

Summary
Associations between human genetic variation and clinical phenotypes have become a foundation of biomedical research. Most repositories of these data seek to be disease-agnostic and therefore lack disease-focused views. The Type 2 Diabetes Knowledge Portal (T2DKP) is a public resource of genetic datasets and genomic annotations dedicated to type 2 diabetes (T2D) and related traits. Here, we seek to make the T2DKP more accessible to prospective users and more useful to existing users. First, we evaluate the T2DKP’s comprehensiveness by comparing its datasets to those of other repositories. Second, we describe how researchers unfamiliar with human genetic data can begin using and correctly interpreting them via the T2DKP. Third, we describe how existing users can extend their current workflows to use the full suite of tools offered by the T2DKP. We finally discuss the lessons offered by the T2DKP toward the goal of democratizing access to complex disease genetic results.
Graphical Abstract
Introduction
Genome wide association studies (GWAS)1 have elucidated complex trait genetic architectures2, improved disease risk prediction3, established causal relationships among traits4, and identified potential therapeutic targets5. Many such insights, however, are not apparent from published results and require downstream analyses of the full set of GWAS associations6. These analyses often incorporate auxiliary data to help interpret the molecular and cellular effects of variants within noncoding regions of the genome, which constitute the vast majority of statistically significant associations7.
Downstream analyses of GWAS results predominantly draw from four types of resource. First, full sets of GWAS summary statistics – that is, p-values, allele frequencies, and effect sizes and their standard errors for every variant in a GWAS – are increasingly available for download from consortium websites or other public resources (e.g., the NHGRI-EBI GWAS Catalog8, GRASP9, the GWAS Atlas10, the Global Biobank Engine11, genebass12, PheGenI13, and the IEU OpenGWAS project14). Second, genomic annotations – which help interpret variant effects on molecular or cellular processes – include genomic features (e.g., the ENSEMBL15 and UCSC genome browsers16); epigenomic annotations (e.g., from the ENCODE17 and Roadmap Epigenomics18 Projects); transcriptomic annotations (e.g., the GEO19, GTEx20 portal, or SCAN database21); proteomic, protein-protein interaction, and pathway datasets (e.g., UniProt22, STRING23, and MSigDB24); and perturbational datasets (e.g., the IMPC25 and MGI26 databases). Third, many bioinformatic methods integrate summary statistics and genomic annotations to draw insights from GWAS associations6: some methods have dedicated web portals (e.g., LD Hub27, MR-Base28, PredictDB29) and some have been integrated within more general “post-GWAS” analysis platforms (e.g., FUMA30, Open Targets31). Fourth, expert knowledge about genetic associations or their biology is often extractable from the literature through manual curation (e.g., OMIM32, ClinGen33, HGMD34), automated text mining (e.g., PubTator35, SemMedDB36), or a combination of the two (e.g., GWASkb37).
Beginning in 2015, we developed the Type 2 Diabetes Knowledge Portal (T2DKP) to address two gaps in the capabilities of these resources from the perspective of type 2 diabetes (T2D) researchers. First, many disease-specific datasets – for example, GWAS of disease-specific endophenotypes38 or genomic annotations in highly specific cell populations39 – are too narrow in scope to be identified and included by disease-agnostic resources. Second, many analyses can only be conducted after data from disparate resources are combined – for example, decisions to experimentally characterize variants at a GWAS locus are influenced not only by GWAS association strength but also by genomic annotations of variant regulatory effects and bioinformatic predictions of downstream “effector” genes40. The T2DKP addresses these gaps by aggregating and integrating data and methods of many types, while focusing on only one disease area (T2D and related traits).
The T2DKP was the main output of the Accelerating Medicines Partnership® in Type 2 Diabetes (AMP®-T2D), a five-year public-private partnership launched in 2014 to generate, analyze, and “democratize” access to genetic and genomic data for T2D-relevant traits. The ~100 scientists within AMP-T2D – many of whom have led advances in genetic mapping and functional study of T2D over the past two decades41–43 – have collectively defined the scope and data of the T2DKP and guided its evolution throughout its existence. Initially, the T2DKP provided a simple website to access association statistics from T2D GWAS and whole exome sequencing (WES) studies, and it gradually expanded to include genomic annotations, bioinformatic method results, and data from additional traits. In 2020, the T2DKP saw a major revision to its user interface that created both an opportunity and a challenge with regards to its accessibility. On the one hand, the new interface provides simple visualizations that enable even non-geneticists to incorporate human genetic data into their research – which, if prominent publications are any guide44, they infrequently do today. On the other hand, the T2DKP’s new interface offers an increasingly complex suite of tools that even genetic experts may not know how to fully exploit.
Here we seek to increase the accessibility of the T2DKP to both non-geneticists and genetic experts. We first document the comprehensiveness of the T2DKP by comparing its datasets and genetic associations to those in other major GWAS resources. We then describe the recent expansion of T2DKP data and tools and evaluate – based on an analysis of usage statistics and citations – its current usage patterns. We use these patterns to define two classes of potential T2DKP users and suggest new ways for each to make fuller use of the T2DKP.
Results
Overview of the T2DKP
The T2DKP contains genetic association summary statistics, genomic annotations, bioinformatic method results, and expert knowledge for T2D and related traits – which include T2D complications (e.g., cardiovascular, hepatic, ocular, and renal traits), glycemic and anthropometric traits, and metabolites (including lipids). Dataset scope is defined by the AMP-T2D consortium, which represents scientists from academia, the pharmaceutical industry, the government, and non-profit organizations45. We maintain these datasets through a three-step process (STAR Methods). First, we combine manual and automated approaches to identify datasets of interest, collaborate with the communities that generated each dataset, and aggregate them within the T2DKP “software platform”. Second, we subject these datasets to quality control (QC) and automated downstream bioinformatic analyses. In the final step, we communicate results of these analyses through the T2DKP’s web-interface and programmatic APIs (Figure 1). At all steps, we ensure that we respect data use restrictions and make only analytical results – and never sensitive individual-level data – publicly available.
Figure 1: Data are collected, processed by the T2DKP platform, and provided through the T2DKP web-interface via a multi-step process.
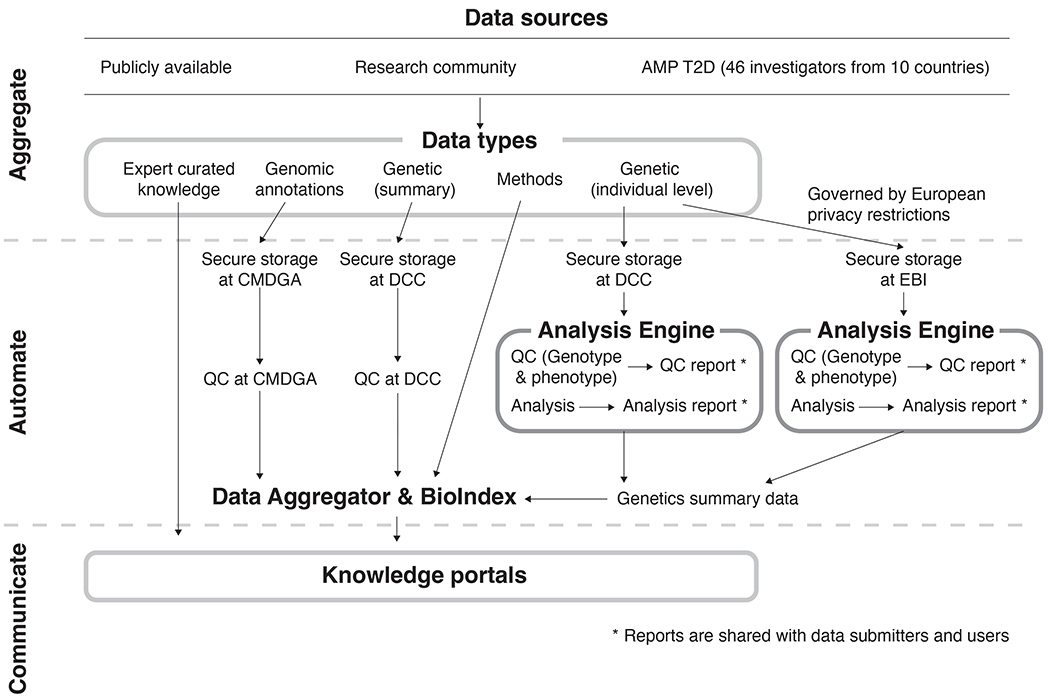
Data sources for the T2DKP are of varied origin and of multiple Data types. Summary-level genetic datasets are transferred to the Data Coordinating Center (DCC) at the Broad Institute, while genomic annotations are transferred to the Common Metabolic Diseases Genome Atlas (CMDGA). Individual-level genetic datasets are transferred to the DCC or European Bioinformatics Institute (EBI) depending on permissions, and the Analysis Engine processes them through a common analytical workflow to produce summary-level associations. The Data Aggregator then analyzes summary-level genetic datasets and genomic annotations with a series of bioinformatic methods, the results of which are stored in the BioIndex. The Knowledge portals access the data within the BioIndex and present them via a web-interface.
The T2DKP web interface includes a set of “core pages” for browsing these results in the vicinity of a gene, variant, or region of interest, and it also offers a suite of more complex tools for expert users (Figure 2). The T2DKP has steadily expanded in its data and functionality over its lifetime, with an accelerating rate of updates since a major redesign in 2020 (Figure 3). This growing complexity and scope of the T2DKP motivates the current article, which is targeted at researchers who may not be fully aware of the T2DKP’s capabilities – or who may not know how to use it at all.
Figure 2: Overview of the T2DKP web-interface.
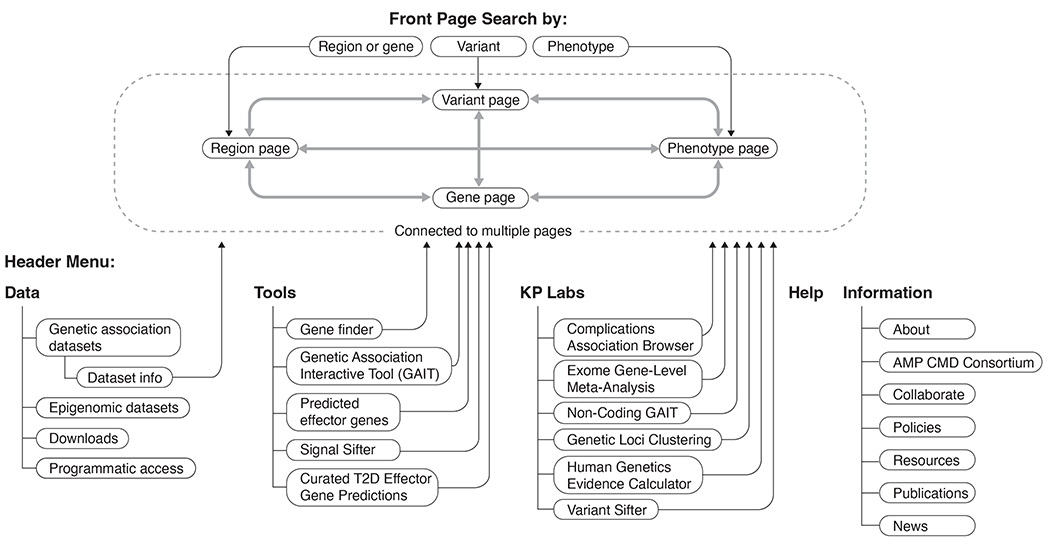
Users of the T2DKP can browse its data by searching for a phenotype, gene, variant, or region. A phenotype search allows views of all associations and datasets for a trait. A region or gene search directs users to a summary of associations within the region (or nearby the gene). Users can select a gene in the region to navigate to the gene page, which shows a summary of gene-level associations for the gene. The variant page shows a summary of associations for a selected variant. The T2DKP also contains a header menu with information about the data in the resource as well as a suite of tools and visualizations.
Figure 3: The T2DKP has added datasets and features over time.
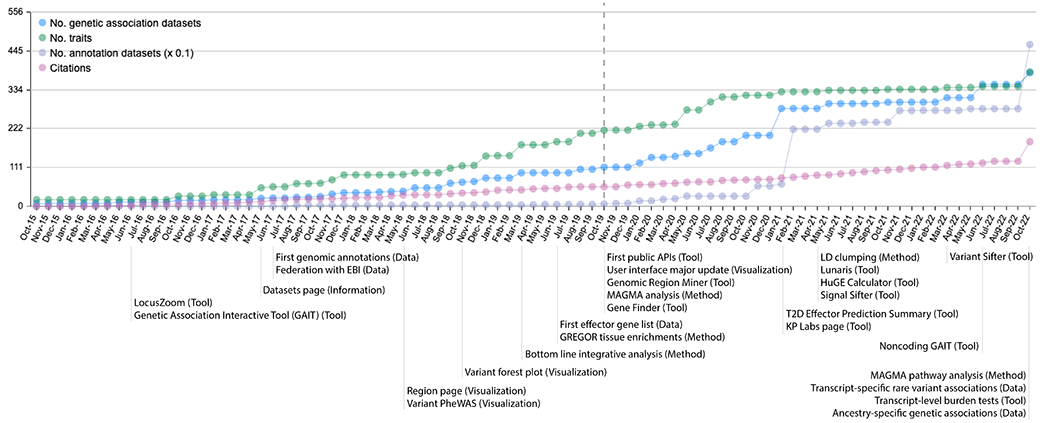
On a regular basis, we update the T2DKP with new genetic association datasets (blue dots) for one or more traits (green dots), genomic annotation datasets (purple dots; represented as one-tenth of the actual number), and tools and visualizations (text on bottom of the plot). T2DKP citations (pink dots) have also increased over time. In 2020, the T2DKP received a major update (vertical dashed line) that significantly changed its user interface.
Analysis of datasets in the T2DKP
Genetic datasets.
As of October 2022, the T2DKP contained 382 GWAS full summary statistic datasets (Table S1), each consisting of association results (across every SNP in the GWAS) for one or more traits. To evaluate the comprehensiveness of these datasets for T2D and related glycemic traits (fasting glucose (FG), fasting insulin (FI), and HbA1c; Table S2), we compared datasets for these four traits to those within the GWAS Catalog8, GWAS Atlas10, and OpenGWAS project14, three other widely used GWAS association resources (STAR Methods). The T2DKP included 107 datasets across these traits, compared to 155 in the GWAS Catalog, 18 in the GWAS Atlas, and 35 in the OpenGWAS project (Figure 4a). Considering only full summary statistic datasets for these traits (Figure 4b), the T2DKP contained most (107 of 138) of the datasets across the resources, including 71 unavailable elsewhere (Figure 4cd). The 31 summary statistic datasets not included in the T2DKP were either small, old, or largely overlapping with other studies in the T2DKP (Figure 4ef; Table S2). T2DKP datasets skew heavily toward European samples, due to the ethnic and sociodemographic biases of GWAS46, although ethnic diversity has increased somewhat over time (Figure 4g).
Figure 4: The T2DKP emphasizes genetic datasets for T2D and related traits.
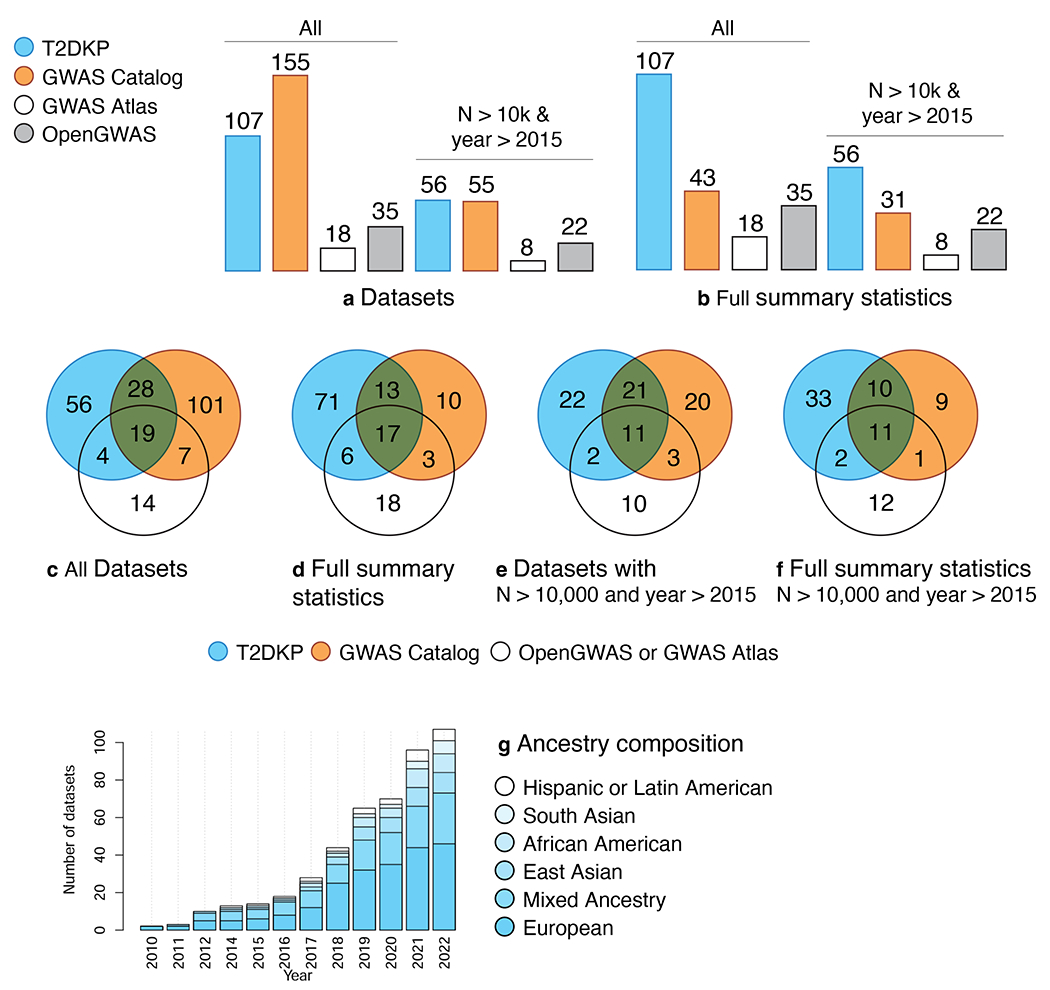
We compared genetic datasets for glycemic traits (T2D, fasting glucose, fasting insulin, and HbA1C) in the T2DKP (blue) to those in the GWAS Catalog (orange), the GWAS Atlas (white), and the OpenGWAS project (gray), in October 2022 (Table S2). We conducted an analysis of all datasets and an analysis of datasets newer than 2015 and with >10K samples. a. Considering all datasets in each resource, including those without full summary statistics available, the GWAS Catalog contains the most glycemic trait genetic datasets. b. When only datasets with full summary statistics are considered, the T2DKP contains the most glycemic trait genetic datasets. c. Both the T2DKP and the GWAS Catalog contain datasets unavailable through other resources. d. When only genetic datasets with full summary statistics are considered, the T2DKP contains many more datasets unavailable through other resources. e. Most of the datasets unique to the GWAS Catalog are either from prior to 2015 or contain fewer than 10K samples. f. The T2DKP contains nearly all datasets newer than 2015 and with more than 10K samples. g. Datasets in the T2DKP are predominantly from analyses of European samples, but the ethnic diversity it captures has increased over time.
To obtain associations between each variant and each trait in the T2DKP, we employ a “bottom-line” approach (STAR Methods) that meta-analyzes the datasets in which a variant is observed and statistically adjusts for the (usually unknown) degree of sample overlap among them47. To evaluate the comprehensiveness of both T2DKP bottom-line associations (p<5×10−8 in the meta-analysis) and “dataset-level” associations (p<5×10−8 in one or more datasets), we compared the loci with T2D, FG, FI, or HbA1C associations in the T2DKP to those in the GWAS Catalog and the OpenGWAS project (STAR Methods). As of October 2022, the T2DKP contained 1900 such loci, 1209 (64%) of which were significant at both the dataset-level and in the bottom-line analysis (Figure 5a). Most of the loci unique to the bottom-line analysis had moderate (but not genome-wide significant) evidence of association in multiple datasets (Figure 5b) – such loci are thus only revealed after meta-analysis of T2DKP datasets. Conversely, loci significant only at the dataset-level included those observed only in older or smaller datasets (likely association artifacts) and those unique to an ancestry or analytical approach (true associations, but not significant in the transethnic bottom-line meta-analysis).
Figure 5: The T2DKP both adds and omits glycemic trait associations relative to the GWAS Catalog.
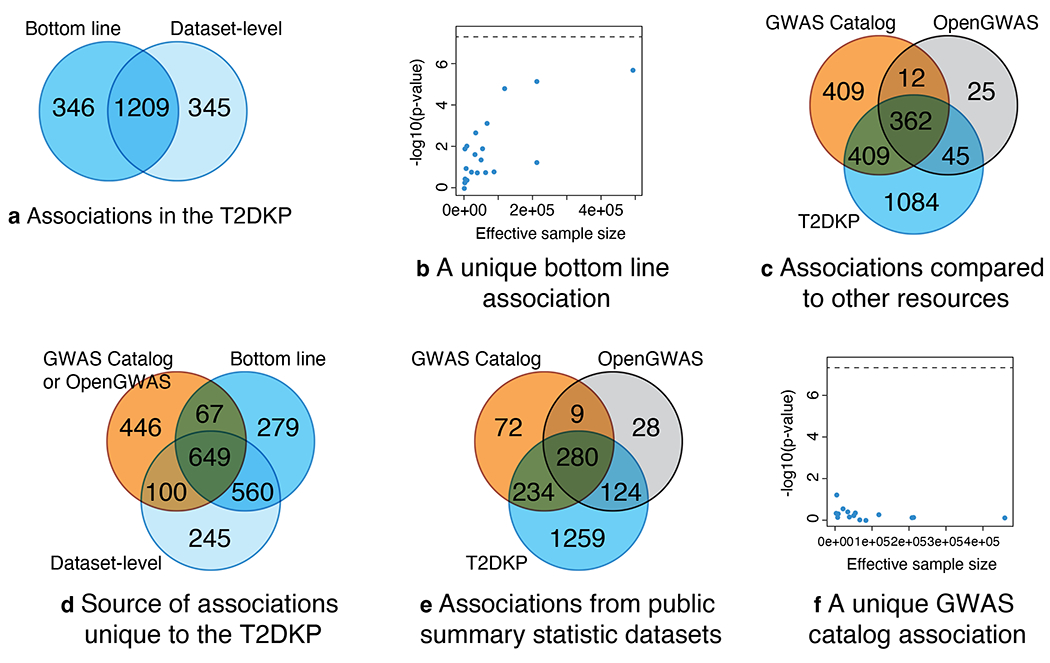
We evaluated the glycemic trait genetic associations (for T2D, fasting insulin, fasting glucose, and HbA1C) in the T2DKP. We compared genetic associations produced by the T2DKP’s overlap-aware meta-analysis (Bottom line, STAR Methods) to genetic associations reported by individual genetic datasets (Dataset-level). a. The bottom-line and dataset- level associations largely overlap, but the bottom-line analysis both adds and removes associations. b. Associations added by the bottom-line analysis have suggestive associations across many datasets. An example association unique to the bottom-line analysis (rs1000237) has moderate p-values (y-axis) in numerous datasets (points), including nominally significant but not genome-wide significant associations in the datasets with the largest effective sample sizes (x-axis). The horizontal line indicates genome-wide significance. c. Comparing the glycemic trait associations in the T2DKP to those in the GWAS Catalog and the OpenGWAS project, each resource contains unique associations. d. Associations unique to the T2DKP are a mixture of associations due to datasets unique to it (Dataset-level associations) and its bottom-line analysis (Bottom line). e. Most associations unique to the GWAS Catalog are due to studies without summary statistics publicly available. f. An example association unique to the GWAS Catalog (rs10932672) is unsupported by larger, more recent datasets in the T2DKP (points on the right side of the plot).
Many of these loci (771, 41%) also had associations in the GWAS Catalog, and a smaller number (407, 21%) had associations in the OpenGWAS project. The 1084 loci unique to the T2DKP included 805 loci attributable to T2DKP-exclusive datasets (dataset-level associations in Figure 5d). Conversely, of the 446 loci not in the T2DKP, the majority (364, 81.6%) were due to studies without publicly available association summary statistics (Figure 5e). The other 96 loci were mostly due to old, small, or non-standard studies inconsistent with the larger studies meta-analyzed in the T2DKP (51 out of 96 lead SNPs had p>0.01 in the T2DKP bottom line analysis; e.g., Figure 5f). This analysis demonstrates the T2DKP as a comprehensive resource of associations extractable from publicly available summary statistics for T2D and related traits, and the GWAS Catalog as a complementary resource of all published associations (including those from older and smaller studies).
Genomic annotations.
The genetic datasets in the T2DKP are augmented by (as of October 2022) 5,418 genomic annotations, including 304 (5.6%) unique to it (Table S3). The vast majority (5073; 93.6%) of these annotations describe the location of cis-regulatory elements (Figure S1a), which include accessible chromatin sites from ATAC-seq or DNase-seq (814, 15.0%), transcription factor binding sites from ChIP-seq (720, 13.3%), and computationally predicted candidate regulatory elements (2504, 46.2%) or chromatin states (1035, 19.1%) from ChIP-seq and DNase-seq. The remaining 345 datasets contain predicted linkages between cis-regulatory elements and target genes using data from single-cell co-accessibility (scATAC-seq or snATAC-seq39), 3D physical interactions (Hi-C or promoter capture Hi-C), or activity-by-contact (ABC)48. These datasets are stored in the Common Metabolic Diseases Genome Atlas (CMDGA)49, a sister resource connected to the T2DKP through a series of REST APIs.
Each genomic annotation is described with a high-level tissue of origin and a finer-grained biosample (Table S3). As many genomic annotations in the T2DKP are drawn from the ENCODE project17, the distribution of datasets across tissues is roughly similar to ENCODE. However, because the T2DKP includes additional genomic annotations from T2D-relevant tissues, there are some tissues for which annotations are over-represented relative to ENCODE. For example, as of October 2022, 173 (3.2%) datasets were from whole pancreas or pancreatic islets, compared to 1.5% of datasets in ENCODE for Homo sapiens cell lines or tissues (fisher p=9.1×10−13). (Figure S1b). Other over-represented tissues included muscle (6.4% in the T2DKP vs. 1.3% in ENCODE; fisher p=6.1×10−66), adipose (1.6% vs. 0.009%; fisher p=2.6×10−4), and kidney (7.4% vs. 3.1%; fisher p=3.8×10−32).
T2DKP usage patterns
To understand T2DKP usage patterns and how we might improve them, we analyzed page accesses (using Google Analytics from October 2021 through March 2022) and the 129 T2DKP citations. Both measures suggest that existing users focus on simple result summaries: 49.3% of page accesses (31,558 out of a total 64,005 accesses) were for the core “region”, “variant”, “gene”, and “phenotype” summary pages (Figure 2), 48.8% of page accesses (31,230) were for informational pages, and only 3.9% of page accesses (1,217) were for more complex tools. Similarly, 81 (86.2%) of the 94 citations used the T2DKP core pages for simple queries.
These usage patterns are consistent with two major categories of current and prospective T2DKP users. First, non-geneticists who primarily work with experimental models likely browse the T2DKP core pages (if they use the site at all44) but do not fully understand how to interpret the data within them. Second, researchers who regularly analyze genetic associations likely understand the data on the T2DKP core pages but are mostly unaware of (or do not understand) the more complex tools on the T2DKP – possibly due to the pace with which these tools have been added in recent years (Figure 3). To increase the accessibility of the T2DKP for both non-geneticists and genetic experts, below we describe the T2DKP features most useful to each category of user, along with “best-practices” for their use.
T2DKP usage and best-practices for non-geneticist users
Genetic support workflow.
For non-geneticists, the T2DKP is likely most useful to evaluate “genetic support”5 for a gene suspected, based on experimental work, to be relevant to disease (Figure 6). Searching a gene name on the T2DKP home page brings the user to the region page, showing genetic associations within 50kb of the gene boundaries. The region page contains a “PheWAS”50 plot of traits with genome-wide significant associations (filterable to those observed for a chosen ancestry) and (for one or more selected traits) a LocusZoom51,52 plot or table of associations across the region. Genes with human genetic support usually have at least one associated variant in the region.
Figure 6: We recommend non-geneticists follow a “genetic support” workflow within the T2DKP.
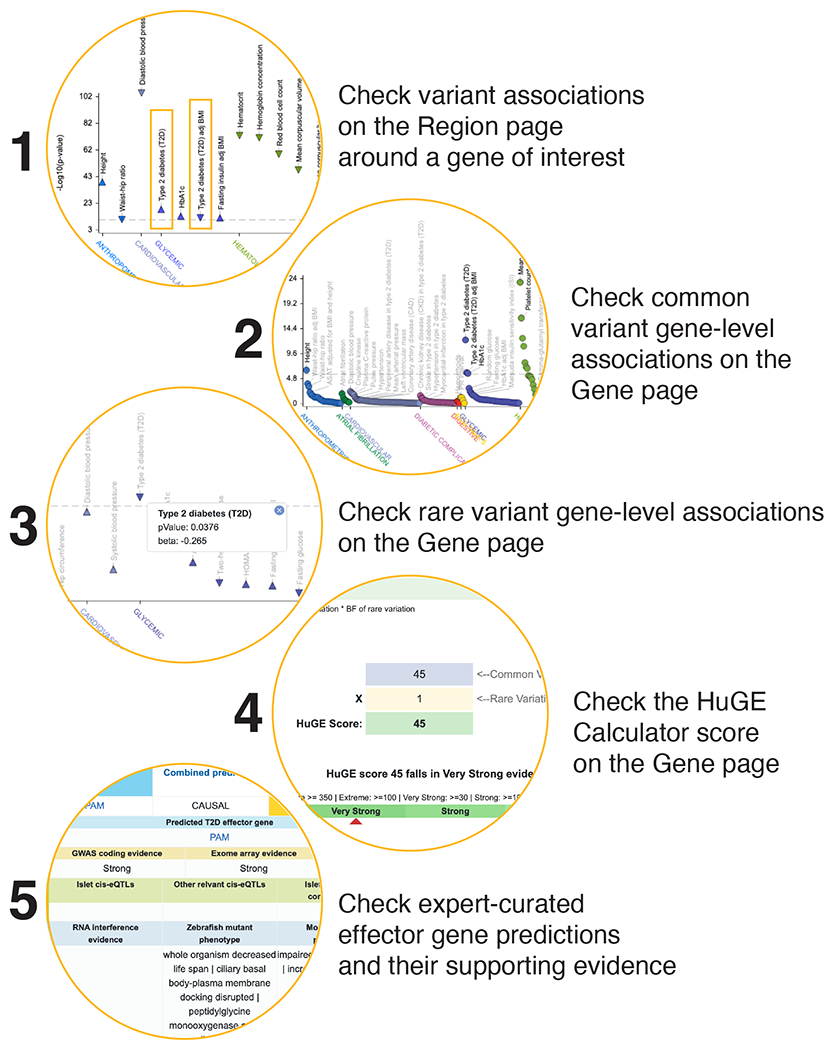
To evaluate whether human genetic associations support the involvement of a gene of interest in human disease, users can first use the “region page” to see if the gene lies nearby associations (1), then use the “gene page” to view a distillation of these associations into a gene-level score (2) and also view complementary rare variant associations for the gene (3). The HuGE calculator, also on the gene page, summarizes these two gene-level associations into a single score for the gene (4). The T2DKP effector gene list contains a curated set of genes suggested from genome-wide analyses to be involved in disease (5). Table S4 contains information on these and other modules of the T2DKP.
A more complete summary of a gene’s genetic support is available on the “gene page”, accessible by clicking a gene symbol at the top of the region page. PheWAS plots and tables show gene-level associations for both common variants (calculated by MAGMA53 and viewable across ancestries or specific to a chosen ancestry; STAR Methods) and rare variants (optionally viewable for a specific transcript of the gene; STAR Methods) within the T2DKP. Both classes of association are summarized on the gene page by the Human Genetic Evidence (HuGE) Calculator, which implements previously outlined44 probabilistic calculations of genetic support for a gene.
If a gene of interest does have genetic support, the “variant page” displays additional provenance underlying each variant association nearby the gene. The variant page contains a PheWAS plot, forest plot, and a table of the bottom-line associations for a variant across traits in the T2DKP. In the table, users can see the dataset-level associations that contributed to the bottom-line analysis for the variant – this feature is useful to evaluate the consistency of the association across studies. All tables and visualizations on the variant page can be filtered to associations specific to a chosen ancestry.
As an example of this “genetic support” workflow, a recent study proposed a novel mode of insulin action based on an interaction between the SIN3A and FOXO1 proteins54, leading to the hypothesis that selective modulation of FOXO1 could treat hyperglycemia in humans55. A researcher wondering whether there is evidence for involvement of SIN3A in T2D would enter the gene name on the T2DKP home page to browse the SIN3A region page, which indeed shows a T2D GWAS association near the gene (p=9.21×10−16). Browsing further, the SIN3A gene page shows that its common variant association for T2D is genome-wide significant (MAGMA p=6.78×10−13) and the HuGE Calculator shows a “Very Strong” level of evidence for association of SIN3A with T2D. Similarly, while the region page shows no T2D GWAS association nearby FOXO1, the gene page shows a nominally significant (burden test p=0.038) rare variant gene-level association for it, leading to a “Moderate” level of evidence according to the HuGE Calculator. Although neither of these associations unequivocally implicates SIN3A or FOXO1 in T2D, they do add support to the experimental data of the original study54.
Caveats when using the genetic support workflow.
Several best-practices are important for this “genetic support” workflow. First, while a significant association near a gene supports its involvement in disease, the absence of significant associations does not preclude the gene’s involvement in disease – a gene will only produce disease associations if a genetic variant affecting its function is sufficiently frequent in the population. When no associations exist nearby a gene, users can employ the T2DKP PheWAS plots to examine not only the primary trait of interest but also related traits; evaluating associations nearby interacting partners of the gene or for genes within shared pathways can also increase sensitivity in some cases56. Second, and conversely, users should not engage in “data dredging”57 and consider a gene to have support because a single variant nearby it shows association in a single dataset; instead, association evidence for a gene should be considered across all variants, corrected for the number of portal queries (e.g., via Bonferroni correction), and integrated across all datasets – the bottom-line association analysis is our preferred statistical method for evaluating association strength across all T2DKP datasets, but it is an approximation and can also hide population-specific effects or gene-by-environment interactions. The T2DKP HuGE calculator is our preferred tool on the portal to estimate genetic support accounting for these and other caveats, albeit with some simplifying assumptions.
Effector gene lists.
If users would like a list of genes with the strongest genetic support for T2D or related traits, then they should use the “Predicted Effector Genes” tool (STAR Methods). This tool contains four lists of genes predicted (based on different methodologies) to mediate GWAS associations in the T2DKP, categorizes genes based on the strength of these predictions, and allows users to see the evidence underlying each prediction. It is useful for analyses of genes identified from unbiased “forward genetic” approaches, such as the construction of potential protein interaction networks for diabetes-related endophenotypes58.
To illustrate another application of the effector gene list, we investigated the extent to which genes identified from T2D GWAS also harbor rare coding variant associations, of relevance to the debate59 regarding the ability of GWAS to identify genes central to disease pathogenesis as opposed to genes on the periphery of dense regulatory networks60. We downloaded the list of 132 T2D effector genes produced by a curation-based methodology (under the “Curated T2D Effector Gene Predictions” page on the T2DKP “Tools” menu), in which genes are assigned to one of five evidence categories from “Causal” (containing a causal coding variant) to “Weak” (prioritized based on a single line of regulatory or perturbational evidence; STAR Methods). Using a Wilcox test and data from the AMP-T2D-GENES WES study43 (STAR Methods), we found these genes to be significantly enriched for rare coding variant associations (p=0.015; Figure S2), with this enrichment due mostly to genes in the “Causal” set (p=0.0023 for “Causal” genes, p=0.21 for all other genes). Conversely, the 132 genes with the highest T2D MAGMA scores were not enriched for rare coding variant associations (p=0.27). This example illustrates the intended use of the effector gene list as a more effective starting point (compared to genes simply nearby GWAS associations) for downstream investigation of candidate disease genes.
T2DKP usage and best-practices for genetic expert users
Our usage and citation statistics indicate that most users – even genetic experts – do not routinely use the more advanced T2DKP tools. Many of these tools are complex, and we have therefore developed complete guides for them (Table S4) accessible under the T2DKP “Help” menu. Here, we motivate and provide examples of three advanced analyses that can be conducted with these tools.
Analysis 1: associations across multiple traits.
A variant’s or gene’s pattern of associations across a range of traits can be useful in basic (e.g., identifying cases of pleiotropy61), clinical (e.g., clustering patients into disease subgroups62), and translational (e.g., predicting if modulating a candidate drug target will have adverse side effects63) research. To visualize a variant’s association pattern across traits in the T2DKP, the variant page contains both a PheWAS plot, highlighting association significance and directionality, and a forest plot, highlighting relative effect sizes. Conversely, the “Signal Sifter” identifies association signals following a user-specified association pattern (i.e., positive or negative effects) across a user-specified collection of traits (STAR Methods). For example, we filtered p<5×10−8 fasting insulin associations adjusted for BMI (FIadjBMI) to those matching an “insulin resistance signature” of p<0.005 associations with triglyceride levels (TG; same direction of effect as FIadjBMI) and high-density lipoprotein levels (HDL; opposite direction of effect). The resulting 46 associations (Table S5) included 29 previously identified by a 2016 analysis64, as well as 17 now significant for the insulin resistance signature in larger GWAS conducted since 2016.
Similar to the Signal Sifter is the “Gene Finder”, which identifies genes with a MAGMA p-value below a user-specified threshold for each of a user-specified set of traits. For example, the Gene Finder identified 11 genes with p<2.5×10−6 for FIadjBMI, TG, HDL, T2D and Waist-hip ratio adjusted for BMI (Figure 7a). These genes were significantly enriched for expression specific to adipose tissue, a major site of insulin action (subcutaneous adipose tissue-specific expression Wilcox p=7.8×10−4, effect=20.02 “t-stat”65 units; visceral adipose tissue-specific expression Wilcox p=1.2×10−3, effect=16.1 t-stat units; Figure S2ef; STAR Methods). By comparison, the 11 genes with the lowest T2D MAGMA p-values were not as strongly enriched for adipose-specific expression (subcutaneous Wilcox p=0.045, effect=4.9 t-stat units; visceral adipose Wilcox p=0.32, effect=−0.12 t-stat units; Figure S2gh). These results demonstrate how – through filters on gene-level association p-values alone – the Gene Finder can be used as a starting point to prioritize genes that act through a pathway or mechanism of interest.
Figure 7: The T2DKP enables exploratory and interactive analyses for the genetic expert user.

a. A Gene Finder search for genes associated with Fasting Insulin adjusted for BMI, Triglycerides, HDL cholesterol, T2D, and Waist-hip ratio adjusted for BMI returns 11 genes that have MAGMA p<2.5×10−6 for each trait. These genes were significantly enriched for adipose tissue-specific expression (Figure S2ef) b. A query of ‘CDC123’ on the Variant Sifter shows a regional plot of the T2D association. Tracks below the plot show the locations of variants in the credible set and genomic annotations for transcription factor binding sites within the pancreas. A table lists the variants within the credible set that overlap the displayed genomic annotations. The actual Variant Sifter page contains more visualizations than those shown in the figure; because of space limitations we have spliced the LocusZoom plot, credible sets plot, annotations plot, and variant table together. c. An association analysis in GAIT between rare MC4R variants (in the 5/5 mask) and T2D shows the impact of p.I269N on the association signal – after removing the variant from the analysis, the T2D association p-value is increased by nine orders of magnitude and the BMI association is ablated. Table S4 contains information on these and other modules of the T2DKP.
Caveats for interpreting associations across multiple traits.
As the T2DKP does not yet support formal co-localization analysis66, a variant associated with multiple traits on the PheWAS plot is not necessarily causal for each trait, and signals identified by the Signal Sifter or Gene Finder may have different causal variants across traits. Genes identified by the Gene Finder are also not necessarily the effector genes for the nearby association signals.
Analysis 2: prioritizing variants at a GWAS locus.
A second advanced use case of the T2DKP is to explore the variants, cell types, regulatory elements, and causal genes underlying GWAS associations1,67. The “Variant Sifter” enables users to filter variants within a region based on available credible set(s) and genomic annotations; users can manually select annotations and tissues of interest or use GREGOR enrichments to choose globally disease-relevant annotations (more sophisticated predictions of locus-specific tissues of action68 are a potential future addition). As one example, a Variant Sifter query of the T2D associations within 50kb of CDC123 shows 282 variants achieving p<5×10−8, which can be filtered to only one variant (rs11257655) in the most recent credible set69. Next, adding genomic annotations for enhancers and transcription factor binding sites, and filtering these to tissues in which each annotation has a GREGOR enrichment of p<0.05 and fold-enrichment>2, shows that rs11257655 is contained in a pancreatic enhancer and a pancreatic islet transcription factor binding site in pancreatic islets (Figure 7b). Indeed, rs11257655 has been shown to affect binding of the FOXA1 and FOXA2 transcription factors – which are essential for pancreas and liver development – to an active pancreatic enhancer region70.
After filtering variants based on credible sets and annotations, the Variant Sifter shows genes linked to any of the remaining variants. For example, the credible sets and genomic annotations nearby KCNQ1 indicate that rs231361 has a posterior probability of T2D association >0.99 and overlaps accessible chromatin annotations in pancreatic islets (as well as other pancreatic cell types). Viewing genes linked to rs231361 in the pancreas shows connections (via chromatin co-accessibility) to INS and IGF2 – 500kb away – but not KCNQ1. Experiments have in fact showed INS levels to be affected by genome-editing of rs231361 in stem-cell derived pancreatic beta cells39.
Caveats when prioritizing variants at a GWAS locus.
When prioritizing variants with the Variant Sifter, the strongest evidence of causality is a high posterior probability within a credible set (above 80% or even 95%), and presence within a genomic annotation in a disease-relevant tissue provides further71 – although not conclusive72 – evidence. Inferences about cell types and genes that mediate an association should be made with care, as genomic annotations are often correlated across tissues, and variants are often linked to multiple genes. Finally, while global enrichments are a useful starting point for selecting genomic annotations, relevant annotations will vary by locus and (ideally) should be chosen based on the pattern of trait associations observed at the locus62. Any hypotheses generated from the Variant Sifter should be replicated experimentally.
Analysis 3: interactive coding variant analyses.
A third advanced tool on the T2DKP, the Genetic Association Interactive Tool (GAIT), supports rare coding variant analyses of WES data from the AMP-T2D-GENES43 (24 traits) and TOPMed73 (T2D, FG, and FI) studies. Users can select a gene (or transcript), choose among seven pre-defined variant groupings based on functional annotation (“masks”)43, optionally de-select variants from the mask (according to their displayed protein position and bioinformatic annotations), and conduct an on-the-fly aggregate association analysis using one or more methods (a burden test74, SKAT75, or SKAT-O76 analysis; STAR Methods). One use of GAIT is to conduct association analyses of custom collections of coding variants (perhaps those assayed as “functional”77,78). Another is to refine association signals to determine the variants most important to the association. For example, a GAIT burden test of MC4R variants in AMP-T2D-GENES and the 5/5 mask43 produces p=3.2×10−11 (odds ratio=1.20) for T2D and p=9.4×10−3 (beta = 0.042 kg/m2) for BMI, consistent with published results43. As previously reported43, much of this signal is due to the p.I269N MC4R variant in the 5/5 mask. With p.I269N excluded, GAIT reports p=0.018 (odds ratio=1.12) for T2D and p=0.90 (beta=−0.003 kg/m2) for BMI (Figure 7c). These results suggest that nearly all of the BMI signal for the 5/5 mask – and most but not all of the T2D signal – is due to p.I269N, illustrating how users can dissect the contribution of rare variants to aggregate association signals using GAIT.
Caveats when conducting interactive coding variant analyses.
GAIT is useful primarily for exploratory association analyses where instantaneous results are important. However, it employs a straightforward analysis procedure (STAR Methods) that may not be optimal for all genes or traits. Users should also be careful to correct GAIT p-values for the number of analyses they perform. Like those for all T2DKP tools, the results of GAIT should ideally be further investigated or replicated before using them in a publication.
Support for additional analyses.
All T2DKP tools are listed in Table S4. Over time we plan to add additional tools such as analysis of user-specified or predefined pathways79, automated credible set and co-localization calculations80, analysis of association heterogeneity across phenotypic subgroups or ancestries (including via random-effects meta-analyses), exploration of richer phenotypes including those measured longitudinally, investigation of genetic or environmental interaction terms81, and calculations of SNP posterior effect sizes for use in polygenic risk scores or instrumental variable analysis3. For now, users who wish to use T2DKP datasets in such analyses can download many of the original datasets directly from the T2DKP. Processed data can also be accessed programmatically via REST services82. In the future we plan to provide additional programmatic access mechanisms, including direct import of T2DKP data into Python or R data structures.
Additional resources
For a more detailed description of T2DKP workflows and visualizations, we have provided several sources of documentation. Links to a step-by-step guide and a tutorial video for the “genetic support” workflow are prominent on the home page and on the pages included within the workflow. These provide a starting point for first-time portal users. Each of the more advanced T2DKP tools has a graphic at the top that illustrates the conceptual steps in the tool, with links shown between each conceptual step and a corresponding user interface component on the page. The graphic contains links to an expanded documentation page and video tutorial for the tool and its underlying data. All portal documentation is organized under its “Help” page, which has sections for Data, Methods, Pages/Tools, and Workflows. Documentation can also be navigated through a full-text search option in the upper right corner of each page.
Discussion
A human genetic association supports a gene’s role in disease and increases its viability as a therapeutic target5. The T2DKP aims to make such supporting evidence readily available and interpretable in a T2D-specific resource, thereby promoting the understanding and treatment of T2D and its complications. The T2DKP focuses on recent, large, and community-nominated datasets for T2D-related traits and in T2D-relevant tissues, which our analysis suggests form a more comprehensive resource for T2D genetics compared to resources that represent genomic data without regard to a specific disease area. In this article, we have focused on increasing the T2DKP’s usability in two respects. First, to increase the reach of the T2DKP to non-geneticists who do not yet incorporate human genetics into their research44, we have detailed a simple “genetic support” workflow. Second, to inspire expert geneticists to expand their use of the portal to include more complex tools, we have detailed three scientific questions and how they map to tools in the T2DKP. Our hope is that this information will increase both the breadth and the depth of the T2DKP user-base.
Beyond its role as a resource for T2D researchers, the T2DKP provides one potential model for sharing information derived from sensitive datasets in a manner that is broadly accessible but still useful to disease experts. Compared to “data commons”83 such as the NHLBI BioData Catalyst84 or the NHGRI AnVIL85, which provide analytical tools to analyze large or sensitive datasets within a cloud-based workspace, the model of the T2DKP is to delineate individual-level genetic datasets, which in general cannot be made publicly accessible, from summary representations of them, which support the needs of the vast majority of users. This paradigm – in which a public web-interface displays summary statistics automatically calculated from community-maintained private individual-level data – could be an effective path to make other types of information in these “data commons” more readily available to users who do not need or desire direct data access.
The T2DKP also suggests a means to encourage researchers to share data and results in settings where they have traditionally been hesitant. It serves as a dedicated portal with community-designed content and branding, providing researchers with an incentive to share data and contribute expertise to manual parts of the data curation process. The data underlying the portal, however, are served through a general and scalable software platform, and improvements in automation of the data discovery process accrue to any other portals built upon the same platform. The potential of “community portals” like the T2DKP has been recognized by other disease communities that have used its platform to build portals for cardiovascular and cerebrovascular disease, type 1 diabetes, and sleep disorders, all of which are now collected with the T2DKP under the broader banner of the Common Metabolic Diseases Knowledge Portal (CMDKP) – the current focus of the AMP-T2D (now AMP®-CMD) partnership following its initial funding period. The growth of these resources illustrates a potentially cost-effective strategy to democratize access to expert-generated genomic data and accelerate the discovery, validation, and interpretation of genetic and genomic results.
Limitations of the study
In the Results section, we have discussed caveats and limitations of key T2DKP workflows and tools. More generally, while the T2DKP aims to be an “authoritative” resource of genetic and genomic data for T2D and related traits, incompleteness and bias in its datasets and functionality are inevitable. We have recently added more automated capture of datasets (STAR Methods), but manual review of datasets has been dominant historically and will remain necessary to some extent in the future. The datasets in the T2DKP are in particular biased toward individuals of European ancestry, as are most GWAS86, and a major focus in the future will be to address this with studies of more diverse ancestries. Additionally, the T2DKP presents summary association statistics to users, which necessarily discard information available in the original genetic and genomic datasets; in some cases, the summary association statistics have been combined with those of other datasets and therefore may not match those in the original manuscript describing a dataset.
STAR Methods
Resource Availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Dr. Jason Flannick (jason.flannick@childrens.harvard.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
All data reported in this paper are included in the main text, figures, and supplementary files.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
All values used to generate the graphs of the paper can be found in the file Data S1 – Source Data. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Method details
GWAS summary statistics
As of October 2022, the T2DKP contained 382 GWAS summary statistic datasets (Table S1), each of which consists of association results (across every SNP in the GWAS) for one or more traits. We identified these GWAS datasets – and regularly identify additional ones to include in the T2DKP – via three mechanisms. First, we monitor the new literature via weekly PubMed searches and by monitoring Twitter to find relevant new preprints and papers. We review these papers to determine whether the summary statistics are publicly available; if they are not, we contact authors to ask whether they would like to contribute their results to the T2DKP. Second, we work with scientists within AMP-T2D and the T2D genetics community to obtain datasets they generate or identify. Often, authors provide a final version of their summary statistics as they prepare their manuscripts, so that the results can be released on the T2DKP immediately upon publication. Third, we work with scientists (funded as part of AMP-T2D) from the European Bioinformatics Institute, home of the GWAS Catalog, to identify new association datasets for T2D-relevant traits for which full summary statistics are available in the GWAS Catalog; we review each of these datasets for eligibility and prioritize those of most interest to the community targeted by the T2DKP. This more automated and systematic data discovery approach complements the other two manual curation approaches that we employ. In these analyses, the standardized ontology terms to which GWAS Catalog traits are mapped are employed to interrogate the Catalog REST-API for traits of relevance to the T2DKP. Datasets for which the specific author-reported trait curated by the GWAS Catalog align to T2DKP traits of interest are then assessed for the availability of full summary statistics and compatibility of the file contents with T2DKP ingest requirements. Finally, datasets are harmonized via the standard pipeline87 used at the GWAS Catalog before transfer to the T2DKP for loading.
We focus on phenotypes related to T2D, which include T2D complications (e.g., cardiovascular, hepatic, ocular, and renal traits), glycemic and anthropometric traits, and metabolites (including lipids). We prioritize datasets with disease-relevant phenotypes not yet included in the T2DKP, with sample sizes larger than any study yet in the T2DKP, or with subjects of non-European ancestry. While most investigators are enthusiastic about contributing their data to the T2DKP, some have concerns about data sharing or lack resources to do so. To address potential data sharing concerns, we have developed a set of policies (informed by our engagement with these communities over time) for responsible data stewardship88. To address potential resource limitations, AMP-T2D has in some cases38,89 provided funding for investigators to contribute their data.
If a summary statistic dataset is publicly available, we download it and record its original URL for provenance. If the dataset is not publicly available, we obtain it directly from the investigators who produced it and record the citation describing it for provenance. We ensure that each summary statistic dataset has for each variant at minimum a chromosome, position, genome build, effect allele, and p-value; we also obtain effect sizes, standard errors, and effect allele frequencies for each variant when available. When a GWAS has calculated credible sets42, we accept the posterior probabilities of association for each variant. We document the dataset with the genotyping technology used, sample size, and study ancestry. In alignment with the GWAS Catalog, we record study ancestry as one of nine terms: African American or Afro-Caribbean, African unspecified, East Asian, European, Greater Middle Eastern, Hispanic or Latin American, South Asian, Sub-Saharan African, or Mixed Ancestry90. We assign ancestry descriptions as they are stated in published papers or in communications with data generators. Due to a lack of reference genetic data for some of these ancestries, in downstream analyses we consider only six ancestries: African American (which includes Sub-Saharan African and African unspecified), East Asian, European (which includes Greater Middle Eastern), Hispanic or Latin American, South Asian, and Mixed Ancestry.
We conduct a four-step QC process for each summary statistic dataset (Figure S3), which are originally provided to the T2DKP in tab delimited format. In the first step, we assign the summary statistic input file standardized column headings and filter out any rows with incompatible chromosomes, reference alleles, or alternate alleles. Additionally, as needed we set optional columns (e.g., odds ratio) to null and infer non-optional columns (e.g., effect size is inferred from odds ratio if it is missing but odds ratio exists). We do not follow any special procedure for different classes of variants; indel variants and SNPs are treated identically. Datasets are also lifted over to GRCh37, if they are specified relative to a different genome build, and assigned identifiers in the format chromosome:position:reference_allele:other_allele.
In the second step, we align all effect sizes to the alternate allele (relative to the GRCh37 reference genome). We first analyze strand-unambiguous variants, detecting whether the effect allele (and consequently sign of effect and odds ratio) must be flipped for the non-effect allele to align to the GRCh37 reference genome – we strand-complement reference alleles and alternate alleles if necessary. We then analyze strand-ambiguous variants. We first determine if each variant should be complemented, which we do if (a) >10% of unambiguous variants are complemented, (b) the variant has minor allele frequency (MAF) between 30–70%, and (c) the frequency of the effect allele is further from its frequency in the 1000G project (AF1000g) than it is from 1-AF1000g. After complementing (or not complementing) the variant, we then flip its effect, odds ratio, and effect allele as needed to align the non-effect allele to the GRCh37 reference genome.
The third QC step filters out variants with summary statistics incompatible with the analyses that will be performed on them in subsequent steps of our analysis pipeline. For example, it excludes variants with negative MAF, standard error set to infinity, or odds ratio less than zero.
The fourth and final QC step is effect size scaling for quantitative phenotypes. The scaling process performs linear regression with intercept pinned to zero on se2 ~ MAF * (1 − MAF) * N, where se is the variant’s reported standard error and N is its reported sample size. The regression only uses variants with MAF > 0.05 and within the 25%−75% percentiles of standard error. If fewer than 1000 such variants exist, typical for datasets without MAF reported, we instead use a proxy MAF=0.25 for every variant. We divide all variant effects and standard errors by the regression slope if (a) MAF is available and the slope is greater than 2 or less than 0.5 or (b) MAF is unavailable and the slope is greater than 5 or less than 0.2. Otherwise, no scaling is performed.
Whole exome sequencing (WES) summary statistics
As of October 2021, the T2DKP contained associations from two WES datasets: AMP-T2D-GENES43, containing 45,231 individuals and analyzed for T2D and 24 related quantitative traits, and TOPMed73, containing associations for T2D, FG, and FI on 23,211 to 44,083 individuals (the TOPMed WES data consist of whole genome sequencing data subset to the exome). For each dataset, the T2DKP includes single-variant association statistics, which we processed following the same procedure that we use for GWAS summary statistics. For the AMP-T2D-GENES dataset, the T2DKP also includes pre-computed gene-level association statistics for seven variant “masks”43 (annotation categories).
Individual-level genetic data
In addition to datasets with pre-computed GWAS summary statistics, the T2DKP contained (as of October 2022) 38 datasets with individual-level genotypes and phenotypes (Table S1). Most of these datasets include SNP array data from samples measured for T2D-related complications or samples studied longitudinally. Most were collected by researchers in the AMP-T2D consortium and were prioritized to fill gaps in publicly available datasets.
To obtain individual-level datasets for the T2DKP, we collaborate directly with the contributing investigators. After signing a data transfer agreement (DTA), the contributor securely transfers their dataset to one of two sites depending on data use restrictions: the Broad Institute (for datasets allowed to be stored in the United States) or the European Bioinformatics Institute (for datasets required to remain in Europe). Each site stores datasets behind a firewall that prevents public access. We record all phenotypes in a template developed by AMP-T2D to support flexibility in capturing T2D-related traits. Regulations and policies regarding data transfer and appropriate use of the T2DKP are described on an extensive “Policies” page under the T2DKP “Information” menu (https://t2d.hugeamp.org/policies.html).
We then analyze each individual-level dataset using an association analysis pipeline that we developed and used as part of several large-scale T2D association studies41,43 (Figures S4 and S5). We apply the same pipeline regardless of whether datasets are stored at the Broad Institute or the European Bioinformatics Institute. The pipeline conducts cleaning and normalization of phenotype values, harmonization of genotypes to a modern imputation reference panel91, sample and variant QC, genetic ancestry and sex determination, measurement and correction for population structure via genetic principal components or genetic relationship matrices, single-variant joint and meta-analysis, and (for WES data) gene-level analysis of various variant masks. In addition to widely used QC techniques (e.g., for sample call rate or variant departures from Hardy-Weinberg equilibrium), our pipeline includes a novel sample QC protocol43 which, for each sample, calculates a series of ~10 metrics indicative of sequencing or genotyping quality (e.g., number of called variants, heterozygosity), adjusts these metrics for ancestry, identifies outlier individuals according to either a single metric or principal components of all metrics, and excludes these individuals from analysis.
We upload the association statistics produced by this pipeline to the T2DKP, following the same procedure as used for pre-computed GWAS summary statistics (Figure S3). We do not upload any individual-level genotypes and phenotypes to the T2DKP.
Genomic annotations
As of October 2022, the T2DKP included 5,418 genomic annotation datasets derived from molecular assays such as RNA-seq, ChIP-seq, ATAC-seq, DNase-seq, and Hi-C of human cell lines, stem cell-derived models, and primary tissue (Table S3). These annotations inform on the location of cis-regulatory elements, target genes of these elements, the expression levels of genes, and genetic variant effects on elements and genes (Figure S6). As we do for GWAS datasets, we identify genomic annotation datasets to include in the T2DKP based on the feedback of AMP-T2D scientists and T2DKP users. We focus on genomic annotations derived from tissues most relevant to T2D92, notably pancreatic islets, all types of adipose tissue, liver, and skeletal muscle. For comparison, we also include annotations derived from other tissues if they are collected and processed via the same methods.
We obtain genomic annotation datasets either by download from public repositories (e.g., GEO, EGA, SRA), download from resources such as ENCODE, or by direct collaboration with a contributing investigator. We assign to each dataset a unique accession number and permanent URL and then document the dataset with metadata describing donor information, cell type or tissue, molecular assay, experimental conditions, protocols, software tools, external references, and publications. We obtain raw experimental assay data (e.g., ATAC-seq, ChIP-seq, Hi-C reads) when available. We standardize cell types and tissue names to the Uber-anatomy ontology for tissues93, Cell Ontology94 for cells, and Cell Line Ontology95 or Experimental Factor Ontology96 for cell lines. We assign tissues and cells to one of 29 high-level categories describing broad tissue and anatomical system groupings (Table S3). When summary-level annotations derived from these assays (e.g., chromatin state, accessible chromatin sites, target gene predictions) have not been pre-computed, we compute them from the raw data. When summary-level annotations have been pre-computed, we obtain them as well. Prior to including pre-computed annotations in the T2DKP, we perform several QC checks. First, we check the file formats to ensure that they contain the requisite number of fields and the correct type of value per field. Second, we check the contents of pre-computed annotations to ensure that they meet minimum standards such as number of records, distribution of records across the genome, and degree of overlap with blacklisted regions of the genome. While the methods, software tools, and filters used to create pre-computed annotations may in some cases differ from those used to create T2DKP computed annotations, detailed data processing and analysis steps used to create the pre-computed annotations are included as metadata with the annotation record.
We store all genomic annotation datasets in the Common Metabolic Diseases Genome Atlas49 (CMDGA), a sister resource to the T2DKP. The CMDGA makes summary-level annotations of each dataset (represented as a set of genomic regions with relevant metadata; e.g., BED files97) accessible by the T2DKP software platform through a series of REST APIs. Data that could be subject to data use restrictions (e.g., raw reads) are maintained at the CMDGA and not made available for public access.
Quantification and statistical analysis
Statistical and bioinformatic methods
We process GWAS associations and genomic annotations through a series of five statistical and bioinformatic methods (Figure S7). Each method integrates one or more datasets to make predictions regarding variant associations or their functional effects (e.g., on disease-susceptibility genes, disease-relevant tissues, or regulatory annotations). We select methods to implement based on the following factors (in rough order of priority): ease of implementation, expected impact (based on citations and use of the method), requests by the AMP-T2D and the T2D genetics communities, and requests by users. We apply the methods within an automated analysis pipeline that tracks provenance of the calculations and regularly updates results as new data become available. Wherever possible, we consult and collaborate with the original developers of the methods when encoding them in our analysis pipeline.
The first step in the pipeline is a “bottom-line” association meta-analysis. The goal of this analysis is to calculate an integrated measure of association between each variant in the T2DKP and each trait, accounting for all datasets in which the variant is observed. As many of the larger datasets may have a (usually unknown) degree of sample overlap, the bottom-line analysis statistically infers and adjusts for the degree of overlap47 by estimating the covariance between effect sizes across studies and then inflating the variance of the test statistic (under the null) to correct for any observed non-zero covariance. This calculation has limitations but offers a compromise between presenting multiple association values for each variant (which are challenging to interpret) and conducting a proper combined analysis of the original genetic data across each study (which is impractical). Because the statistical methodology of the bottom-line method is limited to common (MAF > 5%) variants and a single ancestry, we implement it by first partitioning datasets by ancestry and then separating common and rare variants within each dataset. We next, for each ancestry, (a) conduct an overlap-aware fixed-effect meta-analysis of all common variants across all datasets for the ancestry; (b) assign each rare variant the association statistic from the largest dataset for the ancestry; and (c) combine the common and rare variant associations to produce ancestry-specific bottom-line results. We finally conduct a final fixed-effect meta-analysis (without overlap-detection) of the ancestry-specific results to obtain transethnic association statistics. The assumption of fixed variant effects across ancestries is a simplification and may reduce power to detect ancestry-specific associations; a random-effects meta-analysis is a potential future approach to add to the portal. We retain both the ancestry-level association statistics, and the transethnic association statistics, for downstream analyses.
The second step in the pipeline is to annotate all variants using the Variant Effect Predictor98 (VEP). We run VEP with its standard command line options and the LofTee and dbNSFP plugins, “picking” the annotation in the transcript determined by previously described criteria43.
Third, we run LD-clumping on the bottom-line associations to produce, for each trait, a set of “clumps”, each consisting of a lead variant and a set of variants in LD with the lead variant. We perform LD-clumping by first running the PLINK99 clump command with its default parameters. We conduct this analysis separately for each ancestry-level set of associations, estimating LD using an appropriate subset of samples from the 1000G Project; we use African 1000G samples for the African American ancestry, East Asian 1000G samples for the East Asian ancestry, European 1000G samples for the European ancestry, Middle/South American 1000G samples for the Hispanic or Latin American ancestry, and South Asian 1000G samples for the South Asian ancestry. We also conduct a transethnic LD-clumping analysis by clumping the transethnic bottom-line associations five times, once per 1000G ancestry, and then merging clumps across ancestries that have at least one variant in common.
Fourth, for each trait, we calculate the enrichment of each genomic annotation for bottom-line associations. For each trait and each annotation, we apply the GREGOR method100 to compare the observed proportion of genome-wide significant associations within annotated regions to the expected proportion based on the total size of annotated regions. GREGOR produces a fold-enrichment of each annotation for significant associations, as well as a p-value of statistical significance. We compute these enrichments within each ancestry, using LD estimates from the 1000G project in the same manner as we do for the ancestry-specific clumping analysis. If there does not exist a Mixed Ancestry dataset for the trait that is larger than the combination of the ancestry-level datasets, we then meta-analyze the ancestry-specific enrichments to obtain a transethnic enrichment. If there does exist a larger Mixed Ancestry dataset, then we conduct a GREGOR analysis of the largest Mixed Ancestry dataset to produce a transethnic enrichment; since most GWAS (even those of Mixed Ancestry) are skewed heavily toward European populations, we use LD estimates from the 1000G European samples for this analysis.
Fifth, we calculate gene-level association statistics using MAGMA53. We run MAGMA on the bottom-line association results for each trait with its default parameters and a window size of 50kb. We calculate MAGMA associations within each ancestry, as well as at the transethnic level; the choice of samples for LD estimates is analogous to the choice for GREGOR, as is the combination of ancestry-level and Mixed Ancestry datasets to produce transethnic results.
We load the results of these analyses into cloud-based storage buckets (currently housed on Amazon S3) and index them with custom software that we term “BioIndex”101. BioIndex uses a MySQL database to map genomic coordinates to storage bucket locations, enabling random access to data for any variant or genomic region directly from cloud storage. Alongside these data we also store references to the original GWAS association statistics (which are also stored in the cloud) or to the original genomic annotations in CMDGA.
The results of these analyses are integrated into many tools within the T2DKP, and their most significant results for each phenotype are available on the “phenotype page” (Figure 2). All tables and visualizations on the phenotype page can display results calculated across all datasets in the portal or results calculated only for a chosen ancestry.
Pre-computed statistical and bioinformatic method results
In addition to the statistical bioinformatic methods that we automatically apply to every dataset, the T2DKP also includes pre-computed statistical and bioinformatic method results. These include: (a) credible sets (for 40 traits42,102); (b) association by contact (ABC) predictions, which integrate ATAC-seq, H3K27ac, and Hi-C data to predict linkages between regions and gene promoters103; and (c) predictions of GWAS effector genes (for 11 traits) from a machine learning algorithm104. For each pre-computed method result, we record provenance as either a publication or online resource describing the method and data used by it.
Curated knowledge
The T2DKP includes a curated list of putative T2D GWAS effector genes. The goal of this list is to represent the current consensus predictions of the AMP-T2D consortium regarding the genes that mediate T2D GWAS associations, together with the confidence behind each prediction. The effector gene predictions have limitations, as they are based on a series of heuristics rather than a formal statistical methodology. Genes in a region harboring a genome-wide significant T2D association are assigned to one of five tiers (Causal, Strong, Moderate, Possible, Weak) based on three types of evidence: genetic (whether a coding variant in the gene is causal for the association), regulatory (whether genomic annotations support a link between the causal variant and the gene), and perturbational (whether animal or cellular studies of the gene have linked it to T2D or related traits). Each piece of evidence is determined based on a literature review by AMP-T2D scientists, and we record the publication describing it as provenance supporting the effector gene prediction. We note that the effector gene list is unpublished and thus has not undergone peer review, and it is subject to the biases of the scientists who created it; however, it represents (to our knowledge) the best available distillation of curated knowledge regarding T2D effector genes. Over time, as this and other lists are improved, we will update the T2DKP with them and offer users the ability to compare different lists.
In addition to putative T2D effector genes, this gene list also includes putative effector genes for T2D-related diseases and traits. These include genes implicated in monogenic conditions with presentations similar to T2D (e.g., Maturity Onset Diabetes of the Young, Lipodystrophies, or Neonatal Diabetes Mellitus) and putative effector genes for glycemic traits (e.g., fasting glucose or HbA1c).
Tool implementation
To implement the Signal Sifter, we first find all SNPs associated at p<5×10–8 with a lead phenotype and then expand each “index” SNP into a clump using the results of LD clumping. We next filter the clumps to those that contain at least one SNP associated (at a user-specified p-value threshold below 0.05) with each of a set of secondary phenotypes. Next, we set the index SNP for each secondary phenotype to the SNP with the strongest association for that phenotype; the index SNPs may thus differ across phenotypes. Finally, we align the direction of effect for each secondary phenotype index SNP to the direction of effect to the lead phenotype index SNP: we switch the secondary phenotype direction of effect for the secondary phenotype index SNP as needed to match secondary phenotype direction of effect for the lead phenotype index SNP.
To implement the Gene Finder, we identify all genes with that have MAGMA p-value below a user-specified threshold for each of the selected traits. We sort the resulting list of genes according to the p-value produced by a meta-analysis of their trait-level p-values (according to Fisher’s method).
To implement the Variant Sifter, we identify all variants that are (a) within a user-selected credible set that also (b) overlap any of the user-selected annotations (users can alter this default behavior to retain only variants that overlap all user-selected annotations) and finally (c) are linked to any of the selected genes. We show annotation enrichments as calculated by GREGOR.
To implement the GAIT module for individual-level WES data in the T2DKP (currently the AMP-T2D-GENES and TOPMed WES datasets), we use LDserver105 to calculate score statistics and covariance matrices106 from the individual genotypes and phenotypes. LDserver responds to a REST query (specifying a list of variants) from the T2DKP and either computes these statistics on-demand (for the AMP-T2D-GENES dataset) or accesses pre-computed files of genome-wide statistics (for the TOPMed dataset). It then converts these statistics to an aggregate association statistic106. Individual-level data stored within LDserver are not directly accessible by the T2DKP.
Analyses conducted for this manuscript
To compare GWAS summary statistic datasets across the T2DKP, GWAS Catalog, OpenGWAS, and GWAS Atlas, we identified the datasets in each resource for T2D, fasting glucose (FG), fasting insulin (FI), or HbA1c. For the T2DKP, we defined these as all datasets listed on the T2DKP “Genetic association datasets” page that included associations for at least one of the four phenotypes. For the GWAS Catalog, OpenGWAS, and GWAS Atlas, we defined these as the datasets listed in the results for searches performed for each of the four phenotypes. We conducted all searches for this analysis on October 16, 2022; the datasets included in our analysis are shown in Table S2. We defined datasets as having “full” summary statistics accessible if either (a) summary statistic files for every variant in the study were available for public download or (b) associations for every variant in the study were represented in the resource through genome-wide query interfaces or genome-wide visualizations (e.g., Manhattan plots on the GWAS Atlas). We defined “partial” datasets as those for which some associations are available, as in the tables of top associations provided by the GWAS Catalog, but for which full summary statistics are not accessible through that resource.
To define associated loci for each trait, we first downloaded association p-values from each resource. We then ran a clumping analysis across all associations (using the PLINK 2 software package with parameters –clump-p1 5e-8 –clump-p2 5e-8 –clump-r2 0 –clump-kb 250) to merge genome-wide significant associations within 250kb of each other into clumps (i.e., associated loci). For all calculations comparing the loci included by each resource, we then defined a resource to include a locus if it contained at least one associated variant (p<5×10−8) within the clump.
To evaluate rare coding variant association enrichments for T2D effector genes, we first downloaded the effector gene list from the T2DKP. Following a previously described procedure43, we then used a Wilcoxon Rank Sum Test to compare the gene-level association p-values from the AMP-T2D-GENES study for these genes to those of a set of genes matched based on various features (such as the number of variants and aggregate allele count within each gene); we removed genes from this analysis which are present on the effector gene list due to rare variant associations (PAM, SLC30A8, MC4R, and BCL11A). For comparison, we repeated this analysis for the same number of genes with the lowest MAGMA T2D p-values.
To evaluate the enrichment of the set of genes returned by the Gene Finder for adipose-specific expression, we first obtained tissue-specific expression values for each gene across each tissue, based on a previous analysis of samples from the GTEx project65. Tissue-specific expression is represented as a “t-stat” for each gene in each tissue. We used a Wilcox test (for each tissue) to compare the t-stat values for genes in the Gene Finder list to the other genes in the genome. For comparison, we also conducted this analysis for the same number of genes with the lowest MAGMA T2D p-values.
Supplementary Material
The T2DKP contains 382 genetic datasets (October 2022), Related to Figure 4.
Compared to the GWAS Catalog and GWAS Atlas, the T2DKP contains unique genetic datasets for T2D, fasting glucose (FG), fasting insulin (FI), and HbA1c levels (October, 2022), Related to Figure 4.
The T2DKP contains thousands of genomic annotations drawn from the Common Metabolic Diseases Genome Atlas (October, 2022), Related to STAR Methods.
SNPs matching an insulin resistance signature in the Signal Sifter, Related to STAR Methods
Figures S1-S7
Source data.
Acknowledgments
This paper is dedicated to the memories of two colleagues who were instrumental in developing the T2DKP: Todd Green and Julia Brosnan.
This work was supported predominantly by 2UM1DK105554. Other funding that supported this work includes: R01HG009976 (M.B.); NHGRI grant FAIN# U01HG011723 (J.M.M.); NIDDK U01DK105535 (M.I.M.); and RFP awards 1, 2, 3, 4, 7, 8a, 8b, 9, 10, 11, 13, 14, 15, and 16 from the Foundation for the National Institutes of Health. J.M.M. was supported by American Diabetes Association Innovative and Clinical Translational Award 1-19-ICTS-068. M.I.M. was a Wellcome Investigator and an NIHR Senior Investigator (Wellcome: 090532, 098381, 106130, 203141, 212259). P.D. was supported by R01DK125490.
Declaration of interests
A.C. is a Sanofi employee and holds shares and stock options in the company. M.I.M. has served on advisory panels for Pfizer, Novo Nordisk and Zoe Global, has received honoraria from Merck, Pfizer, Novo Nordisk and Eli Lilly, and research funding from Abbvie, Astra Zeneca, Boehringer Ingelheim, Eli Lilly, Janssen, Merck, Novo Nordisk, Pfizer, Roche, Sanofi Aventis, Servier, and Takeda. As of June 2019, M.I.M. is an employee of Genentech, and a holder of Roche stock. M.R.M. is a Pfizer employee and holds shares of stock in the company. M.K.T. is an employee and shareholder of Eli Lilly and Company. As of April 2022, P.D. is an employee and stockholder of Regeneron Pharmaceuticals.
Consortia
The AMP-T2D Consortium: Gonçalo Abecasis, Beena Akolkar, Benjamin R. Alexander, Nicholette D. Allred, David Altshuler, Jennifer E. Below, Richard Bergman, Joline W.J. Beulens, John Blangero, Michael Boehnke, Krister Bokvist, Erwin Bottinger, Andrew P. Boughton, Donald Bowden, M Julia Brosnan, Christopher Brown, Kenneth Bruskiewicz, Noël P. Burtt, Mary Carmichael, Lizz Caulkins, Inês Cebola, John Chambers, Yii-Der Ida Chen, Andriy Cherkas, Audrey Y. Chu, Christopher Clark, Melina Claussnitzer, Maria C. Costanzo, Nancy J. Cox, Marcel den Hoed, Duc Dong, Marc Duby, Ravindranath Duggirala, Josée Dupuis, Petra J.M. Elders, Jesse M. Engreitz, Eric Fauman, Jorge Ferrer, Jason Flannick, Paul Flicek, Matthew Flickinger, Jose C. Florez, Caroline S. Fox, Timothy M. Frayling, Kelly A. Frazer, Kyle J. Gaulton, Clint Gilbert, Anna L. Gloyn, Todd Green, Craig L. Hanis, Robert Hanson, Andrew T. Hattersley, Quy Hoang, Hae Kyung Im, Sidra Iqbal, Suzanne B.R. Jacobs, Dong-Keun Jang, Tad Jordan, Tania Kamphaus, Fredrik Karpe, Thomas M. Keane, Seung K. Kim, Alexandria Kluge, Ryan Koesterer, Parul Kudtarkar, Kasper Lage, Leslie A. Lange, Mitchell Lazar, Donna Lehman, Ching-Ti Liu, Ruth J.F. Loos, Ronald Ching-wan Ma, Patrick MacDonald, Jeffrey Massung, Matthew T. Maurano, Mark I. McCarthy, Gil McVean, James B. Meigs, Josep M. Mercader, Melissa R. Miller, Braxton Mitchell, Karen L. Mohlke, Samuel Morabito, Claire Morgan, Shannon Mullican, Sharvari Narendra, Maggie C.Y. Ng, Lynette Nguyen, Colin N.A. Palmer, Stephen C.J. Parker, Antonio Parrado, Afshin Parsa, Aaron C. Pawlyk, Ewan R. Pearson, Andrew Plump, Michael Province, Thomas Quertermous, Susan Redline, Dermot F. Reilly, Bing Ren, Stephen S. Rich, J. Brent Richards, Jerome I. Rotter, Oliver Ruebenacker, Hartmut Ruetten, Rany M. Salem, Maike Sander, Michael Sanders, Dharambir Sanghera, Laura J. Scott, Sebanti Sengupta, David Siedzik, Xueling Sim, Xueling Sim, Preeti Singh, Robert Sladek, Kerrin Small, Philip Smith, Peter Stein, Dylan Spalding, Heather M. Stringham, Ying Sun, Katalin Susztak, Leen M. ‘t Hart, Daniel Taliun, Kent Taylor, Melissa K. Thomas, Jennifer A. Todd, Miriam S. Udler, Miriam S. Udler, Benjamin Voight, Marcin von Grotthuss, Andre Wan, Ryan P. Welch, David Wholley, Kaan Yuksel, Norann A. Zaghloul
References
- 1.Claussnitzer M, Cho JH, Collins R, Cox NJ, Dermitzakis ET, Hurles ME, Kathiresan S, Kenny EE, Lindgren CM, MacArthur DG, et al. (2020). A brief history of human disease genetics. Nature 577, 179–189. 10.1038/s41586-019-1879-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang Y, Qi G, Park J-H, and Chatterjee N (2018). Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat. Genet 50, 1318–1326. 10.1038/s41588-018-0193-x. [DOI] [PubMed] [Google Scholar]
- 3.Vilhjálmsson BJ, Yang J, Finucane HK, Gusev A, Lindström S, Ripke S, Genovese G, Loh P-R, Bhatia G, Do R, et al. (2015). Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am. J. Hum. Genet 97, 576–592. 10.1016/j.ajhg.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davey Smith G, and Hemani G (2014). Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet 23, R89–R98. 10.1093/hmg/ddu328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.King EA, Davis JW, and Degner JF (2019). Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval.: Supplementary Methods And Results (Genetics) 10.1101/513945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasaniuc B, and Price AL (2017). Dissecting the genetics of complex traits using summary association statistics. Nat. Rev. Genet 18, 117–127. 10.1038/nrg.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cano-Gamez E, and Trynka G (2020). From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases. Front. Genet 11, 424. 10.3389/fgene.2020.00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sollis E, Mosaku A, Abid A, Buniello A, Cerezo M, Gil L, Groza T, Güneş O, Hall P, Hayhurst J, et al. (2023). The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res 51, D977–D985. 10.1093/nar/gkac1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eicher JD, Landowski C, Stackhouse B, Sloan A, Chen W, Jensen N, Lien J-P, Leslie R, and Johnson AD (2015). GRASP v2.0: an update on the Genome-Wide Repository of Associations between SNPs and phenotypes. Nucleic Acids Res. 43, D799–804. 10.1093/nar/gku1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian D, Wang P, Tang B, Teng X, Li C, Liu X, Zou D, Song S, and Zhang Z (2020). GWAS Atlas: a curated resource of genome-wide variant-trait associations in plants and animals. Nucleic Acids Res. 48, D927–D932. 10.1093/nar/gkz828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McInnes G, Tanigawa Y, DeBoever C, Lavertu A, Olivieri JE, Aguirre M, and Rivas MA (2019). Global Biobank Engine: enabling genotype-phenotype browsing for biobank summary statistics. Bioinforma. Oxf. Engl 35, 2495–2497. 10.1093/bioinformatics/bty999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karczewski KJ, Solomonson M, Chao KR, Goodrich JK, Tiao G, Lu W, Riley-Gillis BM, Tsai EA, Kim HI, Zheng X, et al. (2022). Systematic single-variant and gene-based association testing of thousands of phenotypes in 394,841 UK Biobank exomes. Cell Genomics 2. 10.1016/j.xgen.2022.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramos EM, Hoffman D, Junkins HA, Maglott D, Phan L, Sherry ST, Feolo M, and Hindorff LA (2014). Phenotype-Genotype Integrator (PheGenI): synthesizing genome-wide association study (GWAS) data with existing genomic resources. Eur. J. Hum. Genet. EJHG 22, 144–147. 10.1038/ejhg.2013.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elsworth B, Lyon M, Alexander T, Liu Y, Matthews P, Hallett J, Bates P, Palmer T, Haberland V, Smith GD, et al. (2020). The MRC IEU OpenGWAS data infrastructure. 10.1101/2020.08.10.244293. [DOI] [Google Scholar]
- 15.Howe KL, Achuthan P, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, Bhai J, et al. (2021). Ensembl 2021. Nucleic Acids Res. 49, D884–D891. 10.1093/nar/gkaa942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Navarro Gonzalez J, Zweig AS, Speir ML, Schmelter D, Rosenbloom KR, Raney BJ, Powell CC, Nassar LR, Maulding ND, Lee CM, et al. (2021). The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 49, D1046–D1057. 10.1093/nar/gkaa1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kundaje Anshul, Meuleman Wouter, Ernst Jason, Bilenky Misha, Yen Angela, Alireza Heravi-Moussavi Pouya Kheradpour, Zhang Zhizhuo, Wang Jianrong, Ziller Michael J., et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clough E, and Barrett T (2016). The Gene Expression Omnibus database. Methods Mol. Biol. Clifton NJ 1418, 93–110. 10.1007/978-1-4939-3578-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The GTEx Consortium, Ardlie KG, Deluca DS, Segrè AV, Sullivan TJ, Young TR, Gelfand ET, Trowbridge CA, Maller JB, Tukiainen T, et al. (2015). The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660. 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Gamazon ER, Zhang X, Konkashbaev A, Liu C, Szilágyi KL, Dolan ME, and Cox NJ (2015). SCAN database: facilitating integrative analyses of cytosine modification and expression QTL. Database J. Biol. Databases Curation 2015, bav025. 10.1093/database/bav025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.UniProt: a worldwide hub of protein knowledge (2019). Nucleic Acids Res. 47, D506–D515. 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al. (2021). The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, D605–D612. 10.1093/nar/gkaa1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muñoz-Fuentes V, Cacheiro P, Meehan TF, Aguilar-Pimentel JA, Brown SDM, Flenniken AM, Flicek P, Galli A, Mashhadi HH, Hrabě de Angelis M, et al. (2018). The International Mouse Phenotyping Consortium (IMPC): a functional catalogue of the mammalian genome that informs conservation. Conserv. Genet. Print 19, 995–1005. 10.1007/s10592-018-1072-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eppig JT, Smith CL, Blake JA, Ringwald M, Kadin JA, Richardson JE, and Bult CJ (2017). Mouse Genome Informatics (MGI): Resources for Mining Mouse Genetic, Genomic, and Biological Data in Support of Primary and Translational Research. Methods Mol. Biol. Clifton NJ 1488, 47–73. 10.1007/978-1-4939-6427-7_3. [DOI] [PubMed] [Google Scholar]
- 27.Zheng J, Erzurumluoglu AM, Elsworth BL, Kemp JP, Howe L, Haycock PC, Hemani G, Tansey K, Laurin C, Early Genetics and Lifecourse Epidemiology (EAGLE) Eczema Consortium, et al. (2017). LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinforma. Oxf. Engl 33, 272–279. 10.1093/bioinformatics/btw613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, et al. (2018). The MR-Base platform supports systematic causal inference across the human phenome. eLife 7, e34408. 10.7554/eLife.34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, Eyler AE, Denny JC, Consortium, Gte., Nicolae DL, et al. (2015). A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet 47, 1091–1098. 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Watanabe K, Taskesen E, van Bochoven A, and Posthuma D (2017). Functional mapping and annotation of genetic associations with FUMA. Nat. Commun 8. 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carvalho-Silva D, Pierleoni A, Pignatelli M, Ong C, Fumis L, Karamanis N, Carmona M, Faulconbridge A, Hercules A, McAuley E, et al. (2019). Open Targets Platform: new developments and updates two years on. Nucleic Acids Res. 47, D1056–D1065. 10.1093/nar/gky1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, and Hamosh A (2015). OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 43, D789–798. 10.1093/nar/gku1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, et al. (2015). ClinGen--the Clinical Genome Resource. N. Engl. J. Med 372, 2235–2242. 10.1056/NEJMsr1406261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, and Cooper DN (2017). The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet 136, 665–677. 10.1007/s00439-017-1779-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei C-H, Kao H-Y, and Lu Z (2013). PubTator: a web-based text mining tool for assisting biocuration. Nucleic Acids Res. 41, W518–522. 10.1093/nar/gkt441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kilicoglu H, Shin D, Fiszman M, Rosemblat G, and Rindflesch TC (2012). SemMedDB: a PubMed-scale repository of biomedical semantic predications. Bioinforma. Oxf. Engl 28, 3158–3160. 10.1093/bioinformatics/bts591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuleshov V, Ding J, Vo C, Hancock B, Ratner A, Li Y, Ré C, Batzoglou S, and Snyder M (2019). A machine-compiled database of genome-wide association studies. Nat. Commun 10, 3341. 10.1038/s41467-019-11026-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salem RM, Todd JN, Sandholm N, Cole JB, Chen W-M, Andrews D, Pezzolesi MG, McKeigue PM, Hiraki LT, Qiu C, et al. (2019). Genome-Wide Association Study of Diabetic Kidney Disease Highlights Biology Involved in Glomerular Basement Membrane Collagen. J. Am. Soc. Nephrol. JASN 30, 2000–2016. 10.1681/ASN.2019030218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiou J, Zeng C, Cheng Z, Han JY, Schlichting M, Miller M, Mendez R, Huang S, Wang J, Sui Y, et al. (2021). Single-cell chromatin accessibility identifies pancreatic islet cell type– and state-specific regulatory programs of diabetes risk. Nat. Genet 53, 455–466. 10.1038/s41588-021-00823-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grotz AK, Gloyn AL, and Thomsen SK (2017). Prioritising Causal Genes at Type 2 Diabetes Risk Loci. Curr. Diab. Rep 17. 10.1007/s11892-017-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, Ma C, Fontanillas P, Moutsianas L, McCarthy DJ, et al. (2016). The genetic architecture of type 2 diabetes. Nature 536, 41–47. 10.1038/nature18642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. (2018). Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet 50, 1505. 10.1038/s41588-018-0241-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flannick J, Mercader JM, Fuchsberger C, Udler MS, Mahajan A, Wessel J, Teslovich TM, Caulkins L, Koesterer R, Barajas-Olmos F, et al. (2019). Exome sequencing of 20,791 cases of type 2 diabetes and 24,440 controls. Nature 570, 71–76. 10.1038/s41586-019-1231-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dornbos P, Singh P, Jang D-K, Mahajan A, Biddinger SB, Rotter JI, McCarthy MI, and Flannick J (2022). Evaluating human genetic support for hypothesized metabolic disease genes. Cell Metab. 34, 661–666. 10.1016/j.cmet.2022.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Type 2 Diabetes Knowledge Portal - AMP T2D Partnership. https://t2d.hugeamp.org/ampt2dpartnership.html.
- 46.Fitipaldi H, and Franks PW (2022). Ethnic, gender and other sociodemographic biases in genome-wide association studies for the most burdensome non-communicable diseases: 2005–2022. Hum. Mol. Genet, ddac245. 10.1093/hmg/ddac245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.METAL Documentation - Genome Analysis Wiki. https://genome.sph.umich.edu/wiki/METAL_Documentation#Sample_Overlap_Correction.
- 48.Nasser J, Bergman DT, Fulco CP, Guckelberger P, Doughty BR, Patwardhan TA, Jones TR, Nguyen TH, Ulirsch JC, Lekschas F, et al. (2021). Genome-wide enhancer maps link risk variants to disease genes. Nature 593, 238–243. 10.1038/s41586-021-03446-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Common Metabolic Diseases Genome Atlas. https://cmdga.org/.
- 50.Bush WS, Oetjens MT, and Crawford DC (2016). Unravelling the human genome–phenome relationship using phenome-wide association studies. Nat. Rev. Genet 17, 129–145. 10.1038/nrg.2015.36. [DOI] [PubMed] [Google Scholar]
- 51.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, and Willer CJ (2010). LocusZoom: regional visualization of genome-wide association scan results. Bioinforma. Oxf. Engl 26, 2336–2337. 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boughton AP, Welch RP, Flickinger M, VandeHaar P, Taliun D, Abecasis GR, and Boehnke M (2021). LocusZoom.js: interactive and embeddable visualization of genetic association study results. Bioinformatics 37, 3017–3018. 10.1093/bioinformatics/btab186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leeuw C.A. de, Mooij JM, Heskes T, and Posthuma D (2015). MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLOS Comput. Biol 11, e1004219. 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Langlet F, Haeusler RA, Lindén D, Ericson E, Norris T, Johansson A, Cook JR, Aizawa K, Wang L, Buettner C, et al. (2017). Selective Inhibition of FOXO1 Activator/Repressor Balance Modulates Hepatic Glucose Handling. Cell 171, 824–835.e18. 10.1016/j.cell.2017.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Haeusler RA, Han S, and Accili D (2010). Hepatic FoxO1 Ablation Exacerbates Lipid Abnormalities during Hyperglycemia. J. Biol. Chem 285, 26861–26868. 10.1074/jbc.M110.134023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gustafsson M, Gawel DR, Alfredsson L, Baranzini S, Björkander J, Blomgran R, Hellberg S, Eklund D, Ernerudh J, Kockum I, et al. (2015). A validated gene regulatory network and GWAS identifies early regulators of T cell–associated diseases. Sci. Transl. Med 7, 313ra178–313ra178. 10.1126/scitranslmed.aad2722. [DOI] [PubMed] [Google Scholar]
- 57.Smith GD, and Ebrahim S (2002). Data dredging, bias, or confounding. BMJ 325, 1437–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saxena A, Wahi N, Kumar A, and Mathur SK (2020). Functional Interactomes of Genes Showing Association with Type-2 Diabetes and Its Intermediate Phenotypic Traits Point towards Adipo-Centric Mechanisms in Its Pathophysiology. Biomolecules 10, 601. 10.3390/biom10040601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wray NR, Wijmenga C, Sullivan PF, Yang J, and Visscher PM (2018). Common Disease Is More Complex Than Implied by the Core Gene Omnigenic Model. Cell 173, 1573–1580. 10.1016/j.cell.2018.05.051. [DOI] [PubMed] [Google Scholar]
- 60.Boyle EA, Li YI, and Pritchard JK (2017). An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186. 10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hackinger S, and Zeggini E (2017). Statistical methods to detect pleiotropy in human complex traits. Open Biol. 7, 170125. 10.1098/rsob.170125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Udler MS, Kim J, Grotthuss M, von, Bonàs-Guarch S, Cole JB, Chiou J, Isgc CDA. on behalf of M. and the, Boehnke M, Laakso M, Atzmon G, et al. (2018). Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: A soft clustering analysis. PLOS Med. 15, e1002654. 10.1371/journal.pmed.1002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nguyen PA, Born DA, Deaton AM, Nioi P, and Ward LD (2019). Phenotypes associated with genes encoding drug targets are predictive of clinical trial side effects. Nat. Commun 10, 1579. 10.1038/s41467-019-09407-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, Gaulton KJ, Eicher JD, Sharp SJ, Luan J, et al. (2016). Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat. Genet 10.1038/ng.3714. [DOI] [PubMed] [Google Scholar]
- 65.Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, Gazal S, Loh P-R, Lareau C, Shoresh N, et al. (2018). Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet 50, 621–629. 10.1038/s41588-018-0081-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Foley CN, Staley JR, Breen PG, Sun BB, Kirk PDW, Burgess S, and Howson JMM (2021). A fast and efficient colocalization algorithm for identifying shared genetic risk factors across multiple traits. Nat. Commun 12, 764. 10.1038/s41467-020-20885-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lappalainen T, and MacArthur DG (2021). From variant to function in human disease genetics. Science 373, 1464–1468. 10.1126/science.abi8207. [DOI] [PubMed] [Google Scholar]
- 68.Torres JM, Abdalla M, Payne A, Fernandez-Tajes J, Thurner M, Nylander V, Gloyn AL, Mahajan A, and McCarthy MI (2020). A Multi-omic Integrative Scheme Characterizes Tissues of Action at Loci Associated with Type 2 Diabetes. Am. J. Hum. Genet 107, 1011–1028. 10.1016/j.ajhg.2020.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mahajan A, Spracklen CN, Zhang W, Ng MCY, Petty LE, Kitajima H, Yu GZ, Rüeger S, Speidel L, Kim YJ, et al. (2022). Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet 54, 560–572. 10.1038/s41588-022-01058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fogarty MP, Cannon ME, Vadlamudi S, Gaulton KJ, and Mohlke KL (2014). Identification of a Regulatory Variant That Binds FOXA1 and FOXA2 at the CDC123/CAMK1D Type 2 Diabetes GWAS Locus. PLoS Genet 10, e1004633. 10.1371/journal.pgen.1004633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gaulton KJ, Ferreira T, Lee Y, Raimondo A, Mägi R, Reschen ME, Mahajan A, Locke A, Rayner NW, Robertson N, et al. (2015). Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat. Genet 47, 1415–1425. 10.1038/ng.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mahajan A, Wessel J, Willems SM, Zhao W, Robertson NR, Chu AY, Gan W, Kitajima H, Taliun D, Rayner NW, et al. (2018). Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat. Genet 50, 559–571. 10.1038/s41588-018-0084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sarnowski C, Leong A, Raffield LM, Wu P, de Vries PS, DiCorpo D, Guo X, Xu H, Liu Y, Zheng X, et al. (2019). Impact of Rare and Common Genetic Variants on Diabetes Diagnosis by Hemoglobin A1c in Multi-Ancestry Cohorts: The Trans-Omics for Precision Medicine Program. Am. J. Hum. Genet 105, 706–718. 10.1016/j.ajhg.2019.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Goldstein DB, Allen A, Keebler J, Margulies EH, Petrou S, Petrovski S, and Sunyaev S (2013). Sequencing studies in human genetics: design and interpretation. Nat. Rev. Genet 14, 460–470. 10.1038/nrg3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu MC, Lee S, Cai T, Li Y, Boehnke M, and Lin X (2011). Rare-Variant Association Testing for Sequencing Data with the Sequence Kernel Association Test. Am. J. Hum. Genet 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee S, Wu MC, and Lin X (2012). Optimal tests for rare variant effects in sequencing association studies. Biostat. Oxf. Engl 13, 762–775. 10.1093/biostatistics/kxs014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Majithia AR, Flannick J, Shahinian P, Guo M, Bray M-A, Fontanillas P, Gabriel SB, Consortium G, Project NJAS, Consortium ST, et al. (2014). Rare variants in PPARG with decreased activity in adipocyte differentiation are associated with increased risk of type 2 diabetes. Proc. Natl. Acad. Sci 111, 13127–13132. 10.1073/pnas.1410428111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lotta LA, Mokrosiński J, Mendes de Oliveira E, Li C, Sharp SJ, Luan J, Brouwers B, Ayinampudi V, Bowker N, Kerrison N, et al. (2019). Human Gain-of-Function MC4R Variants Show Signaling Bias and Protect against Obesity. Cell 177, 597–607.e9. 10.1016/j.cell.2019.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Leeuw CA, Neale BM, Heskes T, and Posthuma D (2016). The statistical properties of gene-set analysis. Nat. Rev. Genet 17, 353–364. 10.1038/nrg.2016.29. [DOI] [PubMed] [Google Scholar]
- 80.Hormozdiari F, van de Bunt M, Segrè AV, Li X, Joo JWJ, Bilow M, Sul JH, Sankararaman S, Pasaniuc B, and Eskin E. (2016). Colocalization of GWAS and eQTL Signals Detects Target Genes. Am. J. Hum. Genet 99, 1245–1260. 10.1016/j.ajhg.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Westerman KE, Majarian TD, Giulianini F, Jang D-K, Miao J, Florez JC, Chen H, Chasman DI, Udler MS, Manning AK, et al. (2022). Variance-quantitative trait loci enable systematic discovery of gene-environment interactions for cardiometabolic serum biomarkers. Nat. Commun 13, 3993. 10.1038/s41467-022-31625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Metabolic Disorders Knowledge Portal - Project. https://hugeamp.org/project.html?project=lunaris.
- 83.Jensen MA, Ferretti V, Grossman RL, and Staudt LM (2017). The NCI Genomic Data Commons as an engine for precision medicine. Blood 130, 453–459. 10.1182/blood-2017-03-735654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.BioData Catalyst: Home https://biodatacatalyst.nhlbi.nih.gov/.
- 85.NHGRI Analysis Visualization and Informatics Lab-space The AnVIL. https://anvilproject.org/.
- 86.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, and Daly MJ (2019). Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet 51, 584–591. 10.1038/s41588-019-0379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.gwas-sumstats-harmoniser (2022). [Google Scholar]
- 88.AMP T2DKP Policies. https://t2d.hugeamp.org/policies.html.
- 89.Laakso M, Kuusisto J, Stančáková A, Kuulasmaa T, Pajukanta P, Lusis AJ, Collins FS, Mohlke KL, and Boehnke M (2017). The Metabolic Syndrome in Men study: a resource for studies of metabolic and cardiovascular diseases. J. Lipid Res 58, 481–493. 10.1194/jlr.O072629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Morales J, Welter D, Bowler EH, Cerezo M, Harris LW, McMahon AC, Hall P, Junkins HA, Milano A, Hastings E, et al. (2018). A standardized framework for representation of ancestry data in genomics studies, with application to the NHGRI-EBI GWAS Catalog. Genome Biol. 19, 21. 10.1186/s13059-018-1396-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, et al. (2016). Next-generation genotype imputation service and methods. Nat. Genet 48, 1284–1287. 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Galicia-Garcia U, Benito-Vicente A, Jebari S, Larrea-Sebal A, Siddiqi H, Uribe KB, Ostolaza H, and Martín C (2020). Pathophysiology of Type 2 Diabetes Mellitus. Int. J. Mol. Sci 21, 6275. 10.3390/ijms21176275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mungall CJ, Torniai C, Gkoutos GV, Lewis SE, and Haendel MA (2012). Uberon, an integrative multi-species anatomy ontology. Genome Biol. 13, R5. 10.1186/gb-2012-13-1-r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Diehl AD, Meehan TF, Bradford YM, Brush MH, Dahdul WM, Dougall DS, He Y, Osumi-Sutherland D, Ruttenberg A, Sarntivijai S, et al. (2016). The Cell Ontology 2016: enhanced content, modularization, and ontology interoperability. J. Biomed. Semant 7, 44. 10.1186/s13326-016-0088-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sarntivijai S, Lin Y, Xiang Z, Meehan TF, Diehl AD, Vempati UD, Schürer SC, Pang C, Malone J, Parkinson H, et al. (2014). CLO: The cell line ontology. J. Biomed. Semant 5, 37. 10.1186/2041-1480-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Malone J, Holloway E, Adamusiak T, Kapushesky M, Zheng J, Kolesnikov N, Zhukova A, Brazma A, and Parkinson H (2010). Modeling sample variables with an Experimental Factor Ontology. Bioinformatics 26, 1112–1118. 10.1093/bioinformatics/btq099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. 10.1093/bioinformatics/btq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, and Cunningham F (2016). The Ensembl Variant Effect Predictor. Genome Biol. 17, 122. 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. (2007). PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet 81, 559–575. 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schmidt EM, Zhang J, Zhou W, Chen J, Mohlke KL, Chen YE, and Willer CJ (2015). GREGOR: evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics 31, 2601–2606. 10.1093/bioinformatics/btv201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bio-Index (2021). [Google Scholar]
- 102.UKBB Fine-mapping README Google Docs. https://docs.google.com/document/u/3/d/14LWxqlSC6hl9FtA984CQjUdFcgQQkXuffYcbXaUoqGM/edit?usp=embed_facebook.
- 103.Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA, et al. (2019). Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nat. Genet 51, 1664–1669. 10.1038/s41588-019-0538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Forgetta V, Jiang L, Vulpescu NA, Hogan MS, Chen S, Morris JA, Grinek S, Benner C, Jang D-K, Hoang Q, et al. (2022). An effector index to predict target genes at GWAS loci. Hum. Genet 141, 1431–1447. 10.1007/s00439-022-02434-z. [DOI] [PubMed] [Google Scholar]
- 105.LDServer (2021). [Google Scholar]
- 106.Feng S, Liu D, Zhan X, Wing MK, and Abecasis GR (2014). RAREMETAL: fast and powerful meta-analysis for rare variants. Bioinformatics 30, 2828–2829. 10.1093/bioinformatics/btu367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The T2DKP contains 382 genetic datasets (October 2022), Related to Figure 4.
Compared to the GWAS Catalog and GWAS Atlas, the T2DKP contains unique genetic datasets for T2D, fasting glucose (FG), fasting insulin (FI), and HbA1c levels (October, 2022), Related to Figure 4.
The T2DKP contains thousands of genomic annotations drawn from the Common Metabolic Diseases Genome Atlas (October, 2022), Related to STAR Methods.
SNPs matching an insulin resistance signature in the Signal Sifter, Related to STAR Methods
Figures S1-S7
Source data.
Data Availability Statement
All data reported in this paper are included in the main text, figures, and supplementary files.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
All values used to generate the graphs of the paper can be found in the file Data S1 – Source Data. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.