering position in MDM2 ligand, based upon a co‐crystal structure of the MDM2 inhibitor nutlin‐3 and MDM2 protein (PDB ID: 4J3E).
4.2. The utilizations in PROTACs
4.2.1. AR
Nutlin‐3 (Figure 4; MDM2‐1) is a potent MDM2 inhibitor that bind to the p53‐binding pocket of MDM2. 348 , 349 In 2008, Nutlin‐3 were also used for the design of the first all‐small‐molecule type PROTAC‐12 to recruit endogenous MDM2 for targeting AR by Crews group (Table 1). 5
4.2.2. BRD4
In 2019, The Crews group developed an MDM2/nutlin‐based BRD4 degrader A1874 with potent degradation on BRD4 protein and inhibition on the growth of cancer cells harboring a wild type p53 (Table 1 and Figure 4; MDM2‐2). 350
4.2.3. PARP1
The nuclear protein PARP1 has a clear role in DNA damage response and repair and is an effective therapeutic target for human cancers and other human diseases. 351 , 352 Although some small‐molecule PARP1 inhibitors, such as Olaparib and Rucarparib, have been approved by the US FDA to treat ovarian and breast cancer with BRCA mutations, there are still significant challenges such as developed drug resistance limiting their efficacy. TPD is a promising strategy to solve this problem.
In 2018, The Rao group developed the first‐in‐class PARP1‐targeting PROTAC‐13 (Table 1) by connecting the PARP1 inhibitor niraparib to the MDM2 ligand nutlin‐3 (Figure 4; MDM2‐3). 353 Specifically, Compound PROTAC‐13 selectively and significantly induced PARP1 degradation and cell apoptosis in MDA‐MB‐231 cells with a fivefold increased potency in antiproliferative activity, as compared with the PARP1 inhibitors niraparib, olaparib, or veliparib alone, while showing no cytotoxicity in normal cells. 353
4.2.4. MDM2
In 2021, Sheng group developed the first‐in‐class homo‐PROTAC‐14, targeting MDM2 by inducing its self‐degradation (Table 1 and Figure 4; MDM2‐1/MDM2‐4). The compound PROTAC‐14 efficiently induced the dimerization of MDM2 in A549 NSCLC cells, thereby inducing the self‐degradation of MDM2 through the proteasome system, harboring a wild‐type p53. 278
Given an extensive list of small molecule MDM2 inhibitors, the development of MDM2‐based PROTAC degraders should have more opportunity for clinical application.
5. IAP LIGANDS AND THEIR UTILIZATIONS IN PROTACS
5.1. Common IAP ligands
cIAP1, cIAP2, and X‐chromosome‐linked IAP (XIAP) belong to the family of antiapoptotic proteins that play a critical role in the control of apoptotic machinery. 354 , 355 , 356 , 357 , 358 , 359 , 360 , 361 The widespread use of IAPs as an E3 ligase in TPD has drawn much attention from scientists in both academia and industry. 362 , 363 , 364 , 365 , 366 , 367 , 368 Correspondingly, many IAPs inhibitors were discovered and applied in the design of PROTAC degraders. 284 Specifically, the pink moiety is exposed to the solvent as a potentially ideal linking position in the design of PROTAC degraders, based on the crystal structure of IAPs inhibitor with IAPs protein (Figure 5; PDB ID: 5M6H). 369 , 370
FIGURE 5.
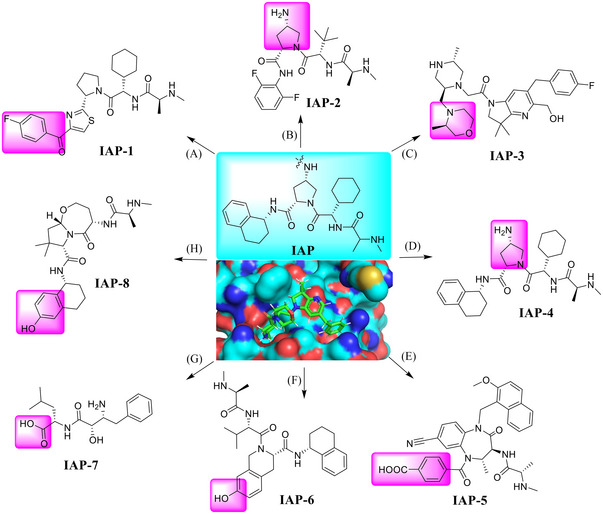
A variety of small molecules that serve as the ligands for IAP E3 ligase. (A–H) Various small molecules (IAP‐1–8) acting as IAP E3 ligase ligands (PDB ID: 5M6H).
5.2. The utilizations in PROTACs
5.2.1. AR
IAP‐targeting heterobifunctional degraders were also named as specific and nongenetic IAP‐dependent protein erasers (SNIPERs). In 2010, Hashimoto described the first concept of SNIPER. 363 In 2018, the Naito group reported AR degrader PROTAC‐15, which were named SNIPERs (Table 1). Specifically, compound PROTAC‐15 was designed using an AR antagonist and a ligand IAP‐1 (Figure 5) for the cellular inhibitor of apoptosis protein 1 (cIAP1) as the E3 ligase. 371
5.2.2. RIPK2
Receptor interaction serine/threonine kinase 2 (RIPK2) is a single‐channel transmembrane protein receptor with intracellular serine/threonine protein kinase activity. 372 In 2021, the Harling group reported a series of PROTAC RIPK2 degraders which recruit members of the inhibitor IAP‐2 (Figure 5) of apoptosis family of E3 ligases. 373 Specifically, the authors identified a series of IAP binding compounds and have successfully applied to the new and highly effective RIPK2 PROTACs PROTAC‐16 and PROTAC‐17 (Table 1).
5.2.3. BTK
Bruton's tyrosine kinase (BTK), nonreceptor cytoplasmic tyrosine kinase, is a key regulator in the B‐cell receptor (BCR) signaling pathway. 374 , 375 BTK plays a key role in B‐cell lymphomas and is a validated target for mantle cell lymphoma (MCL). Although BTK inhibitors are clinically effective in cancer therapy, drug resistance remains a major challenge. 374 , 376 For example, ibrutinib, a covalent inhibitor approved by the US FDA in 2013, was used to treat MCL and ABC‐DLBCL in patients who have developed resistance due to a C481S missense BTK mutation. 377
Currently, a variety of noncovalent degraders have been developed that can effectively degrade wild‐type and Ibrutinib‐resistant C481S BTKS, which may be an effective solution to the problem of serious resistance to BTK inhibitors. IAP‐4 (Figure 5) was used in the discovery of PROTAC BTK degraders by the Harling group in 2019. Reversible compound PROTAC‐18 (Table 1) caused BTK degradation effectively and reduced cIAP1 levels at concentrations of >30 nM. 378 In 2020, Calabrese group designed and synthesized a potent BTK degrader BCPyr (Table 1) by linking amino‐pyrazole derivatives to IAP ligand IAP‐6 (Figure 5) with a DC50 of 800 nM. 379
5.2.4. ER
IAP‐5 (Figure 5), another type of IAP ligand, was used in efforts by Genentech Inc. to develop a series of potent degraders of the ERα, as exemplified by PROTAC‐19 (Table 1). In this report, the authors described a novel application of antibody‐drug coupling technologies that effectively delivered chimeric ERα degrader molecules to targeted MCF7 cells. 380
5.2.5. BCR–ABL
In 2016, compound SNIPER‐2 (Table 1) was reported as a degrader targeting BCR–ABL by linking a BCL‐ABL inhibitor imatinib derivative to IAP‐7 (Figure 5) that binds to cIAP147. 381
5.2.6. BCL‐XL
BCL‐XL is an antiapoptotic BCL2 (B‐cell lymphoma) family member and its overexpression is a key marker of partial escape from apoptosis in cancer. 382 Studies have shown that BCL‐XL is a very effective cancer target. Inhibition of BCL‐XL protein has been widely recognized as a promising strategy for cancer therapy, and several representative anticancer drug candidates in the BCL‐XL inhibitor class have been produced. 383 Although these inhibitors are effective in the treatment of certain hematological malignancies such as CLL and AML, BCL‐XL inhibitors still present significant challenges, such as developed resistance and dose limitations that limit their clinical efficacy. 384 Thus, there is an urgent need to develop a new approach to the development of BCL‐XL targeted drugs, and PROTAC technology comes to the stage. In 2020, the Zheng group developed a series of PROTACs by recruiting IAP E3 ligase (Figure 5; IAP‐8) for BCL‐XL degradation, as exemplified by compound PROTAC‐20 (Table 1). 385 BCL‐XL can be degraded by compound PROTAC‐20 powerfully in multiple cancer cells.
Future development of IAP‐based PROTAC degraders targeting other disease‐associated proteins is expected.
6. KEAP1 LIGANDS AND THEIR UTILIZATIONS IN PROTACS
6.1. Common KEAP1 ligands
The KEAP1 is a substrate‐recognizing subunit of cullin RING ligase (CRL), responsible for ubiquitylation and subsequent degradation of antioxidant transcription factor nuclear factor erythroid 2‐related factor 2 (Nrf2). 26 , 386 , 387 , 388 , 389 Structurally, KEAP1 mainly consists of an N‐terminal bric‐a‐brac of tramtrack, a broad complex (BTB) domain for cullin 3 binding, and a C‐terminal Kelch domain for substrate recruitment. 390 , 391 , 392 , 393 Recently, a number of small molecules have been shown to modulate the KEAP1/NRF2 axis (Figure 6). 284 , 394 , 395 , 396 , 397 , 398 The BTB domain inhibitors, including natural products KEAP1‐1 (a semi‐synthetic oleanolic acid derivative bardoxolone) and KEAP1‐2 (piperlongumine), were discovered as a promising NRF2 modulators utilized in TPD applications by covalently interacting with cysteine residues of KEAP1. 399 On the other hand, a Kelch domain reversible inhibitor, designated as KEAP1 KEAP1‐3, was also discovered as an E3 ligand. 284 The pink portion with exposure to the solvent zone is proposed as a potentially ideal linking position in the design of KEAP1‐based PROTACs (Figure 6; PDB ID: 6QMD). 400
FIGURE 6.
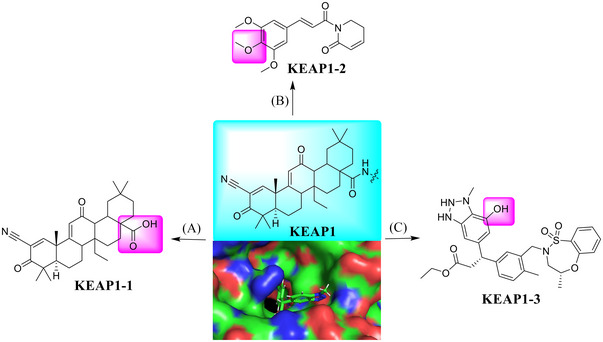
A variety of small molecules that serve as the ligands for KEAP1 E3 ligase. (A–C) Various small molecules (KEAP1‐1–3) acting as KEAP1 E3 ligase ligands (PDB ID: 6QMD).
6.2. The utilizations in PROTACs
6.2.1. BRD4
In 2018, Lu group developed the first‐in‐class KEAP1‐dependent peptide PROTAC. 401 However, the peptide binding component of KEAP1 greatly limits its potential in TPD applications. In 2020, the Daniel group discovered a BRD4‐targeting PROTAC based on the BTB ligand KEAP1‐1, as exemplified by compound CDDO‐JQ1 (Table 1 and Figure 6; KEAP1‐1). 399 In 2021, the Jin group reported that the KEAP1‐1 E3 ligase ligand (Figure 6) can be designed for TPD of BRD2/3/4 proteins, as exemplified by compound MS‐83 (Table 1). 402
6.2.2. CDK
Recently, the Lv group developed a type of PROTAC CDK9 degrader PROTAC‐21 on preprint bioRxiv via hijacking piperlongumine KEAP1‐2 (Figure 6) as a new type of E3 ligand to induce TPD (Table 1). 403 The optimized compound PROTAC‐21 could effectively induce the degradation of CDK9 with a good DC50 value of 9 nM in MOLT4 cell line. Currently, the reported KEAP1‐based PROTAC degraders are still very limited and more studies are needed to evaluate its potential in clinical application.
7. OTHER TYPES OF E3 LIGASE LIGANDS
7.1. DCAF ligands and their utilizations in PROTACs
7.1.1. Common DCAF ligands
DCAF is located in the cell nucleus and represents a substrate recognizing subunit of the CRL4 E3 ligases, including DCAF11, DCAF15, DCAF1, and DCAF16. 404 , 405 , 406 Although the lack of structural information makes it difficult for further optimization of DCAF derivatives, the corresponding ligands have been successfully used in PROTAC (Figure 7A).
FIGURE 7.
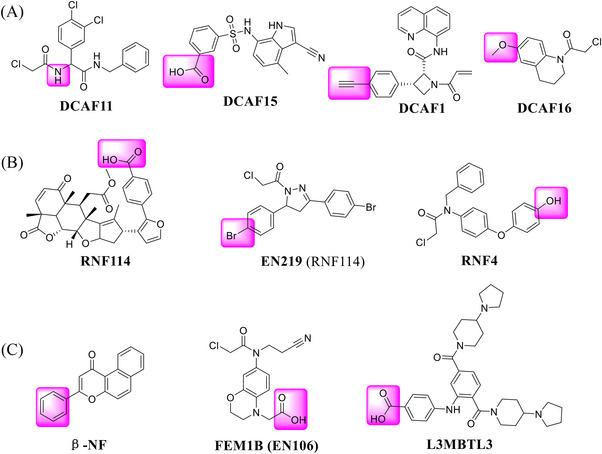
Other types of the ligands for few E3 ligases. (A) Novel DCAF CRL4 E3 ligase ligands used in PROTACs. (B) Novel RNF SUMO E3 ligase ligands used in PROTACs. (C) Novel FEM1B CRL2 E3 ligand, L3MBTL3 Cul4DCAF5 E3 ligand, and β‐NF E3 ligand used in PROTACs.
7.1.2. The utilizations in PROTACs
FKBP12: FK506 binding protein 12 kDa (FKBP12) is a evolutionarily conserved protein with peptidyl prolinase (PPIase) activity existing in eukaryotes. 407 FKBP12 is an effective target for drug development, including immunosuppressive drugs tacrolimus (FK506) and Sirolimus (rapamycin). 408 In 2019, the Cravatt group first reported a new E3 ligase ligand identified as DCAF16 (Figure 7A) and applied it in the discovery of PROTAC FKBP12 degrader KB02‐SLF (Table 1). 406 The same group reported a functional screening strategy performed in a focused library for candidate electrophilic PROTACs, and discovered bifunctional compounds that degrade proteins in human cells via covalently engaging DCAF11 ligand (Figure 7A), as exemplified by compound 21‐SLF in 2021 (Table 1). 409
BRD4: Most recently, Cravatt group continued to expand the scope of DCAF E3 ligands and discovered a new type of E3 ligand reacting with a cysteine (C1113) in the E3 ligase substrate receptor DCAF1 (Figure 7A), leading to identification of the new DCAF1 E3 ligand for targeted BRD4 degradation with compound YT117R (Table 1). 410 On the other hand, in 2020, Chen and Li group developed a new type of DCAF15‐hijacking BRD4 PROTAC DP‐1, based on DCAF15 ligand sulfonamide E7820 that was linked to a pan‐BET inhibitor JQ‐1 (Table 1 and Figure 7A). 411
However, the studies engaging DACF family E3 ligands are very limited at the present time.
7.2. RNF ligands and their utilizations in PROTACs
7.2.1. Common RNF ligands
RNF114 contains a C‐terminal ubiquitin interaction motif and an N‐terminal RING domain, both of which are associated with its E3 ubiquitin ligase activity, 412 , 413 whereas RNF4 is a small ubiquitin‐like modifier (SUMO)‐targeted ubiquitin ligase. 414 , 415 , 416 Nimbolide, a derivative of natural product, was identified as an effective E3 ligand targeting RNF114 protein. 413 Since then, EN219, a fully synthetic RNF114 binder, was also employed as the RNF114 recruiter in the design of PROTAC degraders. 417 , 418 , 419 , 420 , 421 , 422 Recently, the covalent binder, compound RNF4 was discovered as a new RNF4 E3 ligase recruiter (Figure 7B). 423
7.2.2. The utilizations in PROTACs
BRD: The studies using RNF114 or RNF4 as the E3 ligases for PROTAC degraders were exclusively from the Nomura group. In 2019, the group identified a terpenoid natural product Nimbolide as a novel RNF114 E3 ligase ligand (Figure 7B) to recruit RNF114 for targeted degradation of BRD4 protein with compound XH‐2 (Table 1). 413 In 2021, again the same group identified a fully synthetic covalent ligand EN219 (Figure 7B) that targets RNF114 E3 ligase to effectively degrade BRD4 protein with compound ML 2−14 (Table 1). 417 Furthermore, the Nomura group developed cysteine‐reactive small‐molecules that react with the E3 ubiquitin ligase RNF4 by utilizing activity‐based protein profiling (ABPP)‐based covalent ligand screening approaches, and demonstrated that the BRD4 degrader CCW 28‐3 could effectively induce the degradation of BRD4 protein in a proteasome‐ and RNF4‐dependent manner (Table 1 and Figure 7B). 423 This series of exploratory findings have expanded the range of E3 ligase recruiters for PROTAC‐based TPD. In 2022, Nomura group reported a cysteine‐reactive covalent ligand, EN106, that recruited FEM1B, an CRL2 substrate‐recognizing subunit responsive to the cellular reductive stress (Figure 7C), 424 and showed that a PROTAC NJH‐1‐106 (Table 1) connecting EN106 with the pan‐BET inhibitor JQ1 or the kinase inhibitor dasatinib triggered the degradation of BRD4 and BCR–ABL, respectively. 425 Very recently, Crews group reported a novel approach to target protein degradation by hijacking a methyl‐lysine reader protein L3MBTL3, which binds to the Cul4DCAF5 E3 ligase complex (Figure 7C). 426 Here, L3MBTL3 E3 ligand was employed as the L3MBTL3 E3 ligase recruiter in the design, synthesis and biological evaluation of KL‐7 and KL‐4 of BRD2 and FKBP12 proteins, respectively (Table 1). The authors proposed this approach as a general way to extend the E3 ligase toolbox and to explore the full potential of TPD by utilizing E3 ligase complexes associated with other reader proteins in PROTAC designs. 426 This will make already large‐sized PROTAC degrader even bigger.
BCR–ABL: In 2020, the Nomura group also applied RNF114 E3 ligase ligand (Figure 7C) to discover PROTAC BCR–ABL degrader BT‐1, and demonstrated that nimbolide‐recruited BCR–ABL bifunctional compound selectively degraded BCR–ABL over c‐ABL in leukemia cancer cells (Table 1). 427 Compound BT‐1 induced the degradation of BCR–ABL with a DC50 value of 1 μM in K562 cell line.
CRABPs: Cellular retinoic acid binding proteins (CRABPs) are a class of high‐affinity retinoid‐binding proteins mainly found in the cytoplasm. 428 Two members of CRABPs, CRABP‐I, and CRABP‐II, are found in all vertebrates and conserved between species. 429 Research has linked CRABP‐I/II to Alzheimer's disease and many cancers. CRABPs may be promising targets for these diseases. 430 However, direct inhibition of CRABPs' function by small molecule inhibitors remains challenging. In 2019, the Naito group reported an AhR ligand (β‐NF) (Figure 7C) recruiting the AhR E3 ligase complex to develop a novel PROTAC β‐NF‐ATRA (Table 1). β‐NF‐ATRA, a PROTAC that recruits CRABPs, induces CRABPI and CRABPII degradation in an AhR‐dependent manner via the UPS pathway, which may lead to potential synergistic antitumor activity. 431
8. CONCLUSION AND PROSPECTS
As a revolutionary technology in the drug discovery and development, TPD, mainly by the PROTAC and molecular glue, has been utilized to successfully degrade a variety of pathogenic proteins, including oncogenic proteins, viral and bacterial proteins, and has been widely used for potential targeted therapy of a variety of human diseases such as cancers, infectious diseases, neurodegenerative diseases, autoimmune diseases, among others. At present, ARV‐471, a PROTAC degrader of ER for the treatment of breast cancer, is at the phase III clinical trial, along with few dozens of degraders, having entered or about to enter the early phase of clinical trials. The field is blooming with many PROTAC degraders competing for the clinical market.
In the past two decades, although scientists have developed few E3 ligase ligands and successfully applied them to the discovery and development of PROTAC degraders, the highly utilized E3 ligands were mainly limited to VHL and CRBN E3s, a family of multicomponent cullin‐RING ligases. In this review, we summarized approximate 10 reported E3 ligases and a variety of paired small molecule ligands currently used in PROTAC degraders and their applications for degradation of different oncogenic targets. A major challenge for further extension of PROTAC technology in drug discovery is to discover and develop more potent and selective small molecule ligands to couple with a variety of E3 ubiquitin ligases with a family of more than 600 members in the human genome. To this end, two feasible approaches should be fully employed to identify and characterize more potent and selective small molecule ligands for additional E3 ubiquitin ligases: (1) the AI‐based structure prediction of E3 ligases, coupled with computer‐based docking/virtue‐screening; (2) the DNA‐encoded library screening of novel E3 ligase ligands. The breakthrough in discovery of more E3 ligands will certainly expand current scope in the development of effective PROTAC degraders for eventual treatment of various human diseases.
AUTHOR CONTRIBUTION
X. H. drafted and Y. S. revised/finalized the manuscript. All authors have read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflicts of interest in this work.
ETHICS STATEMENT
No ethical approval is required.
ACKNOWLEDGMENTS
This work is supported by the grants from the National Key R&D Program of China (2022YFC3401500 and 2021YFA1101000 to Y. S.) and the Zhejiang Provincial Natural Science Foundation of China (LD22H300003 to Y. S.), and the National Natural Science Foundation of China (92253203 and U22A20317 to Y. S., and 82272637, 82204429 to X. H.).
Han X, Sun Y. PROTACs: A novel strategy for cancer drug discovery and development. MedComm. 2023;4:e290. 10.1002/mco2.290
Contributor Information
Xin Han, Email: xinhan@zju.edu.cn.
Yi Sun, Email: yisun@zju.edu.cn.
DATA AVAILABILITY STATEMENT
All data are freely available from the corresponding author upon request.
REFERENCES
- 1. Humphreys PG, Atkinson SJ, Bamborough P, et al. Design, synthesis, and characterization of I‐BET567, a Pan‐Bromodomain and Extra Terminal (BET) bromodomain oral candidate. J Med Chem. 2022;65(3):2262‐2287. [DOI] [PubMed] [Google Scholar]
- 2. Zhao Y, Murciano‐Goroff YR, Xue JY, et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature. 2021;599(7886):679‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bacterial drug resistance overcome by synthetic restructuring of antibiotics. Nature. 2021; [DOI] [PubMed] [Google Scholar]
- 4. Rodriguez‐Gonzalez A, Cyrus K, Salcius M, et al. Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer. Oncogene. 2008;27(57):7201‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg Med Chem Lett. 2008;18(22):5904‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Edmondson SD, Yang B, Fallan C. Proteolysis targeting chimeras (PROTACs) in ‘beyond rule‐of‐five’ chemical space: recent progress and future challenges. Bioorg Med Chem Lett. 2019;29(13):1555‐1564. [DOI] [PubMed] [Google Scholar]
- 7. Ermondi G, Jimenez DG, Sebastiano MR, Caron G. Rational control of molecular properties is mandatory to exploit the potential of PROTACs as oral drugs. Acs Med Chem Lett. 2021;12(7):1056‐1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lu B, Ye JP. Commentary: PROTACs make undruggable targets druggable: challenge and opportunity. Acta Pharmacol Sin B. 2021;11(10):3335‐3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Poongavanam V, Kihlberg J. PROTAC cell permeability and oral bioavailability: a journey into uncharted territory. Future Med Chem. 2022;14(3):123‐126. [DOI] [PubMed] [Google Scholar]
- 10. Sakamoto KM, Kim KB, Verma R, et al. Development of Protacs to target cancer‐promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003;2(12):1350‐8. [DOI] [PubMed] [Google Scholar]
- 11. Schneekloth JS Jr., Fonseca FN, Koldobskiy M, et al. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004;126(12):3748‐54. [DOI] [PubMed] [Google Scholar]
- 12. Powell CE, Gao Y, Tan L, et al. Chemically induced degradation of Anaplastic Lymphoma Kinase (ALK). J Med Chem. 2018;61(9):4249‐4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol Ther. 2017;174:138‐144. [DOI] [PubMed] [Google Scholar]
- 14. Burslem GM, Crews CM. Small‐molecule modulation of protein homeostasis. Chem Rev. 2017;117(17):11269‐11301. [DOI] [PubMed] [Google Scholar]
- 15. Itoh Y. Chemical protein degradation approach and its application to epigenetic targets. Chem Rec. 2018;18(12):1681‐1700. [DOI] [PubMed] [Google Scholar]
- 16. Paiva SL, Crews CM. Targeted protein degradation: elements of PROTAC design. Curr Opin Chem Biol. 2019;50:111‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pei HX, Peng YR, Zhao QH, Chen YH. Small molecule PROTACs: an emerging technology for targeted therapy in drug discovery. Rsc Adv. 2019;9(30):16967‐16976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xiao M, Zhao J, Wang Q, Liu J, Ma L. Recent advances of degradation technologies based on PROTAC mechanism. Biomolecules. 2022;12(9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu J, Ma J, Liu Y, et al. PROTACs: a novel strategy for cancer therapy. Semin Cancer Biol. 2020;67(Pt 2):171‐179. [DOI] [PubMed] [Google Scholar]
- 20. Garber K. The PROTAC gold rush. Nat Biotechnol. 2022;40(1):12‐16. [DOI] [PubMed] [Google Scholar]
- 21. Bekes M, Langley DR, Crews CM. PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov. 2022; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ha S, Luo G, Xiang H. A comprehensive overview of small‐molecule androgen receptor degraders: recent progress and future perspectives. J Med Chem. 2022;65(24):16128‐16154. [DOI] [PubMed] [Google Scholar]
- 23. Bian Y, Alem D, Beato F, et al. Development of SOS1 inhibitor‐based degraders to target KRAS‐mutant colorectal cancer. J Med Chem. 2022;65(24):16432‐16450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tran NL, Leconte GA, Ferguson FM. Targeted protein degradation: design considerations for PROTAC development. Curr Protoc. 2022;2(12):e611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sinatra L, Yang J, Schliehe‐Diecks J, et al. Solid‐phase synthesis of cereblon‐recruiting selective histone deacetylase 6 degraders (HDAC6 PROTACs) with antileukemic activity. J Med Chem. 2022;65(24):16860‐16878. [DOI] [PubMed] [Google Scholar]
- 26. Chen H, Nguyen NH, Magtoto CM, et al. Design and characterization of a heterobifunctional degrader of KEAP1. Redox Biol. 2023;59:102552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Krasavin M, Adamchik M, Bubyrev A, et al. Synthesis of novel glutarimide ligands for the E3 ligase substrate receptor Cereblon (CRBN): Investigation of their binding mode and antiproliferative effects against myeloma cell lines. Eur J Med Chem. 2023;246:114990. [DOI] [PubMed] [Google Scholar]
- 28. Radhakrishnan S, Hoff O, Muellner MK. Current challenges in small molecule proximity‐inducing compound development for targeted protein degradation using the Ubiquitin proteasomal system. Molecules. 2022;27(23):8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nath D, Shadan S. The ubiquitin system. Nature. 2009;458(7237):421. [DOI] [PubMed] [Google Scholar]
- 30. Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin‐proteasome system. J Biosci. 2006;31(1):137‐155. [DOI] [PubMed] [Google Scholar]
- 31. Xiong Y, Yu C, Zhang Q. Ubiquitin‐proteasome system‐regulated protein degradation in spermatogenesis. Cells. 2022;11(6):1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yadav D, Lee JY, Puranik N, et al. Modulating the ubiquitin‐proteasome system: a therapeutic strategy for autoimmune diseases. Cells. 2022;11(7):1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marini E, Marino M, Gionfriddo G, et al. Investigation into the use of Encorafenib to develop potential PROTACs directed against BRAF(V600E) protein. Molecules. 2022;27(23):8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Si R, Liu N, Wang J, et al. Discovery of selective platelet‐derived growth factor receptor‐beta (PDGFR‐beta) bifunctional small‐molecule degraders. Bioorg Med Chem. 2023;77:117115. [DOI] [PubMed] [Google Scholar]
- 35. Jia X, Han X. Targeting androgen receptor degradation with PROTACs from bench to bedside. Biomed Pharmacother. 2023;158:114112. [DOI] [PubMed] [Google Scholar]
- 36. Jin J, Wu Y, Zhao Z, et al. Small‐molecule PROTAC mediates targeted protein degradation to treat STAT3‐dependent epithelial cancer. JCI Insight. 2022;7(22):e160606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Do TC, Lau JW, Sun C, et al. Hypoxia deactivates epigenetic feedbacks via enzyme‐derived clicking proteolysis‐targeting chimeras. Sci Adv. 2022;8(50):eabq2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kargbo RB. Potent PROTACs targeting EGFR mutants in drug discovery. Acs Med Chem Lett. 2022;13(12):1835‐1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kargbo RB. Discovery of potent PROTACs for the potential treatment of leukemia. Acs Med Chem Lett. 2022;13(12):1831‐1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu Z, Hu M, Yang Y, et al. An overview of PROTACs: a promising drug discovery paradigm. Mol Biomed. 2022;3(1):46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yang J, Ruan Y, Wang D, et al. VHL‐recruiting PROTAC attenuates renal fibrosis and preserves renal function via simultaneous degradation of Smad3 and stabilization of HIF‐2alpha. Cell Biosci. 2022;12(1):203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yu F, Cai M, Shao L, Zhang JH. Targeting protein kinases degradation by PROTACs. Front Chem. 2021;9:679120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li H, Dong J, Cai M, Xu Z, Cheng XD, Qin JJ. Protein degradation technology: a strategic paradigm shift in drug discovery. J Hematol Oncol. 2021;14(1):138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li X, Song Y. Proteolysis‐targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J Hematol Oncol. 2020;13(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. He Y, Khan S, Huo Z, et al. Proteolysis targeting chimeras (PROTACs) are emerging therapeutics for hematologic malignancies. J Hematol Oncol. 2020;13(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Snyder LB, Flanagan JJ, Qian Y, et al. The discovery of ARV‐471, an orally bioavailable estrogen receptor degrading PROTAC for the treatment of patients with breast cancer. Cancer Research. 2021;81(13):44. [Google Scholar]
- 47. Neklesa T, Snyder LB, Willard RR, et al. ARV‐110: An oral androgen receptor PROTAC degrader for prostate cancer. Journal of Clinical Oncology. 2019;37(7):259. [Google Scholar]
- 48. Neklesa T, Snyder LB, Willard RR, et al. ARV‐110: an androgen receptor PROTAC degrader for prostate cancer. Cancer Research. 2018;78(13). [Google Scholar]
- 49. Mullard A. First targeted protein degrader hits the clinic. Nature Reviews Drug Discovery. 2019;18(4):237‐239. [DOI] [PubMed] [Google Scholar]
- 50. Zou Y, Ma D, Wang Y. The PROTAC technology in drug development. Cell Biochem Funct. 2019;37(1):21‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gao HY, Sun XY, Rao Y. PROTAC technology: opportunities and challenges. Acs Med Chem Lett. 2020;11(3):237‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shergalis AG, Marin VL, Rhee DY, et al. CRISPR screen reveals BRD2/4 molecular glue‐like degrader via recruitment of DCAF16. ACS Chem Biol. 2023;18(2):331‐339. [DOI] [PubMed] [Google Scholar]
- 53. Liang D, Jiang L, Bhat SA, et al. Palmitoylation and PDE6delta regulate membrane‐compartment‐specific substrate ubiquitylation and degradation. Cell Rep. 2023;42(1):111999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li N, Zhou Q, Yi Z, Zhang H, Zhou D. Ubiquitin protein E3 ligase ASB9 suppresses proliferation and promotes apoptosis in human spermatogonial stem cell line by inducing HIF1AN degradation. Biol Res. 2023;56(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tseng C, Han Y, Lv Z, et al. The CRL4(DCAF6) E3 ligase ubiquitinates CtBP1/2 to induce apoptotic signalling and promote intervertebral disc degeneration. J Mol Med (Berl). 2023;101(1‐2):171‐181. [DOI] [PubMed] [Google Scholar]
- 56. Ming H, Li B, Jiang J, et al. Protein degradation: expanding the toolbox to restrain cancer drug resistance. J Hematol Oncol. 2023;16(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Villanueva MT. A PROTAC for ALL. Nat Rev Drug Discov. 2022;21(10):713. [DOI] [PubMed] [Google Scholar]
- 58. Sun N, Kabir M, Lee Y, et al. Discovery of the first lactate dehydrogenase proteolysis targeting chimera degrader for the treatment of pancreatic cancer. J Med Chem. 2023;66(1):596‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang R, Zhong T, Bian Q, et al. PROTAC degraders of androgen receptor‐integrated dissolving microneedles for androgenetic alopecia and recrudescence treatment via single topical administration. Small Methods. 2023;7(1):e2201293. [DOI] [PubMed] [Google Scholar]
- 60. He Y, Ju Y, Hu Y, et al. Brd4 proteolysis‐targeting chimera nanoparticles sensitized colorectal cancer chemotherapy. J Control Release. 2023;354:155‐166. [DOI] [PubMed] [Google Scholar]
- 61. Yang C, Yang Y, Li Y, Ni Q, Li J. Radiotherapy‐triggered proteolysis targeting chimera prodrug activation in tumors. J Am Chem Soc. 2023;145(1):385‐391. [DOI] [PubMed] [Google Scholar]
- 62. Cross JM, Coulson ME, Smalley JP, et al. A ‘click’ chemistry approach to novel entinostat (MS‐275) based class I histone deacetylase proteolysis targeting chimeras. RSC Med Chem. 2022;13(12):1634‐1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rishfi M, Krols S, Martens F, et al. Targeted AURKA degradation: Towards new therapeutic agents for neuroblastoma. Eur J Med Chem. 2023;247:115033. [DOI] [PubMed] [Google Scholar]
- 64. Wang P, Zhu H, Liu J, et al. Design, synthesis, and biological evaluation of novel protopanoxadiol derivatives based PROTACs technology for the treatment of lung cancer. Bioorg Chem. 2022;131:106327. [DOI] [PubMed] [Google Scholar]
- 65. Venkatesan J, Murugan D, Rangasamy L. A perspective on newly emerging proteolysis‐targeting strategies in antimicrobial drug discovery. Antibiotics (Basel). 2022;11(12):1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Qi SM, Dong J, Xu ZY, Cheng XD, Zhang WD, Qin JJ. PROTAC: An effective targeted protein degradation strategy for cancer therapy. Front Pharmacol. 2021;12:692574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Inuzuka H, Liu J, Wei W, Rezaeian A‐H. PROTAC technology for the treatment of Alzheimer's disease: advances and perspectives. Acta Materia Medica. 2022;1(1):24‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yokoo H, Naganuma M, Oba M, Demizu Y. Recent advances in PROTAC technology toward new therapeutic modalities. Chem Biodivers. 2022;19(11):e202200828. [DOI] [PubMed] [Google Scholar]
- 69. Loren G, Espuny I, Llorente A, et al. Design and optimization of oestrogen receptor PROTACs based on 4‐hydroxytamoxifen. Eur J Med Chem. 2022;243:114770. [DOI] [PubMed] [Google Scholar]
- 70. Ma L, Wang J, Zhang Y, et al. BRD4 PROTAC degrader MZ1 exerts anticancer effects in acute myeloid leukemia by targeting c‐Myc and ANP32B genes. Cancer Biol Ther. 2022;23(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhu S, Liu J, Xiao D, et al. Design, synthesis, and biological evaluation of Wee1 kinase degraders. Eur J Med Chem. 2022;243:114786. [DOI] [PubMed] [Google Scholar]
- 72. Crunkhorn S. Developing antibody‐based PROTACs. Nat Rev Drug Discov. 2022;21(11):795. [DOI] [PubMed] [Google Scholar]
- 73. Saraswat AL, Vartak R, Hegazy R, Patel A, Patel K. Drug delivery challenges and formulation aspects of proteolysis targeting chimera (PROTACs). Drug Discov Today. 2023;28(1):103387. [DOI] [PubMed] [Google Scholar]
- 74. Burslem GM, Crews CM. Proteolysis‐targeting Chimeras as therapeutics and tools for biological discovery. Cell. 2020;181(1):102‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Reznickova E, Krajcovicova S, Perina M, Kovalova M, Soural M, Krystof V. Modulation of FLT3‐ITD and CDK9 in acute myeloid leukaemia cells by novel proteolysis targeting chimera (PROTAC). Eur J Med Chem. 2022;243:114792. [DOI] [PubMed] [Google Scholar]
- 76. Zimprich A. LRRK2 PROTAC degraders as a potential novel targeting strategy for Parkinson's disease? Mov Disord. 2022;37(11):2193. [DOI] [PubMed] [Google Scholar]
- 77. Liu M, Martyn AP, Quinn RJ. Natural product‐based PROteolysis TArgeting Chimeras (PROTACs). Nat Prod Rep. 2022;39(12):2292‐2307. [DOI] [PubMed] [Google Scholar]
- 78. Zhou XL, Zhao F, Xu YT, et al. A comprehensive review of BET‐targeting PROTACs for cancer therapy. Bioorg Med Chem. 2022;73:117033. [DOI] [PubMed] [Google Scholar]
- 79. Li L, Xu J. The androgen receptor‐targeted proteolysis targeting chimera and other alternative therapeutic choices in overcoming the resistance to androgen deprivation treatment in prostate cancer. Clin Transl Oncol. 2023;25(2):352‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li R, Liu M, Yang Z, Li J, Gao Y, Tan R. Proteolysis‐Targeting Chimeras (PROTACs) in cancer therapy: present and future. Molecules. 2022;27(24):8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sachkova AA, Andreeva DV, Tikhomirov AS, et al. Design, Synthesis and in vitro investigation of cabozantinib‐based PROTACs to target c‐Met kinase. Pharmaceutics. 2022;14(12);2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Murakami Y, Osawa H, Kurohara T, et al. Structure‐activity relationship study of PROTACs against hematopoietic prostaglandin D(2) synthase. RSC Med Chem. 2022;13(12):1495‐1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ghosh S, Ramadas B, Manna D. Targeted protein degradation using the lysosomal pathway. RSC Med Chem. 2022;13(12):1476‐1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tang K, Wang S, Gao W, Song Y, Yu B. Harnessing the cyclization strategy for new drug discovery. Acta Pharm Sin B. 2022;12(12):4309‐4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ding M, Shao Y, Sun D, et al. Design, synthesis, and biological evaluation of BRD4 degraders. Bioorg Med Chem. 2023;78:117134. [DOI] [PubMed] [Google Scholar]
- 86. Yang F, Tan Y, Wu C, et al. dSTORM‐based single‐cell protein quantitative analysis can effectively evaluate the degradation ability of PROTACs. Chembiochem. 2022; [DOI] [PubMed] [Google Scholar]
- 87. Zhou QQ, Xiao HT, Yang F, Wang YD, Li P, Zheng ZG. Advancing targeted protein degradation for metabolic diseases therapy. Pharmacol Res. 2022;188:106627. [DOI] [PubMed] [Google Scholar]
- 88. Wang YW, Lan L, Wang M, et al. PROTACS: A technology with a gold rush‐like atmosphere. Eur J Med Chem. 2023;247:115037. [DOI] [PubMed] [Google Scholar]
- 89. Koroleva OA, Dutikova YV, Trubnikov AV, et al. PROTAC: targeted drug strategy. Principles and limitations. Russ Chem Bull. 2022;71(11):2310‐2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Li Z, Ma S, Zhang L, et al. Targeted protein degradation induced by HEMTACs based on HSP90. J Med Chem. 2023;66(1):733‐751. [DOI] [PubMed] [Google Scholar]
- 91. Ji W, Wang ES, Manz TD, et al. Development of potent and selective degraders of PI5P4Kgamma. Eur J Med Chem. 2023;247:115027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Li J, Cai Z, Li XW, Zhuang C. Natural product‐inspired targeted protein degraders: advances and perspectives. J Med Chem. 2022;65(20):13533‐13560. [DOI] [PubMed] [Google Scholar]
- 93. Hu M, Li Y, Li J, et al. Discovery of potent and selective HER2 PROTAC degrader based Tucatinib with improved efficacy against HER2 positive cancers. Eur J Med Chem. 2022;244:114775. [DOI] [PubMed] [Google Scholar]
- 94. Li Y, Lin S, Gu Z, Chen L, He B. Zinc‐dependent deacetylases (HDACs) as potential targets for treating Alzheimer's disease. Bioorg Med Chem Lett. 2022;76:129015. [DOI] [PubMed] [Google Scholar]
- 95. Ding Y, Xing D, Fei Y, Lu B. Emerging degrader technologies engaging lysosomal pathways. Chem Soc Rev. 2022;51(21):8832‐8876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhou Q, Wu W, Jia K, Qi G, Sun XS, Li P. Design and characterization of PROTAC degraders specific to protein N‐terminal methyltransferase 1. Eur J Med Chem. 2022;244:114830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Guedeney N, Cornu M, Schwalen F, Kieffer C, Voisin‐Chiret AS. PROTAC technology: a new drug design for chemical biology with many challenges in drug discovery. Drug Discov Today. 2023;28(1):103395. [DOI] [PubMed] [Google Scholar]
- 98. Pettersson M, Crews CM. PROteolysis TArgeting Chimeras (PROTACs) ‐ past, present and future. Drug Discov Today Technol. 2019;31:15‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lai AC, Crews CM. Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov. 2017;16(2):101‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Neklesa TK, Winkler JD, Crews CM. Targeted protein degradation by PROTACs. Pharmacol Therapeut. 2017;174:138‐144. [DOI] [PubMed] [Google Scholar]
- 101. Li L, Mi DZ, Pei HX, et al. In vivo target protein degradation induced by PROTACs based on E3 ligase DCAF15. Signal Transduct Tar. 2020;5(1):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ishikawa M, Tomoshige S, Demizu Y, Naito M. Selective degradation of target proteins by chimeric small‐molecular drugs, PROTACs and SNIPERs. Pharmaceuticals‐Base. 2020;13(4):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Dale B, Cheng M, Park KS, Kaniskan HU, Xiong Y, Jin J. Advancing targeted protein degradation for cancer therapy. Nature Reviews Cancer. 2021;21(10):638‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mukhamejanova Z, Tong YC, Xiang Q, Xu F, Pang JY. Recent advances in the design and development of anticancer molecules based on PROTAC technology. Current Medicinal Chemistry. 2021;28(7):1304‐1327. [DOI] [PubMed] [Google Scholar]
- 105. Wang XR, Wang S, Mu HX, et al. Discovery of novel VEGFR‐2‐PROTAC degraders based on the localization of lysine residues via recruiting VHL for the treatment of gastric cancer. Eur J Med Chem. 2022;244:114821. [DOI] [PubMed] [Google Scholar]
- 106. Darwish S, Heimburg T, Ridinger J, et al. Synthesis, biochemical, and cellular evaluation of HDAC6 targeting proteolysis targeting Chimeras. Methods Mol Biol. 2023;2589:179‐193. [DOI] [PubMed] [Google Scholar]
- 107. Lim YS, Yoo SM, Patil V, et al. Orally bioavailable BTK PROTAC active against wild‐type and C481 mutant BTKs in human lymphoma CDX mouse models. Blood Adv. 2023;7(1):92‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Shah VJ, Dikic I. Localization matters in targeted protein degradation. Cell Chem Biol. 2022;29(10):1465‐1466. [DOI] [PubMed] [Google Scholar]
- 109. Li D, Yu D, Li Y, Yang R. A bibliometric analysis of PROTAC from 2001 to 2021. Eur J Med Chem. 2022;244:114838. [DOI] [PubMed] [Google Scholar]
- 110. Li Z, Bai H, Xi X, et al. PROTAC vaccine: a new way to live attenuated vaccines. Clin Transl Med. 2022;12(10):e1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lei T, Xiao Z, Bi W, Cai S, Yang Y, Du H. Targeting small heat shock proteins to degrade aggregates as a potential strategy in neurodegenerative diseases. Ageing Res Rev. 2022;82:101769. [DOI] [PubMed] [Google Scholar]
- 112. Lu D, Yu X, Lin H, et al. Applications of covalent chemistry in targeted protein degradation. Chem Soc Rev. 2022;51(22):9243‐9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Soares P, Gadd MS, Frost J, et al. Group‐based optimization of potent and cell‐active inhibitors of the von hippel‐lindau (VHL) E3 ubiquitin ligase: structure‐activity relationships leading to the chemical probe (2S,4R)‐1‐((S)‐2‐(1‐cyanocyclopropanecarboxamido)‐3,3‐dimethylbutanoyl)‐4‐hydroxy ‐N‐(4‐(4‐methylthiazol‐5‐yl)benzyl)pyrrolidine‐2‐carboxamide (VH298). J Med Chem. 2018;61(2):599‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Frost J, Rocha S, Ciulli A. Von Hippel‐Lindau (VHL) small‐molecule inhibitor binding increases stability and intracellular levels of VHL protein. J Biol Chem. 2021;297(2):100910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bhat SA, Vasi Z, Adhikari R, et al. Ubiquitin proteasome system in immune regulation and therapeutics. Curr Opin Pharmacol. 2022;67:102310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wang J, Park KS, Yu X, et al. A cryptic transactivation domain of EZH2 binds AR and AR's splice variant, promoting oncogene activation and tumorous transformation. Nucleic Acids Res. 2022;50(19):10929‐10946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Weng G, Cai X, Cao D, et al. PROTAC‐DB 2.0: an updated database of PROTACs. Nucleic Acids Res. 2023;51(D1):D1367‐D1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lee B, Kim DG, Lee A, et al. Synthesis and discovery of the first potent proteolysis targeting chimaera (PROTAC) degrader of AIMP2‐DX2 as a lung cancer drug. J Enzyme Inhib Med Chem. 2023;38(1):51‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Frost J, Galdeano C, Soares P, et al. Potent and selective chemical probe of hypoxic signalling downstream of HIF‐alpha hydroxylation via VHL inhibition. Nat Commun. 2016;7:13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Crew AP, Raina K, Dong H, et al. Identification and characterization of Von Hippel‐Lindau‐Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK‐binding kinase 1. J Med Chem. 2018;61(2):583‐598. [DOI] [PubMed] [Google Scholar]
- 121. Buckley DL, Van Molle I, Gareiss PC, et al. Targeting the von Hippel‐Lindau E3 Ubiquitin Ligase using small molecules to disrupt the VHL/HIF‐1 alpha interaction. Journal of the American Chemical Society. 2012;134(10):4465‐4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Galdeano C, Gadd MS, Soares P, et al. Structure‐guided design and optimization of small molecules targeting the protein protein interaction between the von Hippel‐ Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. Journal of Medicinal Chemistry. 2014;57(20):8657‐8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Sun D, Zhang J, Dong G, He S, Sheng C. Blocking non‐enzymatic functions by PROTAC‐mediated targeted protein degradation. J Med Chem. 2022;65(21):14276‐14288. [DOI] [PubMed] [Google Scholar]
- 124. Liu H, Mi Q, Ding X, et al. Discovery and characterization of novel potent BCR‐ABL degraders by conjugating allosteric inhibitor. Eur J Med Chem. 2022;244:114810. [DOI] [PubMed] [Google Scholar]
- 125. Chatterjee DR, Kapoor S, Jain M, Das R, Chowdhury MG, Shard A. PROTACting the kinome with covalent warheads. Drug Discov Today. 2023;28(1):103417. [DOI] [PubMed] [Google Scholar]
- 126. Li G, Lin SS, Yu ZL, et al. A PARP1 PROTAC as a novel strategy against PARP inhibitor resistance via promotion of ferroptosis in p53‐positive breast cancer. Biochem Pharmacol. 2022;206:115329. [DOI] [PubMed] [Google Scholar]
- 127. Marcellino BK, Yang X, Umit Kaniskan H, et al. An MDM2 degrader for treatment of acute leukemias. Leukemia. 2022;37(2):370‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Khan S, Kellish P, Connis N, et al. Co‐targeting BCL‐X(L) and MCL‐1 with DT2216 and AZD8055 synergistically inhibit small‐cell lung cancer growth without causing on‐target toxicities in mice. Cell Death Discov. 2023;9(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Wang C, Zheng C, Wang H, et al. Dual degradation mechanism of GPX4 degrader in induction of ferroptosis exerting anti‐resistant tumor effect. Eur J Med Chem. 2023;247:115072. [DOI] [PubMed] [Google Scholar]
- 130. Zhao HY, Xin M, Zhang SQ. Progress of small molecules for targeted protein degradation: PROTACs and other technologies. Drug Dev Res. 2023;84(2):337‐394. [DOI] [PubMed] [Google Scholar]
- 131. Chen Q, Liu C, Wang W, et al. Optimization of PROTAC ternary complex using DNA encoded library approach. ACS Chem Biol. 2023;18(1):25‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Mai H, Zimmer MH, Miller TF 3rd., Exploring PROTAC cooperativity with coarse‐grained alchemical methods. J Phys Chem B. 2023;127(2): 446‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Chen M, Zhou P, Kong Y, et al. Inducible degradation of oncogenic nucleolin using an aptamer‐based PROTAC. J Med Chem. 2023;66(2):1339‐1348. [DOI] [PubMed] [Google Scholar]
- 134. Zhang X, Zhao T, Sun M, et al. Design, synthesis and biological evaluation of KRAS(G12C)‐PROTACs. Bioorg Med Chem. 2023;78:117153. [DOI] [PubMed] [Google Scholar]
- 135. Galdeano C, Gadd MS, Soares P, et al. Structure‐guided design and optimization of small molecules targeting the protein‐protein interaction between the von Hippel‐Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J Med Chem. 2014;57(20):8657‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Takwale AD, Jo SH, Jeon YU, et al. Design and characterization of cereblon‐mediated androgen receptor proteolysis‐targeting chimeras. Eur J Med Chem. 2020;208:112769. [DOI] [PubMed] [Google Scholar]
- 137. Chen LR, Han LQ, Mao SJ, et al. Discovery of A031 as effective proteolysis targeting chimera (PROTAC) androgen receptor (AR) degrader for the treatment of prostate cancer. European Journal of Medicinal Chemistry. 2021;216:113307. [DOI] [PubMed] [Google Scholar]
- 138. Da Y, Liu SD, Lin P, et al. Design, synthesis, and biological evaluation of small molecule PROTACs for potential anticancer effects. Med Chem Res. 2020;29(2):334‐340. [Google Scholar]
- 139. Kregel S, Wang C, Han X, et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia. 2020;22(2):111‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Shibata N, Nagai K, Morita Y, et al. Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands. J Med Chem. 2018;61(2):543‐575. [DOI] [PubMed] [Google Scholar]
- 141. Itoh Y, Ishikawa M, Kitaguchi R, Sato S, Naito M, Hashimoto Y. Development of target protein‐selective degradation inducer for protein knockdown. Bioorg Med Chem. 2011;19(10):3229‐41. [DOI] [PubMed] [Google Scholar]
- 142. Coleman KG, Crews CM. Proteolysis‐targeting chimeras: harnessing the ubiquitin‐proteasome system to induce degradation of specific target proteins. Annu Rev Canc Biol. 2018;2:41‐58. [Google Scholar]
- 143. Ohoka N, Shibata N, Hattori T, Naito M. Protein knockdown technology: application of ubiquitin ligase to cancer therapy. Curr Cancer Drug Tar. 2016;16(2):136‐146. [DOI] [PubMed] [Google Scholar]
- 144. Wang Y, Jiang X, Feng F, Liu W, Sun H. Degradation of proteins by PROTACs and other strategies. Acta Pharm Sin B. 2020;10(2):207‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. An S, Fu L. Small‐molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. Ebiomedicine. 2018;36:553‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Ma D, Yuan Q, Peng F, et al. Engineered PROTAC‐CID systems for mammalian inducible gene regulation. J Am Chem Soc. 2023;145(3):1593‐1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Wang S, Luo D, Pu C, et al. Discovery of the GSH responsive “Y‐PROTACs” targeting ALK and CDK4/6 as a potential treatment for cancer. Eur J Med Chem. 2023;248:115082. [DOI] [PubMed] [Google Scholar]
- 148. Fang Q, Cole RN, Wang Z. Mechanisms and targeting of proteosome‐dependent androgen receptor degradation in prostate cancer. Am J Clin Exp Urol. 2022;10(6):366‐376. [PMC free article] [PubMed] [Google Scholar]
- 149. Kaur SD, Bedi N, Kumar D, Kapoor DN. PROTACs: Promising approach for anticancer therapy. Cancer Lett. 2023;556:216065. [DOI] [PubMed] [Google Scholar]
- 150. Su W, Tan M, Wang Z, et al. Targeted degradation of PD‐L1 and activation of the STING pathway by carbon‐dot‐based PROTACs for cancer immunotherapy. Angew Chem Int Ed Engl. 2023;62(11):e202218128. [DOI] [PubMed] [Google Scholar]
- 151. Tian C, Burgess K. PROTAC compatibilities, degrading cell‐surface receptors, and the sticky problem of finding a molecular glue. ChemMedChem. 2021;16(2):316‐318. [DOI] [PubMed] [Google Scholar]
- 152. Dong G, Ding Y, He S, Sheng C. Molecular glues for targeted protein degradation: from serendipity to rational discovery. J Med Chem. 2021;64(15):10606‐10620. [DOI] [PubMed] [Google Scholar]
- 153. Kim GY, Song CW, Yang YS, et al. Chemical degradation of androgen receptor (AR) using bicalutamide analog‐thalidomide PROTACs. Molecules. 2021;26(9):2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Scott DE, Rooney TPC, Bayle ED, et al. Systematic Investigation of the permeability of androgen receptor PROTACs. Acs Med Chem Lett. 2020;11(8):1539‐1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Xie J, He H, Kong W, et al. Targeting androgen receptor phase separation to overcome antiandrogen resistance. Nat Chem Biol. 2022;18(12):1341‐1350. [DOI] [PubMed] [Google Scholar]
- 156. Katleba KD, Tsamouri MM, Jathal M, et al. Androgen receptor‐dependent regulation of metabolism in high grade bladder cancer cells. Sci Rep. 2023;13(1):1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Marcias G, Erdmann E, Lapouge G, et al. Identification of novel truncated androgen receptor (AR) mutants including unreported pre‐mRNA splicing variants in the 22Rv1 hormone‐refractory prostate cancer (PCa) cell line. Hum Mutat. 2010;31(1):74‐80. [DOI] [PubMed] [Google Scholar]
- 158. Morova T, McNeill DR, Lallous N, et al. Androgen receptor‐binding sites are highly mutated in prostate cancer. Nat Commun. 2020;11(1):832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Hickey TE, Selth LA, Chia KM, et al. The androgen receptor is a tumor suppressor in estrogen receptor‐positive breast cancer. Nat Med. 2021;27(2):310‐320. [DOI] [PubMed] [Google Scholar]
- 160. Yan K. Androgen receptor pathway inhibitor combination in prostate cancer. Nat Rev Urol. 2022;19(10):627. [DOI] [PubMed] [Google Scholar]
- 161. Salami J, Alabi S, Willard RR, et al. Androgen receptor degradation by the proteolysis‐targeting chimera ARCC‐4 outperforms enzalutamide in cellular models of prostate cancer drug resistance. Commun Biol. 2018;1:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Kargbo RB. Treatment of Prostate cancers and Kennedy's disease by PROTAC‐androgen receptor degradation. Acs Med Chem Lett. 2019;10(5):701‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Han X, Zhao LJ, Xiang WG, et al. Discovery of highly potent and efficient PROTAC degraders of Androgen Receptor (AR) by employing weak binding affinity VHL E3 ligase ligands. Journal of Medicinal Chemistry. 2019;62(24):11218‐11231. [DOI] [PubMed] [Google Scholar]
- 164. Zhao LJ, Han X, Lu JF, McEachern D, Wang SM. A highly potent PROTAC androgen receptor (AR) degrader ARD‐61 effectively inhibits AR‐positive breast cancer cell growth in vitro tumor growth in vivo. Neoplasia. 2020;22(10):522‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Kregel S, Wang C, Han X, et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Cancer Research. 2020;80(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Han X, Wang C, Qin C, et al. Discovery of ARD‐69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. Journal of Medicinal Chemistry. 2019;62(2):941‐964. [DOI] [PubMed] [Google Scholar]
- 167. Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16(5):281‐98. [DOI] [PubMed] [Google Scholar]
- 168. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170(1):17‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Thevakumaran N, Lavoie H, Critton DA, et al. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat Struct Mol Biol. 2015;22(1):37‐43. [DOI] [PubMed] [Google Scholar]
- 170. Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF‐mutant melanoma. Nature. 2010;467(7315):596‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Alabi S, Jaime‐Figueroa S, Yao Z, et al. Mutant‐selective degradation by BRAF‐targeting PROTACs. Nat Commun. 2021;12(1):920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169‐81. [DOI] [PubMed] [Google Scholar]
- 173. Bai Y, Liu X, Zheng L, et al. Comprehensive profiling of EGFR mutation subtypes reveals genomic‐clinical associations in non‐small‐cell lung cancer patients on first‐generation EGFR inhibitors. Neoplasia. 2023;38:100888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Huntington SF, Schuster SJ, Ding W, et al. DTRMWXHS‐12, a novel Bruton tyrosine kinase inhibitor, in combination with everolimus and pomalidomide in patients with relapsed/refractory lymphomas: an open‐label, multicenter, phase 1a/1b study. Am J Hematol. 2023;98(5):739‐749. [DOI] [PubMed] [Google Scholar]
- 175. Cheng M, Yu X, Lu K, et al. Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small‐molecule degraders. J Med Chem. 2020;63(3):1216‐1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Zhang X, Xu F, Tong L, et al. Design and synthesis of selective degraders of EGFR(L858R/T790M) mutant. Eur J Med Chem. 2020;192:112199. [DOI] [PubMed] [Google Scholar]
- 177. Zhao HY, Yang XY, Lei H, et al. Discovery of potent small molecule PROTACs targeting mutant EGFR. Eur J Med Chem. 2020;208:112781. [DOI] [PubMed] [Google Scholar]
- 178. Zhang H, Zhao HY, Xi XX, et al. Discovery of potent epidermal growth factor receptor (EGFR) degraders by proteolysis targeting chimera (PROTAC). Eur J Med Chem. 2020;189:112061. [DOI] [PubMed] [Google Scholar]
- 179. Scheffzek K, Ahmadian MR, Kabsch W, et al. The Ras‐RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science. 1997;277(5324):333‐338. [DOI] [PubMed] [Google Scholar]
- 180. Tabernero J, Van Cutsem E, Garralda E, et al. A phase Ib/II study of WNT974 + Encorafenib + cetuximab in patients with BRAF V600E‐mutant KRAS wild‐type metastatic colorectal cancer. Oncologist. 2023;28(3):230‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Bian Y, Bi G, Shan G, et al. Characterization of infiltrating immune cells and secretory or membrane‐associated proteins in KRAS lung adenocarcinoma. J Immunol Res. 2023;2023:4987832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Wang X, Allen S, Blake JF, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRAS(G12D) inhibitor. J Med Chem. 2022;65(4):3123‐3133. [DOI] [PubMed] [Google Scholar]
- 183. Kemp SB, Cheng N, Markosyan N, et al. Efficacy of a small‐molecule inhibitor of KrasG12D in immunocompetent models of pancreatic cancer. Cancer Discov. 2023;13(2):298‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Hallin J, Bowcut V, Calinisan A, et al. Anti‐tumor efficacy of a potent and selective non‐covalent KRAS(G12D) inhibitor. Nat Med. 2022;28(10):2171‐2182. [DOI] [PubMed] [Google Scholar]
- 185. Ostrem JM, Shokat KM. Direct small‐molecule inhibitors of KRAS: from structural insights to mechanism‐based design. Nat Rev Drug Discov. 2016;15(11):771‐785. [DOI] [PubMed] [Google Scholar]
- 186. Bond MJ, Chu L, Nalawansha DA, Li K, Crews CM. Targeted degradation of oncogenic KRAS(G12C) by VHL‐recruiting PROTACs. ACS Cent Sci. 2020;6(8):1367‐1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Yang F, Wen Y, Wang C, et al. Efficient targeted oncogenic KRAS(G12C) degradation via first reversible‐covalent PROTAC. Eur J Med Chem. 2022;230:114088. [DOI] [PubMed] [Google Scholar]
- 188. Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib‐resistant Philadelphia chromosome‐positive leukemias. N Engl J Med. 2006;354(24):2531‐41. [DOI] [PubMed] [Google Scholar]
- 189. Reddy EP, Aggarwal AK. The ins and outs of bcr‐abl inhibition. Genes Cancer. 2012;3(5‐6):447‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Rittavee Y, Artus J, Desterke C, et al. miR‐495‐3p sensitizes BCR‐ABL1‐expressing leukemic cells to tyrosine kinase inhibitors by targeting multidrug resistance 1 gene in T315I mutated cells. Exp Hematol. 2023;118:40‐52. [DOI] [PubMed] [Google Scholar]
- 191. Yin Z, Liao M, Yan R, et al. Transcriptome‐ and metabolome‐based candidate mechanism of BCR‐ABL‐independent resistance to olverembatinib in Philadelphia chromosome‐positive acute lymphoblastic leukemia. Funct Integr Genomics. 2023;23(1):53. [DOI] [PubMed] [Google Scholar]
- 192. Pophali PA, Patnaik MM. The role of new tyrosine kinase inhibitors in chronic myeloid leukemia. Cancer J. 2016;22(1):40‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Lai AC, Toure M, Hellerschmied D, et al. Modular PROTAC design for the degradation of oncogenic BCR‐ABL. Angew Chem Int Ed Engl. 2016;55(2):807‐810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Burslem GM, Bondeson DP, Crews CM. Scaffold hopping enables direct access to more potent PROTACs with in vivo activity. Chem Commun (Camb). 2020;56(50):6890‐6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Liu H, Ding X, Liu L, et al. Discovery of novel BCR‐ABL PROTACs based on the cereblon E3 ligase design, synthesis, and biological evaluation. Eur J Med Chem. 2021;223:113645. [DOI] [PubMed] [Google Scholar]
- 196. Barton GM, Medzhitov R. Toll‐like receptor signaling pathways. Science. 2003;300(5625):1524‐1525. [DOI] [PubMed] [Google Scholar]
- 197. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88‐IRAK4‐IRAK2 complex in TLR/IL‐1R signalling. Nature. 2010;465(7300):885‐890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Ferrao R, Zhou H, Shan Y, et al. IRAK4 dimerization and trans‐autophosphorylation are induced by Myddosome assembly. Mol Cell. 2014;55(6):891‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Cohen P. The TLR and IL‐1 signalling network at a glance. J Cell Sci. 2014;127(Pt 11):2383‐2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Wee ZN, Yatim SM, Kohlbauer VK, et al. IRAK1 is a therapeutic target that drives breast cancer metastasis and resistance to paclitaxel. Nat Commun. 2015;6:8746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Chen Y, Bai G, Ning Y, et al. Design and synthesis of Imidazo[1,2‐b]pyridazine IRAK4 inhibitors for the treatment of mutant MYD88 L265P diffuse large B‐cell lymphoma. Eur J Med Chem. 2020;190:112092. [DOI] [PubMed] [Google Scholar]
- 202. Fu L, Zhang J, Shen B, et al. Discovery of highly potent and selective IRAK1 degraders to probe scaffolding functions of IRAK1 in ABC DLBCL. J Med Chem. 2021;64(15):10878‐10889. [DOI] [PubMed] [Google Scholar]
- 203. Nunes J, McGonagle GA, Eden J, et al. Targeting IRAK4 for degradation with PROTACs. Acs Med Chem Lett. 2019;10(7):1081‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Wnuk A, Przepiorska K, Pietrzak BA, Kajta M. Emerging evidence on membrane estrogen receptors as novel therapeutic targets for central nervous system pathologies. Int J Mol Sci. 2023;24(4):4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Shen K, Yu H, Xie B, et al. Anticancer or carcinogenic? The role of estrogen receptor beta in breast cancer progression. Pharmacol Ther. 2023;242:108350. [DOI] [PubMed] [Google Scholar]
- 206. Choupani E, Mahmoudi Gomari M, Zanganeh S, et al. Newly developed targeted therapies against the androgen receptor in triple‐negative breast cancer: A review. Pharmacol Rev. 2023;75(2):309‐327. [DOI] [PubMed] [Google Scholar]
- 207. Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem. 2006;6(3):181‐202. [PubMed] [Google Scholar]
- 208. Yoh K, Ikeda K, Horie K, Inoue S. Roles of estrogen, estrogen receptors, and estrogen‐related receptors in skeletal muscle: regulation of mitochondrial function. Int J Mol Sci. 2023;24(3):1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Mohammed Alwan A, Tavakol Afshari J, Afzaljavan F. Significance of the estrogen hormone and single nucleotide polymorphisms in the progression of breast cancer among female. Arch Razi Inst. 2022;77(3):943‐958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Zlotos DP, Kronenberger T, Laufer SA. Anticancer drug conjugates incorporating estrogen receptor ligands. Pharmaceutics. 2022;15(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Hu J, Hu B, Wang M, et al. Discovery of ERD‐308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER). J Med Chem. 2019;62(3):1420‐1442. [DOI] [PubMed] [Google Scholar]
- 212. Roberts BL, Ma ZX, Gao A, et al. Two‐stage strategy for development of proteolysis targeting chimeras and its application for estrogen receptor degraders. ACS Chem Biol. 2020;15(6):1487‐1496. [DOI] [PubMed] [Google Scholar]
- 213. Disch JS, Duffy JM, Lee ECY, et al. Bispecific estrogen receptor alpha degraders incorporating novel binders identified using DNA‐encoded chemical library screening. J Med Chem. 2021;64(8):5049‐5066. [DOI] [PubMed] [Google Scholar]
- 214. Torrente E, Fodale V, Ciammaichella A, et al. Discovery of a novel series of imidazopyrazine derivatives as potent SHP2 allosteric inhibitors. Acs Med Chem Lett. 2023;14(2):156‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Tang T, Zhou J, Zhang LX, et al. Targeting SHP2 reverses BRAF inhibitor tolerance in anaplastic thyroid carcinoma. Anticancer Agents Med Chem. 2023; [DOI] [PubMed] [Google Scholar]
- 216. Mi D, Li Y, Chen Y. Small‐molecule modulators targeting SHP2 for cancer therapy. Anticancer Agents Med Chem. 2023;23(5):498‐504. [DOI] [PubMed] [Google Scholar]
- 217. Wang M, Lu J, Wang M, Yang CY, Wang S. Discovery of SHP2‐D26 as a first, potent, and effective PROTAC degrader of SHP2 protein. J Med Chem. 2020;63(14):7510‐7528. [DOI] [PubMed] [Google Scholar]
- 218. Winter GE, Buckley DL, Paulk J, et al. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348(6241):1376‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Qin C, Hu Y, Zhou B, et al. Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra‐terminal (BET) proteins capable of inducing complete and durable tumor regression. J Med Chem. 2018;61(15):6685‐6704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Raina K, Lu J, Qian Y, et al. PROTAC‐induced BET protein degradation as a therapy for castration‐resistant prostate cancer. Proc Natl Acad Sci U S A. 2016;113(26):7124‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Gadd MS, Testa A, Lucas X, et al. Structural basis of PROTAC cooperative recognition for selective protein degradation. Nat Chem Biol. 2017;13(5):514‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Bond AG, Craigon C, Chan KH, et al. Development of BromoTag: a “Bump‐and‐Hole”‐PROTAC system to induce potent, rapid, and selective degradation of tagged target proteins. J Med Chem. 2021;64(20):15477‐15502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Thoma C. Kidney cancer: combination of HDAC inhibitor with IL‐2 promising. Nat Rev Urol. 2017;14(11):639. [DOI] [PubMed] [Google Scholar]
- 224. Liu G, Barczak W, Lee LN, et al. The HDAC inhibitor zabadinostat is a systemic regulator of adaptive immunity. Commun Biol. 2023;6(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Zhang X, Yuan Z, Zhang Y, et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27(2):197‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2017;18(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Melesina J, Simoben CV, Praetorius L, Bulbul EF, Robaa D, Sippl W. Strategies to design selective histone deacetylase inhibitors. ChemMedChem. 2021;16(9):1336‐1359. [DOI] [PubMed] [Google Scholar]
- 228. Patel P, Wahan SK, Vishakha S, et al. Recent progress in histone deacetylase (HDAC) 1 inhibitors as anticancer agent. Curr Cancer Drug Targets. 2022;23(1):47‐70. [DOI] [PubMed] [Google Scholar]
- 229. Yang H, Lv W, He M, et al. Plasticity in designing PROTACs for selective and potent degradation of HDAC6. Chem Commun (Camb). 2019;55(98):14848‐14851. [DOI] [PubMed] [Google Scholar]
- 230. An Z, Lv W, Su S, Wu W, Rao Y. Developing potent PROTACs tools for selective degradation of HDAC6 protein. Protein Cell. 2019;10(8):606‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Yang K, Wu H, Zhang Z, et al. Development of selective histone deacetylase 6 (HDAC6) degraders recruiting Von Hippel‐Lindau (VHL) E3 ubiquitin ligase. Acs Med Chem Lett. 2020;11(4):575‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Xiong Y, Donovan KA, Eleuteri NA, et al. Chemo‐proteomics exploration of HDAC degradability by small molecule degraders. Cell Chem Biol. 2021;28(10):1514‐1527 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Wang C, Zhang YJ, Wang J, Xing DM. VHL‐based PROTACs as potential therapeutic agents: recent progress and perspectives. European Journal of Medicinal Chemistry. 2022;227. [DOI] [PubMed] [Google Scholar]
- 234. Diehl CJ, Ciulli A. Discovery of small molecule ligands for the von Hippel‐Lindau (VHL) E3 ligase and their use as inhibitors and PROTAC degraders. Chem Soc Rev. 2022; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Ito T, Ando H, Handa H. Teratogenic effects of thalidomide: molecular mechanisms. Cell Mol Life Sci. 2011;68(9):1569‐1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Chamberlain PP, Lopez‐Girona A, Miller K, et al. Structure of the human Cereblon‐DDB1‐lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nat Struct Mol Biol. 2014;21(9):803‐809. [DOI] [PubMed] [Google Scholar]
- 237. Farrell BM, Gerth F, Yang CR, Yeh JT. A synthetic KLHL20 ligand to validate CUL3(KLHL20) as a potent E3 ligase for targeted protein degradation. Genes Dev. 2022;36(17‐18):1031‐1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Dai MY, Shi YY, Wang AJ, Liu XL, Liu M, Cai HB. High‐potency PD‐1/PD‐L1 degradation induced by Peptide‐PROTAC in human cancer cells. Cell Death Dis. 2022;13(11):924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Zhai J, Li C, Sun B, et al. Sunitinib‐based Proteolysis Targeting Chimeras (PROTACs) reduced the protein levels of FLT‐3 and c‐KIT in leukemia cell lines. Bioorg Med Chem Lett. 2022;78:129041. [DOI] [PubMed] [Google Scholar]
- 240. Jaiswal A, Jaiswal A, Williamson EA, et al. Resistance to the BCL‐XL degrader DT2216 in T‐cell acute lymphoblastic leukemia is rare and correlates with decreased BCL‐XL proteolysis. Cancer Chemother Pharmacol. 2023;91(1):89‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Cantley J, Ye X, Rousseau E, et al. Selective PROTAC‐mediated degradation of SMARCA2 is efficacious in SMARCA4 mutant cancers. Nat Commun. 2022;13(1):6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Kazantsev A, Krasavin M. Ligands for cereblon: 2017–2021 patent overview. Expert Opin Ther Pat. 2022;32(2):171‐190. [DOI] [PubMed] [Google Scholar]
- 243. Alcock LJ, Chang Y, Jarusiewicz JA, et al. Development of potent and selective Janus kinase 2/3 directing PG‐PROTACs. Acs Med Chem Lett. 2022;13(3):475‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Heim C, Pliatsika D, Mousavizadeh F, et al. De‐novo design of cereblon (CRBN) effectors guided by natural hydrolysis products of thalidomide derivatives. J Med Chem. 2019;62(14):6615‐6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Heim C, Spring AK, Kirchgassner S, Schwarzer D, Hartmann MD. Identification and structural basis of C‐terminal cyclic imides as natural degrons for cereblon. Biochem Biophys Res Commun. 2022;637:66‐72. [DOI] [PubMed] [Google Scholar]
- 246. Heim C, Spring AK, Kirchgassner S, Schwarzer D, Hartmann MD. Cereblon neo‐substrate binding mimics the recognition of the cyclic imide degron. Biochem Biophys Res Commun. 2023;646:30‐35. [DOI] [PubMed] [Google Scholar]
- 247. Loos H. [Clinical experience with the sedative contergan‐forte & the sedative contergan which is administered daily]. Medizinische. 1958;12(12):482‐483. [PubMed] [Google Scholar]
- 248. Zhang Y, Mi D, Chen Y. An instructive attempt on developing aptamer‐constructed PROTAC for breast cancer treatment. Mol Ther Nucleic Acids. 2022;30:351‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Wu Y, Yang Y, Wang W, et al. PROTAC technology as a novel tool to identify the target of lathyrane diterpenoids. Acta Pharm Sin B. 2022;12(11):4262‐4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Potts MB. How to kill an ERKsome target: PROTACs deliver the deathblow. Cell Chem Biol. 2022;29(11):1569‐1571. [DOI] [PubMed] [Google Scholar]
- 251. Zhang X, Zhang Z, Xue X, et al. PROTAC degrader of estrogen receptor alpha targeting DNA‐binding domain in breast cancer. ACS Pharmacol Transl Sci. 2022;5(11):1109‐1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Park S, Kim D, Lee W, et al. Discovery of pan‐IAP degraders via a CRBN recruiting mechanism. Eur J Med Chem. 2023;245(Pt 2):114910. [DOI] [PubMed] [Google Scholar]
- 253. Kato JY, Korenaga S, Iwakura M. Discovery of a potent and subtype‐selective TYK2 degrader based on an allosteric TYK2 inhibitor. Bioorg Med Chem Lett. 2023;79:129083. [DOI] [PubMed] [Google Scholar]
- 254. Gordon G. The mechanism of thalidomide deformities correlated with the pathogenic effects of prolonged dosage in adults. Dev Med Child Neurol. 1966;8(6):761‐7. [DOI] [PubMed] [Google Scholar]
- 255. Fletcher I. Review of the treatment of thalidomide children with limb defeciency in Great Britain. Clin Orthop Relat Res. 1980;(148):18‐25. [PubMed] [Google Scholar]
- 256. Grosshans E, Illy G. Thalidomide therapy for inflammatory dermatoses. Int J Dermatol. 1984;23(9):598‐602. [DOI] [PubMed] [Google Scholar]
- 257. Sheskin J. The treatment of lepra reaction in lepromatous leprosy. Fifteen years' experience with thalidomide. Int J Dermatol. 1980;19(6):318‐22. [DOI] [PubMed] [Google Scholar]
- 258. Quach H, Ritchie D, Stewart AK, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia. 2010;24(1):22‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Hu Z, Crews CM. Recent developments in PROTAC‐mediated protein degradation: from bench to clinic. Chembiochem. 2022;23(2):e202100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Bondeson DP, Crews CM. Targeted protein degradation by small molecules. Annu Rev Pharmacol Toxicol. 2017;57:107‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Dong L, Zieren RC, Xue W, de Reijke TM, Pienta KJ. Metastatic prostate cancer remains incurable, why? Asian J Urol. 2019;6(1):26‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Wang ML, Lu JF, Wang M, Yang CY, Wang SM. Discovery of SHP2‐D26 as a first, potent, and effective PROTAC degrader of SHP2 protein. Journal of Medicinal Chemistry. 2020;63(14):7510‐7528. [DOI] [PubMed] [Google Scholar]
- 263. Zhang X, Thummuri D, Liu XG, et al. Discovery of PROTAC BCL‐X‐L degraders as potent anticancer agents with low on‐target platelet toxicity. European Journal of Medicinal Chemistry. 2020;192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Khan S, Zhang X, Lv DW, et al. A selective BCL‐X‐L PROTAC degrader achieves safe and potent antitumor activity. Nat Med. 2019;25(12):1938‐+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Maniaci C, Hughes SJ, Testa A, et al. Homo‐PROTACs: bivalent small‐molecule dimerizers of the VHL E3 ubiquitin ligase to induce self‐degradation. Nat Commun. 2017;8(1):830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Ishida T, Ciulli A. E3 ligase ligands for PROTACs: how they were found and how to discover new ones. Slas Discov. 2021;26(4):484‐502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Bricelj A, Steinebach C, Kuchta R, Gutschow M, Sosic I. E3 ligase ligands in successful PROTACs: an overview of syntheses and linker attachment points. Front Chem. 2021;9:707317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Nguyen TV, Li J, Lu CJ, et al. p97/VCP promotes degradation of CRBN substrate glutamine synthetase and neosubstrates. Proc Natl Acad Sci U S A. 2017;114(14):3565‐3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Okuhira K, Shoda T, Omura R, et al. Targeted degradation of proteins localized in subcellular compartments by hybrid small molecules. Mol Pharmacol. 2017;91(3):159‐166. [DOI] [PubMed] [Google Scholar]
- 270. Shibata N, Miyamoto N, Nagai K, et al. Development of protein degradation inducers of oncogenic BCR‐ABL protein by conjugation of ABL kinase inhibitors and IAP ligands. Cancer Sci. 2017;108(8):1657‐1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. An SA, Fu LW. Small‐molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. Ebiomedicine. 2018;36:553‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Du Y, Chen Y, Wang Y, et al. HJM‐561, a potent, selective and orally bioavailable EGFR PROTAC that overcomes osimertinib‐resistant EGFR triple mutations. Mol Cancer Ther. 2022; [DOI] [PubMed] [Google Scholar]
- 273. Golovine KV, Makhov PB, Teper E, et al. Piperlongumine induces rapid depletion of the androgen receptor in human prostate cancer cells. Prostate. 2013;73(1):23‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1‐Cullin‐F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA. 2001;98(15):8554‐8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Nishiguchi G, Keramatnia F, Min J, et al. Identification of potent, selective, and orally bioavailable small‐molecule GSPT1/2 degraders from a focused library of cereblon modulators. J Med Chem. 2021;64(11):7296‐7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Pei J, Wang G, Feng L, et al. Targeting lysosomal degradation pathways: new strategies and techniques for drug discovery. J Med Chem. 2021;64(7):3493‐3507. [DOI] [PubMed] [Google Scholar]
- 277. Steinebach C, Lindner S, Udeshi ND, et al. Homo‐PROTACs for the chemical knockdown of cereblon. ACS Chem Biol. 2018;13(9):2771‐2782. [DOI] [PubMed] [Google Scholar]
- 278. He S, Ma J, Fang Y, et al. Homo‐PROTAC mediated suicide of MDM2 to treat non‐small cell lung cancer. Acta Pharm Sin B. 2021;11(6):1617‐1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. He S, Dong G, Cheng J, Wu Y, Sheng C. Strategies for designing proteolysis targeting chimaeras (PROTACs). Med Res Rev. 2022; [DOI] [PubMed] [Google Scholar]
- 280. Ambrosini M, Fuca G, Duca M, et al. Targeted protein degraders from an oncologist point of view: The Holy Grail of cancer therapy? Crit Rev Oncol Hematol. 2022;169:103532. [DOI] [PubMed] [Google Scholar]
- 281. Fischer ES, Bohm K, Lydeard JR, et al. Structure of the DDB1‐CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512(7512):49‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Zhai F, Wang J, Yang W, Ye M, Jin X. The E3 ligases in cervical cancer and endometrial cancer. Cancers (Basel). 2022;14(21). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Yang S, Wang C, Shi L, et al. Design, synthesis and biological evaluation of novel diarylpyridine derivatives as tubulin polymerisation inhibitors. J Enzyme Inhib Med Chem. 2022;37(1):2755‐2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Sosic I, Bricelj A, Steinebach C. E3 ligase ligand chemistries: from building blocks to protein degraders. Chem Soc Rev. 2022;51(9):3487‐3534. [DOI] [PubMed] [Google Scholar]
- 285. He M, Cao C, Ni Z, et al. PROTACs: great opportunities for academia and industry (an update from 2020 to 2021). Signal Transduct Target Ther. 2022;7(1):181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Han X, Zhao L, Xiang W, et al. Strategies toward discovery of potent and orally bioavailable proteolysis targeting chimera degraders of androgen receptor for the treatment of prostate cancer. J Med Chem. 2021;64(17):12831‐12854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Xiang WG, Zhao LJ, Han X, et al. Discovery of ARD‐2585 as an exceptionally potent and orally active PROTAC degrader of androgen receptor for the treatment of advanced prostate cancer. J Med Chem. 2021;64(18):13487‐13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Borcherding DC, Amin NV, He K, et al. MEK inhibition synergizes with TYK2 inhibitors in NF1‐associated malignant peripheral nerve sheath tumors. Clin Cancer Res. 2023; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Gurbi B, Brauswetter D, Penzes K, et al. MEK is a potential indirect target in subtypes of head and neck cancers. Int J Mol Sci. 2023;24(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Fasolino G, Awada G, Moschetta L, et al. Assessment of retinal pigment epithelium alterations and chorioretinal vascular network analyses in patients under treatment with BRAF/MEK inhibitor for different malignancies: a pilot study. J Clin Med. 2023;12(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Hu J, Wei J, Yim H, et al. Potent and selective mitogen‐activated protein kinase kinase 1/2 (MEK1/2) heterobifunctional small‐molecule degraders. J Med Chem. 2020;63(24):15883‐15905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Tadesse S, Caldon EC, Tilley W, Wang S. Cyclin‐dependent kinase 2 inhibitors in cancer therapy: an update. J Med Chem. 2019;62(9):4233‐4251. [DOI] [PubMed] [Google Scholar]
- 293. Ying M, Shao X, Jing H, et al. Ubiquitin‐dependent degradation of CDK2 drives the therapeutic differentiation of AML by targeting PRDX2. Blood. 2018;131(24):2698‐2711. [DOI] [PubMed] [Google Scholar]
- 294. Adon T, Shanmugarajan D, Kumar HY. CDK4/6 inhibitors: a brief overview and prospective research directions. Rsc Adv. 2021;11(47):29227‐29246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Wang L, Shao X, Zhong T, et al. Discovery of a first‐in‐class CDK2 selective degrader for AML differentiation therapy. Nat Chem Biol. 2021;17(5):567‐575. [DOI] [PubMed] [Google Scholar]
- 296. Teng M, Jiang J, He Z, et al. Development of CDK2 and CDK5 dual degrader TMX‐2172. Angew Chem Int Ed Engl. 2020;59(33):13865‐13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Wei MM, Zhao R, Cao YT, et al. First orally bioavailable prodrug of proteolysis targeting chimera (PROTAC) degrades cyclin‐dependent kinases 2/4/6 in vivo. European Journal of Medicinal Chemistry. 2021;209. [DOI] [PubMed] [Google Scholar]
- 298. Schneider JL, Lin JJ, Shaw AT. ALK‐positive lung cancer: a moving target. Nat Cancer. 2023; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Ando K, Manabe R, Kishino Y, et al. Comparative efficacy of ALK inhibitors for treatment‐naive ALK‐positive advanced non‐small cell lung cancer with central nervous system metastasis: a network meta‐analysis. Int J Mol Sci. 2023;24(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Katayama R. Drug resistance in anaplastic lymphoma kinase‐rearranged lung cancer. Cancer Sci. 2018;109(3):572‐580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Goyal S, Jamal S, Shanker A, Grover A. Structural basis for drug resistance mechanisms against anaplastic lymphoma kinase. J Cell Biochem. 2019;120(1):768‐777. [DOI] [PubMed] [Google Scholar]
- 302. Ren C, Sun N, Kong Y, et al. Structure‐based discovery of SIAIS001 as an oral bioavailability ALK degrader constructed from Alectinib. Eur J Med Chem. 2021;217:113335. [DOI] [PubMed] [Google Scholar]
- 303. Xie S, Sun Y, Liu Y, et al. Development of alectinib‐based PROTACs as novel potent degraders of anaplastic lymphoma kinase (ALK). J Med Chem. 2021;64(13):9120‐9140. [DOI] [PubMed] [Google Scholar]
- 304. Shangary S, Wang S. Targeting the MDM2‐p53 interaction for cancer therapy. Clin Cancer Res. 2008;14(17):5318‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Shangary S, Wang S. Small‐molecule inhibitors of the MDM2‐p53 protein‐protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol. 2009;49:223‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Zhao Y, Aguilar A, Bernard D, Wang S. Small‐molecule inhibitors of the MDM2‐p53 protein‐protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J Med Chem. 2015;58(3):1038‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Wang Z, Sun Y. Targeting p53 for novel anticancer therapy. Transl Oncol. 2010;3(1):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308. Aguilar A, Wang S. Therapeutic strategies to activate p53. Pharmaceuticals (Basel). 2022;16(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Traweek RS, Cope BM, Roland CL, Keung EZ, Nassif EF, Erstad DJ. Targeting the MDM2‐p53 pathway in dedifferentiated liposarcoma. Front Oncol. 2022;12:1006959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Li Y, Yang J, Aguilar A, et al. Discovery of MD‐224 as a first‐in‐class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J Med Chem. 2019;62(2):448‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Wang B, Liu J, Tandon I, et al. Development of MDM2 degraders based on ligands derived from Ugi reactions: lessons and discoveries. Eur J Med Chem. 2021;219:113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Lin HY, Chen YS, Wang K, Chien HW, Hsieh YH, Yang SF. Fisetin inhibits epidermal growth factor‐induced migration of ARPE‐19 cells by suppression of AKT activation and Sp1‐dependent MMP‐9 expression. Mol Vis. 2017;23:900‐910. [PMC free article] [PubMed] [Google Scholar]
- 313. Waniczek D, Snietura M, Lorenc Z, Nowakowska‐Zajdel E, Muc‐Wierzgon M. Assessment of PI3K/AKT/PTEN signaling pathway activity in colorectal cancer using quantum dot‐conjugated antibodies. Oncol Lett. 2018;15(1):1236‐1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. You I, Erickson EC, Donovan KA, et al. Discovery of an AKT degrader with prolonged inhibition of downstream signaling. Cell Chem Biol. 2020;27(1):66‐73 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Yu X, Xu J, Xie L, et al. Design, synthesis, and evaluation of potent, selective, and bioavailable AKT kinase degraders. J Med Chem. 2021;64(24):18054‐18081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Li YJ, Zhang C, Martincuks A, Herrmann A, Yu H. STAT proteins in cancer: orchestration of metabolism. Nat Rev Cancer. 2023; [DOI] [PubMed] [Google Scholar]
- 317. Zhang W, Li D, Li B, Chu X, Kong B. STAT3 as a therapeutic target in the metformin‐related treatment. Int Immunopharmacol. 2023;116:109770. [DOI] [PubMed] [Google Scholar]
- 318. Smith MR, Satter LRF, Vargas‐Hernandez A. STAT5b: A master regulator of key biological pathways. Front Immunol. 2022;13:1025373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Huang B, Lang X, Li X. The role of IL‐6/JAK2/STAT3 signaling pathway in cancers. Front Oncol. 2022;12:1023177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Purohit M, Gupta G, Afzal O, et al. Janus kinase/signal transducers and activator of transcription (JAK/STAT) and its role in Lung inflammatory disease. Chem Biol Interact. 2023;371:110334. [DOI] [PubMed] [Google Scholar]
- 321. Dong J, Cheng XD, Zhang WD, Qin JJ. Recent update on development of small‐molecule STAT3 inhibitors for cancer therapy: from phosphorylation inhibition to protein degradation. J Med Chem. 2021;64(13):8884‐8915. [DOI] [PubMed] [Google Scholar]
- 322. Yang L, Lin S, Xu L, Lin J, Zhao C, Huang X. Novel activators and small‐molecule inhibitors of STAT3 in cancer. Cytokine Growth Factor Rev. 2019;49:10‐22. [DOI] [PubMed] [Google Scholar]
- 323. Orlova A, Wagner C, de Araujo ED, et al. Direct targeting options for STAT3 and STAT5 in cancer. Cancers (Basel). 2019;11(12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Zhou H, Bai L, Xu R, et al. SD‐91 as a potent and selective STAT3 degrader capable of achieving complete and long‐lasting tumor regression. Acs Med Chem Lett. 2021;12(6):996‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Zhou H, Bai L, Xu R, et al. Structure‐based discovery of SD‐36 as a potent, selective, and efficacious PROTAC degrader of STAT3 protein. J Med Chem. 2019;62(24):11280‐11300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Bai L, Zhou H, Xu R, et al. A potent and selective small‐molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell. 2019;36(5):498‐511 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Kaneshige A, Bai L, Wang M, et al. Discovery of a potent and selective STAT5 PROTAC degrader with strong antitumor activity in vivo in acute myeloid leukemia. J Med Chem. 2023;66(4):2717‐2743. [DOI] [PubMed] [Google Scholar]
- 328. Kaneshige A, Bai L, Wang M, et al. A selective small‐molecule STAT5 PROTAC degrader capable of achieving tumor regression in vivo. Nat Chem Biol. 2023; [DOI] [PubMed] [Google Scholar]
- 329. Qu X, Liu H, Song X, et al. Effective degradation of EGFR(L858R+T790M) mutant proteins by CRBN‐based PROTACs through both proteosome and autophagy/lysosome degradation systems. Eur J Med Chem. 2021;218:113328. [DOI] [PubMed] [Google Scholar]
- 330. Lei B, Liu H, Han S, Wang Z, inventor; Preparation of novel bifunctional compounds by conjugation of EGFR inhibitors with E3 ligase ligands and methods of use thereof for degradation of EGFR. patent WO2022012622. 2022.
- 331. Kim SA, Go A, Jo SH, et al. A novel cereblon modulator for targeted protein degradation. Eur J Med Chem. 2019;166:65‐74. [DOI] [PubMed] [Google Scholar]
- 332. Wang Q, Zhou X, Yang L, et al. The natural compound notopterol binds and targets JAK2/3 to ameliorate inflammation and arthritis. Cell Rep. 2020;32(11):108158. [DOI] [PubMed] [Google Scholar]
- 333. Sanachai K, Mahalapbutr P, Tabtimmai L, et al. Discovery of JAK2/3 inhibitors from quinoxalinone‐containing compounds. ACS Omega. 2022;7(37):33587‐33598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. de Wispelaere M, Du G, Donovan KA, et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat Commun. 2019;10(1):3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Shaikh MF, Morano WF, Lee J, et al. Emerging role of MDM2 as target for anti‐cancer therapy: a review. Ann Clin Lab Sci. 2016;46(6):627‐634. [PubMed] [Google Scholar]
- 336. Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small‐molecule antagonists of MDM2. Science. 2004;303(5659):844‐8. [DOI] [PubMed] [Google Scholar]
- 337. Li F, Hu Q, Zhang X, et al. DeepPROTACs is a deep learning‐based targeted degradation predictor for PROTACs. Nat Commun. 2022;13(1):7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Li JW, Zheng G, Kaye FJ, Wu L. PROTAC therapy as a new targeted therapy for lung cancer. Mol Ther. 2022; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Chen Y, Yang Q, Xu J, et al. PROTACs in gastrointestinal cancers. Mol Ther Oncolytics. 2022;27:204‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Zhang C, Xu M, He S, Huang J, Xu C, Pu K. Checkpoint nano‐PROTACs for activatable cancer photo‐immunotherapy. Adv Mater. 2022:e2208553. [DOI] [PubMed] [Google Scholar]
- 341. Palomba T, Tassone G, Vacca C, et al. Exploiting ELIOT for scaffold‐repurposing opportunities: TRIM33 a possible novel E3 ligase to expand the toolbox for PROTAC design. Int J Mol Sci. 2022;23(22). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Negi A, Kesari KK, Voisin‐Chiret AS. Estrogen receptor‐alpha targeting: PROTACs, SNIPERs, peptide‐PROTACs, antibody conjugated PROTACs and SNIPERs. Pharmaceutics. 2022;14(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Han X, Wei W, Sun Y. PROTAC degraders with ligands recruiting MDM2 E3 ubiquitin ligase: an updated perspective. Acta Materia Medica. 2022;1(2):244‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Munisamy M, Mukherjee N, Thomas L, et al. Therapeutic opportunities in cancer therapy: targeting the p53‐MDM2/MDMX interactions. Am J Cancer Res. 2021;11(12):5762‐5781. [PMC free article] [PubMed] [Google Scholar]
- 345. Yee‐Lin V, Pooi‐Fong W, Soo‐Beng AK. Nutlin‐3, a p53‐Mdm2 antagonist for nasopharyngeal carcinoma treatment. Mini‐Rev Med Chem. 2018;18(2):173‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Ding Q, Zhang Z, Liu JJ, et al. Discovery of RG7388, a potent and selective p53‐MDM2 inhibitor in clinical development. J Med Chem. 2013;56(14):5979‐83. [DOI] [PubMed] [Google Scholar]
- 347. Vu B, Wovkulich P, Pizzolato G, et al. Discovery of RG7112: a small‐molecule MDM2 inhibitor in clinical development. Acs Med Chem Lett. 2013;4(5):466‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Secchiero P, di Iasio MG, Gonelli A, Zauli G. The MDM2 inhibitor nutlins as an innovative therapeutic tool for the treatment of haematological malignancies. Curr Pharm Design. 2008;14(21):2100‐2110. [DOI] [PubMed] [Google Scholar]
- 349. Vassilev LT. MDM2 inhibitors for cancer therapy. Trends Mol Med. 2007;13(1):23‐31. [DOI] [PubMed] [Google Scholar]
- 350. Hines J, Lartigue S, Dong H, Qian Y, Crews CM. MDM2‐recruiting PROTAC offers superior, synergistic antiproliferative activity via simultaneous degradation of BRD4 and stabilization of p53. Cancer Res. 2019;79(1):251‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10(4):293‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Kleine H, Poreba E, Lesniewicz K, et al. Substrate‐assisted catalysis by PARP10 limits its activity to mono‐ADP‐ribosylation. Mol Cell. 2008;32(1):57‐69. [DOI] [PubMed] [Google Scholar]
- 353. Zhao Q, Lan T, Su S, Rao Y. Induction of apoptosis in MDA‐MB‐231 breast cancer cells by a PARP1‐targeting PROTAC small molecule. Chem Commun (Camb). 2019;55(3):369‐372. [DOI] [PubMed] [Google Scholar]
- 354. Carneiro BA, El‐Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. 2020;17(7):395‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Huang Y, Park YC, Rich RL, Segal D, Myszka DG, Wu H. Structural basis of caspase inhibition by XIAP: differential roles of the linker versus the BIR domain. Cell. 2001;104(5):781‐790. [PubMed] [Google Scholar]
- 356. Zhang Q, Yan P, Zhao P, et al. Design, synthesis, and biological evaluation of mTOR‐targeting PROTACs based on MLN0128 and pomalidomide. Chem Pharm Bull (Tokyo). 2022; [DOI] [PubMed] [Google Scholar]
- 357. Jia SQ, Zhuo R, Zhang ZM, et al. The BRD4 inhibitor dBET57 exerts anticancer effects by targeting superenhancer‐related genes in neuroblastoma. J Immunol Res. 2022;2022:7945884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Zhang S, Chen Y, Xu Z, et al. The PROTAC selectively degrading Bcl‐x(L) represents a novel Hedgehog pathway inhibitor with capacity of combating resistance to Smoothened inhibitors while sparing bone growth. Theranostics. 2022;12(17):7476‐7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Zhan Q, Zhang H, Wu B, Zhang N, Zhang L. E3 ubiquitin ligases in the acute leukemic signaling pathways. Front Physiol. 2022;13:1004330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Cheng J, He S, Xu J, Huang M, Dong G, Sheng C. Making protein degradation visible: discovery of theranostic PROTACs for detecting and degrading NAMPT. J Med Chem. 2022;65(23):15725‐15737. [DOI] [PubMed] [Google Scholar]
- 361. Xiong Y, Zhong Y, Yim H, et al. Bridged proteolysis targeting chimera (PROTAC) enables degradation of undruggable targets. J Am Chem Soc. 2022;144(49):22622‐22632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Naito M, Ohoka N, Shibata N. SNIPERs‐hijacking IAP activity to induce protein degradation. Drug Discov Today Technol. 2019;31:35‐42. [DOI] [PubMed] [Google Scholar]
- 363. Itoh Y, Ishikawa M, Naito M, Hashimoto Y. Protein knockdown using methyl bestatin‐ligand hybrid molecules: design and synthesis of inducers of ubiquitination‐mediated degradation of cellular retinoic acid‐binding proteins. Journal of the American Chemical Society. 2010;132(16):5820‐5826. [DOI] [PubMed] [Google Scholar]
- 364. Akizuki Y, Morita M, Mori Y, et al. cIAP1‐based degraders induce degradation via branched ubiquitin architectures. Nat Chem Biol. 2022; [DOI] [PubMed] [Google Scholar]
- 365. Bala R, Sindhu R, Madaan R, Shantanu. PROTAC: A Novel drug delivery technology for targeting proteins in cancer cells. Curr Drug Discov Technol. 2022; [DOI] [PubMed] [Google Scholar]
- 366. Zhu X, Liu H, Chen L, et al. Addressing the Enzyme‐independent tumor‐promoting function of NAMPT via PROTAC‐mediated degradation. Cell Chem Biol. 2022;29(11):1616‐1629 e12. [DOI] [PubMed] [Google Scholar]
- 367. Bhela IP, Ranza A, Balestrero FC, et al. A versatile and sustainable multicomponent platform for the synthesis of protein degraders: proof‐of‐concept application to BRD4‐degrading PROTACs. J Med Chem. 2022;65(22):15282‐15299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368. Peng X, Pan W, Jiang F, et al. Selective PARP1 inhibitors, PARP1‐based dual‐target inhibitors, PROTAC PARP1 degraders, and prodrugs of PARP1 inhibitors for cancer therapy. Pharmacol Res. 2022;186:106529. [DOI] [PubMed] [Google Scholar]
- 369. Tamanini E, Buck IM, Chessari G, et al. Discovery of a potent nonpeptidomimetic, small‐molecule antagonist of cellular inhibitor of apoptosis protein 1 (cIAP1) and X‐linked inhibitor of apoptosis protein (XIAP). J Med Chem. 2017;60(11):4611‐4625. [DOI] [PubMed] [Google Scholar]
- 370. Wang C, Zhang YJ, Shi LY, et al. Recent advances in IAP‐based PROTACs (SNIPERs) as potential therapeutic agents. J Enzym Inhib Med Ch. 2022;37(1):1437‐1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Shibata N, Nagai K, Morita Y, et al. Development of protein degradation inducers of androgen receptor by conjugation of androgen receptor ligands and inhibitor of apoptosis protein ligands. Journal of Medicinal Chemistry. 2018;61(2):543‐575. [DOI] [PubMed] [Google Scholar]
- 372. Honjo H, Watanabe T, Kamata K, Minaga K, Kudo M. RIPK2 as a new therapeutic target in inflammatory bowel diseases. Front Pharmacol. 2021;12:650403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Miah AH, Smith IED, Rackham M, et al. Optimization of a series of RIPK2 PROTACs. J Med Chem. 2021;64(17):12978‐13003. [DOI] [PubMed] [Google Scholar]
- 374. Garg N, Padron EJ, Rammohan KW, Goodman CF. Bruton's tyrosine kinase inhibitors: the next frontier of B‐cell‐targeted therapies for cancer, autoimmune disorders, and multiple sclerosis. J Clin Med. 2022;11(20). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Shen J, Liu J. Bruton's tyrosine kinase inhibitors in the treatment of primary central nervous system lymphoma: a mini‐review. Front Oncol. 2022;12:1034668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Nakhoda S, Vistarop A, Wang YL. Resistance to Bruton tyrosine kinase inhibition in chronic lymphocytic leukaemia and non‐Hodgkin lymphoma. Br J Haematol. 2023;200(2):137‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Das D, Wang J, Hong J. Next‐generation Bruton's tyrosine kinase (BTK) inhibitors potentially targeting BTK C481S mutation‐ recent developments and perspectives. Curr Top Med Chem. 2022;22(20):1674‐1691. [DOI] [PubMed] [Google Scholar]
- 378. Tinworth CP, Lithgow H, Dittus L, et al. PROTAC‐mediated degradation of Bruton's tyrosine kinase is inhibited by covalent binding. Acs Chemical Biology. 2019;14(3):342‐347. [DOI] [PubMed] [Google Scholar]
- 379. Schiemer J, Horst R, Meng Y, et al. Snapshots and ensembles of BTK and cIAP1 protein degrader ternary complexes. Nat Chem Biol. 2021;17(2):152‐160. [DOI] [PubMed] [Google Scholar]
- 380. Dragovich PS, Adhikari P, Blake RA, et al. Antibody‐mediated delivery of chimeric protein degraders which target estrogen receptor alpha (ER alpha). Bioorg Med Chem Lett. 2020;30(4). [DOI] [PubMed] [Google Scholar]
- 381. Demizu Y, Shibata N, Hattori T, et al. Development of BCR‐ABL degradation inducers via the conjugation of an imatinib derivative and a cIAP1 ligand. Bioorg Med Chem Lett. 2016;26(20):4865‐4869. [DOI] [PubMed] [Google Scholar]
- 382. Negi A, Voisin‐Chiret AS. Strategies to reduce the on‐target platelet toxicity of Bcl‐x(L) inhibitors: PROTACs, SNIPERs and prodrug‐based approaches. Chembiochem. 2022;23(12):e202100689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Pawlak A, Henklewska M. The role of Bcl‐xL protein research in veterinary oncology. Int J Mol Sci. 2020;21(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Takei Y. siRNA‐based drug targeting human Bcl‐xL against cancers. Methods Mol Biol. 2019;1974:31‐40. [DOI] [PubMed] [Google Scholar]
- 385. Zhang X, He Y, Zhang P, et al. Discovery of IAP‐recruiting BCL‐XL PROTACs as potent degraders across multiple cancer cell lines. Eur J Med Chem. 2020;199:112397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Zhu L, He S, Huang L, et al. Chaperone‐mediated autophagy degrades Keap1 and promotes Nrf2‐mediated antioxidative response. Aging Cell. 2022;21(6):e13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Xie T, Zahid H, Ali AR, et al. Inhibitors of Keap1‐Nrf2 protein‐protein interaction reduce estrogen responsive gene expression and oxidative stress in estrogen receptor‐positive breast cancer. Toxicol Appl Pharmacol. 2023;460:116375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Tossetta G, Marzioni D. Targeting the NRF2/KEAP1 pathway in cervical and endometrial cancers. Eur J Pharmacol. 2023;941:175503. [DOI] [PubMed] [Google Scholar]
- 389. Qi Z, Tong Y, Luo H, Chen M, Zhou N, Chen L. Neuroprotective effect of a Keap1‐Nrf2 protein‐protein inter‐action inhibitor on cerebral ischemia/reperfusion injury. Bioorg Chem. 2023;132:106350. [DOI] [PubMed] [Google Scholar]
- 390. Chatterjee D, Costa CAM, Wang XF, Jevitt A, Huang YC, Deng WM. Single‐cell transcriptomics identifies Keap1‐Nrf2 regulated collective invasion in a Drosophila tumor model. Elife. 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. He L, Huan P, Xu J, et al. Hedysarum polysaccharide alleviates oxidative stress to protect against diabetic peripheral neuropathy via modulation of the keap1/Nrf2 signaling pathway. J Chem Neuroanat. 2022;126:102182. [DOI] [PubMed] [Google Scholar]
- 392. Hassanein EHM, Althagafy HS, Atwa AM, Kozman MR, Kotb El‐Sayed MI, Soubh AA. Taurine attenuated methotrexate‐induced intestinal injury by regulating NF‐kappaB/iNOS and Keap1/Nrf2/HO‐1 signals. Life Sci. 2022;311(Pt A):121180. [DOI] [PubMed] [Google Scholar]
- 393. Qi X, Zhang X, Meng J, et al. Briarane‐type diterpenoids, the inhibitors of osteoclast formation by interrupting Keap1‐Nrf2 interaction and activating Nrf2 pathway. Eur J Med Chem. 2023;246:114948. [DOI] [PubMed] [Google Scholar]
- 394. Boyenle ID, Divine UC, Adeyemi R, et al. Direct Keap1‐kelch inhibitors as potential drug candidates for oxidative stress‐orchestrated diseases: A review on In silico perspective. Pharmacol Res. 2021;167:105577. [DOI] [PubMed] [Google Scholar]
- 395. Pouremamali F, Pouremamali A, Dadashpour M, Soozangar N, Jeddi F. An update of Nrf2 activators and inhibitors in cancer prevention/promotion. Cell Commun Signal. 2022;20(1):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Crisman E, Duarte P, Dauden E, et al. KEAP1‐NRF2 protein‐protein interaction inhibitors: design, pharmacological properties and therapeutic potential. Med Res Rev. 2023;43(1):237‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Davinelli S, Medoro A, Intrieri M, Saso L, Scapagnini G, Kang JX. Targeting NRF2‐KEAP1 axis by Omega‐3 fatty acids and their derivatives: emerging opportunities against aging and diseases. Free Radic Biol Med. 2022;193(Pt 2):736‐750. [DOI] [PubMed] [Google Scholar]
- 398. Wu KC, McDonald PR, Liu JJ, Chaguturu R, Klaassen CD. Implementation of a high‐throughput screen for identifying small molecules to activate the Keap1‐Nrf2‐ARE pathway. PLoS One. 2012;7(10):e44686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Tong B, Luo M, Xie Y, et al. Bardoxolone conjugation enables targeted protein degradation of BRD4. Sci Rep. 2020;10(1):15543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400. Heightman TD, Callahan JF, Chiarparin E, et al. Structure‐activity and structure‐conformation relationships of aryl propionic acid inhibitors of the Kelch‐like ECH‐associated protein 1/nuclear factor erythroid 2‐related factor 2 (KEAP1/NRF2) protein‐protein interaction. J Med Chem. 2019;62(9):4683‐4702. [DOI] [PubMed] [Google Scholar]
- 401. Lu M, Liu T, Jiao Q, et al. Discovery of a Keap1‐dependent peptide PROTAC to knockdown Tau by ubiquitination‐proteasome degradation pathway. Eur J Med Chem. 2018;146:251‐259. [DOI] [PubMed] [Google Scholar]
- 402. Wei J, Meng F, Park KS, et al. Harnessing the E3 ligase KEAP1 for targeted protein degradation. J Am Chem Soc. 2021;143(37):15073‐15083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Pei J, Xiao Y, Liu X, W Hu, A Sobh, Y Yuan, et al. Identification of Piperlongumine (PL) as a new E3 ligase ligand to induce targeted protein degradation. bioRxiv. 2022;
- 404. Zhang XY, Luukkonen LM, Eissler CL, et al. DCAF11 supports targeted protein degradation by electrophilic proteolysis‐targeting chimeras. Journal of the American Chemical Society. 2021;143(13):5141‐5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Han T, Goralski M, Gaskill N, et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science. 2017;356(6336). [DOI] [PubMed] [Google Scholar]
- 406. Zhang XY, Crowley VM, Wucherpfennig TG, Dix MM, Cravatt BF. Electrophilic PROTACs that degrade nuclear proteins by engaging DCAF16. Nature Chemical Biology. 2019;15(7):737‐+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Kasahara K. Physiological function of FKBP12, a primary target of rapamycin/FK506: a newly identified role in transcription of ribosomal protein genes in yeast. Curr Genet. 2021;67(3):383‐388. [DOI] [PubMed] [Google Scholar]
- 408. Annett S, Moore G, Robson T. FK506 binding proteins and inflammation related signalling pathways; basic biology, current status and future prospects for pharmacological intervention. Pharmacol Ther. 2020;215:107623. [DOI] [PubMed] [Google Scholar]
- 409. Zhang X, Luukkonen LM, Eissler CL, et al. DCAF11 supports targeted protein degradation by electrophilic proteolysis‐targeting chimeras. J Am Chem Soc. 2021;143(13):5141‐5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Tao YF, Remillard D, Vinogradova EV, et al. Targeted protein degradation by electrophilic PROTACs that stereoselectively and site‐specifically engage DCAF1. Journal of the American Chemical Society. 2022; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Li L, Mi D, Pei H, et al. In vivo target protein degradation induced by PROTACs based on E3 ligase DCAF15. Signal Transduct Target Ther. 2020;5(1):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Han J, Kim YL, Lee KW, et al. ZNF313 is a novel cell cycle activator with an E3 ligase activity inhibiting cellular senescence by destabilizing p21(WAF1.). Cell Death Differ. 2013;20(8):1055‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Spradlin JN, Hu X, Ward CC, et al. Harnessing the anti‐cancer natural product nimbolide for targeted protein degradation. Nat Chem Biol. 2019;15(7):747‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Kumar R, Sabapathy K. RNF4‐a paradigm for SUMOylation‐mediated ubiquitination. Proteomics. 2019;19(21‐22):e1900185. [DOI] [PubMed] [Google Scholar]
- 415. Abu Ahmad Y, Oknin‐Vaisman A, Bitman‐Lotan E, Orian A. From the evasion of degradation to ubiquitin‐dependent protein stabilization. Cells. 2021;10(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Wang C, Zhang Y, Wu Y, Xing D. Developments of CRBN‐based PROTACs as potential therapeutic agents. Eur J Med Chem. 2021;225:113749. [DOI] [PubMed] [Google Scholar]
- 417. Luo M, Spradlin JN, Boike L, et al. Chemoproteomics‐enabled discovery of covalent RNF114‐based degraders that mimic natural product function. Cell Chem Biol. 2021;28(4):559–566 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Kiely‐Collins H, Winter GE, Bernardes GJL. The role of reversible and irreversible covalent chemistry in targeted protein degradation. Cell Chem Biol. 2021;28(7):952‐968. [DOI] [PubMed] [Google Scholar]
- 419. Girardini M, Maniaci C, Hughes SJ, Testa A, Ciulli A. Cereblon versus VHL: hijacking E3 ligases against each other using PROTACs. Bioorg Med Chem. 2019;27(12):2466‐2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420. Ito T, Yamaguchi Y, Handa H. Exploiting ubiquitin ligase cereblon as a target for small‐molecule compounds in medicine and chemical biology. Cell Chem Biol. 2021;28(7):987‐999. [DOI] [PubMed] [Google Scholar]
- 421. Bondeson DP, Smith BE, Burslem GM, et al. Lessons in PROTAC design from selective degradation with a promiscuous warhead. Cell Chem Biol. 2018;25(1):78–87 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Wan Y, Yan C, Gao H, Liu T. Small‐molecule PROTACs: novel agents for cancer therapy. Future Med Chem. 2020;12(10):915‐938. [DOI] [PubMed] [Google Scholar]
- 423. Ward CC, Kleinman JI, Brittain SM, et al. Covalent ligand screening uncovers a RNF4 E3 ligase recruiter for targeted protein degradation applications. ACS Chem Biol. 2019;14(11):2430‐2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424. Jevtic P, Haakonsen DL, Rape M. An E3 ligase guide to the galaxy of small‐molecule‐induced protein degradation. Cell Chem Biol. 2021;28(7):1000‐1013. [DOI] [PubMed] [Google Scholar]
- 425. Henning NJ, Manford AG, Spradlin JN, et al. Discovery of a covalent FEM1B recruiter for targeted protein degradation applications. J Am Chem Soc. 2022;144(2):701‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Nalawansha DA, Li K, Hines J, Crews CM. Hijacking methyl reader proteins for nuclear‐specific protein degradation. J Am Chem Soc. 2022;144(12):5594‐5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Tong B, Spradlin JN, Novaes LFT, et al. A nimbolide‐based kinase degrader preferentially degrades oncogenic BCR‐ABL. ACS Chem Biol. 2020;15(7):1788‐1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Wei LN. Cellular retinoic acid binding proteins: genomic and non‐genomic functions and their regulation. Subcell Biochem. 2016;81:163‐178. [DOI] [PubMed] [Google Scholar]
- 429. Wolf G. Cellular retinoic acid‐binding protein II: a coactivator of the transactivation by the retinoic acid receptor complex RAR.RXR. Nutr Rev. 2000;58(5):151‐153. [DOI] [PubMed] [Google Scholar]
- 430. Napoli JL. Cellular retinoid binding‐proteins, CRBP, CRABP, FABP5: effects on retinoid metabolism, function and related diseases. Pharmacol Ther. 2017;173:19‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Ohoka N, Tsuji G, Shoda T, et al. Development of small molecule chimeras that recruit AhR E3 ligase to target proteins. Acs Chemical Biology. 2019;14(12):2822‐2832. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are freely available from the corresponding author upon request.