

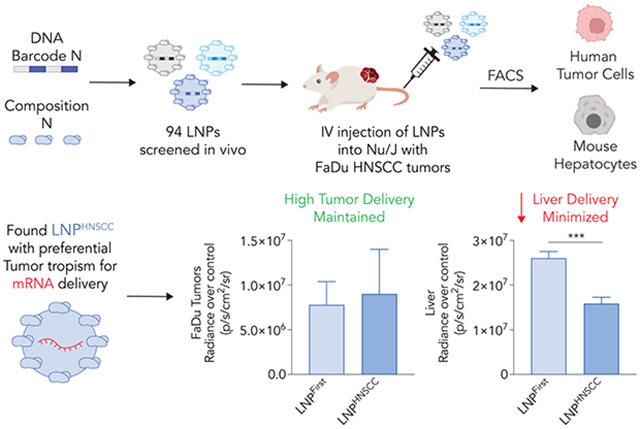
Abstract
Lipid nanoparticles (LNPs) are a clinically relevant way to deliver therapeutic mRNA to hepatocytes in patients. However, LNP-mRNA delivery to end-stage solid tumors such as head and neck squamous cell carcinoma (HNSCC) remains more challenging. While scientists have used in vitro assays to evaluate potential nanoparticles for HNSCC delivery, high-throughput delivery assays performed directly in vivo have not been reported. Here we use a high-throughput LNP assay to evaluate how 94 chemically distinct nanoparticles delivered nucleic acids to HNSCC solid tumors in vivo. DNA barcodes were used to identify LNPHNSCC, a novel LNP for systemic delivery to HNSCC solid tumors. Importantly, LNPHNSCC retains tropism to HNSCC solid tumors while minimizing off-target delivery to the liver.
Keywords: DNA barcode, mRNA delivery, lipid nanoparticles, cancer
Graphical Abstract
1. Introduction
The efficacy of mRNA vaccines [1, 2] and the speed of their regulatory approval [3] have established lipid nanoparticle (LNP)-mRNA drugs as useful medicines. Complementing these approvals are data suggesting that systemically administered LNP-mRNA drugs can deliver Cas9-based gene editors in patients [4, 5]. Taken together, these clinical data hint at the potential impact of RNA therapeutics that can be delivered to a target tissue.
One tissue that, if targeted, could lead to important clinical advances is head and neck squamous cell carcinoma (HNSCC). Head and neck cancers are the sixth most common cancer worldwide [6] and account for 3–4% of cancers in the United States. A subset of these malignancies, end-stage HNSCC, can afflict otherwise healthy young patients infected by human papillomavirus, and is characterized by chronic pain, uncontrolled bleeding, and death. Unfortunately, there are few treatment options available besides chemotherapy or external beam radiation therapy, which lead to severe toxicity [7]. Scientists have therefore pursued gene-based oncolytic therapies that drive cytoreduction in HNSCC; these have worked in pre-clinical models as well as a Phase 1 clinical trial [8-14]. Notably, such efforts have used viral vectors to express the therapeutic gene. One alternative approach is the use of LNPs to deliver therapeutic nucleic acids. LNP-mediated nucleic acid delivery could circumvent challenges associated with viral vectors including constrained payload size [15], pre-existing immunity [16], inability to redose [17], and complicated manufacturing at human scale [18, 19].
One acknowledged limitation of LNPs is their tendency to accumulate in the liver [20, 21]. We therefore sought an LNP that would deliver nucleic acid to HNSCC while minimizing off-target delivery to hepatic tissues. Given the fact that in vitro (i.e., cell culture) nanoparticle delivery can poorly predict in vivo (i.e., in an adult animal) nanoparticle delivery [22], we reasoned that the best approach would be to perform a direct in vivo high-throughput screen of many chemically diverse LNPs. Here we report how 94 LNPs delivered nucleic acids to HNSCC tumors derived from cancer cell lines or patient-derived tumors as well as hepatocytes, all in vivo. We also report the subsequent LNP named LNPHNSCC, which delivers nucleic acids to tumor cells and minimizes delivery to liver hepatocytes in vivo, without the use of active targeting ligands such as antibodies or aptamers. These data suggest that high-throughput in vivo screens may identify nanoparticles with tumor tropism and de-targeted liver delivery following systemic administration.
2. Materials and Methods
Nanoparticle formulation.
7C1 was synthesized as described previously [23]. Briefly, C15 lipids were combined with PEI600 and heated to 90 °C in 100% ethanol for 48-72 hours. Products were characterized with MALDI-TOF and 1H-NMR. Nanoparticles were formulated using a microfluidic device as previously described [24]. Specifically, citrate and ethanol phases were combined in a microfluidic device using syringes (Hamilton Company) at a flow rate of 3:1. DNA barcodes were diluted in 10 mM citrate buffer (Teknova) and purchased from Integrated DNA Technologies. Poly(ethylene glycol) (PEG)-lipids, cholesterols, and helper lipids were diluted in 100% ethanol and purchased from Avanti Lipids.
Nanoparticle characterization.
The diameter and polydispersity of formulated LNPs were measured using dynamic light scattering (DLS) (DynaPro Plate Reader II, Wyatt). LNPs were diluted in sterile 1X PBS and sterile purified using a 0.22 μm filter. LNPs were included only if they met three criteria: diameter >20 nm and <200 nm, polydispersity index (PDI) <0.5, and correlation function with 1 inflection point. For screens, particles that met these criteria were pooled. Particles were dialyzed in 20 kD dialysis cassettes (Thermo Scientific). Nucleic acid concentration was determined using NanoDrop (Thermo Scientific).
Cell culture.
FaDu and Cal-27 cells, human head and neck squamous cell carcinoma cells, were purchased from ATCC. FaDu cells were maintained with Dulbecco’s modified Eagle’s medium (DMEM) and 10% FBS. Cal-27 cells were maintained with Minimum Essential Medium Eagle (EMEM) and 10% FBS. Cell-related experiments were performed with cells from passage 5–20. Patient-derived mixed/crude (non-clonal) tumor cells (328373-195-R-J1-PDC) were extracted from a lateral neck soft tissue mass obtained according to leading-edge National Cancer Institute (NCI) Patient-Derived Models Repository (PDMR) protocols (https://pdmr.cancer.gov/) based on banking of cryopreserved patient tumor samples. Cancer models from the PDMR undergo rigorous quality control of their profiling, which makes the PDX model used here ideal in terms of doubling time and carcinogenic pathology to assess antitumor activity in preclinical studies with reliable fidelity. HEK-293 cells (ATCC, CRL-1573™) were cultured and transfected in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% Primocin. AML-12 hepatocytes (ATCC, CRL-2254™) were cultured and transfected in clear DMEM supplemented with 10% FBS, 1% Primocin, and Cell Maintenance Cocktail B (Thermo Fisher Scientific).
Animal experiments.
All animal experiments were performed in accordance with the Georgia Institute of Technology and Emory University School of Medicine IACUC. All animals were housed in the Georgia Institute of Technology or Emory University animal facilities. NU/J mice (00201) and NOD.Cg-Prkdscid IL2rgtm1Wjl/SzJ (NSG) mice were purchased from Jackson Laboratories (or Charles River). At least N = 3–4 mice/group were used for all studies, unless noted otherwise.
Xenograft models.
HNSCC tumors were inoculated in mice by subcutaneous injection of the cancer cells: 0.05 mL aliquots of sterilized saline containing 1 × 106 FaDu cells or Cal-27 cells were injected into both flanks of NuJ mice; 0.1 mL aliquots of sterilized saline containing 5 × 106 patient derived PDX cells were administered into the right flanks of NSG mice. Mice were evaluated daily and tumor measurements taken several times a week using calipers until up to size.
Cell isolation & staining for fluorescence-activated cell sorting (FACS).
When screening particles, tumors were harvested 24h after systemic injection with LNPs, unless otherwise noted. Mice were perfused with 20 mL of 1X PBS through the right atrium. Tumors or livers were finely cut and then placed in a digestive enzyme solution with collagenase type I (Sigma-Aldrich), collagenase XI (Sigma-Aldrich) and hyaluronidase (Sigma-Aldrich) at 37 °C at 550 rpm for 45 minutes. Cell suspension was filtered through 70 μm mesh and red blood cells lysed. Specific cell populations were sorted using the BD FacsFusion equipment housed at the Georgia Institute of Technology Cellular Analysis Core. Antibody clones used for these studies include anti-CD31 (390, BioLegend) for murine endothelial cells, anti-CD45.2 (104 BioLegend) for murine immune cells, and anti-hCD47 (CC2C6, BioLegend) for the human cancer cells. Representative flow gates are shown in Supplementary Fig. S2A,B.
PCR amplification.
Samples were amplified and prepared for sequencing using a one-step PCR protocol as previously described [25]. More specifically, 1 μL of primers (5 uM for final reverse/forward, 0.5 uM for base forward) were added to 5 μL of Kapa HiFi 2X master mix and 4 μL template DNA/water. If the PCR reaction did not produce clear bands in the gel, the primer concentrations, DNA template input, PCR temperature, and number of cycles were optimized for individual samples.
Deep sequencing.
Deep sequencing was performed on an Illumina MiniSeq™. Primers were designed based on Nextera XT adapter sequences.
Data normalization.
Counts for each particle, per tissue, were normalized to the barcoded LNP mixture injected into each mouse. This “input” DNA provided the DNA barcode counts, which were used to normalize DNA barcode counts from each of the cell types and tissues as shown in Fig. 2D-F.
Fig. 2. A second high-throughput LNP screen was designed to optimize LNPs that preferentially deliver to human tumor cells in vivo.

(A) Enrichment in the top 5% of LNPs, subdivided by the PEG type and (B) mole ratio of 7C1 suggests that C18PEG2000 and mole ratio 50 were enriched in human tumor tissues in vivo. (C) A second screen was designed based on enriched characteristics. (D) As expected, normalized delivery of the negative control (unencapsulated DNA, shown in blue) was much lower than normalized delivery for all LNPs. Correlation between delivery of LNPs in mouse hepatocytes and human tumor cells in (E) FaDu and (F) Cal-27 tumor cell lines. LNPHNSCC is highlighted in red.
Data analysis & statistics.
Sequencing results were processed using a custom Python-based tool to extract raw barcode counts for each tissue. These raw counts were then normalized with an R script prior to further analysis. Statistical analysis was done using GraphPad Prism 8. Data is plotted as mean ± standard error of mean unless otherwise stated. Statistical tests and p-value significance associated with each figure are reported in the respective figure caption.
TNS assay.
The pKa of the LNPHNSCC nanoparticle was measured as previously described [26]. Briefly, a stock solution of 10 mM HEPES (Sigma-Aldrich), 10mM MES (Sigma-Aldrich), 10 mM sodium acetate (Sigma), and 140 mM sodium chloride (Sigma-Aldrich) was prepared and pH adjusted with hydrogen chloride and sodium hydroxide to a range of pH between 4 and 10. Using 4 replicates for each nanoparticle at each pH, 140 μL pH-adjusted buffer was added to a 96-well plate, followed by the addition of 5 μL of 2-(p-toluidino)-6-naphthalene sulfonic acid (60 μg/mL). Five microliters of each nanoparticle were added to each well. After 5 min of incubation under gentle shaking, fluorescence absorbance was measured using excitation wavelengths of 325 nm and emission wavelengths of 435 nm.
Zeta potential.
The zeta potential of LNPs was measured using a Malvern Zetasizer Nano Z. Eight hundred microliters of the LNPs were loaded into a Malvern disposable folded capillary cell and the following settings were executed: material refractive index of 1.4, absorbance of 0.01, dispersant viscosity of 0.882 cP, refractive index of 1.33, and dielectric constant of 79.
Anchored nanoluciferase-encoding and anchored VHH-encoding mRNA.
Messenger RNA encoding anchored nanoluciferase and camelid-derived VHH were synthesized as described previously [27, 28]. Briefly, the GPI-anchored VHH or NanoLuc sequence was ordered as a DNA gBlock from IDT (Integrated DNA Technologies)] with certain modifications including extensions to allow for Gibson assembly, a 3’ UTR derived from the mouse alpha-globin sequence, and a 5’ UTR with Kozak sequence. The IDT website was used to human codon optimize the sequence. A NEBuilder with 3 molar excesses of insert was then used to clone the gBlock into a PCR amplified pMA7 vector through Gibson assembly. Prior to assembly reaction, Gibson assembly reaction transcripts were purified using a 0.8% agarose gel. Sanger sequencing of subsequent plasmids from each colony confirmed sequence identity. Plasmids were digested into a linear template using NotI-HF (New England Biolabs (NEB)) overnight at 37 °C. Linearized templates underwent purification by ammonium acetate (Thermo Fisher Scientific) precipitation and were resuspended with nuclease-free water. In vitro transcription was achieved overnight at 37 °C using the HiScribe T7 kit (NEB) following the manufacturer’s directions. RNA product was then treated with Dnase I (Aldevron) and purified via lithium chloride precipitation (Thermo Fisher Scientific). RNA transcripts were heat-denatured at 65 °C for 10 min and capped with a Cap1 structure using guanylyl transferase (Aldevron) and 2’-O-methyltransferase (Aldevron). Transcripts were then polyadenylated enzymatically (Aldevron). Finally, mRNA was purified by lithium chloride precipitation, treated with alkaline phosphatase (NEB), and purified once more. Concentrations were measured using a NanoDrop, and stock concentrations were between 4 and 5 mg/mL. mRNA stocks were stored at −80 °C. Purity levels of RNA products were confirmed by gel electrophoresis.
Immunohistochemical staining of aVHH-transfected cells in vitro.
FaDu human cancer cells were plated at 80,000 cells per well, suspended in 0.5 mL of EMEM, in a coverglass-bottom 24-well plate (Cellvis). A day later, the wells were transfected with LNPHNSCC carrying aVHH-encoding mRNA at 1 and 2 μg of RNA at 50 μL/well. On the day following the transfection, the FaDu cells were fixed with 4% PFA (in 1x PBS) for 10 min at room temperature, washed with PBS, permeabilized with 0.2% Triton X-100 in PBS for 5 min at room temperature, blocked with 5% BSA in PBS for 30 min at 37 °C, and incubated in rabbit anti-VHH primary (GenScript) diluted (1:250 dilution) in PBS for 30 min at 37 °C; 250 μL/well was used to cover the entire well. Next, the cells were washed twice with PBS, incubated in fluorescent donkey anti-rabbit secondary antibody diluted (1:250 dilution) in PBS for 30 min at 37 °C; 250 μL/well was used to cover the entire well. Cells were then washed twice with PBS, incubated in DAPI diluted in PBS for 10 min at room temperature with some shaking, and washed once with PBS, and a drop of Prolong Gold was placed onto the cells under a 12 mm circular coverslip (Electron Microscopy Sciences) on top. Finally, the stained aVHH-transfected cells were imaged using a PerkinElmer UltraVIEW spinning disk confocal microscope with a Zeiss ×63 numerical aperture (NA) 1.4 Plan-Apochromat objective lens with Volocity software. The images were linearly contrast-enhanced equally across all images in Volocity.
Whole organ imaging.
Tissues were isolated 24 h after administration of LNPs to measure bioluminescence. Harvested organs were submerged in Nano-Glo Luciferase Assay Substrate (Promega) for 5 minutes before being placed on solid black paper for imaging. Luminescence was measured using an IVIS imaging system (PerkinElmer, Waltham, MA) and quantified using Living Image software (PerkinElmer).
3. Results & Discussion
To explore the delivery of LNPs to human tumor models in vivo, we first performed high-throughput in vivo DNA barcoding screens [29]. We formulated 64 chemically distinct LNPs by mixing four components: the oligomer-lipid 7C1, cholesterol, 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), and PEG-lipids with varying molar amounts, alkyl lengths, and molecular weights (Fig. 1A). 7C1 was chosen as the ionizable or cationic lipid due to evidence that it can deliver siRNA to solid tumors in vivo [23]. We chose DSPC given that it is the only helper lipid used in all FDA-approved LNPs (Onpattro®, Spikevax®, and Comirnaty®). Given the important role of PEG in controlling LNP opsonization and clearance [30], we tested LNPs containing four different PEG-lipids of compositions between 1% and 15% molar. Each LNP carried a unique DNA barcode (Fig. 1B): LNP 1, with chemical structure 1, was formulated with barcode 1, whereas LNP N, with chemical structure N, was formulated with barcode N. The formulation details and size for each LNP are detailed in Supplementary Figure S1. Quality control was conducted for each LNP using DLS. LNPs with hydrodynamic diameters between 20 and 200 nm and monodisperse DLS spectra were considered to pass quality control. Of the 64 LNPs formulated, 41 were deemed small and monodisperse; these were pooled together and filtered through a 0.22 μm pore. The diameter of the LNP pool was monodisperse and under 200 nm, providing one line of evidence that the LNPs did not come out of solution after pooling. Next, the pool was injected intravenously (IV) into two groups of immunocompromised Nu/J mice at a total dose of 0.5 mg/kg of DNA barcode (Fig. 1C) (i.e., 0.012 mg/kg/barcode on average, for all 41 barcodes). The two animal cohorts were previously inoculated with two different human cancer cell lines (FaDu or Cal-27 head and neck tumor models) to establish hind-leg xenograft tumors. FaDu cells are an immortalized line established from a punch biopsy of a hypopharyngeal tumor removed from a 56-year-old white male patient with squamous cell carcinoma in 1968. This model is commonly used in HNSCC research [31, 32]. Each mouse carried two tumors. Twenty-four hours following LNP pool administration, human tumor cells and liver hepatocytes were isolated via FACS (Supplementary Fig. S2). DNA barcodes were isolated and quantified via next-generation sequencing (NGS). Normalized DNA delivery (%) from tumor cells and hepatocytes for each tumor type was plotted (Fig. 1D). Normalized delivery was chosen since it allows scientists to quantify how many different LNPs work relative to one another in a single experiment. However, one limitation is that its readout could be different than the functional delivery of mRNA. As a negative control, we measured delivery of an unencapsulated barcode; as expected, this was delivered less efficiently than barcodes in LNPs (Fig. 1D). To identify LNP traits with a maximum ratio of tumor to liver delivery, we plotted hepatocyte delivery against human tumor cell delivery across both cancer models. We identified three LNPs (highlighted in red) with higher ratios of tumor-to-liver delivery than other LNPs in both tumor models (Fig. 1E,F). Importantly, none of these LNPs were the winner candidate of this first screen, termed LNPFirst, with the highest delivery to tumor cells but also the highest delivery to hepatocytes, given that we were trying to identify properties that minimized hepatic delivery.
Fig. 1. High-throughput DNA barcoding can be used to study the delivery of LNPs to solid tumors in vivo.

(A) A diverse library of LNPs was formulated using 7C1, cholesterol, DSPC, and PEG-lipid variants at various mole ratios. (B) Each LNP was formulated to carry a distinct DNA barcode. (C) Small, monodisperse LNPs were pooled together. Human head and neck cancer cells (FaDu or Cal-27) were subcutaneously injected into Nu/J mice to establish xenograft hind-leg tumors. Pooled LNP libraries were intravenously administered to mice at a dose of 0.5 mg/kg of DNA barcode. Twenty-four hours later, human cancer cells and liver hepatocytes were isolated for sequencing. (D) Normalized delivery of all LNPs in host tumor cells and hepatocytes. To find an LNP that preferentially delivered to human cancer cells, we quantified the correlation between delivery for all LNPs in mouse hepatocytes and human tumor cells. Normalized delivery in (E) FaDu and (F) Cal-27 tumor cells was plotted. Red-highlighted dots indicate an LNP that preferentially delivered to human cancer cells. The LNP with highest delivery to tumors but also to hepatocytes is represented by the green-highlighted dot, and termed LNPFirst as the winner of this first screen. The blue dot represents unencapsulated DNA barcode, which did not deliver well to either cell type and acted as a control establishing that LNP encapsulation increases delivery of barcodes tested here.
We then examined the chemical composition of three lead LNPs. All three contained 50% molar ratio of 7C1 and C18PEG2000 at 3%, 7%, and 11% molar ratio (Supplementary Fig. S1C), all ratios larger than the traditional 1.5-1.6% molar ratio of PEG used in the FDA-approved LNPs (Onpattro®, Spikevax®, and Comirnaty®) [33]. Next, we characterized traits within the top five LNPs of the whole screen that had the highest delivery to tumors, irrespective of their tumor/liver delivery ratio, to identify traits associated with successful systemic delivery to HNSCC tumors among the LNPs screened. We found that most of them used C18PEG2000 (Fig. 2A) and all of them had a 50% 7C1 mole ratio (Fig. 2B). Based on these findings, we designed a second, iterative screen around C18PEG2000 at 1–8.5% molar ratio and 7C1 at 50% molar ratio. We also included an additional helper lipid, DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) based on data suggesting it could improve tumor delivery [34, 35]. In this second screen, 64 different 7C1-based LNPs were barcoded using the same methodology employed in the first screen (Fig. 2C). Of these 64 LNPs, 53 were considered small and stable enough to be pooled (Supplementary Fig. S1D). Once again, two groups of immunocompromised mice were implanted with either Cal-27 or FaDu human tumor cells to establish hind-leg xenograft tumors (two per animal). Mice were injected with the stable LNP pool at a dose of 0.5 mg/kg of DNA barcode. Human tumor cells and hepatocytes were then isolated via FACS, and barcodes were sequenced. DNA barcodes across tumor cells and hepatocytes in both cell lines were quantified (Fig. 2D). From this experiment, we plotted the normalized delivery to hepatocytes against normalized delivery to human tumor cells (Fig. 2E,F). We identified an LNP, highlighted in red, that delivered nucleic acid selectively to tumor cells while minimizing hepatocyte delivery in both tumor models (Fig. 2E,F).
These iterative screens allowed us to identify an LNP, termed LNPHNSCC, that delivers nucleic acid to human tumor cells in vivo (Fig. 3A). We characterized the size, dispersity, pKa, and zeta potential of LNPHNSCC to validate its stability (Fig. 3B). LNPHNSCC had a relatively neutral zeta potential and a pKa of 4.8, as well as a low PDI indicating a monodisperse diameter. Interestingly, LNPHNSCC had a bigger diameter and PDI than LNPFirst (Fig. 3C), the winner of the first screen with the highest delivery to tumor cells and liver hepatocytes. These data present one line of evidence that PEG content of the LNPs may contribute to their diameter.
Fig. 3. LNPHNSCC efficiently delivers mRNA to cancer cells in vitro.

(A) A lead LNP from screen 2, LNPHNSCC, was identified and characterized. (B) Diameter (nm), polydispersity index (PDI), pKa, and zeta potential of LNPHNSCC. (C) Composition, diameter (nm), and PDI of LNPFirst, the winner of the first screen with highest tumor and hepatocyte delivery. (D) LNPHNSCC was formulated with a GFP mRNA and an anchored VHH (aVHH) mRNA and used to transfect FaDu cancer cells in vitro at 0.50, 1, and 2 μg/80K cells. Sixteen hours later, (E) GFP and (F) aVHH expression (% transfected and MFI) were quantified via flow cytometry. (G) aVHH transfection of FaDu cells was also validated via immunohistochemical staining and microscopy. LNPHNSCC and LNPFirst were formulated with aVHH mRNA and used to transfect (H) FaDu human cancer cells, (I) murine AML-12 hepatocytes, and (J) human embryonic kidney (HEK-293) cells in vitro at 0.10, 0.25, and 0.50 μg/80K cells. LNPHNSCC was better at transfecting human cell lines, while LNPFirst showed superior transfection of murine hepatocytes. MFI: Mean fluorescence intensity. Two-way ANOVA, Tukey’s multiple comparisons test, *P < 0,05, **P < 0.01, ***P < 0.001, ****P < 0.001, average ± S.D.
Next, we sought to demonstrate that LNPHNSCC can deliver mRNA to human head and neck cancer cells. We first encapsulated GFP-encoding mRNA in LNPHNSCC and transfected FaDu cancer cells in vitro (Fig. 3D). We administered the LNPs at a dose of 0.5, 1, and 2 μg/well seeded at 80K cells/well in 24-well plates. Sixteen hours later, we measured GFP protein using flow cytometry and observed that 60% of the cells expressed GFP compared to untreated cells at 0.5 μg/well doses (Fig. 3E). To ensure the results were not GFP-specific, we repeated the experiment using a different mRNA. We formulated LNPHNSCC with mRNA encoding an anchored VHH (aVHH) [27, 28, 36] and observed that 80% of the cells expressed aVHH at the 0.5 μg/well dose (Fig. 3F) using flow cytometry. We confirmed aVHH transfection using immunohistochemical staining (Fig. 3G), which showed functional aVHH protein expression throughout FaDu cell membranes.
We also tested whether the transfection efficacy of LNPHNSCC was cell line-dependent and compared it against LNPFirst. We formulated LNPHNSCC and LNPFirst carrying aVHH mRNA and used them to transfect FaDu human cancer cells (Fig. 3H), AML-12 murine hepatocytes (Fig. 3I), and human embryonic kidney (HEK-293) cells (Fig. 3J) in vitro at 0.1, 0.25, and 0.5 μg/80K cells. LNPHNSCC yielded superior transfection of both human cell lines (FaDu and HEK-293), while LNPFirst showed superior transfection of AML-12 murine hepatocytes. While the percent transfected levels of LNPFirst were high for both FaDu and HEK-293 cells, their mean fluorescence intensity (MFI) was lower than the MFI observed in FaDu and HEK-293 cells transfected with LNPHNSCC (Fig. 3H,J). MFI differences were not as pronounced for the AML-12 murine hepatocytes, where LNPFirst yielded superior transfection versus LNPHNSCC (Fig. 3I). One limitation to these data is that in vitro nucleic acid delivery does not predict in vivo delivery for LNPs [22]. Nevertheless, this suggested that LNPHNSCC may have better affinity for human cell lines, particularly FaDu cancer cells, while LNPFirst may have better affinity for murine hepatocytes.
We then evaluated whether LNPHNSCC delivered mRNA in vivo and compared it with LNPFirst to verify whether the sequential high-throughput screens yielded an LNP with preferential tumor tropism. Once more, we established two FaDu xenograft tumors in Nu/J mice. We then formulated LNPHNSCC and LNPFirst with an mRNA that encoded for an anchored nanoluciferase (anNanoLuc) [27]. Mice with FaDu flank tumors were intravenously administered with LNPHNSCC or LNPFirst at a dose of 2 mg/kg anNanoLuc mRNA; control mice for background luminescence were injected with 1X PBS. Thirty-six hours later, we harvested tumors and livers and used an in vivo imaging system (IVIS) to quantify the luciferase expression by measuring whole-organ bioluminescence (Fig. 4A). FaDu tumors transfected with LNPHNSCC had comparable bioluminescence to those transfected with LNPFirst, as measured by the fold change in total flux compared to the control tumors and by the radiance after subtracting the controls’ background radiance (Fig. 4B). When imaging the livers, LNPHNSCC yielded significantly lower bioluminescence than LNPFirst (Fig. 4C). To quantify this preferential tumor tropism, we computed the tumor-to-liver ratios using fold change and radiance over control for both LNPs and observed that LNPHNSCC had higher ratios than LNPFirst under both measurement criteria (Fig. 4D). These data were consistent with the goal of maximizing tumor delivery and minimizing liver delivery.
Fig. 4. LNPHNSCC shows preferential systemic delivery of mRNA to human solid tumors while de-targeting the liver in vivo.

(A) LNPHNSCC and LNPFirst, the winner LNP of the first screen, were formulated with an anchored NanoLuc mRNA and administered intravenously to Nu/J mice carrying FaDu xenograft tumors at 2 mg of RNA/kg. Thirty-six hours later, luciferase expression was quantified via whole-organ imaging in (B) FaDu tumors and (C) livers. Fold change total flux and radiance over control organs’ background (p/s/cm2/sr) in tumors were comparable while LNPHNSCC had lower NanoLuc expression in the livers, demonstrating de-targeted delivery to the liver. (D) Tumor tropism was quantified by computing the tumor-to-liver ratio using fold change and radiance over control for both LNPs, demonstrating LNPHNSCC had superior tumor preferential delivery. (E) LNPHNSCC and LNPFirst were formulated with an aVHH mRNA and administered intravenously to Nu/J mice carrying FaDu xenograft tumors at 2 mg of RNA/kg. Sixteen hours later, aVHH protein expression (% transfected and MFI) was quantified via flow cytometry in (F) FaDu tumors (human FaDu cells, ECs, and immune cells) and (G) livers (immune cells, ECs, and hepatocytes). LNPHNSCC and LNPLlver both functionally delivered aVHH mRNA to tumor cell types comparably while LNPHNSCC had lower aVHH expression in liver ECs and hepatocytes, demonstrating de-targeted delivery to the liver. (H) Tumor tropism was quantified by computing the tumor-to-liver ECs and FaDu-to-hepatocyte ratios using % transfected and MFI for both LNPs, demonstrating LNPHNSCC had superior tumor preferential delivery ECs: Endothelial cells, MFI: Mean fluorescence intensity. Two-way ANOVA, Tukey’s multiple comparisons test, *P < 0,05, **P < 0.01, ***P < 0.001, ****P < 0.001, average ± S.D.
To test whether LNPHNSCC functionally delivered mRNA independently of the HNSCC tumor model, we injected anNanoLuc-carrying LNPHNSCC intravenously at 2 mg/kg to NSG mice inoculated with HNSCC patient-derived xenograft (PDX) tumors. Patient-derived cells (328373-195-R-J1-PDC) obtained from the National Cancer Institute Patient-Derived Models Repository protocols were subcutaneously administered into hind legs to create one xenograft per mouse. When the tumors reached 150–250 mm3, they were treated; thirty-six hours later, tissues were isolated (Supplementary Fig. S3A). PDX tumors again displayed significant bioluminescence compared to PBS-treated mice, with some delivery to the liver (Supplementary Fig. S3B), providing another line of evidence that LNPHNSCC effectively transfects HNSCC tumor masses in vivo. We did not observe acute systemic toxicity from LNPHNSCC in NSG mice with PDX tumors, which we measured with animal weights and histological staining (Supplementary Fig. S4). We also quantified delivery in the spleen, which is another common off-target organ. We observed higher transfection in the spleen than in the liver (Supplementary Fig. S3C). This made us wonder whether this was also the case in the Nu/J mice inoculated with FaDu tumors. Thus, we re-confirmed the results by injecting LNPHNSCC carrying NanoLuc mRNA to Nu/J mice bearing FaDu tumors (Supplementary Fig. S5A). We found that the tumors were still transfected while liver delivery decreased (Supplementary Fig. S5B). However, the spleen was transfected at higher levels than the liver (Supplementary Fig. S5C). This suggests spleen de-targeting could also be required.
As a final control, we formulated LNPHNSCC and LNPFirst carrying aVHH mRNA and injected them intravenously into Nu/J mice with bilateral FaDu flank tumors at a dose of 2 mg/kg mRNA (Fig. 4E). Sixteen hours later, we harvested tumors and livers, digested them into single-cell suspensions, stained them with cell marker antibodies and anti-VHH antibody, and quantified the aVHH protein expression via flow cytometry (Fig. 4E). Within FaDu tumors, we looked at aVHH transfection levels (% transfected and MFI) in human FaDu HNSCC cells (anti-human CD47+), murine ECs (CD31+), and murine immune cells (CD45+). Both LNPFirst and LNPHNSCC transfected ECs with highest efficiency in the FaDu tumors, as expected given the physiological fate of LNPs traveling through vasculature after systemic administration (Fig. 4F). FaDu HNSCC and immune cells were transfected at lower efficacy, but significantly higher MFI was observed for both LNPs compared to PBS-treated control tumors across all cell types. Within the livers, we looked at aVHH transfection levels in murine ECs (CD31+), murine immune cells (CD45+), and murine hepatocytes (CD31−, CD45−) after gating for apoptotic cells and red blood cells. LNPHNSCC had significantly lower aVHH transfection levels than LNPFirst for both ECs and hepatocytes (Fig. 4G), further validating the liver de-targeting effect observed with the bioluminescence measurements. To quantify this preferential tumor tropism, we computed the tumor-to-liver ECs and FaDu-to-hepatocyte ratios using % transfected and MFI for both LNPs and observed that LNPHNSCC had higher ratios than LNPFirst under both measurement criteria (Fig. 4H). When looking at aVHH transfection levels in the spleen, off-target delivery was high, consistent with the bioluminescence measurements in the spleen (Supplementary Fig. S6). We noted that LNPHNSCC had significantly lower spleen delivery than LNPFirst (Supplementary Fig. S6B), similar to observations in the liver. Overall, these data further support the goal of maximizing tumor delivery and minimizing liver delivery, now validated with a second mRNA and quantified via flow cytometry. Sequential in vivo barcoded screens guided us to an LNP that de-targeted the liver while still maintaining optimal mRNA delivery to human tumor models after systemic administration, as demonstrated in these studies.
4. Conclusion
LNPs are clinically relevant delivery vehicles for mRNA. While intramuscular vaccines and systemic mRNA drugs that target hepatocytes are already in the clinic, delivery to other tissues remains an important challenge. Given the increasing research on mRNA-LNP therapies for oncology [3, 37, 38], identifying LNPs with reduced off-target delivery could be important. To maximize tumor delivery while minimizing liver delivery, we performed two iterative, high-throughput in vivo screens in mice inoculated with different HNSCC tumor models. This direct in vivo approach identified LNPHNSCC, a novel LNP. Notably, when we evaluated LNPHNSCC against LNPFirst, the winner of the first screen with the highest tumor delivery but also highest liver delivery, the results were consistent with the barcode data: high FaDu transfection was maintained while significantly reducing liver transfection. Even though LNPHNSCC reduced liver delivery in vivo, we found evidence for the importance of considering the spleen. These data provide one line of evidence that future barcoding screens could efficiently identify LNPs with improved on- and off-target delivery ratios.
It is important to recognize several limitations of this work. First, we screened and confirmed LNPHNSCC in mice, which may exhibit delivery profiles different from non-human primates and humans [28]. Second, our screens were performed using DNA barcodes only, which may not represent functional mRNA delivery. Third, we did not identify the molecular mechanism driving LNP delivery into the human cancer cells, or extracellular drug interactions with the heterogeneous tumor microenvironment [39-41], or the effects of immune system competency in systemic delivery of LNPs to solid tumors; this may require more complex datasets that combine delivery and RNA sequencing [42-44] and immunocompetent preclinical cancer models [45]. Fourth, the hydrodynamic diameter of LNPHNSCC was inconsistent at times. Future work will be needed to optimize the consistency of the formulation as well as the robustness of relationships between LNP diameter and delivery. Despite these limitations, we believe the approach described here supports future efforts to more efficiently identify LNPs that can reach solid tumors while minimizing off-target delivery to the liver.
Supplementary Material
Acknowledgments.
The authors thank Karen Tiegren at Georgia Tech for copyediting the manuscript.
Funding.
This research was funded by the National Institutes of Health (R01DE026941, awarded to E.J.S. and J.E.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest. J.E.D. is an advisor to GV. E.J.S. has ownership interest in PNP Therapeutics, Inc. and serves on the Board of Directors for the company, which develops products used in cancer research. Dr. Sorscher is also an inventor of technology being evaluated in studies described by this report. The terms of this arrangement for Dr. Sorscher have been reviewed and approved by Emory University in accordance with its conflict-of-interest policies. Dr. Jeong Hong also has minor equity interest in this company. All other authors declare no conflict of interest.
CReDit Author Statement
Sebastian G. Huayamares: Conceptualization, Methodology, Validation, Investigation, Writing – Original Draft. Melissa P. Lokugamage: Conceptualization, Methodology, Investigation, Writing – Original Draft. Regina Rab: Methodology, Investigation. Alejandro Da Silva: Investigation. Hyejin Kim: Investigation. Afsane Radmand: Investigation. David Loughrey: Investigation. Liming Lian: Investigation. Yuning Hou: Investigation. Bhagelu R. Achyut: Investigation. Annette Ehrhardt: Investigation. Jeong S. Hong: Investigation. Cory D. Sago: Investigation. Kalina Paunovska: Investigation. Elisa Schrader Echeverri: Investigation. Daryll Vanover: Investigation. Philip J. Santangelo: Supervision. Eric J. Sorscher: Conceptualization, Methodology, Supervision, Writing – Review & Editing, Project Administration. James E. Dahlman: Conceptualization, Methodology, Supervision, Writing – Review & Editing.
References.
- [1].Baden LR, El Sahly HM, Essink B, Kotloff K, Frey S, Novak R, Diemert D, Spector SA, Rouphael N, Creech CB, McGettigan J, Khetan S, Segall N, Solis J, Brosz A, Fierro C, Schwartz H, Neuzil K, Corey L, Gilbert P, Janes H, Follmann D, Marovich M, Mascola J, Polakowski L, Ledgerwood J, Graham BS, Bennett H, Pajon R, Knightly C, Leav B, Deng W, Zhou H, Han S, Ivarsson M, Miller J, Zaks T, Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine, New England Journal of Medicine, 384 (2020) 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Polack FP, Thomas SJ, Kitchin N, Absalon J, Gurtman A, Lockhart S, Perez JL, Pérez Marc G, Moreira ED, Zerbini C, Bailey R, Swanson KA, Roychoudhury S, Koury K, Li P, Kalina WV, Cooper D, Frenck RW, Hammitt LL, Türeci Ö, Nell H, Schaefer A, Ünal S, Tresnan DB, Mather S, Dormitzer PR, Üahin U, Jansen KU, Gruber WC, Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine, New England Journal of Medicine, 383 (2020) 2603–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Barbier AJ, Jiang AY, Zhang P, Wooster R, Anderson DG, The clinical progress of mRNA vaccines and immunotherapies, Nature Biotechnology, (2022). [DOI] [PubMed] [Google Scholar]
- [4].IntelliaTherapeutics, Intellia and Regeneron Announce Initial Data from the Cardiomyopathy Arm of Ongoing Phase 1 Study of NTLA-2001, an Investigational CRISPR Therapy for the Treatment of Transthyretin (ATTR) Amyloidosis, in, https://ir.intelliatx.com/news-releases/news-release-details/intellia-and-regeneron-announce-initial-data-cardiomyopathy-arm, 2022.
- [5].IntelliaTherapeutics, Study to Evaluate Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of NTLA-2001 in Patients With Hereditary Transthyretin Amyloidosis With Polyneuropathy (ATTRv-PN) and Patients With Transthyretin Amyloidosis-Related Cardiomyopathy (ATTR-CM) - NCT04601051, in, clinicaltrials.gov, 2021.
- [6].Johnson DE, Burtness B, Leemans CR, Lui VWY, Bauman JE, Grandis JR, Head and neck squamous cell carcinoma, Nature Reviews Disease Primers, 6 (2020) 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Carvalho AL, Nishimoto IN, Califano JA, Kowalski LP, Trends in incidence and prognosis for head and neck cancer in the United States: a site-specific analysis of the SEER database, Int J Cancer, 114 (2005) 806–816. [DOI] [PubMed] [Google Scholar]
- [8].Chisholm E, Bapat U, Chisholm C, Alusi G, Vassaux G, Gene therapy in head and neck cancer: a review, Postgrad Med J, 83 (2007) 731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Behbahani TE, Rosenthal EL, Parker WB, Sorscher EJ, Intratumoral generation of 2-fluoroadenine to treat solid malignancies of the head and neck, Head Neck, 41 (2019) 1979–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Suominen E, Toivonen R, Grenman R, Savontaus M, Head and neck cancer cells are efficiently infected by Ad5/35 hybrid virus, J Gene Med, 8 (2006) 1223–1231. [DOI] [PubMed] [Google Scholar]
- [11].van Zeeburg HJT, Huizenga A, Brink A, van den Doel PB, Zhu ZB, McCormick F, Brakenhoff RH, van Beusechem VW, Comparison of oncolytic adenoviruses for selective eradication of oral cancer and pre-cancerous lesions, Gene Therapy, 17 (2010) 1517–1524. [DOI] [PubMed] [Google Scholar]
- [12].Reddy PS, Ganesh S, Yu DC, Enhanced gene transfer and oncolysis of head and neck cancer and melanoma cells by fiber chimeric oncolytic adenoviruses, Clin Cancer Res, 12 (2006) 2869–2878. [DOI] [PubMed] [Google Scholar]
- [13].Fukuhara H, Ino Y, Todo T, Oncolytic virus therapy: A new era of cancer treatment at dawn, Cancer Sci, 107 (2016) 1373–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].GeoVax, Safety and Efficacy of Repeat Administration of Ad/PNP and Fludarabine Phosphate in Patients With Local Head/Neck Cancer - NCT03754933, in, clinicaltrials.gov, 2019.
- [15].Wu Z, Yang H, Colosi P, Effect of genome size on AAV vector packaging, Mol Ther, 18 (2010) 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Aronson SJ, Veron P, Collaud F, Hubert A, Delahais V, Honnet G, de Knegt RJ, Junge N, Baumann U, Di Giorgio A, D'Antiga L, Ginocchio VM, Brunetti-Pierri N, Labrune P, Beuers U, Bosma PJ, Mingozzi F, Prevalence and Relevance of Pre-Existing Anti-Adeno-Associated Virus Immunity in the Context of Gene Therapy for Crigler-Najjar Syndrome, Hum Gene Ther, 30 (2019) 1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ertl HCJ, High KA, Impact of AAV Capsid-Specific T-Cell Responses on Design and Outcome of Clinical Gene Transfer Trials with Recombinant Adeno-Associated Viral Vectors: An Evolving Controversy, Hum Gene Ther, 28 (2017) 328–337. [DOI] [PubMed] [Google Scholar]
- [18].Chandler M, Panigaj M, Rolband LA, Afonin KA, Challenges to optimizing RNA nanostructures for large scale production and controlled therapeutic properties, Nanomedicine (Lond), 15 (2020) 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Paunovska K, Loughrey D, Dahlman JE, Drug delivery systems for RNA therapeutics, Nature Reviews Genetics, 23 (2022) 265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Loughrey D, Dahlman JE, Non-liver mRNA Delivery, Accounts of Chemical Research, 55 (2022) 13–23. [DOI] [PubMed] [Google Scholar]
- [21].Da Silva Sanchez AJ, Zhao K, Huayamares SG, Hatit MZC, Lokugamage MP, Loughrey D, Dobrowolski C, Wang S, Kim H, Paunovska K, Kuzminich Y, Dahlman JE, Substituting racemic ionizable lipids with stereopure ionizable lipids can increase mRNA delivery, J Control Release, 353 (2023) 270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Paunovska K, Sago CD, Monaco CM, Hudson WH, Castro MG, Rudoltz TG, Kalathoor S, Vanover DA, Santangelo PJ, Ahmed R, Bryksin AV, Dahlman JE, A Direct Comparison of in Vitro and in Vivo Nucleic Acid Delivery Mediated by Hundreds of Nanoparticles Reveals a Weak Correlation, Nano Letters, 18 (2018) 2148–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dahlman JE, Barnes C, Khan OF, Thiriot A, Jhunjunwala S, Shaw TE, Xing Y, Sager HB, Sahay G, Speciner L, Bader A, Bogorad RL, Yin H, Racie T, Dong Y, Jiang S, Seedorf D, Dave A, Singh Sandhu K, Webber MJ, Novobrantseva T, Ruda VM, Lytton-Jean AKR, Levins CG, Kalish B, Mudge DK, Perez M, Abezgauz L, Dutta P, Smith L, Charisse K, Kieran MW, Fitzgerald K, Nahrendorf M, Danino D, Tuder RM, von Andrian UH, Akinc A, Panigrahy D, Schroeder A, Koteliansky V, Langer R, Anderson DG, In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight, Nature Nanotechnology, 9 (2014) 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen D, Love KT, Chen Y, Eltoukhy AA, Kastrup C, Sahay G, Jeon A, Dong Y, Whitehead KA, Anderson DG, Rapid Discovery of Potent siRNA-Containing Lipid Nanoparticles Enabled by Controlled Microfluidic Formulation, Journal of the American Chemical Society, 134 (2012) 6948–6951. [DOI] [PubMed] [Google Scholar]
- [25].Dahlman JE, Kauffman KJ, Xing Y, Shaw TE, Mir FF, Dlott CC, Langer R, Anderson DG, Wang ET, Barcoded nanoparticles for high throughput in vivo discovery of targeted therapeutics, Proceedings of the National Academy of Sciences, 114 (2017) 2060–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Carrasco MJ, Alishetty S, Alameh M-G, Said H, Wright L, Paige M, Soliman O, Weissman D, Cleveland TE, Grishaev A, Buschmann MD, Ionization and structural properties of mRNA lipid nanoparticles influence expression in intramuscular and intravascular administration, Communications Biology, 4 (2021) 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lindsay KE, Vanover D, Thoresen M, King H, Xiao P, Badial P, Araínga M, Park SB, Tiwari PM, Peck HE, Blanchard EL, Feugang JM, Olivier AK, Zurla C, Villinger F, Woolums AR, Santangelo PJ, Aerosol Delivery of Synthetic mRNA to Vaginal Mucosa Leads to Durable Expression of Broadly Neutralizing Antibodies against HIV, Mol Ther, 28 (2020) 805–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hatit MZC, Lokugamage MP, Dobrowolski CN, Paunovska K, Ni H, Zhao K, Vanover D, Beyersdorf J, Peck HE, Loughrey D, Sato M, Cristian A, Santangelo PJ, Dahlman JE, Species-dependent in vivo mRNA delivery and cellular responses to nanoparticles, Nat Nanotechnol, 17 (2022) 310–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lokugamage MP, Sago CD, Dahlman JE, Testing thousands of nanoparticles in vivo using DNA barcodes, Current Opinion in Biomedical Engineering, 7 (2018) 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Suk JS, Xu Q, Kim N, Hanes J, Ensign LM, PEGylation as a strategy for improving nanoparticle-based drug and gene delivery, Adv Drug Deliv Rev, 99 (2016) 28–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Cho KJ, Park EJ, Kim MS, Joo YH, Characterization of FaDu-R, a radioresistant head and neck cancer cell line, and cancer stem cells, Auris Nasus Larynx, 45 (2018) 566–573. [DOI] [PubMed] [Google Scholar]
- [32].Parker WB, Allan PW, Waud WR, Hong J, Gilbert-Ross M, Achyut BR, Joshi D, Behbahani T, Rab R, Ealick SE, Sorscher EJ, The use of Trichomonas vaginalis purine nucleoside phosphorylase to activate fludarabine in the treatment of solid tumors, Cancer Chemother Pharmacol, 85 (2020) 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schoenmaker L, Witzigmann D, Kulkarni JA, Verbeke R, Kersten G, Jiskoot W, Crommelin DJA, mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability, International Journal of Pharmaceutics, 601 (2021) 120586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cheng X, Lee RJ, The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery, Adv Drug Deliv Rev, 99 (2016) 129–137. [DOI] [PubMed] [Google Scholar]
- [35].Zhang R, El-Mayta R, Murdoch TJ, Warzecha CC, Billingsley MM, Shepherd SJ, Gong N, Wang L, Wilson JM, Lee D, Mitchell MJ, Helper lipid structure influences protein adsorption and delivery of lipid nanoparticles to spleen and liver, Biomaterials Science, 9 (2021) 1449–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Tiwari PM, Vanover D, Lindsay KE, Bawage SS, Kirschman JL, Bhosle S, Lifland AW, Zurla C, Santangelo PJ, Engineered mRNA-expressed antibodies prevent respiratory syncytial virus infection, Nat Commun, 9 (2018) 3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fiedler K, Lazzaro S, Lutz J, Rauch S, Heidenreich R, mRNA Cancer Vaccines, Recent Results Cancer Res, 209 (2016) 61–85. [DOI] [PubMed] [Google Scholar]
- [38].Huayamares SG, Lokugamage MP, Da Silva Sanchez AJ, Dahlman JE, A systematic analysis of biotech startups that went public in the first half of 2021, Current Research in Biotechnology, 4 (2022) 392–401. [Google Scholar]
- [39].Huayamares SG, Song JY, Huang A, Crowl SR, Groer CE, Forrest ML, Berkland CJ, Constructing a Biomaterial to Simulate Extracellular Drug Transport in Solid Tumors, Macromolecular Bioscience, 20 (2020) 2000251. [DOI] [PubMed] [Google Scholar]
- [40].Huayamares Moreno SG, A Foe-Inspired Friend: Constructing a Tumor Tissue-Mimicking Biomaterial to Study Intratumoral Drug Transport, in, University of Kansas, United States -- Kansas, 2020, pp. 57. [Google Scholar]
- [41].Huang A, Pressnall MM, Lu R, Huayamares SG, Griffin JD, Groer C, DeKosky BJ, Forrest ML, Berkland CJ, Human intratumoral therapy: Linking drug properties and tumor transport of drugs in clinical trials, Journal of Controlled Release, 326 (2020) 203–221. [DOI] [PubMed] [Google Scholar]
- [42].Radmand A, Lokugamage MP, Kim H, Dobrowolski C, Zenhausern R, Loughrey D, Huayamares SG, Hatit MZC, Ni H, Del Cid A, Da Silva Sanchez AJ, Paunovska K, Schrader Echeverri E, Shajii A, Peck H, Santangelo PJ, Dahlman JE, The Transcriptional Response to Lung-Targeting Lipid Nanoparticles in Vivo, Nano Letters, 23 (2023) 993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Paunovska K, Loughrey D, Sago CD, Langer R, Dahlman JE, Using Large Datasets to Understand Nanotechnology, Adv Mater, 31 (2019) e1902798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hatit MZC, Dobrowolski CN, Lokugamage MP, Loughrey D, Ni H, Zurla C, Da Silva Sanchez AJ, Radmand A, Huayamares SG, Zenhausern R, Paunovska K, Peck HE, Kim J, Sato M, Feldman JI, Rivera M-A, Cristian A, Kim Y, Santangelo PJ, Dahlman JE, Nanoparticle stereochemistry-dependent endocytic processing improves in vivo mRNA delivery, Nature Chemistry, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chuprin J, Buettner H, Seedhom MO, Greiner DL, Keck JG, Ishikawa F, Shultz LD, Brehm MA, Humanized mouse models for immuno-oncology research, Nature Reviews Clinical Oncology, 20 (2023) 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.