

SUMMARY
Epigenetic alterations are a key hallmark of aging but have been limitedly explored in tissues. Here, using naturally aged murine liver as a model and extending to other quiescent tissues, we find that aging is driven by temporal chromatin alterations that promote a refractory cellular state and compromise cellular identity. Using an integrated multi-omics approach and the first direct visualization of aged chromatin, we find that globally, old cells show H3K27me3-driven broad heterochromatinization and transcriptional suppression. At the local level, site-specific loss of H3K27me3 over promoters of genes encoding developmental transcription factors leads to expression of otherwise non-hepatocyte markers. Interestingly, liver regeneration reverses H3K27me3 patterns and rejuvenates multiple molecular and physiological aspects of the aged liver.
Graphical Abstract
In brief
Yang et al. find that the histone modification H3K27me3 accumulates in many tissues with age, due to increased time in quiescence. H3K27me3 is gained over lamin-associated domains but reduced at developmental gene promoters with consequent changes in global and local transcription. Liver regeneration breaks quiescence and partially restores youthful patterns.
INTRODUCTION
The epigenome is responsible for connecting the genetic information in our chromosomes to functional outcomes; i.e., it links the genotype with the phenotype.1 Age-related epigenomic changes drive organ dysfunction through increased inflammation, fibrosis, reduced regeneration, and incidence of disease.2–5 Given the reversible nature of epigenomic changes, they are attractive therapeutic targets.
Aside from DNA methylation, age-related chromatin mechanisms have been investigated limitedly, assayed primarily in model organisms or cellular models.2 Consequently, this has restricted our understanding of cellular aging in quiescent mammalian tissues. Quiescence refers to a reversible G0 state of differentiated cells required to achieve and maintain optimal organ mass, geometry, and homeostatic function. Quiescent cells do not allocate resources to cell division or growth but remain metabolically active and responsive to environmental stresses.6 Aged tissue thus contains cells that have remained quiescent for prolonged periods but whether prolonged quiescence (a state we call hyper-quiescence) incurs temporal epigenomic alterations and impacts organ function is unknown. Alternatively, aged tissues also accumulate senescent cells7 distinctly from classic quiescence. Senescent cells in most tissues are rare and organ dysfunction is evident prior to detection of senescent cells, suggesting that prolonged quiescence may dominate age-associated functional decline.
One chromatin mechanism that has been implicated but not investigated deeply in quiescence and aging is lysine trimethylation on histone H3 (H3K27me38). H3K27me3 induces compaction and gene silencing. This repressive modification is deposited by polycomb repressive complex 2 (PRC2), which assembles with one of two paralogous catalytic subunits: enhancer of zeste homolog 1 (EZH1) or EZH2 to form canonical and non-canonical complexes.9–11 PRC2, along with PRC1, represses key developmental gene promoters required to establish a specific cell lineage. Unlike most repressors, PRC2 is not recruited to these sites by any transcription factor (TF) but rather by the unique chromatin environment. Interestingly, transcription itself can evict PRC2, suggesting that transcriptionally dormant states (such as prolonged quiescence) can perpetuate modifications such as H3K27me3.10,12
We use the liver as a model organ to investigate chromatin changes in aging given its pivotal role in metabolism and evidence of clear age-related decline. Furthermore, the liver has remarkable regenerative capacity,13 which provides an opportune rejuvenation system to test the reversibility of age-associated chromatin states. Our findings reveal H3K27me3 is a mediator of the aging process and further suggest that regeneration partially mitigates age-related H3K27me3 patterns. Analogous chromatin changes occur in other organs (kidney, heart, and muscle).
RESULTS
Increased H3K27me3 is a signature of aging
We used several chromatin-based assays to evaluate the altered epigenome in the aging liver (Figure 1A). Using liquid chromatography-tandem mass spectrometry (LC-MS/MS) we surveyed the histone post-translational modification (hPTM) landscape in the aging liver (Tables S1 and S2, MS raw data at chorusproject: chorusproject.org/1769). As proof-of-principle, the ratio of variant H3.3 to canonical H3.1/3.2 (hereon H3) was increased in old livers (Figure S1A), consistent with previous reports.14 There was no significant difference in monomethyl (me1), dimethyl (me2), trimethyl (me3) or acetyl (ac)-ated histone abundance with age (Figure S1B). However, quantification of single hPTMs revealed some striking changes with age.
Figure 1. H3K27me3 increases during aging and forms age-domains.
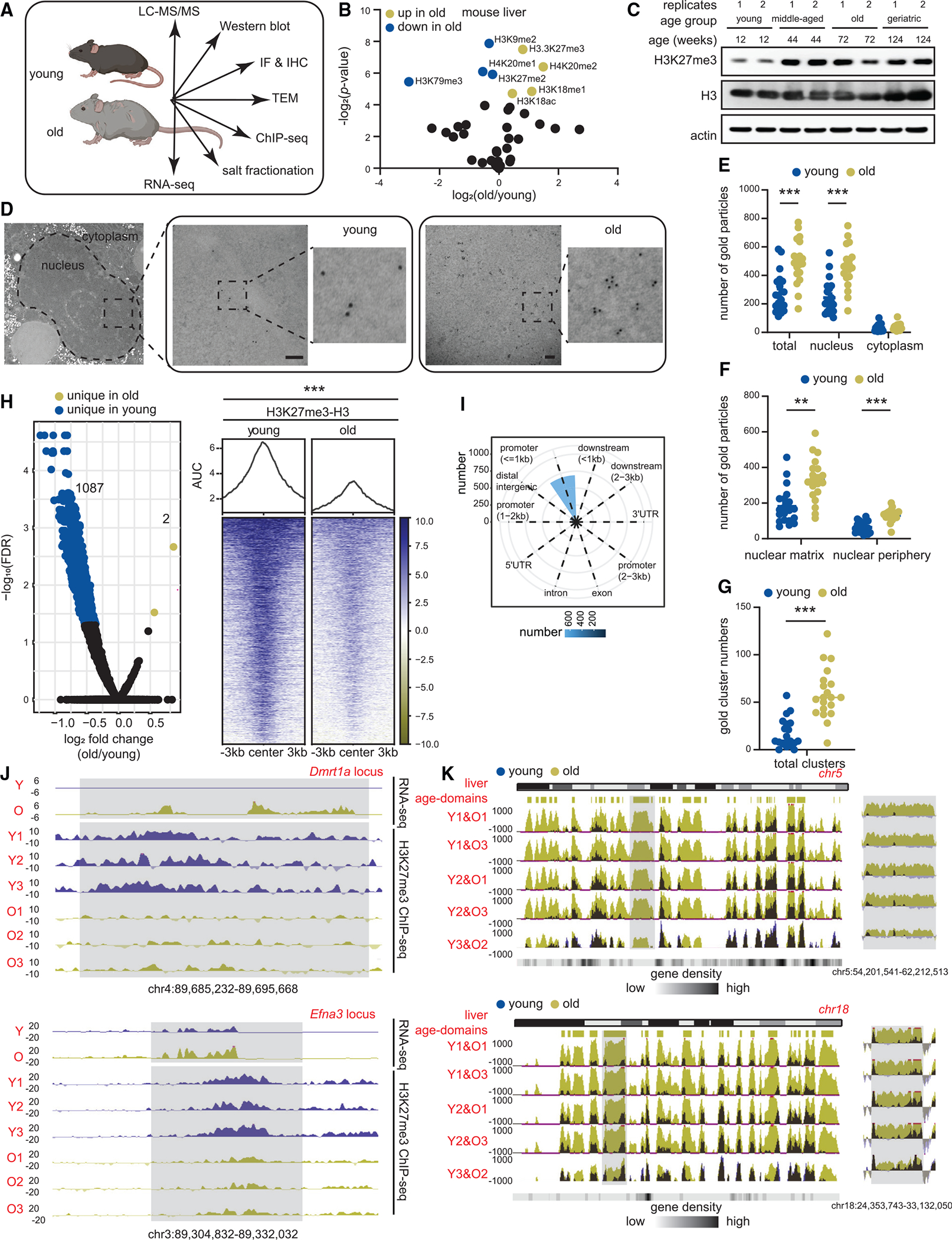
(A) Schematic of chromatin assays performed.
(B) Volcano plot of single hPTMs in old vs. young livers (young 11–12 weeks and old 79–95 weeks, n = 3 biological replicates per group). The significantly increased (green) and decreased (blue) hPTMs are labeled (two-tailed unpaired t test).
(C) Western blot of H3K27me3, H3 and β-actin in liver lysates from mice of indicated ages (young 11–12 weeks, middle-aged 44 weeks, old 79–95 weeks, and geriatric 118 weeks, n = 2 biological replicates per group).
(D) Representative TEM images of young and old hepatocytes with immunogold labeling of H3K27me3 (young 12 weeks and old 91–99 weeks, n = 2 biological replicates per group). Scale bar is 100 nm.
(E–G) (E) Subcellular location, (F) subnuclear location, and (G) cluster (i.e., ≥3) quantification of gold particles. For (E)–(G), data are summarized as mean ± SEM with each dot representing one cell. ** p < 0.01 and *** p < 0.001 from a Kolmogorov-Smirnov test with corrections for multiple comparisons at 1% FDR (two-stage step-up Benjamini, Krieger, and Yekutieli).
(H) Volcano plot (left) and heatmap (right) of DiffBind output (young 10–12 weeks and old 79–95 weeks, n = 3 biological replicates per group). Significantly enriched H3K27me3 peaks in old (FDR < 0.05) are in green and those depleted are in blue.
(I) Annotation of differential peaks identified in (H).
(J) Genome browser snapshots of two differentially enriched peak regions from (H). Gray regions show age-related loss of H3K27me3, and de-repression of genes.
(K) Genome browser snapshot of overlaid H3K27me3 ChIP signal (E. coli spike-in normalized) over chr5 (top) and chr18 (bottom) in sex-matched pairs of young and old mouse livers. Location of age-domains are shown on the top and gene density at the bottom. Gray area is expanded on the right of each chromosome.
In pairwise comparisons of young and old livers, the repressive hPTMs H3K27me3, H3K18me1, H4K20me2, and a single active mark, H3K18ac, were significantly enriched in old livers (Figure 1B, green circles). We were intrigued by the increase of H3K27me3 in aging, which has been noted (but not always emphasized) in several reports including those in aged Drosophila head and muscle,15 killifish brain,16 killifish muscle,17 aged murine muscle stem cells18,19 and aged human post-mortem brains.20 As examples, we mined the published LC-MS/MS data from murine muscle stem cells and human brains. Much like the murine liver, these tissues showed significant increase in H3K27me3 with age (Figures S1C and S1D). We used orthogonal methods such as western blot (Figure 1C; Table S1), immunofluorescence (IF, Figure S1E; Table S1) and chromatin immunoprecipitation sequencing (ChIP-seq) (Figure S1F, explained in the next section; Table S1) to validate the increase of H3K27me3 in mouse livers. Furthermore, we carefully examined the hepatocyte ultrastructure by transmission electron microscopy (TEM) coupled to immunogold labeling to visualize the spatial location of H3K27me3 in young and old livers (Table S1). Most gold particles were detected in the nucleus (Figures 1D and 1E) with significantly higher numbers in old hepatocytes (Figure 1E). Within the nucleus, there was a clear preference for localization in the nuclear matrix (>200 nm from the nuclear membrane) over nuclear periphery (<200 nm of the nuclear membrane, Figure 1F). Interestingly, the immunogold particles in the old hepatocyte nuclei formed clusters (i.e., 3 or more gold particles in proximity, Figures 1D and 1G), suggesting an interaction between H3K27me3-rich regions in the aged cell.
Given that aging is likely to change cell composition of the liver, we tested the abundance of major cell types in young and old livers (Figures S1K–S1M; Table S1) and wondered whether the global age-related increase of H3K27me3 occurred in all or specific cell types. Using the PhenoImager platform, we imaged liver sections with a 6-panel cocktail: CD31 (endothelial cells), CK19 (cholangiocytes), F4/80 (Kupffer cells), HNFα (hepatocytes), H3K27me3 (histone modification) and DAPI (DNA). We noted a significant increase in Kupffer cells and decrease in hepatocyte numbers (Figure S1L). Importantly, H3K27me3 was increased in every cell type we tested except for Kupffer cells (Figure S1M). Together, our results show that a global increase in H3K27me3 is a common epigenetic signature of aging.
H3K27me3 shows a unique genomic pattern in aged tissue: Loss of peaks and gain of domains (age-domains)
We next performed H3K27me3 ChIP-seq in young and old livers with total H3 and IgG as controls (Table S1; quality control (QC) in Figures S2A and S2B). Two % exogenous human chromatin from HeLa cells was spiked in before immunoprecipitation for quantitative comparisons (ChIP-Rx21). A lower % of human reads were recovered in old H3K27me3 ChIP samples (1.7% in old vs. 2.1% in young), suggesting proportionally higher mouse H3K27me3 in old livers (Figure S1F). In contrast, the recovery of human reads in H3 and input was comparable between the two groups.
A principal-component analysis (PCA) of both genome coverage (Figure S1G) and H3K27me3 peaks (Figure S1H) clearly segregated young and old samples, suggesting distinct genome-enrichment patterns. Interestingly, there were fewer H3K27me3 peaks in the old samples (mean in young 5,211 vs. old 3,354) although the differences were not significant (Figure S1I). Curiously, the peaks identified in old samples were generally broader, had higher tag density and lower peak height (Figure S1J), suggesting a “spreading” of the signal. Differential peak analysis revealed only 2 peaks with higher H3K27me3 enrichment and 1,087 peaks with lower H3K27me3 enrichment in old livers (FDR < 0.05, Figure 1H; Table S3). These differential peaks were annotated primarily to promoters (Figure 1I). Example genome browser views of two annotated peaks and related gene expression change are shown in Figure 1J. These findings however were inconsistent with our LC-MS/MS, IF, TEM, western blot and ChIP-seq spike-in results that showed a global accumulation of H3K27me3 with age.
This inconsistency prompted us to look deeper into H3K27me3 genome distribution patterns. The full extent of H3K27me3 signal was captured in the whole chromosome view. At this scale, old livers showed strong enrichment across multi-megabase sized regions across all chromosomes (chr5 and chr18 shown as examples in Figures S3A–S3E). We call this type of enrichment “age-domains.” Age-domains were found in all 3 old biological samples (Figures S3C and S3E) and were more prominent when we scaled the coverage maps to spike-in reads (Figure 1K). Furthermore, we noted that age-domains occurred over gene-poor regions (note gene density, Figure 1K, bottom). In sum, our ChIP-seq data identified two different chromatin states in young and old based on H3K27me3 genome-enrichment patterns: young (more peaks, less age-domains) and old (less peaks, more age-domains).
Age-domains lose H3K9me3 and are lamina associated
To further characterize age-domains (schematic in Figure 2A), we assayed the epigenetic landscape at these sites. We “called” age-domains in old samples with the enriched domain detector (EDD) algorithm, which is proficient in detecting wide genomic enrichments with high sensitivity and robustness against local variations.22 EDD accurately identified age-domains as visualized on the genome browser (Figure 1K, note bars on top). We surveyed multiple hPTMs (H3K27me3, H3K9me3, H2A119ub, H3K36me3) as well as total RNA polymerase II (RNAPII) levels at age-domains by ChIP-seq (Table S1). As expected, age-domains showed strong enrichment of H3K27me3 in old samples (Figure 2B). We further mined lamin B1 ChIP-seq data from Whitton et al.23 to profile lamina-associated domains (LADs) and performed DNA immunoprecipitation with anti-5-methylcytosine (5mC) antibody followed by sequencing (meDIP-seq) to identify regions of methylated DNA enrichment (Table S1). These data revealed age-domains were enriched with lamin B1 in young cells with significant loss with age (Figure 2C), suggesting that these are classic LADs.24 Furthermore, as reported before for LADs, these regions underwent significant loss of H3K9me3 with age25 (Figure 2D). Age-domains were depleted for H2A119ub, H3K36me3, 5mC and RNAPII signal (Figures 2E–2H).
Figure 2. Age-domains lose H3K9me3 and are lamina associated.
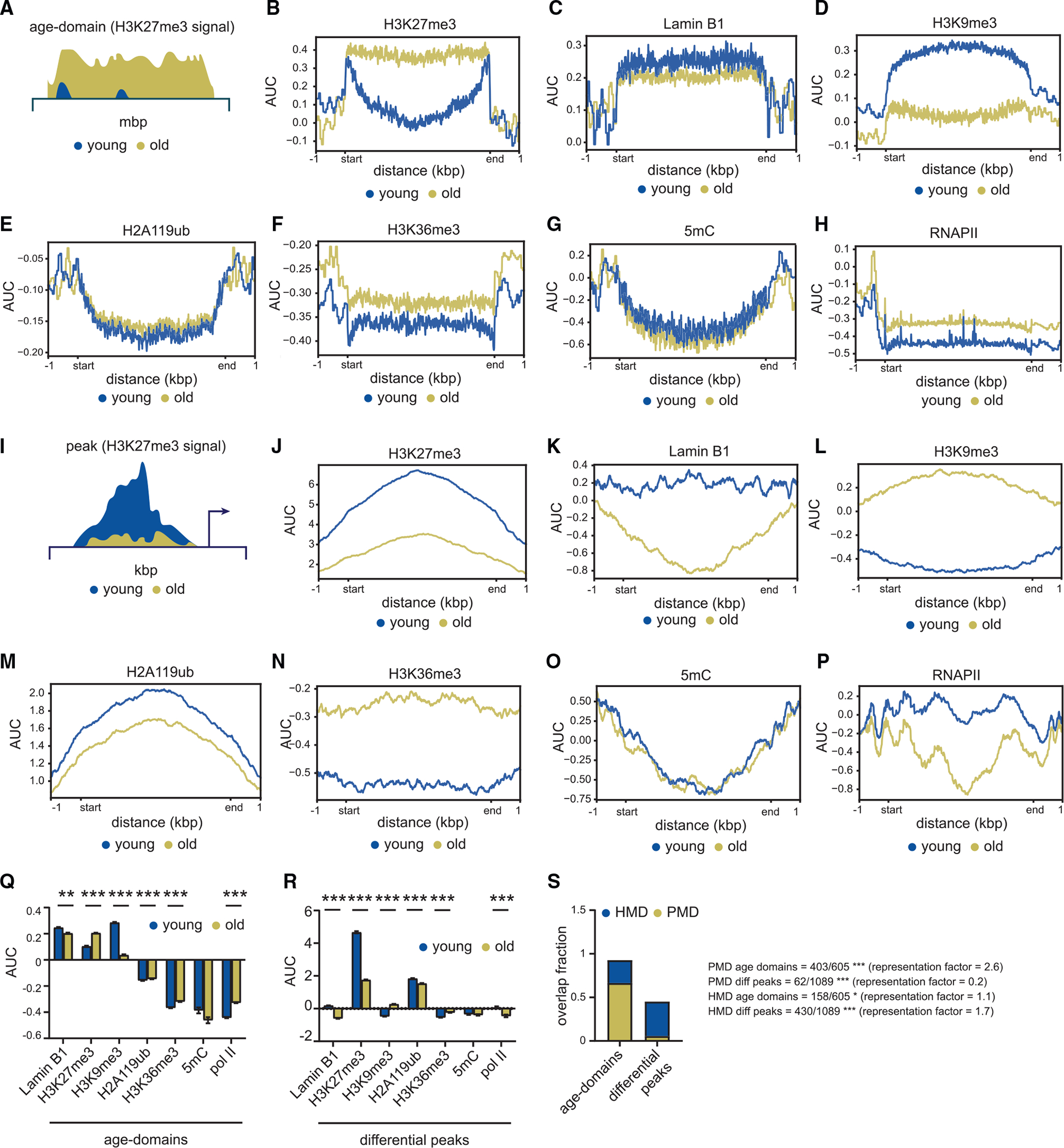
(A) Schematic of an age-domain.
(B–H) (B) Metaplot of H3K27me3, (C) lamin B1, (D) H3K9me3, (E) H2A119ub, (F) H3K36me3, (G) 5mC, and (H) RNAPII signal over age-domains specific to old livers.
(I) Schematic of an H3K27me3 peak region.
(J–P) (J) Metaplot of H3K27me3, (K) lamin B1, (L) H3K9me3, (M) H2A119ub, (N) H3K36me3, (O) 5mC, and (P) RNAPII signal over differential peak regions. For (G) and (O), young 12–18 weeks and old 81–90 weeks, n = 4 biological replicates per group. For all other panels except (C) and (K) (published data), young 12–14 weeks and old 80 weeks, n = 4 biological replicates per group.
(Q and R) (Q) Chromatin alterations at age-domains and (R) H3K27me3 differential peaks. For (Q) and (R), data are summarized as mean ± SEM. ** p < 0.01 and *** p < 0.001 from a Welch’s two-tailed unpaired t test.
(S) Stacked bar plots showing overlap of PMDs and HMDs with age-domains and H3K27me3 differential peaks.
* p < 0.05 and *** p < 0.001 from a hypergeometric test.
In contrast to age-domains, H3K27me3 peak regions (schematic in Figure 2I) showed low enrichment of lamin B1 which was further reduced with age. H3K27me3 and H3K9me3 showed opposite enrichments (as in age-domains), with the latter gaining modest signal and the former losing signal in old samples. H2A119ub also decreased at peaks similar to H3K27me3, suggesting that these sites undergo PRC1/2 mediated repression in young cells. Peak regions were generally depleted of H3K36me3, 5mC and RNAPII (Figures 2J–2P). The findings at age-domains and peak regions are summarized in Figures 2Q and 2R.
In keeping with age-domains being LADs, we observed they significantly overlapped with partially methylated domains (PMDs) that are sites of frequent DNA methylation loss in cancer genomes, as has been reported previously26 (Figure 2S). In contrast, differential peak regions overlapped more with highly methylated domains (HMDs).
Age-domains are heterochromatinized while H3K27me3 peak regions are euchromatinized during aging
To assess whether the H3K27me3 peaks and age-domains segregate into different fractions of chromatin, we measured the salt solubility of mononucleosomes from these regions by extraction under different salt concentrations (Figure 3A). The extracted DNA was purified and analyzed by sequencing. The final pellet (insoluble chromatin) was dissolved in a solubilization buffer and analyzed in a similar fashion (Table S1; QC in Figure S2C). As reported previously,27 we noted that low salt concentrations (0–250 mM) extracted few histones in the supernatant (Figure 5H) and the associated DNA mapped to active regions of the genome (Figures 3B, inset 1, and S4A, inset 1). In the young samples, the euchromatin eluted in the 150 and 250 mM fractions. Euchromatic regions in the old liver showed relatively lower enrichment (Figures 3B, inset 1, and S4A, inset 1), suggesting a decrease in its proportion with aging. Importantly, the old euchromatin fraction eluted at lower salt concentrations (0, 67.5, and 150 mM salt), suggesting a decondensation of this fraction with aging. H3K27me3 peaks in young that were lost in the old (Figure 1H) were among those that showed chromatin decondensation (Figure 3C). Remarkably, we observed that H3K27me3 age-domains in old livers overlapped almost perfectly with the 350 mM and pellet fraction (Figures 3B, inset 2, 3D, and S4A, inset 2), suggesting that these regions are packed into insoluble heterochromatin.
Figure 3. Age-domains are heterochromatinized while H3K27me3 peak regions are euchromatinized during aging.
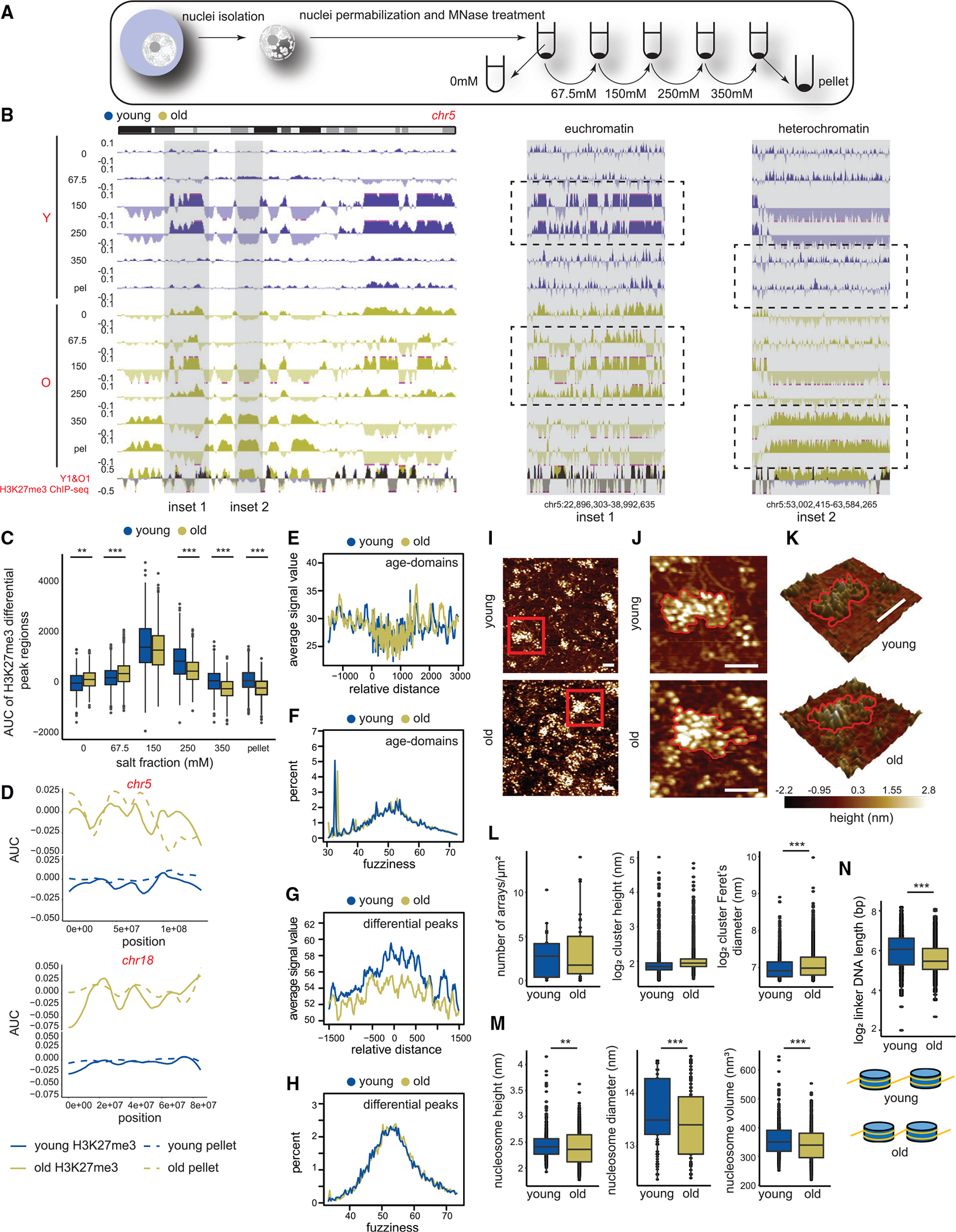
(A) Schematic of salt fractionation experiment (young 11 weeks and old 81 weeks, n = 2 replicates per group).
(B) Genome browser snapshot of salt fraction enrichments on chr5. Inset 1 is euchromatinized and inset 2 is heterochromatinized with age and expanded on the right.
(C) The salt elution profile of differential peaks in Figure 1H. ** p < 0.01, *** p < 0.001 from a Welch’s two-tailed unpaired t test.
(D) H3K27me3 area under the curve (AUC) (solid line) and salt fraction (dotted line) over chr5 and chr18 depicted as fraction of maximum.
(E and F) (E) Nucleosome occupancy and (F) nucleosome fuzziness score distribution at age-domains.
(G and H) (G) Nucleosome occupancy and (H) nucleosome fuzziness score distribution at differential peaks from Figure 1H.
(I) Representative AFM images of extracted chromatin arrays (young 13–14 weeks and old 78–79 weeks, n = 3 biological replicates per group).
(J) The red boxed region in (I) is magnified. An array in each sample is highlighted with a red contour.
(K) Same as (J) but represented as a 3D image. For (I)–(K), scale bar is 100 nm.
(L and M) (L) Chromatin array number per μm2, cluster height and Feret’s diameter and (M) nucleosomal height, diameter, and volume from young and old livers.
(N) NRL (top) and schematic showing NRL changes in young and old (bottom). ** p < 0.01, *** p < 0.001 from a Welch’s two-tailed unpaired t test. For (C) and (L)–(N), the boxes are bounded by the 25th and 75th percentile values with the median represented as the bar in the middle. The whiskers extend to 1.5× the interquartile length.
Figure 5. H3K27me3 patterns in aging are mimicked by deep quiescence cultures (hyper-quiescence).
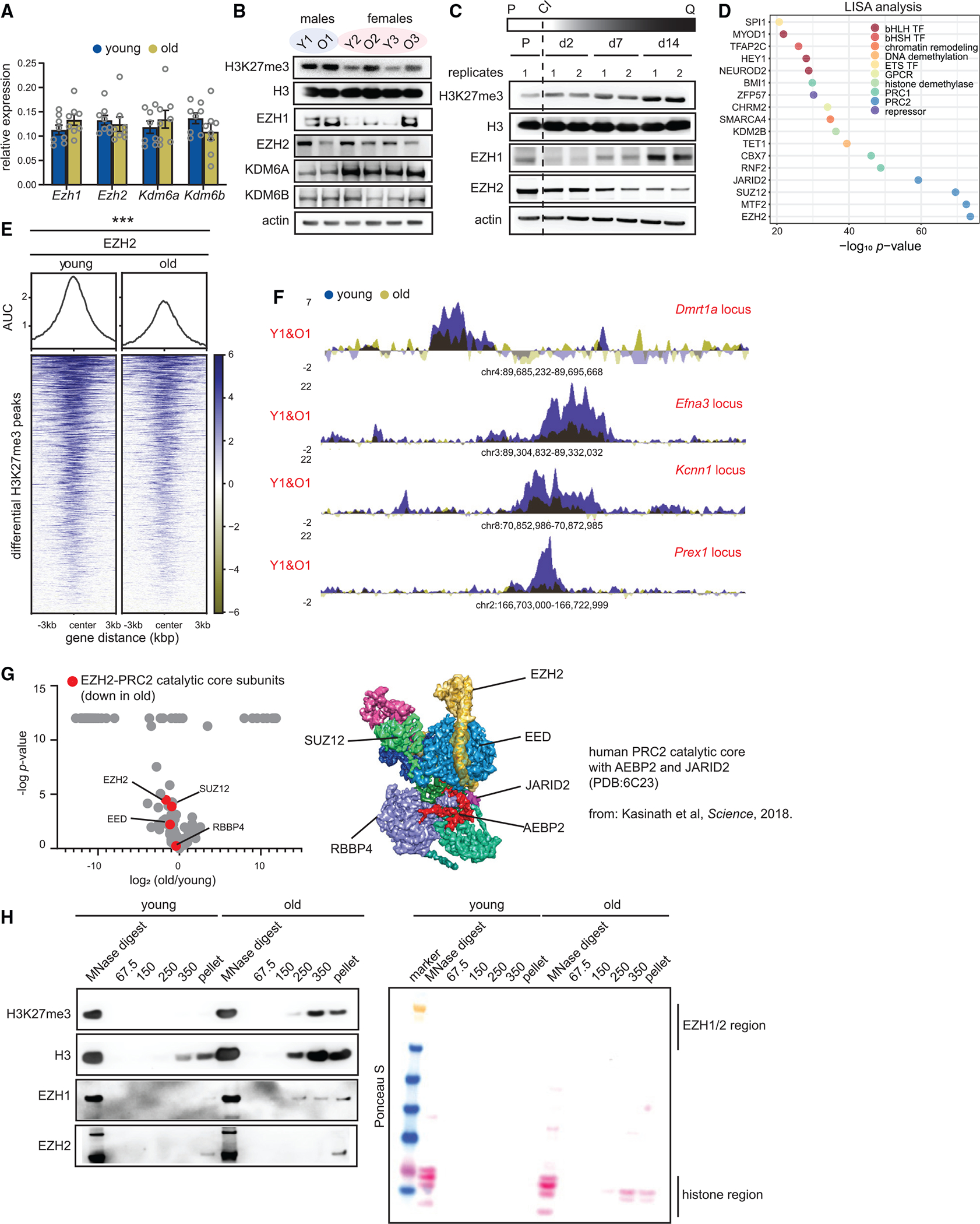
(A) Reverse-transcriptase quantitative PCR (RT-qPCR) analysis of Ezh1, Ezh2, Kdm6a, and Kdm6b mRNA levels in young and old livers. Data are summarized as mean ± SEM (young 12 weeks and old 80 weeks, n = 8 biological replicates per group). A Welch’s two-tailed unpaired t test did not yield any significant results.
(B) Western blot of H3K27me3, H3, EZH1, EZH2, KDM6A, and KDM6B protein levels in young and old livers (young 11–12 weeks and old 79–95 weeks, n = 3 biological replicates per group).
(C) Western blot of H3K27me3, H3, EZH1, and EZH2 in proliferating and quiescent cells over 14 days. Quiescence was induced by contact inhibition (n = 2 biological replicates per group). P is proliferating, Q is quiescence, and CI is contact inhibition.
(D) LISA analysis of H3K27me3 differential peak regions from Figure 1H. p value is calculated using the Wilcoxon rank test comparison of the query and background.
(E) Heatmap showing EZH2 signal at differential peaks from Figure 1H (young 11 weeks and old 80 weeks, n = 4 biological replicates per group). *** p < 0.001 from a Welch’s two-tailed unpaired t test.
(F) Genome browser snapshots of EZH2 enrichment over 4 peak regions from (E).
(G) Volcano plot of EZH2 interactors in old vs. young livers (young 20 weeks and old 86 weeks, n = 4 biological replicates per group). The PRC2 catalytic core subunits are in red. p values are from a two-tailed unpaired t test. The published cryo-EM structure of the PRC2 catalytic core (with AEBP2 and JARID2) is shown on the right.
(H) Western blot of H3K27me3, H3, EZH1, and EZH2 from different salt fractions of chromatin from Figure 3B in young and old livers, Ponceau S-stained membrane on the right.
Additionally, the input samples from salt fractionation experiments afforded us the opportunity to assess nucleosome positions similar to a micrococcal nuclease sequencing (MNase-seq) experiment. Using dynamic analysis of nucleosome position (DANPOS),28 we quantified nucleosome occupancy and fuzziness and did not find any differences at age-domains (Figures 3E and 3F). In contrast, at H3K27me3 differential peak regions, nucleosome occupancy was reduced, concordant with de-repression of these genes, without changes in fuzziness (Figures 3G and 3H).
To physically assess whether differences exist between chromatin structures in young and old livers, we purified chromatin using a very mild MNase digestion protocol followed by extraction from nuclei in buffer containing 67.5 mM NaCl. We analyzed these samples (Table S1) by single-molecule atomic force microscopy (AFM). By very mildly digesting chromatin, only the most accessible (hyper-sensitive) DNA is cut, thereby, preserving the more compacted chromatin arrays. This allowed us to quantify how many distinct chromatin arrays were isolated from young and old liver nuclei under identical conditions (Figures 3I–3K). Qualitatively, chromatin obtained from young livers possessed distinct well-organized clusters. In contrast, chromatin from old livers appeared less coherent, with an overall increase in nucleosome number per field and a “carpet-like” appearance (Figure 3I). This result agrees with the observed increase in solubility of old chromatin at 67.5 mM salt (Figures 3B and 3C). Next, we quantified the number of distinct chromatin arrays (clusters) isolated, as well as the height and Feret’s diameter of these arrays. Despite a trend for larger number of arrays and cluster height in the old livers, these results were not statistically significant (Figure 3L, panels 1 and 2). However, in chromatin obtained from old livers, the Feret’s diameter was broader (Figure 3L, panel 3), showing that arrays from old liver nuclei are larger. To delve deeper into nucleosome-level differences, we next quantified the dimensions of individual nucleosomes (height, diameter, and volume) in arrays obtained from young and old livers. The height of nucleosomes, diameter, and volume were significantly smaller in old livers (Figure 3M, panels 1–3). We further probed the nucleosome repeat length (NRL) of the chromatin arrays. We determined linker lengths by measuring the contour length from the center of one nucleosome to the center of the neighboring nucleosome, as it was not always possible to identify the exact entry and exit location of the DNA strand. Old chromatin fibers had significantly lower NRLs compared to young (Figure 3N). Taken together, these data suggest that in older samples, chromatin arrays are larger and more compact. Individual nucleosomes are also more compacted, possibly reflecting a less accessible state of site exposure and that chromatin arrays from older livers occupy larger areas, suggesting either an increased number of nucleosomes per array and/or a loss in higher order coherence.
Consequences of H3K27me3 genomic redistribution in aging
We wanted to further probe the functional consequences of H3K27me3 peak loss and age-domain gain in old livers. Interestingly, genes near H3K27me3 peaks that were lost in the old livers were related to development (particularly nervous system and cardiac) and cell differentiation (Figure 4A). Loss of H3K27me3 de-repressed these genes (Figure 4B). In support, the H3K36me3 signal also increased over these genes with age (Figure 4C). Ontology terms associated with neuronal function were also seen in the genes upregulated with age (Figure S5D). Most of the genes that lost H3K27me3 signal with age encode developmental TFs, such as Hox, Ascl1, Sox2, Neurog1, Gata4, etc. (Figure 4D; Table S3), that are known to play important roles in neuronal/cardiac lineage specification. Indeed, expression of only 3 TFs, one of which is Ascl1, can reprogram hepatocytes to neurons29
Figure 4. Consequences of H3K27me3 genomic redistribution in aging.
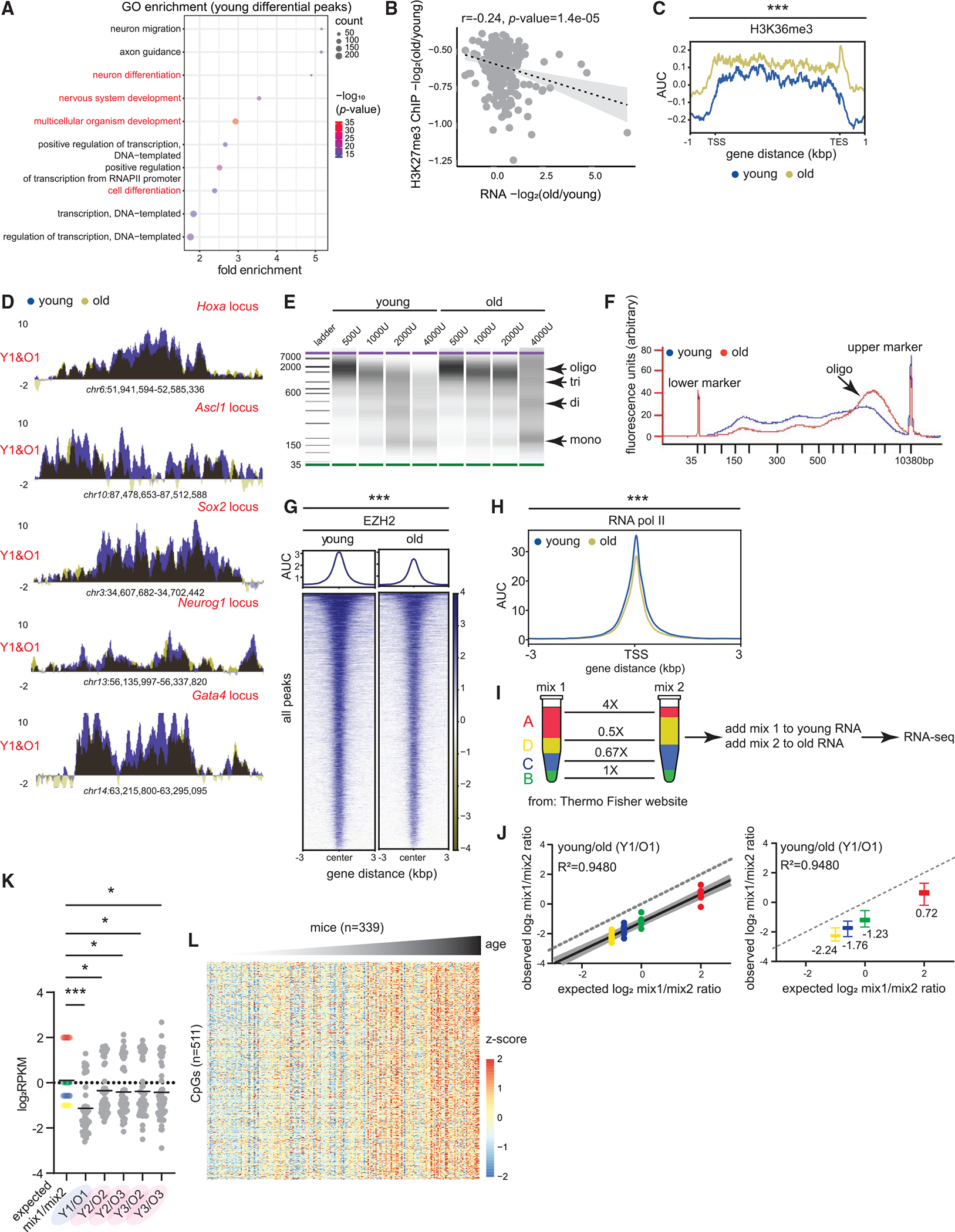
(A) Gene ontology (GO) terms associated with differential peaks in Figure 1H. Development and differentiation genes are in red. p values are from Fisher’s exact test.
(B) Correlation between H3K27me3 and gene expression change in old vs. young. Pearson r and p values are reported.
(C) Metaplot of H3K36me3 signal over gene bodies near differential peaks from Figure 1H (young 12–14 weeks and old 80 weeks, n = 4 biological replicates per group). *** p < 0.001 from a Welch’s two-tailed unpaired t test.
(D) H3K27me3 ChIP-seq profiles at indicated loci in young and old livers.
(E) BioAnalyzer profiles of MNase digest from young and old livers.
(F) The overlaid digestion profile with 2,000 U of MNase.
(G) Heatmap showing EZH2 signal at all called peaks in young and old livers (young 11 weeks and old 80 weeks, n = 4 biological replicates per group).
(H) Metaplot of RNAPII signal (young 12–14 weeks and old 80 weeks, n = 4 biological replicates per group) at all annotated TSSs. For (G)–(H), *** p < 0.001 from a Welch’s two-tailed unpaired t test.
(I) ERCC Exfold transcript abundance and RNA-seq spike-in strategy (young 10–12 weeks and old 79–95 weeks, n = 3 biological replicates per time point). (A)–(D) represent the 4 groups of transcripts present in mix 1 and 2.
(J) Observed vs. expected plot of mix1/mix2 log ratio for one pair of sex-matched young and old animals before resection. The dotted line represents a hypothetical experiment where the observed mix1/mix2 ratio is the same as expected. On the right, same data shown as boxplots with median values indicated. The boxes are bounded by the 25th and 75th percentile values with the median represented as the bar in the middle. The whiskers extend to 1.5× the interquartile length.
(K) Expected and observed mix1/mix2 ratios plotted for all animals. *p < 0.05 and *** p < 0.001 from a one-way ANOVA with corrections for multiple comparisons (FDR method of Benjamini and Hochberg).
(L) Liver DNA methylation signal plotted across different ages from Mozhui et al.30
To assess whether H3K27me3-mediated regulation of developmental genes was conserved in aging human livers, we performed cleavage under targets and tagmentation (CUT&Tag)31 with H3K27me3 antibody on human liver core-needle biopsies (Table S1; QC in Figures S2A and S2B). These human subjects were hospitalized for either cholecystectomy, bariatric procedures, or gastroesophageal reflux disease, and had otherwise normal livers. Given the limited sample amount, we were unable to perform ChIP-seq. Nevertheless, from CUT&Tag data, we identified age-related differential sites in the young (1,117 regions) and old (62 regions) samples (Figure S3F), which was numerically similar to the changes seen in mice. As in mice, genes annotated regions with lower H3K27me3 signal in the old were linked to neuronal and cardiac development (Figure S3G). Of note, we could not visualize age-domains of H3K27me3 in our human samples due to the limitation of CUT&Tag in capturing domain-like enrichments.
However, our peak level analyses in both mice and humans suggest a major loss of H3K27me3-mediated suppression of cell identity genes.
While local loss of H3K27me3 compromises cellular identity, we surmised that broad heterochromatinization may have more global effects such as reduction of chromatin accessibility and global transcription. In concordance, old chromatin was more resistant to MNase digestion compared to young, suggesting the formation of a more condensed structure with age (Figures 4E and 4F). This in turn, likely reduces the genome-wide binding of EZH2 to its target regions as assessed by ChIP-seq (Figure 4G; Table S1). In addition, RNAPII ChIP-seq (Figure 4H; Table S1) showed reduced signal at all annotated TSSs in old samples. To ascertain global transcription levels during aging, we prepared RNA-seq libraries (Table S1, see “_bef” samples) with Exfold External RNA Control Consortium (ERCC) spike-in controls.32 ERCC spike-ins allow for comparisons across samples with different global RNA levels. We added mix 1 to young and mix 2 to old samples prior to ribo-depletion and library generation (Figure 4I). The data were normalized to reads per kilobase per million mapped reads (RPKM) and filtered to remove transcripts < 1 RPKM. The log2RPKM (mix1/mix2) for each ERCC transcript (observed ratio) and the log2 ratio of the known attomolar concentrations of the two mixes (expected ratio) were plotted (Figure 4J). If global transcription is unaffected, the observed ratio (black solid line with gray shaded 95% confidence bands) of mix1/mix2 ERCC transcripts is predicted to mirror the expected ratios (dotted line). Our analysis, however, showed that mix1/mix2 ratios were lower than expected, suggesting a strong genome-wide transcription suppression in old livers. This global transcription suppression was observed for every sex-matched young and old animal we compared (Figure 4K), with males showing stronger suppression than females.
Several reports have suggested an epigenetic switch mechanism operating at developmental gene promoters where loss of H3K27me3 is concomitant with gain of DNA methylation.33,34 Our meDIP-seq data failed to capture any changes at these peak regions (Figure 2O), likely due to the fact that meDIP is biased toward genomic regions with low cytosine-phosphate-guanine (CpG) density.35 Indeed, a significant proportion (422/1,089) of H3K27me3 differential peaks occur at gene promoters with CpG islands (CGI+), while only 53/1,089 occur at CGI-genes. As an alternative, we mined published pan-mammalian bead-based methylation array data (HorvathMammalMethylChip40, ~37K CpGs) from 339 predominantly female genetically diverse C57BL/6J and DBA/2J cross (BXD) mice.30 We overlapped the CpGs from the array data with our 1,089 H3K27me3 peak regions and traced the methylation status of the 511 overlapped CpGs across different ages. The results indicate a progressive increase in methylation over these CpGs (Figure 4L), thus confirming that loss of H3K27me3 at developmental gene promoters is concomitant with an increase in DNA methylation. Complementarily, a recent study shows that many clock CpGs that become hypermethylated with age are PRC2 targets.36
Together, our data show that H3K27me3 redistribution during aging has consequences, including a suggested loss of cell identity, decreased accessibility to genome-cutting enzymes, global transcription suppression, and CpG hypermethylation.
H3K27me3 patterns in aging are mimicked by deep quiescence cultures (hyper-quiescence)
While EZH2 plays a dominant role in H3K27me3-mediated gene repression in actively dividing cells, the paralogous EZH1 is highly expressed in post-mitotic tissue.11 We hypothesized that as tissues spend more time in the post-mitotic stage, i.e., with age, they switch to a more EZH1-dependent mode of repression. To test this, we measured the levels of enzymes affecting H3K27me3 in young and old livers. At the RNA level there was no age-associated change in expression of EZH1/2 or demethylases KDM6A/6B (Figure 5A; Table S1). However, at the protein level, EZH2 levels declined while EZH1 showed increase with age (Figure 5B). Protein levels of KDM6A/6B remained unaltered with age (Figure 5B; Table S1). Of note, KDM6A is encoded by the X chromosome and hence is about half the level in males compared to females. KDM6C was not tested as it is a male-specific inactive paralog. Our results support an EZH2-to-EZH1 switch mechanism in aging.
We reasoned that this switch toward EZH1 occurs due to hyper-quiescence i.e., a state of deep quiescence achieved in aged solid tissues due to prolonged stay in a post-mitotic state. To test this directly, we induced quiescence in Hep-G2 hepatoblastoma cell lines by contact inhibition (Figure 5C). Contact inhibition-induced quiescence best mimics the state of cells in normal tissue homeostasis without nutrient deprivation. We saw an increase in H3K27me3 upon entry into quiescence with concomitant loss of EZH2 and gain of EZH1, a condition that is mimicked in aging (Figure 5B). We thus propose that aged livers are in a hyper-quiescent state with a global gain in EZH1-mediated H3K27me3 and loss of EZH2.
We next wanted to understand the mechanism of H3K27me3 loss at peak regions and gain at age-domains. Epigenetic landscape in silico deletion analysis (LISA) is an algorithm that leverages H3K27ac ChIP-seq, transcription regulator ChIP-seq, imputed binding sites and chromatin accessibility profiles to predict transcriptional regulators of a list of genes.37 LISA analysis of our differential genes predicted that the H3K27me3 peaks at promoters were mostly regulated by EZH2, and other polycomb group proteins (Figure 5D; Table S4). Indeed, EZH2 ChIP-seq in old livers showed reduced binding at differentially regulated H3K27me3 peak regions (Figures 5E and 5F; Table S1; QC in Figures S2A and S2B). Additionally, we performed EZH2 IP followed by mass spectrometry from young and old livers (Figure 5G; Table S2) to determine if the PRC2 composition was altered with age. Concordant with the overall loss of EZH2 expression with age (Figure 5B) and the loss of EZH2 binding at developmental gene promoters (Figures 5E and 5F), peptides from the core catalytic components of PRC2 were significantly lower in old samples. We conclude that local loss of H3K27me3 is in part, mediated by the global loss of EZH2.
Converse to the loss of signal at H3K27me3 peaks, age-domains gain signal. We wondered whether this gain of H3K27me3 signal at age-domains was due to EZH1 which increased in expression during aging (Figure 5B). Given that commercial ChIP-grade antibodies detecting the mouse endogenous EZH1 protein are lacking, we blotted for EZH1/2 in the different salt fractions of chromatin in young and old livers from Figures 3A–3D. As expected, H3K27me3 was detected in the high salt fractions and old livers had stronger signal.
Interestingly, EZH1 was detected in the high salt and pellet fraction in old and to a much lesser extent in young livers. EZH2 was detected mainly in the pellet fraction.
We thus conclude that both EZH1 and EZH2 associate with age-domains, however, since EZH1 is expressed more in aging and detected over several high salt fractions in the old, it is speculated to play a more prominent role.
Liver regeneration dilutes H3K27me3 and rejuvenates old tissue
We wondered whether disrupting hyper-quiescence through liver regeneration can epigenetically and functionally rejuvenate aged livers. Besides, regeneration is hypothesized to reduce H3K27me3 by replication dilution.38
The multi-lobular structure of the rodent liver enables removal of specific lobes (partial hepatectomy [PH]) without necrosis,39 allowing for the study of pure regenerative events unlike chemically induced models of regeneration. We used the 70% PH model40 in young and old mice (Figures S5A–S5C; Table S1) and archived “before” and “after” resection livers; 48, 72, 96, 120, and 240 h post-surgery. The 240 h time point represents fully regenerated livers.
To assay regeneration at the transcriptome level, we analyzed our ribo-depleted bulk RNA-seq libraries. In pairwise comparisons before resection, few mRNAs were differentially regulated between young and old (Figure S5D; Table S5). However, 48 h post-resection, there was strong induction of 379 mRNAs associated with cell division in the young liver (Figure 6A, left; Table S5). Congruently, LISA identified E2F factors, known to regulate cell cycle entry into S-phase on the top 341 (Table S4). The 177 mRNAs that were upregulated in the old livers 48 h after surgery instead were related to cell adhesion, inflammation, and response to glucose and insulin (Figure 6A, right) but unrelated to cell proliferation. In contrast, at the later 96 h time point, 199 mRNAs were upregulated in the old livers and were associated with cell division (Figure 6B, right). Sixty of the 199 mRNAs overlapped with the set of 379 mRNAs upregulated at 48 h in young livers (p value = 1.04e–65, representation factor 23.9; hypergeometric test). Contrary to the robust response noted in young livers at 48 h, the response in old livers at 96 h was modest with higher p values and lower gene counts. Additionally, unlike the narrow time window of response in the young (48 h), the proliferative response in old livers spread over 96–120 h post-resection (Figures 6B and S5E). The top candidate factor in LISA analysis for mRNAs upregulated at 96 h (Table S4) and 120 h (Table S4) in the old was also an E2F factor, E2F4. The 80 mRNAs that were upregulated in the young at 96 h were related to circadian regulation (Figure 6B, left), suggesting a return to homeostasis. Of note, the 72 and 240 h time points did not show many differentially expressed mRNAs (Figures S5F and S5G). The 72 h time point likely represents a quiet phase when the young liver has mostly completed its proliferation, but the old livers have not yet induced cell cycle. In contrast, the 240 h time point may represent a return to quiescence for both young and old livers. We validated the RNA-seq results by tracing the cell proliferation marker Ki67 by immunohistochemistry (IHC) (Figures S5H and S6A; Table S1), IF (Figures S5I and S6B; Table S1) and RT-qPCR analyses (Figure 6C; Table S1) across the regeneration time course. We further measured the expression of additional cell cycle markers by RT-qPCR (Figure 6C). All proliferation markers peaked at 48 h post-resection in young animals, while those in old animals showed an attenuated peak at ~72–96 h. Figure 6D summarizes the dynamics of cell proliferation mRNAs across age and regeneration. These findings were validated by an independent method, ImpulseDE2,42 designed for high-throughput time series datasets (Figures S5J–S5L; Table S6).
Figure 6. Transcriptomic signatures of aging and regeneration.
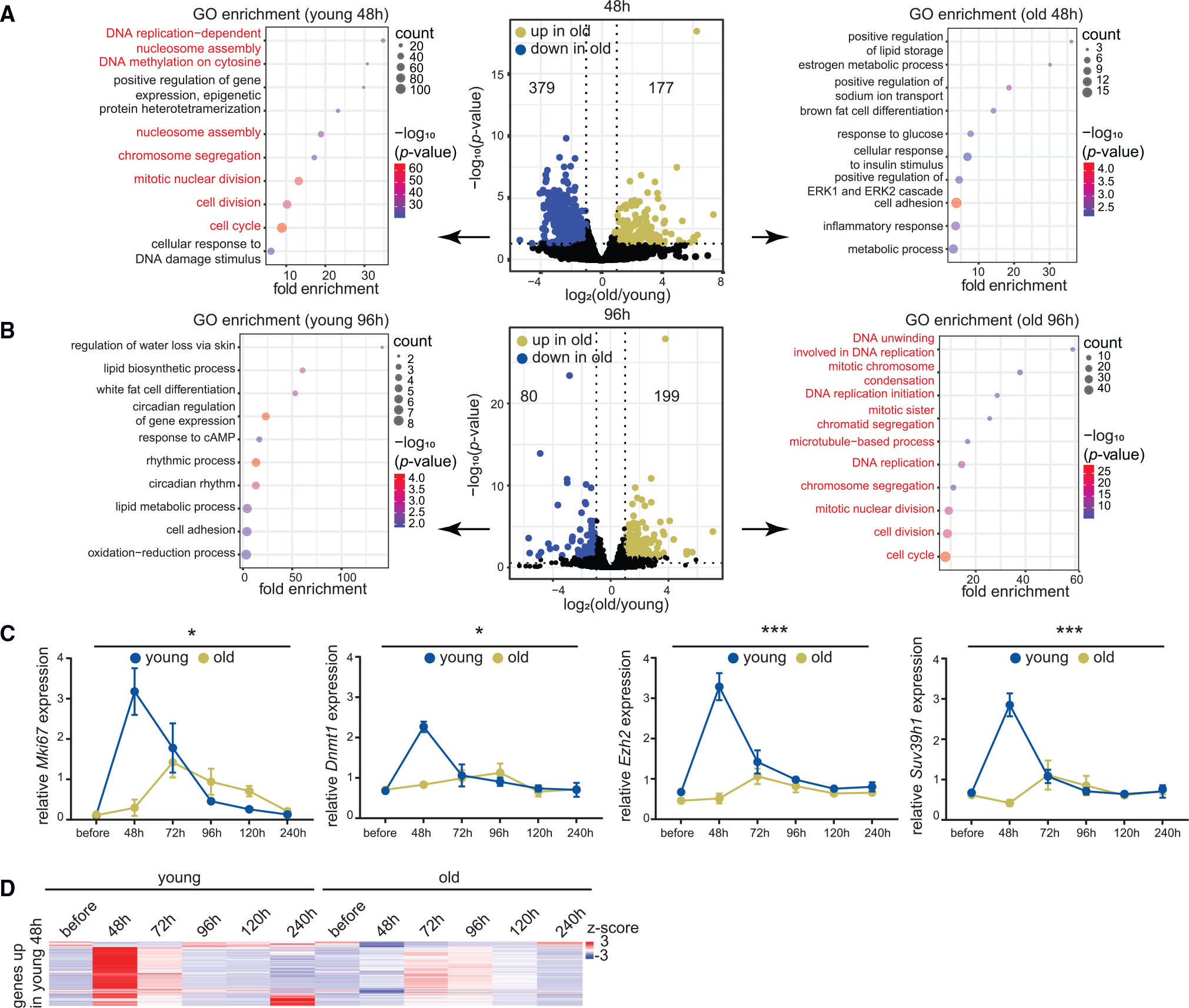
(A) Volcano plot of differentially expressed mRNAs 48 h post-resection in young and old livers (young 10–15 weeks and old 79–83 weeks, n = 3 biological replicates per time point). mRNAs significantly upregulated (p < 0.05 from Benjamini-Hochberg procedure) in old are in green and those downregulated are in blue. Biological process GO terms are indicated for genes downregulated (left) or upregulated (right) in the old. Cell proliferation genes are in red. p values are from Fisher’s exact test.
(B) Same as (A) except samples are 96 h post-resection (young 10–12 weeks and old 79–83 weeks, n = 3 biological replicates per time point).
(C) RT-qPCR analysis of Mki67, Dnmt1, Ezh2, and Suv39h1 expression relative to Actb across the regeneration time course (young 10–16 weeks and old 79–98 weeks, n = 3 biological replicates per time point). Data are summarized as mean ± SEM (n = 3 biological replicates per group per time point). * p < 0.05 and *** p < 0.001 from a two-way ANOVA with Tukey’s multiple comparisons test.
(D) Heatmap of cell proliferation gene counts across the regeneration time course.
Consistent with previous reports,43–45 our results showed delayed and subdued regenerative response with age.
Despite a reduction in regenerative response, a proportion of old hepatocytes entered cell cycle (Figures S5H, S5I, and S6). We reasoned that this mild proliferative event in old livers would be sufficient to dilute H3K27me3 and allow us to test epigenomic rejuvenation. Indeed, old regenerated livers (240 h post-surgery, Figure 7A) showed reduction in H3K27me3 signal after regeneration (Figure 7B, compare before and after regeneration samples; Table S1). We also noted that the differences in expression of EZH1/2 between young and old livers (Figure 5B) were abolished post-regeneration (Figure 7C). We confirmed that regeneration reversed multiple molecular aspects of aging. For example, in regenerated livers, there was an increase in H3K27me3 at developmental gene promoters by ChIP-seq (Figure 7D) which consequentially decreased expression of developmental genes (Figure 7E) restoring cell identity. We also observed there was a marked reduction in age-domains (Figure 7F). Only the global transcriptome suppression failed to be reversed (compare Figures 4K and 7G), likely because age-domains, though reduced, persist post-regeneration (Figure 7F). Nonetheless, PCA plots of both H3K27me3 patterns (Figure 7H) and transcriptome (Figure 7I) confirmed that the old regenerated livers were rejuvenated. To determine restoration of youthful function in old regenerated livers, we analyzed the expression profile of liver-enriched genes (Table S7) obtained from the Human Protein Atlas.46 Liver-enriched genes in old regenerated livers showed expression patterns that were similar to young livers (Figure 7J). Furthermore, we investigated if the genes that were upregulated with age (Figure S5D; Table S5) were downregulated by regeneration. Indeed, for a major subset of genes, this was true (Figure 7K). A minor subset of age-upregulated genes was further upregulated upon regeneration (Figure 7K). These genes are related to DNA damage-repair and acute phase response, suggesting either ongoing repair in response to acute injury (PH) or chronic injury (aging). We next inquired whether the regeneration conditions could be modeled in human HepG2 cells by simply splitting cells after contact inhibition. Indeed, H3K27me3 levels were significantly reduced, as in the liver (Figure 7L). Taken together, our data show that liver regeneration, and the ensuing replication dilution of H3K27me3, reverses multiple aspects of aging at the molecular level.
Figure 7. Liver regeneration dilutes H3K27me3 and rejuvenates old tissue.
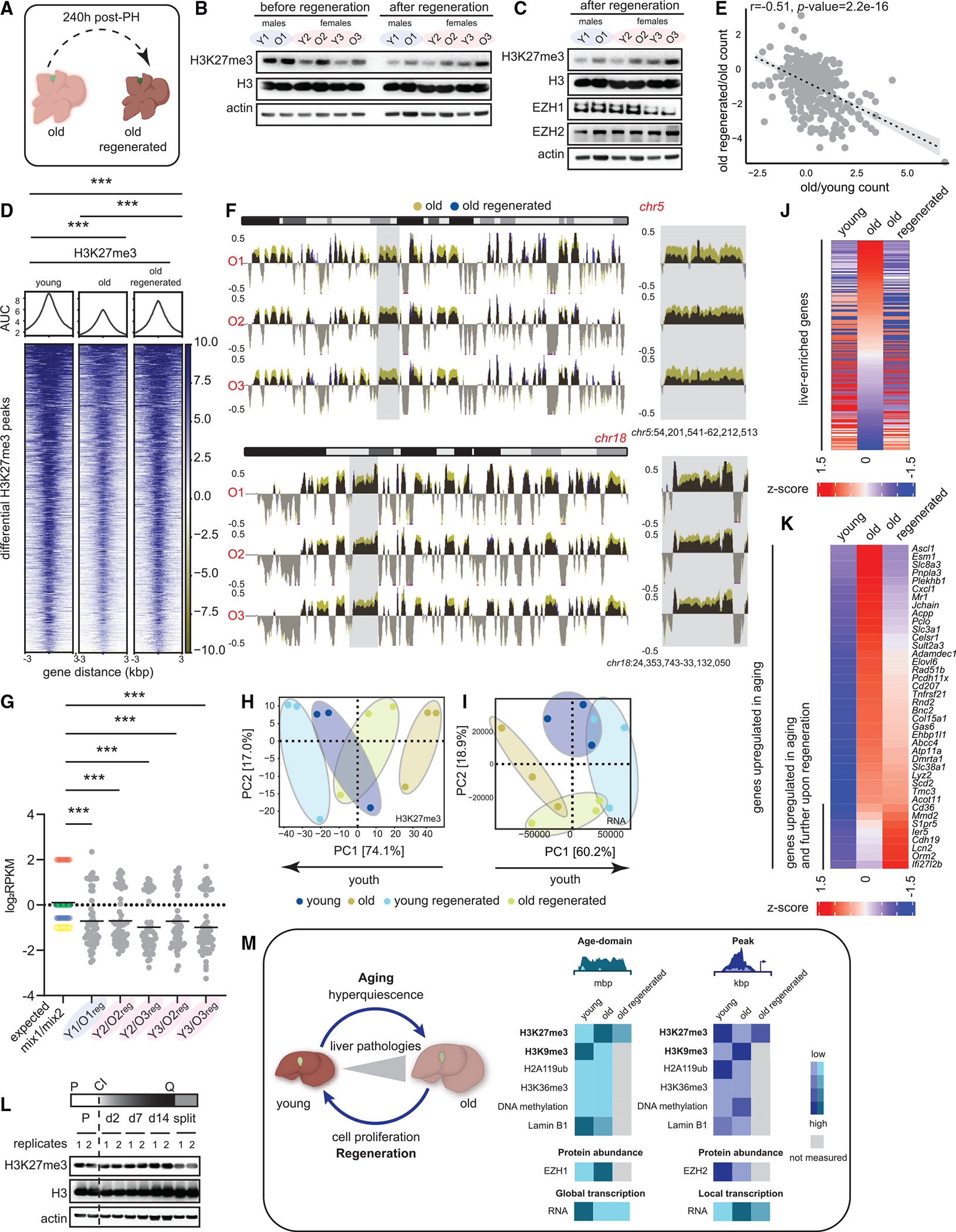
(A) Schematic of complete liver regeneration.
(B) Western blot of H3K27me3, H3, and β-actin showing replication dilution of H3K27me3 post-regeneration. The “before regeneration” H3K27me3 blot is same as in Figure 5B.
(C) Western blot of H3K27me3, H3, EZH1, EZH2, and β-actin post-regeneration. The “after regeneration” H3K27me3, H3, and actin blots are same as in (B).
(D) Heatmap of H3K27me3 signal at differential peaks from Figure 1H before and after regeneration. *** p < 0.001 from a Welch’s two-tailed unpaired t test.
(E) Correlation of gene expression between old vs. young and old regenerated vs. old. The gene set corresponds to common genes that were de-repressed in aging and re-repressed post-regeneration. Pearson r and p values are reported.
(F) Genome browser snapshot of overlaid H3K27me3 signal over chr5 (top) and chr18 (bottom) before and after regeneration. Gray area is expanded on the right.
(G) Expected and observed mix1/mix2 ratios plotted for all animals. *** p < 0.001 from a one-way ANOVA with corrections for multiple comparisons (FDR method of Benjamini and Hochberg).
(H) PCA plot of H3 subtracted H3K27me3 genome coverage from young, young regenerated, old, and old regenerated livers.
(I) Same as (H) except PCA is from RNA-seq data.
(J) Heatmap of liver-enriched gene counts in young, old, and old regenerated livers sorted on the old sample.
(K) Heatmap of age-upregulated (from Figure S5D) gene counts in young, old, and old regenerated livers sorted on the old sample. For (B)–(J), young 11–12 weeks and old 79–95 weeks, n = 3 biological replicates per group.
(L) Western blot of H3K27me3, H3, and β-actin from HepG2 lysates prepared from cells establishing and exiting quiescence. P is proliferating, Q is quiescence, and CI is contact inhibition.
(M) Overview of H3K27me3 changes in aging and regeneration.
Multiple tissues show features of hyper-quiescent chromatin during aging
Finally, we investigated whether age-domains were evident in aged quiescent tissues other than the liver, thus presenting a common epigenetic mechanism of tissue aging. We performed ChIP-seq of H3K27me3 in young and old kidney (Table S1; QC in Figures S2A and S2B). As in the aged liver, we observed age-domains of H3K27me3 across chromosomes in the aged kidney (this study), aged mouse heart and quadriceps muscle.47 The results showed evidence of H3K27me3 age-domains as in liver (Figures S7A and S7B). Furthermore, there was striking overlap in the locations of the age-domains in these tissues, although the heart was somewhat dissimilar (Figure S7C). Taken together, our results indicate that accumulation of H3K27me3 across large regions of the genome might be a universal molecular phenotype of aged quiescent tissues.
DISCUSSION
In this study, we dissected the fundamental properties of aged chromatin using multi-omics and direct imaging. In the quiescent liver, hepatocytes stay viable and non-mitotic over a long lifespan (200–400 days). During the long post-mitotic phase of aged hepatocytes, we noted a paralog switching event in the enzymes catalyzing H3K27 methylation (Figure 5B). There was a decline in EZH2 expression with age which coincided with the age-related loss of H3K27me3 and EZH2 peaks at developmental gene promoters (Figures 1H, 4D, 5E, and 5F). Conversely, EZH1 protein levels increased (Figure 5B) and co-eluted with age-domains of H3K27me3 along with remnant EZH2 (Figure 5H). Histological and biochemical experiments have previously reported that EZH1 is expressed more in post-mitotic cells, has weaker methyltransferase activity, and represses transcription both in vitro and in vivo.11 Additionally, H3K27me3 is known to deposit slowly on newly replicated DNA48,49 and is maintained more on H3.3, which accumulates with age (Figure S1A).50 These properties of H3K27me3 and EZH1 agree with our observations of EZH1 expression, H3K27me3 accumulation and global transcription suppression with age.
Quiescence cultures induced by contact inhibition were able to model the accumulation of H3K27me3 and the antagonistic behavior of EZH1 and EZH2 observed in aging (Figure 5C). We thus propose that aged cells are in a state of prolonged quiescence i.e., hyper-quiescence.
Our work identifies a unique H3K27me3 enrichment pattern in aged tissues, megabase-scale heterochromatin domains that we call age-domains (Figures 1K, 3B, 3D, and S3). These age-domains are identified by specialized domain calling algorithms22 and are visible in chromosome-wide views. Age-domains cannot be identified in CUT&Tag type experiments that use antibody-tethered genome-cutting enzymes without sonication and efficient exposure of epitopes in heterochromatin. The size of age-domains, and the fact that they were relatively gene poor (Figure 1K), a feature of LADs,24 prompted us to probe their genomic context further. We observed that age-domains were indeed LADs (Figure 2C) and showed loss of H3K9me3 with age (Figure 2D). They were depleted of H2A119ub, H3K36me3, 5mC, and RNAPII (Figures 2E–2H). Age-domains were extracted at high salt concentrations from old samples (Figure 3B). The loss of H3K9me3-rich, lamina-associated heterochromatin domains in senescence, aging, and premature aging diseases is well established.25,51,52 Our work shows a compensatory replacement of the H3K9me3 (constitutive) with H3K27me3 (facultative) heterochromatin (Figure 2). This switch may in turn affect genome folding, a question we wish to address in the future.
Upon injury, hepatocytes enter cell cycle and repopulate the liver with new cells although this function declines with age. Knowing that H3K27me3 can be reduced by replication dilution,38 we inspected the old regenerated liver (240 h post-surgery) for signs of epigenetic rejuvenation. Importantly, at 240 h post-regeneration, the liver was fully quiescent representing a state where some cells have reduced their “post-mitotic age” (Figure S6). We found that even limited proliferation was able to abolish differences in EZH1/2 expression (Figure 7C), increase H3K27me3 signals at developmental genes (Figure 7D), silence these genes (Figure 7E), reduce age-domains (Figure 7F), and partially correct age-related transcriptomic changes (Figures 7I–7K). Overall, our results demonstrate that removing H3K27me3 by controlled cell division can reset the epigenetic landscape (Figure 7M). For the same reasons, age-domains are unlikely to be seen in actively dividing cells in the body such as stem cells.
We further show that many organs remain in a repressed hyper-quiescent state in vivo and form age-domains (Figure S7). Keeping tissues in a repressed, non-dividing state, may confer anti-tumor properties. Our study highlights the importance of monitoring chromatin in its native in vivo context as growth factor stimulation in cell culture may prompt cell cycle entry and replication dilution of epigenetic marks such as H3K27me3. In C. elegans, the knockdown or heterozygous mutation of the H3K27me3 demethylase, utx-1, increases H3K27me3 and extends lifespan53 further suggesting a beneficial role for this histone modification.
Based on our observations, we speculate that some of the benefits of interventions such as Yamanaka factor-based reprogramming or parabiosis could be partially due to cell proliferation although they involve additional dedifferentiation events. Curiously, several reprogramming strategies have reported proliferation events in reprogrammed tissues.54–56 Similarly, heterochronic parabiosis has reported increased cell proliferation in the aged partner with reduction in repressive complexes known to inhibit proliferation.57
Overall, our study suggests cell proliferation-mediated global reduction in H3K27me3 levels could be a potential strategy to ameliorate age-relate decline and disease.
Limitations of the study
Although our study identifies facultative heterochromatin domains (age-domains) in old mice, our mice are only 18 months old. Age-domains may or may not exist in geriatric mice (>28 months old) or may be differently regulated. Furthermore, due to limitations with existing antibodies, the exact mechanism of how EZH1 establishes age-domains could not be investigated. The relationship between age-domains and transcriptional output has also not been directly assessed in our study. Several other outstanding questions remain: (1) what the connection between gain of age-domains and loss of H3K27me3 at peak regions is, (2) why is PRC2 binding lost at developmental promoters with age and (3) whether longevity interventions modulate age-domains.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Payel Sen (payel.sen@nih.gov).
Materials availability
This study did not generate new unique reagents. Materials used in this study are listed in the key resources table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mo/Rt Ki67 | Invitrogen | 14–5698-82; RRID:AB_10854564 |
| Rabbit IgG, polyclonal-Isotype Control(ChIP Grade) | Abcam | ab171870; RRID:AB_2687657 |
| Normal Rabbit IgG | Cell Signaling Technology | 2729s; RRID:AB_1031062 |
| pAb Histone H3K27me3 | Active Motif | 39155; RRID:AB_2561020 |
| Tri-Methyl-Histone H3K27 (C36B11) Rabbit | Cell Signaling Technology | 9733S; RRID:AB_2616029 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | A-11008; RRID:AB_143165 |
| Goat anti-rabbit IgG (H+L) HRP conjugated | Bio-Rad | 170–6515; RRID:AB_11125142 |
| Goat anti-mouse IgG (H+L) HRP conjugated | Bio-Rad | 172–1011; RRID:AB_11125936 |
| EZH1 antibody | Active Motif | 61583; RRID:AB_2793687 |
| EZH2 antibody | Cell Signaling Technology | 5246S; RRID:AB_10694683 |
| KDM6A antibody | Cell Signaling Technology | 33510S; RRID:AB_2721244 |
| KDM6B antibody | Active Motif | 61387; RRID:AB_2793615 |
| Guinea Pig anti-Rabbit IgG (Heavy & Light Chain) Antibody - Preadsorbed | antibodies-onlines.com | ABIN101961; RRID:AB_10775589 |
| Anti-Histone H3 (tri methyl K9) antibody - ChIP Grade | Abcam | ab8898; RRID:AB_306848 |
| Histone H3K36me3 antibody (pAb) | Active Motif | 61902; RRID: AB_2615073 |
| Ubiquityl-Histone H2A (Lys119) (D27C4) XP® Rabbit mAb | Cell Signaling Technology | 8240; RRID: AB_10891618 |
| Rpb1 NTD (D8L4Y) Rabbit mAb | Cell Signaling Technology | 14958; RRID: AB_2687876 |
| Rb pAb to Histone H3 | Abcam | ab1791; RRID:AB_302613 |
| Human HNF-4 alpha/NR2A1 MAb | R&D systems | PP-H1415–0C; |
| F4/80 antibody | Cl:A3–1 | BIO-RAD | MCA497GA; RRID:AB_323806 |
| Anti-CD31 antibody | Abcam | ab124432; RRID: AB_2802125 |
| Keratin 17/19 (D4G2) XP® Rabbit mAb | Cell Signaling Technology |
12434; RRID: AB_2797912 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Human liver | Saint Agnes Healthcare, Department of Surgery | Supp. Table 1 |
| Mouse liver, kidney | NIA Rodent Colony | Supp. Table 1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 16% Formaldehyde (w/v), Methanol-free | Thermo Fisher Scientific | 28908 |
| Q5 Hot Start High-Fidelity 2X Master Mix | New England Biolabs | M0494S |
| MNase (in salt fraction studies) | New England Biolabs | M0247S |
| MNase (in AFM studies) | Sigma | N3755–500UN |
| SPRI beads | Beckman coulter | B23318 |
| TriPure Isolation Reagent | Sigma Aldrich | 11667157001 |
| 2.5% normal goat serum | Vector Biolabs | S-1012–50 |
| DPX mountant for histology | Sigma Aldrich | 06522 |
| Antigen Unmasking Solution, Citrate-Based | Vector Biolabs | H-3300–250 |
| ProLong Gold Antifade reagent | Invitrogen | P36934 |
| Epredia Lab Vision PermaFluor Aqueous Mounting Medium | Fisher Scientific | TA006FM |
| Critical Commercial Assays | ||
| Zymo-Seq RiboFree Total RNA Library Kit | Zymo Research | R3000 |
| ERCC ExFold RNA Spike-In Mixes | Thermo Fisher Scientific | 4456739 |
| SimpleChIP® Plus Sonication Chromatin IP Kit | Cell Signaling Technology |
56383 |
| Nuclei Isolation Kit | Invent Biotechnologies | NI-024 |
| RNeasy Mini Kit | Qiagen | 74104 |
| QIAshredder | Qiagen | 79656 |
| QIAquick Gel Extraction Kit | Qiagen | 28706 |
| RNase-Free DNase Set | Qiagen | 79254 |
| Dovetail Micro-C Kit | Dovetail Genomics | 21006 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | E7645L |
| NEBNext Library Quant Kit for Illumina | New England Biolabs | E7630L |
| NEBNext Multiplex Oligos for Illumina (96 Unique Dual Index Primer Pairs) | New England Biolabs | E6440L |
| MagMeDIP Kit | Diagenode | C02010021 |
| High-capacity cDNA to RNA kit | Thermo Fisher Scientific | 4387406 |
| Qubit 1X dsDNA, High Sensitivity Assay Kit | Thermo Fisher Scientific | Q33231 |
| Qubit RNA, High Sensitivity Assay Kit | Thermo Fisher Scientific | Q32852 |
| Qubit RNA, Broad Range Assay Kit | Thermo Fisher Scientific | Q10210 |
| Qubit RNA Integrity and Quality (IQ) Assay Kit | Thermo Fisher Scientific | Q33221 |
| DNA 1000 kit | Agilent | 5067–1504 |
| DNA HS kit | Agilent | 5067–4626 |
| Liquid DAB+, 2-component system, Immunohistochemistry Visualization | Agilent | K346711–2 |
| Deposited Data | ||
| All sequencing data (GEO super series) | This paper | GEO: GSE185708 |
| RNA-seq | This paper | GEO: GSE185705 |
| ChIP-seq | This paper | GEO: GSE185704 |
| Salt fraction DNA-seq | This paper | GEO: GSE185707 |
| CUT&Tag | This paper | GEO: GSE185703 |
| Histone modification ChIP-seq data of mouse heart and quadriceps muscle |
Sleiman et al, 2020 47 | SRA: SRP162386 |
| meDIP | This paper | GEO: GSE223480 |
| Mass spec histone modifications | Chorus | chorusproject.org/1736 |
| Western blot | MendeleyData | doi: 10.17632/7wkwn9vw3f.1 |
| Immunofluorescence (IF) | MendeleyData | doi: 10.17632/7wkwn9vw3f.1 |
| Immunohistochemistry (IHC) | MendeleyData | doi: 10.17632/7wkwn9vw3f.1 |
| Transmission Electron Microscopy (TEM) | MendeleyData | doi: 10.17632/7wkwn9vw3f.1 |
| PhenoImager | MendeleyData | doi: 10.17632/7wkwn9vw3f.1 |
| Atomic Force Microscopy (AFM) | MendeleyData | doi: 10.17632/7wkwn9vw3f.1 |
| Experimental Models: Cell Lines | ||
| HepG2 | ATCC | HB-8065 |
| HeLa | ATCC | CCL-2 |
| Experimental Models: Organisms/Strains | ||
| Mouse (C56BL/6JN) | NIA Rodent Colony | Supp. Table 1 |
| Oligonucleotides | ||
| Primers for Ezh1 | GGTTCCTGCTGTCCTCTATTT | TCCACAGCACCTCACTTTATC |
| Primers for Ezh2 | GCTGATGCCCTGAAGTATGT | GGCAGCTGTTTCAGAGAGAA |
| Primers for Kdm6a | CATTACCATCCGCCTCATCTT | GGTGCAGAACTGGGTTACTT |
| Primers for Kdm6b | GCTAGCTGTGATCCCTGTATATG | AGAGGTGAGTGGGACTGTTA |
| Primers for Dnmt1 | AGGCTGCTACCAAGGACTA | CACAGACACTGAGCACAAGA |
| Primers for Mki67 | CAGGGATCTCAGCGCAATTA | GAGCTTTATTGGATAGGACAGAGG |
| Primers for Suv39h1 | TTGCAGGCCTCTTCATTACTC | GCTTGATGTTAGGCTGGTATCT |
| Recombinant DNA | ||
| Software and Algorithms | ||
| GO | Huang da et al, 200978 | https://david.ncifcrf.gov/summary.jsp; RRID:SCR_001881 |
| GraphPad Prism/9.0.0 (121) | Dotmatics | https://www.graphpad.com/ |
| DRAGEN FASTQ Generation/3.7.4 | Illumina | https://support.illumina.com/downloads/dragen-workflow-fastq-installer.html |
| bowtie/2–2.4.2 | Langmead and Salzberg, 201279 | http://bowtie-bio.sourceforge.net/index.shtml; RRID:SCR_016368 |
| trimgalore/0.6.6 | N/A |
https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/; doi 10.5281/zenodo.5127898 |
| STAR/2.7.5b | Dobin et al, 201364 | https://github.com/alexdobin/STAR; RRID:SCR_004463 |
| samtools/1.10 | Li et al., 200965 | http://www.htslib.org/doc/samtools.html; RRID:SCR_002105 |
| picard/2.20.8 | N/A | https://broadinstitute.github.io/picard/; RRID:SCR_006525 |
| Rsubread/2.6.4 | Liao et al, 201966 |
https://bioconductor.org/packages/release/bioc/html/Rsubread.html; RRID:SCR_009803 |
| DESeq2/1.30.1 | Love et al, 201467 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html; RRID:SCR_015687 |
| ImpulseDE2/0.99.10 | Fischer et al, 201842 | https://bioconductor.riken.jp/packages/3.9/bioc/html/ImpulseDE2.html |
| Gene Set Enrichment Analysis (GSEA/4.1.0) | Subramanian et al, 200580 | https://www.gseamsigdb.org/gsea/msigdb/index.jsp; RRID:SCR_003199 |
| Complex Heatmap/2.6.2 | Gu et al, 201669 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| Sambamba/0.7.1 | Tarasov et al, 201572 | https://github.com/biod/sambamba/releases |
| deepTools/3.5.0 | Ramirez et al. 201473 | https://deeptools.readthedocs.io/en/develop/; RRID:SCR_016366 |
| peakranger/1.18 | Feng et al, 201181 | http://ranger.sourceforge.net/; RRID:SCR_010863 |
| DiffBind/3.2.6 | Stark et al, 201174 |
https://bioconductor.org/packages/release/bioc/html/DiffBind.html; RRID:SCR_012918 |
| diffReps/1.55.6 | Shen et al, 201375 | https://github.com/shenlab-sinai/diffreps; RRID:SCR_010873 |
| EDD | Lund et al, 201422 | https://github.com/CollasLab/edd; RRID:SCR_001693 |
| DANPOS | Chen et al, 201328 | https://sites.google.com/site/danposdoc/; RRID:SCR_015527 |
| BEDTools | Quinlan et al, 201082 | https://bedtools.readthedocs.io/en/latest/; RRID:SCR_006646 |
| All code in this paper (Github) | This paper | https://doi.org/10.5281/zenodo.7789081 |
| Other | ||
| DMEM (4.5g/L glucose) | Thermo Fisher Scientific | 11965126 |
| PBS | Thermo Fisher Scientific | 14190136 |
| Trypsin (0.25%) | Thermo Fisher Scientific | 25200056 |
| FBS | Thermo Fisher Scientific | 16000044 |
| Penicillin-Streptomycin Solution, 100X | Thermo Fisher Scientific | 15140122 |
Data and code availability
All genome-wide datasets have been submitted to the Gene Expression Omnibus portal (GEO: GSE185708) and will be publicly available as of the date of publication. Raw mass spec data are deposited at chorusproject: chorusproject.org/1769. Accession numbers and DOI are also listed in the key resources table.
All original code has been deposited at Github and Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines and culture conditions
Hep-G2 (human male; RRID:CVCL_0027) cells were cultured in a 37°C 5% CO2 and 20% O2 humidified incubator with Dulbecco’s Modified Eagles Medium (DMEM, Gibco) supplemented with 1% penicillin/streptomycin (Thermo Fisher) and 10% Fetal Bovine Serum (Thermo Fisher). For ChIP-Rx experiments, HeLa (human female; RRID:CVCL_0030) cells cultured in the same way as HepG2 cells were used.
Animals
This study was approved by the Animal Care and Use Committee of the NIA in Baltimore, MD under Animal Study Protocol numbers 481-LGG-2022 and 481-LGG-2025. Young and old inbred C57BL6/JN mice of both sexes were acquired from the NIA aged rodent colony (https://ros.nia.nih.gov/) and housed in rooms that were maintained at 22.2 ± 1 °C and 30–70% humidity. Routine tests are performed to ensure that mice are pathogen-free and sentinel cages are maintained and tested according to American Association for Accreditation of Laboratory Animal Care (AAALAC) criteria. The age and sex information are available in Table S1.
Human liver samples
Core-needle biopsies of the liver were obtained from consenting patients undergoing cholecystectomy, bariatric procedures, or gastroesophageal reflux disease with mostly normal livers. Exclusion criteria included known liver disease and elevated liver enzymes. Approval was obtained by the Institutional Review Board of Ascension Saint Agnes Hospital, Baltimore (protocol number RPN 2019–016) prior to initiation of the study. Informed consent was obtained from all patients.
METHOD DETAILS
Induction of quiescence
To induce quiescence by contact inhibition, cells were allowed to grow until they reached 100% confluency. The cultures were maintained for 2, 7 or 14 days with media change (with serum) every two days.
Partial hepatectomy
70% partial hepatectomy was performed following guidelines in Mitchell and Willenbring.40 The removed liver lobes were archived as “before resection” samples. Following specified times after surgery, the animals were sacrificed by carbon dioxide asphyxiation and cervical dislocation. The liver was dissected and either frozen in isopentane chilled with liquid nitrogen and stored in −80°C or processed for paraffin embedding and/or OCT (Tissue-Tek) encapsulation. All surgeries were performed at approximately the same time of day to prevent confounding variables such as circadian rhythms.
Immunohistochemistry
6 μm sections of liver were cut from a paraffin block onto positively charged glass slides. Sections were deparaffinized, rehydrated and autoclaved in citrate buffer (Thermo Fisher) for antigen retrieval. Following several washes in Tris Buffered Saline with or without 0.1% Tween-20 (TBS or TBST; Pierce), the sections were blocked for 30 min with 2.5% normal goat serum (Vector Biolabs) and incubated with primary antibody overnight at 4°C in a humidified chamber. After washing with TBST, endogenous peroxidase was blocked using the Peroxidase and Alkaline Phosphatase Blocking Reagent (Agilent Dako) and the Dual-Link Envision system (Agilent Dako) was applied for 30 min at room temperature. The sections were then incubated with chromogenic substrate DAB+ (Agilent Dako) for 2 min followed by washing in deionized water, staining with Mayer’s hematoxylin (Vector Laboratories) and treatment with Scott’s tap water (Sigma). Following further washing, the sections were dehydrated and cleared with xylene before mounting with DPX mountant (Sigma Aldrich). Images were taken on a Zeiss Axiovert 200 microscope using brightfield settings. Antibodies are listed in key resources table.
Immunofluorescence
Liver sections (10–12 μm) were cut from an OCT block onto positively charged glass slides using a cryostat. The sections were permeabilized with 0.2% Triton X-100 at room temperature for 5 min. Antigen retrieval was performed by heating to 95°C for 30 min. Blocking and primary antibody incubation was performed as in IHC. The sections were then incubated with secondary antibody conjugated to a fluorescent dye for 1 h at room temperature followed by washes with TBST and staining with 5 μg/ml DAPI for 15 min at room temperature. After two washes with TBS, the sections were mounted with Epredia Lab Vision PermaFluor Aqueous Mounting Medium (Fisher Scientific). Photographs were taken using a Zeiss LSM 710 confocal microscope. Intensities were quantified using Image J (https://imagej.nih.gov/ij/). Antibodies are listed in key resources table
RNA isolation and RT-qPCR
RNA was isolated from frozen tissue by homogenization in Trizol followed by isopropanol precipitation. The RNA was further purified using RNeasy columns (Qiagen). An on-column DNase I digestion was performed during the purification step to remove genomic DNA. The RNA amount and integrity were confirmed using the Qubit RNA HS Assay Kit and RNA IQ Assay (Thermo Fisher) respectively. Total RNA (~500 ng) was converted to cDNA using the High Capacity RNA to cDNA kit (Thermo Fisher) for RT-qPCR analysis. 1 μl of 1:10 dilution of cDNA was analyzed by RT-qPCR using PowerUp SYBR Green Master Mix (Thermo Fisher) following the standard curve method on a QuantStudio 7 Flex machine (Thermo Fisher). A “minus RT” control was used to confirm removal of genomic DNA. Primers are listed in key resources table.
RNA sequencing with ERCC Ex-Fold spike-in
Total RNA (~1 μg) was used to make RNA-seq libraries following the Zymo-Seq Ribo-free Total RNA Library Kit (Zymo Research) instructions with dual indexing. Prior to ribo-depletion, total RNA from young livers were spiked with 2 μl of a 1:100 dilution of ERCC Ex-fold Mix 1 while that from old livers were spiked with 2 μl of 1:100 dilution of ERCC Ex-fold Mix 2 (Thermo Fisher). Exfold ERCC spike-ins are provided in two mixes, mix 1 and mix 2, that have the same pre-formulated blend of 92 transcripts but at different amounts such that a group of transcripts in Mix 1 are always at a defined fold difference from the same group in Mix 2 (Figure 1H). There are four such groups with log2(mix1/mix2) ratios of 2 (4X), 0 (1X), -0.58 (0.67X) and -1 (0.5X). Library quality and quantity was confirmed on a BioAnalyzer (Agilent) DNA HS chip. Equimolar amounts of each library were combined, and the pooled library was further quantified using a NEBNext Library Quant Kit (New England Biolabs). The RNA-seq libraries were subjected to two rounds of 75bp paired end sequencing on a NextSeq 550 platform using a 150-cycle kit (Illumina).
Bottom-up nanoLC-MS/MS and data analysis
Histone extraction and digestion
Histone proteins were extracted from nuclei pellet as previously described by Sidoli et al.72 to ensure good-quality identification and quantification of single histone marks. Briefly, nuclei were isolated by douncing ~20 mg tissue in 1ml nuclei isolation buffer (15 mM Tris–HCl pH 7.5, 15 mM NaCl, 60 mM KCl, 5 mM MgCl2, 1 mM CaCl2, 250 mM sucrose, 0.2% NP-40) supplemented with 1 mM DTT, 1X Halt protease and phosphatase inhibitor cocktail (Thermo Fisher), 1 mM PMSF and 10 mM sodium butyrate. Nuclei were pelleted by centrifugation at 700 g for 5 min. Histones were acid-extracted with chilled 0.2 M sulfuric acid (5:1, sulfuric acid:pellet) and incubated with constant rotation for 4 h at 4°C, followed by precipitation with 33% trichloroacetic acid (TCA) overnight at 4°C. Then, the supernatant was removed, and the tubes were rinsed with ice-cold acetone containing 0.1% HCl, centrifuged and rinsed again using 100% ice-cold acetone. After the final centrifugation, the supernatant was discarded, and the pellet was dried using a vacuum centrifuge. The pellet was dissolved in 50 mM ammonium bicarbonate, pH 8.0, and histones were subjected to derivatization using 5 μl of propionic anhydride and 14 μl of ammonium hydroxide (Sigma Aldrich) to balance the pH at 8.0. The mixture was incubated for 15 min and the procedure was repeated. Histones were then digested with 1 μg of sequencing grade trypsin (Promega) diluted in 50 mM ammonium bicarbonate (1:20, enzyme:sample) overnight at room temperature. Derivatization reaction was repeated to derivatize peptide N-termini. The samples were dried using a vacuum centrifuge.
Sample desalting
Prior to mass spectrometry analysis, samples were desalted using a 96-well plate filter (Orochem) packed with 1mg of Oasis HLB C-18 resin (Waters). Briefly, the samples were resuspended in 100 μl of 0.1% TFA and loaded onto the HLB resin, which was previously equilibrated with 100 μl of the same buffer. After washing with 100 μl of 0.1% TFA, the samples were eluted with a buffer containing 70 μl of 60% acetonitrile and 0.1% TFA and then dried in a vacuum centrifuge.
LC-MS/MS acquisition and analysis
Samples were resuspended in 10 μl of 0.1% TFA and loaded onto a Dionex RSLC Ultimate 300 (Thermo Scientific), coupled online with an Orbitrap Fusion Lumos (Thermo Scientific).
Chromatographic separation was performed with a two-column system, consisting of a C-18 trap cartridge (300 μm ID, 5 mm length) and a picofrit analytical column (75 μm ID, 25 cm length) packed in-house with reversed-phase Repro-Sil Pur C18-AQ 3 μm resin. Histone peptides were separated using a 30 min gradient from 1–30% buffer B (buffer A: 0.1% formic acid, buffer B: 80% acetonitrile + 0.1% formic acid) at a flow rate of 300 nl/min. The mass spectrometer was set to acquire spectra in a data-independent acquisition (DIA) mode. Briefly, the full MS scan was set to 300–1100 m/z in the orbitrap with a resolution of 120,000 (at 200 m/z) and an AGC target of 5×10e5. MS/MS was performed in the orbitrap with sequential isolation windows of 50 m/z with an AGC target of 2×10e5 and an HCD collision energy of 30.
Western blot
Tissues (~20 mg) or cells were homogenized in RIPA buffer containing 50 mM Tris pH 7.5, 0.5 mM EDTA, 150 mM NaCl, 1% NP40, 1% SDS, supplemented with 1X Halt protease and phosphatase inhibitor cocktail, 1 mM PMSF and 10 mM sodium butyrate. The lysates were briefly sonicated with a Bioruptor (Diagenode), and cleared by centrifugation at max speed for 10 min at 4°C. In some cases, nuclear lysates were used for EZH1 western; lysates were prepared as described previously.73 The supernatants were quantified using the BCA kit (Pierce) and ~10– 30 μg total protein subjected to electrophoresis using NuPAGE 12% Bis-Tris gel in MES buffer (Thermo Fisher). The proteins were transferred to a 0.2-micron nitrocellulose membrane using a XCell II blot module (Thermo Fisher) for 1 h at 30V surrounded by ice. Proper transfer was verified by Ponceau S staining. 5% milk in TBST was used to block the membrane at room temperature for 1 h followed by primary antibody incubation at 4°C overnight. The membrane was washed and incubated with HRP-conjugated secondary antibodies (BioRad) at room temperature for 1 h. The membrane was washed again 3 times, developed, and imaged by a KwikQuant imager (Kindle Biosciences). Intensities were quantified using Image J (https://imagej.nih.gov/ij/). Antibodies are listed in key resources table.
Transmission electron microscopy with immunogold labeling
Sample processing
Animals were perfused with freshly made EM grade 100 mM Sorensen’s phosphate buffer pH 7.4 with 2% paraformaldehyde, 1% glutaraldehyde and 5 mM MgCl2 at 4°C. All subsequent steps were done at 4°C until 70% ethanol dehydration. Livers were carefully dissected in fixative and cut into pieces measuring no more than 2 mm3. Samples were rinsed thrice in buffer containing 3% sucrose for 15 min each and then osmicated in 1% osmium tetroxide (1.5% potassium ferrocyanide reduced) in 100 mM phosphate buffer, containing 5 mM MgCl2 for 2 hrs on ice. Tissue was put back in phosphate/sucrose buffer overnight on a cold room rocker.
Samples were rinsed thrice in 100 mM maleate buffer containing 3% sucrose for 5 min each and then en-bloc stained with 2% filtered uranyl acetate in the same buffer for 1 hr. Samples were dehydrated at 4°C up to 70% ethanol after which they were brought to room temperature and further dehydrated to 100% ethanol. Liver pieces were embedded with Eponate 12 after propylene oxide transition, and finally cured in a 60°C oven for two days.
Immunogold labeling
Ultra-thin [60 nm (grey)] sections were picked up on glow discharged formvar coated 200 mesh nickel grids. Sections were floated on all subsequent steps. Anti-capillary tweezers were used to transfer grids and then placed on 3% sodium meta periodate (aq) twice for 15 min each. After a 15 min rinse in distilled water, grids were floated on 10 mM citrate buffer pH 6.2 for 20 min at 95°C for antigen retrieval. After cooling down, grids were placed on 0.1 M glycine in TBS for 10 min, followed by 30 min incubation in blocking buffer (1% BSA in TBST) and primary antibody (1:100) incubation overnight. Samples with no primary antibody added served as negative controls. Next day, grids were equilibrated to room temperature for 1 h and placed in blocking solution for 10 min, followed by a 10 min rinse in TBS. Gold conjugated secondary antibodies were diluted 1:40 (6 nm goat anti-rabbit, Jackson Immunochemicals) in TBS and grids were incubated for 2 h at room temperature in a humidified chamber. After a 10 min TBS incubation followed by a quick distilled water rinse, grids were hard fixed in 2% glutaraldehyde in 100 mM sodium cacodylate buffer for 5 min. After a brief distilled water rinse, grids were stained with 2% uranyl acetate in 50% methanol for 10 min, rinsed again with distilled water, blot dried and allowed to sit in grid boxes overnight before viewing. Sections were viewed on a Hitachi H 7600 TEM operating at 90 kV and digital images captured with an ER-80 (8 megapixel) CCD camera, by AMT. Antibodies are listed in key resources table.
Immunogold particle quantification
Ten cells were randomly chosen from each sample (60 nm sections). In each cell, 5 non-overlapping regions of interest (ROI) in the nucleus were identified. For each ROI, the following were counted: total gold number, gold number in cytoplasm, gold number at nuclear periphery (within 200 nm), and gold number in nuclear matrix. Clusters (≥3 gold particles) in the nuclear matrix, nuclear periphery and cytoplasm for each ROI were also quantified.
Chromatin immunoprecipitation and sequencing
Crude nuclei preparations were made from ~100 mg frozen tissue by douncing in nuclei preparation buffer (10 mM Tris pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween 20, 0.1% NP-40, 0.01% digitonin and 1 mM BSA, supplemented with protease inhibitors and 1 mM sodium butyrate). After centrifugation, the nuclei pellet was crosslinked with 1% formaldehyde in 2 ml PBS by constant rotation at room temperature for 10 min followed by quenching with 125 mM glycine and two PBS washes. The nuclei were lysed with nuclei lysis buffer (10 mM Tris-HCl pH 7.4, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% sodium-deoxycholate, 0.5% N-lauroylsarcosine supplemented with protease inhibitors and 1mM sodium butyrate) and sheared to <500bp using a Covaris S220 Ultrasonicator. An aliquot of the sheared chromatin sample was removed to check sonication success and to quantify the amount of chromatin. The immunoprecipitation, wash and elution steps were performed as reported previously73 except ~4% HeLa chromatin was spiked in to 2 μg mouse chromatin prior to immunoprecipitation (only for H3K27me3 liver samples). A second round of H3K27me3, EZH2, H3K9me3, H3K36me3, H2A119ub, and total RNAPII ChIP was performed using the SimpleChIP plus sonication kit (CST #56383). ChIP enrichment was verified by qPCR around the Cdkn2a (p16) promoter (-200bp, -1000bp and -5000bp upstream of TSS for H3K27me3 or around the HoxA1 and HoxD10 locus for EZH2), actin promoter as the negative control locus and IgG as specificity control for the antibody. DNA (~5 ng) was used to prepare libraries with the NEBNext Ultra II library preparation kit with unique dual index primers (New England Biolabs). The library quality and quantity were verified by BioAnalyzer DNA 1000 (Agilent) run and qPCR with NEBNext Library Quant kit (New England Biolabs) respectively. The liver libraries were pooled and paired-end sequenced on the NextSeq 2000 platform (Illumina) using the P2 or P3 100 cycle kit. Antibodies are listed in key resources table.
Cell composition determination by multispectral imaging using PhenoImager
Dissected liver tissue from mice was fixed in 10% formaldehyde for 48 h and were subsequently dehydrated and embedded in paraffin. The fresh blocks were then resected until the tissue was exposed and the top 40 μm of tissue removed. Tissue was sectioned at 5 μm on positively charged slides (Superfrost Plus, Fisher Scientific) at 2 specimens/slide. The tissue was then dried and baked at 55°C for 3 h in a slide oven. The slides were then stained using the automated BondRx autostainer (Leica) using a custom protocol. Staining utilized the Opal protocol (Akoya Biosciences). The following antibodies were utilized as follows [Ab:Dilution:AssignedWavelength]: [HNF4a:(1:200):480], [CD31:(1:150):570], [H2K27me3:(1:150):620], [F4/80:(1:150):690], [Cytokeratin:(1:150):780]. Epitope retrieval employed heat-induced epitope retrieval (HIER) with Epitope Retrieval solution 1 (ER1) on all steps at atmospheric pressure for 20 min.
Following staining, the slides were dried and mounted using the ProLong AntiFade Diamond (Fisher Scientific) mountant. Slides were imaged using the PhenoImager HT (Akoya Biosciences) at 20X magnification. The central area of each specimen was then selected for further processing using the Inform tissue analysis platform (Akoya Biosciences). Using an unstained control slide, autofluorescence was deducted and then trainable cell and tissue segmentation was used to identify cells and phenotype them. Antibodies are listed in key resources table.
Methyl DNA immunoprecipitation sequencing (meDIP-seq)
The meDIP assay was performed using the MagMeDIP-seq Package (Diagenode) following the manufacturer’s protocol with a mouse antibody against 5mC (Diagenode, 33D3 clone, C15200081). Briefly, 1.2 μg of gDNA isolated from liver was sonicated into ~200 bp fragments using the S220 focused ultrasonicator (Covaris). Prior to immunoprecipitation, samples were spiked with methylated and unmethylated internal DNA controls. IP efficiency and success was verified by qPCR targeting spike-in DNA controls and endogenous controls (the CpG region of the mouse Tsh2b gene as positive control and mouse Gapdh as negative control). The DNA amount was quantified by Qubit HS DNA kit and the fragment size was assessed on a 2100 BioAnalyzer using a DNA HS kit (Agilent). Individual libraries for immunoprecipitated DNA and 10% input were dual indexed (NEBNext Multiplex Oligos, unique dual indices, New England Biolabs), PCR amplified and then pooled into equimolar amounts and further quantified using the NEBNext Library Quant Kit (New England Biolabs). The pooled library was subjected to 50 bp paired-end sequencing on the Illumina NextSeq 2000 platform. Antibodies are listed in key resources table.
Cut&Tag
Cut&Tag was performed following guidelines in the CUT&Tag@home v.1 protocol by Henikoff et al.31 Briefly, nuclei were isolated from ~20mg frozen human liver tissue using Minute Detergent-free Nuclei Isolation Kit (Invent Biotechnologies). The nuclei were permeabilized with 0.1% Triton X-100 and their integrity and number was checked under a microscope using a hemocytometer. ~10,000 nuclei per sample were bound to Concanavalin A beads (Bangs Laboratories). The bead-bound nuclei were incubated with primary antibody (1 h at room temperature), secondary antibody (0.5 h at room temperature) and home-made pA-Tn5 transposome (1 h at room temperature). Targeted tagmentation was then performed by addition of buffer containing MgCl2 for 1 h at 37°C. The tagmented DNA was washed with a TAPS-EDTA wash buffer (10 mM TAPS, pH 8.5, 0.2 mM EDTA) to remove excess salt and Mg2+ ions followed by targeted release with 0.1% SDS and neutralization of SDS by Triton X-100. The released DNA was amplified using dual-indexing primers following a PCR protocol that biases amplification of short DNA fragments. The excess primers were removed by a 1.3X SPRI bead purification (Beckman Coulter). Library quality and quantity was confirmed on a BioAnalyzer (Agilent) DNA HS chip. Equimolar amounts of each library were combined, and the pooled library was further quantified using a NEBNext Library Quant Kit (New England Biolabs). The CUT&Tag libraries were subjected to 50bp paired end sequencing on a NextSeq 2000 platform using a P2 100-cycle kit (Illumina). Antibodies are listed in key resources table.
Salt fractionation of chromatin
Nuclei was released from frozen tissue by douncing in TM2 buffer (10 mM Tris HCl, pH 7.4, 2 mM MgCl2 supplemented with 1X Halt protease and phosphatase inhibitor and 0.5 mM PMSF), pelleted by centrifugation and washed once to remove debris. The washed pellet was resuspended in TM2 buffer with 1 mM CaCl2 and 12000 U of MNase (New England Biolabs), incubated at 23°C for 15 minutes and the reactions stopped by addition of 0.5 mM EGTA. Approximately 30% was saved for analysis (“0”) and the rest was pelleted by centrifugation. The supernatant was removed and saved as “Supernatant” fraction after clearing. The nuclei were washed once with TM2 buffer and then resuspended in 70 μl Triton buffer (10 mM Tris–HCl pH 7.4, 2 mM MgCl2, 2 mM EGTA, 0.1% Triton X-100 supplemented with 1X Halt protease and phosphatase inhibitor and 0.5 mM PMSF) and evenly divided. The first aliquot was saved as “Input” while the second was used for sequential salt extraction by using Triton buffer with either 67.5, 150, 250 or 350 mM NaCl. Each extraction was done by incubating the resuspended pellet at 4°C for 2 hours. After each extraction, the nuclei were pelleted by centrifugation and the supernatant saved as “67.5”, “150”, “250” or “350” mM fraction. Finally, the remainder pellet which corresponds to ~5–10% of chromatin, was resuspended in 35 μl of TNE buffer (10 mM Tris–HCl pH 7.4, 200 mM NaCl, 1 mM EDTA supplemented with 1X Halt protease and phosphatase inhibitor) and labeled as “pellet” fraction. All extractions were performed under physiological concentrations of Mg2+ to preserve nuclear and chromatin integrity. DNA was purified from each of these fractions using DNA clean and concentrator columns (Zymo Research). Purified DNA (~10 ng) was used to make libraries using the NEBNext Ultra II kit (New England Biolabs) and sequenced on the NextSeq 2000 (Illumina) with a 51PE format.
Atomic force microscopy
Nuclei was extracted from livers74 and chromatin was digested with 1 U MNase (Sigma) for 2 minutes in 2 ml 0.1 M TE (10 mM Tris pH 6.8, 0.2 mM EDTA, 100 mM NaCl) supplemented with 1.5 mM CaCl2. MNase reactions were quenched with 10 mM EGTA and centrifuged at 94 g at 4°C. Supernatant was removed and chromatin was extracted overnight at 4°C in 0.5X PBS (67.5 mM NaCl) with a protease inhibitor cocktail (Roche) on an end-over-end rotator. Extracted chromatin was subsequently diluted 800-fold to create a single layer of chromatin when deposited on mica. Mica was freshly cleaved and functionalized with aminopropyl-silantrane (APS) before chromatin was deposited.75 Images were obtained with commercial AFM equipment (Oxford Instruments, Asylum Research’s Cypher S AFM) with silicon cantilevers (OTESPA-R3 from Olympus with nominal resonances of ~300 kHz, stiffness of ~42 N/m) in non-contact tapping mode in air. Images were processed as previously reported.75 Image analysis was performed using Image J (https://imagej.nih.gov/ij/) and statistical analysis and visualization were done in R/4.1.1.
MNase titration
An MNase titration was performed with 20 mg frozen liver tissue. Briefly, the frozen tissue was ground to a fine powder using a liquid nitrogen cooled mini mortar and pestle set (Bel-Art). The ground tissue was crosslinked first with 3 mM DSG and then with 1% formaldehyde, each for 10 min at room temperature. The crosslinked sample was centrifuged, and the pellet washed twice with wash buffer and sequentially filtered through 200 μm and 50 μm filters. Following centrifugation, the pellet was resuspended in nuclease digestion buffer with MNase and incubated at 22°C for exactly 15 min. The reaction was stopped by addition of 50 mM EGTA and 1% SDS. To check MNase digestion, ~2.5 μof the sample was treated with proteinase K, reverse crosslinked and purified using the DNA Clean and Concentrator kit (Zymo Research).
The fragment size distribution was checked on a BioAnalyzer using a DNA HS kit (Agilent).
EZH2 IP-MS
Immunoprecipitation
Nuclei were isolated by douncing frozen liver tissue in nuclei preparation buffer containing 10mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% Tween 20, 0.1% NP-40, 0.01% digitonin, 1 mM BSA, supplemented with 1X Halt protease and phosphatase inhibitor cocktail and 1 mM sodium butyrate. The resulting homogenate was filtered through a 30 μm cell strainer and washed using wash buffer containing 10 mM Tris-HCl (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.1% Tween 20, 1% BSA, supplemented with 1X Halt protease and phosphatase inhibitor cocktail and 1 mM sodium butyrate to stop lysis. After centrifugation, the nuclei pellet was washed thrice in nuclei suspension buffer containing PBS, 2% BSA, 3 mM MgCl2, supplemented with 1X Halt protease and phosphatase inhibitor cocktail and 1 mM sodium butyrate. The resulting nuclei pellet was then lysed on ice for 90 minutes in 2 volumes of nuclei lysis buffer containing 420 mM NaCl, 20 mM HEPES, 20% v/v glycerol, 2 mM MgCl2, 0.2 mM EDTA, 0.1% NP40, 0.5 mM DTT, supplemented with 1X Halt protease and phosphatase inhibitor cocktail and 1 mM sodium butyrate. The lysate was centrifuged at max speed at 4°C for 10 min and the supernatant transferred to new tubes. Total protein concentration was determined using the BCA kit (Pierce). EZH2 pull-downs were performed with 5 μg EZH2 antibody and 3.5 mg of total nuclear protein. 10% of the lysate was saved as “input”. The lysate and antibody-bound protein G Dynabeads (Thermo Fisher) were incubated at 4°C overnight with rotation in the presence of 250U/ml of benzonase (Millipore). The beads were washed 5 times with immunoprecipitation wash buffer (150 mM NaCl, 20 mM HEPES (pH 7.5), 10% glycerol, 2 mM MgCl2, 0.2 mM EDTA, 0.02% NP-40. 0.5 mM DTT) supplemented with 1X Halt protease and phosphatase inhibitor cocktail and 1 mM sodium butyrate. The immunoprecipitated material was eluted from the Dynabeads by adding an elution buffer containing 1% SDS, 5mM DTT and 50 mM ammonium bicarbonate (pH 8.0) for 1 h at room temperature.
Sample processing
The reduced proteins were alkylated with 20 mM iodoacetamide for 30 min in the dark followed by addition of phosphoric acid to a final concentration of 1.2%. The protein pellet was resuspended in 165 μL of loading buffer (90% methanol and 10 mM sodium bicarbonate (pH 8.0)) and loaded onto an S-Trap Micro Spin Column (Protifi). The column was washed twice with 150 μL of loading buffer and gently centrifuged before overnight digestion at 37°C with 0.1 μg/μL of trypsin (Promega) in 50 mM ammonium bicarbonate. After a gentle centrifuge, peptides were first eluted with 40 μL of 0.2% aqueous formic acid and then eluted again with 35 μL of 50% acetonitrile containing 0.2% formic acid. Eluted peptides were dried in DNA120 SpeedVac (Thermo Fisher) with no heat.
LC-MS/MS acquisition and analysis
Samples were reconstituted in 5–10 μl of HPLC solvent A (2.5% acetonitrile, 0.1% formic acid). A nano-scale reverse-phase HPLC capillary column was created by packing 2.6 μm C18 spherical silica beads into a fused silica capillary (100 μm inner diameter × ~30 cm length) with a flame-drawn tip. After equilibrating the column each sample was loaded via a Famos auto sampler (LC Packings) onto the column. A gradient was formed, and peptides were eluted with increasing concentrations of solvent B (97.5% acetonitrile, 0.1% formic acid).
As peptides eluted, they were subjected to electrospray ionization and then entered into an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Fisher). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences (and hence protein identity) were determined by matching protein databases with the acquired fragmentation pattern by the software program, Sequest (Thermo Fisher). All databases include a reversed version of all the sequences and the data was filtered to between a one and two percent peptide false discovery rate.
Bioinformatic analysis
RNA-seq analysis
Illumina sequencing reads (~32 million paired-end reads per sample) were de-multiplexed using bcl2fastq/2.20.0. Reads were trimmed to remove adapter sequences using trimmomatic/0.39.76 The quality of the resulting FASTQs was assessed using FASTQC/0.11.977 and reads were aligned to the mouse reference genome (assembly GRCm38/mm10, concatenated with the ERCC transcripts) using STAR/2.7.5b.60 BAM files were sorted and indexed using samtools/1.10,61 and duplicates were removed using picard/2.20.8. The BAM files were then filtered to retain alignments with a minimum mapping quality of 10 using samtools/1.10.61 The featureCounts function of the Rsubread R package/2.6.462 was used to estimate counts for all mRNA and ERCC transcripts.
Counts for the ERCC transcripts were RPKM normalized then filtered for 1 RPKM. ERCC transcripts were then plotted against their known molecular concentration. Sex- and time-matched fold-change ratios (young vs old) were calculated for each ERCC transcript and plotted against their known fold-change ratio. Linear regression analyses were performed to assess dose-response relationships using GraphPad Prism/9.0.0 (121).
Differential gene expression analysis (excluding ERCC transcripts) between old and young samples was performed separately for each timepoint using the R Bioconductor package, DESeq2/1.30.1.63 Additionally, since pairwise comparison approaches may fail to account for temporal dependencies in time-course experiments,78 the ImpulseDE2 R package/0.99.1042 was used to also assess transient (temporal) and monotonous (permanent) transcriptional changes occurring during the liver regenerative process. Heatmaps of the impulse-fitted data by time point were created using the R Bioconductor package, ComplexHeatmap/2.6.2.65 Both analyses were conducted at a Benjamini-Hochberg corrected p-value (false discovery rate, FDR) significance threshold of 0.05.79
Comparisons between RNA-seq and ChIP-seq data was performed by correlating the fold-change ratio in gene expression (old vs young) with the fold-change ratio in enrichment (old vs young) for the corresponding gene.
LC-MS/MS analysis for histone peptides
Histone peptides raw files were imported into EpiProfile 2.0 software.80 From the extracted ion chromatogram, the area under the curve was obtained and used to estimate the abundance of each peptide. To calculate the relative abundance of hPTMs, the sum of all the different modified forms of a histone peptide was considered as 100% and the area of the particular peptide was divided by the total area for that histone peptide in all of its modified forms. The relative ratio of two isobaric forms was estimated by averaging the ratio for each fragment ion with different mass between the two species. The resulting peptide lists generated by EpiProfile were exported to Microsoft Excel and further processed for a detailed analysis. Differences between conditions were assessed using t-test statistics; significant changes were considered at the heteroscedastic t-test p<0.05.
ChIP-seq analysis
Illumina sequencing reads (~50 million paired end reads per sample) were de-multiplexed generating compressed FASTQ files by the on-board DRAGEN informatics pipeline (Illumina DRAGEN FASTQ Generation – 3.7.4) on the NextSeq 2000. The FASTQ files were trimmed to remove adapter sequences with trimgalore/0.6.6 and the qualities of the FASTQs assessed using FASTQC/0.11.9.77 The reads were aligned to the GRCm38/mm10 and GRCh38/hg38 genome assemblies using bowtie/2–2.4.2 end-to-end parameter. Sam output files were then filtered to retain alignments with a minimum mapping quality of 10 using samtools/1.961 and PCR duplicates were removed using picard/2.23.7. Sambamba/0.7.166 was used to retain only uniquely mapping reads. Reads mapping to the Encyclopedia of DNA Elements (ENCODE) blacklisted regions were also removed from the analysis. There were no statistically significant differences in sequencing depth, alignment rate or alignable fragments per million across the young and old groups in any tissue. The bamCoverage function in deepTools/3.5.067 was used to generate RPKM normalized bigWig files. H3 and input reads were subtracted from H3K27me3 and IgG respectively with bigwigCompare. Peaks were called using peakranger/1.18 with the bcp parameter using H3 and input as background for H3K27me3 and IgG respectively from the same animal. Differential peak analysis was performed using DiffBind/3.2.6.69 Age-domains were called using the Enriched Domain Detector (EDD/1.1.19) algorithm22 with slight adjustments to the configuration file. The parameter “required_fraction_of_informative_bins” was reduced to 0.97 and the gap penalty was automatically calculated.
Scale factor for spike-in normalization was calculated as follows. For each sample, is defined as the scale factor, is the percentage mapped to GRCm38/mm10 genome in input, and is the percentage mapped to GRCh38/hg38 genome (spike-in). Based on our experimental design, the fraction of the spike-in human genome over the total mouse genome in each sample should be the same as in input and similar across all samples.
For visualization, bigWig files were generated using deepTools/3.5.0 bamCoverage with the scale parameter set as the calculated scale factor for each sample.
CUT&Tag analysis
CUT&Tag analysis with E. coli spike-in normalization was performed as outlined in Zheng et al.,81 following the protocols.io tutorial (https://dx.doi.org/10.17504/protocols.io.bjk2kkye). Briefly, Illumina sequencing reads (~6 million paired end reads per sample) were de-multiplexed generating compressed FASTQ files by the on-board DRAGEN informatics pipeline (Illumina DRAGEN FASTQ Generation – 3.7.4) on the NextSeq 2000. The qualities of the FASTQs were assessed using FASTQC/0.11.9.77 The reads were aligned to the GRCh38/hg38 genome with bowtie/2–2.4.2 using the end-to-end parameter. Sam output files were then filtered to retain alignments with a minimum mapping quality of 2 using samtools/1.9.61 Reads mapping to Encyclopedia of DNA Elements (ENCODE) blacklist regions were removed from the analysis. There were no statistically significant differences in sequencing depth, alignment rate or alignable fragments per million across the young and old groups in any tissue. The bamCoverage function in deepTools/3.5.0 was used to generate RPKM (reads per kilobase per million mapped reads) normalized bigWig files. H3 was subtracted from H3K27me3 and IgG with bigwigCompare function of deepTools/3.5.0. Differentially enriched sites were identified using the R Bioconductor package diffReps/1.55.6.70
Salt fractionation analysis
Illumina sequencing reads (~54 million paired end reads per sample) were de-multiplexed generating compressed FASTQ files by the on-board DRAGEN informatics pipeline (Illumina DRAGEN FASTQ Generation – 3.7.4) on the NextSeq 2000. The FASTQ files were trimmed to remove adapter sequences with cutadapt/3.0 and the qualities of the FASTQs assessed using FASTQC/0.11.9.77 The reads were aligned to the GRCm38/mm10 genome using bowtie/2–2.4.2 using the end-to-end parameter. Sam output files were then filtered to keep alignments with a minimum mapping quality of 10 using samtools/1.1361 and PCR duplicates removed with picard/2.23.7. Sambamba/0.7.1 was used to retain only uniquely aligned reads. Reads mapping to the Encyclopedia of DNA Elements (ENCODE) blacklisted regions were also removed from the analysis. There were no statistically significant differences in sequencing depth, alignment rate or alignable fragments per million across the young and old groups in any tissue. The bamCoverage function in deepTools/3.5.1 was used to generate RPKM (reads per kilobase per million mapped reads) normalized bigWig files. Input reads were subtracted from the different salt fractions and pellet with bigwigCompare.
The input samples were further used to calculate nucleosome positions using DANPOS/3.1.1.28 The samples were read normalized using the fold change normalization method and occupancy, signal periodicity, fuzziness profiles were plotted using the profile or stat function.
LISA
Epigenetic Landscape In Silico deletion Analysis (LISA) was performed with the genes annotated to the top 500 differential H3K27me3 peaks using the webtool at http://lisa.cistrome.org/.
PCA plots
RNA-seq PCA plot was generated in R/4.0 with the DESeq2 output and plotted with ggplot2. ChIP-seq peak PCA was generated with DiffBind.69
Area under the curve (AUC) calculation
bwtool/1.082 was used to get genome coverage information (AUC) across regions of interest and then plotted with ggplot2 in R/4.0.
Annotation
Genomics regions were annotated using ChIPseeker/1.26 annotatePeak function in R/4.0.83
Correlation matrix
Correlation matrices were generated using the plotCorrelation function of deepTools/3.5.1 using the multibigWig summary files and a Spearman correlation.
Heatmaps
Heatmaps were made with deepTools/3.5.0. The computeMatrix function was first used to calculate the signal intensity on the H3K27me3 differential peak regions and then heatmap drawn with the plotHeatmap function.
Venn diagrams
Venn diagrams were constructed using Intervene/0.6.5.
Gene Ontology (GO) analysis
GO analysis of the differentially expressed mRNAs, or differential peaks (old vs young) was performed using DAVID/6.858 with Mus musculus genes as background. The top 10–20 significant GO terms (reported as p-values using a Fisher’s exact test) for the Biological Process category (sorted by fold enrichment) are reported.
Genome browser tracks
Genome browser tracks were created for individual and pooled (across replicates) samples by converting the BAM files to bigWig files using the bamCoverage function of deepTools/3.5.0 then uploaded on the University of California, Santa Cruz Genome Browser using either custom tracks or track hubs.
Hypergeometric test
Hypergeometric tests were performed with the online web calculator http://nemates.org/MA/progs/overlap_stats.cgi.
IP-MS analysis
Total peptide counts were used to perform downstream analysis. We filtered peptides in each sample using the following parameters: only enriched in EZH2 IP sample compared to IgG, and EZH2/Input fold change >3. The filtered peptides were listed as IP-enriched peptides.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses for all experiments were performed in GraphPad Prism/9.0.0 or R Stats/4.0.5. Statistical data are presented as mean ± S.E.M or S.D. Sample size (n), statistical tests used, and p-values are specified in the figure legends.
Supplementary Material
Highlights.
Global increase of H3K27me3 is a common signature of aged liver and other tissues
H3K27me3 is lost at peak regions and gained at lamin-associated domains
Liver regeneration reverses age-related H3K27me3 and transcriptomic patterns
ACKNOWLEDGMENTS
We wish to acknowledge the NIA IRP for financial support. We thank the Comparative Medicine Section at NIA for providing support with animal experiments. We thank Steven Henikoff and Kaifu Chen for discussions, Aubrey Mwinyogle and Artem Shmelev for human-liver biopsies, Elin Lehrmann for GEO upload, and Myriam Gorospe and Shelley Berger for critical feedback. We thank the Taplin Biological Mass Spectrometry Facility and Spatial Technologies Unit for IP-MS and PhenoImager experiments, respectively. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). Some figures were made using BioRender and the model by Arne Fabritius and Radhika Patnala. The work was funded by NIH ZIA AG000679 to P.S., NIH ZIA BC011206 to Y.D., NIH S10 OD030286–01, NIH P30 CA01333047, Leukemia Research Foundation, AFAR, The Japan Agency for Medical Research and Development, Merck, and Deerfield to S. Stransky and S. Sidoli.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2023.04.005.
REFERENCES
- 1.Berger SL, Kouzarides T, Shiekhattar R, and Shilatifard A (2009). An operational definition of epigenetics. Genes Dev. 23, 781–783. 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sen P, Shah PP, Nativio R, and Berger SL (2016). Epigenetic mechanisms of longevity and aging. Cell 166, 822–839. 10.1016/j.cell.2016.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pal S, and Tyler JK (2016). Epigenetics and aging. Sci. Adv. 2, e1600584. 10.1126/sciadv.1600584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benayoun BA, Pollina EA, and Brunet A (2015). Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 16, 593–610. 10.1038/nrm4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang N, and Sen P (2018). The senescent cell epigenome. Aging (Albany, NY) 10, 3590–3609. 10.18632/aging.101617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fiore APZP, Ribeiro PF, and Bruni-Cardoso A (2018). Sleeping beauty and the microenvironment enchantment: microenvironmental regulation of the proliferation-quiescence decision in normal tissues and in cancer development. Front. Cell Dev. Biol. 6, 59. 10.3389/fcell.2018.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, Lahat A, Day CP, Burt A, Palmer A, Anstee QM, et al. (2017). Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun. 8, 15691. 10.1038/ncomms15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kane AE, and Sinclair DA (2019). Epigenetic changes during aging and their reprogramming potential. Crit. Rev. Biochem. Mol. Biol. 54, 61–83. 10.1080/10409238.2019.1570075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Croce L, and Helin K (2013). Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 20, 1147–1155. 10.1038/nsmb.2669. [DOI] [PubMed] [Google Scholar]
- 10.Yu JR, Lee CH, Oksuz O, Stafford JM, and Reinberg D (2019). PRC2 is high maintenance. Genes Dev. 33, 903–935. 10.1101/gad.325050.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, and Reinberg D (2008). Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 32, 503–518. 10.1016/j.molcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riising EM, Comet I, Leblanc B, Wu X, Johansen JV, and Helin K (2014). Gene silencing triggers polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol. Cell 55, 347–360. 10.1016/j.molcel.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Forbes SJ, and Newsome PN (2016). Liver regeneration - mechanisms and models to clinical application. Nat. Rev. Gastroenterol. Hepatol. 13, 473–485. 10.1038/nrgastro.2016.97. [DOI] [PubMed] [Google Scholar]
- 14.Tvardovskiy A, Schwämmle V, Kempf SJ, Rogowska-Wrzesinska A, and Jensen ON (2017). Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res. 45, 9272–9289. 10.1093/nar/gkx696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma Z, Wang H, Cai Y, Wang H, Niu K, Wu X, Ma H, Yang Y, Tong W, Liu F, et al. (2018). Epigenetic drift of H3K27me3 in aging links glycolysis to healthy longevity in Drosophila. eLife 7, e35368. 10.7554/eLife.35368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baumgart M, Groth M, Priebe S, Savino A, Testa G, Dix A, Ripa R, Spallotta F, Gaetano C, Ori M, et al. (2014). RNA-seq of the aging brain in the short-lived fish N. furzeri - conserved pathways and novel genes associated with neurogenesis. Aging Cell 13, 965–974. 10.1111/acel.12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cencioni C, Heid J, Krepelova A, Rasa SMM, Kuenne C, Guenther S, Baumgart M, Cellerino A, Neri F, Spallotta F, and Gaetano C (2019). Aging Triggers H3K27 trimethylation Hoarding in the chromatin of Nothobranchius furzeri skeletal muscle. Cells 8. 10.3390/cells8101169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schwörer S, Becker F, Feller C, Baig AH, Köber U, Henze H, Kraus JM, Xin B, Lechel A, Lipka DB, et al. (2016). Epigenetic stress responses induce muscle stem-cell ageing by Hoxa9 developmental signals. Nature 540, 428–432. 10.1038/nature20603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu L, Cheung TH, Charville GW, Hurgo BM, Leavitt T, Shih J, Brunet A, and Rando TA (2013). Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 4, 189–204. 10.1016/j.celrep.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nativio R, Lan Y, Donahue G, Sidoli S, Berson A, Srinivasan AR, Shcherbakova O, Amlie-Wolf A, Nie J, Cui X, et al. (2020). An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat. Genet. 52, 1024–1035. 10.1038/s41588-020-0696-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 9, 1163–1170. 10.1016/j.celrep.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 22.Lund E, Oldenburg AR, and Collas P (2014). Enriched domain detector: a program for detection of wide genomic enrichment domains robust against local variations. Nucleic Acids Res. 42, e92. 10.1093/nar/gku324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whitton H, Singh LN, Patrick MA, Price AJ, Osorio FG, López-Otín C, and Bochkis IM (2018). Changes at the nuclear lamina alter binding of pioneer factor Foxa2 in aged liver. Aging Cell 17, e12742. 10.1111/acel.12742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Steensel B, and Belmont AS (2017). Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell 169, 780–791. 10.1016/j.cell.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, et al. (2006). Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 103, 8703–8708. 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou W, Dinh HQ, Ramjan Z, Weisenberger DJ, Nicolet CM, Shen H, Laird PW, and Berman BP (2018). DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet. 50, 591–602. 10.1038/s41588-018-0073-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henikoff S, Henikoff JG, Sakai A, Loeb GB, and Ahmad K (2009). Genome-wide profiling of salt fractions maps physical properties of chromatin. Genome Res. 19, 460–469. 10.1101/gr.087619.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen K, Xi Y, Pan X, Li Z, Kaestner K, Tyler J, Dent S, He X, and Li W (2013). DANPOS: dynamic analysis of nucleosome position and occupancy by sequencing. Genome Res. 23, 341–351. 10.1101/gr.142067.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marro S, Pang ZP, Yang N, Tsai MC, Qu K, Chang HY, Südhof TC, and Wernig M (2011). Direct lineage conversion of terminally differentiated hepatocytes to functional neurons. Cell Stem Cell 9, 374–382. 10.1016/j.stem.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mozhui K, Lu AT, Li CZ, Haghani A, Sandoval-Sierra JV, Wu Y, Williams RW, and Horvath S (2022). Genetic loci and metabolic states associated with murine epigenetic aging. eLife 11. 10.7554/eLife.75244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henikoff S, Henikoff JG, Kaya-Okur HS, and Ahmad K (2020). Efficient chromatin accessibility mapping in situ by nucleosome-tethered tagmentation. eLife 9. 10.7554/eLife.63274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lovén J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, and Young RA (2012). Revisiting global gene expression analysis. Cell 151, 476–482. 10.1016/j.cell.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jung M, and Pfeifer GP (2015). Aging and DNA methylation. BMC Biol. 13, 7. 10.1186/s12915-015-0118-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, Lin JC, Liang G, Jones PA, and Tanay A (2008). Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc. Natl. Acad. Sci. USA 105, 12979–12984. 10.1073/pnas.0806437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nair SS, Coolen MW, Stirzaker C, Song JZ, Statham AL, Strbenac D, Robinson MD, and Clark SJ (2011). Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics 6, 34–44. 10.4161/epi.6.1.13313. [DOI] [PubMed] [Google Scholar]
- 36.Horvath S, Haghani A, Macoretta N, Ablaeva J, Zoller JA, Li CZ, Zhang J, Takasugi M, Zhao Y, Rydkina E, et al. (2022). DNA methylation clocks tick in naked mole rats but queens age more slowly than non-breeders. Nat Aging 2, 46–59. 10.1038/s43587-021-00152-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qin Q, Fan J, Zheng R, Wan C, Mei S, Wu Q, Sun H, Brown M, Zhang J, Meyer CA, and Liu XS (2020). Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol. 21, 32. 10.1186/s13059-020-1934-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jadhav U, Manieri E, Nalapareddy K, Madha S, Chakrabarti S, Wucherpfennig K, Barefoot M, and Shivdasani RA (2020). Replicational dilution of H3K27me3 in mammalian cells and the role of poised promoters. Mol. Cell 78. 141.e5–151.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Michalopoulos GK, and Bhushan B (2021). Liver regeneration: biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 18, 40–55. 10.1038/s41575-020-0342-4. [DOI] [PubMed] [Google Scholar]
- 40.Mitchell C, and Willenbring H (2008). A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat. Protoc. 3, 1167–1170. 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
- 41.Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, and Dynlacht BD (2002). E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 16, 245–256. 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fischer DS, Theis FJ, and Yosef N (2018). Impulse model-based differential expression analysis of time course sequencing data. Nucleic Acids Res. 46, e119. 10.1093/nar/gky675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bucher NL, Swaffield MN, and Ditroia JF (1964). The influence of age upon the incorporation of Thymidine-2-C14 into the DNA of regenerating rat liver. Cancer Res. 24, 509–512. [PubMed] [Google Scholar]
- 44.Fry M, Silber J, Loeb LA, and Martin GM (1984). Delayed and reduced cell replication and diminishing levels of DNA polymerase-alpha in regenerating liver of aging mice. J. Cell. Physiol. 118, 225–232. 10.1002/jcp.1041180302. [DOI] [PubMed] [Google Scholar]
- 45.Bucher NL, and Glinos AD (1950). The effect of age on regeneration of rat liver. Cancer Res. 10, 324–332. [PubMed] [Google Scholar]
- 46.Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 47.Bou Sleiman M, Jha P, Houtkooper R, Williams RW, Wang X, and Auwerx J (2020). The gene-regulatory footprint of aging highlights conserved central regulators. Cell Rep. 32, 108203. 10.1016/j.celrep.2020.108203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reverón-Gómez N, González-Aguilera C, Stewart-Morgan KR, Petryk N, Flury V, Graziano S, Johansen JV, Jakobsen JS, Alabert C, and Groth A (2018). Accurate recycling of parental histones reproduces the histone modification landscape during DNA replication. Mol. Cell 72. 239.e5–249.e5. 10.1016/j.molcel.2018.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alabert C, Barth TK, Reverón-Gómez N, Sidoli S, Schmidt A, Jensen ON, Imhof A, and Groth A (2015). Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 29, 585–590. 10.1101/gad.256354.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kori Y, Lund PJ, Trovato M, Sidoli S, Yuan ZF, Noh KM, and Garcia BA (2022). Multi-omic profiling of histone variant H3.3 lysine 27 methylation reveals a distinct role from canonical H3 in stem cell differentiation. Mol. Omics 18, 296–314. 10.1039/d1mo00352f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scaffidi P, and Misteli T (2006). Lamin A-dependent nuclear defects in human aging. Science 312, 1059–1063. 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scaffidi P, and Misteli T (2005). Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 11, 440–445. 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maures TJ, Greer EL, Hauswirth AG, and Brunet A (2011). The H3K27 demethylase UTX-1 regulates C. elegans lifespan in a germline-independent, insulin-dependent manner. Aging Cell 10, 980–990. 10.1111/j.1474-9726.2011.00738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ocampo A, Reddy P, Martinez-Redondo P, Platero-Luengo A, Hatanaka F, Hishida T, Li M, Lam D, Kurita M, Beyret E, et al. (2016). In vivo amelioration of age-associated hallmarks by partial reprogramming. Cell 167. 1719.e12–1733.e12. 10.1016/j.cell.2016.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarkar TJ, Quarta M, Mukherjee S, Colville A, Paine P, Doan L, Tran CM, Chu CR, Horvath S, Qi LS, et al. (2020). Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 11, 1545. 10.1038/s41467-020-15174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chondronasiou D, Gill D, Mosteiro L, Urdinguio RG, Berenguer-Llergo A, Aguilera M, Durand S, Aprahamian F, Nirmalathasan N, Abad M, et al. (2022). Multi-omic rejuvenation of naturally aged tissues by a single cycle of transient reprogramming. Aging Cell 21, e13578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, and Rando TA (2005). Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433, 760–764. 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- 58.Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using David bioinformatics resources. Nat. Protoc. 4, 44–57. 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 59.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liao Y, Smyth GK, and Shi W (2019). The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47. 10.1093/nar/gkz114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 66.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, and Prins P (2015). Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034. 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ramírez F, Dündar F, Diehl S, Gru€ning BA, and Manke T. (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191. 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feng X, Grossman R, and Stein L (2011). PeakRanger: a cloud-enabled peak caller for ChIP-seq data. BMC Bioinformatics 12, 139. 10.1186/1471-2105-12-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stark R, and Brown G (2011). DiffBind: differential binding analysis of ChIP-seq peak data R package version 100. http://bioconductor.org/packages/release/bioc/html/DiffBind.html.
- 70.Shen L, Shao NY, Liu X, Maze I, Feng J, and Nestler EJ (2013). diffReps: detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PLoS One 8, e65598. 10.1371/journal.pone.0065598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sidoli S, Bhanu NV, Karch KR, Wang X, and Garcia BA (2016). Complete workflow for analysis of histone post-translational modifications using bottom-up mass spectrometry: from histone extraction to data analysis. J. Vis. Exp. 10.3791/54112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sen P, Lan Y, Li CY, Sidoli S, Donahue G, Dou Z, Frederick B, Chen Q, Luense LJ, Garcia BA, et al. (2019). Histone acetyltransferase p300 induces de novo super-enhancers to drive cellular senescence. Mol. Cell 73. 684.e8–698.e8. 10.1016/j.molcel.2019.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nabbi A, and Riabowol K (2015). Isolation of pure nuclei using a sucrose method. Cold Spring Harb. Protoc. 2015, 773–776. 10.1101/pdb.prot083741. [DOI] [PubMed] [Google Scholar]
- 75.Walkiewicz MP, Bui M, Quénet D, and Dalal Y (2014). Tracking histone variant nucleosomes across the human cell cycle using biophysical, biochemical, and cytological analyses. Methods Mol. Biol 1170, 589–615. 10.1007/978-1-4939-0888-2_34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Andrews S (2017). FastQC: a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- 78.Yuan M, and Kendziorski C (2006). A unified approach for simultaneous gene clustering and differential expression identification. Biometrics 62, 1089–1098. 10.1111/j.1541-0420.2006.00611.x. [DOI] [PubMed] [Google Scholar]
- 79.Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Methodol. 57, 289–300. [Google Scholar]
- 80.Yuan ZF, Sidoli S, Marchione DM, Simithy J, Janssen KA, Szurgot MR, and Garcia BA (2018). EpiProfile 2.0: a computational platform for processing epi-proteomics mass spectrometry data. J. Proteome Res. 17, 2533–2541. 10.1021/acs.jproteome.8b00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zheng Y, Ahmad K, and Henikoff S. CUT&Tag Data Processing and Analysis Tutorial. protocols.io. 10.17504/protocols.io.bjk2kkye. [DOI] [Google Scholar]
- 82.Pohl A, and Beato M (2014). bwtool: a tool for bigWig files. Bioinformatics 30, 1618–1619. 10.1093/bioinformatics/btu056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu G, Wang LG, and He QY (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383. 10.1093/bioinformatics/btv145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genome-wide datasets have been submitted to the Gene Expression Omnibus portal (GEO: GSE185708) and will be publicly available as of the date of publication. Raw mass spec data are deposited at chorusproject: chorusproject.org/1769. Accession numbers and DOI are also listed in the key resources table.
All original code has been deposited at Github and Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.