

Summary
During postnatal development the DNA methyltransferase DNMT3A deposits high levels of non-CG cytosine methylation in neurons. This methylation is critical for transcriptional regulation, and loss of this mark is implicated in DNMT3A-associated neurodevelopmental disorders (NDDs). Here we show in mice that genome topology and gene expression converge to shape histone H3 lysine 36 dimethylation (H3K36me2) profiles, which in turn recruit DNMT3A and pattern neuronal non-CG methylation. We show that NSD1, a H3K36 methyltransferase mutated in NDD, is required for patterning of megabase-scale H3K36me2 and non-CG methylation in neurons. We find that brain-specific deletion of NSD1 causes altered DNA methylation that overlaps with DNMT3A disorder models to drive convergent dysregulation of key neuronal genes that may underlie shared phenotypes in NSD1- and DNMT3A-associated NDD. Our findings indicate that H3K36me2 deposited by NSD1 is important for neuronal non-CG DNA methylation and suggest that the H3K36me2-DNMT3A-non-CG-methylation pathway is likely disrupted in NSD1-associated NDD.
Graphical Abstract
eTOC
Hamagami, Wu et al. find that megabase-scale Histone H3 lysine 36 dimethylation patterns target DNMT3A-mediated non-CG DNA methylation in neurons. They show NSD1 disruption results in altered histone methylation, DNA methylation and transcription that overlaps with DNMT3Adisorder models, uncovering molecular convergence between distinct genetic causes of neurodevelopmental disease.
Introduction
Methylation of cytosines (mC) in genomic DNA is an essential epigenetic mark for transcriptional control in mammalian cells. While all cells methylate CG dinucleotides (mCG), neurons in the brain undergo a unique epigenomic reconfiguration in the early postnatal period1 when the de novo DNA methyltransferase DNMT3A deposits non-CG methylation across the genome. Because the other major de novo DNA methyltransferase, DNMT3B, is not expressed in the postnatal brain,1 and the maintenance DNA methyltransferase, DNMT1, cannot deposit methylation at non-CG sites lacking a hemimethylated template,2 DNMT3A alone is essential for depositing non-CG methylation across the genome.3 Notably, non-CG methylation accumulates primarily at CA dinucleotides (mCA), reaching levels in many adult neuron types similar to that of mCG. These mCA sites are read out by the methyl-binding protein, MeCP2, to downregulate enhancer activity and tune the expression of genes with key functions in the nervous system.4–6
Consistent with a critical role for mCA in the brain, disrupted deposition or readout of this mark has been implicated in neurodevelopmental disorders (NDDs). Mutation of DNMT3A leads to the intellectual disability disorder, Tatton-Brown Rahman Syndrome (TBRS),7 while loss or overexpression of MeCP2 leads to the severe neurological disorders Rett syndrome and MeCP2 duplication syndrome (MDS), respectively.8,9 Epigenomic studies have demonstrated that neuronal mCA levels are globally reduced in mouse models of TBRS, leading to altered gene expression that overlaps expression changes that occur when mCA readout is disrupted in mutant MeCP2 models.10 Notably, exome sequencing studies of NDDs have identified mutations in numerous additional genes that encode epigenetic regulators and could impact neuronal DNA methylation.11–13 In particular, mutation of the histone methyltransferase NSD1 leads to autism and other phenotypes in the NDD Sotos syndrome14,15 that extensively overlap those observed in TBRS,16 but the impact of mutation of NSD1 on neuronal mCA has not been examined.
The level of mCA at gene regulatory elements determines the extent of repression by the mCA-MeCP2 pathway,5 and thus it is critical to understand how genomic mCA profiles are determined. Two independent phenomena have been associated with patterning mCA profiles, but how they interact to determine the ultimate levels of mCA at genomic regions and the molecular mechanisms that drive them are not known. In the first phenomenon, high gene transcription during the early postnatal period (e.g. one to four weeks in mice) correlates with reduced DNMT3A binding within the transcribed region of the gene and results in lower genic mCA levels in the mature brain. The mCA levels determined by this transcription-associated effect can in turn impact whether a gene is subject to subsequent repression at the adult stage.17 At a mechanistic level, an alteration in chromatin structure or steric hinderance by the transcribing polymerase has been proposed to mediate these effects, but no specific chromatin signal that directly affects DNMT3A binding has been defined within genes. Therefore, the mechanism by which this “expression-associated” mCA protection occurs in neurons remains unclear.
Parallel studies have discovered a second, large-scale phenomenon governing genomic mCA levels.5,18 Unlike mCG, which is consistently high across chromosomes, mCA levels can vary substantially in megabase-scale regions of the genome. This “regional mCA” is organized in part by topologically-associating domains (TADs) of chromatin folding. Genes and enhancers found within TADs with a high mCA “set-point” will, on average, be highly bound by DNMT3A postnatally and display higher mCA levels in the mature brain compared to genes and enhancers found within lower mCA set-point TADs. Regional mCA set-points impact gene regulation in the adult brain, as enhancers found within high mCA TADs are reduced in activity, leading to downregulated expression of target genes in that TAD. These genes are subsequently susceptible to dysregulation upon disruption of the mCA-MeCP2 pathway in disease.3 While these findings underscore the importance of regional mCA in gene regulation, the molecular mechanisms that differentially recruit DNMT3A to TADs to determine mCA set-points are not known.
In this study we investigated mechanisms of DNA methylation patterning in mouse cerebral cortex to build an integrated model of neuronal mCA deposition and uncover a role for histone H3 lysine 36 dimethylation (H3K36me2) by the Sotos syndrome protein NSD1 in this process. By analyzing gene expression and mCA levels across TADs and gene bodies, we show that regional mCA set-points and gene expression have independent, equal, and opposing contributions to the mCA profiles observed at genes and enhancers. By systematically profiling histone modifications we find that establishment of regional H3K36me2 levels, and selective depletion of this mark within genes, is a point of convergence that integrates both regional and gene expression-associated influences on mCA patterns. We show that loss of NSD1 disrupts TAD H3K36me2 levels to alter mCA set-points, impacting genes and enhancers inside affected TADs. Notably, we find that alterations in DNA methylation and gene expression that result from disruption of NSD1 in the brain overlap epigenomic and transcriptomic changes in a DNMT3A mutant mouse model of TBRS. Together, our findings present a unifying framework for mCA patterning in the neuronal genome and implicate disruption of neuron-specific DNA methylation as a shared site of molecular etiology across TBRS and Sotos syndrome.
Results
Regional set-point and gene expression additively and independently affect mCA at genes and enhancers
Both TAD-associated regional mCA set-points and gene expression-associated mCA depletion can influence mCA levels across the neuronal genome, but whether they interact and their relative contribution to total mCA levels at genes have not been assessed (Figure 1A). To explore how these phenomena interact, we performed integrated analysis of the mouse cerebral cortex, examining TADs defined by Hi-C, gene expression measured by RNA-seq at two weeks of age when DNMT3A expression is highest, and adult DNA methylation assessed by whole-genome bisulfite sequencing (WGBS) at eight weeks of age after accumulation of adult mCA patterns. Our analysis focused on mCA as the most prevalent form of non-CG methylation, but similar results were obtained in parallel analysis of the more minor forms, mCT and mCC, across the study. As previously reported,5 genome-wide profiles of mCA in the adult exhibited differences in levels in megabase-scale regions, and these fluctuations were far stronger for mCA than for mCG (Figure 1B, S1A). Furthermore, robust depletion of mCA from expressed genes were observed.17,19 Visual inspection of genes in TADs with different mCA set-points (see methods) suggested that regional set-points influence mCA in genes independently of gene expression (Figure 1B–D, S1B–F). These effects can be observed at two genes (Qk, and MeCP2) found in TADs with different mCA set-points but similar expression levels at two weeks (Figure 1C): Qk, which is found in a high mCA TAD, accumulates higher gene body mCA levels than the gene body of MeCP2 which is found in a lower mCA TAD. Notably, the mCA depletion at the gene body for each gene relative to their regional mCA set-point is similar for the two genes, suggesting that gene expression determines the degree of gene body mCA depletion relative to the mCA set-point. Quantification of mCA levels for TADs genome-wide further indicated that regional set-points for mCA are not tightly linked to the expression of genes within each TAD, as gene expression was robustly associated with mCA depletion in each gene but showed little association with mCA in the TAD surrounding the gene (Figure 1D, S1B). These effects were highest for mCA, but mCC and mCT showed similar, lower magnitude trends (Figure S1C,D). Consistent with previous reports,19,20 gene expression showed a modest negative correlation with mCG levels and a lower dynamic range than that for mCA (Figure S1B–E). To quantitatively assess the contributions of regional mCA and gene expression to genic mCA, we constructed a linear model using TAD-mCA levels and gene expression to predict mCA in genes (Figure S1F). This analysis revealed that both mCA set-point levels and gene expression robustly correlate with mCA levels in genes, but there is limited interaction between these two signals (Figure 1D, S1F). Notably, regional mCA and gene expression appeared to make approximately equal quantitative (2-fold) contributions to the level of mCA found within genes, genome wide (Figure 1D, S1B,F). Together these findings suggest that the regional mCA set-point predisposes a gene to high or low levels of mCA, and that the level of gene expression independently influences the degree of mCA depletion below the TAD set-point.
Figure 1.
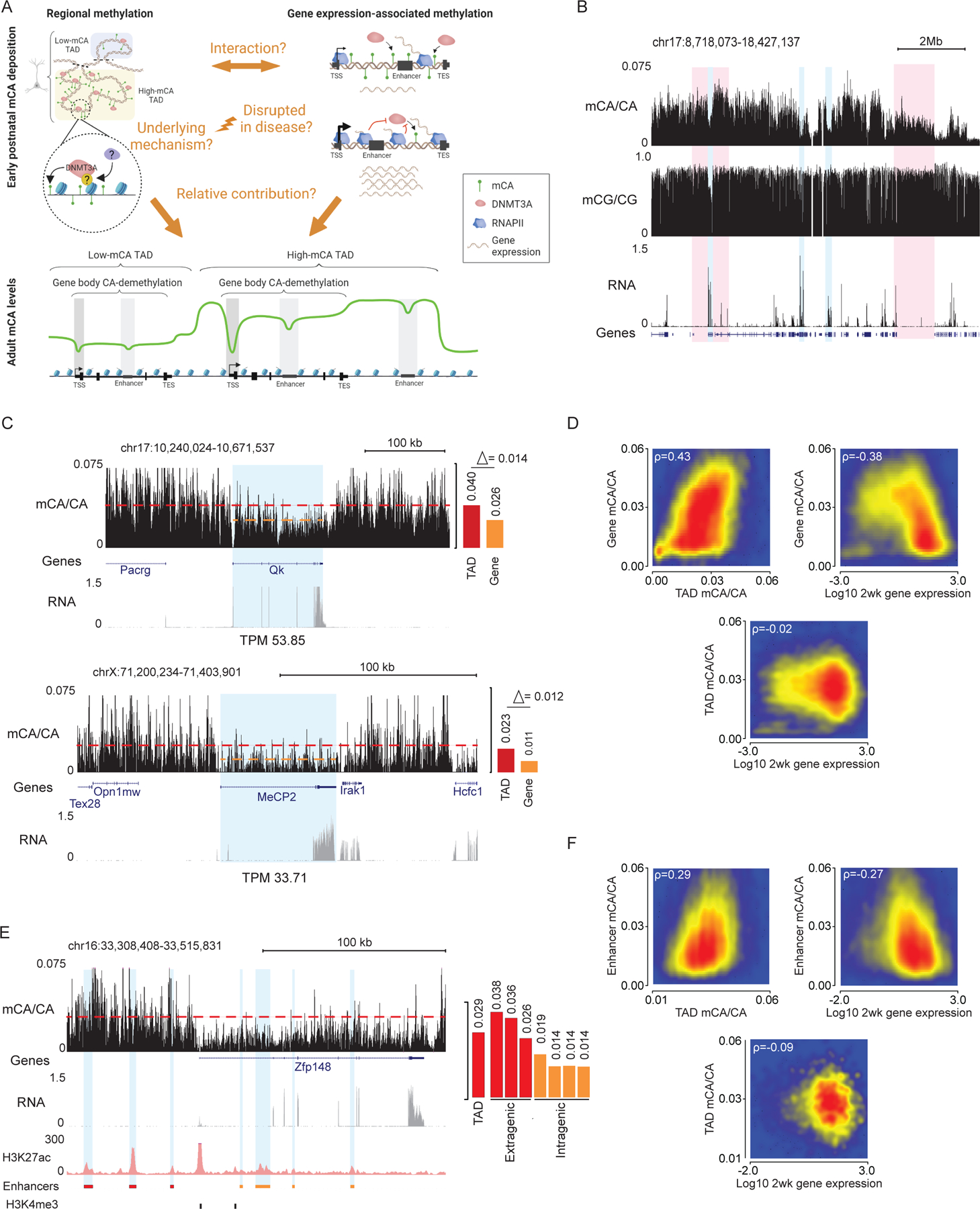
Regional mCA set-points and postnatal gene expression converge to regulate neuronal mCA levels at genes and enhancers.
(A) Left, model of DNMT3A-mediated regional mCA deposition in neurons. Mechanisms guiding DNMT3A to deposit TAD-scale mCA and whether this process is disrupted in disease are not known. Right, postnatal gene expression antagonizes DNMT3A-mediated mCA deposition within gene bodies. The degree to which these phenomena interact and how much each contributes to mCA patterning is not known.
(B) Genome browser view of total RNA-seq and DNA methylation from 2- and 8-week wildtype cerebral cortex, respectively. Red: broad genomic regions with high and low mCA levels. Blue: genes displaying expression-associated mCA depletion. In contrast to mCA, mCG shows little variation on a megabase-scale (see also, Figure S1).
(C) Left, browser view of mCA and RNA-seq from wildtype cerebral cortex at Qk and MeCP2, two genes with similar expression levels but differing TAD mCA setpoints. Right, quantification shows different gene body mCA levels in the two genes despite similar magnitude of gene body depletion relative to mCA levels in the surrounding TAD.
(D) Scatterplots showing correlation between TAD mCA level, gene body mCA, and postnatal gene expression.
(E) Left, browser showing mCA, RNA-seq, and H3K27ac ChIP-seq from wildtype cerebral cortex at the Zfp148 gene. Putative active enhancers (blue) were defined as peaks that contain H3K27ac and do not overlap with promoter H3K4me3 peaks. Right, quantification of TAD mCA level, and extragenic and intragenic enhancer mCA levels. Enhancer mCA depletion relative to the TAD mCA level occurs at intragenic enhancers in this highly expressed gene.
(F) Scatterplots showing correlation between TAD mCA level, intragenic enhancer mCA level, and gene expression.
Data are wild-type 2-week cortex RNA-seq.1 8-week cortex DNA methylation17 and H3K27ac ChIP-seq.5
Given that mCA levels at intragenic enhancers have been tightly linked to regulation of genes by MeCP2,5 we examined these regulatory sequences to determine how regional mCA set-points and early postnatal gene expression impact their ultimate mCA levels. Visual inspection (Figure 1E), quantitative analysis (Figure 1F, S1G), and linear modeling of all enhancers genome-wide (Figure S1H) indicated that intragenic enhancer mCA is associated with both regional mCA set-points and gene expression, with mCA depletion below the set-point being associated with higher gene expression. As observed for gene mCA, regional mCA set-points and gene expression both contribute to intragenic enhancer mCA (Figure S1G,H). Thus, regional mCA and gene expression appear to additively impact the ultimate levels of mCA at intragenic enhancers, which can in turn determine the degree of regulation at these sites by MeCP2 in the mature brain.
Methylation of histone H3 lysine 36 predicts broad mCA patterns
We next investigated the molecular mechanism by which DNMT3A activity could be targeted to broad regions and then tuned at genes and enhancers to determine mCA levels at these sequences. Histone modifications have been shown to influence DNMT3A binding and activity on the genome,21,22 but analysis of multiple histone modifications in the early postnatal brain has not detected any single modification that is a strong candidate for controlling mCA deposition by DNMT3A.17 Notably, outside of the nervous system H3K36me2 has been implicated in recruiting DNMT3A to intergenic regions.23–25 Mutations in the H3K36me2 binding PWWP domain of DNMT3A have been detected in TBRS patients suggesting that disruption of this binding by DNMT3A can drive neurodevelopmental deficits.26 However, H3K36me2 has not been studied for its impact on DNMT3A activity and deposition of mCA in the postnatal brain. We therefore performed ChIP-seq of H3K36 methylation (H3K36me2, and me3) in the cerebral cortex at two weeks of age and assessed the degree to which these marks predict DNMT3A binding and mCA patterns in the mature brain.
Examination of H3K36me2 across the genome showed that this mark fluctuated on a megabase-scale, with profiles mirroring DNMT3A binding across the genome at two weeks and subsequent patterns of mCA in the adult (Figure 2A). Cross-correlation analysis of H3K36me2 indicated that, as previously demonstrated for DNMT3A and mCA,5 this signal is more consistent within TAD structures than across TAD boundaries (Figure S2A). Aggregate (Figure 2B) and quantitative analyses (Figure 2C, S2B–E) revealed a robust correlation between this histone mark, DNMT3A binding, and mCA deposition at TADs, genes, and enhancers genome-wide, indicating that H3K36me2 patterns closely mimic the patterns expected for a signal that drives regional set-point mCA.
Figure 2.
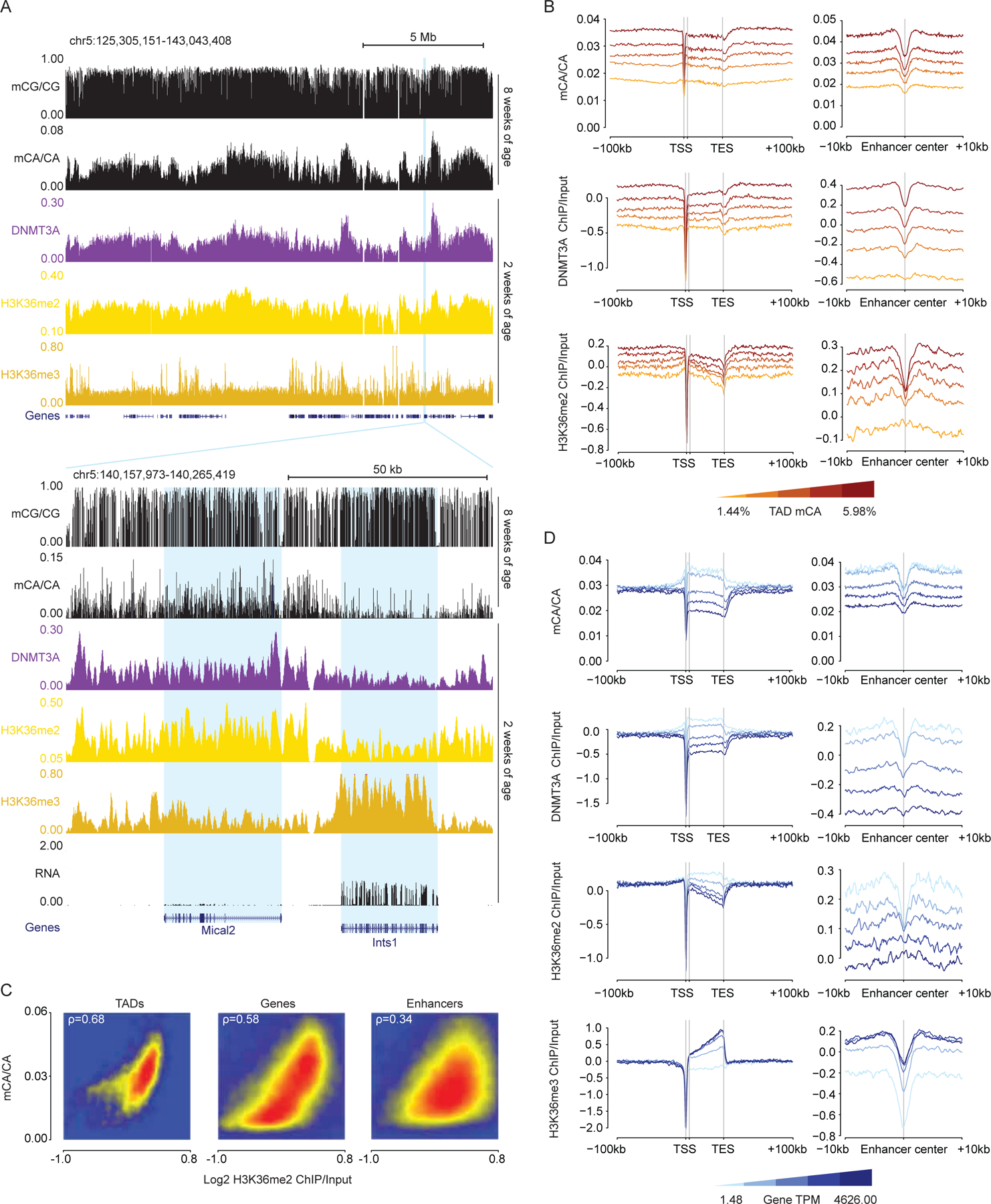
H3K36me2 profiles predict neuronal mCA in the postnatal cerebral cortex.
(A) Top, genome browser view of DNA methylation, ChIP-seq, and RNA-seq from wildtype cerebral cortex illustrating megabase-scale fluctuations in H3K36me2, DNMT3A binding, and mCA accumulation. Bottom, browser view of two differentially expressed genes showing distinct H3K36me2 and H3K36me3 signals corresponding to differences in DNMT3A binding and mCA.
(B) Aggregate mCA, DNMT3A, and H3K36me2 levels at genes and enhancers across quintiles of TADs sorted by regional mCA levels.
(C) Comparison of H3K36me2 and mCA/CA at TADs, enhancers, and genes, showing genome-wide correlations for these signals.
(D) Aggregate mCA, DNMT3A, H3K36me2, H3K36me3 levels at genes and intragenic enhancers across genes sorted by RNA expression quintiles.
Data are from wildtype cerebral cortex; n = 2–3 bioreplicates for 2-week H3K36me2 and DNMT3A ChIP-seq. 2-week RNA-seq data.1 2-week H3K36me3 and 8-week DNA methylation data.17
The conversion of H3K36me2 to H3K36me3 in gene bodies is associated with the level of transcription through the gene.27 Given the concordance between H3K36me2 and DNMT3A recruitment we considered that high gene expression in the postnatal period may increase H3K36me3 in genes while depleting H3K36me2. Some studies have reported H3K36me2 as a higher affinity binding site for DNMT3A than H3K36me3,28,29 and this conversion could explain the reduction in DNMT3A recruitment to highly expressed genes. Visual inspection (Figure 2A) and aggregate analysis of genes as a function of expression levels (Figure 2D) supported this model, as highly expressed genes showed increased H3K36me3, depleted H3K36me2, and lower DNMT3A binding, with reduced subsequent mCA deposition at these sites in the adult brain. Notably, the expression-associated depletion of H3K36me2 impacted mCA deposition at intragenic enhancers as well, with enhancers found within highly expressed genes showing similar depletion of this mark and subsequently lower mCA levels (Figure 2D). These results for TADs, genes, and enhancers were further corroborated by identical trends detected using a second H3K36me2 antibody (Figure S2I–M). These findings support a model in which H3K36me2 is first deposited broadly across the genome, and then depletion of this mark by conversion to the lower affinity DNMT3A binding site, H3K36me3, drives subsequent protection of gene bodies from methylation.
The association between H3K36me2 and DNA methylation in the brain was far stronger for mCA than for mCG, with mCG showing limited fluctuations on a megabase-scale across the genome (Figure 1B, 2A). We observed weaker correlations between mCG levels and H3K36me2 in TADs, genes, and enhancers genome-wide (Figure S2F,M). Similarly, mCG levels showed limited dynamic range in genes as a function of their TAD mCA and gene expression levels (Figure S2G,H). Thus, in the context of the brain, H3K36me2 likely has its greatest impact on deposition of mCA by DNMT3A rather than on mCG, which does not depend as strongly on DNMT3A activity for patterning.
Integration of chromatin signatures accurately predicts mCA levels genome-wide
Given the robust association of H3K36me2 with DNMT3A-deposited mCA, we sought to compare the relative strength of these relationships to signals for other previously profiled chromatin states in the early postnatal brain.17 We therefore carried out integrated analysis of this mark with multiple ChIP-seq datasets measured in the cerebral cortex at two weeks of age. We also performed analysis of all three states of histone H3 lysine 4 methylation, marks which have been linked by biochemical and epigenomic profiling studies outside of the nervous system to inhibition of DNMT3A at regulatory elements,30,31 but which have not been fully assessed for their contribution to DNMT3A activity in the brain.
Having compiled this compendium of datasets that includes the most extensive set of DNMT3A-relevant chromatin signals to date, we assessed the correlation of each chromatin signal with gene expression, DNMT3A binding, and mCA deposition, examining this relationship at regulatory elements, genes, and intergenic/intragenic regions (Figure 3A). This analysis detected a strong negative correlation of H3K4 di- and tri-methylation with DNMT3A and mCA at regulatory elements (Figure 3A, S3A–C) that resembled similar relationships previously detected for Pol II binding at promoters.17 In contrast, H3K4me1 showed little association with mCA depletion compared to H3K4me2/3 (Figure 3A, S3B,C), suggesting that this mark does not strongly block DNMT3A activation. mCG profiles showed similar relationships with H3K4me2 and -me3 as mCA, but H3K4me1 was more strongly associated with mCG depletion compared to mCA (Figure S3A,D,E), suggesting that there may be a differential effect of H3K4me1 on DNMT3A activity at these two dinucleotides. Intragenic signals associated with high gene expression (Pol II pS2, H3K36me3) showed negative correlations with DNMT3A and mCA. Notably, at all sites examined, H3K36me2 consistently showed a positive correlation with DNMT3A and mCA deposition and a negative correlation with gene expression. To examine the degree to which these chromatin signatures can explain mCA deposition patterns, we trained a random forest algorithm to predict mCA levels with these data using 5-fold cross-validation.32–34 The algorithm accurately predicted mCA for both intergenic and intragenic regions genome-wide (Figure 3B). Furthermore, this random forest approach outperformed a simple linear model (Figure 3B), suggesting that DNMT3A binding could not be predicted solely by any one or simple combination of factors and implying a more complex, nonlinear relationship between these factors and mCA deposition (e.g. H3.3 is negatively correlated with mCA at intragenic regions, but positively correlated at intergenic regions, Figure 3A). To assess the contribution of each ChIP signal to algorithm prediction accuracy we performed feature importance and feature elimination analyses (Figure 3C,D). This revealed H3K36me2 as one of the strongest contributors to accuracy across regions genome-wide. Analysis of regulatory regions alone (Figure S3F,G) revealed a larger signal for H3K4me3 at promoters, further implicating this mark as a determinant of mCA at focal regulatory sites of active genes. Together these findings support key roles for H3K4me3 and H3K36me2 in patterning neuronal methylation, with H3K36me2 showing a particularly strong signal across the genome regardless of location.
Figure 3.
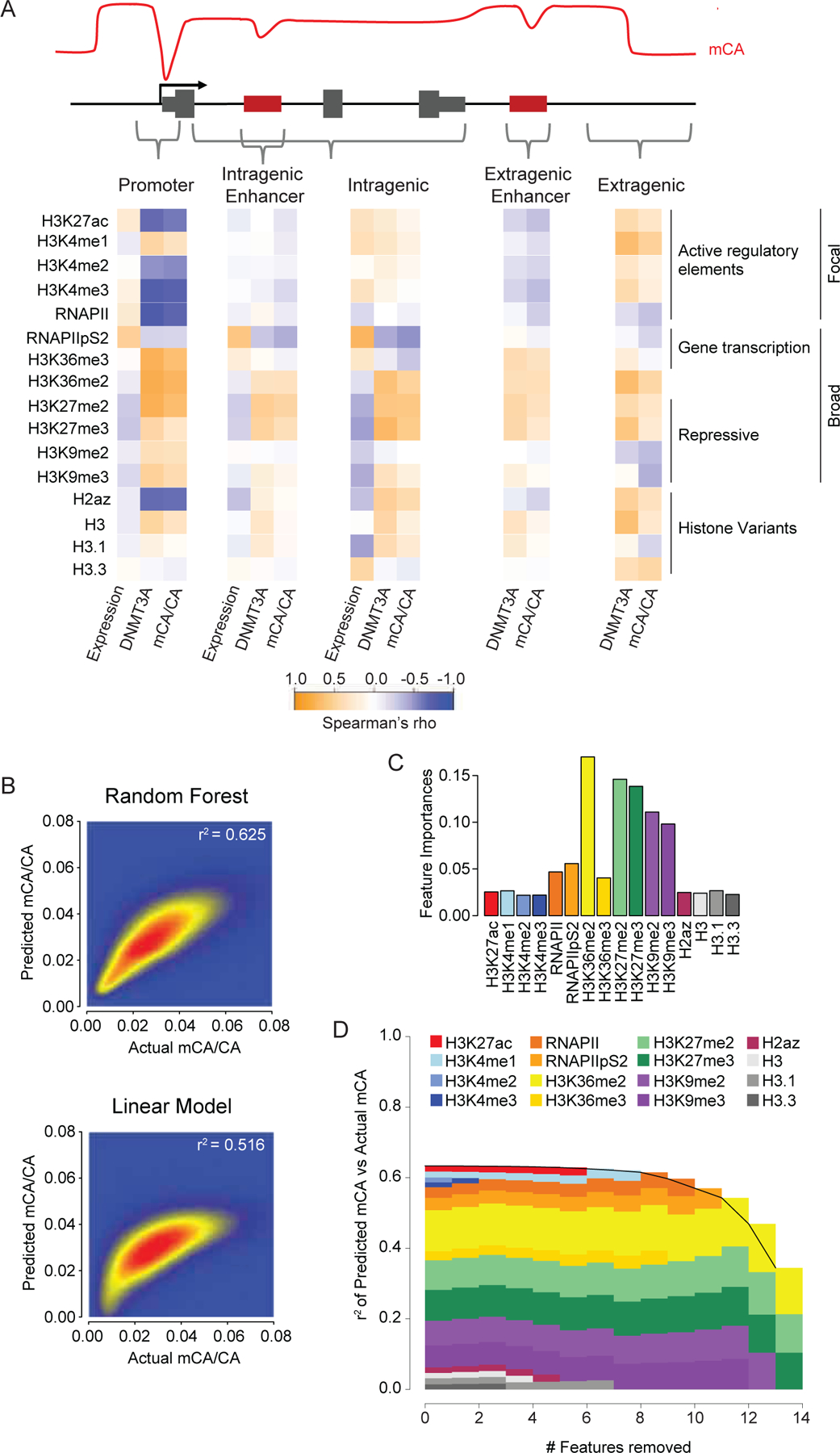
Comparative analysis of chromatin states, DNMT3A binding, and mCA deposition across the neuronal genome.
(A) Correlation of histone and RNA polymerase II modification states with DNMT3A binding, mCA, and gene expression across kilobase-scale genomic regions.
(B) Smoothscatter showing predictive accuracy of ChIP signals for mCA using a Random Forest algorithm (top) or linear model (bottom) in all 5kb windows genome-wide.
(C) Feature importance analysis (see methods) showing relative contribution of chromatin signatures to random forest classifier’s mCA prediction accuracy.
(D) Feature elimination analysis (see methods) shows key marks required for accurate random forest classifier mCA prediction.
Data are n = 2–3 bioreplicates for 2-week H3K36me2, H3K4me1, H3K4me2, and DNMT3A ChIP-seq from this study. 2-week RNA-seq.1 2-week ChIP-seq for all other chromatin marks and 8-week DNA methylation data.17
Disruption of NSD1 in neurons leads to reduction of H3K36me2 and loss of mCA in broad domains
To directly test the importance of H3K36me2 in DNMT3A targeting and mCA patterning, we sought to disrupt H3K36me2 deposition and assess its impact on neuronal DNA methylation. Previously, we have utilized primary cultures from embryonic cortex to observe mCA accumulation in postmitotic neurons and study the effects of NDD-associated DNMT3A mutations on mCA deposition.10 We therefore employed this system to manipulate H3K36me2 profiles and evaluate the effects on DMMT3A-mediated mCA patterning. Given the shared cognitive and behavioral deficits upon NSD1 mutation in Sotos syndrome and DNMT3A mutation in TBRS we selected NSD1 for disruption in our cultures by lentiviral-mediated RNAi knockdown. Two validated NSD1 knockdown shRNAs or control shRNA (Figure S4A) were transduced at Day in vitro (DIV) 1, followed by ChIP-seq of H3K36me2 and DNMT3A binding at DIV 12 and WGBS analysis of DNA methylation at DIV 18. ChIP-Seq and DNA methylation profiles in control neurons further supported a strong association between H3K36me2, DNMT3A binding, and mCA deposition (Figure S4B–D). To examine the effects of NSD1 knockdown on H3K36me2 genome-wide, we performed edgeR differential count analysis35 on H3K36me2. This analysis identified 20,203 10kb windows with significantly altered H3K36me2 levels. These H3K36me2 changes were highly concordant with changes in DNMT3A binding and mCA (Figure 4A,B). Analysis of all 10kb windows genome-wide revealed a strong global concordance between changes in H3K36me2 signal, DNMT3A signal, and mCA (Figure 4C,S4E), demonstrating the role of H3K36me2 in targeting DNMT3A to the neuronal genome.
Figure 4.
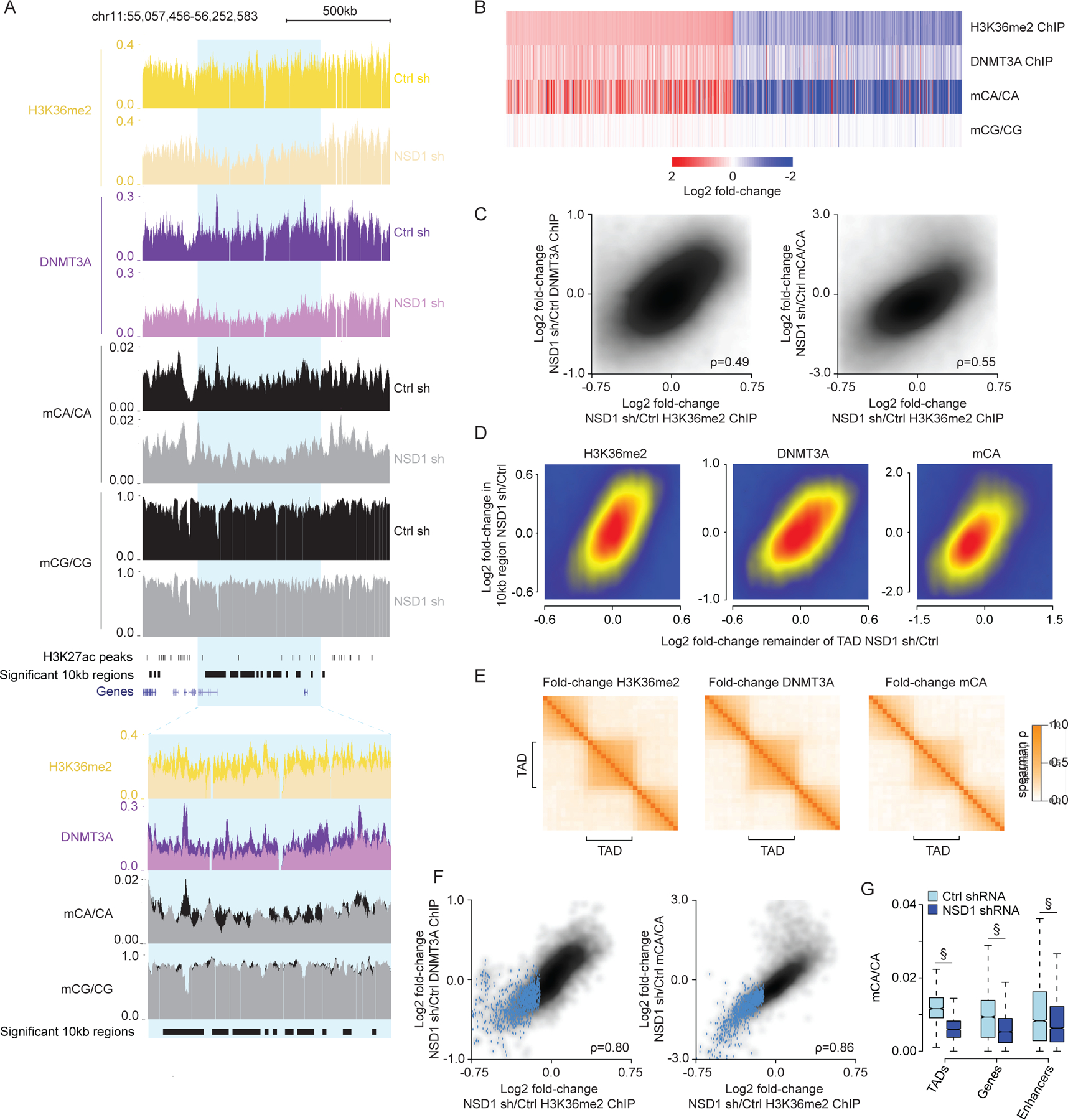
NSD1-mediated H3K36me2 is required for TAD-scale DNMT3A targeting and mCA deposition in postmitotic neurons.
(A) Genome browser view of DIV 12 ChIP-seq and DIV 18 DNA methylation from primary cortical neurons (PCN). Representative TAD with significantly reduced H3K36me2 upon NSD1 knockdown in blue. Overlap of multiple 10kb bins with significantly reduced H3K36me2 in this significantly altered TAD illustrates concordance of changed signals within TADs.
(B) Fold-changes of H3K36me2, DNMT3A, and DNA methylation in shNSD1 transduced PCNs at all 10kb regions identified by edgeR (FDR<0.1) as significantly altered for H3K36me2.
(C) Comparison of changes in H3K36me2 to changes in DNMT3A (left) or mCA (right) for 10kb regions genome-wide.
(D) Comparison of changes in H3K36me2, DNMT3A, or mCA between 10kb genomic regions and the TAD in which they reside.
(E) Cross-correlation analysis of fold-changes in H3K36me2, DNMT3A, and mCA upon NSD1 knockdown for regions inside and outside of TADs across the genome (see methods). Higher correlation of regions found within the same TAD compared to regions across TAD boundaries indicates regions within the same TAD are concordantly affected upon NSD1 loss.
(F) Comparison of changes in H3K36me2 to changes in DNMT3A or mCA for each TAD in the genome. TADs with significantly reduced H3K36me2 by edgeR (FDR<0.1) in blue.
(G) Boxplots of mCA levels in shCtrl and shNSD1 PCNs at TADs with significantly reduced H3K36me2 (edgeR, FDR <0.1) and kilobase-scale genomic elements that reside within these TADs. (§p < 10−15, Wilcoxon test).
Data are from PCNs transduced with shCtrl or shNSD1 on DIV 1 and collected at DIV 12 and DIV 18 for ChIP-seq and WGBS, respectively. Per time point: n = 2–4 bioreplicates for H3K36me2, DNMT3A ChIP, and DNA methylation. TADs are from Hi-C analysis of cortical neurons.59
Given our observations that H3K36me2, DNMT3A, and mCA are organized by TADs, we assessed if NSD1-mediated H3K36me2 deposition impacts mCA deposition across entire TADs. We observed clustering of significantly changed 10kb windows in the genome (Figure 4A) and robust associations between the change in each 10kb window and the change in mCA in the remainder of the TAD surrounding that window (Figure 4D), suggesting that 10kb window changes reflect a larger scale effect on the TAD. Furthermore, cross-correlation analysis showed that alterations in H3K36me2, DNMT3A, and mCA upon NSD1 knockdown are organized by TADs (Figure 4E), and genome-wide comparisons of changes in TAD H3K36me2 levels with changes in DNMT3A and mCA showed strong concordance (Figure 4F). These findings suggest that the intrinsic scale of H3K36me2 targeting by NSD1 is at the level of TADs.
Our analysis above (Figure 2) suggests that the level of H3K36me2 in TADs can influence the level of this mark and subsequent mCA deposition in smaller scale genes and regulatory elements that reside in each TAD. We therefore assessed how mCA is affected at enhancers and genes in TADs with reduced H3K36me2 signal upon NSD1 knockdown. We observe reduced mCA at these regions when found within TADs that display significantly reduced H3K36me2 signal, indicating that NSD1-mediated H3K36me2 deposition in TADs helps to determine the level of mCA at these important sites of gene regulation (Figure 4G). Together these analyses suggest NSD1 activity is required to maintain TAD H3K36me2 patterns in postmitotic neurons, and that the H3K36me2 profile directs regional mCA set-points to impact mCA levels at regulatory elements and genes across the genome.
Shared epigenomic and transcriptomic effects of NSD1 and DNMT3A disruption in vivo
Having detected disruption of H3K36me2 and mCA upon acute depletion of NSD1 in cultured neurons, we next sought to assess how loss of NSD1-mediated H3K36me2 deposition in the brain affects the neuronal methylome and manifests alterations in chromatin structure and gene expression. In particular, our in vitro findings that loss of NSD1 leads to altered DNMT3A binding and mCA deposition suggest that there might be direct mechanistic overlap between the effects of NSD1 disruption and DNMT3A mutation. Such shared changes would be candidates to explain the phenotypic overlap between Sotos syndrome and TBRS.
To carry out in vivo studies, we generated and analyzed NSD1 conditional knockout (cKO) mice, in which deletion of exon 3 in the brain was driven by the expression of Nestin-cre in neural progenitors (Figure S5A). Validation of the NSD1 cKO mouse showed complete depletion of the targeted exon selectively in the brain at two weeks (Figure S5B-D). Reduced NSD1 immunofluorescence signal from NeuN+ cortical nuclei was also observed in these mice (Figure S5F,G), suggesting robust depletion of full length NSD1. However, while deletion of exon 3 results in disrupted splicing of the NSD1 mRNA and early truncation of the coding sequence, we observed only modest reduction of mRNA transcripts downstream of exon 3 by RT-PCR and RNA-seq (Figure S5D,E). Thus, some NSD1 gene product could be produced in these mice due to internal ribosome initiation. Nonetheless, conditional deletion of exon 3 allowed us to assess the effects of at least partial disruption of NSD1 in the brain.
To determine if disruption of NSD1 in vivo alters the neuronal DNA methylation pathway, we first assessed changes in H3K36me2 and DNMT3A at two weeks of age. We performed ChIP-Rx36 on the cerebral cortex of NSD1 cKO and control mice for this analysis to assess if global levels of these signals are altered in the mutant (Figure S5I). Differential count analysis by edgeR revealed TAD-level changes in H3K36me2 and DNMT3A like those observed in culture (Figure 5A–B, S5J), with reductions in TAD H3K36me2 correlating with loss of DNMT3A binding. Base-pair resolution WGBS analysis in adult cortex revealed concordant changes between adult mCA and two-week H3K36me2, including lower mCA levels in TADs showing reduced H3K36me2 (Figure 5B, S5J,L). A concomitant mCA reduction at genes and enhancers found within affected TADs was also observed (Figure 5C). In addition to a significant reduction of mCA at an array of TADs and genomic elements across the neuronal genome, we noted a trend toward lower mCA levels globally, with total mCA levels showing a reduction by approximately 14%. Analysis of global H3K36me2 changes by ChIP-Rx normalization and western blot indicated a subtle trend toward global reduction in H3K36me2 upon loss of NSD1 (Figure S5H,I). Like mCA, analysis of mCT and mCC showed concordance between changes in H3K36me2 and altered methylation (Figure S5K). In contrast, mCG levels were largely maintained globally and across broad genomic elements where H3K36me2 was lost, with only subtle effects mirroring changes in mCA (Figure 5A, S5L). Together these results indicate that mCA levels in a subset of TADs in the genome are particularly sensitive to loss of NSD1 in the brain, and suggest that NSD1 contributes to global H3K36me2 and subsequent DNMT3A-mediated mCA levels genome-wide.
Figure 5.
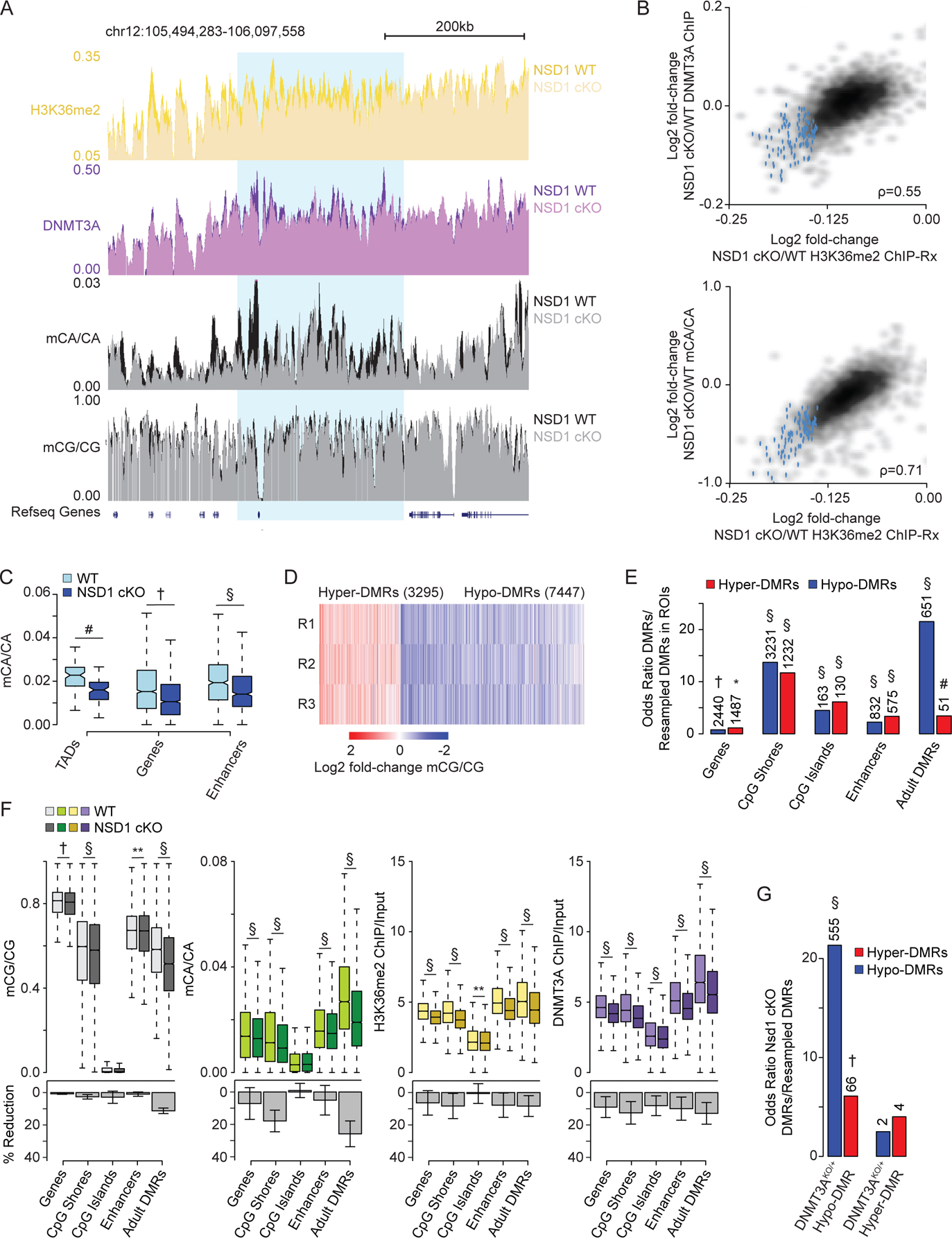
Brain-specific loss of NSD1 in vivo leads to epigenetic dysregulation that overlap DNMT3A mutants.
(A) Genome browser view of 2-week ChIP-Rx and 8-week DNA methylation from NSD1 cKO and wildtype cerebral cortex. A representative TAD with significantly reduced H3K36me2 in blue.
(B) Comparison of changes in H3K36me2 to changes in DNMT3A or mCA for each TAD. TADs with significantly reduced H3K36me2 by edgeR (FDR<0.1) in blue.
(C) Boxplots showing mCA levels at TADs with significantly reduced H3K36me2 (edgeR, FDR <0.1) and kilobase-scale genomic elements that reside within these TADs for NSD1 cKO and wildtype cortex. (Wilcoxon test).
(D) Fold-change of mCG in three independent replicate pairs at CG-DMRs called in the NSD1 cKO cortex.
(E) Odds ratio of overlap of NSD1 cKO CG-DMRs with kilobase-scale genomic regions. Observed number of overlapping regions denoted above (Fisher’s exact test, observed versus resampled DMRs, see methods).
(F) Top, DNA methylation, H3K36me2 levels, and DNMT3A binding in NSD1 cKO versus wildtype cortex across kilobase-scale genomic regions. Bottom, mean and SEM percent reduction. (Wilcoxon test).
(G) Odds ratio of overlap of NSD1 cKO CG-DMRs with DNMT3AKO/+ CG-DMRs.10 Observed number of overlapping DMRs denoted above (Fisher’s exact test, observed versus resampled DMRs, see methods).
Data are from NSD1 cKO and wildtype cortex. n = 5 for H3K36me2, H3K36me3, DNMT3A ChIP-Rx at 2-weeks. n = 3 for DNA methylation, n = 6 for total RNA-seq at 8-weeks. TADs derived from Hi-C analysis of cortex.5,60
*p < 0.05, **p < 0.01, #p < 10−5, †p < 10−10, §p < 10−15.
In addition to genome-wide deposition of mCA, DNMT3A also deposits de novo mCG at kilobase-scale regions across the neuronal genome during postnatal brain maturation.1,10 Notably, heterozygous disruption of DNMT3A in a mouse model of TBRS (DNMT3AKO/+) leads to loss of mCG at differentially methylated regions (DMRs) that overlap with these sites.10 We therefore sought to assess the impact of NSD1 loss on mCG levels at discrete regions across the genome and interrogate if these sites overlap with regions disrupted in models of DNMT3A disorders. De novo identification of mCG-DMRs in the NSD1 cKO (see methods) led to the detection of 3295 hyper-DMRs and 7447 hypo-DMRs (Figure 5D). The identification of a preponderance of hypo-DMRs further supports a role for H3K36me2 in targeting deposition of DNA methylation in the neuronal genome. Analysis of the locations of these DMRs showed that loss of NSD1 leads to changes in mCG at important regulatory sites including genes, promoters, CpG shores, CpG islands, and enhancers (Figure 5E,F). Notably, we found that adult DMRs, regions of the genome that acquire mCG during postnatal development in a DNMT3A-dependent manner,1 were most enriched for overlap with NSD1 cKO DMRs (Figure 5E). Further quantitative analysis of changes in H3K36me2, DNMT3A, and DNA methylation levels revealed concordant reductions across these signals at important regulatory sites, with adult DMRs exhibiting the largest reductions in mCA and mCG (Figure 5F).
Notably, adult DMRs are susceptible to CG hypo-methylation in DNMT3AKO/+ mice,10 suggesting similar losses of mCG between TBRS models and NSD1 cKO mice. To assess if the effects on mCG are similar between NSD1 and DNMT3A mutant models, we determined the degree of overlap between DMRs called in the NSD1 cKO versus those called in the DNMT3AKO/+ and found significant overlap between hypo-DMRs called in each model (Figure 5G). These results indicate that specific mCG sites are sensitive to loss of NSD1 and overlap with aberrantly methylated sites in DNMT3A mutant models. Our findings further support a model in which NSD1-mediated H3K36me2 deposition functions as a direct upstream regulator of DNMT3A to put down large-scale mCA and site-specific mCG in the postnatal brain.
We next investigated whether the epigenetic changes we observe in the NSD1 cKO result in altered gene expression in the brain. We performed differential gene expression analysis by total RNA-seq in control and NSD1 cKO adult cortex, identifying 217 (177 up, 40 down) significantly dysregulated genes (FDR<0.1) (Figure 6A). Epigenomic analysis of these genes revealed reduced H3K36me2, DNMT3A, and mCA levels at aberrantly upregulated genes (Figure 6B) that is consistent with an increase in transcription due to loss of NSD1 activity. Paradoxically, the small number of down-regulated genes detected in the NSD1 cKO displayed variable reductions in DNMT3A binding and DNA methylation. To investigate these trends with higher statistical power, we analyzed a larger group of the top 500 significantly up- and down-regulated genes in the genome. This analysis revealed a robust reduction of H3K36me2, DNMT3A, and DNA methylation at upregulated genes, but did not replicate the loss of methylation at downregulated genes (Figure S6A). Analysis of levels of these chromatin and DNA modifications for the TADs in which the significantly called dysregulated genes were found (Figure S6B,C) revealed similar changes in these modifications at the TAD-level. Together, these results suggest that the gene dysregulation observed in the NSD1 cKO is due in part to changes in H3K36me2 and subsequent mCA levels, with upregulated genes showing the closest association with H3K36me2 loss and reduced DNA methylation.
Figure 6.
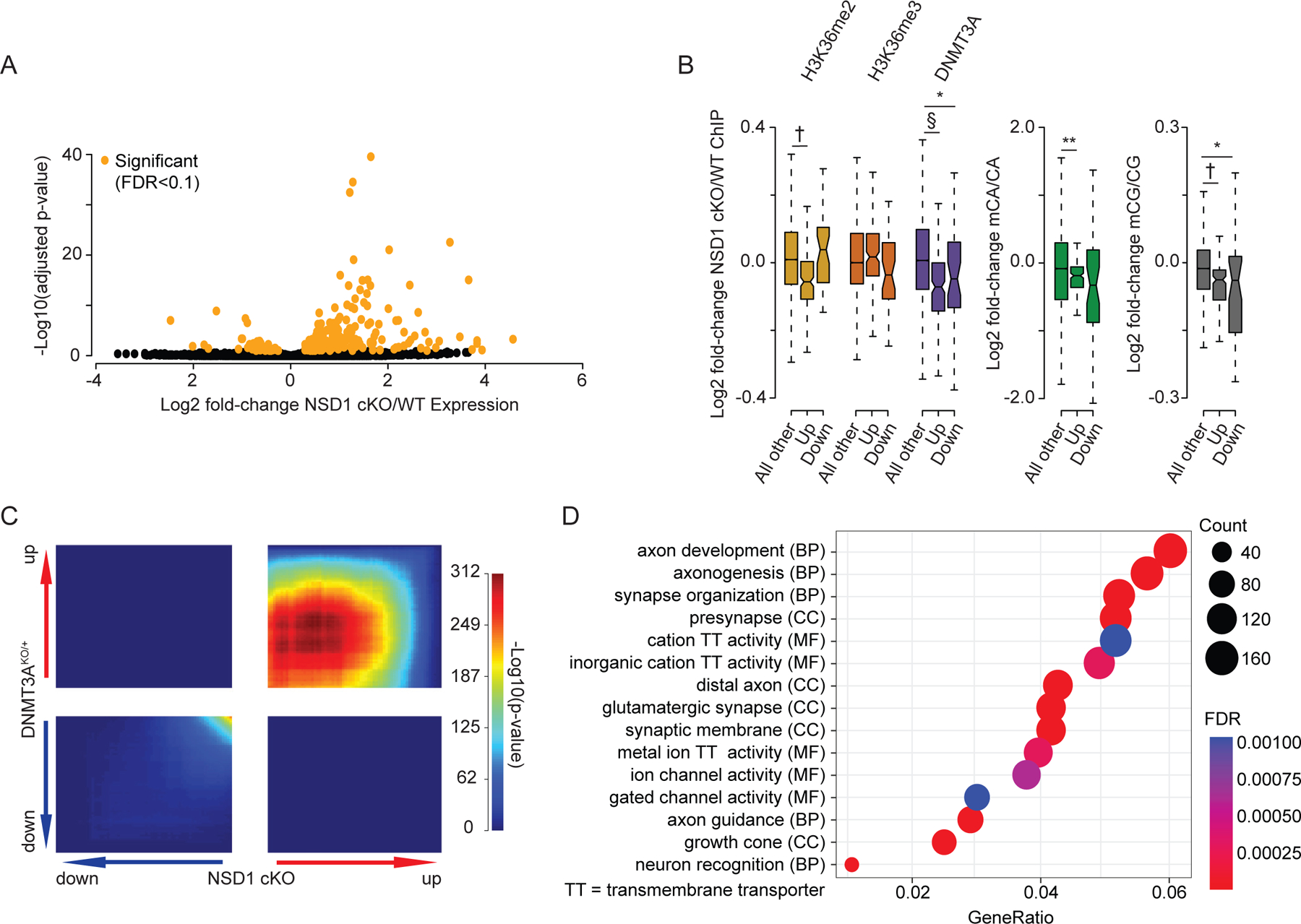
Brain-specific loss of NSD1 in vivo leads to transcriptional changes that overlap DNMT3A mutants.
(A) Volcano plot of fold-changes in gene expression of NSD1 cKO versus wildtype cortex RNA-seq. Significant genes by DESeq2 (FDR <0.1) in orange.
(B) Fold-changes of H3K36me2, H3K36me3, DNMT3A, and DNA methylation at genes significantly dysregulated (DESeq2, FDR <0.1) in NSD1 cKO RNA-seq. (Wilcoxon test).
(C) Rank-rank hypergeometric overlap (RRHO)38,61 of transcriptome-wide gene expression changes in the cortex of NSD1 cKO versus DNMT3AKO/+ mice.10
(D) Top five Gene Ontology terms from “Biological Process”, “Molecular Function”, and “Cellular Components” from RRHO-determined “uu” (upregulated-upregulated) genes from NSD1 cKO and DNMT3AKO/+ mouse cortex using clusterProfiler.39,40
Data are from NSD1 cKO and wildtype cortex: 2-weeks, n = 5 for H3K36me2, H3K36me3, DNMT3A ChIP-Rx. 8-weeks, n = 3 for DNA methylation, n = 6 for total RNA-seq. TADs derived from Hi-C analysis of cortex.5,60
*p < 0.05, **p < 0.01, #p < 10−5, †p < 10−10, §p < 10−15.
In light of the overlapping epigenetic disruption we detect in NSD1 and DNMT3A mutant mice, we investigated if shared gene expression effects may occur between these mutant models. Identification of commonly dysregulated genes in these mice would provide candidates for drivers of shared pathology in disease. We therefore examined the extent of overlap in gene dysregulation between NSD1 cKO and DNMT3A mutant models. Both models display a small number of genes that reach statistical significance for dysregulation. Despite this fact, overlap analysis of significantly dysregulated genes in the NSD1 cKO (217 genes) and the DNMT3AKO/+ (65 genes) revealed significant enrichment of co-upregulated genes across the two mouse models (Figure S6D).
Analysis of multiple models of NDD has demonstrated that gene expression effects in the brain caused by these mutations result in widespread but subtle dysregulation of many genes that is not captured by analysis of statistically significant genes alone.10,18,37 Therefore, to further assess transcriptome-wide overlap in gene dysregulation between these two genotypes, we performed Rank-Rank Hypergeometric Overlap (RRHO) analysis38 of gene expression changes in the NSD1 cKO and DNMT3A mutant models. RRHO revealed widespread and significant concordance for upregulated genes between the NSD1 cKO and DNMT3A mutant models, indicating global similarity between gene de-repression effects caused by disruption of these genes (Figure 6C, S6E). Such broad, shared de-repression is consistent with the loss of the repressive mCA mark across the genome, and points to substantial overlap in gene dysregulation in these models.
We next sought to investigate the functional pathways impacted by the shared transcriptional effects observed in the NSD1 cKO and DNMT3A mutant models. We therefore performed Gene Ontology analysis39,40 of concordantly dysregulated genes identified by RRHO. This analysis detected terms associated with key neurodevelopmental processes during postnatal brain development, including synapse organization and axon guidance (Figure 6D, S6F). Identification of terms such as glutamatergic synapse and ion channel activity further suggests disrupted neurotransmission manifesting due to transcriptional dysregulation upon DNMT3A and NSD1 mutation. Thus, the role of NSD1 as a direct upstream regulator of DNMT3A binding and mCA deposition may influence critical pathways involved in developing and mature neural circuits. Overall, these findings identify key functional pathways in the brain with shared disruption upon NSD1 or DNMT3A loss and provide evidence of etiological convergence at the molecular level for NDDs caused by mutation of these genes.
Discussion
In this study we have interrogated the mechanisms of DNMT3A targeting in the brain to uncover a major role of H3K36me2 in dictating neuronal mCA accumulation. Our findings are consistent with a model in which NSD1 activity is directed by genome topology to broadly pattern H3K36me2 across the neuronal genome in the developing nervous system. This regional H3K36me2 can then be modified through depletion at genes when H3K36me2 is converted to H3K36me3 by active transcription. During the postnatal period DNMT3A is recruited to H3K36me2 sites through its PWWP domain to establish mCA patterns. mCA is then read out by MeCP2 and possibly other proteins to repress regulatory elements and tune transcription of essential gene programs in the mature brain. Mutation of NSD1, DNMT3A, or MeCP2 can disrupt the establishment or read out of this critical H3K36me2-mCA pathway, contributing to nervous system dysfunction in human NDDs.
By integrating analyses of gene expression and regional methylation together and exploring underlying chromatin marks associated with these phenomena, our study reconciles two distinct mechanisms of mCA patterning that had been independently described.5,17 Rather than working through completely independent mechanisms or interacting through complex non-linear processes, we find that regional set-points and gene-expression-associated depletion of mCA can be explained by two steps in H3K36me2 patterning: regional deposition of the mark followed by selective conversion to H3K36me3. Thus, our findings provide a unifying framework to explain how neurons acquire their unique methylomes.
Our comprehensive analysis of chromatin marks with DNMT3A binding and mCA deposition across gene regulatory elements establishes H3K36me2 as a strong predictor of DNMT3A-mediated mCA deposition genome-wide. Histone variant H2Az also displayed a strong negative relationship with DNMT3A binding and mCA deposition at promoters (Figure 3A), which is supported by previous studies with similar results between H2Az and mCG.41 Our analysis also demonstrates that deposition of H3K4me2/3 at active regulatory sites is associated with blocking of DNMT3A activity, presumably by abrogating the capacity of the ADD domain to activate the enzyme and driving focal depletion of mCA.30,42 This blocking of mCA is consistent with regulation of DNMT3A described for mCG deposition in non-neural systems41,43,44 and is likely an important additional determinant of mCA-directed repression of enhancers and gene expression in the mature brain. Notably, disruption of both H3K4 methyltransferases (KMT2A, KMT2C, KMT2D) and demethylases (LSD1, KDM5C) have been observed in individuals with NDD, suggesting additional disorders in which the neuronal DNA methylome may be impacted.45,46 Future studies can dissect the role of H3K4 methylation in patterning mCA and examine if this process contributes to these NDDs.
Interestingly, our random forest algorithm also uncovered H3K27 and H3K9 methylation as predictors of DNMT3A-mediated mCA deposition (Figure 3C,D, S3G). While previous studies have detected a positive association between H3K9 methylation and DNA methylation,47,48 the interplay between H3K27 methylation and DNA methylation is complex and remains incompletely defined. However, recent studies have suggested that H3K36me2 may coordinate with H3K27 methylation and other chromatin marks to dynamically regulate DNMT3A binding and DNA methylation patterning.49–51 Studies investigating the contribution of these other chromatin-associated marks will be important to comprehensively understand the complex role and predictive capacity of H3K36me2 on DNMT3A-mediated mCA patterning in the brain and in neurodevelopmental disease.
Our analysis in cerebral cortex tissue establishes a model for how targeting of mCA is carried out during postnatal development. Notably however, mCA exhibits highly cell-type specific patterns across different types of neurons in the brain.19,52,53 Thus, the patterns we observe here reflect the averages across many cell types. While the mechanisms we uncover provide conceptual insight into how DNMT3A is targeted, these mechanisms are likely to be differentially utilized in cell types to create unique mCA patterns. For example, differential activation of genes can drive expression-associated protection from mCA at different gene sets across cell types.17,54 It will be important to explore how H3K36me2 and regional set-point methylation contribute to shared and distinct mCA patterns across neuronal cell-types.
TBRS and Sotos syndrome are marked by multiple overlapping phenotypes including overgrowth, intellectual disability, and a high penetrance of autism. Our findings suggest that disrupted mCA deposition in broad domains and loss of discrete regions of mCG targeting upon NSD1 or DNMT3A mutation may contribute to shared cognitive and behavioral phenotypes across these disorders. Our analysis indicates that these overlapping epigenetic effects in NSD1 and DNMT3A mutant mice result in concordant gene dysregulation that can drive these nervous system deficits. Overgrowth in these disorders is likely due to epigenetic alterations in dividing cells during early development. The convergence of epigenetic disruption across these disorders both inside and outside the nervous system may present targets for therapies. Notably, in addition to specific megabase-scale domains being affected by NSD1 deletion in the brain, there is a trend toward reduced global mCA upon NSD1 mutation that mimics global reductions observed in TBRS models (Figure S5I). Thus, augmenting existing DNMT3A activity in neurons with genetic approaches or enzyme agonists may ameliorate epigenetic anomalies in both disorders, and may represent a feasible strategy for shared therapeutic development.
While knockdown or conditional deletion of NSD1 alters broad H3K36me2 patterns and mCA in neurons, loss of these marks is incomplete in these conditions. Partial disruption of H3K36me2 could be due, in part, to the acute nature of NSD1 inactivation combined with the long half-life of histones and histone modifications in postmitotic neurons.55 Incomplete loss of NSD1 upon knockdown or potential residual NSD1 catalytic activity due to internal translation initiation of the deleted transcript in our NSD1 cKO mutant may also contribute to the small changes in H3K36me2 we observe. However, given the high expression of other NSD family members in the developing nervous system, it is likely that other H3K36 methyltransferases, including ASH1L and NSD2, can deposit and maintain this mark. Notably, these genes are also mutated in NDD with autistic features or cognitive deficits.11,56 Disruption of these modifiers may lead to both overlapping or distinct effects on H3K36me2 and DNA methylation in the brain compared to loss of NSD1 and DNMT3A. Furthermore, SETD2, the methyltransferase that converts H3K36me2 to H3K36me3 in expressed genes is also mutated in NDD,57 raising the possibility that, in addition to direct effects of loss of H3K36me3, expression-associated mCA depletion in genes may be affected in individuals with SETD2 mutations. Thus, multiple additional genetic causes of NDD may impact the mCA pathway, and alterations in mCA may, in turn, contribute to disease pathology across these additional disorders. Future studies can examine the degree to which H3K36me2 and mCA are altered in models of these disorders, mapping the unique neuronal epigenome as a growing point of convergent disruption across autism and related NDDs.
Limitations of the study
Our study links NSD1 knockdown in neurons and conditional deletion of NSD1 in vivo to alterations in H3K36me2 and resulting disruption of neuronal DNA methylation. However, RNA-seq and RT-PCR quantification of NSD1 exons suggest stable transcript expression after targeted exon deletion (Figure S5E). Furthermore, due to the limitations of anti-rodent NSD1 antibodies including high background signal and the potential for a truncated c-terminal fragment to be expressed but not detected (see methods, Figure S5F,G), it remains possible that low levels of residual NSD1 persist upon knockdown, or that expression of a truncated protein due to an internal ribosome initiation event may maintain some NSD1 activity in vivo. Therefore, our study may underestimate the disruption of H3K36me2 that occurs upon complete loss of NSD1, and subsequent effects on DNMT3A recruitment and mCA deposition may be more substantial under these conditions than our study currently suggests.
Our data support a model in which conversion of H3K36me2 to H3K36me3 causes reduction of DNMT3A targeting and mCA deposition within gene bodies of highly expressed genes due to DNMT3A’s preferential binding affinity for H3K36me2 over H3K36me3. These findings are supported by our genomic analysis and biochemical studies of DNMT3A binding to modified H3 peptides.23,28 However, DNMT3A displays stronger affinity to H3K36me3 compared to unmodified histones in some studies,58 and thus it remains unclear if this additional affinity plays a role in DNMT3A targeting. Future studies can directly disrupt H3K36me3 to test if this mark plays an independent role in DNMT3A targeting.
STAR Methods
Resource Availability
Contact for Reagent and Resource Sharing
Requests for reagents and resources should be directed towards the Lead Contact, Harrison Gabel (gabelh@wustl.edu).
Data and Code Availability
All sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources Table. All unprocessed imaging data have been deposited at Mendeley and are publicly available as of the date of publication. DOI is listed in the Key Resources Table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ab1 = Di-Methyl-Histone H3 (Lys36) (C75H12) Rabbit mAb | Cell Signaling | Cat# 2901; RRID:AB_1030983 |
| ab2 = Anti-dimethyl Histone H3 Antibody (Lys36) | Millipore | Cat# 07–369 RRID:AB_310559 |
| Rabbit polyclonal anti-Dnmt3a | Abcam | Cat# ab2850; RRID:AB_303355 |
| Rabbit polyclonal anti-Histone (tri methyl K36) | Active Motif | Cat# 61101; RRID:AB_2615073 |
| Rabbit polyclonal anti-Histone H3 (mono methyl K4 | Abcam | Cat# ab8895; RRID:AB_306847 |
| Rabbit polyclonal anti-Histone H3 (di methyl K4) | Abcam | Cat# ab7766; RRID:AB_2560996 |
| Rabbit polyclonal anti-Histone H3 (acetyl K27) | Abcam | Cat# ab4729; RRID:AB_2118291 |
| Rabbit polyclonal anti-Histone H3 | Abcam | Cat# ab1791; RRID:AB_302613 |
| Rabbit polyclonal anti-Nuclear Receptor Binding SET Domain Protein 1 (NSD1) | Abbexa | Cat# abx135901 |
| Mouse monoclonal anti-NeuN, clone A60 | Millipore | Cat# MAB377; RRID:AB_2298772 |
| Goat Anti-Rabbit IgG (H+L) Antibody, Alexa Fluor 488 | ThermoFisher | Cat# A-11008; RRID:AB_143165 |
| Goat Anti-Mouse IgG (H+L) Antibody, Alexa Fluor 568 | ThermoFisher | Cat# A-11031; RRID:AB_144696 |
| IRDye 800CW Donkey anti-Rabbit IgG antibody | LI-COR Biosciences | Cat# 926–32213; RRID:AB_621848 |
| Critical Commercial Assays | ||
| Allprep DNA/RNA Mini Kit | Qiagen | Cat#: 80204 |
| NEBNext Ultra Directional RNA Library Prep Kit for Illumina | NEB | Cat#: E7420S |
| NEBNext Mulitplex Oligos for Illumina (Index Primers Set 1) | NEB | Cat#: E7335S |
| NEBNext rRNA Depletion Kit v2 (Human/Mouse/Rat) | NEB | Cat#: E7400X |
| Accel-NGS 2S Plus DNA Library Kit | Swift Biosciences | Cat#: 21024 |
| Accel-NGS 2S Set A Indexing Kit | Swift Biosciences | Cat#: 26148 |
| Accel-NGS 2S Set B Indexing Kit | Swift Biosciences | Cat#: 26248 |
| EZ DNA Methylation-Direct Kit | Zymo Research Corporation | Cat#: D5020 |
| Accel-NGS Methyl-Seq DNA Library Kit | Swift Biosciences | Cat#: 30024 |
| Methyl-Seq Combinatorial Dual Indexing Kit | Swift Biosciences | Cat#: 38096 |
| High-Capacity cDNA Reverse Transcription Kit | ThermoFisher | Cat#: 4368814 |
| Power SYBR Green PCR Master Mix | ThermoFisher | Cat#: 4368577 |
| Deposited Data | ||
| RNA-sequencing data | This paper | GEO: GSE212847 |
| ChIP-sequencing data | This paper | GEO: GSE212847 |
| ChIP-Rx data | This paper | GEO: GSE212847 |
| Bisulfite sequencing data | This paper | GEO: GSE212847 |
| ChIP-sequencing data (H3K27ac) | Clemens et al., 20195 | GEO: GSE123373 |
| ChIP-, Bisulfite-sequencing data | Stroud et al., 201717 | GEO: GSE104298 |
| RNA-sequencing data | Lister et al., 20131 | GEO: GSE47966 |
| Imaging data | This paper | Mendeley DOI: 10.17632/h9393xvpy5.1 |
| Mus musculus mm9 genome assembly | UCSC | http://hgdownload.soe.ucsc.edu/goldenPath/mm9/ |
| Ensembl gene models | UCSC | https://genome.ucsc.edu/cgi-bin/hgTables |
| Experimental Models: Organisms/Strains | ||
| Primary cortical neurons | Derived from E14.5 cortex dissected from pure C57BL6 mice (IMSR Cat# JAX:000664, RRID: IMSR_JAX: 000664) obtained from the Jackson Laboratory. | JAX:000664, RRID: IMSR_JAX:000664 |
| Mouse: NSD1 cKO | Derived from NSD1 fl/fl strain obtained from Shanghai Model Organisms and Nes-Cre62, mice obtained from The Jackson Laboratory (IMSR Cat# JAX: 003771; RRID: IMSR_JAX:003771). | JAX: 003771; RRID:IMSR_JAX:003771 |
| Oligonucleotides | ||
| Actb Forward: AAGGCCAACCGTGAAAAGAT | IDT | N/A |
| Actb Reverse: GTGGTACGACCAGAGGCATAC | IDT | N/A |
| NSD1_exon3–4 Forward: CCCTGCAGGATCTGTTCTGA | IDT | N/A |
| NSD1_exon3–4 Reverse: AAGGGTCTCCAAAAGCCTCT | IDT | N/A |
| NSD1_exon5 Forward: AGAGCTCCCTGCTTGTGGTA | IDT | N/A |
| NSD1_exon5 Reverse: CATCTGGGCTGAAATGGTTT | IDT | N/A |
| NSD1_exon11–12 Forward: CGGCACTCAAGGAGAATGTG | IDT | N/A |
| NSD1_exon11–12 Reverse: CAAGGCACTCCAAGTGGAAA | IDT | N/A |
| NSD1_GT Forward: TATGCAGGCAGAATGCTGTA | IDT | N/A |
| NSD1_GT Reverse: GGGGCCAGAGGGAGTTCAATAC | IDT | N/A |
| NSD1_splicePCR Forward: TCACCATCCACTTCACAGGA | IDT | N/A |
| NSD1_splicePCR Reverse: CACAGGGCTTTTCCTTTTCA | IDT | N/A |
| Recombinant DNA | ||
| pLKO.TRC1.shmNsd1_1, puro | Sigma-Aldrich | TRCN0000123379 |
| pLKO.TRC2.shmNsd1_2, puro | Sigma-Aldrich | TRCN0000441097 |
| pLKO.1-puro Non-Target shRNA Control Transduction Particles | Sigma-Aldrich | SHC016H-1EA |
| Software and Algorithms | ||
| DESeq2 (v1.14.1) | Love et al., 201463 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| edgeR (v3.16.5) | Robinson et al., 201064 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| SAMtools (v1.3) | Li and Durbin, 200965 | https://sourceforge.net/projects/samtools/files/ |
| BEDtools2 (v2.25.0) | Quinlan and Hall, 201066 | https://github.com/arq5x/bedtools2 |
| Bowtie2 (v2.2.5) | Langmead and Salzberg, 201267 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR | Dobin et al., 201368 | https://github.com/alexdobin/STAR |
| fastQC | https://www.bioinformatics.babraham.ac.uk/proiects/fastqc/ | |
| MACS2 (v2.1.0) | Zhang et al., 200869 | https://github.com/taoliu/MACS |
| Trim galore | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ | |
| BS-seeker2 | Guo et al., 201370 | https://github.com/BSSeeker/BSseeker2 |
| Bsmooth | Hansen et al., 201271 | https://github.com/kasperdanielhansen/bsseq |
| RRHO2 | Cahill et al., 2018,38 Plaisier et al., 201061 | https://github.com/RRHO2/RRHO2 |
| clusterProfiler | Wu et al., 2021,39 Yu et al., 201240 | https://git.bioconductor.org/packages/clusterProfiler |
Experimental Model and Subject Details
Mice
Male and female heterozygous Nsd1flx/+ mice were generated by Shanghai Model Organisms. Nsd1flx/flx mice were generated by crossing Nsd1flx/+ males to Nsd1flx/+ females. Female Nsd1flx/flx mice were crossed to male B6.Cg-Tg(Nes-cre)1Kln/J (Nestin-Cre+/−) to generate Nsd1flx/+; Nestin-Cre+/−. Male Nsd1flx/+; Nestin-Cre+/− were then crossed to female Nsd1flx/flx to generate Nsd1flx/flx Tg(Nes-cre)1Kln/J conditional knockout mice (NSD1 Nestin-cKO) at 2 and 8 weeks of age. Genotypes of each mouse was confirmed using primers surrounding floxed region (F: TATGCAGGCAGAATGCTGTA, R: GGGGCCAGAGGGAGTTCAATAC). Relative abundance of NSD1 long isoforms in NSD1 control and cKO mice were assess using primers targeting exons 2 through 5 (F: TCACCATCCACTTCACAGGA, R: CACAGGGCTTTTCCTTTTCA). Validation of reduction in NSD1 protein upon NSD1 loss in culture and in our NSD1 cKO mouse model was attempted via western blot and immunohistochemistry across eight different NSD1 antibodies, but only one antibody appeared to detect specific signal in the nucleus by immunohistochemistry (anti-NSD1, Abbexa abx135901, Figure S5F,G).
Method Details
Total DNA/RNA isolation
Cerebral cortex was dissected on ice in phosphate buffered saline (PBS) from NSD1 Nestin-cko and wild-type litter mates at 8 weeks old. Total DNA and RNA were extracted from 300,000 primary cultured neurons or 1/10 of a whole cortex using RLT+ buffer following the Allprep DNA/RNA Mini Kit (QIAGEN).
qRT-PCR
RNA isolated from neuronal cortical culture or mouse cerebral cortex tissue was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Nsd1 and Actb were measured by qPCR using the Power SYBR Green PCR Master Mix and primers Actb (F: AAGGCCAACCGTGAAAAGAT, R: GTGGTACGACCAGAGGCATAC) and Nsd1 (PNCs - F: CGGCACTCAAGGAGAATGTG, R: CAAGGCACTCCAAGTGGAAA; Cortex tissue - F: CCCTGCAGGATCTGTTCTGA, R: AAGGGTCTCCAAAAGCCTCT). Relative quantity of Actb to Nsd1 cDNA was determined by comparing the Ct of each primer set in each sample to a standard curve and then normalizing the Nsd1 signal by the Actb signal.
Western blotting
Western blotting was performed as previously described.10 Brain tissue samples were homogenized with a dounce homogenizer in lysis buffer (10mM HEPES pH 7.9, 10mM KCl, 1.5mM MgCl2, 1mM DTT, 10mM EDTA). 1% SDS was added to a portion of the lysate. Samples were boiled for 10 minutes, followed by a 10-minute spin at 15,000 g. Supernatant was collected and run through a Wizard Column (Fisher), then diluted in LDS sample buffer with 5% b-mercaptoethanol. Samples were boiled for 5 minutes, 30ug per sample was loaded and run on a 13% acrylamide gel for 90 minutes at 85–100V and transferred to a nitrocellulose membrane at 100V for 60 minutes. Membrane was blocked with 3% bovine serum albumin in TBS-T for 1 hour at room temperature and immunostained with anti-H3K36me2 (1:1000, Cell Signaling 2901) for 12–16 hours at 4 °C. All primary and secondary antibodies were diluted in 3% Bovine Serum Albumin in TBS-T. Membrane was subsequently washed three times with TBS-T and incubated for 1 hour at room temperature with IR-dye secondary antibody (1:15000, IRDye 800CW Donkey anti-Rabbit, LI-COR Biosciences 926–32213). Blot was then washed three times with TBS-T followed by one wash with PBS. Membrane was dried for 20 minutes and then imaged using the LiCOR Odyssey XCL system. Fluorescence was quantified using Image Studio Lite software (LICOR Biosciences). Membrane was subsequently stripped with a mild stripping buffer (1.5% glycine, 0.1% SDS, 1% Tween20, pH 2.2), re-blocked, and immunostained with anti-Histone H3 (1:5000, Abcam ab1791). H3K36me2 levels were normalized to total H3 levels per sample and global loss of H3K36me2 was calculated relative to wildtype per bioreplicate.
Immunofluorescence
Tissue preparation
2-week old wildtype and NSD1 cKO mice were anesthetized and perfused with ice cold PBS and 4% paraformaldehyde (PFA). Brains were dissected and postfixed in 4% PFA overnight at 4 °C. Brains were then dehydrated in 30% sucrose overnight at 4 °C. Brains were placed in cryomold filled with Scigen Tissue-Plus O.C.T. Compound (Fisher, product number: 23–730-571) and flash frozen on dry ice. Embedded tissue was subsequently placed in a precooled cryostat (Leica CM1860 UV) and allowed to equilibrate to −20 °C. All chambers and tools were cleaned with RNase-away and 70% ethanol. Slices were cut coronally at 10 μm.
Staining
Frozen brain sections were washed in PBS, permeabilized with 0.2% Triton X-100 in PBS (PBST) for 5 minutes, and blocked with 10% goat serum in PBS-T for 1 hour at room temperature. Sections were incubated in primary antibodies anti-NSD1 (1:250, Abbexa 135901) and anti-Neun (1:500, Millipore MAB377) overnight at 4 °C. All primary and secondary antibodies were diluted in 1% goat serum in PBS-T. Sections were subsequently washed three times in PBS-T and incubated with secondary antibodies Alexa-Fluor 488 anti-rabbit and Alexa-Fluor 568 anti-mouse (1:500, ThermoFisher A-11008 and A-11031, respectively) at room temperature for 2 hours. Sections were subsequently washed two times with PBS-T and one time with PBS and mounted in DAPI-included VECTASHIELD Antifade Mounting Medium (Vector Laboratories, product number: H-1200–10).
Imaging and Quantification
Sections were imaged using a Zeiss LSM 980 with Airyscan 2 confocal microscope with a 40x water immersion objective. Laser settings were kept constant for each image. Regions of interest (ROI) were defined by a blinded observer using an automatic threshold (Huang, B/W) in FIJI on DAPI signal and the following particle analysis settings - Size: 25-infinity; Circularity: 0.5–1; Show: outlines; Check: Display results, Clear results, Add to manager, Exclude on edges. NeuN signal and NSD1 signal were quantified within all ROIs. A NeuN threshold of >=5000 was used to filter for NeuN+ nuclei across conditions and replicates. 2 separate immunostaining experiments were run, with the upper cortical quadrant of each condition (WT or NSD1 cKO) being quantified across 3 images per experiment. Sample sizes are as follows: WT - 6 images, 374 nuclei (R1: 178 nuclei, R2: 196 nuclei); NSD1 cKO - 6 images, 423 cells (R1: 187 nuclei, R2: 236 nuclei).
Chromatin immunoprecipitation from primary neuron cultures
Cross-linking ChIP in primary neuron cultures was performed using 700,000 cells per immunoprecipitation. Medium was aspirated and cells were cross-linked directly on the plate using 1% paraformaldehyde for 10 min at room temperature with gentle shaking. Glycine was added to quench (final concentration 125 mM, incubated for 5 min at room temperature), then cells were washed twice with cold PBS (with PMSF). To obtain soluble chromatin extract, cells were resuspended in 1ml L1 (50 mM HEPES, 140 mM NaCl, 1 mM EDTA, 1mM EGTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, 1% Na butyrate and 1× Complete protease inhibitor), scraped off the plates, and incubated on ice for 5 min with occasional mixing. Cells were centrifuged and resuspended again in L1. Samples were centrifuged and resuspended in 1ml L2 (10 mM Tris-HCl pH 8.0, 200 mM NaCl, 1% Na butyrate, 1% Triton X-100, 0.5% SDS, and 1× Compete protease inhibitor). Finally, samples were centrifuged and resuspended in 300μl L3 (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 1 mM EGTA, 1% Na butyrate, and 1× Complete protease inhibitor). Chromatin extracts were then sonicated for 30 min (30 cycles: 30s on, 30s off) using a Diagenode Bioruptor sonicator. The lysates were incubated with 15 μl protein A Dynabeads (Invitrogen) bound to anti-H3K36me2 (0.1μg, Cell Signaling 2901) or anti-DNMT3A (4μg, Abcam ab2850) and incubated overnight at 4 °C with 20μl kept as input DNA. Magnetic beads were sequentially washed with low-salt buffer (150 mM NaCl; 0.1% SDS; 1% Triton X-100; 2 mM EDTA and 20 mM Tris-HCl), high salt buffer (500 mM NaCl; 0.1% SDS; 1% Triton X-100; 2 mM EDTA and 20 mM Tris-HCl), LiCl buffer (150 mM LiCl; 0.5% Na deoxycholate; 1% NP-40; 1 mM EDTA and 10 mM Tris-HCl) and TE buffer (1 mM EDTA and 10 mM Tris-HCl). Beads were eluted in TE + 1%SDS and incubated for 30 min at 65 °C for two rounds. The eluate was reverse cross-linked overnight at 65 °C. The eluate was then treated with RNase A for 1 h at 37 °C and with Proteinase K (Roche) for 2 h at 55 °C and DNA was recovered using a Qiagen PCR purification kit.
Chromatin immunoprecipitation from tissue
Chromatin immunoprecipitation was performed as previously described.5,72 Cerebral cortex was dissected on ice in PBS from NSD1 Nestin-cKO and control littermates at 2- and 8-weeks old. The tissue was flash-frozen in liquid nitrogen and stored at −80°C. Chromatin was fragmented with the Covaris S220 sonicator (5% Duty Factory, 140Peak Incidence Power, 200 cycles per burst, milliTUBE 1mL AFA Fiber). After centrifugation, samples were spiked with soluble chromatin from Drosophila S2 cells to comprise 5% of total chromatin in the lysate. ChIP was performed with H3K36me2 antibody (0.1 μg, Cell Signaling 2901), H3K36me3 antibody (0.2 μg, Active motif pAb), H3K27me2 antibody (0.2 μg, Cell Signaling 9728), H3K27me3 antibody (0.5 μg, Active motif pAb), H3K27ac antibody (0.1μg; Abcam ab4729), and DNMT3A antibody (4 μg, Abcam ab2850). Libraries were generated using Accel-NGS 2S Plus DNA Library Kit (Swift Biosciences). Libraries were pooled to a final concentration of 10nM and sequenced using Illumina HiSeq 3000 with the Genome Technology Access Center at Washington University in St. Louis, typically yielding 15–40 million single-end reads per sample.
Whole genome bisulfite sequencing
DNA was isolated from primary neuron cultures or mouse brain tissue using the Allprep DNA/RNA Mini Kit (QIAGEN). Libraries were prepared for sequencing using the Accel-NGS Methyl-Seq DNA Library Kit (Swift, 30024) and the EZ DNA Methylation-Direct Kit (Zymo, D5020) was used for bisulfite conversion. For these samples, 50 ng of DNA was fragmented for 45 s with the Covaris S220 sonicator (10% Duty Factory, 175 Peak Incidence Power, 200 cycles per burst, microTUBE 200μL AFA Fiber). DNA was then purified using 0.7 volumes of SPRISelect Beads (Beckman Coulter Life Sciences) to select for long DNA inserts for sequencing. Samples underwent bisulfite conversion under the following cycling conditions: 98°C, 8 min; 64°C, 4.25 h, 4°C hold. Libraries were PCR-amplified for 10 cycles. Libraries were then pooled to a final concentration of 5nM and sequenced using Illumina HiSeq 3000 with the Genome Technology Access Center at Washington University in St. Louis, typically yielding 100 million paired-end reads per sample.
Quantification and Statistical Analysis
Machine Learning
Random forest models were built with python sklearn with the parameters n_estimators=100, min_samples_split=2, criterion=‘mse’. 5-fold cross-validation was used to evaluate performance. Individual models were built and assessed for each set of regions, using input-normalized values for the ChIP-signal in each region. Region orders were randomized before cross-validation, using random.seed(9115).
Feature importances of recursive feature elimination were plotted as a series of stacked barplots, with their heights scaled to the accuracy of the model with the given number of features removed. Linear models were built with R using input-normalized data.
Whole-genome bisulfite analysis
Bisulfite sequencing analysis was performed as previously described,5 with the addition of nonconversion correction for regional %mC assessment. Briefly, data were adaptor-trimmed, mapped to mm9, then deduplicated and called for methylation using BS-seeker2. Methylation levels across regions were assessed using bedtools map -o sum, summing the number of reads mapping to Cs (supporting mC) and the number of reads mapping to Cs + Ts (supporting C) in the region, then dividing those two numbers.66 The coverage from split sequencing runs were pooled together before assessing %mC, while the %mC values from biological replicates were averaged together.
Regions were adjusted for nonconversion rate as measured by % methylation in Lambda spike-ins per-sample, as in Lister et al., 2013.1 Briefly, following %mC calculation, the lambda % methylation value was subtracted from the calculated %mC value. If the corrected value was below 0, the %mC value was brought up to 0.
RNA sequencing analysis
RNA sequencing analysis was performed as previously described.5 Briefly, FASTQ files were adapter and quality-trimmed with Trim Galore and filtered for rRNA sequences with Bowtie. The remaining reads were aligned to the mm9 genome with STAR.68 Reads mapping to multiple regions were removed, and the remaining uniquely mapping reads were converted to BED files and separated by intronic and exonic reads. These BED files were then used to assess coverage in genes using bedtools coverage -counts.
DESeq2 was used to identify significant differentially expressed genes (FDR<0.1) in the control versus NSD1 cKO cerebral cortex.
Chromatin immunoprecipitation analysis
ChIP sequencing analysis was performed as previously described.5 Briefly, reads were mapped to mm9 with bowtie2, then deduplicated with picardtools MarkDuplicates. bedtools coverage - counts was used to assess ChIP signal at the various genomic regions examined.
The global change in ChIP enrichment was calculated using ChIP-Rx, using an exogenously added Drosophila chromatin (5% of sample) as an internal control.36 For each ChIP-seq sample, the ChIP-Rx ratio was calculated as follows:
in which SampleP and SampleDP refer to the percentages of reads that aligned to the mouse (mm9) and Drosophila (dm6) genome, respectively (likewise, InputP and InputDP refer to the percentage of input that aligned to each genome). H3K36me2 ChIP-seq track (Figure 5A) was normalized using bedtools genomecov -scale with the scale value equal to the percent global change in H3K36me2 enrichment in the NSD1 cKO per bioreplicate (5 bioreplicates from ab1).
Controlled resampling
A similar resampling approach was used as previously described.5 Briefly, genes were sorted based on some criteria (e.g. expression). Then, for every entry in a set of interest (e.g. NSD1 cKO upregulated genes), an entry from a control set (e.g. all other genes) with a similar rank as the entry of interest was taken, generating a control set of the same size and variable distribution as the set of interest.
TAD cross-correlation
Cross-correlation matrices (e.g., Figure 4E, Figure S2A) were generated as previously described.5 Briefly, each TAD was divided into 10 equally-sized bins. 10 identically sized bins were subsequently appended up- and downstream of the TAD, resulting in a single matrix of 30 bins. Each column of this matrix was then correlated against each other, making a 30 × 30 correlation matrix, which was plotted in heatmap form.
To generate heatmaps of correlations of fold-changes of H3K36me2, DNMT3A, and mCA of regions inside versus outside of TADs (Figure 4E) as well as H3K36me2 levels in and around all TADs (Figure S2A), each element was paired to each other element on the same chromosome. Each pair was then assessed if they paired within or between TADs. Because H3K36me2, DNMT3A, and mCA vary with genomic distance, each intra-TAD pair was matched to the inter-TAD pair with the most similar distance between elements. Spearman correlations were then calculated on the two distance-matched sets.
Identification of NSD1 changed regions
Regions (10kb bins across the genome, TADs) were assessed for ChIP signal in control and NSD1 knockdown or NSD1 cKO samples. edgeR was used to identify regions with significantly changed enrichment of H3K36me2 (FDR <0.1).
Tiling of genome into 5kb and 10kb bins
Genome was divided into 5kb and 10kb bins with a method similar to Stroud et al., 2017.17 Briefly, bins were placed throughout the genome and labeled in a gene-centric manner – using a sorted annotation file of genes, bins were iteratively placed at the center of a gene’s TSS, then downstream of the TSS to the TES if the gene was long enough to allow at least one bin inside it. Bins were then placed immediately downstream of the gene’s TES until the bins ran into the TSS of another gene, at which point the process repeats itself until the chromosome ends.
Rank-rank hypergeometric overlap (RRHO) analysis
For each genotype, an input gene list was created, such that the first column contained the gene name and the second column contained the gene score. The gene score was calculated as −log10(pvalue) * sign(log2FC) using values obtained from the DESeq2 output for each gene. RRHO2_initialize() was used to generate RRHO object. RRHO2_heatmap() was used to generate a heatmap of overlapping genes between two genotypes.
Over-representation analysis with clusterProfiler
A list of genes with overlapping effects (“dd” = gene in both genotypes is downregulated, “uu” gene in both genotypes is upregulated) were extracted from the RRHO object generated using RRHO2_initialize(). enrichGO() with parameters pAdjustMethod = “fdr”, ont = “ALL”, and default minGSSize and maxGSSize was used to obtain significant GO terms in each category (“BP”, “MF”, “CC”). dotplot() with parameters showCategory=5 and split = “ONTOLOGY” was used to visualize the top five significantly enriched GO terms in each category.
Supplementary Material
Table S1. Table of in vitro primary neuron culture edgeR ChIP signal outputs at each genomic region. Related to Figure 4.
Table S2. Table of in vitro primary neuron culture DNA methylation levels at each genomic region. Related to Figure 4.
Table S3. Table of in vivo NSD1 control and cKO cerebral cortex edgeR ChIP signal outputs at each genomic region. Related to Figure 5.
Table S4. Table of in vivo NSD1 control and cKO cerebral cortex DNA methylation levels at each genomic region. Related to Figure 5.
Table S5. Table of in vivo NSD1 control and cKO cerebral cortex differential gene expression and DESeq2 outputs. Related to Figure 6.
Highlights:
Genome topology and gene expression independently pattern neuronal non-CG DNA methylation
Histone H3 lysine 36 methylation targets neuronal non-CG DNA methylation by DNMT3A
NSD1 loss causes shared epigenetic and transcriptomic changes with DNMT3A disorder models
Acknowledgements
We thank members of the Gabel lab, as well as J. Edwards, N. Mosammaparast, and J. Yi, for providing support and feedback on the manuscript. Next-Generation-Sequencing was carried out through the Genome Technology Access Center at the McDonnell Genome Institute and The Edison Family Center for Genome Sciences and Systems Biology at Washington University in St. Louis. This work was supported by NIH NICHD 1F30HD110156–01 to N.H. and NIMH R01MH117405 and NINDS R01NS041021 to H.W.G.
Diversity Statement
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper self-identifies as a gender minority in their field of research. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References Cited
- 1.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. (2013). Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 341, 1237905. 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gowher H, and Jeltsch A (2001). Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpA sites1 1Edited by J. Karn. J Mol Biol 309, 1201–1208. 10.1006/jmbi.2001.4710. [DOI] [PubMed] [Google Scholar]
- 3.Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, and Greenberg ME (2015). Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93. 10.1038/nature14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lagger S, Connelly JC, Schweikert G, Webb S, Selfridge J, Ramsahoye BH, Yu M, He C, Sanguinetti G, Sowers LC, et al. (2017). MeCP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. Plos Genet 13, e1006793. 10.1371/journal.pgen.1006793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clemens AW, Wu DY, Moore JR, Christian DL, Zhao G, and Gabel HW (2019). MeCP2 Represses Enhancers through Chromosome Topology-Associated DNA Methylation. Mol Cell 77, 279–293.e8. 10.1016/j.molcel.2019.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boxer LD, Renthal W, Greben AW, Whitwam T, Silberfeld A, Stroud H, Li E, Yang MG, Kinde B, Griffith EC, et al. (2020). MeCP2 Represses the Rate of Transcriptional Initiation of Highly Methylated Long Genes. Mol Cell 77, 294–309.e9. 10.1016/j.molcel.2019.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Duarte S del V, Zachariou A, Hanks S, O’Brien E, Aksglaede L, et al. (2014). Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet 46, 385–388. 10.1038/ng.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amir RE, Veyver I.B.V. den, Wan M, Tran CQ, Francke U, and Zoghbi HY (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 23, 185–188. 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 9.Esch HV, Bauters M, Ignatius J, Jansen M, Raynaud M, Hollanders K, Lugtenberg D, Bienvenu T, Jensen LR, Gécz J, et al. (2005). Duplication of the MECP2 Region Is a Frequent Cause of Severe Mental Retardation and Progressive Neurological Symptoms in Males. Am J Hum Genetics 77, 442–453. 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christian DL, Wu DY, Martin JR, Moore JR, Liu YR, Clemens AW, Nettles SA, Kirkland NM, Papouin T, Hill CA, et al. (2020). DNMT3A Haploinsufficiency Results in Behavioral Deficits and Global Epigenomic Dysregulation Shared across Neurodevelopmental Disorders. Cell Reports 33, 108416. 10.1016/j.celrep.2020.108416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Satterstrom FK, Kosmicki JA, Wang J, Breen MS, Rubeis SD, An J-Y, Peng M, Collins R, Grove J, Klei L, et al. (2020). Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584.e23. 10.1016/j.cell.2019.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. (2014). The contribution of de novo coding mutations to autism spectrum disorder. Nature 515, 216–221. 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, Nagai T, Ohashi H, Naritomi K, Tsukahara M, Makita Y, et al. (2002). Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet 30, 365–366. 10.1038/ng863. [DOI] [PubMed] [Google Scholar]
- 15.Douglas J, Hanks S, Temple IK, Davies S, Murray A, Upadhyaya M, Tomkins S, Hughes HE, Cole RPT, and Rahman N (2003). NSD1 Mutations Are the Major Cause of Sotos Syndrome and Occur in Some Cases of Weaver Syndrome but Are Rare in Other Overgrowth Phenotypes. Am J Hum Genetics 72, 132–143. 10.1086/345647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A, Elliott A, Wylie H, Ardissone A, Rittinger O, et al. (2017). Mutations in Epigenetic Regulation Genes Are a Major Cause of Overgrowth with Intellectual Disability. Am J Hum Genetics 100, 725–736. 10.1016/j.ajhg.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stroud H, Su SC, Hrvatin S, Greben AW, Renthal W, Boxer LD, Nagy MA, Hochbaum DR, Kinde B, Gabel HW, et al. (2017). Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 171, 1151–1164.e16. 10.1016/j.cell.2017.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clemens AW, and Gabel HW (2020). Emerging Insights into the Distinctive Neuronal Methylome. Trends Genet. 10.1016/j.tig.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, et al. (2015). Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 86, 1369–1384. 10.1016/j.neuron.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Zhong C, Hu S, Le T, Fan G, et al. (2014). Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci 17, 215–222. 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, Chen X, and Lu C (2021). The interplay between DNA and histone methylation: molecular mechanisms and disease implications. Embo Rep 22, e51803. 10.15252/embr.202051803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu K, Bonora G, and Pellegrini M (2019). Interactions between core histone marks and DNA methyltransferases predict DNA methylation patterns observed in human cells and tissues. Epigenetics 15, 1–11. 10.1080/15592294.2019.1666649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, Horth C, McGuire JT, Xu X, Nikbakht H, et al. (2019). The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281–286. 10.1038/s41586-019-1534-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinberg DN, Rosenbaum P, Chen X, Barrows D, Horth C, Marunde MR, Popova IK, Gillespie ZB, Keogh M-C, Lu C, et al. (2021). Two competing mechanisms of DNMT3A recruitment regulate the dynamics of de novo DNA methylation at PRC1-targeted CpG islands. Nat Genet, 1–7. 10.1038/s41588-021-00856-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu H, Coskun V, Tao J, Xie W, Ge W, Yoshikawa K, Li E, Zhang Y, and Sun YE (2010). Dnmt3a-Dependent Nonpromoter DNA Methylation Facilitates Transcription of Neurogenic Genes. Science 329, 444–448. 10.1126/science.1190485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tatton-Brown K, Zachariou A, Loveday C, Renwick A, Mahamdallie S, Aksglaede L, Baralle D, Barge-Schaapveld D, Blyth M, Bouma M, et al. ; Clinical Assessment of the Utility of Sequencing and Evaluation as a Ser-vice (CAUSES) Research Study; Deciphering Developmental Disorders (DDD) Study (2018). The Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants. Wellcome Open Research, 3:46. 10.12688/wellcomeopenres.14430.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner EJ, and Carpenter PB (2012). Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Bio 13, 115–126. 10.1038/nrm3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dukatz M, Holzer K, Choudalakis M, Emperle M, Lungu C, Bashtrykov P, and Jeltsch A (2019). H3K36me2/3 binding and DNA binding of the DNA methyltransferase DNMT3A PWWP domain both contribute to its chromatin interaction. J Mol Biol 431, 5063–5074. 10.1016/j.jmb.2019.09.006. [DOI] [PubMed] [Google Scholar]
- 29.Xu W, Li J, Rong B, Zhao B, Wang M, Dai R, Chen Q, Liu H, Gu Z, Liu S, et al. (2020). Correction to: DNMT3A reads and connects histone H3K36me2 to DNA methylation. Protein Cell 11, 230–230. 10.1007/s13238-019-00678-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo X, Wang L, Li J, Ding Z, Xiao J, Yin X, He S, Shi P, Dong L, Li G, et al. (2015). Structural insight into autoinhibition and histone H3-induced activation of DNMT3A. Nature 517, 640–644. 10.1038/nature13899. [DOI] [PubMed] [Google Scholar]
- 31.Noh K-M, Wang H, Kim HR, Wenderski W, Fang F, Li CH, Dewell S, Hughes SH, Melnick AM, Patel DJ, et al. (2015). Engineering of a Histone-Recognition Domain in Dnmt3a Alters the Epigenetic Landscape and Phenotypic Features of Mouse ESCs. Mol Cell 59, 89–103. 10.1016/j.molcel.2015.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dietterich TG (1998). Approximate Statistical Tests for Comparing Supervised Classification Learning Algorithms. Neural Comput 10, 1895–1923. 10.1162/089976698300017197. [DOI] [PubMed] [Google Scholar]
- 33.Liaw A, and Wiener M (2002). Classification and Regression by randomForest. R News 2/3, 18–22. [Google Scholar]
- 34.Stone M (1974). Cross‐Validatory Choice and Assessment of Statistical Predictions. J Royal Statistical Soc Ser B Methodol 36, 111–133. 10.1111/j.2517-6161.1974.tb00994.x. [DOI] [Google Scholar]
- 35.Nikolayeva O, and Robinson MD (2014). Stem Cell Transcriptional Networks, Methods and Protocols. Methods Mol Biology 1150, 45–79. 10.1007/978-1-4939-0512-6_3. [DOI] [PubMed] [Google Scholar]
- 36.Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq Normalization Reveals Global Modulation of the Epigenome. Cell Reports 9, 1163–1170. 10.1016/j.celrep.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 37.Nord AS, and West AE (2020). Neurobiological functions of transcriptional enhancers. Nat Neurosci 23, 5–14. 10.1038/s41593-019-0538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cahill KM, Huo Z, Tseng GC, Logan RW, and Seney ML (2018). Improved identification of concordant and discordant gene expression signatures using an updated rank-rank hypergeometric overlap approach. Sci Rep-uk 8, 9588. 10.1038/s41598-018-27903-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu T, Hu E, Xu S, Chen M, Guo P, Dai Z, Feng T, Zhou L, Tang W, Zhan L, et al. (2021). clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2, 100141. 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. Omics J Integr Biology 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edwards JR, O’Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, Chanrion B, Fu Y, Su T, Hibshoosh H, et al. (2010). Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res 20, 972–980. 10.1101/gr.101535.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li B-Z, Huang Z, Cui Q-Y, Song X-H, Du L, Jeltsch A, Chen P, Li G, Li E, and Xu G-L (2011). Histone tails regulate DNA methylation by allosterically activating de novo methyltransferase. Cell Res 21, 1172–1181. 10.1038/cr.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ooi SKT, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin S-P, Allis CD, et al. (2007). DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448, 714–717. 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Jurkowska R, Soeroes S, Rajavelu A, Dhayalan A, Bock I, Rathert P, Brandt O, Reinhardt R, Fischle W, et al. (2010). Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res 38, 4246–4253. 10.1093/nar/gkq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Faundes V, Newman WG, Bernardini L, Canham N, Clayton-Smith J, Dallapiccola B, Davies SJ, Demos MK, Goldman A, Gill H, et al. (2018). Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am J Hum Genetics 102, 175–187. 10.1016/j.ajhg.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vallianatos CN, and Iwase S (2015). Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics-uk 7, 503–519. 10.2217/epi.15.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. (2008). Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 454, 766–770. 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Epsztejn-Litman S, Feldman N, Abu-Remaileh M, Shufaro Y, Gerson A, Ueda J, Deplus R, Fuks F, Shinkai Y, Cedar H, et al. (2008). De novo DNA methylation promoted by G9a prevents reprogramming of embryonically silenced genes. Nat Struct Mol Biol 15, 1176–1183. 10.1038/nsmb.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen H, Hu B, Horth C, Bareke E, Rosenbaum P, Kwon SY, Sirois J, Weinberg DN, Robison FM, Garcia BA, et al. (2022). H3K36 dimethylation shapes the epigenetic interaction landscape by directing repressive chromatin modifications in embryonic stem cells. Genome Res 32, gr.276383.121. 10.1101/gr.276383.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Streubel G, Watson A, Jammula SG, Scelfo A, Fitzpatrick DJ, Oliviero G, McCole R, Conway E, Glancy E, Negri GL, et al. (2018). The H3K36me2 Methyltransferase Nsd1 Demarcates PRC2-Mediated H3K27me2 and H3K27me3 Domains in Embryonic Stem Cells. Mol Cell 70, 371–379.e5. 10.1016/j.molcel.2018.02.027. [DOI] [PubMed] [Google Scholar]
- 51.Gu T, Hao D, Woo J, Huang T-W, Guo L, Lin X, Guzman AG, Tovy A, Rosas C, Jeong M, et al. (2022). The disordered N-terminal domain of DNMT3A recognizes H2AK119ub and is required for postnatal development. Nat Genet 54, 625–636. 10.1038/s41588-022-01063-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu H, Zhou J, Tian W, Luo C, Bartlett A, Aldridge A, Lucero J, Osteen JK, Nery JR, Chen H, et al. (2021). DNA methylation atlas of the mouse brain at single-cell resolution. Nature 598, 120–128. 10.1038/s41586-020-03182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luo C, Keown CL, Kurihara L, Zhou J, He Y, Li J, Castanon R, Lucero J, Nery JR, Sandoval JP, et al. (2017). Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604. 10.1126/science.aan3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stroud H, Yang MG, Tsitohay YN, Davis CP, Sherman MA, Hrvatin S, Ling E, and Greenberg ME (2020). An Activity-Mediated Transition in Transcription in Early Postnatal Neurons. Neuron. 10.1016/j.neuron.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maze I, Wenderski W, Noh K-M, Bagot RC, Tzavaras N, Purushothaman I, Elsässer SJ, Guo Y, Ionete C, Hurd YL, et al. (2015). Critical Role of Histone Turnover in Neuronal Transcription and Plasticity. Neuron 87, 77–94. 10.1016/j.neuron.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bergemann AD, Cole F, and Hirschhorn K (2005). The etiology of Wolf–Hirschhorn syndrome. Trends Genet 21, 188–195. 10.1016/j.tig.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 57.Luscan A, Laurendeau I, Malan V, Francannet C, Odent S, Giuliano F, Lacombe D, Touraine R, Vidaud M, Pasmant E, et al. (2014). Mutations in SETD2 cause a novel overgrowth condition. J Med Genet 51, 512. 10.1136/jmedgenet-2014-102402. [DOI] [PubMed] [Google Scholar]
- 58.Xu W, Li J, Rong B, Zhao B, Wang M, Dai R, Chen Q, Liu H, Gu Z, Liu S, et al. (2020). DNMT3A reads and connects histone H3K36me2 to DNA methylation. Protein Cell 11, 150–154. 10.1007/s13238-019-00672-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonev B, Cohen NM, Szabo Q, Fritsch L, Papadopoulos GL, Lubling Y, Xu X, Lv X, Hugnot J-P, Tanay A, et al. (2017). Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 171, 557–572.e24. 10.1016/j.cell.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, and Ren B (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Plaisier SB, Taschereau R, Wong JA, and Graeber TG (2010). Rank–rank hypergeometric overlap: identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res 38, e169–e169. 10.1093/nar/gkq636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, and Schütz G (1999). Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet 23, 99–103. 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- 63.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9, R137. 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo W, Fiziev P, Yan W, Cokus S, Sun X, Zhang MQ, Chen P-Y, and Pellegrini M (2013). BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. Bmc Genomics 14, 774. 10.1186/1471-2164-14-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hansen KD, Langmead B, and Irizarry RA (2012). BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol 13, R83. 10.1186/gb-2012-13-10-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cohen S, Gabel HW, Hemberg M, Hutchinson AN, Sadacca LA, Ebert DH, Harmin DA, Greenberg RS, Verdine VK, Zhou Z, et al. (2011). Genome-Wide Activity-Dependent MeCP2 Phosphorylation Regulates Nervous System Development and Function. Neuron 72, 72–85. 10.1016/j.neuron.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Table of in vitro primary neuron culture edgeR ChIP signal outputs at each genomic region. Related to Figure 4.
Table S2. Table of in vitro primary neuron culture DNA methylation levels at each genomic region. Related to Figure 4.
Table S3. Table of in vivo NSD1 control and cKO cerebral cortex edgeR ChIP signal outputs at each genomic region. Related to Figure 5.
Table S4. Table of in vivo NSD1 control and cKO cerebral cortex DNA methylation levels at each genomic region. Related to Figure 5.
Table S5. Table of in vivo NSD1 control and cKO cerebral cortex differential gene expression and DESeq2 outputs. Related to Figure 6.
Data Availability Statement
All sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the Key Resources Table. All unprocessed imaging data have been deposited at Mendeley and are publicly available as of the date of publication. DOI is listed in the Key Resources Table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ab1 = Di-Methyl-Histone H3 (Lys36) (C75H12) Rabbit mAb | Cell Signaling | Cat# 2901; RRID:AB_1030983 |
| ab2 = Anti-dimethyl Histone H3 Antibody (Lys36) | Millipore | Cat# 07–369 RRID:AB_310559 |
| Rabbit polyclonal anti-Dnmt3a | Abcam | Cat# ab2850; RRID:AB_303355 |
| Rabbit polyclonal anti-Histone (tri methyl K36) | Active Motif | Cat# 61101; RRID:AB_2615073 |
| Rabbit polyclonal anti-Histone H3 (mono methyl K4 | Abcam | Cat# ab8895; RRID:AB_306847 |
| Rabbit polyclonal anti-Histone H3 (di methyl K4) | Abcam | Cat# ab7766; RRID:AB_2560996 |
| Rabbit polyclonal anti-Histone H3 (acetyl K27) | Abcam | Cat# ab4729; RRID:AB_2118291 |
| Rabbit polyclonal anti-Histone H3 | Abcam | Cat# ab1791; RRID:AB_302613 |
| Rabbit polyclonal anti-Nuclear Receptor Binding SET Domain Protein 1 (NSD1) | Abbexa | Cat# abx135901 |
| Mouse monoclonal anti-NeuN, clone A60 | Millipore | Cat# MAB377; RRID:AB_2298772 |
| Goat Anti-Rabbit IgG (H+L) Antibody, Alexa Fluor 488 | ThermoFisher | Cat# A-11008; RRID:AB_143165 |
| Goat Anti-Mouse IgG (H+L) Antibody, Alexa Fluor 568 | ThermoFisher | Cat# A-11031; RRID:AB_144696 |
| IRDye 800CW Donkey anti-Rabbit IgG antibody | LI-COR Biosciences | Cat# 926–32213; RRID:AB_621848 |
| Critical Commercial Assays | ||
| Allprep DNA/RNA Mini Kit | Qiagen | Cat#: 80204 |
| NEBNext Ultra Directional RNA Library Prep Kit for Illumina | NEB | Cat#: E7420S |
| NEBNext Mulitplex Oligos for Illumina (Index Primers Set 1) | NEB | Cat#: E7335S |
| NEBNext rRNA Depletion Kit v2 (Human/Mouse/Rat) | NEB | Cat#: E7400X |
| Accel-NGS 2S Plus DNA Library Kit | Swift Biosciences | Cat#: 21024 |
| Accel-NGS 2S Set A Indexing Kit | Swift Biosciences | Cat#: 26148 |
| Accel-NGS 2S Set B Indexing Kit | Swift Biosciences | Cat#: 26248 |
| EZ DNA Methylation-Direct Kit | Zymo Research Corporation | Cat#: D5020 |
| Accel-NGS Methyl-Seq DNA Library Kit | Swift Biosciences | Cat#: 30024 |
| Methyl-Seq Combinatorial Dual Indexing Kit | Swift Biosciences | Cat#: 38096 |
| High-Capacity cDNA Reverse Transcription Kit | ThermoFisher | Cat#: 4368814 |
| Power SYBR Green PCR Master Mix | ThermoFisher | Cat#: 4368577 |
| Deposited Data | ||
| RNA-sequencing data | This paper | GEO: GSE212847 |
| ChIP-sequencing data | This paper | GEO: GSE212847 |
| ChIP-Rx data | This paper | GEO: GSE212847 |
| Bisulfite sequencing data | This paper | GEO: GSE212847 |
| ChIP-sequencing data (H3K27ac) | Clemens et al., 20195 | GEO: GSE123373 |
| ChIP-, Bisulfite-sequencing data | Stroud et al., 201717 | GEO: GSE104298 |
| RNA-sequencing data | Lister et al., 20131 | GEO: GSE47966 |
| Imaging data | This paper | Mendeley DOI: 10.17632/h9393xvpy5.1 |
| Mus musculus mm9 genome assembly | UCSC | http://hgdownload.soe.ucsc.edu/goldenPath/mm9/ |
| Ensembl gene models | UCSC | https://genome.ucsc.edu/cgi-bin/hgTables |
| Experimental Models: Organisms/Strains | ||
| Primary cortical neurons | Derived from E14.5 cortex dissected from pure C57BL6 mice (IMSR Cat# JAX:000664, RRID: IMSR_JAX: 000664) obtained from the Jackson Laboratory. | JAX:000664, RRID: IMSR_JAX:000664 |
| Mouse: NSD1 cKO | Derived from NSD1 fl/fl strain obtained from Shanghai Model Organisms and Nes-Cre62, mice obtained from The Jackson Laboratory (IMSR Cat# JAX: 003771; RRID: IMSR_JAX:003771). | JAX: 003771; RRID:IMSR_JAX:003771 |
| Oligonucleotides | ||
| Actb Forward: AAGGCCAACCGTGAAAAGAT | IDT | N/A |
| Actb Reverse: GTGGTACGACCAGAGGCATAC | IDT | N/A |
| NSD1_exon3–4 Forward: CCCTGCAGGATCTGTTCTGA | IDT | N/A |
| NSD1_exon3–4 Reverse: AAGGGTCTCCAAAAGCCTCT | IDT | N/A |
| NSD1_exon5 Forward: AGAGCTCCCTGCTTGTGGTA | IDT | N/A |
| NSD1_exon5 Reverse: CATCTGGGCTGAAATGGTTT | IDT | N/A |
| NSD1_exon11–12 Forward: CGGCACTCAAGGAGAATGTG | IDT | N/A |
| NSD1_exon11–12 Reverse: CAAGGCACTCCAAGTGGAAA | IDT | N/A |
| NSD1_GT Forward: TATGCAGGCAGAATGCTGTA | IDT | N/A |
| NSD1_GT Reverse: GGGGCCAGAGGGAGTTCAATAC | IDT | N/A |
| NSD1_splicePCR Forward: TCACCATCCACTTCACAGGA | IDT | N/A |
| NSD1_splicePCR Reverse: CACAGGGCTTTTCCTTTTCA | IDT | N/A |
| Recombinant DNA | ||
| pLKO.TRC1.shmNsd1_1, puro | Sigma-Aldrich | TRCN0000123379 |
| pLKO.TRC2.shmNsd1_2, puro | Sigma-Aldrich | TRCN0000441097 |
| pLKO.1-puro Non-Target shRNA Control Transduction Particles | Sigma-Aldrich | SHC016H-1EA |
| Software and Algorithms | ||
| DESeq2 (v1.14.1) | Love et al., 201463 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html |
| edgeR (v3.16.5) | Robinson et al., 201064 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| SAMtools (v1.3) | Li and Durbin, 200965 | https://sourceforge.net/projects/samtools/files/ |
| BEDtools2 (v2.25.0) | Quinlan and Hall, 201066 | https://github.com/arq5x/bedtools2 |
| Bowtie2 (v2.2.5) | Langmead and Salzberg, 201267 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| STAR | Dobin et al., 201368 | https://github.com/alexdobin/STAR |
| fastQC | https://www.bioinformatics.babraham.ac.uk/proiects/fastqc/ | |
| MACS2 (v2.1.0) | Zhang et al., 200869 | https://github.com/taoliu/MACS |
| Trim galore | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ | |
| BS-seeker2 | Guo et al., 201370 | https://github.com/BSSeeker/BSseeker2 |
| Bsmooth | Hansen et al., 201271 | https://github.com/kasperdanielhansen/bsseq |
| RRHO2 | Cahill et al., 2018,38 Plaisier et al., 201061 | https://github.com/RRHO2/RRHO2 |
| clusterProfiler | Wu et al., 2021,39 Yu et al., 201240 | https://git.bioconductor.org/packages/clusterProfiler |