

Abstract
Purpose:
We investigated why three patient-derived xenograft (PDX) childhood BRAFV600E-mutant brain tumor models are highly sensitive to trametinib. Mechanisms of acquired resistance selected in situ, and approaches to prevent resistance were also examined, which may translate to both low-grade glioma (LGG) molecular subtypes.
Experimental Design:
Sensitivity to trametinib [MEK inhibitor (MEKi)] alone or in combination with rapamycin (TORC1 inhibitor), was evaluated in pediatric PDX models. The effect of combined treatment of trametinib with rapamycin on development of trametinib resistance in vivo was examined. PDX tissue and tumor cells from trametinib-resistant xenografts were characterized.
Results:
In pediatric models TORC1 is activated through ERK-mediated inactivation of the tuberous sclerosis complex (TSC): consequently inhibition of MEK also suppressed TORC1 signaling. Trametinib-induced tumor regression correlated with dual inhibition of MAPK/TORC1 signaling, and decoupling TORC1 regulation from BRAF/MAPK control conferred trametinib resistance. In mice, acquired resistance to trametinib developed within three cycles of therapy in all three PDX models. Resistance to trametinib developed in situ is tumor-cell–intrinsic and the mechanism was tumor line specific. Rapamycin retarded or blocked development of resistance.
Conclusions:
In these three pediatric BRAF-mutant brain tumors, TORC1 signaling is controlled by the MAPK cascade. Trametinib suppressed both MAPK/TORC1 pathways leading to tumor regression. While low-dose intermittent rapamycin to enhance inhibition of TORC1 only modestly enhanced the antitumor activity of trametinib, it prevented or retarded development of trametinib resistance, suggesting future therapeutic approaches using rapamycin analogs in combination with MEKis that may be therapeutically beneficial in both KIAA1549::BRAF- and BRAFV600E-driven gliomas.
Introduction
Brain tumors constitute about 21% of pediatric cancers, and of these low-grade gliomas (LGG) occur most frequently. While the survival for these patients is relatively good, even at 5 years, these indolent tumors eventually progress (1–3). Current treatment for LGG includes intensive chemo-radiation therapy that leads to cognitive decline, malignant transformation, and other life-debilitating or –threatening sequelae (2). The 15-year incidence of adverse outcomes such as blindness, hearing loss, obesity, hormonal imbalance respectively was 18%, 22%, 53%, and approximately 25% respectively (4–7). Among survivors assessed for intellectual function, 34% had an intelligence quotient (IQ) below average. Treatment-resistant progressive disease is the most common cause of death (8).
Genomic studies have shown that in low-grade tumors (juvenile pilocytic astrocytoma) approximately 90% have a tandem duplication involving the KIAA1549 and BRAF genes that generate KIAA1549: BRAF constitutively active fusions (9, 10). Higher grade tumors tend to have activating point mutations of BRAF, most frequently the V600E variant. Such activating mutations have been identified in diffuse astrocytomas (23%), gangliogliomas (33%), pleomorphic xanthoastrocytoma (PXA; 70%), and glioblastoma (10%–15%; refs. 9–12). Drawing from available databases (ACS and CBTRUS), approximately 1,400 new pediatric BRAF mutant brain tumors are diagnosed annually in the United States (13–15).
The characteristic of these BRAF-fusion, BRAFV600E tumors, and those in NF1 patients, is activation of the MAPK pathway (16). However, the response to BRAF, MEK, or combinations of MAPK inhibitors (MAPKi) is context specific. Selumetinib (AZD6244) an allosteric inhibitor of MEK1/2 is highly active in KIAA1549::BRAF, BRAFV600E, and NF1-driven tumors pediatric LGGs (17, 18). Dabrafenib, a BRAF-selective inhibitor has shown activity in epitheloid glioblastoma (19) and BRAF-mutation positive LGG (20). However, BRAF inhibitors are not active in KIAA1549::BRAF LGG, the major molecular subtype (21). We previously reported that selumetinib caused regression of an astrocytoma patient-derived xenograft (PDX) model with BRAFV600E but not in a PDX model with wild-type BRAF (22). However, compared with the pediatric PDX, BRAFV600E glioblastoma cell lines and their xenografts displayed much less sensitivity to selumetinib (23). The basis for differential sensitivity to selumetinib observed in different BRAFV600E cell lines remains poorly understood.
While selumetinib has demonstrated clinical activity in KIAA1549::BRAF, BRAFV600E, and NF1-driven pediatric brain tumors (18), patients with BRAFV600E mutations have a higher rate of progression on treatment (i.e., become resistant) or progress rapidly when treatment is interrupted or dose-reduced (24, 25). Thus, while selumetinib causes tumor shrinkage, it is not curative. Preventing resistance and enhancing the cytotoxicity of MEK-targeted [MEK inhibitor (MEKi)] therapy thus remains the clinical challenge (24). Unfortunately, there are no xenograft models of fusion-driven LGG to use in development of more effective MEKi-based therapies. To overcome this limitation, we have used three models driven by BRAFV600E point mutations as surrogates to improve MEKi-based therapies.
Here we have attempted to identify the mechanism/s that define sensitivity to trametinib of three PDX models derived from BRAFV600E pediatric brain tumors. Using these models we have explored drug combinations that may retard or prevent emergence of trametinib resistance in animal models. Importantly, maintenance of TORC1 inhibition prevents or retards emergence of trametinib resistance in each PDX model. Although these data were developed using BRAFV600E mutant PDX models, the results may translate to improved therapies for tumors driven by the KIAA1549::BRAF and potentially NF1-driven LGG.
Materials and Methods
Reagents
RPMI1640 medium, FBS, Crystal Violet, and Alamar Blue were purchased from Invitrogen. Primary antibodies, if not indicated, were all purchased from Cell Signaling Technology. Selumetinib (Selleck Chemicals) was dissolved in DMSO at concentration of 10 mmol/L; trametinib was purchased from MedChem Express and dissolved in DMSO at 10 mmol/L. Rapamycin and dabrafenib were purchased from LC Laboratories, and dissolved in DMSO at concentration of 20 mmol/L. Details of antibodies, their source, and plasmids used are given in Supplementary Table S1. Details of plasmids and other constructs used are also given in Supplemental Methods.
Childhood brain tumor PDX models
BT-40 was established at diagnosis from a leptomenigial mass in a 14-year–old male (22). NCH-MN-1 was established in mice from an occipital mass in a 16-year–old female at diagnosis. IC-3635 was established from a cerebral mass diagnosed as a pleomorphic xanthoastrocytoma (PXA) from a 10-year–old female (26). All models were directly implanted into mice without culture on plastic. Each PDX model was confirmed as having BRAFV600E mutation by sequencing (27, 28).
Cell culture
The BT40 cell line, which harbors heterozygous BRAFV600E mutation, was developed from the PDX model established in this laboratory (23), AM38 and DBTRG-05MG glioblastoma cell lines with BRAF(V600E) mutation were generously provided by T. Nicolaides [University of California San Francisco (UCSF), San Francisco, CA]. All the cells were maintained in antibiotic-free RPMI 1640 medium (Invitrogen) supplemented with 10% FBS and 2 mmol/L glutamine at 37°C with 5% CO2. All cells were maintained as subconfluent cultures and split 1:3, 24 hours before use. Experiments were carried out within 10 passages of receiving the glioblastoma cells. Cells were subjected to short tandem repeat (STR) DNA Profiling Analysis and BRAFV600E mutation status was verified by restriction analysis and DNA sequencing. HEK293T cells used for lentivirus packaging were from ATCC (CRL-3216) and cultured in DMEM with 10% FBS. One day before transfection, cells were seeded on polylysine coated plates to approximately 70% confluence. All cells were routinely tested for Mycoplasma (MycoAlert, Lonza) and determined to be negative.
Tumor cell isolation and culture from PDX
Primary cultures from BT-40, NCH-MN-1, and IC-3635 and BT40/TramR (trametinib-resistant) tumors resistant to trametinib were developed as described below. Tumor cells were isolated as described (23) with minor modification and grown in Stem Cell culture medium [DMEM/F12 knockout medium supplemented with 20 mL Stem-pro Neural supplement (Gibco™), penicillin–streptomycin, FGF(20ng/mL), and rhEGF (20ng/mL), and transferred to 75 cm2 Corning ultra-low attachment flasks, and cultured at 37°C with 5% CO2 for 3 days. Analysis of cultures using lactate dehydrogenase (LDH) isozyme analysis showed cells to be human without detectable murine contamination.
Clonogenic assays
Clonogenic assay was performed as described (29), with some modification. Briefly, 0.2 to 1 × 104 cells were seeded on 6-well culture plates, and cultured for approximately 24 hours, exposed to increasing concentrations of selumetinib (AZD6244) or trametinib for a further 48 hours. Medium was removed and replaced with drug-free complete medium (RPM1640 with 10% FBS). Cells were allowed to proliferate for another 2 to 3 weeks (37°C, 5% CO2). Colonies were fixed with methanol for 15 minutes and stained with 0.01% (w/v) crystal violet for 10 minutes. Excess crystal violet was washed away with H2O. The stained crystal violet was resolved with 10% acetate solution and colonies (>50 cells) quantified by measuring OD570. Empty wells were subjected to the same staining and washing procedure served as blank controls. Cells treated with DMSO were set as 100%, the cell viability was calculated by comparing the signal from treated cells with that of the cells with control treatment and presented as percent of control.
Cellular viability assay
Cell viability assays were conducted using the Alamar Blue cell viability reagent according to the manufacturer’s directions as previously described (30). Absorbance of a minimum of six replicates per treatment condition was measured.
Western blotting
Protein extraction from xenograft tissue and in vitro cultured cells, cell lysis, and Western blotting were performed as described (23, 31). Immunoreactive bands were visualized by using Super Signal Chemiluminiscence substrate (Pierce) and Biomax MR and XAR film (Eastman Kodak Co.).
In vivo tumor growth inhibition studies
C.B.17SC scid−/− female mice (C.B-Igh-1b/IcrTac-Prkdcscid, Taconic Farms) were used to propagate subcutaneously implanted tumors as previously described (22). All mice were maintained under barrier conditions and experiments were carried out using protocols and conditions approved by the institutional animal care and use committee of University of Texas Health San Antonio. Mice were randomized into groups of 5 or 10 when tumors were 200 to 400 mm3. Trametinib dissolved in DMSO (5%) was diluted with 0.5% methylcellulose/0.2% Tween-80 solution and administered once daily by oral gavage (PO), for a scheduled 6 weeks at a dose of 1 mg/kg. Rapamycin, dissolved in DMSO, was diluted with 5% Tween-80 in water and administered by intraperitoneal injection, using an administration schedule of once daily for 5 days each week for 6 consecutive weeks at a dose of 5 mg/kg. Dabrafenib was formulated in 5% DMSO/0.5% methyl cellulose/0.2% Tween 80 and administered at 30 mg/kg daily (oral gavage). Selumetinib was dissolved in 0.5% hydroxypropyl methyl cellulose, 0.1% polysorbate 80, and administered orally, using a twice-daily schedule (once daily at weekends) for a scheduled 6 weeks at a dose of 75 mg/kg. For subcutaneous tumors, volumes were determined weekly using digital calipers to measure perpendicular diameters, as previously described (32).
Orthotopic xenograft studies
C.B-Igh-1b/IcrTac-Prkdcscid female mice were implanted with 5 × 104 BT-40/Luc cells intracranially to establish BT-40 tumors as previously described (33). Bioluminescent imaging was used for tumor growth monitoring using PerkinElmer IVIS Spectrum on day 7 postinjection and once weekly thereafter. Mice were injected with 150 mg/kg Potassium Luciferin (Gold Biotechnology) i.p. and anesthetized with isoflurane for imaging inside the IVIS chamber. Living Image 4.5 software (PerkinElmer) was used to determine when the region of interest reached a bioluminescence imaging (BLI) value of >1 × 105 photons/sec/cm3/steradian at which point mice were randomized into control or treatment groups.
Apoptotic threshold determination
Fluorescence-based whole-cell mitochondrial depolarization assays with Bim BH3 peptide over a concentration range were performed as described (34) with slight modification. DMSO was set as negative control, and carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (Tocris Bioscience) at final concentration of 10μmol/L was used as the positive control treatment. To produce a normalized value for each profile to simplify comparison, the area of each curve was calculated in Excel, and then subjected the following equation to get the percentage of mitochondrial depolarization: % depolarization (%Δψm loss) 100 × [1−(BH3−FCCP)/(DMSO−FCCP)].
Supplemental methods
Details for antibodies used, plasmids, lentivirus preparation, infection, construction of the TSC2 GAP mutant, generation of luciferase-tagged BT-40 cells, development of trametinib resistance, orthotopic inoculation, and apoptotic threshold assay are given in the supplemental methods.
Data availability statement
The data generated in this study are available upon request from the corresponding author.
Results
Sensitivity to trametinib correlates with TORC1 inhibition
Each of the PDX (subcutaneous) models was sensitive to trametinib. Trametinib (1 mg/kg daily for 42 days) induced partial regression of BT-40 (Fig. 1A) or complete regressions of NCH-MN-1 (Fig. 1B) and IC-3635 (Fig. 1C) models. However, at the end of treatment tumor regrowth was rapid for each model. We reported previously (23) that in BT-40 xenografts selumetinib induced suppression of both phosphor-ERK and decreased TORC1 signaling as determined by decreased phosphorylation of 4EBP1 and S6 protein. Treatment of mice with trametinib (1 mg/kg/day) also reduced phospho-4EBP1 at days 1 and 5 in IC-3635 whereas this was more pronounced after 5 days’ treatment in NCH-MN-1 tumors (Figs. 1D, E).
Figure 1.
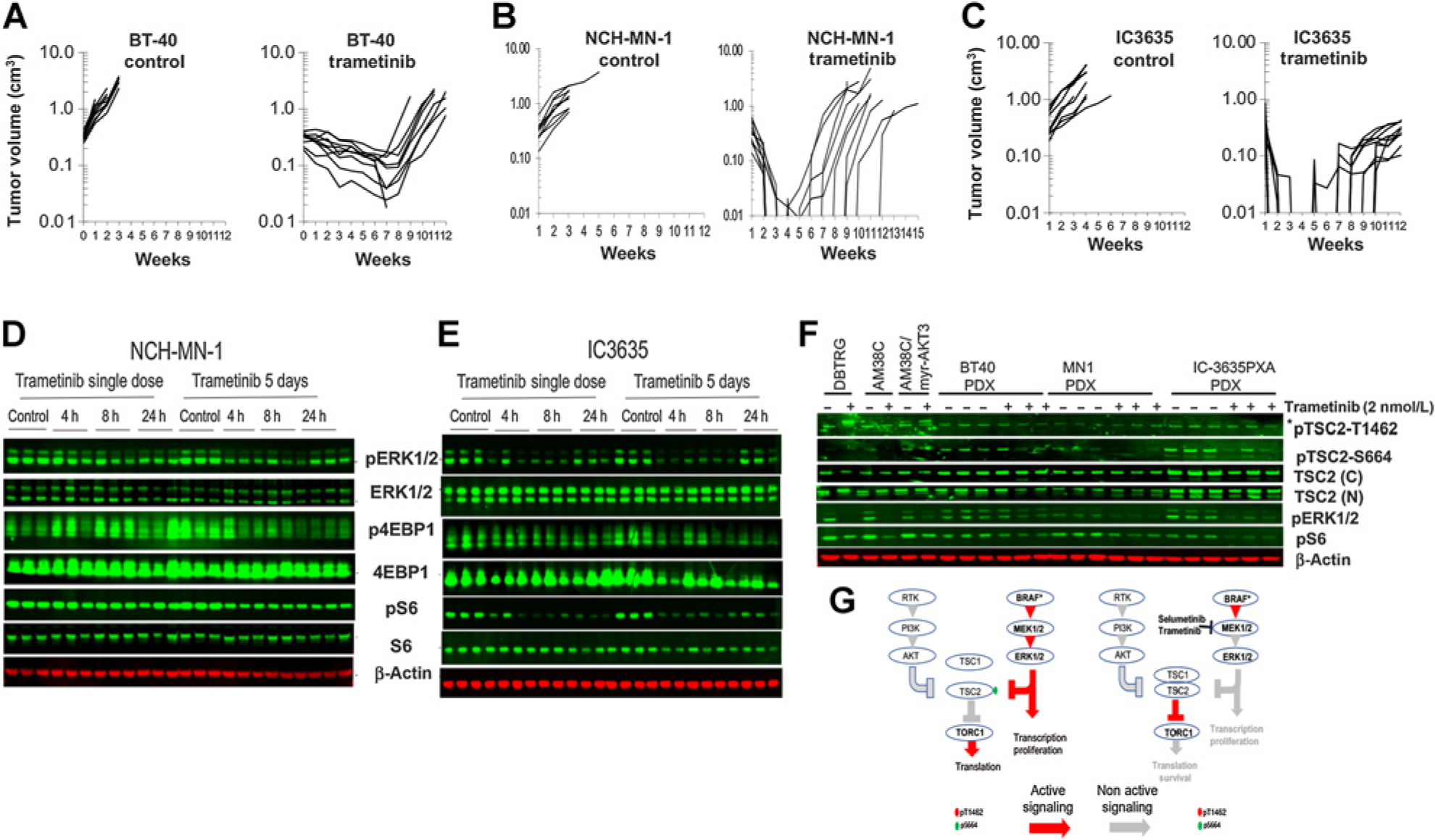
Sensitivity to trametinib of BRAFV600E mutant pediatric brain tumor PDX models correlates with suppression of MAPK and TORC1 signaling. A to C, sensitivity of BT-40, NCH-MN-1, and IC-3635 brain tumor PDX models to trametinib (1 mg/kg daily × 42 days). D to E, Pharmacodynamic changes in MAPK and TORC1 signaling following trametinib (1 mg/kg) administered on day 1 or treatment for 5 consecutive days. Tumors were harvested 4 hours after the final drug administration. F, Mice bearing BT-40, NCH-MN-1, or IC-3635 xenografts were untreated or received trametinib (1 mg/kg/day) for 5 days, and phosphorylation of TSC2 at an Akt site (T1462) and an MAPK site (S664) was determined. No phosphorylation of TSC2 at T1462 was detected in control or treated xenografts, whereas phosphorylation of TSC2 (S664) was decreased in trametinib-treated tumors. Trametinib induces phosphorylation of TSC2(T1462) in DBTRG cells, and expression of Myr-AKT3 induces phosphorylation of TSC2(T1462) in AM38C adult glioblastoma cells. G, Schema of MEKi sensitivity in cells with MAPK mediated inactivation of TSC2 without PI3K/Akt phosphorylation of TSC2. h, hours.
Both selumetinib and trametinib have relatively poor penetration to brain tissue due to efflux mediated by ABCB1 and ABCG2 transporters (35) however trametinib has potential advantages over selumetinib because of increased potency. Comparison of the inhibitory potency of trametinib and selumetinib revealed that trametinib was more than 40-fold more potent than selumetinib, consistent with published literature. For BT-40, IC50 values were for trametinib 0.8 nmol/L a clinically relevant drug exposure (36), and selumetinib 25 nmol/L, respectively (Supplementary Fig. S1A). Consequently, we focused on trametinib to further develop MEKi combinations.
The tuberous sclerosis complex is regulated via MAPK signaling in pediatric BRAF-mutant brain tumor PDX models
The tuberous sclerosis complex (TSC) is inactivated by Akt phosphorylation of TSC2 at several sites (S939, S981, S1130/32 and T1462) (37) and by ERK1/2 at S540 and S664 (38, 39). As shown in Fig. 1F, TSC2 was phosphorylated at S664 in untreated subcutaneous BT-40, NCH-MN-1, and IC-3635 tumors, and this was decreased by trametinib treatment. Phosphorylation at T1462 was not detected in these PDX models, and trametinib treatment did not induce phosphorylation. We next examined the phosphorylation of TSC2 in two adult glioblastoma cell lines. Phosphorylation of TSC2 (S664) was detected in DBTRG-05MG control and trametinib treated cells (Fig. 1F). Phosphorylation of an Akt site on TSC2(T1462), was detected in DBTRG-05MG cells following trametinib treatment consistent with trametinib induced activation of Akt (Supplementary Fig. S1B). Phosphorylation of TSC2(T1462) was observed in AM38 cells engineered to overexpress Myr-Akt3 (Fig. 1F). These data suggest that in some glioblastoma (GBM) lines TSC2 may be inactivated through PI3K/Akt signaling when MEK is inhibited (consistent with Akt phosphorylation in DBTRG-05MG cells), whereas in pediatric BRAFV600E PDX models TSC2 is phosphorylated predominantly or exclusively through the MAPK cascade conferring sensitivity to MEKi (schema Fig. 1G and Supplementary Fig. S2).
Suppression of the tuberous sclerosis complex or activation of PI3K/Akt signaling confers resistance to trametinib
To determine the role of TSC2 in trametinib sensitivity shRNA was used to suppress TSC2, Fig. 2A. Knockdown of TSC2 or expression of TSC2ΔGAP (Fig. 2B) prevented trametinib-induced suppression of phospho-4EBP1. Importantly, knockdown of TSC2, or overexpression of Myr-AKT1 or Myr-AKT3 (Fig. 2C) or PTEN knockdown (Fig. 2D) conferred trametinib resistance. A schematic model of how inhibition of MEK regulates TORC1 is presented in Supplementary Fig. S2. Consistent with the conjecture that sensitivity to trametinib requires inhibition of TORC1 signaling, rapamycin partially restored trametinib sensitivity to BT-40 cells in which TSC2 had been suppressed using short hairpin RNA (shRNA; Supplementary Fig. S3A). These results suggested that addition of rapamycin to trametinib treatment may enhance the antitumor activity of trametinib.
Figure 2.
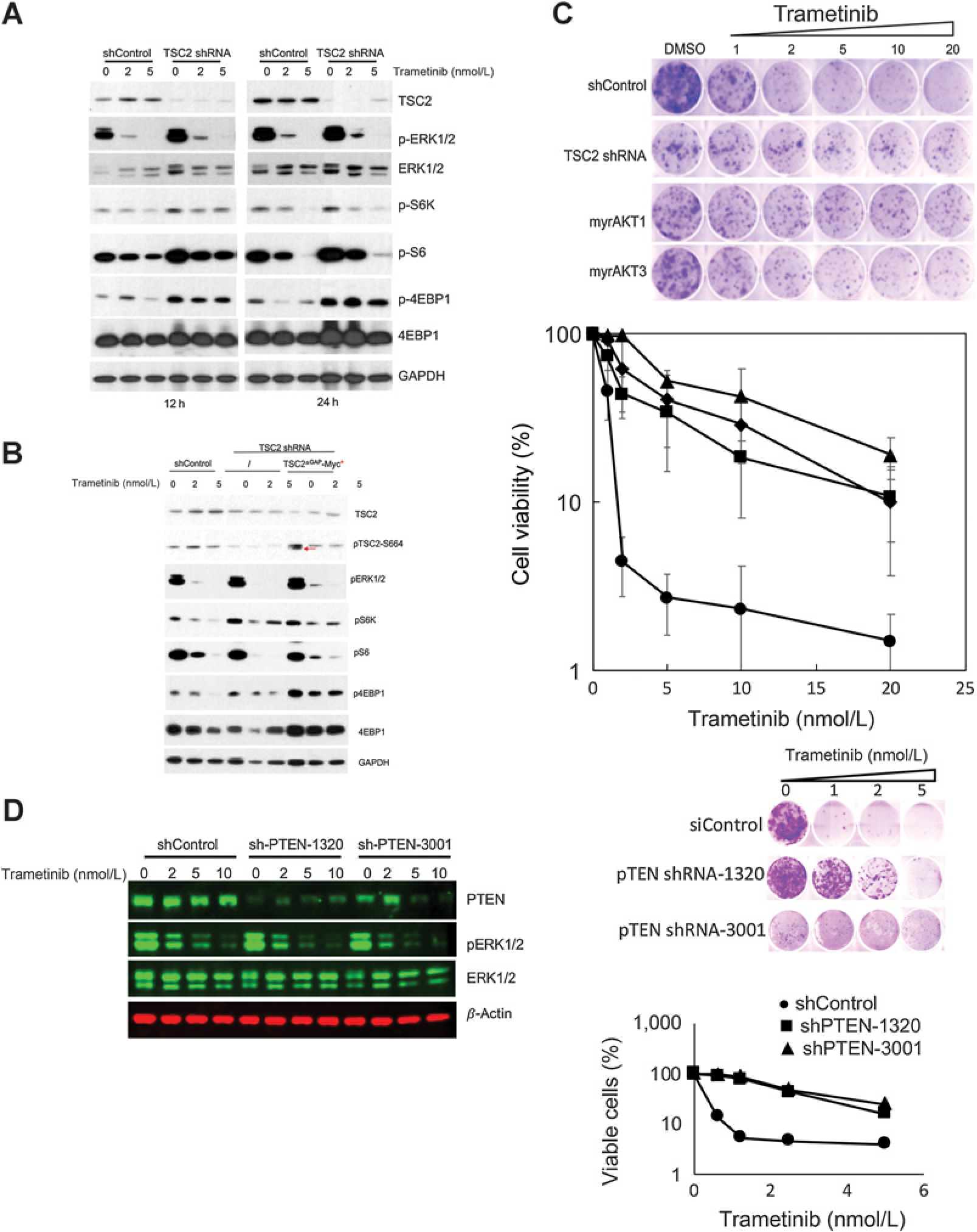
Activating TORC1 signaling confers resistance to trametinib in BT-40 cells. A, TSC2 was suppressed using shRNA. Cells were exposed to trametinib (2 or 5 nmol/L) for 12 or 24 hours. Trametinib inhibited phosphorylation of 4E-BP1 in control transfected cells but not in cells where TSC2 had been down regulated. B, Expression of shTSC2ΔGAP-Myc (red arrow) increases p4E-BP1 that is not inhibited by trametinib. C, Activating TORC1, by knockdown of TSC2, or expression of constitutively active myrAkt1 or myrAkt3 induces trametinib resistance in BT-40 cells. D, Knockdown of PTEN confers trametinib resistance in BT-40 cells. Left, PTEN was suppressed using lentivirus encoded shRNA. After 48 hours cells were exposed to trametinib (2–10 nmol/L) for 24 hours, and lysates probed for PTEN and total and phosphorylated ERK1/2. β-actin was used as a loading control. Right, BT-40 cells were infected with control virus (shControl), or two shPTEN lentiviruses. Cells infected with lentivirus were subjected to 2 days’ selection with 1 ug/mL puromycin, and then used for the subsequent assay. Cells were exposed to trametinib for 48 hours (n = 3 ± SD).
Rapamycin enhances trametinib antitumor activity
The antitumor activity of trametinib, rapamycin or the combination, was next evaluated in subcutaneous BT-40 xenografts. Trametinib (1 mg/kg) and rapamycin (5 mg/kg) or the combination, suppressed phosphorylation of S6 protein within 24 hours (Supplementary Fig. S3B). Trametinib caused partial regression of subcutaneous BT-40 PDX’s, followed by rapid tumor recurrence at the end of treatment. Median time to recurrence (>100 mm3) was at week 8 (Fig. 3A), significantly different from time to event for control tumors (P < 0.001). Rapamycin significantly inhibited growth of BT-40 xenografts (P < 0.001), although it did not cause tumor regressions. Of note, trametinib combined with rapamycin induced more rapid tumor regression, but importantly the median time for tumor regrowth was significantly delayed compared with trametinib alone, (median 10 weeks; P < 0.001), suggesting greater cytotoxic activity of the combination. In a pilot study, the effect of trametinib, rapamycin, or the combination was assessed in the intracranial BT-40 model. As with the subcutaneous BT40 model, trametinib effectively suppressed growth during treatment, whereas rapamycin had less effect on tumor growth. The combination delayed tumor regrowth compared with trametinib alone (Supplementary Fig. S4). In a second study with intracranial BT-40/Luc, where treatment was started when tumors were more advanced, the trametinib–rapamycin combination was effective in causing tumor regression (Fig. 3B, C), and was also more effective than selumetinib against intracranial BT-40/Luc (Supplementary Fig. S5).
Figure 3.
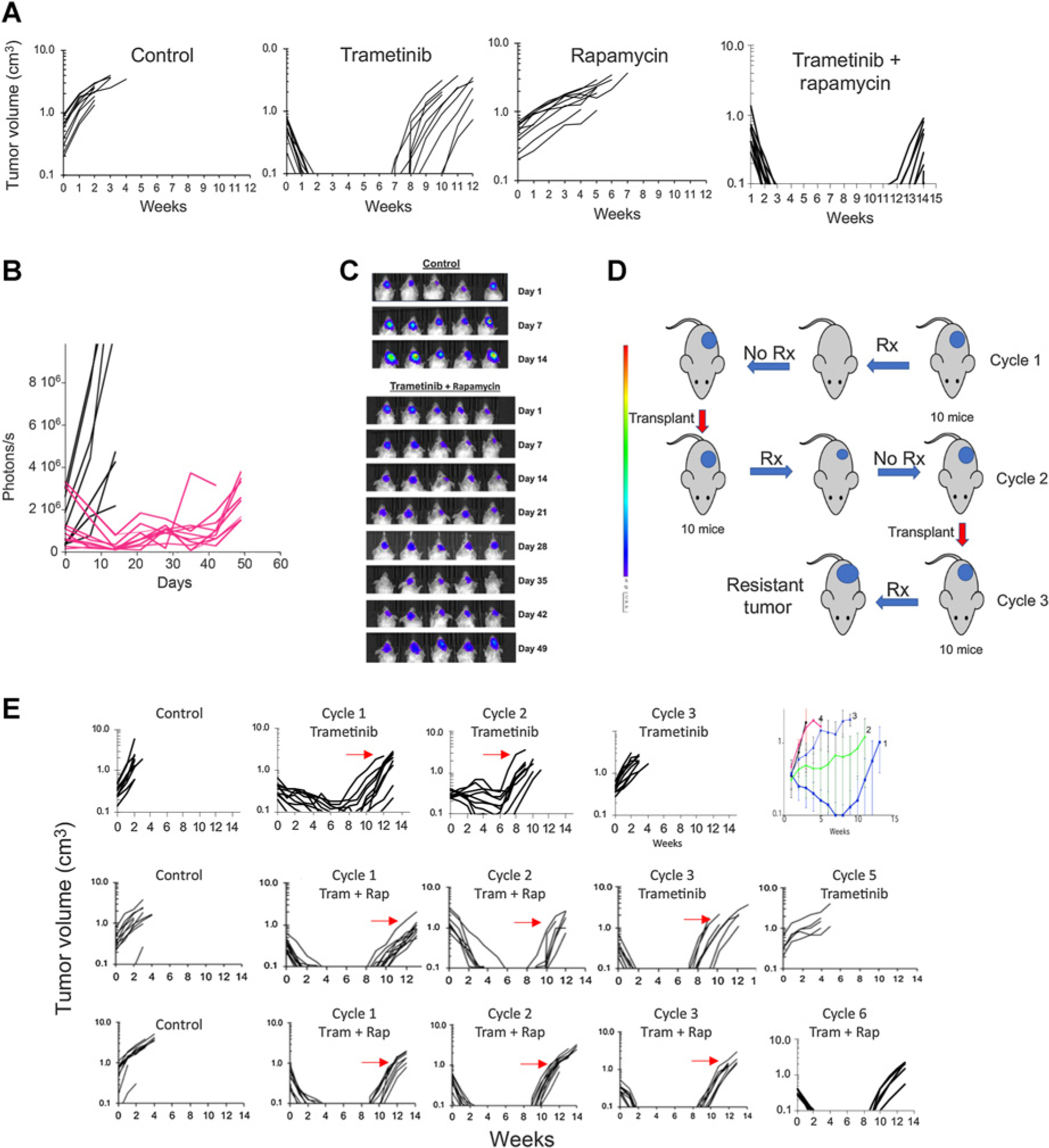
Sensitivity and development of acquired resistance in BT-40 BRAF-mutant xenografts to trametinib, rapamycin, or the combination. A, Responses of BT-40 PDX. Mice bearing subcutaneous BT-40 tumor were randomized to receive no treatment, trametinib (1 mg/kg daily × 42 days), rapamycin (5 mg/kg daily × 5 for 6 consecutive weeks), or the combination. Each curve shows the growth of an individual tumor. B, Activity of the trametinib–rapamycin combination on intracranial BT-40/Luc tumors. 105 BT-40/Luc cells expressing luciferase were implanted intracranially. Mice were randomized into control or treatment groups when the BLI value reached >1 × 105 photons/sec/cm3 and received combination treatment as in (A). Mice were imaged once weekly to ascertain tumor growth. Individual tumor growth curves based on BLI emission. Control (black), treated (Red) BLI values plotted against time for control and treatment groups. C, Luciferase images of cranial tumor growth in control and treatment groups. D, Schema for developing drug resistance in mice. E, Top, Responses of BT-40 xenografts to three cycles of trametinib treatment (1 mg/kg/day for 42 consecutive days). The arrows indicate the tumor that was transplanted into recipient mice for the subsequent cycle of treatment. Top right panel, mean tumor volume (±SD) for cycles 1 to 4 of treatment; Center panels, Responses of BT-40 xenografts for two cycles of trametinib + rapamycin (trametinib 1 mg/kg daily × 42, rapamycin 5 mg/kg daily × 5 for 6 consecutive weeks). After two cycles of treatment, single agent trametinib was administered for three further cycles; Bottom panels, Responses of BT-40 xenografts for six cycles of trametinib + rapamycin (trametinib 1 mg/kg daily × 42, rapamycin 5 mg/kg daily × 5 for 6 consecutive weeks). Each curve represents the growth of a single tumor. The arrows indicate the tumor that was transplanted into recipient mice for the subsequent cycle of treatment. s, second; Rx, treatment; Tram, trametinib; rap, rapamycin.
Development of trametinib resistance
In the recent phase II trial of selumetinib, patients with BRAFV600E tumors had worse outcomes than those with KIAA1549::BRAF-driven tumors, with progression on treatment or rapid progression when treatment was reduced, interrupted, or discontinued at the end of 2 years (24). Thus, although MEK inhibition causes tumor shrinkage, it is not curative, and development of resistance is a significant problem. We therefore investigated whether combination treatment with trametinib and rapamycin could retard or prevent emergence of resistance. To develop trametinib resistance, mice bearing subcutaneous BT-40 xenografts were treated with drug(s) for 42 days, and the first tumor to reach four-fold the volume at initiation of treatment (i.e., the least responsive BT-40 tumor) was transplanted into recipient mice and the treatment repeated (schema Fig. 3D). This treatment and process was repeated until tumors were resistant to trametinib. We defined resistance as more than 25% tumor volume increase while on treatment. Tumors progressed on treatment by Cycle 3 (Fig. 3E, upper row). The rate at which resistance to trametinib developed was consistent in four independent experiments (Fig. 3E right panel; Supplementary Fig. S6). To determine whether combination treatment with rapamycin could delay emergence of trametinib resistance in the BT-40 PDX model we examined the rate at which resistance emerged when trametinib was combined with rapamycin. Two experimental designs were used; in the first study, mice were treated with two cycles of trametinib–rapamycin combination therapy. After cycle 2 transplanted tumors were assessed for their sensitivity to trametinib administered as a single agent. As shown in Fig. 3E (center panels), the response on cycle 3 to trametinib alone was similar to that of drug-naïve BT-40 (see Fig. 3A, trametinib), suggesting that rapamycin had blocked emergence of trametinib resistance. However, two additional cycles of trametinib monotherapy resulted in tumors becoming resistant (Fig. 3E, center panels cycle 5 trametinib).
In the second study, mice were treated with the trametinb–rapamycin combination for six cycles of treatment (Fig. 3B, lower panels). On cycle 6 of combination treatment, BT-40 xenografts remained as sensitive to the combination as were naïve BT-40 xenografts on treatment cycle 1 (P = 0.4538, Supplementary Fig. S7). To address whether addition of rapamycin could resensitize tumors with acquired resistance to trametinib, the combination was tested in subcutaneous BT-40 xenografts that were selected for resistance to single-agent trametinib. The combination slowed tumor progression, but did not induce consistent tumor regressions (Supplementary Fig. S8A). In contrast, a similar study using the combination of dabrafenib (BRAF inhibitor) with trametinib showed that BT-40 tumors progressed during combination treatment by cycle 5 (Supplementary Fig. S8B). Thus, the combination delayed emergence of resistance compared with trametinib, consistent with data from melanoma clinical trials (40).
NCH-MN-1 and IC-3635 subcutaneous PDX models are also highly responsive to trametinib, and like BT-40 xenografts, tumors recur rapidly at the end of treatment. As shown in Fig. 4A, trametinib resistance was acquired rapidly as NCH-MN-1 PDXs showed progressive disease on trametinib treatment on treatment cycle 3. Even by cycle 2 of trametinib treatment, event-free survival (EFS) had significantly shortened (EFS days 71.35 vs. 38.46, for cycle 1 compared with cycle 2, P = 0.0011). In contrast, tumors treated for five cycles of trametinib–rapamycin remained sensitive to treatment with all tumors having at least 50% volume regression [1 complete response (CR)/4 partial response (PR)]. While resistance to trametinib–rapamycin develops more slowly than with trametinib alone, on cycle 5 of trametinib–rapamycin treatment all tumors had at least partial responses although EFS was reduced compared with cycle 1 (112 ± 11 days cycle 1, 66 ± 6 days cycle 5, P = 0.0001; Fig. 4A). NCH-MN-1 tumors were resistant on cycle 6 of trametinib–rapamycin treatment.
Figure 4.
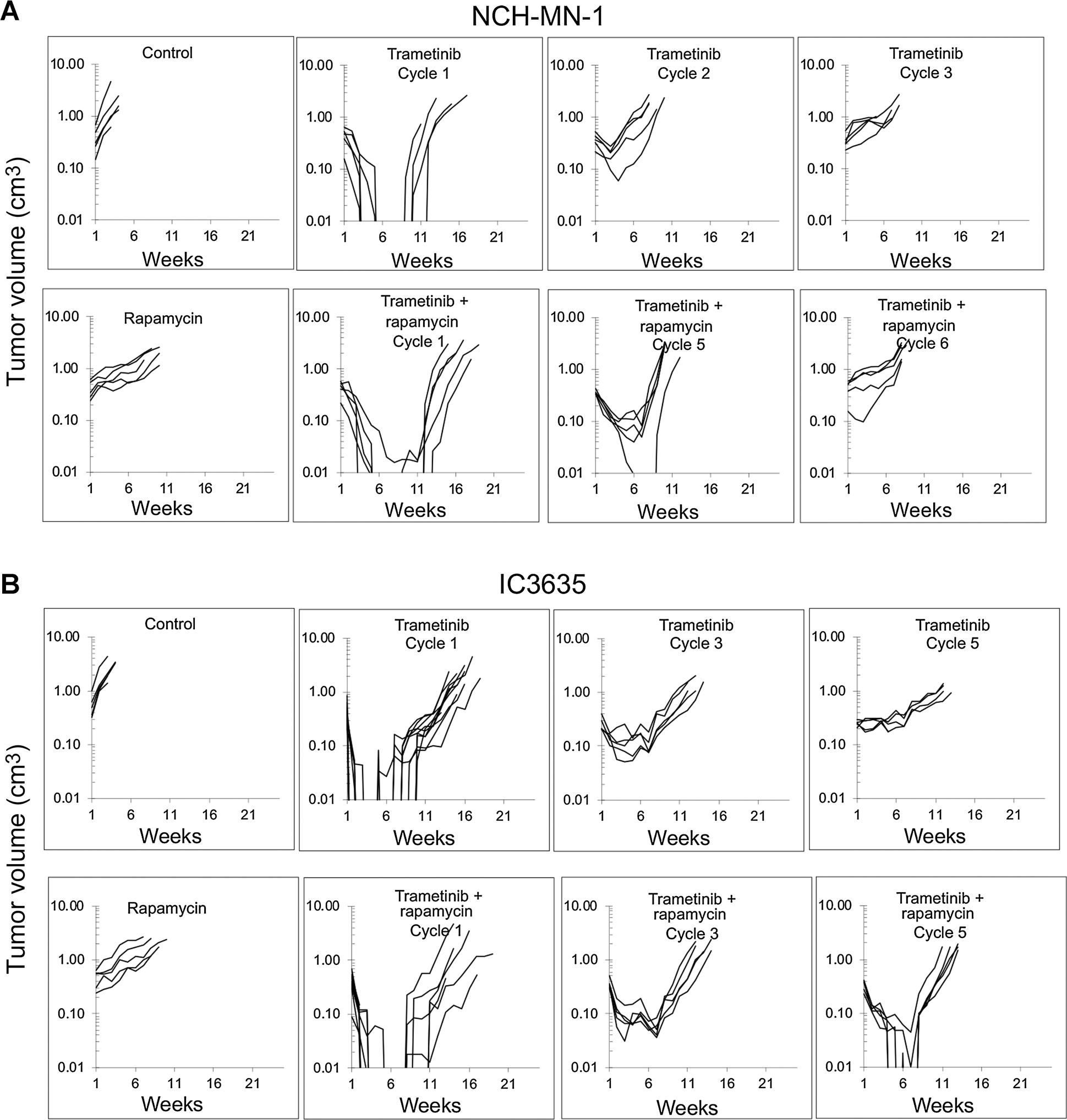
Rapamycin retards development of trametinib resistance in the NCH-MN-1 and IC-3635 PDX models. A, Growth of control NCH-MN-1 and responses of xenografts to treatment with trametinib (1 mg/kg daily × 42 days) on cycles 1 to 3, (top); development of resistance to combination treatment with trametinib-rapamycin (bottom). Mice received trametinib 1 mg/kg daily × 42 days and rapamycin 5 mg/kg daily × 5 for 6 consecutive weeks for five cycles. B, Growth of control IC-3635 and responses of xenografts to treatment with trametinib (1 mg/kg daily × 42 days) on cycles 1 to 5, (top); development of resistance to combination treatment with trametinib-rapamycin (bottom). Mice received trametinib 1 mg/kg daily × 42 days and rapamycin 5 mg/kg daily × 5 for 6 consecutive weeks for five cycles.
For subcutaneous IC-3635 xenografts, development of resistance to trametinib as a single agent took five cycles of treatment, whereas on cycle 5 of trametinib–rapamycin combination treatment tumors were still volume responsive (3 CR/2 PR), although the EFS had shortened from 103.7 ± 6.6 days on cycle 1 to 76.5 ± 4.0 days on cycle 5 (P < 0.0001) of trametinib–rapamycin treatment (Fig. 4B). Thus, rapamycin clearly retards development of trametinib resistance in several PDX models. Hence, the ability of rapamycin to delay or prevent emergence of trametinib resistance seems to be a more general phenomenon.
Pharmacodynamic changes associated with trametinib resistance
Trametinib resistance emerged within 3 to 5 cycles of single-agent treatment in all three subcutaneous PDX models. In contrast, resistance to the combination of trametinib with rapamycin emerged after six cycles of combination treatment in NCH-MN-1 and to a lesser extent in IC3635 PDX’s but not in the BT-40 PDX model. It was therefore of interest to determine how trametinib resistant lines compared with parental lines or after several cycles of combination treatment where tumors remained sensitive.
BT-40 xenografts and primary cultures
Trametinib had essentially similar effects on pERK, pS6, and p4E-BP1 in both parental (sensitive) and trametinib-resistant BT-40 xenografts (Supplementary Fig. S9A), suggesting that resistance was not directly related to MEK inactivation. To determine whether resistance was tumor-cell–intrinsic, cells were isolated from parental BT-40 xenografts, or from two BT-40TramR (trametinib-resistant) sublines where resistance had been developed in independent experiments (designated BT40TramR-A1 and BT40TramR-A3) that had been treated with three cycles of trametinib. In vitro, cells isolated from trametinib resistant xenografts were resistant to trametinib compared with cells isolated from parental BT-40 xenografts indicating that resistance was cell intrinsic (Supplementary Fig. S9B). Trametinib equally inhibited pERK in parental and trametinib resistant cells freshly isolated from xenografts (Supplementary Fig. S9C). Of note, while MEK inhibition was similar, apoptosis, as determined by detection of cleaved PARP1, was observed only in BT-40 parental cells and not in cells isolated from the trametinib-resistant xenografts. Addition of rapamycin to trametinib did not induce PARP cleavage in cells isolated from trametinib-resistant PDX tissue.
Trametinib resistance is associated with altered apoptotic priming threshold.
As BT-40 trametinib-resistant cells failed to show markers of apoptosis, we used a BH3 profiling assay to determine whether cells from resistant tumors had increased resistance to apoptosis (41). Tumor cells freshly isolated from parental BT-40 xenografts or those isolated from trametinib-resistant tumors were exposed to increasing concentrations of Bim BH3 peptide and the loss of mitochondrial potential (Δψ) was determined by decrease in JC1 fluorescence. BT-40 cells were highly sensitive to Bim BH3 peptide, with 50% loss of membrane potential at approximately 0.31 μmol/L BH3 peptide whereas a similar level of depolarization required more than 1.25 μmol/L BH3 peptide in BT-40 trametinib-resistant cells (Fig. 5A). Cells from sensitive or trametinib-resistant tumors were equally sensitive to the uncoupling agent FCCP. In contrast to cells obtained from trametinib-resistant xenografts, cells isolated from BT-40 tumors treated with three cycles of trametinib–rapamycin (BT40Trap), remained equally as sensitive to the Bim BH3 peptide as did cells isolated from naïve BT-40 xenografts (Fig. 5B). To further examine the difference in apoptotic threshold, expression of pro- and antiapoptotic proteins was assessed. In BT-40 trametinib-resistant tumors, Bax levels were significantly reduced compared with levels in BT-40 xenografts whereas levels of Bax were less reduced in tumors after four cycles of trametinib–rapamycin treatment (Fig. 5C). In contrast levels of Bim increased in trametinib—rapamycin–treated xenografts compared with parental BT-40 or the trametinib-resistant derivative (Fig. 5C). BCL-xL levels were not different between parental BT-40 and BT-40 trametinib-resistant xenografts, thus the major difference associated with trametinib resistance was a statistically significant change in the ratio of proapoptotic factors Bax or Bim to the prosurvival factor Bcl-xL (Fig. 5D).
Figure 5.
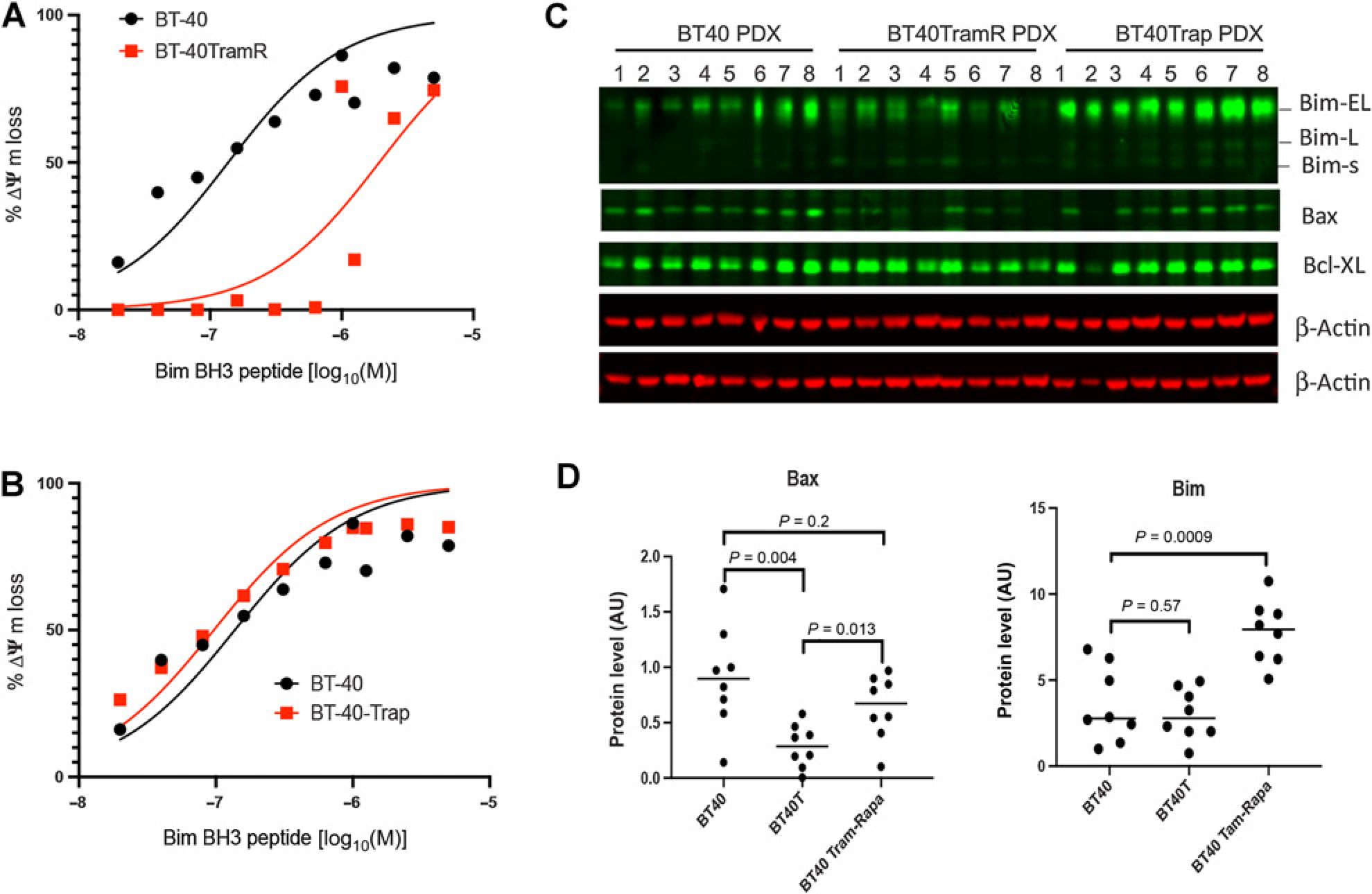
Trametinib resistance is associated with increased apoptotic threshold. A, Sensitivity of freshly isolated BT-40 or BT-40TramR (trametinib-resistant. Eight cycles of treatment in mice) and BT-40 Trap (trametinib–rapamycin—treated; 5 cycles in mice) cells to mitochondrial loss of membrane potential (Δψ) with increasing Bim BH3 peptide concentration; FCCP was used as a control to measure complete loss of membrane potential. ED50: BT-40 0.14 μmol/L [95% confidence interval (CI), 0.08–0.24 μmol/L]; BT-40TramR 1.94 μmol/L (95% CI, 0.91–4.5 μmol/L); BT-40Trap 0.1 μmol/L (95% CI, 0.04–0.15 μmol/L). B, Sensitivity of freshly isolated BT-40 or BT-40Trap cells (three cycles of trametinib + rapamycin in mice) to mitochondrial loss of membrane potential (Δψ) with increasing Bim BH3 peptide concentration; FCCP was used as a control to measure complete loss of membrane potential. C, Western blot for 8 BT-40 tumors and 8 trametinib-resistant PDX (TramR) and 8 trametinib—rapamycin–treated tumors (Trap) for Bax, Bim, and BCLXL; D, Quantitation of Bax and Bim levels from (C).
Downregulation of apoptotic threshold by inhibition of Bcl-xL overcomes the resistance to trametinib
As BCL-xL level was abundant in BT40 and derived tumors (Fig. 5C), we therefore determined whether in trametinib-resistant BT-40 cells survival was dependent on BCL-xL. Knockdown of BCL-xL resulted in increased cleaved PARP, indicating apoptosis, and significant loss of viability in both parental and trametinib-resistant cells (Supplementary Fig. S10A–B). To determine if inhibition of BCL-xL overcomes the resistance to trametinib, we combined suboptimal concentrations of navitoclax that did not induce apoptosis alone with trametinib treatment. Although the viability of BT40TramR cells was not significantly affected by either suboptimal concentration of navitoclax or trametinib alone the combination led to significant decrease of cell viability. In contrast, the combination of trametinib with ABT-199, a specific BCL-2 inhibitor, did not have any enhanced effects on cell killing, indicating BCL-xL was the target protein, which was consistent with the knockdown experiments (Supplementary Fig. S10C).
NCH-MN-1 xenografts and primary cultures
We initially examined MAPK signaling in NCH-MN-1 xenografts that showed progressive disease during cycle 3 of trametinib treatment. As shown in Fig. 6A, pERK was only modestly inhibited in trametinib-resistant NCH-MN-1(NCH-MN-1TramR) xenografts even after treatment daily with trametinib for 5 days. To understand the mechanism of the resistance, primary tumor cells were isolated from xenograft and cultured for in vitro analysis. NCH-MN-1 cells isolated from parental xenografts were sensitive to trametinib, whereas cells from the NCH-MN-1TramR xenograft proliferated slowly in culture and the proliferation was stimulated by trametinib (Supplementary Fig. S11A). In vitro assays of the primary cells showed that MAPK signaling in cells from the NCH-MN-1TramR xenograft (trametinib cycle 3) were resistant to trametinib inhibition relative to parental NCH-MN-1 cells, or cells derived after three cycles of combination treatment (Fig. 6B). Low concentration trametinib (5 nmol/L) combined with a pan-Raf inhibitor completely suppressed ERK1/2 phosphorylation in parental NCH-MN-1 cells, but had modest inhibition on ERK1/2 phosphorylation in NCH-MN-1TramR cells (Fig. 6C; Supplementary Fig. S11B), suggesting the resistance to trametinib may not be from upstream feedback activation of BRAF isoforms. To check if MEK1/2 contribute to the resistance per se, MEK-1 and -2 were knocked down using siRNA in primary cells. In parental NCH-MN-1 cells MEK-1 and -2 knockdown reduced phosphorylation of ERK1/2 dramatically. In contrast, knockdown of MEK-1 and -2 in NCH-MN-1TramR cells did not change ERK1/2 phosphorylation, indicating that maintained MAPK signaling was due to maintained activation of ERK1/2 in the absence of upstream signals, suggesting decreased inactivation of ERK1/2 (Supplementary Fig. S11B–C). ERK1/2 specific dual specificity phosphatase 6 (DUSP6) protein level was indeed decreased only in the cells from the trametinib-resistant model (Fig. 6D). Consistent with the in vitro results, trametinib resistance in NCH-MN-1TramR xenografts corresponded to decreased levels of DUSP6 after two or three cycles of treatment (Fig. 6B). Furthermore, knockdown of DUSP6 in NCH-MN-1 parental cells slowed proliferation and conferred trametinib resistance (Supplementary Fig. S11D–E). In stem cell medium, trametinib treatment caused dramatic cell viability loss of parental NCH-MN-1 cells, but had significantly less effect on the viability of cells from cycle 3 trametinib treatment (Fig. 6E). Results from PDX experiments and in vitro experiments suggest decreased feedback inhibition of the MAPK signaling pathway by downregulation of DUSP6 contributes to trametinib resistance in NCH-MN-1 tumor (42).
Figure 6.
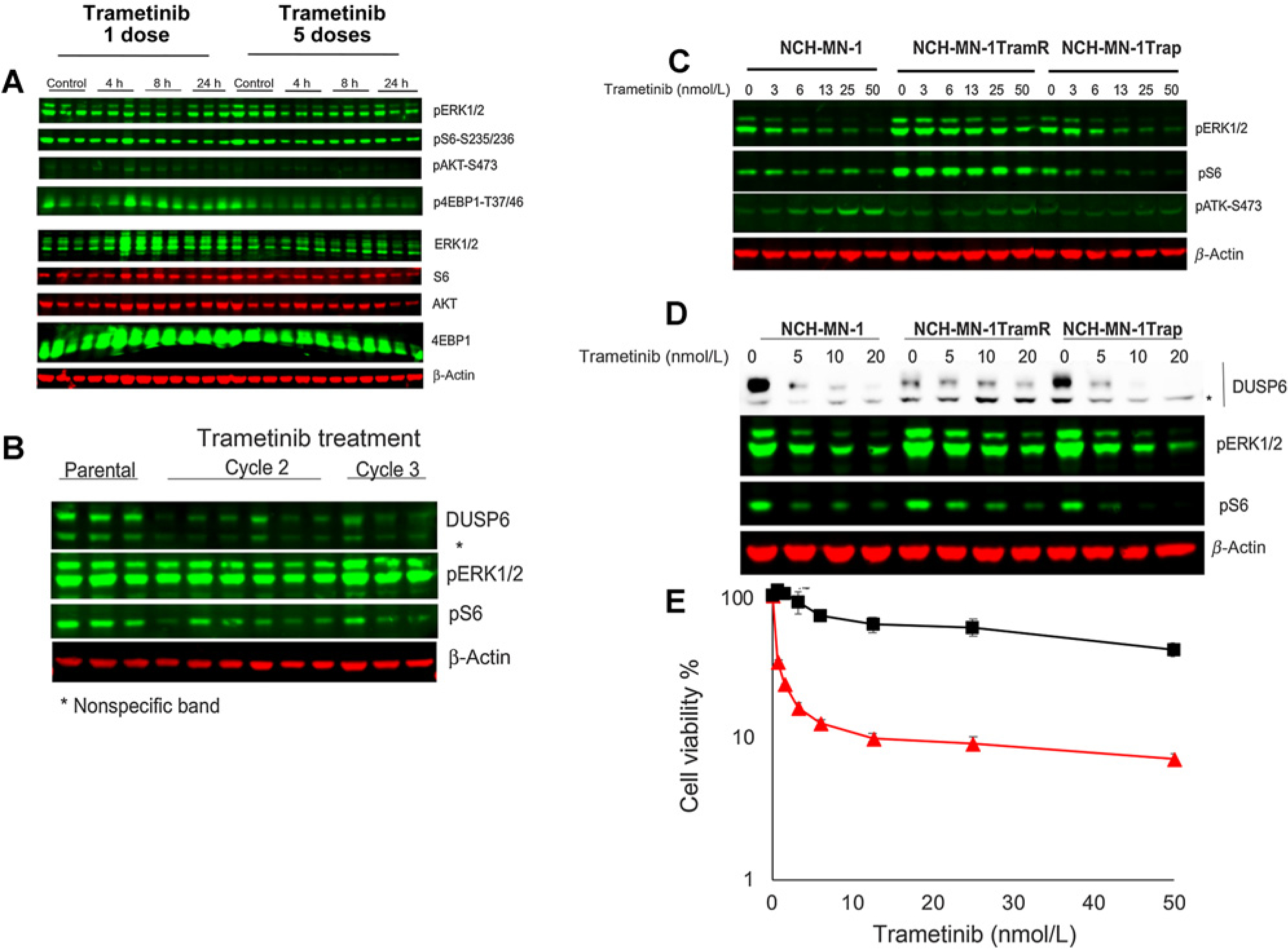
Changes in NCH-MN-1 xenografts with acquired resistance to trametinib. A, Mice bearing NCH-MN-1 tumors resistant to trametinib (cycle 3) were treated with trametinib (1 mg/kg daily for up to 5 days). B, Levels of DUSP6 in parental or trametinib resistant MN-1 xenografts. C, Cells from parental NCH-MN-1 xenografts, trametinib-resistant xenografts (TramR, cycle 3), or cells from NCH-MN-1 tumors treated for three cycles of trametinib—rapamycin (Trap). Trametinib-resistant cells show enhanced MAPK signaling. D, Response to increasing concentrations of trametinib in parental, trametinib-resistant, and cells isolated from tumors after three cycles of combination treatment. E, Response to trametinib of NCH-MN-1 cells from parental PDX tumor ( red line) and trametinib-resistant xenografts (■ black line). Cells cultured in stem cell medium supplemented with FGF and rEGF were exposed to indicated concentration of trametinib for 72 hours. Cell proliferation was measured by Alamar Blue. h, hours.
red line) and trametinib-resistant xenografts (■ black line). Cells cultured in stem cell medium supplemented with FGF and rEGF were exposed to indicated concentration of trametinib for 72 hours. Cell proliferation was measured by Alamar Blue. h, hours.
IC-3635 xenografts and primary cultures
Early-stage cultures from parental subcutaneous IC-3635 xenografts or after four cycles of trametinib or trametinib–rapamycin were next examined. When cells were grown in stem cell medium (serum-free) trametinib suppressed MAPK and TORC1 signaling in each cell type (Fig. 7A). However, when cells were grown in complete medium (RPMI1640 + 10%FBS) trametinib inhibited pERK1/2 in parental cells, and to some extent in cells derived from trametinb—rapamycin–treated tumors, but had less inhibitory effect on cells from trametinib-resistant xenografts. These data could indicate activation of MAPK signaling through increased upstream signaling by activating other RAF isoforms in trametinib-resistant IC-3635 cells. To test whether increased upstream signaling in trametinib resistant cells contributed to reduced activity of trametinib, IC-3635 trametinib-resistant cells were treated with trametinib with or without inhibitors of RTKs (Fig. 7B). Treatment of trametinib-resistant IC-3635 cells with a panRAF inhibitor with minimal paradoxical activation of BRAF [LY3009120 (43), or CCT196969, a panRAF/SRC-LCK/p38MAPK inhibitor (44)] suppressed pERK, as did a panFGFR inhibitor (LY2874455), and high concentrations of the multikinase inhibitor pazotinib. Inhibitors of EGFR (Erlotinib) or BCR-ABL had no effect on pERK levels when cells were maintained in serum containing medium. Thus, maintenance of pERK in this model seemed to be due to increased upstream signaling. We next determined whether combining panRAF inhibition with trametinib blocked proliferation of trametinib-resistant IC-3635 cells (Fig. 7C). IC-3635TramR cells were resistant to trametinib in vitro, but were sensitive to the combination of a panRAF inhibitor (LY3009120) with trametinib, indicating the role of RAF signaling in trametinib resistance for this line.
Figure 7.
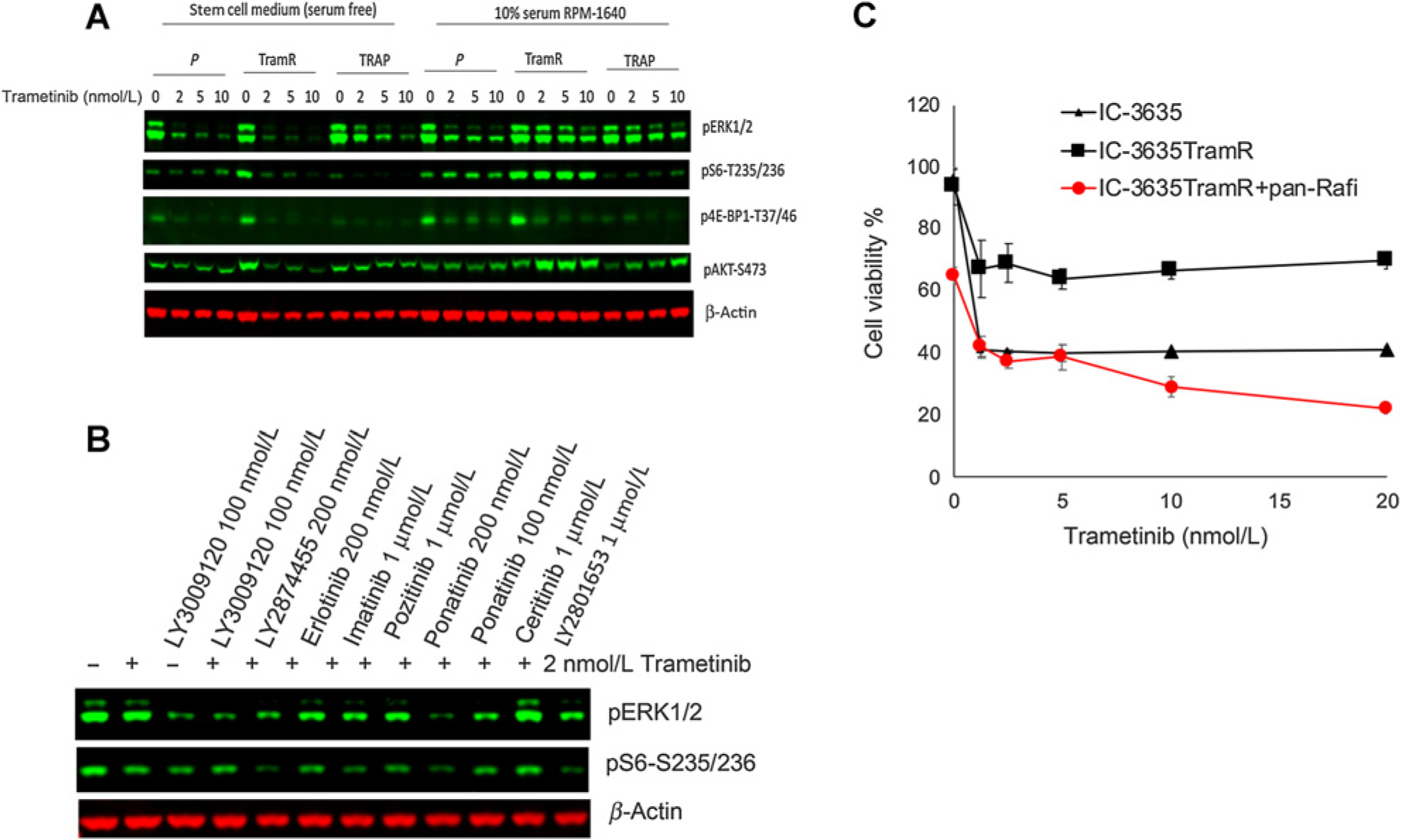
Response of IC-3635 cells derived from trametinib-resistant xenografts. A, Cells were isolated from subcutaneous IC-3635 xenografts [cycle 4 trametinib (TramR) or cycle 4 combination treatment (Trap) treatment] and grown in serum-free conditions (left) or serum supplemented medium. In serum-free medium inhibition of MAPK and TORC1 signaling was similar in all derivatives, whereas in serum containing conditions of growth, inhibition of MAPK and TORC1 signaling was markedly attenuated only in cells derived from parental IC-3635 tumors and from combination treated tumors (Trap). B, IC-3635 trametinib-resistant cells were incubated with trametinib (2 nmol/L) in the absence or presence of receptor tyrosine kinase inhibitors. Phosphorylation of ERK1/2 and S6 were determined at 24 hours. C, Response of parental (▲) or IC-3635TramR (■ trametinib resistant) cells to trametinib without or with the panRAF inhibitor LY3009120 ( ). Cells were cultured in stem cell medium supplemented with FGF and EGF and exposed to the indicated concentration of trametinib with or without combining panRaf inhibitor for 72 hours. Cell viability was measured by Alamar Blue, untreated cells were set as 100%.
). Cells were cultured in stem cell medium supplemented with FGF and EGF and exposed to the indicated concentration of trametinib with or without combining panRaf inhibitor for 72 hours. Cell viability was measured by Alamar Blue, untreated cells were set as 100%.
Discussion
Both MEK and BRAF inhibitors have shown promise for treatment of childhood BRAF-mutant brain tumors, and potentially will provide alternatives to cytotoxic chemotherapy and radiation treatment. However, despite tumor regression or stable disease in response to treatment, in some patients predominantly those where BRAFV600E is the oncogene, resistance occurs either on therapy (acquired resistance) or tumors progress rapidly once therapy is completed or dose-reduced (at 2 years in the recent phase II clinical trial; ref. 25). Thus, while use of MEKis has altered the standard of clinical care for these patients, additional combinations will be required to enhance the cure rate, particularly for tumors harboring BRAFV600E mutations (24)
Here our goal was to focus on pediatric brain tumors with BRAFV600E mutations in order to understand the basis for sensitivity to MEK inhibition, and to build upon targeting MEK to prevent emergence of resistance. We focused on MEKis, rather than combinations with BRAF inhibitors, as LGG with KIAA1549::BRAF fusions do not respond to BRAF inhibitors. Furthermore, no human preclinical models of fusion-driven LGG are available as these cells do not proliferate in culture or in mice. Potentially, results obtained with the current models may be applicable to treatment of fusion-driven LGG.
Our results suggest that intrinsic sensitivity to MEK inhibition in three BRAFV600E-driven pediatric brain tumor PDX models, is through dual inhibition of MAPK and TORC1 signaling that leads to apoptosis, similar to findings in BRAFV600E melanoma (45). In the melanoma study by Corcoran and colleagues (45) it was concluded that MAPK signaling controls TORC1 activity, although the mechanism was not elucidated. Our results support the idea that mutant BRAF signaling in these brain tumor models controls TORC1 activity through regulating the activity of the TSC complex. Downregulation of TSC2, or PTEN, inhibition of TSC2 activity through expression of TSC2ΔGAP, or expression of activated AKT1/3 all conferred resistance to trametinib. Activation of Akt signaling has previously been reported to confer MEKi resistance (46). Phosphorylation of TSC2 (tuberin) by Akt or ERK1/2 leads to dissociation of the TSC, and its inactivation, leading to increased TORC1 signaling (47). TSC2 is phosphorylated by ERK1/2 (Serines 540//664) and by Akt (Serines 939/981/1132 and Threonine1462). We examined phosphorylation of S664 and T1462 as representative of ERK and Akt induced phosphorylation, respectively. In the three pediatric PDX models sensitive to trametinib, phosphorylation of T1462 was not detected. However, phosphorylation at S664 was detected in each line, and was decreased by trametinib treatment leading to inhibition of 4E-BP1 and S6 phosphorylation. These results are consistent with MAPK-signaling–induced phosphorylation and inactivation of the TSC complex.
This explains also, why a MEKi suppresses both MAPK and TORC1 signaling, leading to tumor regression in pediatric LGG. In the adult GBM cell lines examined, inactivation of the TSC complex seems to be regulated partially through PI3K/Akt signaling, explaining why inhibition of the MAPK pathway has a lesser effect on reducing TORC1 signaling. Several reports have suggested that the PI3K/mTOR pathway is activated in pediatric glioma, based on the observation that TORC1 substrates S6 and 4E-BP1 are phosphorylated in 50% of tumors and may play a role in the pathogenesis of these tumors (48). However, unlike primary adult glioblastoma (49), mutations in PTEN have not been reported and activating mutations in receptor tyrosine kinases, such as FGFR1 are rare (12, 50). The data presented here suggest that in the three pediatric PDX models, TORC1 is controlled by BRAF signaling, through suppression of TSC2, rather than activation of the PI3K/Akt pathway.
While each PDX model was initially very sensitive to trametinib, emergence of trametinib resistance was relatively rapid. In BT-40 and MN-1 PDX models, tumors demonstrated progression by treatment cycle 3 and IC-3635 tumors were significantly less sensitive on cycle 3 and showed progressive disease on cycle 5 of trametinib treatment. Mechanisms responsible for acquired trametinib resistance in the models differed. When PDX cells were cultured in vitro, each demonstrated resistance to trametinib relative to cells culltured from the respective parental xenograft indicating tumor-intrinsic resistance. In vitro BT-40 cells demonstrated an increase in apoptotic threshold. There was a clear shift in the concentration of Bim BH3 peptide required to cause mitochondrial depolarization in trametinib-resistant cells. Of note, freshly isolated cells from BT-40 tumors that regrew after cycle 3 of trametinib–rapamycin treatment showed similar sensitivity to parental BT-40 cells in this assay. Together, these results indicate that in this model resistance to trametinib is mediated, at least in part, by an increase in apoptotic threshold. For both NCH-MN-1 and IC-3635 cells acquired resistance to trametinib was associated with decreased inhibition of MAPK signaling by trametinib. In NCH-MN-1 xenografts there was a decrease in the levels of dual specificity phosphatase DUSP6, thus preventing suppression of MAPK signaling. Decreased DUSPs in MEKi-resistant melanoma has been reported previously (51, 52). For IC-3635 the mechanism of trametinib resistance appears to be through increased upstream signaling, and involves activation of RAF isoforms, thus overcoming MEK inhibition, similar to the mechanism of intrinsic resistance to trametinib in RAS-mutant embryonal rhabdomyosarcoma (53), indicating that inhibition of ERK1/2 activity leads to feedback activation of BRAF isoforms. The different mechanisms of resistance to trametinib in these PDX models is consistent with reports indicating that resistance is tumor line-specific and drug-specific (54).
In contrast to the rapid emergence of resistance in mice treated with trametinib monotherapy, resistance to the combination of trametinib-rapamycin was delayed or prevented. BT-40 tumors remained equally as sensitive to trametinib-rapamycin on cycle 6 as were parental tumors. Similarly, IC-3635 xenografts remained sensitive on cycle 5 of combination treatment showing partial or complete tumor regressions, although the EFS was significantly shortentened compared with cycle 1 of trametinib–rapamycin. NCH-MN-1 xenografts remained partially sensitive to the combination on cycle 5, but were resistant on cycle 6 of combination treatment. The mechanism for resistance to the combination of trametinib–rapamycin remains to be elucidated. Thus, combining low-dose intermittent rapamycin with trametinib delayed emergence of trametinib resistance. In mice bearing BT-40 tumors that received two cycles of trametinib-rapamycin, the response to cycle 2 was the same as on cycle 1 (i.e., no indication of resistance). For cycle 3, mice received only trametinib, and the tumor response was similar to that of drug-naïve BT-40 treated with single agent trametinib and it took a further three cycles of single agent trametinib therapy to induce resistance, suggesting that no resistance to trametinib was developed during the two cycles of combination therapy. In contrast, development of resistance to trametinib as a single agent by cycle 3 was consistent in four separate experiments.
The precise mechanism by which rapamycin prevents emergence of trametinib resistance needs further elucidation. However, if as postulated sensitivity to trametinib requires dual MAPK/TORC1 inhibition (45), then a failure to fully inhibit MAPK signaling (NCH-MN-1), or activation of TORC1 signaling through feedback activation of PI3K/AKT would potentially be blocked by rapamycin. How rapamycin blocks the trametinib-induced increase in apoptotic threshold needs further experimentation. However, levels of Bax were only decreased in trametinib but not trametinib—rapamycin–treated BT-40 xenografts, suggesting the combination prevented the reduction of Bax levels. More interestingly, after three cycles of trametinib-rapamycin treatment, levels of Bim were dramaticlly increased whereas they were not significantly changed in the trametinib resistant tumors relative to BT-40 parental tumors. Loss of the proapoptotic protein Bim has previously been reported as a mechanism for trametinib resistance (55).
In summary, these results suggest that the sensitivity of BRAF-mutant pediatric brain tumors to trametinib is due, in part, to MAPK-mediated regulation of TORC1 activity. In each of three pediatric PDX models, TORC1 appears to be regulated via BRAF/ERK-mediated suppression of TSC, hence inhibition of MEK suppresses both MAPK and TORC1 pathways. Our results suggest also, that resistance to trametinib may result from a change in apoptotic threshold, due in part to decreased levels of Bax, or increased MAPK signaling not abrogated by MEK inhibition. Furthermore, rapamycin can retard emergence of resistance (NCH-MN-1, IC3635) or prevent emergence of resistance (BT-40) at least over six cycle of therapy. The current data would suggest that administration of low-dose, intermittent rapamycin analogs may enhance activity of MEKis, and potentially prevent development of drug resistance. These results may be equally applicable to LGG driven by KIAA1549::BRAF fusions. While combined trametinib–rapamycin is tolerated in mice, combining rapamycin with other inhibitors of PI3K/Akt signaling has necessitated dose reductions (56) whether this combination will be tolerated in the context of pediatric brain tumors will have to be assessed clinically.
Supplementary Material
Translational Relevance.
Approximately 1,400 new cases of children with BRAF-activated brain tumors are diagnosed annually in the United States. Inhibitors of MEK1/2 have shown promising antitumor activity in patients with activation of BRAF through tandem duplication (KIAA1549::BRAF) or point mutations that activate BRAF. MEK inhibitors (MEKi) are equally active in both subtypes of glioma, whereas tumors with tandem duplication are not sensitive to BRAF inhibitors. Unfortunately, there are no preclinical PDX models representing BRAF activation due to tandem duplication of KIAA1549::BRAF. To improve on MEKis and potentially develop more effective therapy that would impact tumors driven by the KIAA1549::BRAF duplication, we have characterized the sensitivity to the MEKi, trametinib of three BRAFV600E pediatric brain tumor PDX models. Specifically, we have identified why these PDX models are responsive to treatment and have identified mechanisms for resistance. Based on these findings we have developed a treatment that retards or prevents emergence of trametinib resistance in mice. Potentially, these results could build upon MAPK inhibitor therapy and translate into more effective control of KIAA1549::BRAF driven glioma that represents approximately 90 of low-grade glioma (LGG) as well as tumors driven by BRAF point mutations.
Acknowledgments
This article was supported by NCI (grant nos. CA169368, CA165995, CA199297, CA246807, CA258381, and P30 CA054174), and the Cancer Prevention and Research Institute of Texas (CPRIT; grant no. RP160716).
Footnotes
Authors’ Disclosures
No disclosures were reported.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Note
Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
References
- 1.Gnekow AK, Falkenstein F, von Hornstein S, Zwiener I, Berkefeld S, Bison B, et al. Long-term follow-up of the multicenter, multidisciplinary treatment study HIT-LGG-1996 for low-grade glioma in children and adolescents of the german speaking society of pediatric oncology and hematology. Neuro Oncol 2012;14:1265–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merchant TE, Conklin HM, Wu S, Lustig RH, Xiong X. Late effects of conformal radiation therapy for pediatric patients with low-grade glioma: prospective evaluation of cognitive, endocrine, and hearing deficits. J Clin Oncol 2009;27:3691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ater JL, Zhou T, Holmes E, Mazewski CM, Booth TN, Freyer DR, et al. Randomized study of two chemotherapy regimens for treatment of low-grade glioma in young children: a report from the children’s oncology group. J Clin Oncol 2012;30:2641–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ris MD, Beebe DW, Armstrong FD, Fontanesi J, Holmes E, Sanford RA, et al. Cognitive and adaptive outcome in extracerebellar low-grade brain tumors in children: a report from the children’s oncology group. J Clin Oncol 2008;26:4765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenberger BA, Pulsifer MB, Ebb DH, MacDonald SM, Jones RM, Butler WE, et al. Clinical outcomes and late endocrine, neurocognitive, and visual profiles of proton radiation for pediatric low-grade gliomas. Int J Radiat Oncol Biol Phys 2014;89:1060–8. [DOI] [PubMed] [Google Scholar]
- 6.Perkins SM, Fei W, Mitra N, Shinohara ET. Late causes of death in children treated for CNS malignancies. J Neurooncol 2013;115:79–85. [DOI] [PubMed] [Google Scholar]
- 7.de Blank P, Bandopadhayay P, Haas-Kogan D, Fouladi M, Fangusaro J. Management of pediatric low-grade glioma. Curr Opin Pediatr 2019;31:21–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Upadhyaya SA, Ghazwani Y, Wu S, Broniscer A, Boop FA, Gajjar A, et al. Mortality in children with low-grade glioma or glioneuronal tumors: A single-institution study. Pediatr Blood Cancer 2018;65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 2008;118:1739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sievert AJ, Jackson EM, Gai X, Hakonarson H, Judkins AR, Resnick AC, et al. Duplication of 7q34 in pediatric low-grade astrocytomas detected by high-density single-nucleotide polymorphism-based genotype arrays results in a novel BRAF fusion gene. Brain Pathol 2009;19:449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol 2011; 121:397–405. [DOI] [PubMed] [Google Scholar]
- 12.Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 2013;45:602–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostrom QT, Cioffi G, Gittleman H, Patil N, Waite K, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro Oncol 2019;21:v1–v100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ostrom QT, de Blank PM, Kruchko C, Petersen CM, Liao P, Finlay JL, et al. Alex’s lemonade stand foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol 2015;16 Suppl 10:x1–x36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 16.Forshew T, Tatevossian RG, Lawson AR, Ma J, Neale G, Ogunkolade BW, et al. Activation of the ERK/MAPK pathway: a signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol 2009;218:172–81. [DOI] [PubMed] [Google Scholar]
- 17.Banerjee A, Jakacki RI, Onar-Thomas A, Wu S, Nicolaides T, Young Poussaint T, et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low-grade glioma: a pediatric brain tumor consortium (PBTC) study. Neuro Oncol 2017;19:1135–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fangusaro J, Wu S, MacDonald S, Murphy E, Shaw D, Bartels U, et al. Phase II trial of response-based radiation therapy for patients with localized CNS nongerminomatous germ cell tumors: a children’s oncology group study. J Clin Oncol 2019;37:3283–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burger MC, Ronellenfitsch MW, Lorenz NI, Wagner M, Voss M, Capper D, et al. Dabrafenib in patients with recurrent, BRAF V600E mutated malignant glioma and leptomeningeal disease. Oncol Rep 2017;38:3291–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hargrave DR, Bouffet E, Tabori U, Broniscer A, Cohen KJ, Hansford JR, et al. Efficacy and safety of dabrafenib in pediatric patients with BRAF V600 mutation-positive relapsed or refractory low-grade glioma: results from a phase I/IIa study. Clin Cancer Res 2019;25:7303–11. [DOI] [PubMed] [Google Scholar]
- 21.Sievert AJ, Lang SS, Boucher KL, Madsen PJ, Slaunwhite E, Choudhari N, et al. Paradoxical activation and RAF inhibitor resistance of BRAF protein kinase fusions characterizing pediatric astrocytomas. Proc Natl Acad Sci U S A 2013;110:5957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolb EA, Gorlick R, Houghton PJ, Morton CL, Neale G, Keir ST, et al. Initial testing (stage 1) of AZD6244 (ARRY-142886) by the pediatric preclinical testing program. Pediatr Blood Cancer 2010;55:668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Studebaker A, Bondra K, Seum S, Shen C, Phelps DA, Chronowski C, et al. Inhibition of MEK confers hypersensitivity to X-radiation in the context of BRAF mutation in a model of childhood astrocytoma. Pediatr Blood Cancer 2015;62:1768–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouffet E Selumetinib in paediatric low-grade glioma: a new era? Lancet Oncol 2019;20:900–1. [DOI] [PubMed] [Google Scholar]
- 25.Fangusaro J, Onar-Thomas A, Young Poussaint T, Wu S, Ligon AH, Lindeman N, et al. Selumetinib in paediatric patients with BRAF-aberrant or neurofibromatosis type 1-associated recurrent, refractory, or progressive low-grade glioma: a multicentre, phase 2 trial. Lancet Oncol 2019;20:1011–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kogiso M, Qi L, Lindsay H, Huang Y, Zhao X, Liu Z, et al. Xenotransplantation of pediatric low grade gliomas confirms the enrichment of BRAF V600E mutation and preservation of CDKN2A deletion in a novel orthotopic xenograft mouse model of progressive pleomorphic xanthoastrocytoma. Oncotarget 2017;8:87455–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bid HK, Kibler A, Phelps DA, Manap S, Xiao L, Lin J, et al. Development, characterization, and reversal of acquired resistance to the MEK1 inhibitor selumetinib (AZD6244) in an in vivo model of childhood astrocytoma. Clin Cancer Res 2013;19:6716–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rokita JL, Rathi KS, Cardenas MF, Upton KA, Jayaseelan J, Cross KL, et al. Genomic profiling of childhood tumor patient-derived xenograft models to enable rational clinical trial design. Cell Rep 2019;29:1675–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006;1:2315–9. [DOI] [PubMed] [Google Scholar]
- 30.Yu PY, Gardner HL, Roberts R, Cam H, Hariharan S, Ren L, et al. Target specificity, in vivo pharmacokinetics, and efficacy of the putative STAT3 inhibitor LY5 in osteosarcoma, Ewing’s sarcoma, and rhabdomyosarcoma. PLoS One 2017;12:e0181885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phelps D, Bondra K, Seum S, Chronowski C, Leasure J, Kurmasheva RT, et al. Inhibition of MDM2 by RG7388 confers hypersensitivity to X-radiation in xenograft models of childhood sarcoma. Pediatr Blood Cancer 2015;62:1345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer 2007;49:928–40. [DOI] [PubMed] [Google Scholar]
- 33.Yoo JY, Haseley A, Bratasz A, Chiocca EA, Zhang J, Powell K, et al. Antitumor efficacy of 34.5ENVE: a transcriptionally retargeted and “Vstat120”-expressing oncolytic virus. Mol Ther 2012;20:287–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryan J, Letai A. BH3 profiling in whole cells by fluorimeter or FACS. Methods 2013;61:156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Gooijer MC, Zhang P, Weijer R, Buil LCM, Beijnen JH, van Tellingen O. The impact of P-glycoprotein and breast cancer resistance protein on the brain pharmacokinetics and pharmacodynamics of a panel of MEK inhibitors. Int J Cancer 2018;142:381–91. [DOI] [PubMed] [Google Scholar]
- 36.Leonowens C, Pendry C, Bauman J, Young GC, Ho M, Henriquez F, et al. Concomitant oral and intravenous pharmacokinetics of trametinib, a MEK inhibitor, in subjects with solid tumours. Br J Clin Pharmacol 2014;78:524–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 2002;10:151–62. [DOI] [PubMed] [Google Scholar]
- 38.Ballif BA, Roux PP, Gerber SA, MacKeigan JP, Blenis J, Gygi SP. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proc Natl Acad Sci U S A 2005;102:667–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005;121:179–93. [DOI] [PubMed] [Google Scholar]
- 40.Long GV, Flaherty KT, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, et al. Dabrafenib plus trametinib versus dabrafenib monotherapy in patients with metastatic BRAF V600E/K-mutant melanoma: long-term survival and safety analysis of a phase 3 study. Ann Oncol 2017;28:1631–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 2012;151:344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kun E, Tsang YTM, Ng CW, Gershenson DM, Wong KK. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat Rev 2021;92:102137. [DOI] [PubMed] [Google Scholar]
- 43.Vakana E, Pratt S, Blosser W, Dowless M, Simpson N, Yuan XJ, et al. LY3009120, a panRAF inhibitor, has significant anti-tumor activity in BRAF and KRAS mutant preclinical models of colorectal cancer. Oncotarget 2017;8:9251–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Girotti MR, Lopes F, Preece N, Niculescu-Duvaz D, Zambon A, Davies L, et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell 2015;27:85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Corcoran RB, Rothenberg SM, Hata AN, Faber AC, Piris A, Nazarian RM, et al. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Sci Transl Med 2013;5:196ra98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brighton HE, Angus SP, Bo T, Roques J, Tagliatela AC, Darr DB, et al. New mechanisms of resistance to MEK inhibitors in melanoma revealed by intravital imaging. Cancer Res 2018;78:542–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A 2002;99:13571–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gajjar A, Pfister SM, Taylor MD, Gilbertson RJ. Molecular insights into pediatric brain tumors have the potential to transform therapy. Clin Cancer Res 2014;20:5630–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Appin CL, Brat DJ. Molecular pathways in gliomagenesis and their relevance to neuropathologic diagnosis. Adv Anat Pathol 2015;22:50–8. [DOI] [PubMed] [Google Scholar]
- 50.Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 2013;45:927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hartman ML, Sztiller-Sikorska M, Gajos-Michniewicz A, Czyz M. Dissecting mechanisms of melanoma resistance to BRAF and MEK inhibitors revealed genetic and non-genetic patient- and drug-specific alterations and remarkable phenotypic plasticity. Cells 2020;9:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gupta A, Towers C, Willenbrock F, Brant R, Hodgson DR, Sharpe A, et al. Dual-specificity protein phosphatase DUSP4 regulates response to MEK inhibition in BRAF wild-type melanoma. Br J Cancer 2020;122:506–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia N, Del Pozo V, Yohe ME, Goodwin CM, Shackleford TJ, Wang L, et al. Vertical inhibition of the RAF-MEK-ERK cascade induces myogenic differentiation, apoptosis, and tumor regression in H/NRAS(Q61X) mutant rhabdomyosarcoma. Mol Cancer Ther 2022;21:170–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim SY, Menzies AM, Rizos H. Mechanisms and strategies to overcome resistance to molecularly targeted therapy for melanoma. Cancer 2017;123:2118–29. [DOI] [PubMed] [Google Scholar]
- 55.Sanchez JN, Wang T, Cohen MSBRAF, MEK Inhibitors.: Use and resistance in BRAF-mutated cancers. Drugs 2018;78:549–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tolcher AW, Bendell JC, Papadopoulos KP, Burris HA, 3rd, Patnaik A, Jones SF, et al. A phase IB trial of the oral MEK inhibitor trametinib (GSK1120212) in combination with everolimus in patients with advanced solid tumors. Ann Oncol 2015;26:58–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.