

Abstract
Herein, a series of 11- or 12-substituted benzophenanthridinone derivatives was designed and synthesized for the discovery of dual topoisomerase IB (TOP1) and tyrosyl-DNA phosphodiesterase 1 (TDP1) inhibitors. Enzyme-based assays indicated that two compounds 12 and 38 showed high TOP1 inhibitory potency (+++), and four compounds 35, 37, 39 and 43 showed good TDP1 inhibition with IC50 values ranging from 10 to 18 µM. 38 could induce cellular TOP1cc formation, resulting in the highest cytotoxicity against HCT-116 cells (0.25 µM). The most potent TDP1 inhibitor 43 (10 µM) could induce cellular TDP1cc formation and enhance topotecan-induced DNA damage and showed strong synergistic cytotoxicity with topotecan in both MCF-7 and MCF-7/TDP1 cells.
Keywords: DNA repair, Tyrosyl-DNA phosphodiesterase, Topoisomerase, Anticancer, Inhibitor
Graphical Abstract

A SAR research of 11- or 12-substituted benzophenanthridinone derivatives led to identify potent dual TOP1 and TDP1 inhibitor 43 that can enhance the ability of topotecan to induce DNA damage, shows synergy in combination with topotecan in both MCF-7 and MCF-7/TDP1 cells.
Introduction
DNA topoisomerase IB (TOP1) is ubiquitous and essential enzymes that solve the topological problems that accompany DNA replication, transcription and genomic stability[1–4], and a validated target for the development of anticancer agents[5–8]. TOP1 inhibitors, such as camptothecins and indenoisoquinolines can trap the normally transient TOP1-DNA cleavage complex (TOP1cc) and cause TOP1-mediated DNA damage, resulting in cancer cell death[8–10]. TOP1-mediated DNA damage can be repaired through several pathways, including homologous recombination (high accuracy and low efficiency), cell cycle checkpoint signaling (often absent in cancer cells) and tyrosyl-DNA phosphodiesterase 1 (TDP1)-dependent pathways[11–14].
TDP1 is a member of the phospholipase D superfamily[15]. It can specifically hydrolyze the 3′-phosphotyrosyl bonds of TOP1ccs to generate DNA breaks with 5′-hydroxyl and 3′-phosphate ends for further DNA repair[16,17]. Following the recruitment and activation of poly(ADP-ribose) polymerase (PARP1), the resulting DNA breaks can be repaired by polynucleotide kinase phosphatase (PNKP), DNA polymerase β and DNA ligase III through the (X-ray repair complementation group 1) XRCC-1-dependent pathway[18–23]. In this pathway, TDP1-catalyzed hydrolysis of the 3′-phosphotyrosyl bond is a key step for initiating the repair of TOP1-mediated DNA damage, which can lead to cancer cell resistance to TOP1 inhibitors[24]. Indeed, TDP1-overexpressing cells show resistance to camptothecin (CPT) [25,26]. Conversely, cells with TDP1 deficiency or mutated (SCAN1 cells), and TDP1 knockout mice show hypersensitivity to CPT[27–33]. Recently, several TDP1 inhibitors have been reported to show a synergistic anticancer activity with TOP1 inhibitors in vitro and in vivo[31,34–48]. Therefore, TDP1 is a rational target for the discovery of anticancer drugs[49,50].
As TOP1 is a validated anticancer target, TOP1 inhibitors have been well developed and several camptothecin analogues are extensively used in clinical cancer treatment[5,10,51]. Topotecan (TPT) [52,53], Irinotecan[54], and the recently antibody drug conjugates Trastuzumab deruxtecan and Sacituzumab govitecan have been approved by the Food and Drug Administration (FDA) [10,55,56]. Additionally, 10-hydroxycamptothecin is clinically approved in China[57,58], and Belotecan in Korea[8]. Despite their outstanding efficacy, camptothecin TOP1 inhibitors suffer from several drawbacks including chemical instability, acute and chronic toxicity including drug-related leukemia, and drug resistance. Subsequently, to counter these limitations, much attention is paid to discovery of non-camptothecin inhibitors[59]. In addition, small molecular TDP1 inhibitors have been also developed recently, and several TDP1 scaffold have been reported[60], including steroids[48,61], methyl-3,4-dephostatins[62], arylidenethioxothiazolidinones[63], thioxopyrimidinedione[64], benzopentathiepines[65], indenoisoquinolines[66,67], usnic acid derivatives[39], aminoadamantanes[36], 7-hydroxycoumarin derivatives[35,47], and N,2-diphenylimidazo[1,2-a]amines[43], benzodipyrans[68]. Additionally, recently TDP1 co-crystal structures with inhibitors could provide the structural basis for the rational design of TDP1 inhibitors[43,69].
The physiological function of TDP1 is to specifically hydrolyzes the 3′-phosphotyrosyl bond of TOP1ccs. Based on this point, the dual activities against TOP1 and TDP1 will act on multiple stages of cancer genesis and development, and may play a greater anticancer effect. A few dual TOP1 and TDP1 inhibitors have been identified[37,51,67,70]. The first reported dual TOP1 and TDP1 inhibitors were indenoisoquinoline derivatives such as the representative bisindenoisoquinoline inhibitor 1 is shown in Figure 1[67]. However, further study indicated that several indenoisoquinoline derivatives also inhibited tyrosyl-DNA phosphodiesterase 2[71], a DNA repair enzyme hydrolyzing primarily the 5′-phosphotyrosyl bonds of TOP2 and TOP3 cleavage complexes[72–74], implying that indenoisoquinolines might be promiscuous inhibitors.
Figure 1.

The structures of the reported dual TOP1 and TDP1 inhibitors and the representative oxynitidine derivatives.
In our effort to discover novel dual TOP1 and TDP1 inhibitor chemotypes, two chalcone derivatives 2 and 3 (Figure 1) have been identified from the roots of Isodon ternifolius Kudo as dual inhibitors[42]. However, they just show moderate inhibitory activities for both TOP1 and TDP1. In addition, oxynitidine (4, Figure 1) derivatives have also been identified as dual inhibitors with the ability to trap cellular TDP1-DNA complexes and with anticancer activity in vitro and in vivo[37,46,75]. The representative molecule 5 shows a strong TOP1 inhibition (+++), but a weak TDP1 inhibition (12% enzyme inhibitory activity at 100 μM concentration) [37]. Structure-activity relationship (SAR) analysis has indicated that the lactam skeleton of oxynitidine derivatives is important for TOP1 inhibition. Molecular modeling indicates a hydrogen bond (2.9 Å) formed between the lactam oxygen atom of 5 and the key residue Arg364 possibly contribute to the TOP1 inhibition[37]. And, near the C-ring of 5, there is a hydrophilic binding pocket comprising the key residues Asn352 and Glu356 of TOP1, which could accommodate an additional polar side chain for a TOP1 inhibitor. In addition, the molecular modeling between 5 and TDP1 (PDB 6DJD) indicates that the benzophenanthridinone skeleton binds to the DNA binding region of TDP1 (Figure S1, Supporting Information) [69]. And, there is also a hydrophilic binding pocket comprising the key residues His263, and Ser399, Ser400, Asn516, Ala521 and Glu538 of TDP1 near the C-ring of 5. This observation inspired us to design the 11- or 12-substituted benzophenanthridinone derivatives for the discovery of dual TOP1 and TDP1 inhibitors. In this work, we report the synthesis of the designed compounds, their TOP1 and TDP1 inhibitory activities and anticancer potential.
Results and Discussion
Chemistry
Because of the potential role of the lactam skeleton for TOP1 inhibition, the designed compounds retain the oxynitidine scaffold (Figure 1, N-methylpyridinone structure). The synthesis of the 12-substituted benzophenanthridinone derivatives is illustrated in Scheme 1. According to Cheng’s synthetic method[76], the key intermediate 8 was prepared from the reaction of the Schiff base 6 with 7, prepared through Sonogashira coupling reaction from 5-bromobenzo-[d][1,3]dioxole and 3-butyn-1-ol. Following the Swern oxidation of the hydroxyl group of 8[77], the obtained aldehyde intermediate 9 was further oxidized by NaClO2 under NaH2PO4 condition[78], and subsequently cyclized to give the benzophenanthridinone 10. After hydrolysis of 10, the reaction of 11 with the corresponding chloride material in the presence of K2CO3 and KI gave the 12-substituted benzophenanthridinone derivatives 12-23 (Table 1).
Scheme 1.

Synthesis of 10-23.
Reagents and conditions: (a) NH2Me, MeOH, rt. (b) i) N2, Ni(cod)2, P(o-Tol)3, MeCN; ii) CsOH, K3[Fe(CN)6], MeOH, H2O. (c) (COCl)2, DMSO, TEA, DCM. (d) i) NaClO2, NaH2PO4·2H2O, 2-methyl-2-butene, acetone; ii) Ac2O, AcOK. (e) KOH, H2O, MeOH. (f) R(CH2)nCl, KI, K2CO3, DMF.
Table 1.
The TOP1 and TDP1 Inhibitory Potency, and Cytotoxicity of 10-23.

| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| GI50 ± SD (μM)[c] |
||||||
| Cpd | n | R | TOP1 Inhibition[a] |
IC50 (μM) for TDP1[b] |
HCT-116 | MCF-7 |
| 4 | /d | / | 0/+ | >111 | 75 ± 4.4 | >100 |
| 10 | 0 | Ac | + | >111 | >100 | >100 |
| 11 | 0 | H | 0 | >111 | >100 | >100 |
| 12 | 2 | NHMe | +++ | >111 | 1.3 ± 0.12 | 2.2 ± 0.06 |
| 13 | 3 | + | >111 | 12 ± 1.2 | 33 ± 5.0 | |
| 14 | 2 | NMe2 | + | 37–111 | 4.6 ± 0.41 | 8.5 ± 0.91 |
| 15 | 3 | + | >111 | 36 ± 1.3 | 54 ± 4.9 | |
| 16 | 2 |

|
+ | >111 | >100 | 22 ± 7.7 |
| 17 | 3 | ++ | >111 | 1.8 ± 0.32 | 6.6 ± 1.3 | |
| 18 | 2 |

|
+ | 37–111 | 7.7 ± 0.56 | 7.0 ± 1.3 |
| 19 | 3 | ++ | >111 | 1.5 ± 0.21 | 0.93 ± 0.07 | |
| 20 | 2 |

|
++ | >111 | 2.9 ± 0.45 | 2.7 ± 0.23 |
| 21 | 3 | 0 | >111 | 16 ± 3.6 | 64 ± 3.9 | |
| 22 | 2 | NH(CH2)2NMe2 | + | >111 | 9.1 ± 1.0 | 9.1 ± 2.4 |
| 23 | 1 | COOEt | 0 | >111 | >100 | 91 ± 5.9 |
TOP1 cleavage inhibitory activity of the compounds was semiquantitatively expressed relative to CPT at 1 μM as follows: 0, no inhibition; 0/+, less than 20%; +, between 20% and 50% activity; ++, between 50% and 75% activity; +++, between 75% and 95% activity.
IC50 values for TDP1 inhibition, defined as the concentration of compound that inhibits 50% of enzyme activity, were detected through the gel-based assays.
The cytotoxicity GI50 value was defined as the concentration corresponding to 50% cell growth inhibition and obtained from MTT assay. Every experiment was repeated at least three times independently.
“/” means “inapplicable”.
The synthesis of 11-substituted benzophenanthridinone derivatives is shown in Scheme 2. Using piperonylacetic acid and dimethyloxybenzaldehyde as starting materials, the key intermediate 30 was prepared according to our reported method[79]. The selective demethylation of 30 gave the pyridinone intermediate 31[80]. Following the methylation in the lactam of 31[81], a three-step reaction of 32, including a cyclization under acetic anhydride and AcOK condition, gave the benzophenanthridinone 33. The 11-substituted benzophenanthridinone derivatives 34-45 (Table 2) were prepared from the reaction of 33 with the corresponding chlorides in the presence of K2CO3 and KI.
Scheme 2.

Synthesis of 33-45.
Reagents and conditions: (a) i) ICl, AcOH, rt; ii) MeOH, H2SO4. (b) N2, TMSA, PdCl2(PPh3)2, CuI, NEt3, THF. (c) K2CO3, MeOH. (d) i) AgNO3, I2, MeOH; ii) KMnO4, H2O/MeCN. (e) i) SOCl2, reflux; ii) NH3/H2O, THF. (f) PdCl2(PPh3)2, NEt3, MeCN. (g) i) Me3OBF4, DCM; ii) CF3COOAg, DCM. (h) NaBr, TsOH·H2O, MeOH. (i) MeI, K2CO3, DMF. (j) i) K2CO3, H2O, MeOH; ii) Ac2O, AcOK; iii) K2CO3, H2O, MeOH. (k) R(CH2)nCl, KI, K2CO3, DMF.
Table 2.
The TOP1 and TDP1 Inhibitory Potency, and Cytotoxicity of 33–45.

| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| GI50 ± SD (μM)[c] |
||||||
| Cpd | n | R | TOP1 Inhibition[a] |
IC50 (μM) for TDP1[b] |
HCT-116 | MCF-7 |
| 4 | /d | / | 0/+ | >111 | 75 ± 4.4 | >100 |
| 33 | 0 | H | + | >111 | >100 | >100 |
| 34 | 2 | NMe2 | + | 37–111 | 11 ± 0.48 | 27 ± 1.5 |
| 35 | 3 | 0 | 18 ± 6.9 | 0.55 ± 0.17 | 1.2 ± 0.10 | |
| 36 | 2 | NEt2 | 0 | >111 | 2.1 ± 0.23 | 1.6 ± 0.17 |
| 37 | 3 | + | 15 ± 2.4 | 2.4 ± 0.11 | 2.0 ± 0.10 | |
| 38 | 2 |
|
+++ | 37–111 | 0.25 ± 0.02 | 6.4 ± 1.0 |
| 39 | 3 | + | 15 ± 2.8 | 1.3 ± 0.21 | 1.4 ± 0.13 | |
| 40 | 2 |

|
++ | >111 | 15 ± 1.08 | 18 ± 0.67 |
| 41 | 3 | + | >111 | 3.3 ± 0.73 | 7.9 ± 1.0 | |
| 42 | 3 | OH | + | >111 | 38 ± 2.31 | 51 ± 1.6 |
| 43 | 3 | NH(CH2)2OH | + | 10 ± 3.8 | 0.29 ± 0.19 | 2.2 ± 1.2 |
| 44 | 3 |

|
0/+ | >111 | 10 ± 1.2 | 7.0 ± 0.55 |
| 45 | 3 |

|
0 | >111 | >100 | 85 ± 3.8 |
TOP1 cleavage inhibitory activity of the compounds was semiquantitatively expressed relative to CPT at 1 μM as follows: 0, no inhibition; 0/+, less than 20%; +, between 20% and 50% activity; ++, between 50% and 75% activity; +++, between 75% and 95% activity.
IC50 values for TDP1 inhibition, defined as the concentration of compound that inhibits 50% of enzyme activity, were detected through the gel-based assays.
The cytotoxicity GI50 value was defined as the concentration corresponding to 50% cell growth inhibition and obtained from MTT assay. Every experiment was repeated at least three times independently.
“/” means “inapplicable”.
Finally, twenty-seven benzophenanthridinone derivatives were synthesized and characterized through NMR and HRMS spectra.
TOP1 Inhibition
All the synthesized benzophenanthridinone derivatives were tested for TOP1 inhibition through the TOP1-mediated DNA cleavage assay using a 3′-[32P]-labeled double-stranded DNA oligonucleotide as substrate[82]. The compounds were tested at 100, 10, 1 and 0.1 μM concentrations using CPT and the indenoisoquinoline TOP1 inhibitor LMP744 as positive controls[83].
The TOP1 inhibitory activity was semiquantitatively expressed relative to CPT at 1 μM: 0, no inhibition; 0/+, less than 20%; +, between 20% and 50% activity; ++, between 50% and 75% activity; +++, between 75% and 95% activity, and summarized in Tables 1 and 2.
As shown in Table 1, compared to 4 (0/+), the attachment of an aminoalkoxyl group at 12-position could increase the TOP1 inhibition. All the 12-aminoalkoxyl substituted analogues have TOP1 inhibitory potency ranging from + to +++ except 21. Among them, three analogues 17, 19, and 20 showed moderate TOP1 inhibition (++), and 12 (+++) showed the most potency equal to 5. The attachment of a hydroxy (11) or an alkoxy group without nitrogen atom (23) did not improve the TOP1 inhibition. Similarly, the attachment of an aminoalkoxyl group at 11-position also could increase the TOP1 inhibition (Table 2). And, 38 showed strong TOP1 inhibition of +++, as well as one analogue 40 with moderate inhibition of ++ and six analogues 34, 37, 39 and 41-43 with weak inhibition of +.
Representative cleavage gels of 12, 19, 38 and 40 are shown in Figure 2. The cleavage sites induced by the novel TOP1 inhibitors were similar to that induced by indenoisoquinoline control LMP744 but not to CPT, implying the novel benzophenanthridinone TOP1 inhibitors target the same genome sites as oxynitidine and its analogue 5[37].
Figure 2.
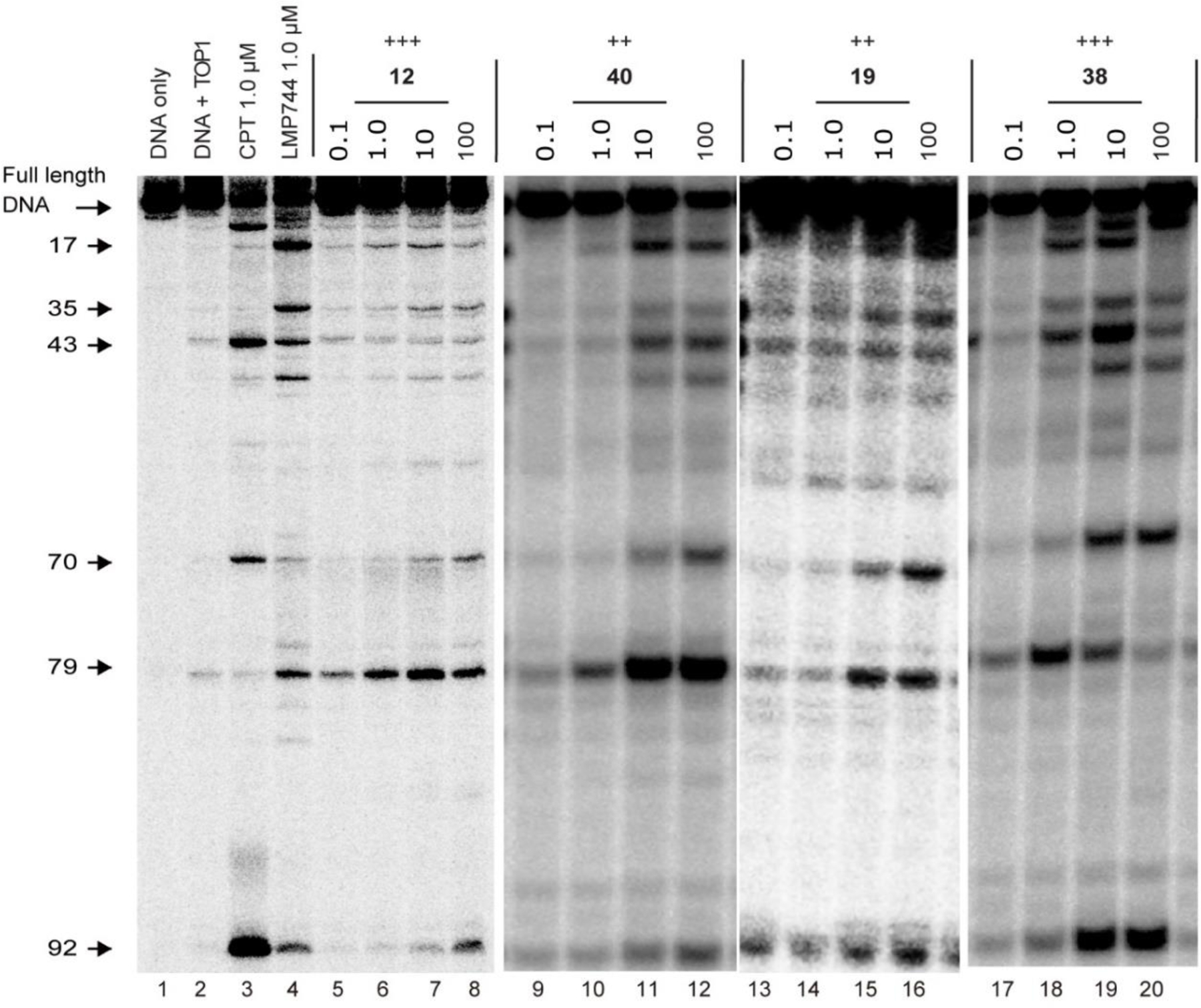
Representative gels of TOP1-mediated DNA cleavage assay. Lane 1, DNA alone; lane 2, DNA and TOP1; lane 3, DNA and TOP1 with CPT (1 μM); lane 4, DNA and TOP1 with LMP744 (1 μM); lanes 5–20, DNA and TOP1 with the tested compounds at 0.1, 1.0, 10, and 100 μM concentration, respectively. The arrows at left indicate the cleavage site positions.
TDP1 Inhibition
All the synthesized benzophenanthridinone derivatives were tested for TDP1 inhibition through a gel-based assay with a Cy5-labeled single-stranded oligonucleotide Cy5–14Y substrate at eight three-fold dilution concentrations (111, 37, 12.3, 4.1, 1.4, 0.46 and 0.051 μM) [43]. The imidazopyridine TDP1 inhibitor XZ664 (Figure S2, Supporting Information) was used as the positive control[43]. As shown in Figure 3, the representative TDP1 inhibition gels indicate that the quantity of TDP1 product Cy5–14P decrease with the addition of increased concentration of XZ664, implying that TDP1 activity is inhibited. The IC50 values, defined as the concentration of compound that inhibits 50% of TDP1 activity, were calculated and summarized in Tables 1 and 2.
Figure 3.

Representative gels of TDP1 assay. Lane 1, DNA alone; lane 2, DNA and recombinant TDP1; lanes 3–26, DNA, recombinant TDP1 and the compounds at the concentrations increasing from 0.051 to 111 μM. The imidazopyridine XZ664 was tested as the positive control at the concentrations increasing from 0.15 to 333 μM. Cy5–14Y and Cy5–14P are the substrate and product of TDP1, respectively.
As shown in Table 1, introduction of a side chain at the 12-position did not enhance the TDP1 inhibitory potency. Only two analogues 14 and 18 showed weak TDP1 inhibition (37–111 μM) for 12-substituted benzophenanthridinone derivatives. For 11-substituted benzophenanthridinone derivatives (Table 2), four analogues 35, 37, 39, and 43 showed good TDP1 inhibitory potency with IC50 values ranging 10 to 18 μM concentration (Figure 3). It seems that the longer side chain at 11-position is favorable for TDP1 inhibitory potency, for example, 35 vs 34, 37 vs 36, 39 vs 38. The most potent TDP1 inhibitor 43 (IC50 = 10 ± 3.8 μM) also showed weak TOP1 inhibition (+). And 38 showing high TOP1 inhibition (+++), only showed weak TDP1 inhibition (37–111 μM).
Cellular TOP1cc and TDP1cc Formation
In previous work, we found benzophenanthridinone TOP1 and TDP1 inhibitors can induce cellular TOP1cc and TDP1cc formation[37,46]. To evaluate the ability of novel inhibitors to trap cellular TOP1ccs, the most potent inhibitor 38 was tested by the immunocomplex of enzyme (ICE) to DNA assay in MCF-7 cells. Lick TPT (Figure 4A), 38 trapped cellular TOP1ccs in MCF-7 cells in a dose-dependent manner, implying that TOP1 is targeted by 38 in cells.
Figure 4.

Cellular TOP1ccs induced by 38 (A) and TDP1cc induced by 43 (B) in MCF-7 cells, detected through ICE assays. (A) Lane 1, untreated control; lanes 2 and 3, cells treated with TPT at 25 and 50 μM, respectively; lanes 4–6, cells treated with 38 at 25, 50, and 100 μM, respectively. (B) Lane 1, untreated control; lanes 2–4, cells treated with TPT (10 μM) and 43 at 10 and 100 μM, respectively; lanes 5 and 6, cells co-treated with TPT (10 μM) and 43 (10 and 100 μM). (C) Hypothetical binding mode of 38 in the ternary TOP1-DNA-drug cleavage complex (PDB 1K4T). 38 is shown as yellow carbon atoms ball and stick representation. Key residues are highlighted in green ball and stick representation. All distances are measured from heavy atom to heavy atom.
ICE assays were also conducted to evaluate whether the most potent TDP1 inhibitor 43 induced the cellular TDP1ccs. As shown in Figure 4B, cellular TDP1ccs significantly enhanced after being treated with high concentration of 43 (100 μM) in MCF-7 cells, but not with TPT (10 μM) and low concentration of 43 (10 μM). After co-treated with TPT and 43 (both at 10 μM and 100 μM), the quantity of cellular TDP1cc significantly enhanced.
These results indicate that the novel benzophenanthridinone TOP1 inhibitors can induce cellular TOP1cc formation in MCF-7 cells, as well as induce cellular TDP1cc formation.
Molecular Modeling
To view the molecular binding mode of novel benzophenanthridinone TOP1 inhibitors within the TOP1cc, the most potent inhibitor 38 (+++) was selected for molecular modeling studies. A hypothetical binding model was built by using in silico docking from the X-ray crystal of the TOP1-DNA-ligand ternary complex (PDB 1K4T) according to our reported method[37,46]. The inhibitor 38 was energy minimized and docked into the binding model. As shown in Figure 4C, the benzophenanthridinone scaffold of 38 intercalates in the DNA break made by TOP1 and readily stacks with the base pairs flanking the DNA cleavage site. Compared to 4[37], the benzophenanthridinone scaffold of 38 is flipped within the TOP1-DNA covalent complex. The A- and B-ring of 38 stack with the scissile stand bases (G and T), while C- and D-ring stack with the noncleaved strand bases (C and A). The 11-side chain extents to the major groove of DNA. A hydrogen bond (2.3 Å) was observed between the lactam oxygen atom and Arg364, implying the importance of the lactam fragment in TOP1 inhibition. Also, two hydrogen bonds between the pyrrolidine nitrogen atom of the side chain and Asn352 (3.3 Å), and between an oxygen atom in the dioxole ring and Glu356 (2.8 Å) might contribute to the trapping of TOP1cc.
Cytotoxicity Assays
The cytotoxicity of the synthesized compounds was tested by MTT assay against two human cancer cell lines, colon cancer (HCT-116) and breast cancer (MCF-7). The cells were treated in a five-dose assay for 72 hours at concentrations ranging from 0.01 to 100 μM. The GI50 values, expressed as the concentrations corresponding to 50% cell growth inhibition, are summarized in Tables 1 and 2.
For 12-substituted benzophenanthridinone derivatives (Table 1), seven analogues 12, 14, 17-20, and 22 displayed significant cytotoxicity against both HCT-116 and MCF-7 cells with GI50 values at micromolar concentrations. The most potent TOP1 inhibitor 12 (+++) showed the highest cytotoxicity for HCT-116 cells (GI50 = 1.3 ± 0.12 µM), as well as 19 (++) for MCF-7 cells (GI50 = 0.93 ± 0.07 µM). For 11-substituted benzophenanthridinone derivatives (Table 2), eight derivatives 35–39, 41, 43, and 44 displayed good cytotoxicity against both HCT-116 and MCF-7 cells with GI50 values at micromolar concentrations. The dual inhibitor 38 with the highest TOP1 inhibition (+++) and low TDP1 inhibition (37–111 μM) showed the highest cytotoxicity for HCT-116 (GI50 = 0.25 ± 0.02 µM), and good cytotoxicity for MCF-7 (GI50 = 6.4 ± 1.0 µM), as well as 43 with the most TDP1 inhibition (10 ± 3.8 µM) and low TOP1 inhibition (+) showed high cytotoxicity for both HCT-116 (GI50 = 0.29 ± 0.19 µM) and MCF-7 (GI50 = 2.2 ± 1.2 µM). Although 35 had no TOP1 inhibition, it showed the highest cytotoxicity for MCF-7 (GI50 = 1.2 ± 0.10 µM) and high cytotoxicity for HCT-116 (GI50 = 0.55 ± 0.17 µM).
Synergistic Effects of 35 and 43 with TPT
In our previous study, the benzophenanthridinone dual TOP1 and/or TDP1 inhibitors showed synergistic effect with TPT in MCF-7 cells[46]. To evaluate the potential synergy of the novel inhibitors in the combination with TPT, a dual TOP1 and TDP1 inhibitor 43 and a selective TDP1 inhibitor 35 were selected for the synergistic test in MCF-7 and MCF-7/TDP1 cells (overexpressing TDP1) using MTT assay. As shown in Figure 5A, after being co-incubated for 96 hours, 43 (1 μM) showed synergistic effect with TPT in MCF-7 cells, and its combination index (CI) values fell in the range of 0.18–0.77 (Figure 5B). Although 35 had weaker TDP1 inhibition then 43, 35 showed a stronger synergistic effect in MCF-7 cells, and its CI values fell in the range of 0.02–0.69, and two doses showed strong synergy (CI < 0.3) and one dose showed very strong synergy (CI < 0.1). In MCF-7/TDP1 cells, both 43 and 35 showed more synergistic effect with TPT than that in MCF-7 cells (Figure 5C). All their CI values fell in 0.003–0.51 (Figure 5D), and two doses of 43 and one dose of 35 showed strong synergy, and two doses of both 43 and 35 showed very strong synergy. These results indicated that the novel benzophenanthridinone inhibitors had synergistic effect with TPT in both MCF-7 and MCF-7/TDP1 cells, and the TDP1 inhibitor with higher potency showed the greater synergistic effect in MCF-7/TDP1 cells.
Figure 5.

The synergistic effects and the combination index (CI) of 35 or 43 with TPT in the MCF-7 (A, B) and MCF-7/TDP1 (C, D) cells. The cells were co-incubated with drugs for 96 h.
DNA Damage Induced by Combined Treatment of 43 and TPT
To evaluate the ability of 43 to enhance TOP1 inhibitor-induced DNA damage, an immunofluorescence assay was conducted to test the γH2AX foci formed in MCF-7 cells after being treated by drugs. As shown in Figure 6A, TPT could obviously induce γH2AX foci formation. And 43 alone had a weak ability to induce DNA damage, about one fifth of that of TPT at 1 μM concentration (Figure 6B), possibly due to its weak TOP1 inhibitory potency (+). However, 43 could increase TPT-induced DNA damage. Compared with TPT treated alone, about 1.9-fold and 2.4-fold γH2AX foci formed after co-treatment with TPT and 43 at 1 and 2 μM, respectively. These results indicated that 43 could effectively enhance the ability of TPT to induce DNA damage in MCF-7 cells, possibly due to its TDP1 inhibitory potency.
Figure 6.

(A) Histone γH2AX foci (red) induced by 43 and TPT alone or combined in MCF-7 cells. The cells were incubated with drugs for 5 h at the indicated concentrations. DNA was stained with DAPI (blue). (B) Quantification of the number of γH2AX foci on the figure (A). P < 0.001 (***) and P < 0.0001 (****). Every experiment was repeated at least three times independently. (C) Flow cytometry histograms of apoptosis in MCF-7 cells induced by 43 at 1, 2, and 4 μM, respectively.
Cancer Cell Apoptosis Induced by 43
Although the dual TOP1 and TDP1 inhibitor 43 exhibited only weak TOP1 inhibitory potency (+), it showed high cytotoxicity against both HCT-116 and MCF-7 cells. To assess the induction of apoptosis by 43, flow cytometry assays were conducted in MCF-7 cells. After being incubated with 43 for 24 hours, the apoptotic MCF-7 cells, especially late apoptotic cells significantly increased in a dose-dependent manner (Figure 6C). At 4 µM concentration, 43 induced approximately late apoptosis in 28.58% cells.
Conclusion
In summary, a series of 11- and 12-substituted benzophenanthridinone derivatives was synthesized and evaluated for TOP1 and TDP1 inhibition against purified TOP1 and TDP1 and in cells, and cytotoxicity against two human cancer cell lines, colon carcinoma HCT116 and breast cancer MCF-7. The TOP1-mediated DNA cleavage assays indicate that the aminoalkoxy displacement at both 11- and 12-position is favorable for TOP1 inhibition. Two derivatives 12 and 38 showed strong TOP1 inhibition (+++), and four compounds 17, 19, 20, and 40 showed moderate potency (++). Furthermore, we demonstrate that the novel TOP1 inhibitor 38 induces TOP1ccs in MCF-7 cells, as detected through ICE assays. TDP1-based gel assays indicate that a longer aminoalkoxy side chain at the 11-position is favorable for TDP1 inhibition. Four compounds 35, 37, 39, and 43 showed TDP1 inhibitory potency with IC50 values ranging from 10 to 18 µM. Three of them 37, 39, and 43 also showed weak TOP1 inhibition (+). And the most potent TOP1 inhibitor 38 (+++) also showed weak TDP1 inhibition (37–111 µM). MTT assay indicated that fifteen compounds 12, 14, 17-20, 23, 35–39, 41, 43, and 44 exhibit high cytotoxicity against both HCT-116 and MCF-7 cells with GI50 values at micromolar concentrations. The dual TOP1 and TDP1 inhibitor 38 showed the highest cytotoxicity for HCT-116 (0.25 ± 0.02 µM), and compound 19 showed the highest cytotoxicity for MCF-7 (0.93 ± 0.07 µM). The combination treatment indicated that both the dual TOP1 and TDP1 inhibitor 43 and the selective TDP1 inhibitor 35 showed strong synergistic cytotoxicity in combination with TPT in both MCF-7 and MCF-7/TDP1 cells. The most potent TDP1 inhibitor 43 was found to induce cellular TDP1ccs, and enhanced TPT-induced DNA damage in MCF-7 cells, resulting in apoptosis. Our previous SAR analysis indicated that the 5-aminoalkyl substituent can increase the dual TOP1 and TDP1 inhibition of benzophenanthridinone derivatives[37,46,75]. Therefore, modifications of benzophenanthridinone derivatives at 5,11-positions fare warranted for the discovery of dual TOP1 and TDP1 inhibitors.
Experimental Section
General experiments
All chemical reagents for synthesis were purchased from local commercial suppliers and were used without further purification unless otherwise indicated. The key intermediates 9 and 30 were prepared in our lab according to the reported method[76,79]. Chemical reaction courses were monitored by silica gel GF254 thin layer chromatography. Melting points were determined in open capillary tubes on a MPA100 Optimelt Automated Melting Point System without being corrected. Nuclear magnetic resonance spectra were recorded on a Bruker AVANCE III 400 MHz or 500 MHz spectrometer using tetramethylsilane as an internal reference. The high-resolution mass spectra (HRMS) were analyzed on a SHIMADZU LCMS-IT-TOF mass spectrometer. All compounds tested for biological activities were analyzed by HPLC and their purities were more than 95%. The analysis condition is: detection at 220 nm, 1.0 mL/min flowrate, a linear gradient of 50%−15% PBS buffer (pH = 3) and 50%−85% MeOH in 30 min.
2,3-Dimethoxy-12-methyl-13-oxo-12,13-dihydro-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-6-yl acetate (10)
To a solution of 9 (260 mg, 0.67 mmol) in freshly acetone (10 mL), 2-methyl-2-butene (375 mg, 5.4 mmol), NaClO2 (182 mg, 2.01 mmol) and NaH2PO4·2H2O (241 mg, 2.01 mmol) were added. The reaction solution was stirred at room temperature for 12 h. After completion of the reaction, the reaction solution was adjusted to pH < 3 with hydrochloride acid (1N). The solution was extracted with ethyl acetate (20 mL x 3). The combined organic layer was concentrated under reduced pressure to give a crude solid, which was used immediately for next synthesis without further purification.
To a solution of the obtained solid (~180 mg, ~0.45 mmol) in acetic anhydride (5 mL), AcOK (89 mg, 0.90 mmol) was added. The suspension was stirred at 140 °C for 3 h. After completion of the reaction, the suspension was cooled to room temperature and added with water (20 mL). Filtration gave a white solid, which was purified by column chromatography on silica gel using methanol/dichloromethane (1/200, v/v) as eluent to give a white solid 10, yield 76%. The NMR spectral data are similar to that reported in the literature[84]. 1H NMR (400 MHz, CDCl3) δ 7.92 (s, 1H), 7.76 (s, 1H), 7.66 (s, 1H), 7.45 (s, 1H), 7.19 (s, 1H), 6.12 (s, 2H), 4.09 (s, 3H), 4.06 (s, 3H), 3.97 (s, 3H), 2.52 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 169.9, 164.3, 153.5, 150.0, 148.2, 147.5, 142.4, 134.1, 128.5, 124.9, 122.3, 119.3, 116.4, 111.0, 108.7, 103.1, 103.0, 101.9, 98.7, 56.4, 56.3, 41.5, 21.2. HRMS (ESI) m/z: 422.1251 [M + H]+, calcd for C23H20NO7 422.1240.
6-Hydroxy-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (11)
To a solution of 10 (90 mg, 0.21 mmol) in methanol (30 mL), an aqueous KOH solution (1N, 2 mL) was added dropwise. The yellow reaction solution was stirred at 80 °C for 3 h. After completion of the reaction, the reaction solution was cooled to room temperature, and water (10 mL) was added, and then adjusted to pH < 3 by addition of hydrochloric acid (2N). The reaction solution was extracted with ethyl acetate (25 mL x 3). The combined organic layer was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using methanol/dichloromethane (1/200, v/v) as eluent to give a white solid 11, yield 85%, mp = 185.2–187.9 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.17 (s, 1H), 7.74 (s, 1H), 7.71 (s, 1H), 7.60 (s, 1H), 7.54 (s, 1H), 7.51 (s, 1H), 6.18 (s, 2H), 4.01 (s, 3H), 3.91 (s, 3H), 3.82 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 162.6, 153.1, 149.3, 148.9, 147.0, 146.8, 128.8, 128.2, 122.9, 121.7, 118.5, 116.3, 108.2, 103.4, 102.6, 101.6, 100.6, 99.1, 55.8, 55.7, 40.8. HRMS (ESI) m/z: 380.1138 [M + H]+, calcd for C21H18NO6 380.1134.
Methyl 2-(6-(6,7-dimethoxy-1-oxo-1,2-dihydroisoquinolin-3-yl)benzo[d][1,3]dioxol-5-yl)acetate (31)
To a solution of 30 (100 mg, 0.25 mmol) in MeOH (30 mL), NaBr (1.55g, 15.0 mmol) and p-toluenesulfonic acid monohydrate (571mg, 3.0 mmol) were added. The solution was stirred at 80 °C for 12 h. And then, the reaction solution was cooled to room temperature and added with NaHCO3 saturated solution (20 mL). The reaction mixture was stirred vigorously for 15 min, and added with ethyl acetate (30 mL). The organic layer was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using methanol/dichloromethane (1/5, V/V) as eluent to give a yellow solid 31, yield 90%. 1H NMR (400 MHz, CDCl3) δ 9.31 (s, 1H), 7.78 (s, 1H), 6.89 (s, 2H), 6.80 (s, 1H), 6.37 (s, 1H), 6.03 (s, 2H), 4.01 (s, 3H), 3.99 (s, 3H), 3.75 (s, 2H), 3.57 (s, 3H). ESI-MS m/z: 398.1 [M + H]+.
Methyl 2-(6-(6,7-dimethoxy-2-methyl-1-oxo-1,2-dihydroisoquinolin-3-yl)benzo[d][1,3]dioxol-5-yl)acetate (32)
Under nitrogen gas, to a solution of 31 (450mg, 1.13 mmol) in dry DMF in seal tube, iodomethane (321mg, 2.26 mmol) and K2CO3 (781mg, 5.65 mmol) were added sequentially. The reaction mixture was stirred at 80 °C for 5 h. After completion of the reaction, water (10 mL) and ethyl acetate (20 mL) were added to the reaction solution. The organic layer was washed with saturated saline (20 mL x 3). The organic layer was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel using ethyl acetate/petroleum ether (1/3, V/V) as eluent to give a yellow solid 32, yield 89%. 1H NMR (400 MHz, CDCl3) δ 7.82 (s, 1H), 6.87 (s, 1H), 6.82 (s, 1H), 6.73 (s, 1H), 6.32 (s, 1H), 6.05 (s, 2H), 4.03 (s, 3H), 3.98 (s, 3H), 3.58 (s, 3H), 3.52–3.45 (m, 2H), 3.30 (s, 3H). ESI-MS m/z: 412.1 [M + H]+.
5-Hydroxy-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (33)
The solution of 32 (140 mg, 0.36 mmol) and K2CO3 (152 mg, 1.08 mmol) in methanol (30 mL) and water (1 mL) was stirred at 80 °C. After completion of the reaction, the solution was cooled to room temperature and added with water (20 mL), and then adjusted with hydrochloride acid (2N) to pH < 3. The reaction solution was extracted with ethyl acetate (25 mL x 3). The combined organic layer was concentrated under reduced pressure to give a white rude solid.
The solution of the obtained white solid (~143 mg, ~0.36 mmol) and AcOK (71 mg, 0.72 mmol) in acetic anhydride (5 mL) was stirred at 140 °C for 3 h. After completion of the reaction, the solution was cooled to room temperature and added with water (20 mL). The suspension was filtrated to give a white solid. The white solid was dissolved in methanol (20 mL) and water (1 mL). K2CO3 (276 mg, 2.0 mmol) was added to the solution. The yellow solution was stirred at 80 °C. After completion of the reaction, the reaction solution was cooled to room temperature and added with water (20 mL), and then extracted with ethyl acetate (25 mL x 3). The combined organic layer was concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using ethyl acetate/petroleum ether (1/3, v/v) as eluent to give a yellow solid 33, yield 52%, mp = 159.2–161.3 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.65 (s, 1H), 9.02 (s, 1H), 7.78 (s, 1H), 7.59 (s, 1H), 7.18 (s, 1H), 7.07 (s, 1H), 6.11 (s, 2H), 3.92 (s, 3H), 3.91 (s, 3H), 3.80 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 163.4, 152.6, 152.4, 148.9, 148.1, 144.9, 138.1, 131.9, 128.9, 119.0, 114.9, 109.6, 109.2, 108.3, 107.0, 103.0, 102.8, 101.7, 55.9, 55.9, 42.4. HRMS (ESI) m/z: 380.1125 [M + H]+, calcd for C21H18NO6 380.1129.
General procedure for the synthesis of compounds 12–23 and 34–45.
Under nitrogen gas, to the solution of 11 (for 12-23) or 33 (for 34-45, 1 mmol) in dry DMF (3 mL), the corresponding chloride material (1.5 mmol), K2CO3 (3 mmol) and KI (0.1 mmol) were added sequentially. The reaction mixture was stirred at 80 °C for 5 h. After completion of the reaction, H2O (10 mL) and ethyl acetate (20 mL) were added. The organic layer was washed with saturated NaCl solution (20 mL x 3), and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel to give the target compounds.
2,3-Dimethoxy-12-methyl-6-(2-(methylamino)ethoxy)-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (12)
White solid, yield 68%, mp = 140.2–140.9 °C. 1H NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 7.97 (s, 1H), 7.84 (s, 1H), 7.79 (s, 1H), 7.75 (s, 1H), 7.67 (s, 1H), 6.22 (s, 2H), 4.60 (t, J= 6.0 Hz, 2H), 4.07 (s, 3H), 3.92 (s, 3H), 3.83 (s, 3H), 3.54 (t, J= 6.0 Hz, 2H), 2.75 (s, 3H). 13C NMR (125 MHz, DMSO-d6) δ 162.7, 153.3, 149.5, 147.3, 147.2, 130.3, 128.4, 122.9, 121.6, 118.6, 115.9, 108.1, 104.4, 102.7, 101.9, 99.9, 98.9, 64.3, 56.5, 55.7, 47.6, 40.9, 33.0. HRMS (ESI) m/z: 437.1732 [M + H]+, calcd for C24H25N2O6 437.1713.
2,3-Dimethoxy-12-methyl-6-(3-(methylamino)propoxy)-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (13)
White solid, yield 40%, mp = 144.2–145.7 °C. 1H NMR (500 MHz, CDCl3) δ 7.94 (s, 1H), 7.65 (s, 1H), 7.60 (s, 1H), 7.49 (s, 1H), 7.30 (s, 1H), 6.11 (s, 2H), 4.33 (t, J = 6.0 Hz, 2H), 4.13 (s, 3H), 4.06 (s, 3H), 3.95 (s, 3H), 2.69 (t, J = 6.0 Hz, 2H), 2.38 (s, 6H), 2.21–2.17 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 164.3, 153.5, 150.8, 149.8, 147.4, 147.3, 130.3, 129.1, 123.8, 122.3, 119.3, 116.9, 108.7, 103.0, 102.7, 101.7, 99.7, 97.6, 66.7, 56.6, 56.3, 56.2, 45.1, 41.4, 27.1. HRMS (ESI) m/z: 451.1781 [M + H]+, calcd for C25H27N2O6 451.1791.
6-(2-(Dimethylamino)ethoxy)-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (14)
White solid, yield 60%, mp = 139.2–139.7 °C. 1H NMR (500 MHz, CDCl3) δ 7.94 (s, 1H), 7.67 (s, 1H), 7.60 (s, 1H), 7.48 (s, 1H), 7.33 (s, 1H), 6.11 (s, 2H), 4.39 (t, J = 6.0 Hz, 2H), 4.11 (s, 3H), 4.05 (s, 3H), 3.96 (s, 3H), 2.96 (t, J = 6.0 Hz, 2H), 2.46 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 164.2, 153.5, 150.8, 149.8, 147.5, 147.5, 130.6, 129.0, 124.0, 122.4, 119.5, 116.7, 108.9, 102.9, 102.7, 101.7, 100.0, 97.9, 67.4, 58.5, 56.3, 46.3, 41.4. HRMS (ESI) m/z: 451.1913 [M + H]+, calcd for C25H27N2O6 451.1864.
6-(3-(Dimethylamino)propoxy)-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (15)
White solid, yield 40%, mp = 144.2–145.7 °C. 1H NMR (500 MHz, CDCl3) δ 7.94 (s, 1H), 7.65 (s, 1H), 7.60 (s, 1H), 7.49 (s, 1H), 7.30 (s, 1H), 6.11 (s, 2H), 4.33 (t, J = 6.0 Hz, 2H), 4.13 (s, 3H), 4.06 (s, 3H), 3.95 (s, 3H), 2.69 (t, J = 6.0 Hz, 2H), 2.38 (s, 6H), 2.21–2.17 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 164.3, 153.5, 150.8, 149.8, 147.4, 147.4, 130.3, 129.1, 123.8, 122.3, 119.3, 116.9, 108.7, 103.0, 102.7, 101.7, 99.7, 97.6, 66.7, 56.6, 56.3, 56.2, 45.1, 41.4, 27.1. HRMS (ESI) m/z: 465.2070 [M + H]+, calcd for C26H29N2O6 465.2020.
2,3-Dimethoxy-12-methyl-6-(2-(piperidin-1-yl)ethoxy)-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (16)
White solid, yield 41%, mp = 175.2–177.7 °C. 1H NMR (400 MHz, CDCl3) δ 7.94 (s, 1H), 7.66 (s, 1H), 7.61 (s, 1H), 7.49 (s, 1H), 7.33 (s, 1H), 6.11 (s, 2H), 4.45 (t, J = 6.0 Hz, 2H), 4.12 (s, 3H), 4.06 (s, 3H), 3.95 (s, 3H), 3.02 (t, J = 6.0 Hz, 2H), 2.72–2.64 (m, 4H), 1.72–1.66 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 164.2, 153.5, 150.7, 149.8, 147.5, 147.4, 130.7, 128.9, 123.9, 122.5, 119.5, 116.8, 108.9, 102.9, 102.8, 101.7, 99.9, 98.1, 67.1, 58.2, 56.5, 56.4, 55.3, 41.4, 25.9, 24.1. HRMS (ESI) m/z: 491.2211 [M + H]+, calcd for C28H31N2O6 491.2177.
2,3-Dimethoxy-12-methyl-6-(3-(piperidin-1-yl)propoxy)-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (17)
White solid, yield 40%, mp = 153.2–155.1°C. 1H NMR (400 MHz, CDCl3) δ 7.95 (s, 1H), 7.67 (s, 1H), 7.61 (s, 1H), 7.49 (s, 1H), 7.29 (s, 1H), 6.11 (s, 2H), 4.31 (t, J = 6.0 Hz, 2H), 4.12 (s, 3H), 4.06 (s, 3H), 3.95 (s, 3H), 2.67–2.62 (m, 2H), 2.53–2.43 (m, 4H), 2.23–2.14 (m, 2H), 1.60–1.42 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 164.1, 153.4, 150.9, 149.7, 147.4, 147.4, 130.4, 128.9, 124.0, 122.4, 119.5, 116.7, 108.9, 102.9, 102.7, 101.6, 99.9, 97.7, 67.3, 56.4, 56.3, 54.8, 41.4, 27.0, 26.0, 24.4. HRMS (ESI) m/z: 505.2393 [M + H]+, calcd for C29H33N2O6 505.2333.
2,3-Dimethoxy-12-methyl-6-(2-(4-methylpiperazin-1-yl)ethoxy)-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (18)
White solid, yield 50%, mp = 152.7–153.4 °C. 1H NMR (400 MHz, CDCl3) δ 7.94 (s, 1H), 7.66 (s, 1H), 7.61 (s, 1H), 7.47 (s, 1H), 7.30 (s, 1H), 6.11 (s, 2H), 4.41 (t, J = 6.0 Hz, 2H), 4.12 (s, 3H), 4.06 (s, 3H), 3.95 (s, 3H), 3.03 (t, J = 6.0 Hz, 2H), 2.81–2.66 (m, 4H), 2.60–2.45 (m, 4H), 2.31 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 164.1, 153.5, 150.7, 149.7, 147.5, 147.5, 130.7, 128.9, 124.0, 122.4, 119.5, 116.7, 108.9, 102.8, 102.7, 101.7, 99.9, 98.0, 67.2, 57.4, 56.3, 55.2, 53.8, 46.1, 41.4. HRMS (ESI) m/z: 506.2297 [M + H]+, calcd for C28H32N3O6 506.2286.
2,3-Dimethoxy-12-methyl-6-(3-(4-methylpiperazin-1-yl)propoxy)-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (19)
White solid, yield 39%, mp = 144.3–145.7 °C. 1H NMR (400 MHz, CDCl3) δ 7.94 (s, 1H), 7.66 (s, 1H), 7.60 (s, 1H), 7.48 (s, 1H), 7.28 (s, 1H), 6.11 (s, 2H), 4.31 (t, J = 6.0 Hz, 2H), 4.12 (s, 3H), 4.06 (s, 3H), 3.95 (s, 3H), 2.69 (t, J = 6.0 Hz, 2H), 2.66–2.54 (m, 4H), 2.55–2.41 (m, 4H), 2.31 (s, 3H), 2.21–2.14 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 164.1, 153.4, 150.8, 149.7, 147.4, 147.4, 130.4, 128.9, 123.9, 122.3, 119.6, 116.6, 108.9, 102.9, 102.7, 101.7, 99.9, 97.7, 67.1, 56.3, 55.5, 55.0, 53.2, 46.0, 41.4, 27.0. HRMS (ESI) m/z: 520.2482 [M + H]+, calcd for C29H34N3O6 520.2442.
6-(2-(1H-Imidazol-1-yl)ethoxy)-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (20)
White solid, yield 41%, mp = 184.2–185.7 °C. 1H NMR (400 MHz, CDCl3) δ 7.93 (s, 1H), 7.73 (s, 1H), 7.60 (s, 1H), 7.50 (s, 1H), 7.39 (s, 1H), 7.18 (s, 1H), 7.16–7.12 (m, 2H), 6.12 (s, 2H), 4.57–4.49 (m, 4H), 4.11 (s, 3H), 4.05 (s, 3H), 3.94 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 164.1, 153.5, 150.0, 149.9, 147.7, 147.7, 137.7, 131.3, 129.9, 128.7, 123.7, 122.5, 119.6, 116.5, 108.9, 102.9, 102.8, 101.9, 99.4, 98.7, 68.7, 56.4, 56.4, 46.8, 41.4. HRMS (ESI) m/z: 474.1696 [M + H]+, calcd for C26H24N3O6 474.1660.
6-(3-(1H-Imidazol-1-yl)propoxy)-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (21)
White solid, yield 67%, mp = 159.2–160.7 °C. 1H NMR (400 MHz, CDCl3) δ 7.94 (s, 1H), 7.63 (s, 2H), 7.53 (s, 1H), 7.41 (s, 1H), 7.21 (s, 1H), 7.08 (s, 1H), 6.97 (s, 1H), 6.13 (s, 2H), 4.36 (t, J = 6.4 Hz, 2H), 4.20 (t, J = 5.1 Hz, 2H), 4.10 (s, 3H), 4.06 (s, 3H), 3.96 (s, 3H), 2.48–2.39 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 164.1, 153.5, 150.2, 149.8, 147.6, 147.6, 137.5, 130.8, 129.8, 128.8, 123.6, 122.5, 119.6, 119.2, 116.6, 108.9, 102.9, 102.8, 101.8, 99.4, 97.8, 64.5, 56.4, 43.7, 41.5, 30.9. HRMS (ESI) m/z: 488.1866 [M + H]+, calcd for C27H26N3O6 488.1816.
6-(2-((2-(Dimethylamino)ethyl)amino)ethoxy)-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (22)
White solid, yield 41%, mp = 227.6–229.7 °C. 1H NMR (400 MHz, CDCl3) δ 7.93 (s, 1H), 7.65 (s, 1H), 7.59 (s, 1H), 7.46 (s, 1H), 7.31 (s, 1H), 6.10 (s, 2H), 4.40–4.34 (m, 2H), 4.11 (s, 3H), 4.05 (s, 3H), 3.94 (s, 3H), 3.25 −3.19 (m, 2H), 2.90–2.84 (m, 2H), 2.54–2.48 (m, 2H), 2.28 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 164.1, 153.5, 150.6, 149.7, 147.4, 130.6, 128.9, 123.9, 122.4, 119.5, 116.6, 108.8, 102.8, 102.8, 101.7, 99.8, 98.1, 68.1, 58.5, 56.3, 48.8, 46.9, 45.3, 41.4. HRMS (ESI) m/z: 494.2342 [M + H]+, calcd for C27H32N3O6 494.2286.
Ethyl 2-((2,3-dimethoxy-12-methyl-13-oxo-12,13-dihydro-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-6-yl)oxy)acetate (23)
White solid, yield 52%, mp = 151.2–152.1 °C. 1H NMR (500 MHz, CDCl3) δ 7.93 (s, 1H), 7.75 (s, 1H), 7.61 (s, 1H), 7.42 (s, 1H), 7.26 (s, 1H), 6.12 (s, 2H), 4.90 (s, 2H), 4.33 (q, J = 7.0 Hz, 2H), 4.11 (s, 3H), 4.06 (s, 3H), 3.95 (s, 3H), 1.33 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 169.0, 164.1, 153.5, 150.0, 149.9, 147.7, 131.4, 128.7, 124.0, 122.6, 119.6, 116.4, 108.9, 102.7, 101.8, 100.1, 98.9, 66.9, 61.7, 56.4, 56.3, 41.4, 14.4. HRMS (ESI) m/z: 466.1522 [M + H]+, calcd for C25H24NO8 466.1496.
5-(2-(Dimethylamino)ethoxy)-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (34)
Yellow solid, yield 49%, mp = 190.8–191.9 °C. 1H NMR (400 MHz, CDCl3) δ 8.88 (s, 1H), 7.99 (s, 1H), 7.47 (s, 1H), 7.06 (s, 1H), 7.01 (s, 1H), 6.07 (s, 2H), 4.35 (t, J = 6.0 Hz, 2H), 4.10 (s, 3H), 4.07 (s, 3H), 3.92 (s, 3H), 2.97 (t, J = 6.0 Hz, 2H), 2.37 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 164.3, 153.8, 152.5, 148.9, 148.0, 145.3, 138.1, 131.1, 128.6, 119.4, 116.2, 110.5, 109.4, 108.3, 103.6, 103.4, 102.8, 101.3, 66.7, 58.4, 56.1, 45.7, 42.2. HRMS (ESI) m/z: 451.1859 [M + H]+, calcd for C25H27N2O6 451.1864.
5-(3-(Dimethylamino)propoxy)-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (35)
Yellow solid, yield 51%, mp = 148.9–150.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 7.99 (s, 1H), 7.47 (s, 1H), 7.05 (s, 1H), 7.02 (s, 1H), 6.06 (s, 2H), 4.31 (t, J = 6.7 Hz, 2H), 4.06 (s, 3H), 4.05 (s, 3H), 3.91 (s, 3H), 2.59 (t, J = 7.2 Hz, 2H), 2.37–2.15 (m, 8H). 13C NMR (100 MHz, CDCl3) δ 164.2, 153.8, 152.4, 148.9, 148.0, 145.3, 138.1, 131.3, 128.6, 119.6, 116.1, 110.4, 109.2, 108.4, 103.7, 103.4, 102.8, 101.3, 67.4, 56.4, 56.1, 45.4, 42.2, 29.7, 27.8. HRMS (ESI) m/z: 465.2020 [M + H]+, calcd for C26H29N2O6 465.2020.
5-(2-(Diethylamino)ethoxy)-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (36)
Yellow solid, yield 67%, mp = 155.8–157.2 °C. 1H NMR (400 MHz, CDCl3) δ 8.73 (s, 1H), 7.91 (s, 1H), 7.40 (s, 1H), 7.00 (s, 1H), 6.96 (s, 1H), 6.00 (s, 2H), 4.33 (t, J = 6.7 Hz, 2H), 4.00 (s, 3H), 3.99 (s, 3H), 3.85 (s, 3H), 3.10 (t, J = 5.8 Hz, 2H), 2.67 (q, J = 7.1 Hz, 4H), 1.05 (t, J = 7.1 Hz, 6H). 13C NMR (100 MHz, CDCl3) δ 164.2, 152.4, 148.9, 148.1, 145.4, 138.2, 131.2, 128.5, 119.6, 116.2, 110.4, 109.2, 108.4, 103.8, 103.5, 102.8, 101.4, 56.1, 52.2, 47.7, 42.2, 11.6. HRMS (ESI) m/z: 479.2170 [M + H]+, calcd for C28H31N2O6 479.2176.
5-(3-(Diethylamino)propoxy)-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (37)
Yellow solid, yield 71%, mp = 237.6–239.6 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.76 (s, 1H), 7.80 (s, 1H), 7.64 (s, 1H), 7.31 (s, 1H), 7.27 (s, 1H), 4.33 (t, J = 6.6 Hz, 2H), 3.96 (s, 3H), 3.93 (s, 3H), 3.80 (s, 3H), 2.52–2.50 (m, 6H), 2.36–2.21 (m, 2H), 1.12 (t, J = 7.0 Hz, 6H). 13C NMR (100 MHz, DMSO-d6) δ 163.1, 153.4, 152.7, 149.0, 148.3, 145.5, 138.1, 131.6, 128.2, 119.2, 115.9, 109.5, 108.5, 104.3, 103.7, 103.2, 101.9, 56.1, 56.0, 47.0, 42.2. HRMS (ESI) m/z: 493.2324 [M + H]+, calcd for C28H33N2O6 493.2333.
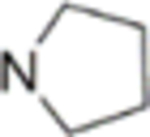
2,3-Dimethoxy-12-methyl-5-(2-(pyrrolidin-1-yl)ethoxy)-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (38)
Yellow solid, yield 66%, mp = 150.1–150.9 °C. 1H NMR (400 MHz, CDCl3) δ 8.87 (s, 1H), 7.97 (s, 1H), 7.44 (s, 1H), 7.04 (s, 1H), 6.99 (s, 1H), 6.06 (s, 2H), 4.38 (t, J = 6.2 Hz, 2H), 4.07 (s, 3H), 4.06 (s, 3H), 3.90 (s, 3H), 3.14 (t, J = 6.2 Hz, 2H), 2.66 (t, J = 6.1 Hz, 4H), 1.82 (t, J = 6.1 Hz, 4H). 13C NMR (100 MHz, CDCl3) δ 164.2, 153.8, 152.5, 148.8, 148.0, 145.3, 138.1, 131.2, 128.6, 119.4, 116.1, 109.4, 108.3, 103.6, 102.8, 101.3, 67.7, 56.1, 56.1, 55.2, 54.5, 42.2, 23.5. HRMS (ESI) m/z: 477.1994 [M + H]+, calcd for C27H29N2O6 477.2020.
2,3-Dimethoxy-12-methyl-5-(3-(pyrrolidin-1-yl)propoxy)-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (39)
Yellow solid, yield 78%, mp = 173.8–175.3 °C. 1H NMR (400 MHz, CDCl3) δ 8.85 (s, 1H), 8.00 (s, 1H), 7.48 (s, 1H), 7.07 (s, 1H), 7.03 (s, 1H), 6.08 (s, 2H), 4.35 (t, J = 7.2 Hz, 2H), 4.08 (s, 3H), 4.06 (s, 3H), 3.93 (s, 3H), 2.81 (t, J = 7.2 Hz, 2H), 2.64 (t, J = 6.0 Hz, 4H), 2.32 (p, J = 7.0 Hz, 2H), 1.86 (t, J = 6.0 Hz, 4H). 13C NMR (100 MHz, CDCl3) δ 164.3, 153.9, 152.4, 148.8, 148.0, 145.2, 138.1, 131.3, 128.6, 119.5, 116.1, 110.4, 109.2, 108.4, 103.6, 103.4, 102.8, 101.3, 67.6, 56.1, 54.3, 53.2, 42.2, 29.1, 23.5. HRMS (ESI) m/z: 491.2163 [M + H]+, calcd for C28H31N2O6 491.2177.
2,3-Dimethoxy-12-methyl-5-(2-morpholinoethoxy)-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (40)
Yellow solid, yield 65%, mp = 182.7–183.5 °C. 1H NMR (500 MHz, CDCl3) δ 8.85 (s, 1H), 7.98 (s, 1H), 7.47 (s, 1H), 7.06 (s, 1H), 7.01 (s, 1H), 6.07 (s, 2H), 4.39 (t, J = 5.6 Hz, 2H), 4.08 (s, 3H), 4.06 (s, 3H), 3.91 (s, 3H), 3.73 (t, J = 4.7 Hz, 4H), 3.03 (t, J = 5.6 Hz, 2H), 2.59 (t, J = 4.7 Hz, 4H). 13C NMR (100 MHz, CDCl3) δ 164.2, 153.7, 152.5, 148.9, 148.1, 145.4, 138.2, 131.2, 128.5, 119.6, 116.3, 110.5, 109.4, 108.4, 103.8, 103.4, 102.8, 101.4, 66.8, 66.2, 57.8, 56.3, 56.1, 54.0, 42.2. HRMS (ESI) m/z: 493.1962 [M + H]+, calcd for C27H29N2O7 493.1969.
2,3-Dimethoxy-12-methyl-5-(3-morpholinopropoxy)-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (41)
Yellow solid, yield 86%, mp = 214.4–215.7 °C. 1H NMR (500 MHz, CDCl3) δ 8.85 (s, 1H), 7.99 (s, 1H), 7.47 (s, 1H), 7.07 (s, 1H), 7.01 (s, 1H), 6.07 (s, 2H), 4.32 (t, J = 6.5 Hz, 2H), 4.07 (s, 3H), 4.05 (s, 3H), 3.92 (s, 3H), 3.73 (t, J = 4.7 Hz, 4H), 2.64 (t, J = 7.1 Hz, 2H), 2.47 (t, J = 4.7 Hz, 4H), 2.23 (p, J = 6.7 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 164.3, 153.9, 152.4, 148.8, 148.0, 145.3, 139.3, 138.1, 131.3, 119.6, 116.1, 110.4, 109.1, 108.4, 103.6, 103.4, 102.8, 101.3, 67.3, 67.0, 56.1, 56.1, 55.6, 53.8, 42.2, 26.9. HRMS (ESI) m/z: 507.2123 [M + H]+, calcd for C28H31N2O7 507.2126.
5-(3-Hydroxypropoxy)-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (42)
Yellow solid, yield 64%, mp = 232.1–233.9 °C. 1H NMR (400 MHz, CDCl3) δ 8.82 (s, 1H), 7.98 (s, 1H), 7.46 (s, 1H), 7.05 (s, 1H), 7.02 (s, 1H), 6.07 (s, 2H), 4.41 (t, J = 6.1 Hz, 2H), 4.19–3.97 (m, 8H), 3.91 (s, 3H), 2.38–2.25 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 163.2, 153.8, 152.6, 149.0, 148.2, 145.4, 138.1, 131.8, 128.3, 119.2, 115.7, 109.4, 108.5, 104.1, 103.7, 103.1, 101.9, 66.5, 58.1, 55.9, 32.8. HRMS (ESI) m/z: 438.1551 [M + H]+, calcd for C24H24NO7 438.1547.
5-(3-((2-Hydroxyethyl)amino)propoxy)-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo [1,2-c]phenanthridin-13(12H)-one (43)
Yellow solid, yield 59%, mp = 124.3– 125.8 °C. 1H NMR (400 MHz, CDCl3) δ 8.82 (s, 1H), 7.97 (s, 1H), 7.44 (s, 1H), 7.03 (s, 1H), 6.97 (s, 1H), 6.06 (s, 2H), 4.32 (t, J = 6.4 Hz, 2H), 4.06 (s, 3H), 4.04 (s, 3H), 3.90 (s, 3H), 3.67 (t, J = 4.0 Hz, 2H), 2.95 (t, J = 6.8 Hz, 2H), 2.81 (t, J = 5.1 Hz, 2H), 2.22 (q, J = 6.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 164.2, 153.8, 152.4, 148.8, 148.0, 145.2, 138.0, 131.2, 128.6, 119.5, 116.1, 110.3, 109.1, 108.4, 103.5, 103.4, 102.8, 101.3, 67.3, 61.1, 56.1, 56.0, 51.3, 46.5, 42.2, 30.3, 29.7. HRMS (ESI) m/z: 481.1952 [M + H]+, calcd for C26H29N2O7 481.1969.
5-(3-(1H-imidazol-1-yl)propoxy)-2,3-dimethoxy-12-methyl-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (44)
Yellow solid, yield 62%, mp = 202.3–204.7 °C. 1H NMR (400 MHz, CDCl3) δ 8.73 (s, 1H), 8.00 (s, 1H), 7.46 (s, 2H), 7.02 (s, 2H), 6.90 (s, 2H), 6.07 (s, 2H), 4.29 (t, J = 6.6 Hz, 2H), 4.19 (t, J = 6.0 Hz, 2H), 4.07 (s, 3H), 4.01 (s, 3H), 3.92 (s, 3H), 2.50 (p, J = 6.3 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 164.1, 153.2, 152.4, 149.0, 148.2, 145.5, 138.3, 131.1, 128.3, 119.7, 116.4, 110.2, 108.8, 108.7, 103.9, 103.5, 102.8, 101.4, 65.3, 56.1, 56.0, 42.2, 30.9. HRMS (ESI) m/z: 488.1813 [M + H]+, calcd for C27H26N3O6 488.1816.
2,3-Dimethoxy-12-methyl-5-(3-(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)propoxy)-[1,3]dioxolo[4’,5’:4,5]benzo[1,2-c]phenanthridin-13(12H)-one (45)
Yellow solid, yield 65%, mp = 176.1–177.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.80 (s, 1H), 7.98 (s, 1H), 7.47 (s, 1H), 7.36 (t, J = 8.0 Hz, 1H), 7.12 (d, J = 7.7 Hz, 1H), 7.07 (d, J = 6.3 Hz, 2H), 7.02 (d, J = 13.2 Hz, 2H), 6.07 (s, 2H), 4.44 (t, J = 6.2 Hz, 2H), 4.37 (t, J = 6.3 Hz, 2H), 4.05 (s, 6H), 3.91 (s, 3H), 3.61 (s, 4H), 3.15–3.13 (m, 4H), 2.43 (p, J = 6.3 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 164.2, 155.1, 153.7, 152.4, 151.2, 148.9, 148.1, 145.4, 138.2, 131.2, 129.7, 128.5, 119.6, 119.4, 116.7 (q, JC-F = 3.6 Hz), 116.2, 112.9 (q, JC-F = 3.8 Hz), 110.3, 108.9, 108.5, 103.6, 103.4, 102.8, 101.4, 66.2, 62.7, 56.1, 56.0, 48.8, 42.2, 29.7, 29.5. HRMS (ESI) m/z: 650.2472 [M + H]+, calcd for C35H34F3N3O6 650.2472.
TOP1-Mediated Cleavage Assay.
DNA cleavage assays were performed according to the previously reported method[82]. A 3′-[32P]-labeled 117-bp DNA oligonucleotide substrate (2 nM) was incubated with recombinant TOP1 in 10 µL of reaction buffer (10 mM Tris-HCl pH 7.5, 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 15 mg/mL BSA) at 25 °C for 20 min in the presence of various concentrations of test compounds. The reactions were terminated by adding SDS (0.5% final concentration), followed by the addition of two volumes of loading dye (80% formamide, 10 mM sodium hydroxide, 1 mM sodium EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue). Aliquots of each reaction mixture were subjected to 20% denaturing PAGE. Gels were dried and visualized by using a phosphoimager and Image Quant software (Molecular Dynamics). Cleavage sites are numbered to reflect actual sites on the 117-bp oligonucleotide.
TDP1 Inhibition Assay
TDP1 inhibition assays were conducted according to the reported method[43]. Briefly, a 5′-Cy5-labeled single-stranded DNA oligonucleotide substrate containing a 3′-phosphotyrosine (Cy5–14Y, 5´-GATCTAAAAGACTTpY-3´) was incubated at 1 nM with 40 pM recombinant human TDP1 in the absence or presence of inhibitor for 15 min at room temperature in buffer containing 50 mM Tris HCl, pH 7.5, 80 mM KCl, 2 mM EDTA, 1 mM DTT, 40 μg/mL BSA, and 0.01% Tween-20. Reactions were terminated by the addition of 1 volume of gel loading buffer [99.5% (v/v) formamide, 5 mM EDTA] and analyzed through a 20% denaturing PAGE. Gel images were scanned using a Typhoon FLA 9500 scanner (GE Healthcare), and densitometry analyses were performed using the ImageQuant software (GE Healthcare).
Cell Culture and MTT Assay
HCT116 cells were cultured on RPMI1640 medium, MCF-7 and MCF-7/TDP1 (overexpression TDP1 in MCF-7 cells) cells were cultured on DMEM medium at 37 °C in a humidified atmosphere with 5% CO2. All cells to be tested in the following assays had a passage number of 3–6. For the cytotoxicity measurements, the cancer cells were treated with the compounds (pre-dissolved in DMSO) at a five-dose assay ranging from 0.01 to 100 μM (0.001–10 μM for topotecan) concentration. After incubation for 72 h at 37 °C, MTT solution (20 μL, 2.5 mg/mL) in PBS (PBS without MTT as the blank) was fed to each well of the culture plate (containing 100 mL medium). After 4 h incubation, the formazan crystal formed in the well was dissolved with 100 µL of DMSO for optical density reading at 570 nm. The GI50 value was calculated by nonlinear regression analysis (GraphPad Prism).
For the drug combination experiments, human breast cancer MCF-7 or MCF-7/TDP1 cells were incubated with topotecan and the tested compounds for 96 h at 37 °C, and then measured by MTT assay.
Immunodetection of Cellular TOP1cc and TDP1cc
The ICE assays for cellular enzyme-DNA adduct was performed according to the reported method[46]. Briefly, mid log phase MCF-7 cells were incubated with drugs at the indicated concentration for 1 h, and then the cells were lysed with DNAzol reagent (1 mL) at 25 °C for 30 min. Ethanol (0.5 mL, 100%) was subsequently added and mixed with the lysate. The solution was incubated at −20 °C overnight. The genomic DNA was collected by centrifugation (12000 rpm) at 25 °C for 10 min and washed with 75% ethanol. The precipitated DNA was dissolved in NaOH (8 mM, 0.2 mL). The pH value was adjusted to 7.2 by adding HEPES (1 M). After centrifugation, DNA (2 μg) in supernatant was dissolved in 30 μL of NaH2PO4 buffer (25 mM, pH 6.5) and then loaded onto nitrocellulose membranes. Membranes were incubated with rabbit monoclonal to human TOP1 (Abcam, 1:1000) or TDP1 (Abcam, 1:1000) at 4 °C overnight, and then incubated with the appropriate HRP conjugated secondary antibodies (Cell Signaling Technology, 1:3000) at room temperature for 1 h. Reactive dots were detected using Immobilon Western Chemiluminescent HRP substrate (Millipore).
Immunofluorescence Assay
Immunofluorescence staining and confocal microscopy were performed according to the reported method[46].Briefly, after being incubated with the tested compounds for 5 h at 37 °C, MCF-7 cells were fixed with 4% paraformaldehyde for 10 min, then permeabilized with 0.5% Triton X-100/PBS at 37 °C for 30 min, and finally blocked with 5% goat serum/PBS at 37 °C for 3 h. The protocol was followed with the γH2AX antibody (no.9718, Cell Signaling Technology) at 4 °C overnight, and the digital images were recorded using an FV3000 microscope (Olympus, Japan) and analyzed with FV31S-SW software.
Flow Cytometry
MCF-7 cells (2×105) were planted in culture dish and treated with 43 at the indicated concentration for 24 h. The apoptosis detection was applied using the FITC Annexin V/PI apoptosis Detection Kit (KeyGEN). The collected cells were resuspended in 500 μL binding buffer and added with 5 μL of FITC Annexin V and PI dyes. Slowly blending the samples and incubating at 25 °C for 15 min away from light. Finally, the fluorescence-positive cells were quantified by flow cytometry (EPICS XL).
Molecular modeling
The TOP1 crystal structure (PDB 1K4T) was obtained and tailored by removing the ligand and water molecules[46], while inspected for errors and missing residues, hydrogens were added, the optimized structure of TOP1 was used for molecular modeling. The ligand centroid coordination of the ternary complex as the center of the binding pocket for molecular modeling. Compounds were constructed using ChemDraw and saved in SDF file formats, which were optimized using Discovery Studio software. Compound 38 was docked 30 times, starting each time from different orientations and the default automatic genetic algorithm parameter settings were used. All torsion angles of compounds were allowed to rotate freely and the results were scored and ranked by using MOE. The selected top ligand-binding pose was merged into the crystal structure. The AMBER force field of the MOE was utilized for energy minimization. The calculation was terminated when the gradient reached a value of 0.05 kcal/mol·Å.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 82173655), Guangdong Basic and Applied Basic Research Foundation (No. 2022A1515010802), Guangzhou Basic and Applied Basic Research Project (No. 202002030312) and by the Intramural Program of the National Cancer Institute (Center for Cancer Research), National Institutes of Health, Bethesda, Maryland, USA (Z01 BC 006150–19).
Reference
- [1].Wang JC, Annu. Rev. Biochem 1996, 65, 635–692. [DOI] [PubMed] [Google Scholar]
- [2].Stewart L, Redinbo MR, Qiu X, Hol WG, Champoux JJ, Science 1998, 279, 1534–1541. [DOI] [PubMed] [Google Scholar]
- [3].Champoux JJ, Annu. Rev. Biochem 2001, 70, 369–413. [DOI] [PubMed] [Google Scholar]
- [4].Pommier Y, Sun Y, Huang SN, Nitiss JL, Nat. Rev. Mol. Cell Biol 2016, 17, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pommier Y, Nat. Rev. Cancer 2006, 6, 789–802. [DOI] [PubMed] [Google Scholar]
- [6].Beretta GL, Perego P, Zunino F, Expert Opin. Ther. Targets 2008, 12, 1243–1256. [DOI] [PubMed] [Google Scholar]
- [7].Pommier Y, Chem. Rev 2009, 109, 2894–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pommier Y, Leo E, Zhang H, Marchand C, Chem. Biol 2010, 17, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pommier Y, Marchand C, Nat. Rev. Drug Discovery 2011, 11, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Thomas A, Pommier Y, Clin. Cancer Res 2019, 25, 6581–6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dasika GK, Lin SCJ, Zhao S, Sung P, Tomkinson A, Lee EY, Oncogene 1999, 18, 7883–7899. [DOI] [PubMed] [Google Scholar]
- [12].Maede Y, Shimizu H, Fukushima T, Kogame T, Nakamura T, Miki T, Takeda S, Pommier Y, Murai J, Mol. Cancer Ther 2014, 13, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sun Y, Saha LK, Saha S, Jo U, Pommier Y, DNA Repair 2020, 94, 102926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sun Y, Saha S, Wang W, Saha LK, Huang SYN, Pommier Y, DNA Repair 2020, 89, 102837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Interthal H, Pouliott JJ, Champoux JJ, Proc. Natl. Acad. Sci. U. S. A 2001, 98, 12009–12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Pouliot JJ, Robertson CA, Nash HA, Genes Cells 2001, 6, 677–687. [DOI] [PubMed] [Google Scholar]
- [17].Debethune L, Kohlhagen G, Grandas A, Pommier Y, Nucleic Acids Res 2002, 30, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yang SW, Burgin AB, Huizenga BN, Robertson CA, Yao KC, Nash HA, Proc. Natl. Acad. Sci. U. S. A 1996, 93, 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang YW, Regairaz M, Seiler JA, Agama KK, Doroshow JH, Pommier Y, Nucleic Acids Res 2011, 39, 3607–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Das BB, Huang SY, Murai J, Rehman I, Christophe Ame J, Sengupta S, Das SK, Majumdar P, Zhang H, Biard D, Majumder HK, Schreiber V, Pommier Y, Nucleic Acids Res 2014, 42, 4435–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Saha LK, Wakasugi M, Akter S, Prasad R, Wilson SH, Shimizu N, Sasanuma H, Huang SYN, Agama K, Pommier Y, Matsunaga T, Hirota K, Iwai S, Nakazawa Y, Ogi T, Takeda S, Proc. Natl. Acad. Sci. U. S. A 2020, 117, 14412–14420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rehman I, Basu SM, Das SK, Bhattacharjee S, Ghosh A, Pommier Y, Das BB, Nucleic Acids Res 2018, 46, 5601–5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bhattacharjee S, Rehman I, Nandy S, Das BB, DNA Repair 2022, 111, 103277. [DOI] [PubMed] [Google Scholar]
- [24].Interthal H, Chen HJ, Champoux JJ, J. Biol. Chem 2005, 280, 36518–36528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Barthelmes HU, Habermeyer M, Christensen MO, Mielke C, Interthal H, Pouliot JJ, Boege F, Marko D, J. Biol. Chem 2004, 279, 55618–55625. [DOI] [PubMed] [Google Scholar]
- [26].Nivens MC, Felder T, Galloway AH, Pena MMO, Pouliot JJ, Spencer HT, Cancer Chemother. Pharmacol 2004, 53, 107–115. [DOI] [PubMed] [Google Scholar]
- [27].Interthal H, Chen HJ, Kehl-Fie TE, Zotzmann J, Leppard JB, Champoux JJ, EMBO J 2005, 24, 2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].El-Khamisy SF, Katyal S, Patel P, Ju L, McKinnon PJ, Caldecott KW, DNA Repair 2009, 8, 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Perego P, Cossa G, Tinelli S, Corna E, Carenini N, Gatti L, Cesare MD, Ciusani E, Zunino F, Luison E, Canevari S, Zaffaroni N, Beretta GL, Biochem. Pharmacol 2012, 83, 27–36. [DOI] [PubMed] [Google Scholar]
- [30].Huang SN, Murai J, Rosa ID, Dexheimer TS, Naumova A, Gmeiner WH, Pommier Y, Nucleic Acids Res 2013, 41, 7793–7803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Leung E, Patel J, Hollywood JA, Zafar A, Tomek P, Barker D, Pilkington LI, van Rensburg M, Langley RJ, Helsby NA, Squire CJ, Baguley BC, Denny WA, Reynisson J, Leung IKH, Oncol. Ther 2021, 9, 541–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ghosh A, Bhattacharjee S, Chowdhuri SP, Mallick A, Rehman I, Basu S, Das BB, Sci. Adv 2019, 5, eaax9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang H, Xiong Y, Su D, Wang C, Srivastava M, Tang M, Feng X, Huang M, Chen Z, Chen J, Nat. Commun 2022, 13, 4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zakharenko A, Luzina O, Koval O, Dmitry N, Gushchina I, Dyrkheeva N, Svedas V, Salakhutdinov N, Lavrik O, J. Nat. Prod 2016, 79, 2961–2967. [DOI] [PubMed] [Google Scholar]
- [35].Khomenko T, Zakharenko A, Odarchenko T, Arabshahi HJ, Sannikova V, Zakharova O, Korchagina D, Reynisson J, Volcho K, Salakhutdinov N, Lavrik O, Bioorg. Med. Chem 2016, 24, 5573–5581. [DOI] [PubMed] [Google Scholar]
- [36].Ponomarev KY, Suslov EV, Zakharenko AL, Zakharova OD, Rogachev AD, Korchagina DV, Zafar A, Reynisson J, Nefedov AA, Volcho KP, Salakhutdinov NF, Lavrik OI, Bioorg. Chem 2018, 76, 392–399. [DOI] [PubMed] [Google Scholar]
- [37].Zhang XR, Wang HW, Tang WL, Zhang Y, Yang H, Hu DX, Ravji A, Marchand C, Kiselev E, Ofori-Atta K, Agama K, Pommier Y, An LK, J. Med. Chem 2018, 61, 9908–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zakharova O, Luzina O, Zakharenko A, Sokolov D, Filimonov A, Dyrkheeva N, Chepanova A, Ilina E, Ilyina A, Klabenkova K, Chelobanov B, Stetsenko D, Zafar A, Eurtivong C, Reynisson J, Volcho K, Salakhutdinov N, Lavrik O, Bioorg. Med. Chem 2018, 26, 4470–4480. [DOI] [PubMed] [Google Scholar]
- [39].Zakharenko AL, Luzina OA, Sokolov DN, Kaledin VI, Nikolin VP, Popova NA, Patel J, Zakharova OD, Chepanova AA, Zafar A, Reynisson J, Leung E, Leung IKH, Volcho KP, Salakhutdinov NF, Lavrik OI , Eur. J. Med. Chem 2019, 161, 581–593. [DOI] [PubMed] [Google Scholar]
- [40].Komarova AO, Drenichev MS, Dyrkheeva NS, Kulikova IV, Oslovsky VE, Zakharova OD, Zakharenko AL, Mikhailov SN, Lavrik OI, Enzyme Inhib J. Med. Chem 2018, 33, 1415–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Xiao LG, Zhang Y, Zhang HL, Li D, Gu Q, Tang GH, Yu Q, An LK, Org. Biomol. Chem 2020, 18(27), 5130–5136. [DOI] [PubMed] [Google Scholar]
- [42].Zhang HL, Zhang Y, Yan XL, Xiao LG, Hu DX, Yu Q, An LK. Bioorg. Med. Chem 2020, 28, 115527. [DOI] [PubMed] [Google Scholar]
- [43].Zhao XZ, Kiselev E, Lountos GT, Wang W, Tropea JE, Needle D, Hilimire TA, Jr JSS, Waugh DS, Pommier Y, Jr TRB, Chem. Sci 2021, 12, 3876–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Dyrkheeva NS, Filimonov AS, Luzina OA, Orlova KA, Chernyshova IA, Kornienko TE, Malakhova AA, Medvedev SP, Zakharenko AL, Ilina ES, Anarbaev RO, Naumenko KN, Klabenkova KV, Burakova EA, Stetsenko DA, Zakian SM, Salakhutdinov NF, Lavrik OI, Int. J. Mol. Sci 2021, 22, 11336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Munkuev AA, Mozhaitsev ES, Chepanova AA, Suslov EV, Korchagina DV, Zakharova OD, Ilina ES, Dyrkheeva NS, Zakharenko AL, Reynisson J, Volcho KP, Salakhutdinov NF, Lavrik OI, Molecules 2021, 26, 3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hu DX, Tang WL, Zhang Y, Yang H, Wang W, Agama K, Pommier Y, An LK, J. Med. Chem 2021, 64, 7617–7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Khomenko TM, akharenko AL, Chepanova AA, Ilina ES, Zakharova OD, Kaledin VI, Nikolin VP, Popova NA, Korchagina DV, Reynisson J, Chand R, Ayine-Tora DM, Patel J, Leung IKH, Volcho KP, Salakhutdinov NF, Lavrik OI, Int. J. Mol. Sci 2019, 21, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Salomatina OV, Dyrkheeva NS, Popadyuk II, Zakharenko AL, Ilina ES, Komarova NI, Reynisson J, Salakhutdinov NF, Lavrik OI, Volcho KP, Molecules 2021, 27, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dexheimer TS, Antony S, Marchand C, Pommier Y, Anti-Cancer Agents Med. Chem 2008, 8, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Comeaux EQ, van Waardenburg RCAM, Drug Metab. Rev 2014, 46, 494–507. [DOI] [PubMed] [Google Scholar]
- [51].Zakharenko A, Dyrkheeva N, Lavrik O, Med. Res. Rev 2019, 39, 1427–1441. [DOI] [PubMed] [Google Scholar]
- [52].Brogden RN, Wiseman LR. Topotecan a review of its potential in advanced ovarian cancer, Drugs 1998, 56, 709–723. [DOI] [PubMed] [Google Scholar]
- [53].Bracher F, Tremmel T, Arch. Pharm. Chem. Life Sci 2017, 350, e1600236. [DOI] [PubMed] [Google Scholar]
- [54].Choia SH, Tsuchidab Y, Yang HW, Cancer Lett 1998, 124, 15–21. [DOI] [PubMed] [Google Scholar]
- [55].Syed YY, Drugs 2020, 80, 1019–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Andrikopoulou A, Zografos E, Liontos M, Koutsoukos K, Dimopoulos MA, Zagouri F, Clin. Breast Cancer 2021, 21, e212–e219. [DOI] [PubMed] [Google Scholar]
- [57].Zhang R, Li Y, Cai Q, Liu T, Sun H, Chambless B, Cancer Chemother. Pharmacol 1998, 41, 257–267. [DOI] [PubMed] [Google Scholar]
- [58].Shao J, Huang F, Fu J, Chin. J. Clin. Oncol 2003, 30, 26–28. [Google Scholar]
- [59].Liang Z, Wang Y, Zhang H, Deng J, Lei F, Li J, Shi T, Wang S, Li R, Wang Z, Eur. J. Med. Chem 2022, 239, 114530. [DOI] [PubMed] [Google Scholar]
- [60].Laev SS, Salakhutdinov NF, Lavrik OI, Bioorg. Med. Chem 2016, 24, 5017–5027. [DOI] [PubMed] [Google Scholar]
- [61].Dexheimer TS, Gediya LK, Stephen AG, Weidlich I, Antony S, Marchand C, Interthal H, Nicklaus M, Fisher RJ, Njar VC, Pommier Y, J. Med. Chem 2009, 52, 7122–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Marchand C, Lea WA, Jadhav A, Dexheimer TS, Austin CP, Inglese J, Pommier Y, Simeonov A, Mol. Cancer Ther 2009, 8, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sirivolu VR, Vernekar SK, Marchand C, Naumova A, Chergui A, Renaud A, Stephen AG, Chen F, Sham YY, Pommier Y, Wang Z, J. Med. Chem 2012, 55, 8671–8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Dean RA, Fam HK, An J, Choi K, Shimizu Y, Jones SJM, Boerkoel CF, Interthal H, Pfeifer TA, J. Biomol. Screening 2014, 19, 1372–1382. [DOI] [PubMed] [Google Scholar]
- [65].Zakharenko A, Khomenko T, Zhukova S, Koval O, Zakharova O, Anarbae R, Lebedeva N, Korchagina D, Komarova N, Vasiliev V, Reynisson J, Volcho K, Salakhutdinov N, Lavrik O, Bioorg. Med. Chem 2015, 23, 2044–2052. [DOI] [PubMed] [Google Scholar]
- [66].Conda-Sheridan M, Reddy PV, Morrell A, Cobb BT, Marchand C, Agama K, Chergui A, Renaud A, Stephen AG, Bindu LK, Pommier Y, Cushman M, J. Med. Chem 2013, 56, 182–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Nguyen TX, Morrell A, Conda-Sheridan M, Marchand C, Agama K, Bermingham A, Bermingam A, Stephen AG, Chergui A, Naumova A, Fisher R, OKeefe BR, Pommier Y, Cushman M, J. Med. Chem 2012, 55, 4457–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wei X, Wang FT, Si-Tu MX, Fan H, Hu JS, Yang H, Guan SY, An LK, Zhang CX, Mar. Drugs 2022, 20, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lountos GT, Zhao XZ, Kiselev E, Tropea JE, Needle D, Pommier Y, Burke TR, Waugh DS, Nucleic Acids Res 2019, 47, 10134–10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Baglini E, Salerno S, Barresi E, Robello M, Settimo FD, Taliani S, Marini AM, Eur. J. Pharm. Sci 2021, 156, 105594. [DOI] [PubMed] [Google Scholar]
- [71].Wang P, Elsayed MSA, Plescia CB, Ravji A, Redon CE, Kiselev E, Marchand C, Zeleznik O, Agama K, Pommier Y, Cushman M, J. Med. Chem 2017, 60, 3275–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ledesma FC, El-Khamisy SF, Zuma MC, Osborn K, Caldecott KW, Nature 2009, 461, 674–678. [DOI] [PubMed] [Google Scholar]
- [73].Pommier Y, Huang SY, Gao R, Das BB, Murai J, Marchand C, DNA Repair 2014, 19, 114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Saha S, Sun Y, Huang SN, Baechler SA, Pongor LS, Agama K, Jo U, Zhang H, Tse-Dinh YC, Pommier Y, Cell Rep 2020, 33, 108569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tang WL, Zhang Y, Hu DX, Yang H, Yu Q, Chen JW, Agama K, Pommie Y, An LK, Eur. J. Med. Chem 2019, 178, 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Korivi RP, Cheng CH, Chem. Eur. J 2010, 16, 282–287. [DOI] [PubMed] [Google Scholar]
- [77].Omura K, Sharma AK, Swern D, J. Org. Chem 1976, 41, 957–961. [Google Scholar]
- [78].Andrus MB, Asgari D, Sclafani JA, J. Org. Chem 1997, 62, 9365–9368. [Google Scholar]
- [79].Yang H, Wang FT, Wu M, Wang W, Agama K, Pommier Y, An LK, Bioorg. Chem 2022, 123, 105789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Yao T, Guo Z, Liang X, Qi L, J. Org. Chem 2018, 83, 13370–13380. [DOI] [PubMed] [Google Scholar]
- [81].Beugelmans R, Bios-Choussy M, Tetrahedron, 1992, 48, 8285–8294. [Google Scholar]
- [82].Dexheimer TS, Pommier Y, Nat. Protoc 2008, 3, 1736–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Cushman M, Jayaraman M, Vroman JA, Fukunaga AK, Fox BM, Kohlhagen G, Strumberg D, Pommier Y, J. Med. Chem 2000, 43, 3688–3698. [DOI] [PubMed] [Google Scholar]
- [84].Wang G, Cushman M, Synth. Commun 1991, 21, 989–996. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.