

Abstract
Rapalogues are powerful therapeutic modalities for breast cancer; however, they suffer from low solubility and dose-limiting side effects. To overcome these challenges, we developed a long-circulating multiheaded drug carrier called 5FA, which contains rapamycin-binding domains linked with elastin-like polypeptides (ELPs). To target these “Hydra-ELPs” toward breast cancer, we here linked 5FA with four distinct peptides which are reported to engage the cell surface form of the 78 kDa glucose-regulated protein (csGRP78). To determine if these peptides affected the carrier solubility, this library was characterized by light scattering and mass spectrometry. To guide in vitro selection of the most potent functional carrier for rapamycin, its uptake and inhibition of mTORC1 were monitored in a ductal breast cancer model (BT474). Using flow cytometry to track cellular association, it was found that only the targeted carriers enhanced cellular uptake and were susceptible to proteolysis by SubA, which specifically targets csGRP78. The functional inhibition of mTOR was monitored by Western blot for pS6K, whereby the best carrier L-5FA reduced mTOR activity by 3-fold compared to 5FA or free rapamycin. L-5FA was further visualized using super-resolution confocal laser scanning microscopy, which revealed that targeting increased exposure to the carrier by ~8-fold. This study demonstrates how peptide ligands for GRP78, such as the L peptide (RLLDTNRPLLPY), may be incorporated into protein-based drug carriers to enhance targeting.
Graphical Abstract
INTRODUCTION
Rapamycin is a macrolide with structural and mechanistic similarities to everolimus, which is approved for combination therapies of hormone-receptor positive breast cancer. The BOLERO-2 clinical trial strongly supported the efficacy of this rapalogue; however, it suffered from significant adverse events.1,2 Postapproval, these issues continue to limit patient drug adherence; furthermore, its low oral bioavailability and solubility have impacted other major clinical studies.2–5 This highlights the need to develop delivery strategies that improve their therapeutic value. One strategy to improve aqueous drug solubility is to hide hydrophobic molecular structures inside an inclusion complex. Rapalogues form a complex with FK506-binding protein (FKBP12), the cognate binding partner of FK506. When bound to FKBP, rapamycin solubility is improved without preventing its inhibition of the mechanistic/mammalian target of rapamycin complex 1 (mTORC1).6,7
MacKay Lab has developed several FKBP-rapalogue formulations fused to elastin-like polypeptides (ELPs), which improve rapalogue solubility and extend the plasma half-life, possibly by reducing glomerular filtration.8 ELPs are biocompatible biopolymers comprising a pentameric repeat of amino acids (VPGXG)n that are soluble below a transition temperature (Tt) but form coacervates upon mild heating. This can be exploited to isolate pure ELPs from cell lysates. This phase change can be modulated by the hydrophobicity of the guest amino acid residue, X, length of the pentameric repeat, n, or concentration.9 MacKay Lab has used these design considerations to engineer FKBP–ELPs as soluble systemic or sustained-release rapamycin formulations. For example, the first-generation FKBP–ELP formulation fused FKBP (F) to a diblock ELP. It comprised hydrophilic (VPGSG)48 adjacent to hydrophobic (VPGIG)48 (FSI) and formed spherical nanoparticles above its Tt.10 This formulation suppressed tumor growth in a breast cancer xenograft model while also reducing side effects compared to a free-rapamycin control.11,12 A next-generation formulation, termed “Berunda polypeptide” (FAF), was engineered to increase rapamycin payload by inserting two FKBP domains at the N- and C- termini of soluble (VPGAG)192 (A192). By retaining drug solubility during subcutaneous administration, it increased tumor accumulation and outperformed the FSI carrier in a breast cancer xenograft model.13 To improve on the FAF design and enhance drug-loading capacity, we next incorporated up to five FKBP repeats per polymer linked by four V24 blocks, (VPGVG)24, to generate the Hydra-ELP known as 5FV. This 5FV formulation formed a depot at the injection site for extended release in a mouse model of Sjögren’s syndrome.14
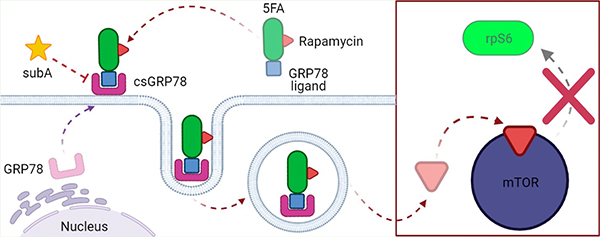
More recently, our team’s FKBP–ELP formulations have improved on previous designs by including a targeting moiety of cell surface motifs. For example, an integrin-targeting moiety was incorporated via an RGD–SI/FSI mixture that improved on the previous FSI formulation via RGD moieties displayed at the nanoparticle corona.15,16 Other formulations fused an A192 to intercellular adhesion molecule 1 (ICAM-1)-targeting peptide, which enhanced accumulation in the inflamed lacrimal glands of non-obese diabetic mice.17 This FKBP–ELP formulation developed in this work now optimizes the targeting of a high-capacity Hydra-ELP called 5FA. To do so, we target the 78 kDa glucose-regulated protein (GRP78), which is a key molecular chaperone. Encoded by the HSPA5 gene, this protein is also referred to as BiP or HSPA5. GRP78 belongs to a class of stress-induced chaperones within the heat shock protein (HSP) family, which primarily reside in the endoplasmic reticulum (ER).18–20 While ER-localized GRP78 is ubiquitous, under ER stress, a fraction translocates to the cell surface, termed cell surface GRP78 (csGRP78). GRP78 is commonly upregulated in cancer cells,21–23 whereby ER stress increases csGRP78.21 Importantly, cancer cells that are aggressive or have acquired resistance to treatment further increase csGRP78, thus making it a valuable target for cancer therapeutics or conduits for cancer-specific drug delivery.24,25 Ligands (W, L, P6, and P13)26–28 targeting csGRP78 have been identified with high binding affinities and represent viable strategies to target therapeutic payloads.16,29,30 Here, we present the development of this small library of ligand-targeted Hydra-ELP carriers (X-5FA). We describe evidence showing expression and purification using the ELP platform, as well as characterization for ligand-dependent internalization and downstream biological effects via mTOR inhibition by rapamycin (Figure 1).
Figure 1.
X-5FA targeting to csGRP78 can deliver rapamycin to BT474 cells. X-5FA targets csGRP78 primarily via the W and L ligands while carrying rapamycin bound to several FKBP domains. Exogenous cleavage by protease SubA blocks csGRP78-dependent cell surface targeting. After ligand binding, X-5FA-rapa is internalized into the cell. Rapamycin reaches its intracellular target mTOR and inhibits downstream signaling via pS6K and rpS6 to inhibit downstream effects (mRNA translation).
MATERIALS AND METHODS
Cloning and Sequencing.
5FA was generated by recursive ligation as previously described,14 which yields an open reading frame expressing the amino acid sequence indicated in Table 1. Forward and reverse 5′ phosphorylated DNA oligonucleotide sequences encoding GRP78-targeting peptides (X = W, L, P6, or P13) were synthesized by Genewiz Inc. (South Plainfield, NJ) and annealed in water at 94 °C for 2 min and cooled to ambient temperature. Prior to ligation, the 5FA plasmid was digested by NdeI [New England BioLabs (NEB), #R0111S], dephosphorylated with alkaline phosphatase (NEB, #M0290), and purified by gel extraction (Qiagen, #). The annealed oligonucleotides inserts were ligated into the linear 5FA vector using T4 DNA ligase (NEB, #M0202) at a 1:3 ratio overnight at 16 °C.
Table 1.
Amino Acid Sequence of csGRP78-Targeted Hydra-ELP
| protein | amino acid sequencea | Tt (°C)b |
|---|---|---|
| 5FA | M-(FKBP (VPGAG24))4 FKBP | 54.1 |
| W-5FA | M-WIFPWIQL-VPGAGM-(FKBP (VPGAG24)4 FKBP | 51 |
| L-5FA | M-RLLDTNRPLLPY-VPGAGM-(FKBP (VPGAG24)4 FKBP | 52.4 |
| P6-5FA | M-RLLDTNRPFLRY-VPGAGM-(FKBP (VPGAG24)4 FKBP | 52.7 |
| P13-5FA | M-RLLDTNRPFLFY-VPGAGM-(FKBP (VPGAG)24)4 FKBP | 50.2 |
FKBP and A24 sequences are available in Table S1 in the Supporting Information.
Tt was experimentally observed at 5 μM in PBS.
Electrocompetent ClearColi BL21 (DE3) cells (Lucigen, #60810-2) were transformed with ligated X-5FA or 5FA plasmid DNA by electroporation in a BTX 600 electroporation system using 1 mm electroporation cuvettes (BTX, #45-0134). Briefly, 25 μL of BL21 cells were aliquoted to electroporation cuvettes and gently mixed with 1 μL of plasmid DNA by tapping. An electroporation pulse was applied at 10 μF, 600 Ω, and 1800 V. Transformed cells were diluted in a 1.5 mL microcentrifuge tube with 975 μL prewarmed Expression Recovery Medium (Lucigen, #80030-1) and incubated at 37 °C for 1 h at 225 RPM. Volumes between 50 and 200 μL of transformed cells were spread on 15 g/L agar (Amresco, #J637) plates supplemented with 25 g/L Luria Broth (LB) (Sigma, #L3522) and 100 μg/mL carbenicillin (Gold Biotechnology, #C-103-100) by the aseptic technique. Plates were incubated at 37 °C until bacterial colonies appeared, which were individually isolated in a 5 mL LB/carbenicillin culture. From each sample, 0.5 mL of turbid culture media was taken, mixed in a 1:1 ratio with glycerol and water, and stored as stocks in −80 °C. The remaining culture was incubated at 37 °C for 18 h at 225 RPM and used to isolate plasmid DNA with a QIAPrep Spin Miniprep kit (Qiagen, #27106). Transformed cell plasmid DNA sequences were verified by Sanger sequencing (Genewiz, South Plainfield, NJ) using T7-F primers. Diagnostic digests with XbaI (NEB, #R0145S) and EcoRI-HF (NEB, #R3101S) were used to evaluate the size of NDA loaded on a 1% agarose gel (Invitrogen, #16500-500). Band sizes of 7900, 5413, and 2487 bp were simulated in SnapGene software (version 5.3) as compared to a 1 kb DNA ladder (NEB, #N3232S). Digests were separated using and confirmed by agarose gel electrophoresis and imaged using an iBright FL1000 system (Thermo Fisher), after which image brightness and contrast were adjusted to aid visibility in the figure. DNA purity was verified by UV–vis spectroscopy for a 260/280 ratio between 1.80 and 2.00 (NanoDrop, Thermo Fisher, Waltham, MA). Stocks meeting quality control criteria above were kept and used for subsequent steps.
X-5FA Protein Production and Purification.
X-5FA-transformed stocks were grown in a starter culture of 50 mL LB supplemented with 1× carbenicillin. After 8 h at 37 °C at 225 RPM, cells were transferred to 950 mL LB and incubated under the same conditions and monitored until reaching an OD600 between 0.6 and 0.8 to start induction with 0.5× IPTG. After culturing at ambient temperature for 18 h and 225 RPM, cells were centrifuged at 4500g for 15 min at 4 °C. Cell pellets were resuspended in 35 mL cold phosphate-buffered saline (PBS) (Genesee Scientific, #25-508) and sonicated on ice for 3 min with a 10 s on and 20 s off cycle. Cell lysate was supplemented with 0.5% polyethylenimine (Sigma, #MKCK1840) and incubated on ice for 20 min with occasional mixing. The cell supernatant was collected after centrifugation at 10Kg for 15 min at 4 °C and purified as previously described by ELP-mediated phase separation.9,16
Briefly, each cycle of ELP phase separation consisted of cell supernatant solution heated to 37 °C and spiked with 5 M NaCl in 1 M increments up to 2.5 M until coacervation. This solution was then centrifuged at 10Kg for 12 min at 37 °C, and the supernatant was discarded. The cell pellet was maintained on ice while resuspended by pipetting magnetic stirring. After coacervate is resuspended, the solution was centrifuged at 16Kg and the cell supernatant was collected to repeat up to three ITC cycles for 95% + purity. Absorbance (OD380) was measured by UV–vis spectrophotometry in a 1:4 mix with 8 M guanidine chloride (6 M final) and used to calculate protein concentration using Beer’s law. Extinction coefficients were calculated from protein sequence using the ExPASy ProtParam Tool (Table 1). After purification, a fraction of X-5FA was labeled using an N-hydroxysuccinimide (NHS)-rhodamine labeling kit (Thermo Fisher, #46406). NHS-rhodamine was dissolved in dimethyl sulfoxide (DMSO) (Invitrogen, #D12345) and incubated with protein at a 3× molar excess overnight at 4 °C. Unbound rhodamine was removed by size-exclusion chromatography (SEC) using 7 K MWCO Zeba Spin Desalting Columns (Thermo Fisher, #89889) following the manufacturer centrifugation protocol. Labeling efficiency was calculated by measuring the UV–vis absorbance of rhodamine (OD555 nm) and protein (OD280 nm). Purified ELP (5FA-Rh and X-5FA-Rh) was confirmed by gel electrophoresis on a 4–20% gradient Mini-Protean TGX precast gel (Bio-Rad Laboratories, #456-1095) loaded with 5 μg from each rhodamine-labeled sample and a Precision Plus Protein Kaleidoscope Protein Standard (Bio-Rad Laboratories, #1610375). Bands were visualized by Bio-Safe Coomassie staining (Bio-Rad Laboratories, #1610786) and fluorescence detection of rhodamine on an iBright FL1000 system (Thermo Fisher). Contrast and brightness were adjusted to aid visibility in the figure.
Characterization of Transition Temperature (Tt).
Purified protein samples (5FA, W-5FA, L-5FA, P6-5FA, and P13-5FA) were diluted to 5, 2.5, 1.25, 0.625, and 0.313 μM in separate quartz Tm microcells (Beckman Instruments, #523878). Absorbance (OD350 nm) was measured on a Beckman Coulter DU800 spectrophotometer at an analytical wavelength of 350 nm while heated from 30 to 75 °C using a temperature controller at a rate of 1 °C/min. OD350 nm was sampled every 0.3 °C to calculate the maximum first derivative of each curve, which was defined as Tt of each sample. Data were transferred to GraphPad Prism software (version 8) (San Diego, CA) for visualization.
Characterization of Nanoparticle Radius by Size Exclusion Chromatography-Multiangle Light Scattering and Dynamic Light Scattering.
For dynamic light scattering (DLS), 50 μL purified samples (5FA and X-5FA) were diluted to 5 μM and filtered through low protein-binding 0.2 μm filters (Pall Life Sciences, #4602) and plated on a clear-bottom 384-well microplate (GreinerOne, # 82051-294) in triplicate. To each well, 15 μL of mineral oil (Ward’s Science, #470301-505) was added to protect from evaporation. Samples were incubated at room temperature and measured on a Wyatt Instruments Inc. (Santa Barbara, CA) DynaPro DLS plate reader. Data were used to measure the hydrodynamic radius (Rh).
For SEC-multiangle light scattering (SEC-MALS), each protein was diluted to 10 μM in 500 μL of PBS and were passed through a 0.2 μm filter. A Shodex column KW-803 (8.0 mm ID × 300 mm) was equilibrated with PBS before introducing a bovine serum albumin (BSA) control at 5 mg/mL or samples above. The control and samples were observed on three in-line detectors for each fraction: a SYC-LC1200 UV detector at 280 nm, a Dawn Heleos MALS detector, and an Optilab rEX differential refractometer.
Exact Mass Determination by Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Analysis.
Purified protein samples were precipitated from a 1:6 acetone mixture at −20 °C for 1 h, followed by centrifugation. The process was repeated 3× before air drying and resuspension in 10 μL water. The sample was mixed with β-mercaptoethanol (Sigma-Aldrich, #M6250) in a 1:3 ratio and boiled at 95 °C for 15 min before spotting with 2′,6′-dihydroxyacetophenone (Sigma-Aldrich, #37468) on a matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) target plate (Bruker Inc., Billerica, MA, #8280781) and air-dried. Samples were measured on a raplifleX MALDI-TOF system. Mass was calculated on Flex Analysis v4.0 software (Bruker Inc., Billerica, MA).
Rapamycin Drug Loading.
Rapamycin powder (LC Laboratories, Woburn, MA, #R-5000) was dissolved in DMSO and slowly added in 10× stoichiometric excess to X-5FA with continuous stirring at 4 °C. After 30 min, samples were centrifuged at 13Kg for 15 min at 4 °C to remove unbound, insoluble rapamycin. The supernatant was subsequently dialyzed against 10,000 MWCO membranes with PBS at 4 °C in a 1:300 volume ratio with buffer replenishment of at least 3× over the course of 24 h. Each rapamycin-loaded Hydra-ELP sample was then passed through Acrodisc Mustang E Membrane filters (Pall Life Sciences, Port Washington, NY, #MSTG25 × 103) at 4 °C. To evaluate the rapamycin-loading efficiency, a standard curve was generated by reverse-phase high-performance liquid chromatography (RT-HPLC) on a C-18 column under a methanol to water gradient ranging from 40 to 90% with 0.1% TFA. Free-rapamycin concentrations ranging between 500 and 1.95 μM. The area under the curve (AUC) for sample peaks was measured and correlated to known rapamycin concentrations and used to evaluate sample loading concentrations (Supporting Information, Figure S1). Protein concentrations were then compared to rapamycin concentrations and used to generate a loading ratio for each sample (rapamycin μM/protein μM).
Mammalian Cell Culture.
Mammary gland breast duct cells (BT474) [American Type Culture Collection (ATCC), Manassas, VA, #HTB-20] were cultured under 37 °C and 5% CO2 conditions in Hybri-Care medium (ATCC, #46-X) supplemented with 10% FBS (Corning Inc., NY, #35-011-CV) and 1.5 g/L NaCHO3 (MilliporeSigma, Burlington, MA, #SX0320-1) adjusted to a pH of 7.2 with HCl. Media were replenished every 2 days and subcultured after reaching 80–90% confluence. Briefly, cells were detached in a minimal volume of prewarmed 0.25% trypsin–EDTA (Thermo Fisher, #25200-056) for 5 min at 37 °C and centrifuged to form a pellet. Cells were resuspended in fresh media and passaged to a 75 cm2 flask, 6-, 12-, or 96-well plates for assaying. Cell line stocks were stored in a 5% anhydrous DMSO (Thermo Fisher, #D12345)/medium solution at −170 °C. Fresh cell stocks from the same lineage were used for each experiment.
Flow Cytometry and Analysis.
BT474 cells were seeded on two six-well plates for cells with and without subtilase cytotoxin A (SubA) treatment; SubA + cells were treated with SubA media (Hybri-Care media, +10% FBS, +0.2 μg/mL SubA) at approximately 50% confluence. Cells were incubated at 37 °C until reaching 70–80% confluence and starved in SubA media (Hybri-Care, FBS, +0.2 μg/mL SubA) 24 h before ELP treatment. SubA- cells were not treated with SubA and were starved in standard Hybri-Care media (Hybri-Care, -FBS, -SubA) for 24 h alongside SubA + cells. Both groups were washed and treated with either 5FA-Rh or X-5FA-Rh at 5 μM, 5FA-Rh at 20 μM, or unstained, then incubated at 37 °C for 3 h. Cells were centrifuged and resuspended in 200 μL FACS buffer (PBS +2% FBS) and passed through a strainer (BD Biosciences, Franklin Lakes, NJ, #352235) before data acquisition on a BD Fortessa X-20. The instrument signal was gated using a positive control (20 μM 5FA-Rh) and an untreated negative control as references. Gating on FlowJo identified positive from the background signal and singlets from doublets. Conditions were applied to all samples and used to extract positive internalization events as a proportion of totally 100,000 sample events were recorded for each treatment condition and analyzed on FlowJo LLC (Ashland, OR) and Graphpad Prism.
Western Blot and Analysis.
BT474 cells were subcultured on six-well plates (Genesee Scientific, El Cajon, CA #25-105) and grown to 80–90% confluence. A dose–response assay was conducted by incubating with either 0, 0.1, 1, 10, 100, or 1000 nM rapamycin, 5FA-R, or X-5FA-R (X = W, L, P6, or P13) for 2 h at 37 °C in a 5% CO2 incubator. A kinetic assay was performed by incubating cells with 1 nM of rapamycin, 5FA-R, or X-5FA for either 0, 15, 30, 60, or 120 min at 37 °C in a 5% CO2 incubator. For both experiments, media were aspirated and washed with 4 °C PBS before incubating with 100 μL RIPA buffer (Thermo Fisher, #89901) spiked with protease inhibitor (Thermo Fisher, #78442) on ice. Cell monolayers were manually removed using a cell scraper (VWR, #10062-904) and vortexed before deep freezing in individual microcentrifuge tubes at −80 °C. After freezing for 1 h, cells were thawed, vortexed, and centrifuged at 13Kg at 4 °C for 15 min. Protein was isolated in the supernatant and quantified using a Pierce BCA Protein Assay Kit (Thermo Fisher, #23227). Briefly, albumin concentrations were generated from a standard (Thermo Fisher, #23209) against protein samples quantified at OD562nm in triplicate. Protein concentrations were quantified and used to calculate 30 μg to load on a 4–20% gradient Mini-Protean TGX precast gel. After separation, blots were transferred to a nitrocellulose membrane using an iBlot2 NC stack (Invitrogen, #IB23001) on an iBlot 2 Dry Blotting System. Membranes were blocked in 5% BSA (Sigma, #A9647) dissolved in 1× tris-buffered saline adjusted to pH 7.4 and spiked with 0.1% tween-20 (TBST) (Santa Cruz Biotechnology, #sc-29113) for 1 h at ambient temperature. Membranes were then incubated in rabbit α-phospho-rpS6 (Ser235/236) (Cell Signaling Technology, #2211) diluted 1:1000 in 5% BSA overnight at 4 °C with gentle shaking. After washing with 1× TBST in triplicate, membranes were incubated with the secondary α-rabbit IgG HRP-linked antibody (Cell signaling, #7074S) diluted to 1:5000 in 5% BSA for 1 h at ambient temperature. Blots were briefly incubated in a 1:1 luminol and peroxide solution (Prometheus Protein Biology, #GSC-925-D10, #GSC-929-D10) and immediately imaged on a ChemiDoc to detect chemiluminescent signal. Membranes were stripped using Restore Western Blot Stripping Buffer (Thermo Fisher, #21059) for 15 min at ambient temperature and washed in 1× TBST. Membranes were reblocked in 5% BSA and incubated in Ms α-GAPDH (Cell Signaling Technology, #97166) and α-Ms IgM HRP (Cell Signaling, #7076S) chemiluminescence repeating the protocol for phospho-rpS6 visualization. Raw data for the integrated density of rpS6 phosphorylation was normalized to GAPDH using FIJI.31 For each lysate, a relative phosphorylation, , was calculated as follows
| (1) |
where , , , , and are the integrated densities from a region of interest (ROI) on each western blot over the rpS6 sample, the sample at zero (concentration or time), the GAPDH for each sample, the GAPDH at zero, and a background ROI on the image, respectively. Relative phosphorylation data was analyzed on GraphPad Prism by comparing AUC of curves for each formulation. Statistical significance was tested using global ANOVA of the log-transformed AUCs, followed by Tukey’s multiple comparisons test.
Confocal Imaging and Analysis.
Poly-D lysine (Sigma, #P0899)-treated cover slips (VWR, #483830-046) were plated with BT474 cells in complete Hybri-Care media and cultured to 70% confluence. Cells were starved without FBS for 24 h before treatment with 5 μM of either 5FA, L-5FA, or neither at 4 °C for 1 h. Cells were either fixed or incubated at 37 °C for 3 h and then fixed as previously described;32,33 cells were fixed in 4% paraformaldehyde (Alfa Aesar, #43368) and neutralized in 50 mM ammonium chloride (Alfa Aesar, #L05181). Cells were permeabilized with triton X-100 (Sigma, #T8787) and then blocked in 1% BSA (Sigma, #A9647) before an overnight incubation in the 1:100 mouse α-ELP primary antibody at 4 °C.34 Cells were then incubated in the 1:100 chicken α-mouse AF647 secondary antibody (Invitrogen, #A21463) overnight at 4 °C before staining with 1x DAPI (Thermo Fisher, #D1306). Cells were mounted in glass slides (VWR, #48311-703) in fluoromount (Diagnostic Biosystems, #K024) and cured overnight protected from light at ambient temperature. Cells were imaged at 630× in super-resolution on a Zeiss confocal microscope in LSMFast mode in three-dimensions using Nyquist sampling. Images were deconvoluted by Airyscan processing in Zen 2 (Black Edition) and visualized in Zen 2 (Blue Edition) by orthogonal projection and three-dimensional mesh reconstruction. Acquisition settings remained consistent between for each temperature condition. Internalization was quantified in FIJI by comparing total signal intensity within cells under each treatment condition for comparison under each temperature condition.
Fluorescence Degradation of 5FA and L-5FA.
BT474 cells were seeded in 10% FBS Hybri–Care media on poly-D lysine-treated 35-mm glass bottom plates (MatTek, #P35GC-0-10-C) and incubated until reaching 70% confluence. Cells were starved for 24 h and treated with 5 μM of either 5FA or L-5FA for imaging by epifluorescence microscopy between 0 and 240 h at different time points. Total rhodamine fluorescence was measured after background subtraction and normalized to the number of cells per frame. Cells were counted as nuclei count, which were automatically counted in FIJI using watershed segmentation from a binary image. Average fluorescence was graphed in GraphPad Prism.
RESULTS AND DISCUSSION
The goal of this study is to optimize the first csGRP78-targeted ELP-based carriers for rapalogues by: (1) utilizing five FKBP domains to increase rapalogue-loading capacity; and (2) identifying a complementary peptide ligand for targeting csGRP78 relevant to breast cancer (Figure 1). Validating these parameters from a library of X-5FA candidates was accomplished after their cloning, expression, purification, and physical characterization by sodium dodecyl sulfate (SDS), DLS, UV–vis, and MALDI-TOF. Their functional targeting was evaluated in vitro by imaging, flow cytometry, and Western blot of downstream of mTORC1 inhibition by rapamycin (rpS6) to assess their dependence on csGRP78 binding.
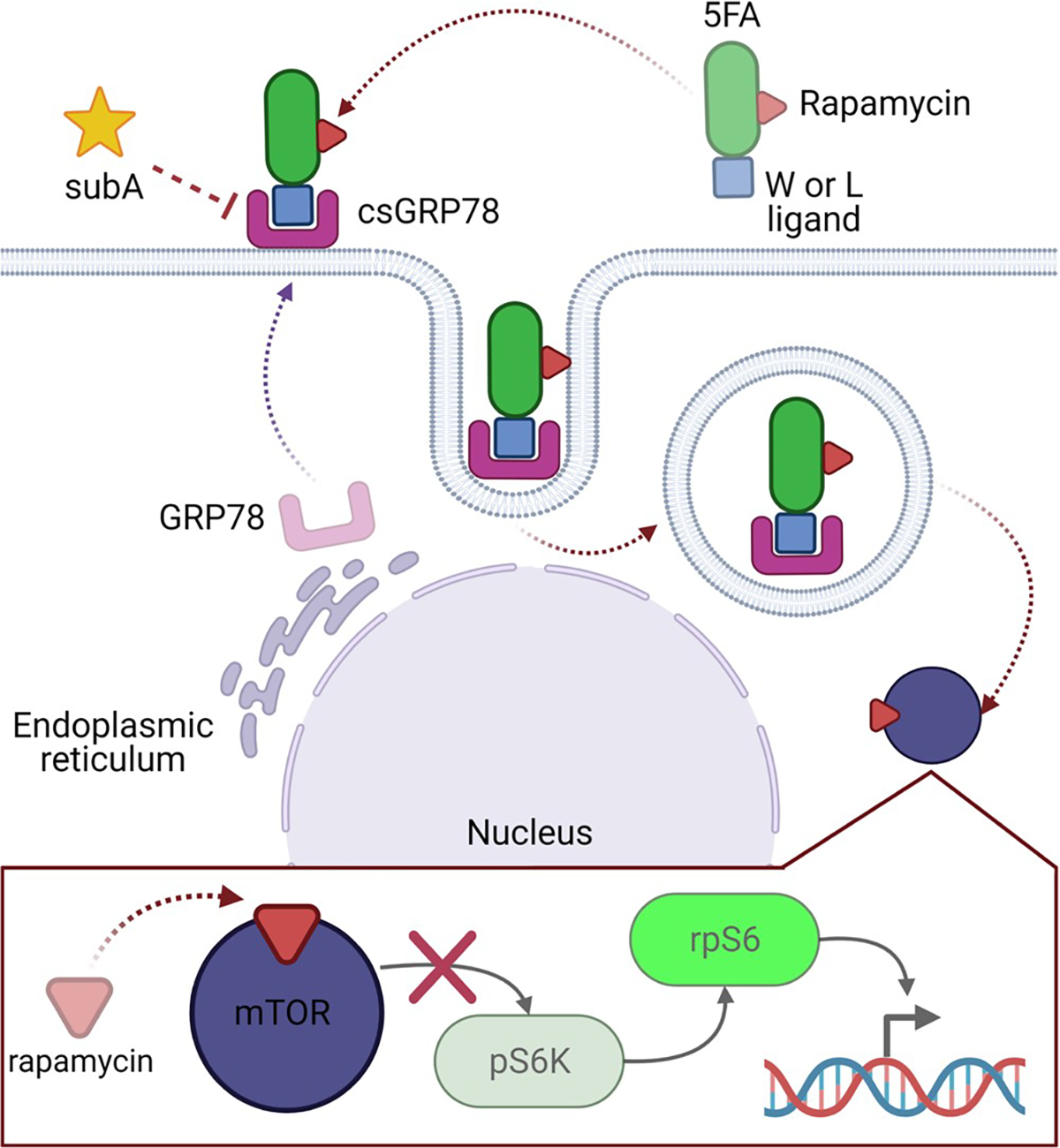
The first step in generating X-5FA constructs relied on a previously reported 5FA plasmid-encoded gene,14 to which oligonucleotide cassettes were ligated that encode several ligands for csGRP78 (W, L, P6, or P13) (Figure 2A). These peptides were reported to have high affinity to csGRP78;27,28 however, it was unclear if fusion to the 5FA backbone would have a detrimental effect on expression, solubility, or ligand binding to cell surface receptors. Plasmid DNA encoding 5FA were quality controlled by UV–vis spectrophotometry, diagnostic restriction digestion, and Sanger sequencing. From successful ligation products, we verified the DNA band size using a double enzymatic digestion to confirm they retain the full sequence for 5FA and X-5FA near 3.2 kbp (Figure 2B). After confirming DNA quality and approximate band size, N-terminal Sanger sequencing was used to verify correct sequence alignment of X-ligands upstream to 5FA (Figure 2C).
Figure 2.
DNA quality control was verified to match by gel electrophoresis and Sanger sequencing. (A) 5FA amino acid sequence is comprised of an FKBP-A24 sequence repeated 4× and capped with a 5th FKBP. The X-ligand at the N-terminus of 5FA makes X-5FA, substituted by either the W-, L-, P6-, or P13-ligands able to bind GRP78. (B) Cloned constructs were verified by gel electrophoresis with fragments matching approximate expected sizes from after digestion by XbaI and EcoRI. As such, the double digests show the isolated 5FA or X-5FA insert (approximate 3200 bp) and an empty plasmid vector (approximate 5500 bp). The size of the vector is confirmed by the inclusion of the empty pET25b vector control. (C) Successful ligation of the X-ligand to the 5FA backbone was further verified by Sanger sequencing using a T7-F primer. Distinct ligands at the N-termini of 5FA were verified successfully with 100% query matching.
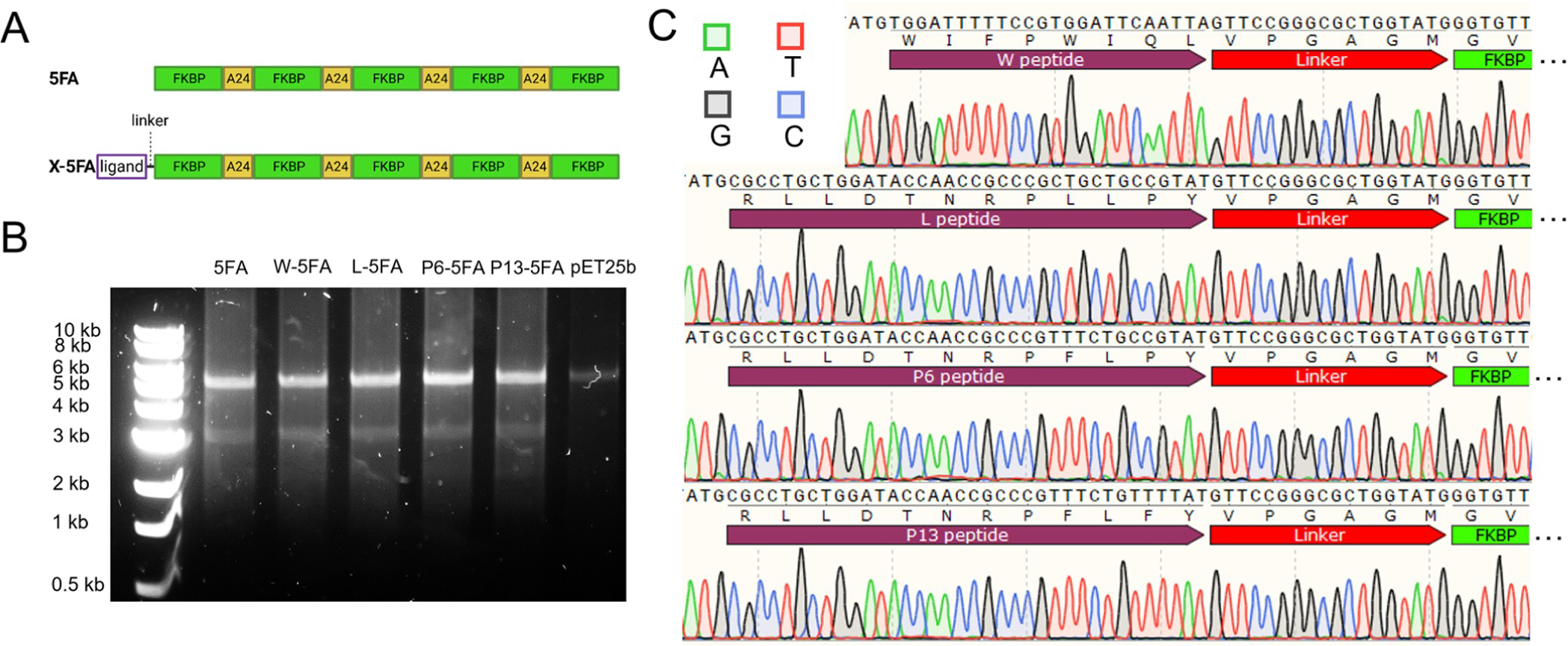
Purified DNA from correctly matched sequences were subsequently cultured for protein expression from ClearColi, which is an engineered cell line that produces a truncated lipopolysaccharide. ELPs display temperature-dependent phase behavior that were used for purification using sequential heating-and-cooling cycles, which yielded a purity range between 90 and 98% with yields between 10 and 25 mg/L. After labeling with NHS-rhodamine, band purity was verified experimentally by SDS-polyacrylamide gel electrophoresis (PAGE) (Figure 3A,B).
Figure 3.
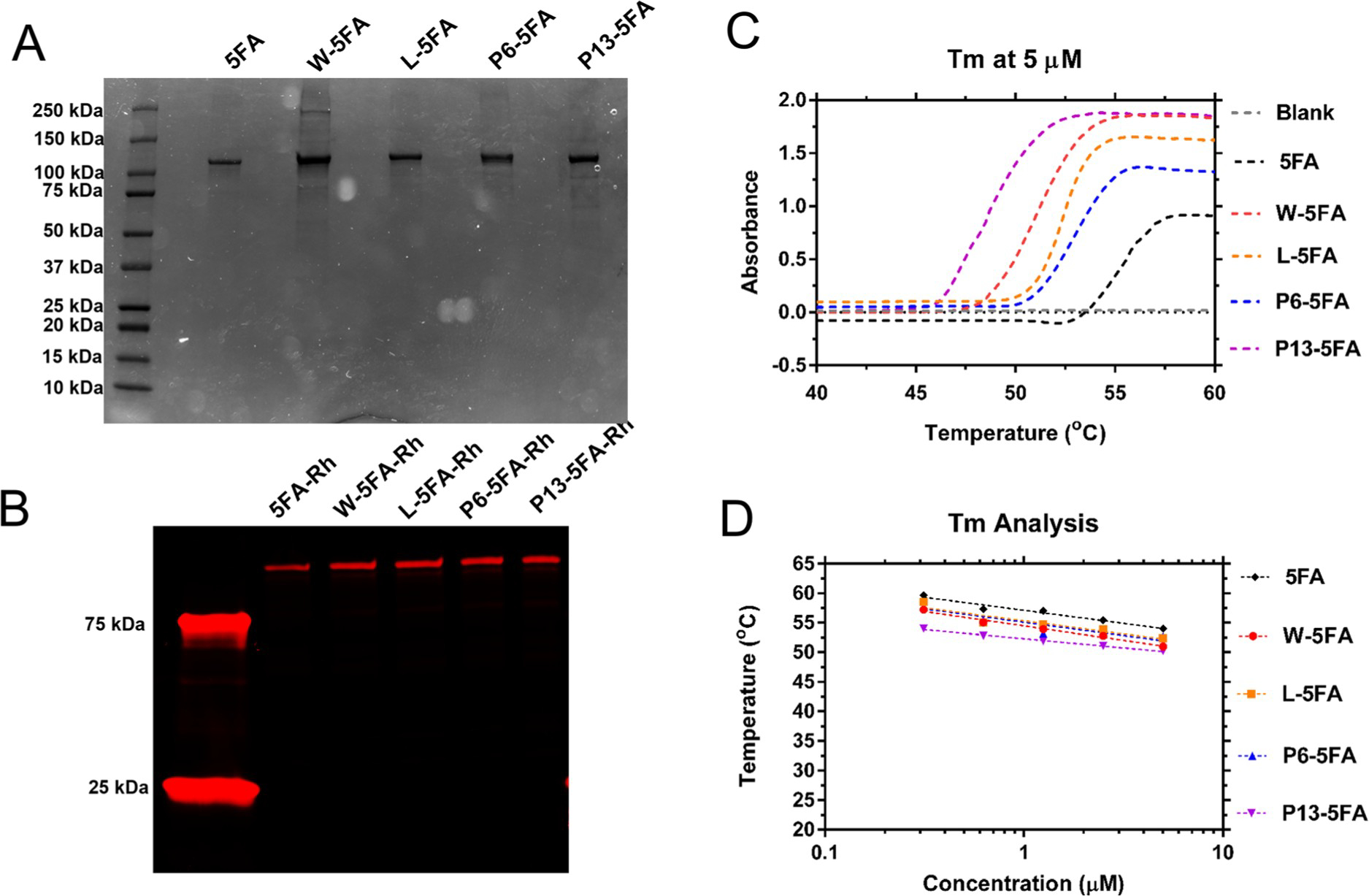
Expression of 5FA and X-5FA was verified along with phase behavior. (A) 5FA and X-5FA constructs were purified by thermal cycling. The approximate size and purity of ELP constructs were verified by SDS–PAGE. (B) NHS-rhodamine consistently labeled these ELPs, enabling fluorescence imaging. (C) The transition characteristics of 5FA and X-5FA were compared at 5 μM showing a slight trend of lower Tt of X-5FA constructs relative to untargeted 5FA. (D) Higher concentration of all ELP constructs indicated an inverse relationship with Tt in a range between 0.3 and 5 μM. Based on the fit to the OD data, Tt was calculated to be above physiological temperatures at all concentrations in the tested range up to 5 μM.
Bands run slightly higher than the expected values predicted in the open reading frame (Table 2). When compared to X-5FA, 5FA runs subtly lower as expected, suggestive of successful ligand expression on 5FA. The W-5FA lane contained a prominent band in the 250 kDa range of an unknown origin; however, it does not represent a significant proportion of impurity relative to the total band intensity (Table 2). Each construct shares sequence similarity through their 5FA domains with the only difference attributed to the X-ligand. Using optical density, their phase diagrams were quantified as a function of temperature and concentration. While there were small differences in Tt between the fusion proteins, they followed no obvious trend, and all constructs are expected to remain soluble within physiological temperatures (Figure 3C). From this range of concentrations, we can extrapolate that X-5FA will remain soluble at concentrations relevant to cellular uptake (5 μM) and inhibition of mTOR (10 nM) (Figure 3D), like previous generations of FKBP–ELPs.15
Table 2.
Biophysical Properties of csGRP78-Targeted Hydra-ELPs
| 5FA | W | L | P6 | P13 | |
|---|---|---|---|---|---|
| expected MW (kDa)a | 95.7 | 97.2 | 97.6 | 97.7 | 97.7 |
| MALDI-TOF MW (kDa)b | 95.5 | 97.5 | 97.7 | 97.6 | 97.9 |
| SEC-MALS MW (kDa)c | 125.7 | 128.9 | 131.8 | 132.7 | 125.6 |
| SDS-PAGE MW (kDa)d | 104.8 | 108.7 | 115.5 | 115.1 | 113.5 |
| SDS-PAGE puritye | 0.98 | 0.93 | 0.98 | 0.98 | 0.90 |
| DLS Rh (nmf | 3.75 ± 0.77 | 3.51 ± 1.13 | 3.83 ± 0.62 | 5.03 ± 0.17 | 3.66 ± 0.27 |
| DLS Rh (nm)g | 5.6 ± 0.83 | 5.4 ± 1.7 | 6.63 ± 1.23 | 5.5 ± 0.67 | 166.9 ± 36.4 |
MW calculated from open reading frame using SnapGene, version 5.3.
MW calculated by MALDI-TOF using FlexAnalysis, version 4.0.
MW calculated by SEC-MALS using Astra, version 6.1.
MW of the SDS band calculated in reference to ladder using ImageJ, version 1.51k.
Protein band purity was quantified on ImageJ, version 1.51k.
Hydrodynamic radius of filtered Hydra-ELPs of the regularization fit of the major peak.
Hydrodynamic radius of filtered Hydra-ELP/Rapa complexes (Supporting Information, Figure S2).
After confirming the expression and purity of each 5FA formulation, we characterized them by SEC-MALS, DLS, and MALDI-TOF (Figure 4). For all five fusions, SEC-MALS revealed that the majority of UV–vis absorbance eluted from the column at a retention time consistent with an ~100 kDa protein. In-line observation of this purified peak revealed an absolute molecular weight on the order of a monomer, albeit slightly larger than expected (Figure 4A). To clarify this ambiguity, MALDI-TOF was employed to measure the exact mass, which clearly displayed clean spectral peaks A, B, and C, representing (M + H), [M + 2H]/2, and [M + 3H]/3 for each formulation, respectively (Figure 4B). Calculated masses corresponded closely to their respective charge states (Table S2). To determine the hydrodynamic radius of these fusions in solution, DLS was employed to confirm that the major species by mass had a radius close to that expected for monomeric proteins, similar to 5FA (X < 10 nm) (Figure 4C). Small peaks near 100 nm were present for L-, W-, and P6-5FA and represent a small fraction of the total. The relative area of the W-5FA peak near 100 nm represents 5.8% of the total, which corresponds to the impurity noted in Figure 3A. Compared to free Hydra-ELPs, there was a slight increase in Rh for samples complexed with rapamycin (Table 2) with the exception of P13-5FA, all formulations containing drug retained a particle size below 10 nm, which is consistent with solubility as free proteins (Supporting Information, Figure S2). Filtration through a 0.02 μm filter does not appear to affect particle size distribution, except for P13-5FA-Rapa, which includes a nanoaggregate population above 100 nm. Except for P13-5FA-Rapa, these data indicate that these constructs remain freely dispersed in solution without forming significant oligomers, which might alter their receptor-binding and cellular-internalization.
Figure 4.
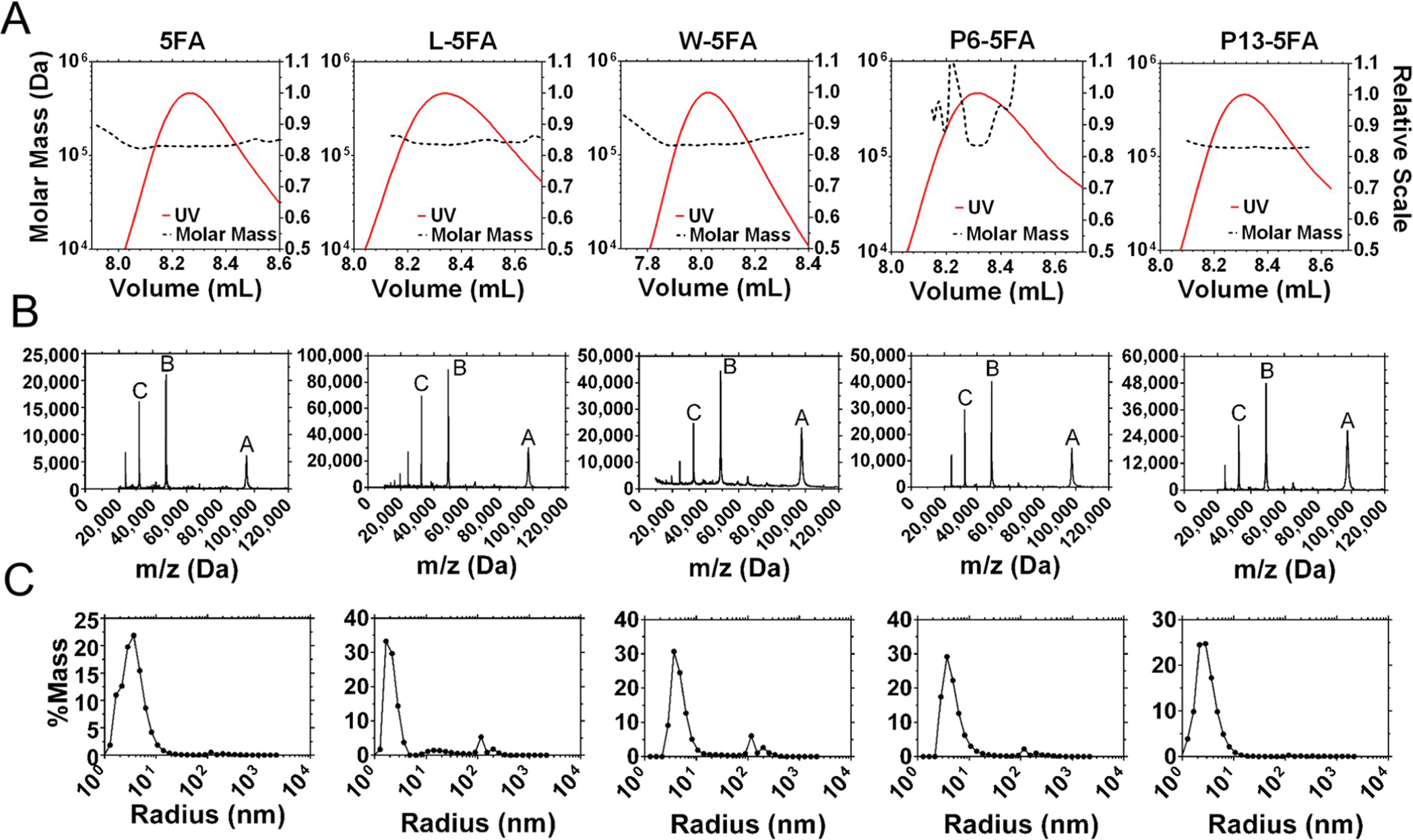
Shape and molecular weight of each expressed 5FA construct was characterized by SEC-MALS, DLS, and MALDI-TOF to corroborate their MW and radii. (A) SEC separation-eluted purified monomer fractions between 8.0 and 8.4 mL. (B) Molecular weights of each construct and purity were examined by MALDI-TOF, which show peaks at the appropriate location for each fusion (Table 2), accurate for m/z for single, double, and triple charge states. (C) Their radii were measured by DLS, which showed most samples falling below 10 nm. This is consistent with their solubility at 37 °C; however, a small peak fraction was detected near 100 nm for W, L, and P6–5FA.
The next series of studies compared the biological functions of this library, with respect to GRP78 targeting and mTOR inhibition. The first step was to determine the relative efficacy of our targeting ligand to each other and to an untargeted control in the absence or presence of SubA,35 a protease known to cleave GRP78 between Leu(416) and Leu(417).36 If targeted X-5FA became dependent on csGRP78 for internalization, this would be revealed by a decline in internalization for cells treated with protease compared to those without treatment (Figure 5). Two internal controls were generated to compare a lower (negative control) and upper signal baseline (untargeted rhodamine-labeled 5FA) for comparison (Figure 5A,B). The difference in internalization between X-5FA constructs with and without SubA is striking. After protease treatment, there was virtually no signal in cells treated with SubA for all X-5FA constructs, which become indistinguishable from the negative control. The proportion of cells with a positive signal above the lower baseline (negative control) was statistically significant for W, L, P6, and P13 after treatment with SubA (Figure 5C–F) but was not the case for untargeted 5FA. Internalization of 5FA was low and minimally affected by SubA compared to any targeted X-5FA (Figure 5G). To assess which csGRP78 targeting ligand is more effective, the signal above the 20 μM 5FA upper baseline was compared (Figure 5B), showing a significant difference between W- and L-5FA compared to other conditions (Figure 5H). Comparing W and L-5FA, we see roughly equivalent internalization.
Figure 5.
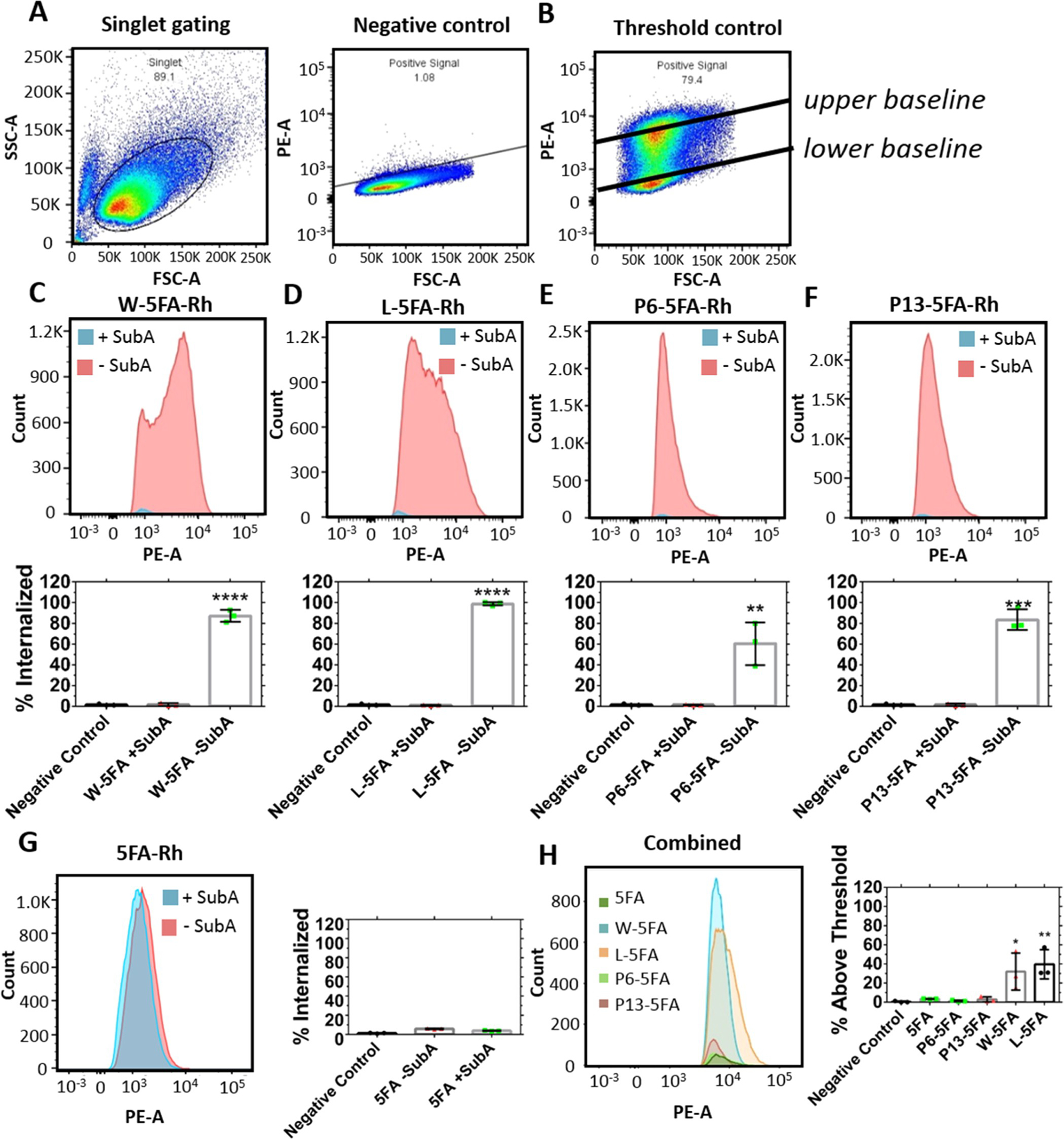
W-5FA and L-5FA undergo csGRP78-dependent cell association by flow cytometry. Internalization of rhodamine-labeled W, L, P6, and P13 fused to 5FA was evaluated in BT474 cells treated with and without SubA (0.2 μg/mL). (A) Unstained negative control was used to define a lower gating baseline for detectable cell association, while an (B) untargeted rhodamine-labeled control (5FA) at 20 μM defined an upper gating baseline for higher cell association. (C–F) Cells treated with or without SubA were treated with X-5FA (5 μM). Signal was cutoff below the lower baseline to show that control cells retained significantly more signal, while cells treated with SubA were indistinguishable from the negative controls. (G) Untargeted 5FA (5 μM) internalization is low relative to targeted X-5FA and was minimally affected by treatment with SubA. (H) To compare between positive internalization data, the signal below the 95th percentile of the upper gating baseline (20 μM 5FA) was cutoff in samples A–G to clarify differences between groups. Internalization of W and L-5FA was significantly higher than that of P6 and P13–5FA. Mean ± SD (n = 3). *p < 0.05, **p < 0.005, ***p < 0.0005, and ****p < 0.00005.
While SubA failed to alter the uptake of unmodified 5FA, it was possible that SubA would induce off-target effects downstream of GRP78 proteolysis. For example, on some cell lines, SubA induces endoplasmic reticular-associated degradation with treatment concentrations of 1 μg/mL for 24 or 48 h, which promoted apoptosis and inhibited global protein synthesis.37 In contrast, another report found no effects to trafficking and toxicity after treatment with 1 μg/mL SubA for 72 h.36 A third report similarly found no toxicity to cells after treatment at 0.1 μg/mL for 48 h.26 Either way, the striking decrease of X-5FA internalization in cells treated with SubA relative to untargeted 5FA internalization suggests that cell association for the entire library is csGRP78-dependent (Figure 5).
After establishing that all X-5FA constructs successfully target csGRP78, we hypothesized that the most effective carrier for rapamycin among them would suppress mTORC1 more potently than free drug. Rapamycin was loaded onto FKBP domains using solvent dialysis as described in the methods. After dialyzing away unencapsulated drug and DMSO, Hydra-ELP and rapamycin levels were independently evaluated using UV–vis and RP-HPLC, respectively (Table S3 and Figure S1 in the Supporting Information). Having five FKBP domains per fusion (Figure 1A), the maximum possible loading ratio is 5 rapamycin per Hydra-ELP, which agreed with observations of 4.5, 3.7, 4.1, 3.3, and 3.4 for 5FA, W-5FA, L-5FA, P6-5FA, and P16-5FA. In addition, DLS assays were performed (Supporting Information, Figure S2) to show that all five formulations retain a hydrodynamic radius that is consistent with that of the free fusion protein (Table 2). Finally, we note that the measured concentrations observed for rapamycin bound to Hydra-ELPs (Supporting Information, Figure S1) exceed the trace water solubility for free rapamycin (<20 μM). Taken together, this data supports the contention that these formulations form stable formulations with Rapa. Rapamycin, either free or bound to 5FA, is already potent at a lower concentration (10 nM) than used above to assess cell association by flow cytometry (~5 μM).
Many reports have described successful inhibition of mTORC1 via binding by rapamycin in diverse animal and cell models.38–40 As such, we first assessed a range of concentrations to find an optimal effective dose before taking a minimum effective dose to assess their kinetics in BT474 cells (Figure 6). Free rapamycin was less effective than targeted W- and L- 5FA-rapamycin, which began to show efficacy at concentrations as low as 0.1–1 nM. Surprisingly, untargeted 5FA-rapamycin was better than two of the targeted carriers (P6- and P13-5FA-rapamycin), with efficacy manifesting only at 10 nM and above (Figure 6A). P6-5FA-rapamycin and free rapamycin were similarly effective at 10 nM and above, while P13-5FA was only effective at 100 nM and above. It is possible that an intermediate concentration below 100 nM would be effective, but this is orders of magnitude above that required for either the W- and the L-5FA-rapamycin formulations (Figure 6B). To quantify this effect, the areas under each curve (AUC) were compared by ANOVA/post-hoc testing to identify significant differences between formulations (Figure 6C). Rapamycin has been implicated in modulating GRP78 via the mTOR axis. One report treated human neuroblastoma cells with 10 nM rapamycin and found a decrease in rpS6 phosphorylation as well as a decrease in GRP78 expression.41 To probe this in the context of our assay, GRP78 was interrogated after treatment with L-5FA-rapamycin at a range of concentrations (0, 1, 10, 100, and 1000 nM). While increasing L-5FA-rapamycin concentrations did have an obvious effect on rpS6 phosphorylation, only a minor effect was noted for GRP78 expression (Supporting Information, Figure S3). At the concentration (1 nM) used in this assay, GRP78 was not attenuated. From these data, we determined that a minimum concentration of 1 nM was sufficient for lead candidates, W and L-5FA-rapamycin; therefore, we further explored the kinetics of their mTOR inhibition by tracking dephosphorylation of rpS6 over time. Because P6 and P13-5FA-rapamycin do not inhibit mTORC1 at this concentration, they were omitted from this kinetic assay (Figure 6D). Consistently with the concentration-based assay, W and L-5FA-rapamycin showed the fastest kinetic effect with 50% attenuation by 30 min. In contrast, free rapamycin and untargeted 5FA-rapamycin required 60 min to reach a similar level. By 2 h, W- and L-5FA-rapamycin were almost fully inhibited, while untargeted 5FA-rapamycin and free Rapamycin showed higher levels of p-rpS6 (Figure 6E). The AUC of each curve was then used to compare their potency in triplicate. Only W- and L-5FA-rapamycin achieved significantly lower AUC compared to free rapamycin. Other comparisons were not significant (Figure 6F). These results mirror the observations in the SubA flow cytometry experiments that show both W- and L-5FA-rhodamine are better at targeting csGRP78 for internalization (Figure 5). These data were also validated with a formazan-based viability assay to demonstrate a concentration dependence, which revealed IC50 values for rapamycin, 5FA-Rapa, W-5FA-Rapa, L-5FA-Rapa, P6-Rapa, and P13-5FA-Rapa at 5.8 (±13.8) nM, 0.17 (±0.70) nM, 0.49 (±1.15) nM, 0.80 (±4.7) nM, 0.94 (±3.9) nM, and 2.8 (±22.6) nM, respectively (Supporting Information, Figure S4). These levels are very consistent with 0.1 and 1.0 nM rapamycin concentrations that inhibit phosphorylation of rpS6 (Figure 6); furthermore, they reflect an ~10-fold improvement of 5FA-Rapa over free drug. Despite this, enhancement due to the GRP78 targeting peptide was not observed in the formazan viability assay. This may be attributed to the relatively long incubation (48 h) for the viability assay compared to the flow cytometry (3 h) and mTORC1 inhibition assays (2 h), during which time a kinetic advantage of the GRP78 peptide (Figure 6E) would be negated.
Figure 6.
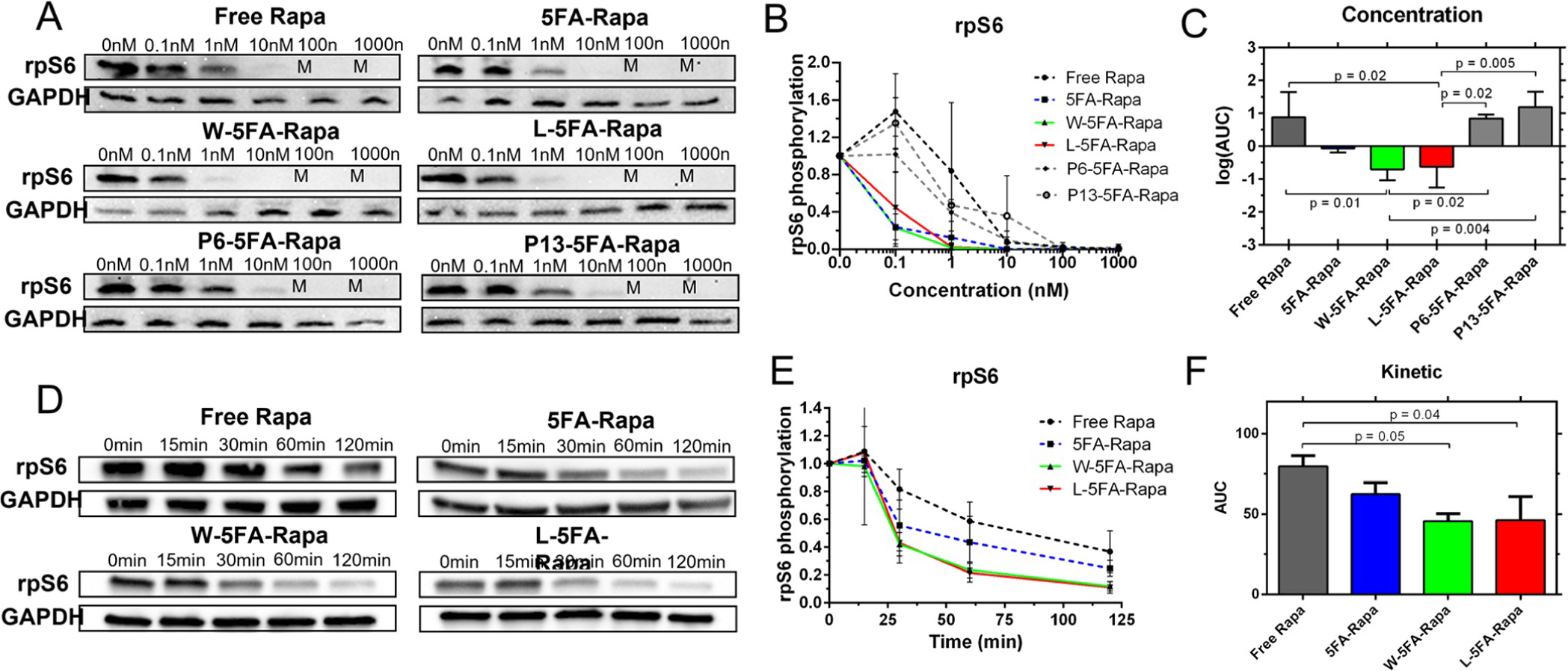
Rapamycin bound to W-5FA and L-5FA inhibits mTORC1 more potently than the untargeted drug. BT474 cells were treated with rapamycin at various concentrations and times. Upon obtaining cell lysates, mTORC1 inhibition was assessed by western blot of rpS6 phosphorylation (upper bands) using GAPDH as a control (lower bands). The relative phosphorylation was estimated (eq 1). (A,B) After treatment at concentrations between 0 and 1000 nM for 120 min at 37 °C, free rapamycin was effective at concentrations above 10 nM. In contrast, untargeted 5FA and W and L-5FA-rapamycin were maximally effective between 1 and 10 nM, an order of magnitude better. P6 and P13–5FA-rapamycin were the least effective, with complete inhibition above 100 nM. (C) Log AUC was quantified for each curve and compared against by ANOVA. Statistical significance was detected between free rapamycin and every other carrier formulation. (D,E) Cells were next treated at 1 nM and lysates were obtained after indicated incubations. W and L-5FA-rapamycin inhibited mTOR almost completely by 120 min. (F) To quantify this difference, the AUC under the relative expression versus time curve was compared, which showed that only W-5FA and L-5FA significantly decreased the AUC compared to free rapamycin (n = 3 per group, **p < 0.005). Mean ± SD.
Despite a lack of similarity between their primary sequence, functional differences between W and L-5FA are minor. For example, physical characterization of both W and L-5FA by DLS and SEC-MALS show very similar particle sizes and similar performance in internalization and mTOR targeting. From these observations, L-5FA is only narrowly better than W-5FA; however, another consideration is that purification of L-5FA by ELP-mediated phase separation delivered a significantly better yield (24 mg/L bacterial culture). While both candidates provided clean final samples (Figure 3B), L-5FA expression was consistently 6-fold higher than W-5FA after several purification cycles. As such, L-5FA was selected as a good candidate to visualize its intracellular fate compared to 5FA (Figure 7). We can infer from flow cytometry and Western blot data that L-5FA is readily internalized compared to untargeted 5FA, so it was not surprising to see a discrepancy in membrane binding by L-5FA-rhodamine at 4 °C compared to untargeted 5FA. An abundance of cell surface L-5FA was detected without a noticeable signal on the inside of cells, indicating that L-5FA was only bound to the membrane without internalization (Figure 7A). At 37 °C, cells treated with 5FA display some internalization, but nothing comparable to the significantly higher amount of L-5FA. Internalization was inferred as a peri-nuclear signal within the cytosol. 5FA staining was modest compared to L-5FA, which appears proportional to levels of bound material observed at 4 °C (Figure 7B). To compare the intracellular fate of each formulation, the duration of cell-associated fluorescence was tracked over 10 days. A signal appears constantly over the first 16 h for both, while displaying the expected differences in the total signal before dropping off after 36 h. The signal then begins to slowly decrease up to the experimental endpoint at 240 h (Figure 7C). These data show that there is a significant effect from the L-ligand over time, which supports the flow cytometry and Western blot experimental data showing that csGRP78 targeting successfully biases this formulation for internalization into BT474. Even after internalization, by comparing the AUC of both formulations, the relative exposure to drug carried by L-5FA was 8-fold greater than that by 5FA (Figure 7D).
Figure 7.
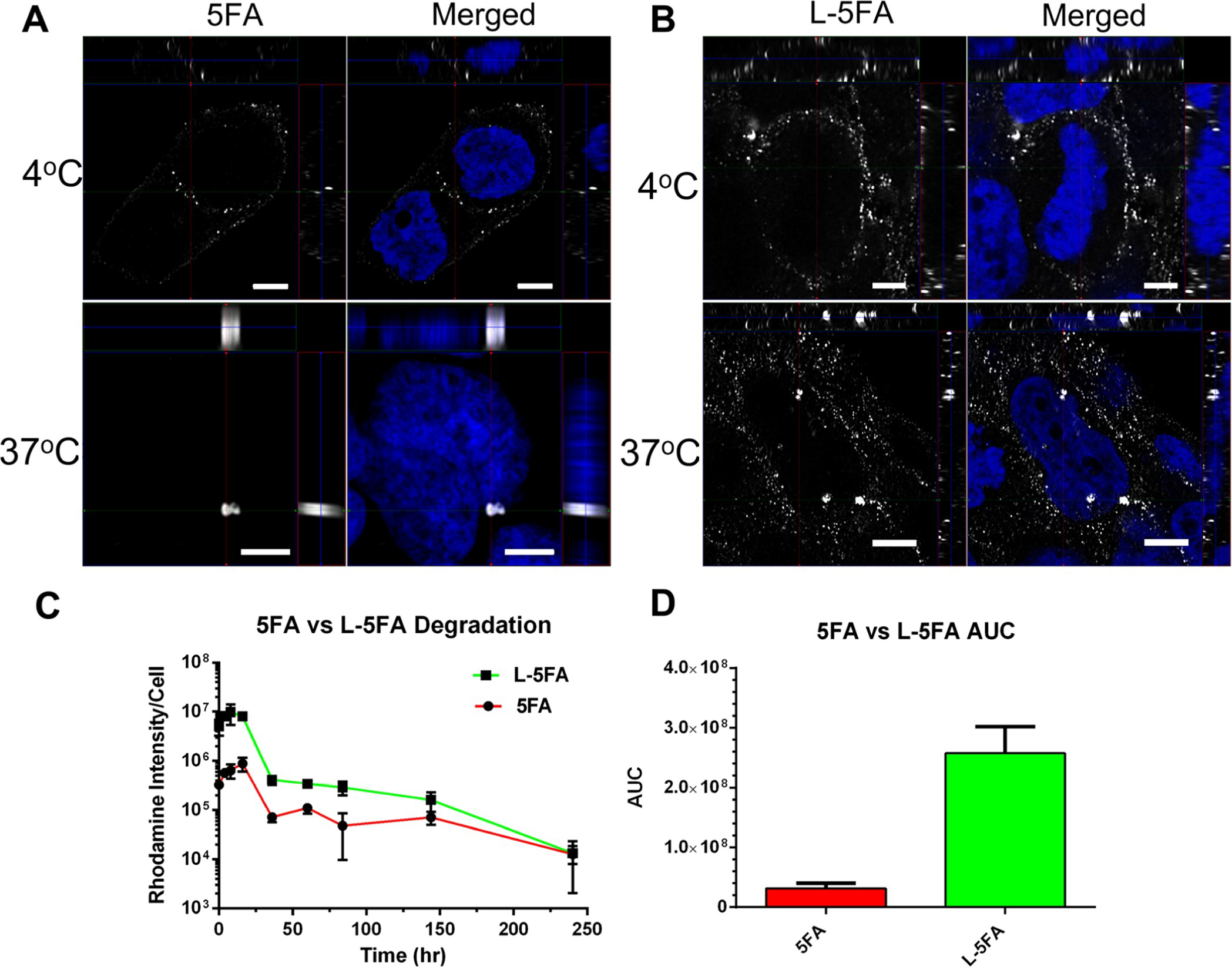
L-5FA significantly increases the cellular exposure compared to untargeted 5FA by fluorescence microscopy. (A,B) BT474 cells were treated with 5 μM of unlabeled 5FA or L-5FA and fixed at 4 and 37 °C to visualize csGRP78-dependent internalization. Cells were visualized by three-dimensional super resolution laser scanning confocal microscopy. Targeted L-5FA was present on the membrane at a higher abundance at 4 °C as well as inside the cell at 37 °C, which are consistent with ligand-dependent enhancement of uptake compared to 5FA. (C) Degradation and/or export of the probe after internalization was characterized in live cells by epifluorescence microscopy, which was quantified by image analysis. Rhodamine-labeled 5FA and L-5FA were visualized in BT474 cells to calculate average fluorescence per cell over time. These data show consistently higher L-5FA cell association; furthermore, they suggest similar degradation kinetics between both constructs. (D) Although degradation kinetics appear to be similar, because L-5FA starts at such an elevated cell association, it yields a significantly higher exposure (AUC) to the rhodamine probe over a 240 h period (n = 3 per group, *p = 0.001). Mean ± SD.
While this data is suggestive of L-5FA endocytosis, its actual mechanism of uptake requires future exploration. Our recent studies show that another untargeted FKBP–ELP undergoes internalization consistent with macropinocytosis; furthermore, it is likely that 5FA follows a similar pathway into these cells.42 In addition, there is evidence for a clathrin-dependent internalization mechanism of csGRP78-targeted peptides. A different small peptide ligand specific to csGRP78 (Pep42) was blocked by a known clathrin-mediated endocytosis inhibitor, chlorpromazine.43 Other reports of csGRP78-ligand complexes include a 60 kDa protein, which is similarly consistent with CME.44 We presume that W- and L-5FA constructs also enter through a CME-dependent process, which may differ from untargeted 5FA.
The identification of csGRP78 has important implications for a broad spectrum of cancers; furthermore, the results herein suggest that it may generally enhance the targeting of small molecules that inhibit mTORC1 or other pathways. This manuscript specifically demonstrates this effect using a protein-based carrier for rapamycin; however, other strategies could also benefit from this approach to develop targeted drug carriers. Recent examples include targeting of cells undergoing high levels of ER stress using a csGRP78 antibody. The PAT-SM6 antibody binds csGRP78, which is tumor-specific and directly induces a modest pro-apoptotic effect.45 This led to a phase I clinical trial in multiple myeloma relapse patients;46 however, the efficacy of PAT-SM6 depends on synergism with multiple cytotoxic agents such as bortezomib and lenalidomide.47 More recently, another antibody formulation, Mab159 IgG, inhibited the PI3K/Akt axis in a murine model, which was associated with caspase 8- and 9-mediated apoptosis.48 Other approaches targeting csGRP78 have relied on small peptides with high binding affinities. One study developed a targeting strategy with the W peptide by fusing it to SubA to cleave csGRP78, which was used as a basis for the flow cytometry work in this study.36 Another study encapsulated doxorubicin in a liposome formulation decorated with W-peptides. This formulation inhibited proliferation of HUVECs activated along the ERK1/2 pathway by vascular endothelial growth factor.49 Other small peptides targeting csGRP78 have been explored to target PEG–PLA nanoparticles that encapsulate paclitaxel. The SNTRVAP peptide successfully targeted glioma cells,50 while the GIRLRG peptide successfully targeted an irradiated breast cancer cell line.51
Here, we have advanced the csGRP78 targeting toolset with an ELP-based system. The flexibility of the ELP backbone has been explored in several therapeutic indications using peptide-based targeting ligands. Notably, these include the Attachment Driven Assembly of Micelles ELPs developed by Chilkoti and co-workers,52 which are assembled around a core of peptides covalently linked with hydrophobic small-molecule chemotherapeutics. Drug conjugation to these ELPs leads to stable colloids, which have can be further enhanced through protein ligands at their exterior, such as an anti-EGFR1 single-chain nanobody called EgA1.53 Improvement of untargeted 5FA by addition of a targeting ligand is supported by enhancements observed in previous generation FKBP–ELP formulations. Inclusion of integrin targeting functionality into the FSI nanoparticle increased its exposure to target tissues and led to successful tumor accumulation in an MDA-MB-468 xenograft model even with a 3-fold less rapamycin dose,16 similar to RGD- and ICAM-targeted formulations.15–17 Here, we provide evidence in support of csGRP78 targeting with a rapamycin payload to a broad population of cancer cell types undergoing ER stress.23 While we provide direct evidence in BT474 cells, the flexibility of the FKBP–ELP system allows it to be engineered for a variety of other therapeutic indications with a diverse set of pharmaceutics.
CONCLUSIONS
This report is the first to confirm the viability of csGRP78-targeted rapamycin and supports further development of protein-based drug carriers containing FKBP complexed with rapamycin. The enhancing effect of csGRP78 ligands play an important role in the targeting of the soluble X-5FA that may further enhance rapamycin delivery in other ELP-based carriers. We have developed a next-generation FKBP–ELP rapamycin carrier that successfully targets breast cancer ductal cells (BT474) in a csGRP78-dependent manner. We show that this high-capacity carrier system can be loaded with rapamycin and delivered to its intracellular target, mTORC1, to inhibit its downstream substrate, rpS6. Incidentally, cancer cells are subjected to both extrinsic and intrinsic ER stress, which constitutively upregulates GRP78 expression and its translocation to the cell surface. This opens up an opportunity to potentially target a broad subset of cancer cells.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank J.W. at the USC School of Pharmacy Translational Research Laboratory, A.A. at the USC School of Pharmacy Mass Spectrometry Core, and S.L. at the USC Center of Excellence in Nanobiophysics, and Richard Van Krieken for his help in methodology development.
Funding
This work was made possible by University of Southern California (USC), the G.S.H. Professorship, National Institutes of Health R01 GM114839 to JAM, R01EY026635 to JAM, R01-CA027607 to ASL, P30 CA014089 to the USC Norris Comprehensive Cancer Center (USC NCCC), and P30 EY029220 to the USC Ophthalmology Center Core Grant for Vision Research. A Programmatic Pilot grant from the USC NCCC Translational and Clinical Sciences Program funded part of this work.
ABBREVIATIONS
- csGRP78
cell surface glucose-regulated protein-78
- DLS
dynamic light scattering
- ELP
elastin-like polypeptide
- ER
endoplasmic reticulum
- FKB
P FK506 binding protein
- HSP
heat shock protein
- MALDI-TOF
matric assisted laser desorption/ionization time-of-flight
- mTORC1
mammalian target of rapamycin complex 1
- PK
pharmacokinetics
- SEC-MALS
size exclusion chromatography-multiangle light scattering
- SubA
subtilase cytotoxin A
- T t
transition temperature
Footnotes
The authors declare the following competing financial interest(s): JA Mackay is an inventor of intellectual property describing commercial applications of elastin-like polypeptides, which are unrelated to this work.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.biomac.2c00048
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.2c00048.
Complete encoded amino acid sequences for A24 and FKBP; MALDI-TOF charge states; molar extinction coefficients for ELPs as evaluated by UV–vis analysis; quantification of rapamycin using analytical RT-HPLC; distribution of hydrodynamic radii of Hydra-ELPs complexed with rapamycin; GRP78 expression Western blot; and cell viability assay of cells treated with Hydra-ELPs loaded with rapamycin (PDF)
Contributor Information
Hugo Avila, Department of Pharmacology and Pharmaceutical Sciences, USC School of Pharmacy, Los Angeles, California 90089, United States.
Jingmei Yu, Department of Pharmacology and Pharmaceutical Sciences, USC School of Pharmacy, Los Angeles, California 90089, United States.
Geetha Boddu, Department of Pharmacology and Pharmaceutical Sciences, USC School of Pharmacy, Los Angeles, California 90089, United States.
Alvin Phan, Department of Pharmacology and Pharmaceutical Sciences, USC School of Pharmacy, Los Angeles, California 90089, United States.
Anh Truong, Department of Pharmacology and Pharmaceutical Sciences, USC School of Pharmacy, Los Angeles, California 90089, United States.
Santosh Peddi, Department of Pharmacology and Pharmaceutical Sciences, USC School of Pharmacy, Los Angeles, California 90089, United States.
Hao Guo, Department of Pharmacology and Pharmaceutical Sciences, USC School of Pharmacy, Los Angeles, California 90089, United States.
Shin-Jae Lee, Department of Pharmacology and Pharmaceutical Sciences, USC School of Pharmacy, Los Angeles, California 90089, United States; Department of Biomedical Engineering, USC Viterbi School of Engineering, Los Angeles, California 90089, United States.
Mario Alba, Department of Pharmacology and Pharmaceutical Sciences, USC School of Pharmacy, Los Angeles, California 90089, United States.
Ethan Canfield, Mass Spectrometry Core, USC School of Pharmacy, Los Angeles, California 90089, United States.
Vicky Yamamoto, Department of Biochemistry and Molecular Medicine, USC Keck School of Medicine, Los Angeles, California 90033, United States.
James C. Paton, Research Centre for Infectious Diseases, Department of Molecular and Biomedical Science, University of Adelaide, Adelaide 5005, Australia
Adrienne W. Paton, Research Centre for Infectious Diseases, Department of Molecular and Biomedical Science, University of Adelaide, Adelaide 5005, Australia
Amy S. Lee, Department of Biochemistry and Molecular Medicine, USC Keck School of Medicine, Los Angeles, California 90033, United States
J. Andrew MacKay, Department of Pharmacology and Pharmaceutical Sciences, USC School of Pharmacy, Los Angeles, California 90089, United States; Department of Biomedical Engineering, USC Viterbi School of Engineering, Los Angeles, California 90089, United States; Department of Ophthalmology, USC Keck School of Medicine, Los Angeles, California 90033, United States.
REFERENCES
- (1).Beck JT; Hortobagyi GN; Campone M; Lebrun F; Deleu I; Rugo HS; Pistilli B; Masuda N; Hart L; Melichar B; Dakhil S; Geberth M; Nunzi M; Heng DYC; Brechenmacher T; El-Hashimy M; Douma S; Ringeisen F; Piccart M Everolimus plus exemestane as first-line therapy in HR(+), HER2(−) advanced breast cancer in BOLERO-2. Breast Cancer Res. Treat. 2014, 143, 459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).André F; O’Regan R; Ozguroglu M; Toi M; Xu B; Jerusalem G; Masuda N; Wilks S; Arena F; Isaacs C; Yap Y-S; Papai Z; Lang I; Armstrong A; Lerzo G; White M; Shen K; Litton J; Chen D; Zhang Y; Ali S; Taran T; Gianni L Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014, 15, 580–591. [DOI] [PubMed] [Google Scholar]
- (3).Demetri GD; Chawla SP; Ray-Coquard I; Le Cesne A; Staddon AP; Milhem MM; Penel N; Riedel RF; Bui-Nguyen B; Cranmer LD; Reichardt P; Bompas E; Alcindor T; Rushing D; Song Y; Lee R.-m.; Ebbinghaus S; Eid JE; Loewy JW; Haluska FG; Dodion PF; Blay J-Y Results of an international randomized phase III trial of the mammalian target of rapamycin inhibitor ridaforolimus versus placebo to control metastatic sarcomas in patients after benefit from prior chemotherapy. J. Clin. Oncol. 2013, 31, 2485–2492. [DOI] [PubMed] [Google Scholar]
- (4).Hess G; Herbrecht R; Romaguera J; Verhoef G; Crump M; Gisselbrecht C; Laurell A; Offner F; Strahs A; Berkenblit A; Hanushevsky O; Clancy J; Hewes B; Moore L; Coiffier B Phase III study to evaluate temsirolimus compared with investigator’s choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J. Clin. Oncol. 2009, 27, 3822–3829. [DOI] [PubMed] [Google Scholar]
- (5).Motzer RJ; Escudier B; Oudard S; Hutson TE; Porta C; Bracarda S; Grünwald V; Thompson JA; Figlin RA; Hollaender N; Kay A; Ravaud A; Group R-S Phase 3 trial of everolimus for metastatic renal cell carcinoma : final results and analysis of prognostic factors. Cancer 2010, 116, 4256–4265. [DOI] [PubMed] [Google Scholar]
- (6).Heitman J; Movva NR; Hall MN Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [DOI] [PubMed] [Google Scholar]
- (7).Banaszynski LA; Liu CW; Wandless TJ Characterization of the FKBP.rapamycin.FRB ternary complex. J. Am. Chem. Soc. 2005, 127, 4715–4721. [DOI] [PubMed] [Google Scholar]
- (8).Kuna M; Mahdi F; Chade AR; Bidwell GL 3rd Molecular Size Modulates Pharmacokinetics, Biodistribution, and Renal Deposition of the Drug Delivery Biopolymer Elastin-like Polypeptide. Sci. Rep. 2018, 8, 7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Despanie J; Dhandhukia JP; Hamm-Alvarez SF; MacKay JA Elastin-like polypeptides: Therapeutic applications for an emerging class of nanomedicines. J. Controlled Release 2016, 240, 93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dhandhukia J; Weitzhandler I; Wang W; MacKay JA Switchable elastin-like polypeptides that respond to chemical inducers of dimerization. Biomacromolecules 2013, 14, 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Shah M; Edman MC; Janga SR; Shi P; Dhandhukia J; Liu S; Louie SG; Rodgers K; Mackay JA; Hamm-Alvarez SF A rapamycin-binding protein polymer nanoparticle shows potent therapeutic activity in suppressing autoimmune dacryoadenitis in a mouse model of Sjogren’s syndrome. J. Controlled Release 2013, 171, 269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shi P; Aluri S; Lin Y-A; Shah M; Edman M; Dhandhukia J; Cui H; MacKay JA Elastin-based protein polymer nanoparticles carrying drug at both corona and core suppress tumor growth in vivo. J. Controlled Release 2013, 171, 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dhandhukia JP; Li Z; Peddi S; Kakan S; Mehta A; Tyrpak D; Despanie J; MacKay JA Berunda Polypeptides: Multi-Headed Fusion Proteins Promote Subcutaneous Administration of Rapamycin to Breast Cancer In Vivo. Theranostics 2017, 7, 3856–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ju Y; Edman MC; Guo H; Janga SR; Peddi S; Louie SG; Junge JA; MacKay JA; Hamm-Alvarez SF Intralacrimal Sustained Delivery of Rapamycin Shows Therapeutic Effects without Systemic Toxicity in a Mouse Model of Autoimmune Dacryoadenitis Characteristic of Sjogren’s Syndrome. Biomacromolecules 2021, 22, 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Peddi S; Roberts SK; MacKay JA Nanotoxicology of an Elastin-like Polypeptide Rapamycin Formulation for Breast Cancer. Biomacromolecules 2020, 21, 1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Dhandhukia JP; Shi P; Peddi S; Li Z; Aluri S; Ju Y; Brill D; Wang W; Janib SM; Lin Y-A; Liu S; Cui H; MacKay JA Bifunctional Elastin-like Polypeptide Nanoparticles Bind Rapamycin and Integrins and Suppress Tumor Growth in Vivo. Bioconjugate Chem. 2017, 28, 2715–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hsueh P-Y; Ju Y; Vega A; Edman MC; MacKay JA; Hamm-Alvarez SF A Multivalent ICAM-1 Binding Nanoparticle which Inhibits ICAM-1 and LFA-1 Interaction Represents a New Tool for the Investigation of Autoimmune-Mediated Dry Eye. Int. J. Mol. Sci. 2020, 21, 2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ni M; Lee AS ER chaperones in mammalian development and human diseases. FEBS Lett. 2007, 581, 3641–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hirsch C; Gauss R; Horn SC; Neuber O; Sommer T The ubiquitylation machinery of the endoplasmic reticulum. Nature 2009, 458, 453–460. [DOI] [PubMed] [Google Scholar]
- (20).Bernasconi R; Galli C; Calanca V; Nakajima T; Molinari M Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J. Cell Biol. 2010, 188, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lee AS GRP78 induction in cancer: therapeutic and prognostic implications. Cancer Res. 2007, 67, 3496–3499. [DOI] [PubMed] [Google Scholar]
- (22).Miao YR; Eckhardt BL; Cao Y; Pasqualini R; Argani P; Arap W; Ramsay RG; Anderson RL Inhibition of established micrometastases by targeted drug delivery via cell surface-associated GRP78. Clin. Cancer Res. 2013, 19, 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lee AS Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tsai Y-L; Zhang Y; Tseng C-C; Stanciauskas R; Pinaud F; Lee AS Characterization and mechanism of stress-induced translocation of 78-kilodalton glucose-regulated protein (GRP78) to the cell surface. J. Biol. Chem. 2015, 290, 8049–8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tseng C-C; Zhang P; Lee AS The COOH-Terminal Proline-Rich Region of GRP78 Is a Key Regulator of Its Cell Surface Expression and Viability of Tamoxifen-Resistant Breast Cancer Cells. Neoplasia 2019, 21, 837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang L; Li Z; Shi T; La X; Li H; Li Z Design, purification and assessment of GRP78 binding peptide-linked Subunit A of Subtilase cytotoxic for targeting cancer cells. BMC Biotechnol. 2016, 16, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Wang S-H; Yu J Structure-based design for binding peptides in anti-cancer therapy. Biomaterials 2018, 156, 1–15. [DOI] [PubMed] [Google Scholar]
- (28).Wang S-H; Lee AC-L; Chen I-J; Chang N-C; Wu H-C; Yu H-M; Chang Y-J; Lee T-W; Yu J-C; Yu AL; Yu J Structure-based optimization of GRP78-binding peptides that enhances efficacy in cancer imaging and therapy. Biomaterials 2016, 94, 31–44. [DOI] [PubMed] [Google Scholar]
- (29).Niu S; Bremner DH; Wu J; Wu J; Wang H; Li H; Qian Q; Zheng H; Zhu L l-Peptide functionalized dual-responsive nanoparticles for controlled paclitaxel release and enhanced apoptosis in breast cancer cells. Drug Deliv. 2018, 25, 1275–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).D’Angelo S; Staquicini FI; Ferrara F; Staquicini DI; Sharma G; Tarleton CA; Nguyen H; Naranjo LA; Sidman RL; Arap W; Bradbury AR; Pasqualini R Selection of phage-displayed accessible recombinant targeted antibodies (SPARTA): methodology and applications. JCI Insight 2018, 3, No. e98305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Schindelin J; Arganda-Carreras I; Frise E; Kaynig V; Longair M; Pietzsch T; Preibisch S; Rueden C; Saalfeld S; Schmid B; Tinevez J-Y; White DJ; Hartenstein V; Eliceiri K; Tomancak P; Cardona A Fiji: an open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tyrpak DR; Wang Y; Avila H; Guo H; Fu R; Truong AT; Park M; Okamoto CT; Hamm-Alvarez SF; MacKay JA Caveolin elastin-like polypeptide fusions mediate temperature-dependent assembly of caveolar microdomains. ACS Biomater. Sci. Eng. 2020, 6, 198–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tyrpak DR; Li Y; Lei S; Avila H; MacKay JA Single-Cell Quantification of the Transition Temperature of Intracellular Elastin-like Polypeptides. ACS Biomater. Sci. Eng. 2021, 7, 428–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Kouhi A; Yao Z; Zheng L; Li Z; Hu P; Epstein AL; MacKay JA Generation of a Monoclonal Antibody to Detect Elastin-like Polypeptides. Biomacromolecules 2019, 20, 2942–2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Paton AW; Srimanote P; Talbot UM; Wang H; Paton JC A new family of potent AB(5) cytotoxins produced by Shiga toxigenic Escherichia coli. J. Exp. Med. 2004, 200, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Paton AW; Beddoe T; Thorpe CM; Whisstock JC; Wilce MCJ; Rossjohn J; Talbot UM; Paton JC AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 2006, 443, 548–552. [DOI] [PubMed] [Google Scholar]
- (37).Lass A; Kujawa M; McConnell E; Paton AW; Paton JC; Wójcik C Decreased ER-associated degradation of alpha-TCR induced by Grp78 depletion with the SubAB cytotoxin. Int. J. Biochem. Cell Biol. 2008, 40, 2865–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Salmond RJ; Mirchandani AS; Besnard A-G; Bain CC; Thomson NC; Liew FY IL-33 induces innate lymphoid cell-mediated airway inflammation by activating mammalian target of rapamycin. J. Allergy Clin. Immunol. 2012, 130, 1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Pearce LR; Alton GR; Richter DT; Kath JC; Lingardo L; Chapman J; Hwang C; Alessi DR Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem. J. 2010, 431, 245–255. [DOI] [PubMed] [Google Scholar]
- (40).Bain J; Plater L; Elliott M; Shpiro N; Hastie CJ; McLauchlan H; Klevernic I; Arthur JSC; Alessi DR; Cohen P The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007, 408, 297–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Thon M; Hosoi T; Yoshii M; Ozawa K Leptin induced GRP78 expression through the PI3K-mTOR pathway in neuronal cells. Sci. Rep. 2014, 4, 7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Peddi S; Pan X; MacKay JA Intracellular Delivery of Rapamycin From FKBP Elastin-Like Polypeptides Is Consistent With Macropinocytosis. Front. Pharmacol. 2018, 9, 1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Liu Y; Steiniger SCJ; Kim Y; Kaufmann GF; Felding-Habermann B; Janda KD Mechanistic studies of a peptidic GRP78 ligand for cancer cell-specific drug delivery. Mol. Pharm. 2007, 4, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Chen M; Zhang Y; Yu VC; Chong Y-S; Yoshioka T; Ge R Isthmin targets cell-surface GRP78 and triggers apoptosis via induction of mitochondrial dysfunction. Cell Death Differ. 2014, 21, 797–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Hensel F; Eckstein M; Rosenwald A; Brändlein S Early development of PAT-SM6 for the treatment of melanoma. Melanoma Res. 2013, 23, 264–275. [DOI] [PubMed] [Google Scholar]
- (46).Rasche L; Duell J; Castro IC; Dubljevic V; Chatterjee M; Knop S; Hensel F; Rosenwald A; Einsele H; Topp MS; Brandlein S GRP78-directed immunotherapy in relapsed or refractory multiple myeloma - results from a phase 1 trial with the monoclonal immunoglobulin M antibody PAT-SM6. Haematologica 2015, 100, 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Rasche L; Menoret E; Dubljevic V; Menu E; Vanderkerken K; Lapa C; Steinbrunn T; Chatterjee M; Knop S; Düll J; Greenwood DL; Hensel F; Rosenwald A; Einsele H; Brändlein S A GRP78-Directed Monoclonal Antibody Recaptures Response in Refractory Multiple Myeloma with Extramedullary Involvement. Clin. Cancer Res. 2016, 22, 4341–4349. [DOI] [PubMed] [Google Scholar]
- (48).Liu R; Li X; Gao W; Zhou Y; Wey S; Mitra SK; Krasnoperov V; Dong D; Liu S; Li D; Zhu G; Louie S; Conti PS; Li Z; Lee AS; Gill PS Monoclonal antibody against cell surface GRP78 as a novel agent in suppressing PI3K/AKT signaling, tumor growth, and metastasis. Clin. Cancer Res. 2013, 19, 6802–6811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Katanasaka Y; Ishii T; Asai T; Naitou H; Maeda N; Koizumi F; Miyagawa S; Ohashi N; Oku N Cancer antineovascular therapy with liposome drug delivery systems targeted to BiP/GRP78. Int. J. Cancer 2010, 127, 2685–2698. [DOI] [PubMed] [Google Scholar]
- (50).Ran D; Mao J; Shen Q; Xie C; Zhan C; Wang R; Lu W GRP78 enabled micelle-based glioma targeted drug delivery. J. Controlled Release 2017, 255, 120–131. [DOI] [PubMed] [Google Scholar]
- (51).Passarella RJ; Spratt DE; van der Ende AE; Phillips JG; Wu H; Sathiyakumar V; Zhou L; Hallahan DE; Harth E; Diaz R Targeted nanoparticles that deliver a sustained, specific release of Paclitaxel to irradiated tumors. Cancer Res. 2010, 70, 4550–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Jenkins IC; Milligan JJ; Chilkoti A Genetically Encoded Elastin-Like Polypeptides for Drug Delivery. Adv. Healthcare Mater. 2021, 10, No. e2100209. [DOI] [PubMed] [Google Scholar]
- (53).Costa SA; Mozhdehi D; Dzuricky MJ; Isaacs FJ; Brustad EM; Chilkoti A Active Targeting of Cancer Cells by Nanobody Decorated Polypeptide Micelle with Bio-orthogonally Conjugated Drug. Nano Lett. 2019, 19, 247–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.