

Abstract
In the last decades, the role of the prion protein (PrP) in neurodegenerative diseases has been intensively investigated, initially in prion diseases of humans (e.g., Creutzfeldt-Jakob disease) and animals (e.g., scrapie in sheep, chronic wasting disease in deer and elk, or “mad cow disease” in cattle). Templated misfolding of physiological cellular prion protein (PrPC) into an aggregation-prone isoform (termed PrP “Scrapie” (PrPSc)), self-replication and spreading of the latter inside the brain and to peripheral tissues, and the associated formation of infectious proteopathic seeds (termed “prions”) are among the essential pathogenic mechanisms underlying this group of fatal and transmissible spongiform encephalopathies. Later, key roles of the correctly folded PrPC were identified in more common human brain diseases (such as Alzheimer’s disease or Parkinson’s disease) associated with the misfolding and/or accumulation of other proteins (such as amyloid-β, tau or α-synuclein, respectively). PrPC has also been linked with neuroprotective and regenerative functions, for instance in hypoxic/ischemic conditions such as stroke. However, despite a mixed “bouquet” of suggested functions, our understanding of pathological and, especially, physiological roles played by PrPC in the brain and beyond is certainly incomplete. Interactions with various other proteins at the cell surface or within intracellular compartments may account for the functional diversity linked with PrPC. Moreover, conserved endogenous proteolytic processing of PrPC generates several defined PrPC fragments, possibly holding intrinsic functions in physiological and pathological conditions, thus making the “true and complete biology” of this protein more complicated to be elucidated. Here, we focus on one of those released PrPC fragments, namely shed PrP (sPrP), generated by a membrane-proximate ADAM10-mediated cleavage event at the cell surface. Similar to other soluble PrPC fragments (such as the N1 fragment representing PrP’s released N-terminal tail upon the major α-cleavage event) or experimentally employed recombinant PrP, sPrP is being suggested to act neuroprotective in Alzheimer’s disease and other protein misfolding diseases. Several lines of evidence on extracellular PrPC (fragments) suggest that induction of PrPC release could be a future therapeutic option in various brain disorders. Our recent identification of a substrate-specific approach to stimulate the shedding by ADAM10, based on ligands binding to cell surface PrPC, may further set the stage for research into this direction.
Key Words: ADAM10, aggregation, Alzheimer’s disease, amyloid, antibodies, Creutzfeldt-Jakob disease, enzymatic cleavage, extracellular vesicles, neurodegeneration, neurotoxicity, proteolytic processing, stroke, transmissible spongiform encephalopathies
Introduction
The cellular prion protein (PrPC) is a ~30 kDa glycosylphosphatidylinositol (GPI)-anchored glycoprotein highly expressed in the nervous system, yet also found in most other tissues (Stahl et al., 1987; Barmada et al., 2004; Adle-Biassette et al., 2006). In prion diseases (or “transmissible spongiform encephalopathies”, short: TSEs), which consist of sporadic (“idiopathic”), genetic (“familial”), and transmitted (including “iatrogenic”) forms, such as sporadic Creutzfeldt-Jakob disease, fatal familial insomnia and variant Creutzfeldt-Jakob disease, respectively, PrPC is transformed into a β-sheet-enriched fibrillizing isoform (PrPSc) (Prusiner et al., 1982; Aguzzi, 2001; Spagnolli et al., 2019; Kamali-Jamil et al., 2021; Kraus et al., 2021; Hallinan et al., 2022; Hoyt et al., 2022; Telling, 2022) causing toxicity and death in cells expressing PrPC, particularly in neurons (Brandner et al., 1996; Lakkaraju et al., 2022).
In the last decade, a dual (i.e., detrimental versus protective) role of PrPC has also been described in Alzheimer’s disease (AD). On one hand, full-length (fl) membrane-bound PrPC mediates toxic signaling as a high-affinity receptor for extracellular amyloid-β (Aβ) and other harmful protein/peptide assemblies associated with neurodegenerative diseases, such as p-Tau and α-synuclein (Lauren et al., 2009; Resenberger et al., 2011; Ferreira et al., 2017; Hu et al., 2018; Ondrejcak et al., 2018; Younan et al., 2018; Gomes et al., 2019; Corbett et al., 2020). On the other hand, PrPC was shown to reduce BACE1-mediated β-amyloid precursor protein cleavage, thus reducing the formation of Aβ (Griffiths et al., 2011). PrPC has also been implicated in the downregulation, phosphorylation, and processing of Tau protein (Vergara et al., 2015).
In line with a beneficial role, PrPC is enriched on many types of extracellular vesicles (EVs; i.e., membrane-enclosed particles released by most cells in the body), where it may sequester toxic Aβ oligomers into larger and possibly less harmful Aβ fibrils, thereby reducing toxicity (Wik et al., 2012; An et al., 2013; Falker et al., 2016). Further neuroprotective roles for PrPC have been described in other pathological conditions, such as oxidative stress (Watt et al., 2005; Mitteregger et al., 2007; Haigh et al., 2009), epilepsy (Walz et al., 1999; Carulla et al., 2011), ischemia and stroke (Shyu et al., 2005; Doeppner et al., 2015; Guitart et al., 2016; Beraldo et al., 2018; Brenna et al., 2020).
This neuroprotection against excitotoxic processes may be performed, at least in part, by PrP’s regulation of cellular Ca2+ homeostasis, protecting the system from intracellular Ca2+ overload (Khosravani et al., 2008; De Mario et al., 2019). Furthermore, depending on cell type and context, surface PrPC may be relevant for the uptake or phagocytosis of toxic protein aggregates, resulting in either harmful cell-to-cell spread of pathology or increased degradation of those conformers, thus highlighting again the potential dual role of this protein (reviewed in Legname and Scialò (2020)).
PrPC has also been related to several physiological functions in the nervous system, such as neuronal differentiation and proliferation (Steele et al., 2006), neuritogenesis, and axonal growth/guidance (Santuccione et al., 2005; Loubet et al., 2011; Amin et al., 2016), myelin maintenance in the peripheral nervous system (Bremer et al., 2010; Kuffer et al., 2016), synapse regulation and long-term potentiation (Maglio et al., 2006; Khosravani et al., 2008), memory acquisition and behavior (Coitinho et al., 2003; Schmitz et al., 2014; Matamoros-Angles et al., 2022), and homeostasis of copper and other divalent metal ions (Brown et al., 1997; Gasperini et al., 2015). Among the many suggested functions attributed to PrPC, only a few have been unambiguously confirmed by several independent groups, yet even in these cases the underlying mechanisms are mostly still not fully understood (Kovač and Čurin Šerbec, 2022). It appears not only conceivable but even likely that some of these roles are mediated by certain constitutively generated PrPC fragments and/or extracellular forms, and an increasing amount of studies are currently supporting this concept.
PrPC undergoes complex posttranslational modifications, including GPI-anchor attachment and N-glycosylation (Puig et al., 2011), and can be subjected to at least four conserved proteolytic processing events (termed α-, β-, γ-cleavage and C-terminal shedding; an overview is exhibited in Figure 1), which lead to the generation of different membrane-bound or released PrPC fragments (reviewed in Linsenmeier et al. (2017)). As mentioned earlier, GPI-anchored PrPC (either fl-PrPC or its C1 fragment resulting from the α-cleavage in the middle of PrPC) can also be released by cells in association with EVs. Recent studies pointed out that this EV-PrPC has an essential regulatory influence on the uptake of EVs by recipient cells (Brenna et al., 2020; D’Arrigo et al., 2021), Aβ fibrillization (Falker et al., 2016), and neuritogenesis in vitro (Gonias et al., 2022).
Figure 1.
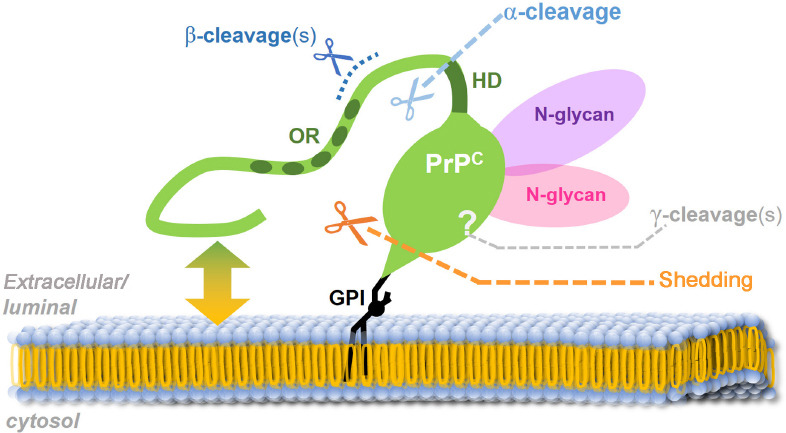
The prion protein (PrPC; green) and its conserved processing.
Mature PrP is attached via a glycosylphosphatidylinositol (GPI) anchor to the luminal (in secretory or endocytic vesicles) / extracellular (at the cell surface or on extracellular vesicles) leaflet of the membrane. The flexible N-terminal tail interacts with lipids of the plasma membrane (green-yellow double arrow) and was described as being able to integrate into or even penetrate the lipid bilayer. Shedding via ADAM10 (orange scissors and dotted line) takes place at the cell surface in the very C-terminal sequence (just a few amino acids away from the GPI anchor). The α-cleavage (bright blue scissors and dotted line), the dominating proteolytic event in many cells/tissues, occurs N-terminally of a hydrophobic domain (HD) and separates the protein into a released flexible N-terminal fragment and a membrane-bound globular C-terminal domain. This latter C1 fragment can also undergo further shedding by ADAM10. The protease(s) responsible for the α-cleavage is/are still a matter of debate. Different cleavages in the vicinity of amino acid position 90 are summarized under the term “β-cleavage” (dark blue) and release N-terminal fragments containing the octameric repeat region (OR). More recently, γ-cleavage (grey) was described to occur within the C-terminal region. Both β- and γ-cleavages are less well understood to date and may primarily be associated with pathological conditions. Likewise, the exact subcellular localization for the α-, β- and γ-cleavages are not unambiguously clarified yet.
The multifunctional character of PrPC, especially its alleged dual role in brain diseases, is likely not restricted to the membrane-anchored full-length form of PrPC, on which the vast majority of research in the prion field has focused so far, but presumably involves proteolytically generated PrPC fragments and/or PrPC forms on the surface of EVs. We will here focus on one of these fragments, termed shed PrP (sPrP), resulting from the membrane-proximate release via the metalloprotease ADAM10 (Taylor et al., 2009; Altmeppen et al., 2011; Figure 1), and discuss new findings and concepts as to its role in neurodegeneration as well as neuroregeneration, and how its production may be stimulated in a substrate-specific manner and despite structural constraints.
Search Strategy and Selection Criteria
This review article was drafted based on relevant literature entries found after performing respective keyword searches (no further limits or filters applied) in PubMed®, NIH/NCBI (https://pubmed.ncbi.nlm.nih.gov) between February 2 and October 31, 2022. We apologize to all colleagues whose important contributions to aspects covered herein were not cited due to space limitations.
Lowering PrPC Expression and PrP-Targeting by Antibodies as Promising Strategies for Prion Disease Treatment and Beyond
Since the discovery of infectious prions (Prusiner et al., 1982), a wealth of studies using different approaches have been performed to cure the associated diseases, so far, unfortunately, without fundamental success. As demonstrated by convincing experimental evidence, prion disease incubation times inversely correlate with PrPC expression levels (Sandberg et al., 2011). Earlier studies demonstrated that, while PrPC knockout (PrP-KO) mice are resistant to prion diseases, mice with heterozygous PrPC expression (+/–) have a prolonged incubation time (Büeler et al., 1994) compared to wild-type controls (+/+), whereas tga20 mice with a severalfold overexpression of PrPC present a drastically shortened time to symptomatic disease and terminal stage (Fischer et al., 1996). In line with this, a conditional PrP-KO mouse model exhibited full recovery after depleting PrPC expression in prion-infected preclinical animals (Mallucci et al., 2003). Moreover, while the presence and formation of PrPSc are essential and defining for prion disease pathogenicity, net amounts of PrPSc seem to be less determining for neurotoxicity and disease tempo than neuronal PrPC surface levels (Altmeppen et al., 2015; Linsenmeier et al., 2021; Lakkaraju et al., 2022). Additionally, based on the knowledge obtained from studying PrP-KO mice (Steele et al., 2007) as well as some goats naturally devoid of PrPC (Benestad et al., 2012), lack of PrPC does not cause deleterious conditions as these animals experience rather normal life span with only subtle phenotypes if any. Thus, when it comes to treating patients with prion diseases or to preventing/delaying the establishment of the disease in confirmed carriers of respective PrP mutations (i.e., genetic cases) by lowering total PrP expression, potential side effects are most likely negligible in view of the anticipated gain. Therefore, reducing or even eliminating cell-associated PrPC is now considered to be one of the most promising therapeutic approaches (Tremblay et al., 1998; Minikel et al., 2020).
The use of oligonucleotides targeting the mRNA to reduce Prnp expression has been studied for several years and this strategy is currently experiencing a straightforward revival and strong experimental support (White et al., 2008; Pulford et al., 2010; Friberg et al., 2012; Lehmann et al., 2014; Raymond et al., 2019; Minikel et al., 2020). For instance, antisense oligonucleotides (ASOs) designed against the Prnp mRNA were shown to not only significantly lower PrP expression, but also to prolong the incubation period for more than two months in prion-infected mice (Friberg et al., 2012; Raymond et al., 2019). In a comprehensive study, Minikel et al. (2020), by using a new set of more advanced ASOs, (i) first of all, confirmed that ASOs are effective in the reduction of PrPC expression, and, more importantly, (ii) showed that a single dose administration of PrP-lowering ASOs in subclinical animals extends the survival time by 61–98%. Moreover, they (iii) highlighted that even neuroinflammatory and neuropathological markers (such as plasma neurofilament light chain levels) in these animals can be reversed, and also (iv) proved that –as expected for targeting PrP as the substrate for any prion conversion– the ASOs work against multiple prion strains (Minikel et al., 2020). These new findings may now pave the way for an effective and causative treatment against prion diseases.
In an alternative approach followed for more than two decades by now, different antibodies directed against PrP have been assessed as potential therapeutic agents against prion diseases (Enari et al., 2001; Peretz et al., 2001; Alexandrenne et al., 2009; Reilly et al., 2022). An early study using chronically prion-infected N2a cells showed that the monoclonal anti-PrP antibody 6H4 abrogated PrPSc accumulation, and a similar effect was also observed after phosphatidylinositol-specific phospholipase C treatment, which cleaves within the GPI anchor structure and releases PrP from the cell surface (Enari et al., 2001). Another study showed that antibodies against PrP inhibit prion propagation and efficiently clear cell cultures from prion infectivity in a dose-dependent manner (Peretz et al., 2001). Transgenic expression of anti-PrP antibodies even blocked prion infection in mice (Heppner et al., 2001). Strikingly, PrP-targeting antibodies are also protective against PrP-mediated neurotoxicity of Aβ oligomers (Lauren et al., 2009; Chung et al., 2010; Freir et al., 2011). For instance, the monoclonal antibodies 6D11 (Lauren et al., 2009) and ICSM35 (Freir et al., 2011), which bind to one of PrP’s Aβ-binding sites, were shown to efficiently block the Aβ-mediated inhibition of LTP. More recently, it was found that peripheral administration of a humanized anti-PrP antibody (PRN100) also interferes with the PrPC-Aβ interaction and prevents the otherwise harmful effects of Aβ derived from Alzheimer’s disease brain extracts when inoculated into rat brains (Klyubin et al., 2014).
Importantly, the same antibody was recently administered in a special treatment program to six Creutzfeldt-Jakob disease patients (Mead et al., 2022). Although all of the patients ultimately died of prion disease and their survival time and disease progression were not significantly different from historical controls, this important study revealed that administration of anti-PrP antibodies might be a suitable future therapeutic option (if given earlier), as an effective concentration of the antibody could reach the brain. Notably, this study also demonstrated that certain (though certainly not any) antibodies against PrPC are safe, as no severe adverse effects were observed (Mead et al., 2022). Moreover, antibody-based inhibition of prion-associated neurotoxicity was also demonstrated in murine hippocampal primary neurons (Reilly et al., 2022). A recent study provided important and detailed structural insight into how certain (rationally designed) ligands may interfere with prion toxicity (Frontzek et al., 2022), yet an effect on shedding was not assessed in that work. Several reports suggesting protective effects of PrP-directed antibodies in neurodegenerative protein misfolding diseases have recently been summarized (Linsenmeier et al., 2021), and it certainly deserves attention that this, more and more, seems to be ageneralized feature not only restricted to prion and Alzheimer`s diseases (Corbett et al., 2020).
A Deeper Look into PrP-Directed Antibody Treatment Reveals Two Overlooked Mechanisms That May Contribute To Protective Effects
In our recent study on PrPC shedding, we further assessed the treatment effects of different anti-PrP antibodies (Linsenmeier et al., 2021). We observed that cell lines or murine brain slice cultures incubated with most of the antibodies and ligands tested (e.g., 6D11, POM1, but also single-chain POM1, single-chain POM2, or a bispecific immunotweezer (Polymenidou et al., 2008; Bardelli et al., 2018)) significantly increase the ADAM10-mediated shedding of PrPC, regardless of the location of their epitopes within PrP. As a consequence, a greater fraction of PrPC is cleaved off from the membrane, and levels of sPrP, which is thought to act protectively against toxic oligomers in the extracellular space, are significantly increased. The exact mechanism of this antibody-mediated and, thus, substrate-specific stimulation of PrPC shedding remains largely unclear and crosslinking does not appear to be a prerequisite. One possible explanation, at least supported by small angle X-ray scattering measurement performed for one of the antibodies (6D11), could be that the flexible N-terminal tail of PrPC somehow “shields” the C-terminal part by spatiotemporally restricting the access of ADAM10 to the cleavage site (yet the complete absence of the N-terminal tail is also disfavored as discussed below). The binding of any of the above-mentioned antibodies to PrPC may slightly modify the “structure” and movements of the N-terminal tail (in relation to the C-terminal domain) to render the cleavage site more accessible for ADAM10, at least transiently. In stark contrast to the majority of antibodies that stimulated shedding, POM2, an antibody with four repetitive epitopes in PrP’s octameric repeat region localized within the flexible N-terminal tail, does not lead to more shedding but, instead, to a strong clustering of PrPC molecules at the cell surface (most likely due to multimolecular crosslinking creating a dense meshwork of PrPC and antibody molecules), which subsequently leads to fast internalization and lysosomal degradation (Linsenmeier et al., 2021). While a degradation-promoting effect towards PrP has been shown earlier for other PrP-directed antibodies (Gilch et al., 2003; Perrier et al., 2004), this fast and remarkable reduction in total PrPC upon treatment with POM2 IgG has not been described before and might contribute to the protective effects attributed to this antibody (Sonati et al., 2013).
Last but not least, it should also be noted that several studies raised awareness that the administration of certain PrP-directed antibodies can also have relevant adverse effects, including severe neuronal loss and tissue damage (Solforosi et al., 2004; Lefebvre-Roque et al., 2007; Sonati et al., 2013; Herrmann et al., 2015; Reimann et al., 2016). From a mechanistic perspective, such harmful effects may involve (but are presumably not restricted to) epitope-dependent structural alterations and subsequent channel-forming insertion of several PrPs’ N-termini causing membrane perturbations (Sonati et al., 2013; Wu et al., 2017; Schilling et al., 2020), crosslinking-induced signaling cascades, impaired physiological trafficking and processing, shifted membrane localization, and/or blockage of PrP’s homeostatic interaction with important physiological binding partners. In conclusion, any therapeutic approaches involving PrP-directed antibodies (and possibly other ligands, too) therefore require careful consideration and experimental testing.
ADAM10-Mediated Shedding of PrPC: Sterical Considerations and Possible Regulatory Roles of Transmembrane Interactors
Several intuitively expectable structural constraints associated with PrPC, surprisingly, do not actually represent sterical hindrance for the access of and cleavage by ADAM10 (Figure 2A). Quite the contrary: the presence of the ever-moving and twisting, flexible, and readily membrane-interacting N-terminal tail (described to be a bona fide intrinsically disordered domain), which creates a spatiotemporal “cloud” or “shield” around PrP’s GPI-anchored and structured C-terminal half (Sonati et al., 2013; Carter et al., 2015; Cheng et al., 2017; Wu et al., 2017; Linsenmeier et al., 2021), seems to be preferred by the protease, since fl-PrPC is more efficiently shed than its N-terminally truncated C1 fragment generated by the physiological α-cleavage (Linsenmeier et al., 2018). Moreover, despite the considerable size of the (up to two) complex-type N-glycans attached to PrPC (Rudd et al., 1999; Demarco and Daggett, 2009; Puig et al., 2011; Yi et al., 2018; Nakić et al., 2021), diglycosylated PrPC is clearly favored by ADAM10 over mono- and nonglycosylated PrPC (Linsenmeier et al., 2018). Lastly, as if these unexpected and somewhat counterintuitive preferences were not enough, our recent study discussed above further revealed that large ligands, such as IgGs (~150 kD), binding to the rather small PrPC (~30 kDa) even stimulate its ADAM10-mediated shedding in an efficient and substrate-specific manner (Linsenmeier et al., 2021). An overview of approximate size relations regarding the aforementioned aspects is depicted in Figure 2A. Increased shedding for other ADAM10 substrates had previously been shown upon dimerization or antibody-mediated crosslinking (Shi et al., 2001; Schelter et al., 2010; Hartmann et al., 2015), yet our study showed that dimerization of PrPC at the cell surface does not seem to be a prerequisite for its stimulated shedding, as single-chain antibodies targeting PrPC also caused this effect. It will now be interesting to design and investigate smaller and therapeutically possibly more favorable ligands of PrPC, such as aptamers (Corda et al., 2018; Murakami et al., 2022) or nanobodies (Abskharon et al., 2019), to stimulate the ADAM10-mediated shedding.
Figure 2.
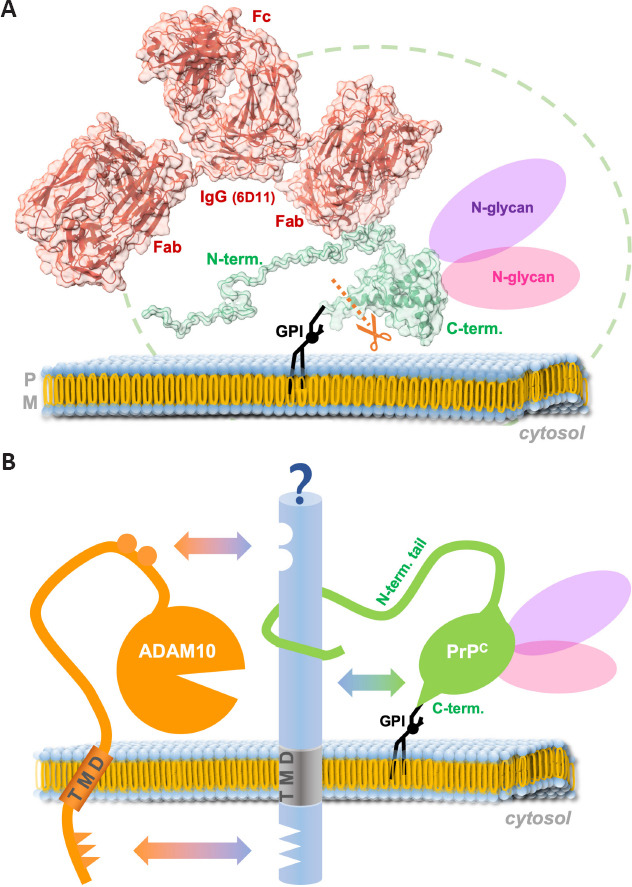
Size relation of modifications supporting the shedding of PrPC and potential influence of binding partners.
(A) PrPC (green) is attached to the outer leaflet of the plasma membrane via a glycosylphosphatidylinositol (GPI)-anchor (black). The flexible N-terminal tail is constantly moving and undergoing interactions with the membrane, thereby creating a spatiotemporal cloud (indicated by the stripped green line) around its globular C-terminal domain. Up to two large N-glycans may be attached to PrPC and this form seems to be preferred by ADAM10 over mono- and nonglycosylated forms. Lastly, binding of large ligands, such as antibodies (6D11 IgG is shown here in red), to cell surface PrPC stimulates its shedding rather than blocking access for ADAM10. The model of the PrP-antibody complex shown here is modified from Linsenmeier et al. (2021) (SASBDB accession ID: SASDLU2). The model was adapted using Chimera X (Pettersen et al., 2021). (B) Common binding partners (blue) may bring together ADAM10 (orange) and PrPC (green) via diverse interaction sites and thereby regulate the shedding event. Fab: Fragment antigen-binding (variable) region; Fc: fragment crystallizable (constant) region; PM: plasma membrane; PrPC: cellular prion protein; TMD: transmembrane domain.
At second glance, however, a possible explanation for why the presence of the N-terminal tail and the N-glycans could be supportive of the shedding process may lie in a putative (and as yet clearly understudied) role of interaction partners. While the importance of N-glycans in protein-protein interactions (Wu et al., 2018) is a well-established biological feature (e.g., in cell adhesion (Zhao et al., 2008) or binding of viral surface proteins to host cell receptors (Cao et al., 2021; Hoffmann et al., 2021)), PrP’s flexible N-terminal tail is a particularly interesting major interaction hub allowing for the binding of various partner molecules (Beland and Roucou, 2012). By physically bridging protease and substrate, certain interactors may indeed warrant the transient yet close encounter between ADAM10 and PrP required for the cleavage process (as suggested in Figure 2B). While, in the case of ADAM10, respective interaction sites may be positioned in both the extracellular part (Janes et al., 2005; Seegar et al., 2017) as well as in the transmembrane and cytosolic domains (Deng et al., 2014; Hitschler and Lang, 2022), the latter does obviously not apply to PrPC with its GPI anchor only sticking into the outer membrane leaflet. The N-terminal tail, however, may well accomplish sufficient interaction with extracellular domains of such putative binding partners at the cell surface.
Strikingly, several transmembrane proteins at the cell surface co-localize or even interact with PrPC, and some of those, such as neural cell adhesion molecule 1 (Schmitt-Ulms et al., 2001; Santuccione et al., 2005; Saftig and Lichtenthaler, 2015; Slapsak et al., 2016), lipoprotein receptor-related protein 1 (Taylor and Hooper, 2007; Parkyn et al., 2008; Liu et al., 2009; Shackleton et al., 2016), cadherins (Morel et al., 2004; Málaga-Trillo et al., 2009; Seipold et al., 2018; Sachs et al., 2021), amyloid precursor protein (Epis et al., 2010; Jorissen et al., 2010; Kuhn et al., 2010; Kaiser et al., 2012), are themselves suggested or proven substrates of ADAM10. This raises the question if shedding might be co-regulated or whether heterophilic interactions of these proteins with PrPC may even provide the impetus for the shedding of both in a (transient) complex. Alternatively, these proteins could also compete with PrPC for the shedding by ADAM10. In this case, exact localization and membrane organization (e.g., center or periphery of lipid rafts versus non-raft regions (Puig et al., 2019)), possibly organized by additional regulators of trafficking and membrane subdomain architecture, such as tetraspanins (Levy and Shoham, 2005; Saint-Pol et al., 2017; Seipold et al., 2018; Koo et al., 2020; Lipper et al., 2022), may determine and modulate the shedding depending on (patho)physiological context and cellular state. In addition to protein-protein binding, interactions of PrP (and in particular its N-terminal tail; Figures 1 and 2A) with membrane lipids and associated effects on membrane subdomain regulation may directly affect its own proteolytic processing as well as effects caused by binding of its released fragments (e.g., N1 or shed PrP) to recipient cells.
As mentioned above, complex N-glycans also play a role in intermolecular crosstalk and this may account for the fact that fully glycosylated PrPC is preferred by ADAM10 over underglycosylated forms. Moreover, full glycan equipment may explain why sPrP seems to interfere with the misfolding process in prion diseases (Altmeppen et al., 2015; Linsenmeier et al., 2021; Mohammadi et al., 2022): Although the conversion of sPrP into an anchorless PrPSc form seems possible (Aguilar-Calvo et al., 2020), the glycans may render sPrP much less prone for this pathogenic event (Winklhofer et al., 2003; Cheng et al., 2017; Camacho et al., 2019; Sevillano et al., 2020), thereby, at least relatively, hindering PrPSc production [in stark contrast to underglycosylated anchorless PrP studied in respective transgenic mice (Chesebro et al., 2005; Race et al., 2017)].
Finally, when it comes to possible (patho)physiological functions of sPrP as a released factor in intercellular communication, both the N-terminal tail and the two glycans may be important features for putative ligand-receptor and other interactions. This should be taken into account when using any proxy, such as (non-glycosylated) recombinant PrP, in experiments aiming at investigating the “real” biological roles of bona fide shed PrP.
Protective Effects of Different PrP Forms and Fragments
While membrane-bound fl-PrPC as a cell surface receptor for toxic protein assemblies plays deleterious roles by inducing neurotoxicity in many neurodegenerative diseases (Lauren et al., 2009; Resenberger et al., 2011; Kostylev et al., 2015; Ferreira et al., 2017; Urrea et al., 2017; De Cecco and Legname, 2018; Corbett et al., 2020; Rösener et al., 2020), once it is cleaved by one of the naturally occurring proteolytic processing events [reviewed in (Altmeppen et al., 2013)], PrP’s neurotoxic potentials may turn into rather neuroprotective ones. For instance, previous studies have shown the protective effects of the soluble N1 fragment against the neurotoxicity of Aβ oligomers (Fluharty et al., 2013; Beland et al., 2014; Scott-McKean et al., 2016). A similar beneficial effect of N1 against toxic phosphorylated tau oligomers was also reported recently (Nieznanska et al., 2021). α-Synuclein oligomers were likewise shown to interact with the N-terminus of PrPC (Ferreira et al., 2017; Rösener et al., 2020) leading to the assumption of a more generalized protective role of N1 against different toxic oligomers. It is expected that other soluble fragments of PrPC (which carry the binding sites for toxic oligomers), such as sPrP, or PrPC forms decorating the surface of EVs, provide similar protective effects as N1.
Physiological roles of soluble PrP forms have been reported in different cellular processes (Martellucci et al., 2019; Gonias et al., 2022; Mantuano et al., 2022). For instance, recombinant PrP (to some extent recapitulating sPrP, yet lacking the important glycosylations) was shown to promote neurite outgrowth in PC12 and N2a cells via activation of N-methyl-D-aspartate-receptor and lipoprotein receptor-related protein 1. In Schwann cells, the interaction of recPrP with the lipoprotein receptor-related protein 1/N-methyl-D-aspartate-receptor complex activates ERK 1/2 and promotes cell migration (Mantuano et al., 2020). Interestingly, the same group later showed a similar effect of EV-associated PrPC using extracellular vesicles derived from human plasma samples (Mantuano et al., 2022).
PrPC promotes post-ischemic protection, (neuro-)regeneration, and angiogenesis (Doeppner et al., 2015), a phenomenon that might actually be mediated by released forms of PrPC. It was also shown that, after oxygen-glucose deprivation (an in vitro model of ischemic stroke), PrPC levels were increased in cultured astrocyte-derived EVs, which then had protective effects on oxygen-glucose deprivation-exposed neurons. This effect was seemingly dependent on EV-associated PrPC, as it was absent when the vesicles were isolated from PrP-KO cells (Guitart et al., 2016). Moreover, a smaller infarction area and faster tissue recovery after stroke was previously reported in PrPC-expressing compared to PrP-KO mice (Mclennan et al., 2004; Weise et al., 2006). A recent study further revealed that PrPC is essential for the regeneration of neurons, as its overexpression promotes early neurogenesis, whereas its absence delays neuronal regeneration after acute nasotoxic injury (Parrie et al., 2020). It certainly deserves additional studies to elucidate the relative contribution and involvement of fl-PrPC in comparison with its diverse fragments in these and other physiological processes linked with PrPC.
Concluding Remarks
The dual role attributed to PrP in neurodegeneration, especially in Alzheimer’s disease and prion diseases, may be explained by considering the conserved proteolytic processing of PrPC, resulting in the production of different fragments with supposedly overlapping but also distinct functions. While membrane-anchored PrPC transduces toxicity of harmful protein assemblies, its extracellular forms (particularly N1, sPrP, and EV-PrP) seem to protect cells by trapping and possibly sequestrating these assemblies and reducing their toxicity. Therefore, any mechanisms capable of reducing fl-PrPC levels at the cell surface while increasing the release of protective PrPC forms into the extracellular space (such as the PrP-directed antibody treatment discussed herein) deserve to be considered and further investigated as potential treatment options against these currently incurable conditions. Moreover, the released PrPC derivatives likely hold functions as ligands in intercellular communication and may regulate diverse physiological processes. Further research into this direction is likewise certainly well justified, especially concerning regenerative processes in the brain.
Footnotes
Author contributions: AMA, BM, and HCA conceptualized and drafted the manuscript. All authors provided critical input and ideas and revised the text carefully. HCA and MS provided the illustrations. AMA, BM, and HCA finalized the manuscript. FS, SB, BP, and MG provided critical input and discussion and/or wrote short paragraphs of the text. All authors checked and approved the final version.
Conflicts of interest: The authors declare no conflicts of interest. No conflicts of interest exist between the Creutzfeldt-Jakob Disease Foundation, Inc. (USA) and publication of this paper.
Data availability statement: No additional data are available.
C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- 1.Abskharon R, Wang F, Wohlkonig A, Ruan J, Soror S, Giachin G, Pardon E, Zou W, Legname G, Ma J, Steyaert J. Structural evidence for the critical role of the prion protein hydrophobic region in forming an infectious prion. PLoS Pathog. 2019;15:e1008139. doi: 10.1371/journal.ppat.1008139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adle-Biassette H, Verney C, Peoc'h K, Dauge MC, Razavi F, Choudat L, Gressens P, Budka H, Henin D. Immunohistochemical expression of prion protein (PrPC) in the human forebrain during development. J Neuropathol Exp Neurol. 2006;65:698–706. doi: 10.1097/01.jnen.0000228137.10531.72. [DOI] [PubMed] [Google Scholar]
- 3.Aguilar-Calvo P, Sevillano AM, Bapat J, Soldau K, Sandoval DR, Altmeppen HC, Linsenmeier L, Pizzo DP, Geschwind MD, Sanchez H, Appleby BS, Cohen ML, Safar JG, Edland SD, Glatzel M, Nilsson KPR, Esko JD, Sigurdson CJ. Shortening heparan sulfate chains prolongs survival and reduces parenchymal plaques in prion disease caused by mobile, ADAM10-cleaved prions. Acta Neuropathol. 2020;139:527–546. doi: 10.1007/s00401-019-02085-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aguzzi A. Peripheral prion pursuit. J Clin Invest. 2001;108:661–662. doi: 10.1172/JCI13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexandrenne C, Hanoux V, Dkhissi F, Boquet D, Couraud JY, Wijkhuisen A. Curative properties of antibodies against prion protein:a comparative in vitro study of monovalent fragments and divalent antibodies. J Neuroimmunol. 2009;209:50–56. doi: 10.1016/j.jneuroim.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 6.Altmeppen HC, Prox J, Puig B, Kluth MA, Bernreuther C, Thurm D, Jorissen E, Petrowitz B, Bartsch U, De Strooper B, Saftig P, Glatzel M. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol Neurodegener. 2011;6:36. doi: 10.1186/1750-1326-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altmeppen HC, Prox J, Puig B, Dohler F, Falker C, Krasemann S, Glatzel M. Roles of endoproteolytic α-cleavage and shedding of the prion protein in neurodegeneration. FEBS J. 2013;280:4338–4347. doi: 10.1111/febs.12196. [DOI] [PubMed] [Google Scholar]
- 8.Altmeppen HC, Prox J, Krasemann S, Puig B, Kruszewski K, Dohler F, Bernreuther C, Hoxha A, Linsenmeier L, Sikorska B, Liberski PP, Bartsch U, Saftig P, Glatzel M. The sheddase ADAM10 is a potent modulator of prion disease. Elife. 2015;4:e04260. doi: 10.7554/eLife.04260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amin L, Nguyen XTA, Rolle IG, D'Este E, Giachin G, Tran TH, Serbec VC, Cojoc D, Legname G. Characterization of prion protein function by focal neurite stimulation. J Cell Sci. 2016;129:3878–3891. doi: 10.1242/jcs.183137. [DOI] [PubMed] [Google Scholar]
- 10.An K, Klyubin I, Kim Y, Jung JH, Mably AJ, O'Dowd ST, Lynch T, Kanmert D, Lemere CA, Finan GM, Park JW, Kim TW, Walsh DM, Rowan MJ, Kim JH. Exosomes neutralize synaptic-plasticity-disrupting activity of Abeta assemblies in vivo. Mol Brain. 2013;6:47. doi: 10.1186/1756-6606-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bardelli M, Frontzek K, Simonelli L, Hornemann S, Pedotti M, Mazzola F, Carta M, Eckhardt V, D'Antuono R, Virgilio T, González SF, Aguzzi A, Varani L. A bispecific immunotweezer prevents soluble PrP oligomers and abolishes prion toxicity. PLOS Pathog. 2018;14:e1007335. doi: 10.1371/journal.ppat.1007335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barmada S, Piccardo P, Yamaguchi K, Ghetti B, Harris DA. GFP-tagged prion protein is correctly localized and functionally active in the brains of transgenic mice. Neurobiol Dis. 2004;16:527–537. doi: 10.1016/j.nbd.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Beland M, Roucou X. The prion protein unstructured N-terminal region is a broad-spectrum molecular sensor with diverse and contrasting potential functions. J Neurochem. 2012;120:853–868. doi: 10.1111/j.1471-4159.2011.07613.x. [DOI] [PubMed] [Google Scholar]
- 14.Beland M, Bedard M, Tremblay G, Lavigne P, Roucou X. Abeta induces its own prion protein N-terminal fragment (PrPN1)-mediated neutralization in amorphous aggregates. Neurobiol Aging. 2014;35:1537–1548. doi: 10.1016/j.neurobiolaging.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Benestad SL, Austbo L, Tranulis MA, Espenes A, Olsaker I. Healthy goats naturally devoid of prion protein. Vet Res. 2012;43:87. doi: 10.1186/1297-9716-43-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beraldo FH, Ostapchenko VG, Xu JZ, Di Guglielmo GM, Fan J, Nicholls PJ, Caron MG, Prado VF, Prado MAM. Mechanisms of neuroprotection against ischemic insult by stress-inducible phosphoprotein-1/prion protein complex. J Neurochem. 2018;145:68–79. doi: 10.1111/jnc.14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- 18.Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka K V, Nave KA, Weis J, Aguzzi A. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13:310–318. doi: 10.1038/nn.2483. [DOI] [PubMed] [Google Scholar]
- 19.Brenna S, Altmeppen HC, Mohammadi B, Rissiek B, Schlink F, Ludewig P, Krisp C, Schlüter H, Failla AV, Schneider C, Glatzel M, Puig B, Magnus T. Characterization of brain-derived extracellular vesicles reveals changes in cellular origin after stroke and enrichment of the prion protein with a potential role in cellular uptake. J Extracell Vesicles. 2020;9:1809065. doi: 10.1080/20013078.2020.1809065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzschmar H. The cellular prion protein binds copper in vivo. Nature. 1997;390:684–687. doi: 10.1038/37783. [DOI] [PubMed] [Google Scholar]
- 21.Büeler H, Raeber A, Sailer A, Fischer M, Aguzzi A, Weissmann C. High prion and PrPSc levels but delayed onset of disease in scrapie-inoculated mice heterozygous for a disrupted PrP gene. Mol Med. 1994;1:19–30. [PMC free article] [PubMed] [Google Scholar]
- 22.Camacho MV, Telling G, Kong Q, Gambetti P, Notari S. Role of prion protein glycosylation in replication of human prions by protein misfolding cyclic amplification. Lab Invest. 2019;99:1741–1748. doi: 10.1038/s41374-019-0282-1. [DOI] [PubMed] [Google Scholar]
- 23.Cao W, Dong C, Kim S, Hou D, Tai W, Du L, Im W, Zhang XF. Biomechanical characterization of SARS-CoV-2 spike RBD and human ACE2 protein-protein interaction. Biophys J. 2021;120:1011–1019. doi: 10.1016/j.bpj.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter L, Kim SJ, Schneidman-Duhovny D, Stöhr J, Poncet-Montange G, Weiss TM, Tsuruta H, Prusiner SB, Sali A. Prion protein - antibody complexes characterized by chromatography-coupled small-angle X-ray scattering. Biophys J. 2015;109:793–805. doi: 10.1016/j.bpj.2015.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carulla P, Bribián A, Rangel A, Gavín R, Ferrer I, Caelles C, Del Río JA, Llorens F. Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol Biol Cell. 2011;22:3041–3054. doi: 10.1091/mbc.E11-04-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng CJ, Koldsø H, Van der Kamp MW, Schiøtt B, Daggett V. Simulations of membrane-bound diglycosylated human prion protein reveal potential protective mechanisms against misfolding. J Neurochem. 2017;142:171–182. doi: 10.1111/jnc.14044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- 28.Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci. 2010;11:130. doi: 10.1186/1471-2202-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coitinho AS, Roesler R, Martins VR, Brentani RR, Izquierdo I. Cellular prion protein ablation impairs behavior as a function of age. Neuroreport. 2003;14:1375–1379. doi: 10.1097/01.wnr.0000078541.07662.90. [DOI] [PubMed] [Google Scholar]
- 30.Corbett GT, Wang Z, Hong W, Colom-Cadena M, Rose J, Liao M, Asfaw A, Hall TC, Ding L, DeSousa A, Frosch MP, Collinge J, Harris DA, Perkinton MS, Spires-Jones TL, Young-Pearse TL, Billinton A, Walsh DM. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 2020;139:503–526. doi: 10.1007/s00401-019-02114-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corda E, Du X, Shim SY, Klein AN, Siltberg-Liberles J, Gilch S. Interaction of peptide aptamers with prion protein central domain promotes α-cleavage of PrP C. Mol Neurobiol. 2018;55:7758–7774. doi: 10.1007/s12035-018-0944-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D'Arrigo G, Gabrielli M, Scaroni F, Swuec P, Amin L, Pegoraro A, Adinolfi E, Di Virgilio F, Cojoc D, Legname G, Verderio C. Astrocytes-derived extracellular vesicles in motion at the neuron surface:involvement of the prion protein. J Extracell vesicles. 2021;10:e12114. doi: 10.1002/jev2.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Cecco E, Legname G. The role of the prion protein in the internalization of alpha-synuclein amyloids. Prion. 2018;12:23–27. doi: 10.1080/19336896.2017.1423186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Mario A, Peggion C, Massimino ML, Norante RP, Zulian A, Bertoli A, Sorgato MC. The link of the prion protein with Ca2+metabolism and ROS production, and the possible implication in Aβtoxicity. Int J Mol Sci. 2019;20:4640. doi: 10.3390/ijms20184640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Demarco ML, Daggett V. Characterization of cell-surface prion protein relative to its recombinant analogue:insights from molecular dynamics simulations of diglycosylated, membrane-bound human prion protein. J Neurochem. 2009;109:60–73. doi: 10.1111/j.1471-4159.2009.05892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng W, Cho S, Su PC, Berger BW, Li R. Membrane-enabled dimerization of the intrinsically disordered cytoplasmic domain of ADAM10. Proc Natl Acad Sci U S A. 2014;111:15987–15992. doi: 10.1073/pnas.1409354111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Doeppner TR, Kaltwasser B, Schlechter J, Jaschke J, Kilic E, Bähr M, Hermann DM, Weise J. Cellular prion protein promotes post-ischemic neuronal survival, angioneurogenesis and enhances neural progenitor cell homing via proteasome inhibition. Cell Death Dis. 2015;6:e2024–2024. doi: 10.1038/cddis.2015.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci U S A. 2001;98:9295–9299. doi: 10.1073/pnas.151242598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Epis R, Marcello E, Gardoni F, Vastagh C, Malinverno M, Balducci C, Colombo A, Borroni B, Vara H, Dell'Agli M, Cattabeni F, Giustetto M, Borsello T, Forloni G, Padovani A, Di Luca M. Blocking ADAM10 synaptic trafficking generates a model of sporadic Alzheimer's disease. Brain. 2010;133:3323–3335. doi: 10.1093/brain/awq217. [DOI] [PubMed] [Google Scholar]
- 40.Falker C, Hartmann A, Guett I, Dohler F, Altmeppen H, Betzel C, Schubert R, Thurm D, Wegwitz F, Joshi P, Verderio C, Krasemann S, Glatzel M. Exosomal cellular prion protein drives fibrillization of amyloid beta and counteracts amyloid beta-mediated neurotoxicity. J Neurochem. 2016;137:88–100. doi: 10.1111/jnc.13514. [DOI] [PubMed] [Google Scholar]
- 41.Ferreira DG, Temido-Ferreira M, Miranda H V, Batalha VL, Coelho JE, Szego EM, Marques-Morgado I, Vaz SH, Rhee JS, Schmitz M, Zerr I, Lopes L V, Outeiro TF. Alpha-synuclein interacts with PrPC to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci. 2017;20:1569–1579. doi: 10.1038/nn.4648. [DOI] [PubMed] [Google Scholar]
- 42.Fischer M, Rulicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- 43.Fluharty BR, Biasini E, Stravalaci M, Sclip A, Diomede L, Balducci C, La Vitola P, Messa M, Colombo L, Forloni G, Borsello T, Gobbi M, Harris DA. An N-terminal fragment of the prion protein binds to amyloid-beta oligomers and inhibits their neurotoxicity in vivo. J Biol Chem. 2013;288:7857–7866. doi: 10.1074/jbc.M112.423954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, Clarke AR, Rowan MJ, Walsh DM, Collinge J. Interaction between prion protein and toxic amyloid beta assemblies can be therapeutically targeted at multiple sites. Nat Commun. 2011;2:336. doi: 10.1038/ncomms1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Friberg KN, Hung G, Wancewicz E, Giles K, Black C, Freier S, Bennett F, DeArmond SJ, Freyman Y, Lessard P, Ghaemmaghami S, Prusiner SB. Intracerebral infusion of antisense oligonucleotides into prion-infected mice. Mol Ther Nucleic Acids. 2012;1:e9. doi: 10.1038/mtna.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frontzek K, Bardelli M, Senatore A, Henzi A, Reimann RR, Bedir S, Marino M, Hussain R, Jurt S, Meisl G, Pedotti M, Mazzola F, Siligardi G, Zerbe O, Losa M, Knowles T, Lakkaraju A, Zhu C, Schwarz P, Hornemann S, et al. ( 2022) A conformational switch controlling the toxicity of the prion protein. Nat Struct Mol Biol. 29:831–840. doi: 10.1038/s41594-022-00814-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gasperini L, Meneghetti E, Pastore B, Benetti F, Legname G. Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal. 2015;22:772–784. doi: 10.1089/ars.2014.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gilch S, Wopfner F, Renner-Müller I, Kremmer E, Bauer C, Wolf E, Brem G, Groschup MH, Schätzl HM. Polyclonal anti-PrP auto-antibodies induced with dimeric PrP interfere efficiently with PrPSc propagation in prion-infected cells. J Biol Chem. 2003;278:18524–18531. doi: 10.1074/jbc.M210723200. [DOI] [PubMed] [Google Scholar]
- 49.Gomes LA, Hipp SA, Rijal Upadhaya A, Balakrishnan K, Ospitalieri S, Koper MJ, Largo-Barrientos P, Uytterhoeven V, Reichwald J, Rabe S, Vandenberghe R, von Arnim CAF, Tousseyn T, Feederle R, Giudici C, Willem M, Staufenbiel M, Thal DR. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019;138:913–941. doi: 10.1007/s00401-019-02053-5. [DOI] [PubMed] [Google Scholar]
- 50.Gonias SL, Banki MA, Azmoon P, Romero HK, Sigurdson CJ, Mantuano E, Campana WM. Cellular prion protein in human plasma-derived extracellular vesicles promotes neurite outgrowth via the NMDA receptor-LRP1 receptor system. J Biol Chem. 2022;298:101642. doi: 10.1016/j.jbc.2022.101642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Griffiths HH, Whitehouse IJ, Baybutt H, Brown D, Kellett KA, Jackson CD, Turner AJ, Piccardo P, Manson JC, Hooper NM. Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J Biol Chem. 2011;286:33489–33500. doi: 10.1074/jbc.M111.278556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guitart K, Loers G, Buck F, Bork U, Schachner M, Kleene R. Improvement of neuronal cell survival by astrocyte-derived exosomes under hypoxic and ischemic conditions depends on prion protein. Glia. 2016;64:896–910. doi: 10.1002/glia.22963. [DOI] [PubMed] [Google Scholar]
- 53.Haigh CL, Drew SC, Boland MP, Masters CL, Barnham KJ, Lawson VA, Collins SJ. Dominant roles of the polybasic proline motif and copper in the PrP23-89-mediated stress protection response. J Cell Sci. 2009;122:1518–1528. doi: 10.1242/jcs.043604. [DOI] [PubMed] [Google Scholar]
- 54.Hallinan GI, Ozcan KA, Hoq MR, Cracco L, Vago FS, Bharath SR, Li D, Jacobsen M, Doud EH, Mosley AL, Fernandez A, Garringer HJ, Jiang W, Ghetti B, Vidal R. Cryo-EM structures of prion protein filaments from Gerstmann-Sträussler-Scheinker disease. Acta Neuropathol. 2022;144:509–520. doi: 10.1007/s00401-022-02461-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hartmann M, Parra LM, Ruschel A, Lindner C, Morrison H, Herrlich A, Herrlich P. Inside-out regulation of ectodomain cleavage of cluster-of-differentiation-44 (CD44) and of neuregulin-1 requires substrate dimerization. J Biol Chem. 2015;290:17041–17054. doi: 10.1074/jbc.M114.610204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heppner FL, Musahl C, Arrighi I, Klein MA, Rülicke T, Oesch B, Zinkernagel RM, Kalinke U, Aguzzi A. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science. 2001;294:178–182. doi: 10.1126/science.1063093. [DOI] [PubMed] [Google Scholar]
- 57.Herrmann US, Sonati T, Falsig J, Reimann RR, Dametto P, O'Connor T. Correction:prion infections and anti-PrP antibodies trigger converging neurotoxic pathways. PLoS Pathog. 2015;11:e1004808. doi: 10.1371/journal.ppat.1004662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hitschler L, Lang T. The transmembrane domain of the amyloid precursor protein is required for antiamyloidogenic processing by α-secretase ADAM10. J Biol Chem. 2022;298:101911. doi: 10.1016/j.jbc.2022.101911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoffmann D, Mereiter S, Jin Oh Y, Monteil V, Elder E, Zhu R, Canena D, Hain L, Laurent E, Grünwald-Gruber C, Klausberger M, Jonsson G, Kellner MJ, Novatchkova M, Ticevic M, Chabloz A, Wirnsberger G, Hagelkruys A, Altmann F, Mach L, et al. Identification of lectin receptors for conserved SARS-CoV-2 glycosylation sites. EMBO J. 2021;40:e108375. doi: 10.15252/embj.2021108375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hoyt F, Standke HG, Artikis E, Schwartz CL, Hansen B, Li K, Hughson AG, Manca M, Thomas OR, Raymond GJ, Race B, Baron GS, Caughey B, Kraus A. Cryo-EM structure of anchorless RML prion reveals variations in shared motifs between distinct strains. Nat Commun. 2022;13:4005. doi: 10.1038/s41467-022-30458-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu NW, Corbett GT, Moore S, Klyubin I, O'Malley TT, Walsh DM, Livesey FJ, Rowan MJ. Extracellular forms of Aβand Tau from iPSC models of Alzheimer's disease disrupt synaptic plasticity. Cell Rep. 2018;23:1932–1938. doi: 10.1016/j.celrep.2018.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB. Adam meets Eph:an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell. 2005;123:291–304. doi: 10.1016/j.cell.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 63.Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, Bartsch U, Weskamp G, Blobel CP, Glatzel M, De Strooper B, Saftig P. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci. 2010;30:4833–4844. doi: 10.1523/JNEUROSCI.5221-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaiser DM, Acharya M, Leighton PLA, Wang H, Daude N, Wohlgemuth S, Shi B, Allison WT. Amyloid beta precursor protein and prion protein have a conserved interaction affecting cell adhesion and CNS development. PLoS One. 2012;7:e51305. doi: 10.1371/journal.pone.0051305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kamali-Jamil R, Ester Vázquez-Fernández, Tancowny B, Rathod V, Amidian S, Wang X, Tang X, Fang A, Senatore A, Hornemann S, Dudas S, Aguzzi A, Young HS, Wille H. The ultrastructure of infectious L-type bovine spongiform encephalopathy prions constrains molecular models. PLoS Pathog. 2021;17:e1009628. doi: 10.1371/journal.ppat.1009628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, Chen L, Villemaire M, Ali Z, Jirik FR, Zamponi GW. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol. 2008;181:551–565. doi: 10.1083/jcb.200711002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Klyubin I, Nicoll AJ, Khalili-Shirazi A, Farmer M, Canning S, Mably A, Linehan J, Brown A, Wakeling M, Brandner S, Walsh DM, Rowan MJ, Collinge J. Peripheral administration of a humanized anti-PrP antibody blocks Alzheimer's disease Abeta synaptotoxicity. J Neurosci. 2014;34:6140–6145. doi: 10.1523/JNEUROSCI.3526-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koo CZ, Harrison N, Noy PJ, Szyroka J, Matthews AL, Hsia HE, Müller SA, Tüshaus J, Goulding J, Willis K, Apicella C, Cragoe B, Davis E, Keles M, Malinova A, McFarlane TA, Morrison PR, Nguyen HTH, Sykes MC, Ahmed H, et al. The tetraspanin Tspan15 is an essential subunit of an ADAM10 scissor complex. J Biol Chem. 2020;295:12822–12839. doi: 10.1074/jbc.RA120.012601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kostylev MA, Kaufman AC, Nygaard HB, Patel P, Haas LT, Gunther EC, Vortmeyer A, Strittmatter SM. Prion-protein-interacting amyloid-beta oligomers of high molecular weight are tightly correlated with memory impairment in multiple Alzheimer mouse models. J Biol Chem. 2015;290:17415–17438. doi: 10.1074/jbc.M115.643577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kovač V, Čurin Šerbec V. Prion protein:the molecule of many forms and faces. Int J Mol Sci. 2022;23:1232. doi: 10.3390/ijms23031232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kraus A, Hoyt F, Schwartz CL, Hansen B, Artikis E, Hughson AG, Raymond GJ, Race B, Baron GS, Caughey B. High-resolution structure and strain comparison of infectious mammalian prions. Mol Cell. 2021;81:4540–4551. doi: 10.1016/j.molcel.2021.08.011. [DOI] [PubMed] [Google Scholar]
- 72.Küffer A, Lakkaraju AKK, Mogha A, Petersen SC, Airich K, Doucerain C, Marpakwar R, Bakirci P, Senatore A, Monnard A, Schiavi C, Nuvolone M, Grosshans B, Hornemann S, Bassilana F, Monk KR, Aguzzi A. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature. 2016;536:464–468. doi: 10.1038/nature19312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010;29:3020–3032. doi: 10.1038/emboj.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lakkaraju AKK, Sorce S, Senatore A, Nuvolone M, Guo J, Schwarz P, Moos R, Pelczar P, Aguzzi A. Glial activation in prion diseases is selectively triggered by neuronal PrPSc. Brain Pathol. 2022;32:e13056. doi: 10.1111/bpa.13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lefebvre-Roque M, Kremmer E, Gilch S, Zou W-Q, Féraudet C, Gilles CM, Salès N, Grassi J, Gambetti P, Baron T, Schätzl H, Lasmézas CI. Toxic effects of intracerebral PrP antibody administration during the course of BSE infection in mice. Prion. 2007;1:198–206. doi: 10.4161/pri.1.3.4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Legname G, Scialò C. On the role of the cellular prion protein in the uptake and signaling of pathological aggregates in neurodegenerative diseases. Prion. 2020;14:257–270. doi: 10.1080/19336896.2020.1854034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lehmann S, Relano-Gines A, Resina S, Brillaud E, Casanova D, Vincent C, Hamela C, Poupeau S, Laffont M, Gabelle A, Delaby C, Belondrade M, Arnaud JD, Alvarez MT, Maurel JC, Maurel P, Crozet C. Systemic delivery of siRNA down regulates brain prion protein and ameliorates neuropathology in prion disorder. PLoS One. 2014;9:e88797. doi: 10.1371/journal.pone.0088797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Levy S, Shoham T. Protein-protein interactions in the tetraspanin web. Physiology (Bethesda) 2005;20:218–224. doi: 10.1152/physiol.00015.2005. [DOI] [PubMed] [Google Scholar]
- 80.Linsenmeier L, Altmeppen HC, Wetzel S, Mohammadi B, Saftig P, Glatzel M. Diverse functions of the prion protein –Does proteolytic processing hold the key? Biochim Biophys Acta Mol Cell Res. 2017;1864:2128–2137. doi: 10.1016/j.bbamcr.2017.06.022. [DOI] [PubMed] [Google Scholar]
- 81.Linsenmeier L, Mohammadi B, Wetzel S, Puig B, Jackson WS, Hartmann A, Uchiyama K, Sakaguchi S, Endres K, Tatzelt J, Saftig P, Glatzel M, Altmeppen HC. Structural and mechanistic aspects influencing the ADAM10-mediated shedding of the prion protein. Mol Neurodegener. 2018;13:18. doi: 10.1186/s13024-018-0248-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Linsenmeier L, Mohammadi B, Shafiq M, Frontzek K, Bär J, Shrivastava AN, Damme M, Song F, Schwarz A, Da Vela S, Massignan T, Jung S, Correia A, Schmitz M, Puig B, Hornemann S, Zerr I, Tatzelt J, Biasini E, Saftig P, et al. Ligands binding to the prion protein induce its proteolytic release with therapeutic potential in neurodegenerative proteinopathies. Sci Adv. 2021;7:1826. doi: 10.1126/sciadv.abj1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lipper CH, Gabriel KH, Seegar TCM, Dürr KL, Tomlinson MG, Blacklow SC. Crystal structure of the Tspan15 LEL domain reveals a conserved ADAM10 binding site. Structure. 2022;30:206–214. doi: 10.1016/j.str.2021.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu Q, Zhang J, Tran H, Verbeek MM, Reiss K, Estus S, Bu G. LRP1 shedding in human brain:roles of ADAM10 and ADAM17. Mol Neurodegener. 2009;4:17. doi: 10.1186/1750-1326-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Loubet D, Dakowski C, Pietri M, Pradines E, Bernard S, Callebert J, Ardila-Osorio H, Mouillet-Richard S, Launay JM, Kellermann O, Schneider B. Neuritogenesis:the prion protein controls beta1 integrin signaling activity. FASEB J. 2011;26:678–690. doi: 10.1096/fj.11-185579. [DOI] [PubMed] [Google Scholar]
- 86.Maglio LE, Martins VR, Izquierdo I, Ramirez OA. Role of cellular prion protein on LTP expression in aged mice. Brain Res. 2006;1097:11–18. doi: 10.1016/j.brainres.2006.04.056. [DOI] [PubMed] [Google Scholar]
- 87.Málaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, Stuermer CA. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 2009;7:e55. doi: 10.1371/journal.pbio.1000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- 89.Mantuano E, Azmoon P, Banki MA, Lam MS, Sigurdson CJ, Gonias SL. A soluble derivative of PrPC activates cell-signaling and regulates cell physiology through LRP1 and the NMDA receptor. J Biol Chem. 2020;295:14178–14188. doi: 10.1074/jbc.RA120.013779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mantuano E, Azmoon P, Banki MA, Sigurdson CJ, Campana WM, Gonias SL. A soluble PrPC derivative and membrane-anchored PrPC in extracellular vesicles attenuate innate immunity by engaging the NMDA-R/LRP1 receptor complex. J Immunol. 2022;208:85–96. doi: 10.4049/jimmunol.2100412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martellucci S, Santacroce C, Santilli F, Piccoli L, Monache SD, Angelucci A, Misasi R, Sorice M, Mattei V. Cellular and molecular mechanisms mediated by recPrP C involved in the neuronal differentiation process of mesenchymal stem cells. Int J Mol Sci. 2019;20:345. doi: 10.3390/ijms20020345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Matamoros-Angles A, Hervera A, Soriano J, Martí E, Carulla P, Llorens F, Nuvolone M, Aguzzi A, Ferrer I, Gruart A, Delgado-García JM, Del Río JA. Analysis of co-isogenic prion protein deficient mice reveals behavioral deficits, learning impairment, and enhanced hippocampal excitability. BMC Biol. 2022;20:17. doi: 10.1186/s12915-021-01203-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mclennan NF, Brennan PM, Mcneill A, Davies I, Fotheringham A, Rennison KA, Ritchie D, Brannan F, Head MW, Ironside JW, Williams A, Bell JE. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am J Pathol. 2004;165:227–235. doi: 10.1016/S0002-9440(10)63291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mead S, Khalili-Shirazi A, Potter C, Mok T, Nihat A, Hyare H, Canning S, Schmidt C, Campbell T, Darwent L, Muirhead N, Ebsworth N, Hextall P, Wakeling M, Linehan J, Libri V, Williams B, Jaunmuktane Z, Brandner S, Rudge P, et al. Prion protein monoclonal antibody (PRN100) therapy for Creutzfeldt–Jakob disease:evaluation of a first-in-human treatment programme. Lancet Neurol. 2022;21:342–354. doi: 10.1016/S1474-4422(22)00082-5. [DOI] [PubMed] [Google Scholar]
- 95.Minikel EV, Zhao HT, Le J, O'Moore J, Pitstick R, Graffam S, Carlson GA, Kavanaugh MP, Kriz J, Kim JB, Ma J, Wille H, Aiken J, McKenzie D, Doh-Ura K, Beck M, O'Keefe R, Stathopoulos J, Caron T, Schreiber SL, et al. Prion protein lowering is a disease-modifying therapy across prion disease stages, strains and endpoints. Nucleic Acids Res. 2020;48:10615–10631. doi: 10.1093/nar/gkaa616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mitteregger G, Vosko M, Krebs B, Xiang W, Kohlmannsperger V, Nolting S, Hamann GF, Kretzschmar HA. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007;17:174–183. doi: 10.1111/j.1750-3639.2007.00061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mohammadi B, Song F, Matamoros-Angles A, Shafiq M, Damme M, Puig B, Glatzel M, Altmeppen HC. Anchorless risk or released benefit?An updated view on the ADAM10-mediated shedding of the prion protein. Cell Tissue Res. 2022 doi: 10.1007/s00441-022-03582-4. doi:10.1007/s00441-022-03582-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morel E, Fouquet S, Chateau D, Yvernault L, Frobert Y, Pincon-Raymond M, Chambaz J, Pillot T, Rousset M. The cellular prion protein PrPc is expressed in human enterocytes in cell-cell junctional domains. J Biol Chem. 2004;279:1499–1505. doi: 10.1074/jbc.M308578200. [DOI] [PubMed] [Google Scholar]
- 99.Murakami K, Izuo N, Bitan G. Aptamers targeting amyloidogenic proteins and their emerging role in neurodegenerative diseases. J Biol Chem. 2022;298:101478. doi: 10.1016/j.jbc.2021.101478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nakić N, Tran TH, Novokmet M, Andreoletti O, Lauc G, Legname G. Correction:site-specific analysis of N-glycans from different sheep prion strains. PLoS Pathog. 2021;17:e1009511. doi: 10.1371/journal.ppat.1009232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nieznanska H, Boyko S, Dec R, Redowicz MJ, Dzwolak W, Nieznanski K. Neurotoxicity of oligomers of phosphorylated Tau protein carrying tauopathy-associated mutation is inhibited by prion protein. Biochim Biophys acta Mol basis Dis. 2021;1867:166209. doi: 10.1016/j.bbadis.2021.166209. [DOI] [PubMed] [Google Scholar]
- 102.Ondrejcak T, Klyubin I, Corbett GT, Fraser G, Hong W, Mably AJ, Gardener M, Hammersley J, Perkinton MS, Billinton A, Walsh DM, Rowan MJ. Cellular prion protein mediates the disruption of hippocampal synaptic plasticity by soluble Tau in vivo. J Neurosci. 2018;38:10595–10606. doi: 10.1523/JNEUROSCI.1700-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Parkyn CJ, Vermeulen EGM, Mootoosamy RC, Sunyach C, Jacobsen C, Oxvig C, Moestrup S, Liu Q, Bu G, Jen A, Morris RJ. LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J Cell Sci. 2008;121:773–783. doi: 10.1242/jcs.021816. [DOI] [PubMed] [Google Scholar]
- 104.Parrie LE, Crowell JAE, Moreno JA, Suinn SS, Telling GC, Bessen RA. The cellular prion protein promotes neuronal regeneration after acute nasotoxic injury. Prion. 2020;14:31–41. doi: 10.1080/19336896.2020.1714373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Peretz D, Williamson RA, Kaneko K, Vergara J, Leclerc E, Schmitt-Ulms G, Mehlhorn IR, Legname G, Wormald MR, Rudd PM, Dwek RA, Burton DR, Prusiner SB. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412:739–743. doi: 10.1038/35089090. [DOI] [PubMed] [Google Scholar]
- 106.Perrier V, Solassol J, Crozet C, Frobert Y, Mourton-Gilles C, Grassi J, Lehmann S. Anti-PrP antibodies block PrPSc replication in prion-infected cell cultures by accelerating PrPC degradation. J Neurochem. 2004;89:454–463. doi: 10.1111/j.1471-4159.2004.02356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, Morris JH, Ferrin TE. UCSF ChimeraX:structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82. doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Polymenidou M, Moos R, Scott M, Sigurdson C, Shi YZ, Yajima B, Hafner-Bratkovic I, Jerala R, Hornemann S, Wuthrich K, Bellon A, Vey M, Garen G, James MN, Kav N, Aguzzi A. The POM monoclonals:a comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS One. 2008;3:e3872. doi: 10.1371/journal.pone.0003872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Prusiner SB, Gajdusek C, Alpers MP. Kuru with incubation periods exceeding two decades. Ann Neurol. 1982;12:1–9. doi: 10.1002/ana.410120102. [DOI] [PubMed] [Google Scholar]
- 110.Puig B, Altmeppen HC, Thurm D, Geissen M, Conrad C, Braulke T, Glatzel M. N-glycans and glycosylphosphatidylinositol-anchor act on polarized sorting of mouse PrP(C) in Madin-Darby canine kidney cells. PLoS One. 2011;6:e24624. doi: 10.1371/journal.pone.0024624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Puig B, Altmeppen HC, Linsenmeier L, Chakroun K, Wegwitz F, Piontek UK, Tatzelt J, Bate C, Magnus T, Glatzel M. GPI-anchor signal sequence influences PrPC sorting, shedding and signalling, and impacts on different pathomechanistic aspects of prion disease in mice. PLoS Pathog. 2019;15:e1007520. doi: 10.1371/journal.ppat.1007520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pulford B, Reim N, Bell A, Veatch J, Forster G, Bender H, Meyerett C, Hafeman S, Michel B, Johnson T, Wyckoff C, Miele G, Julius AC, Kranich J, Schenkel A, Dow S, Zabel MD. Liposome-siRNA-peptide complexes cross the blood-brain barrier and significantly decrease PrP on neuronal cells and PrP in infected cell cultures. PLoS One. 2010;5:e11085. doi: 10.1371/journal.pone.0011085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Race B, Jeffrey M, McGovern G, Dorward D, Chesebro B. Ultrastructure and pathology of prion protein amyloid accumulation and cellular damage in extraneural tissues of scrapie-infected transgenic mice expressing anchorless prion protein. Prion. 2017;11:234–248. doi: 10.1080/19336896.2017.1336274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Raymond GJ, Zhao HT, Race B, Raymond LD, Williams K, Swayze EE, Graffam S, Le J, Caron T, Stathopoulos J, O'Keefe R, Lubke LL, Reidenbach AG, Kraus A, Schreiber SL, Mazur C, Cabin DE, Carroll JB, Minikel EV, Kordasiewicz H, et al. Antisense oligonucleotides extend survival of prion-infected mice. JCI Insight. 2019;5:e131175. doi: 10.1172/jci.insight.131175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Reilly M, Benilova I, Khalili-Shirazi A, Schmidt C, Ahmed P, Yip D, Jat PS, Collinge J. A high-content neuron imaging assay demonstrates inhibition of prion disease-associated neurotoxicity by an anti-prion protein antibody. Sci Rep. 2022;12:9493. doi: 10.1038/s41598-022-13455-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Reimann RR, Sonati T, Hornemann S, Herrmann US, Arand M, Hawke S, Aguzzi A. Differential toxicity of antibodies to the prion protein. PLoS Pathog. 2016;12:e1005401. doi: 10.1371/journal.ppat.1005401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Müller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, Multhaup G, Winklhofer KF, Tatzelt J. The cellular prion protein mediates neurotoxic signalling of β-sheet-rich conformers independent of prion replication. EMBO J. 2011;30:2057–2070. doi: 10.1038/emboj.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rösener NS, Gremer L, Wördehoff MM, Kupreichyk T, Etzkorn M, Neudecker P, Hoyer W. Clustering of human prion protein and α-synuclein oligomers requires the prion protein N-terminus. Commun Biol. 2020;3:365. doi: 10.1038/s42003-020-1085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rudd PM, Endo T, Colominas C, Groth D, Wheeler SF, Harvey DJ, Wormald MR, Serban H, Prusiner SB, Kobata A, Dwek RA. Glycosylation differences between the normal and pathogenic prion protein isoforms. Proc Natl Acad Sci U S A. 1999;96:13044–13049. doi: 10.1073/pnas.96.23.13044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sachs M, Wetzel S, Reichelt J, Sachs W, Schebsdat L, Zielinski S, Seipold L, Heintz L, Müller SA, Kretz O, Lindenmeyer M, Wiech T, Huber TB, Lüllmann-Rauch R, Lichtenthaler SF, Saftig P, Meyer-Schwesinger C. ADAM10-mediated ectodomain shedding is an essential driver of podocyte damage. J Am Soc Nephrol. 2021;32:1389–1408. doi: 10.1681/ASN.2020081213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Saftig P, Lichtenthaler SF. The alpha secretase ADAM10:a metalloprotease with multiple functions in the brain. Prog Neurobiol. 2015;135:1–20. doi: 10.1016/j.pneurobio.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 122.Saint-Pol J, Eschenbrenner E, Dornier E, Boucheix C, Charrin S, Rubinstein E. Regulation of the trafficking and the function of the metalloprotease ADAM10 by tetraspanins. Biochem Soc Trans. 2017;45:937–944. doi: 10.1042/BST20160296. [DOI] [PubMed] [Google Scholar]
- 123.Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature. 2011;470:540–542. doi: 10.1038/nature09768. [DOI] [PubMed] [Google Scholar]
- 124.Santuccione A, Sytnyk V, Leshchyns'ka I, Schachner M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol. 2005;169:341–354. doi: 10.1083/jcb.200409127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schelter F, Kobuch J, Moss ML, Becherer JD, Comoglio PM, Boccaccio C, Kruger A. A disintegrin and metalloproteinase-10 (ADAM-10) mediates DN30 antibody-induced shedding of the met surface receptor. J Biol Chem. 2010;285:26335–26340. doi: 10.1074/jbc.M110.106435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schilling KM, Tao L, Wu B, Kiblen JTM, Ubilla-Rodriguez NC, Pushie MJ, Britt RD, Roseman GP, Harris DA, Millhauser GL. Both N-terminal and C-terminal histidine residues of the prion protein are essential for copper coordination and neuroprotective self-regulation. J Mol Biol. 2020;432:4408–4425. doi: 10.1016/j.jmb.2020.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schmitt-Ulms G, Legname G, Baldwin MA, Ball HL, Bradon N, Bosque PJ, Crossin KL, Edelman GM, DeArmond SJ, Cohen FE, Prusiner SB. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J Mol Biol. 2001;314:1209–1225. doi: 10.1006/jmbi.2000.5183. [DOI] [PubMed] [Google Scholar]
- 128.Schmitz M, Greis C, Ottis P, Silva CJ, Schulz-Schaeffer WJ, Wrede A, Koppe K, Onisko B, Requena JR, Govindarajan N, Korth C, Fischer A, Zerr I. Loss of prion protein leads to age-dependent behavioral abnormalities and changes in cytoskeletal protein expression. Mol Neurobiol. 2014;50:923–936. doi: 10.1007/s12035-014-8655-3. [DOI] [PubMed] [Google Scholar]
- 129.Scott-McKean JJ, Surewicz K, Choi JK, Ruffin VA, Salameh AI, Nieznanski K, Costa ACS, Surewicz WK. Soluble prion protein and its N-terminal fragment prevent impairment of synaptic plasticity by Aβoligomers:implications for novel therapeutic strategy in Alzheimer's disease. Neurobiol Dis. 2016;91:124–131. doi: 10.1016/j.nbd.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Seegar TCM, Killingsworth LB, Saha N, Meyer PA, Patra D, Zimmerman B, Janes PW, Rubinstein E, Nikolov DB, Skiniotis G, Kruse AC, Blacklow SC. Structural basis for regulated proteolysis by the α-secretase ADAM10. Cell. 2017;171:1638–1648. doi: 10.1016/j.cell.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Seipold L, Altmeppen H, Koudelka T, Tholey A, Kasparek P, Sedlacek R, Schweizer M, Bar J, Mikhaylova M, Glatzel M, Saftig P. In vivo regulation of the A disintegrin and metalloproteinase 10 (ADAM10) by the tetraspanin 15. Cell Mol Life Sci. 2018;75:3251–3267. doi: 10.1007/s00018-018-2791-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sevillano AM, Aguilar-Calvo P, Kurt TD, Lawrence JA, Soldau K, Nam TH, Schumann T, Pizzo DP, Nyström S, Choudhury B, Altmeppen H, Esko JD, Glatzel M, Nilsson KPR, Sigurdson CJ. Prion protein glycans reduce intracerebral fibril formation and spongiosis in prion disease. J Clin Invest. 2020;130:1350–1362. doi: 10.1172/JCI131564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Shackleton B, Crawford F, Bachmeier C. Inhibition of ADAM10 promotes the clearance of Aβacross the BBB by reducing LRP1 ectodomain shedding. Fluids Barriers CNS. 2016;13:14. doi: 10.1186/s12987-016-0038-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Shi M, Dennis K, Peschon JJ, Chandrasekaran R, Mikecz K. Antibody-induced shedding of CD44 from adherent cells is linked to the assembly of the cytoskeleton. J Immunol. 2001;167:123–131. doi: 10.4049/jimmunol.167.1.123. [DOI] [PubMed] [Google Scholar]
- 135.Shyu WC, Lin SZ, Chiang MF, Ding DC, Li KW, Chen SF, Yang HI, Li H. Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J Neurosci. 2005;25:8967–8977. doi: 10.1523/JNEUROSCI.1115-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Slapsak U, Salzano G, Amin L, Abskharon RN, Ilc G, Zupancic B, Biljan I, Plavec J, Giachin G, Legname G. The N terminus of the prion protein mediates functional interactions with the neuronal cell adhesion molecule (NCAM) fibronectin domain. J Biol Chem. 2016;291:21857–21868. doi: 10.1074/jbc.M116.743435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Solforosi L, Criado JR, McGavern DB, Wirz S, Sanchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, Masliah E, Gilden D, Oldstone MB, Conti B, Williamson RA. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 2004;303:1514–1516. doi: 10.1126/science.1094273. [DOI] [PubMed] [Google Scholar]
- 138.Sonati T, Reimann RR, Falsig J, Baral PK, O'Connor T, Hornemann S, Yaganoglu S, Li B, Herrmann US, Wieland B, Swayampakula M, Rahman MH, Das D, Kav N, Riek R, Liberski PP, James MN, Aguzzi A. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature. 2013;501:102–106. doi: 10.1038/nature12402. [DOI] [PubMed] [Google Scholar]
- 139.Spagnolli G, Rigoli M, Orioli S, Sevillano AM, Faccioli P, Wille H, Biasini E, Requena JR. Full atomistic model of prion structure and conversion. PLoS Pathog. 2019;15:e1007864. doi: 10.1371/journal.ppat.1007864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 141.Steele A, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci U S A. 2006;103:3416–3421. doi: 10.1073/pnas.0511290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Steele AD, Lindquist S, Aguzzi A. The prion protein knockout mouse:a phenotype under challenge. Prion. 2007;1:83–93. doi: 10.4161/pri.1.2.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Taylor DR, Hooper NM. The low-density lipoprotein receptor-related protein 1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem J. 2007;402:17–23. doi: 10.1042/BJ20061736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Taylor DR, Parkin ET, Cocklin SL, Ault JR, Ashcroft AE, Turner AJ, Hooper NM. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J Biol Chem. 2009;284:22590–22600. doi: 10.1074/jbc.M109.032599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Telling GC. The shape of things to come:structural insights into how prion proteins encipher heritable information. Nat Commun. 2022;13:4003. doi: 10.1038/s41467-022-31460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tremblay P, Meiner Z, Galou M, Heinrich C, Petromilli C, Lisse T, Cayetano J, Torchia M, Mobley W, Bujard H, DeArmond SJ, Prusiner SB. Doxycycline control of prion protein transgene expression modulates prion disease in mice. Proc Natl Acad Sci U S A. 1998;95:12580–12585. doi: 10.1073/pnas.95.21.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Urrea L, Ferrer I, Gavin R, Del Rio JA. The cellular prion protein (PrPC) as neuronal receptor for alpha-synuclein. Prion. 2017;11:226–233. doi: 10.1080/19336896.2017.1334748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vergara C, Ordonez-Gutierrez L, Wandosell F, Ferrer I, del Rio JA, Gavin R. Role of PrP(C) expression in Tau protein levels and phosphorylation in Alzheimer's disease evolution. Mol Neurobiol. 2015;51:1206–1220. doi: 10.1007/s12035-014-8793-7. [DOI] [PubMed] [Google Scholar]
- 149.Walz R, Amaral OB, Rockenbach IC, Roesler R, Izquierdo I, Cavalheiro EA, Martins VR, Brentani RR. Increased sensitivity to seizures in mice lacking cellular prion protein. Epilepsia. 1999;40:1679–1682. doi: 10.1111/j.1528-1157.1999.tb01583.x. [DOI] [PubMed] [Google Scholar]
- 150.Watt NT, Taylor DR, Gillott A, Thomas DA, Perera WS, Hooper NM. Reactive oxygen species-mediated beta-cleavage of the prion protein in the cellular response to oxidative stress. J Biol Chem. 2005;280:35914–35921. doi: 10.1074/jbc.M507327200. [DOI] [PubMed] [Google Scholar]
- 151.Weise J, Sandau R, Schwarting S, Crome O, Wrede A, Schulz-Schaeffer W, Zerr I, Bähr M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke. 2006;37:1296–1300. doi: 10.1161/01.STR.0000217262.03192.d4. [DOI] [PubMed] [Google Scholar]
- 152.White MD, Farmer M, Mirabile I, Brandner S, Collinge J, Mallucci GR. Single treatment with RNAi against prion protein rescues early neuronal dysfunction and prolongs survival in mice with prion disease. Proc Natl Acad Sci U S A. 2008;105:10238–10243. doi: 10.1073/pnas.0802759105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wik L, Klingeborn M, Willander H, Linne T. Separate mechanisms act concurrently to shed and release the prion protein from the cell. Prion. 2012;6:498–509. doi: 10.4161/pri.22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Winklhofer KF, Heller U, Reintjes A, Tatzelt J. Inhibition of complex glycosylation increases the formation of PrPsc. Traffic. 2003;4:313–322. doi: 10.1034/j.1600-0854.2003.00088.x. [DOI] [PubMed] [Google Scholar]
- 155.Wu B, McDonald AJ, Markham K, Rich CB, McHugh KP, Tatzelt J, Colby DW, Millhauser GL, Harris DA. The N-terminus of the prion protein is a toxic effector regulated by the C-terminus. Elife. 2017;6:e23473. doi: 10.7554/eLife.23473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wu D, Struwe WB, Harvey DJ, Ferguson MAJ, Robinson CV. N-glycan microheterogeneity regulates interactions of plasma proteins. Proc Natl Acad Sci U S A. 2018;115:8763–8768. doi: 10.1073/pnas.1807439115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yi CW, Wang LQ, Huang JJ, Pan K, Chen J, Liang Y. Glycosylation significantly inhibits the aggregation of human prion protein and decreases its cytotoxicity. Sci Rep. 2018;8:13486. doi: 10.1038/s41598-018-30770-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Younan ND, Chen KF, Rose RS, Crowther DC, Viles JH. Prion protein stabilizes amyloid-β(Aβ) oligomers and enhances Aβneurotoxicity in a Drosophila model of Alzheimer's disease. J Biol Chem. 2018;293:13090–13099. doi: 10.1074/jbc.RA118.003319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhao Y, Sato Y, Isaji T, Fukuda T, Matsumoto A, Miyoshi E, Gu J, Taniguchi N. Branched N-glycans regulate the biological functions of integrins and cadherins. FEBS J. 2008;275:1939–1948. doi: 10.1111/j.1742-4658.2008.06346.x. [DOI] [PubMed] [Google Scholar]