

Significance
Cells integrate mechanical signals to regulate biological outputs within tissues. These processes are required for tissue function and homeostasis. Here, we show how mechanical cues (e.g., tension) direct tendon cell function at a transcriptional and chromatin accessibility level. Furthermore, we show that disruption of these mechanical cues leads to an aberrant, catabolic cell state, indicating that these mechanosensitive pathways could be important for diseases driven by altered matrix remodeling, such as tendinopathy. Finally, we demonstrate that genetic perturbation of a single protein can preserve cell and chromatin state following a loss of tension.
Keywords: mechanobiology, epigenetics, chromatin
Abstract
Cells integrate mechanical cues to direct fate specification to maintain tissue function and homeostasis. While disruption of these cues is known to lead to aberrant cell behavior and chronic diseases, such as tendinopathies, the underlying mechanisms by which mechanical signals maintain cell function are not well understood. Here, we show using a model of tendon de-tensioning that loss of tensile cues in vivo acutely changes nuclear morphology, positioning, and expression of catabolic gene programs, resulting in subsequent weakening of the tendon. In vitro studies using paired ATAC/RNAseq demonstrate that the loss of cellular tension rapidly reduces chromatin accessibility in the vicinity of Yap/Taz genomic targets while also increasing expression of genes involved in matrix catabolism. Concordantly, the depletion of Yap/Taz elevates matrix catabolic expression. Conversely, overexpression of Yap results in a reduction of chromatin accessibility at matrix catabolic gene loci, while also reducing transcriptional levels. The overexpression of Yap not only prevents the induction of this broad catabolic program following a loss of cellular tension, but also preserves the underlying chromatin state from force-induced alterations. Taken together, these results provide novel mechanistic details by which mechanoepigenetic signals regulate tendon cell function through a Yap/Taz axis.
All tissues, and in particular those that are designed to bear load, sense, and respond to physical forces. Indeed, mechanically mediated tissue remodeling is a hallmark of musculoskeletal development, homeostasis, and disease progression (1). For example, tendons, a dense collagenous connective tissue whose function is to transmit tensile forces from muscle to bone, are subject to thousands of loading cycles each day as a consequence of muscle contraction during activities of daily living (2). Tendons maintain a state of residual tension on which these applied loads are superimposed (3). This “tensional homeostasis” is critical for maintaining tissue properties of tendons. Intrinsic tension arises from both direct cellular contractility and from residual stress in the tendon matrix. Both intrinsic and extrinsic tensile cues are thus central to tissue function, and loss of either can impact tissue homeostasis (4, 5). Loss of tensional homeostasis at the macroscale, as occurs with transection or periods of immobility, or at the microscale, as is the case when microdamage occurs that locally interrupts the fibrous architecture of the tissue, alters load transmission to the endogenous fibroblast cell population. The loss of these tensile cues in turn perturbs the homeostatic function of tendon fibroblasts (5–7). Indeed, tendinopathic tissues show an imbalance in matrix turnover, and at later stages, tendon cells may actually differentiate along aberrant lineages such as cartilage and bone (8).
While the etiology of tendinopathy is multifactorial, most cases are associated with overuse (8). Overuse activities can produce tissue microdamage, disrupting load transmission to the cells (i.e., overuse leads to underuse) (7). The degenerative end stage of tendinopathy is well established given that clinical specimens are available at this later symptomatic stage of pathology (9). End-stage tendinopathy manifests with multiscale changes in matrix properties (e.g., matrix damage, increased vascularity, mucoid degeneration, etc.) and alterations to the proportions and phenotype of resident cells (e.g. catabolic, pro-inflammatory) (6, 8). Preclinical models that induce tendinopathic-like changes via overuse and/or fatigue loading regimens have provided information on the types and magnitudes of loads that produce microtrauma and how cells respond to these disrupted mechanical cues, including the production of catabolic enzymes and inflammatory mediators (9, 10). Nonetheless, the early mechanistic cellular events mediated by changes in mechanical loading are not yet known. Understanding the mechanisms by which tendon cells sense and respond to changing mechanical cues is critical not only to better understand disease pathogenesis but to better treat patients with one of the most effective conservative treatment approaches, exercise physical therapy.
Mesenchymal cells, across tissue types, build and maintain the extracellular matrix which is critical for tissue and organ function (11). Regardless of tissue origin, these cells are universally receptive to mechanical signaling to modulate their respective function (11). Mechanoresponsive signaling pathways are central regulators of fibroblast and other mesenchymal cells, controlling a spectrum of activities from proliferation, contractility, differentiation, and extracellular matrix (ECM) remodeling (12–14). In tensile loading scenarios, mechanical cues are relayed through the contractile cytoskeleton to the nucleus to control transcriptional activities either indirectly through activation of other mechanosignaling pathways (e.g., stretch-activated channels, mechanoresponsive transcriptional co-activators such as Yes-Associated Protein (YAP) and Transcriptional co-activator with PDZ binding motif (TAZ)) or directly (e.g., deformation of the nucleus, nuclear pores, and chromatin) (15–19). Whether through direct chromatin deformation or via mobilization of mechanoactive factors, mechanosignaling pathways ultimately converge onto transcriptional regulators, which possess epigenetic and transcriptional activity to regulate cell function (20). In situ and in vitro studies show that disruption of mechanosignaling in tendon and other connective tissue fibroblasts alters their functionality (4, 5, 21, 22). Other work has shown that mechanical cues, such as matrix stiffness, can regulate chromatin accessibility and other epigenetic features in fibroblasts and other cell types, suggesting a direct role for these cues in regulating the epigenetic state and transcriptional receptiveness of these cells to mechanoactivation (23–26). However, the transcriptional events that dictate how these cells respond to an acute loss of mechanical signals remains unknown.
To address this gap in knowledge, we evaluated tendon cell response to a loss of tensional homeostasis, in vitro and in vivo. Our data show that an acute loss of tension results in an increase in the expression of catabolic gene programs in tendon fibroblasts leading to weakening of the tendon tissue. Subsequent mechanistic chromatin accessibility (Assay for Transposase-Accessible Chromatin followed by sequencing, ATACseq) and transcriptomic (RNA sequencing, RNAseq) analyses identified that cytoskeletal de-tensioning results in a global decrease in chromatin accessibility coincident with an increase in the expression of a matrix catabolic gene program. Unbiased analyses of ATACseq performed on de-tensioned tendon fibroblasts identified Yap/Taz/Tead targets as differentially regulated loci, suggesting that Yap/Taz/Tead may act as a transcriptional repressors of catabolic gene programs under homeostatic conditions. Depletion of Yap/Taz phenocopied the loss of cellular tension, instigating a broad catabolic program in both murine and human tendon fibroblasts. Conversely, overexpression of Yap/Taz reduced chromatin accessibility and downstream expression of genes involved in matrix catabolism, even in the face of a loss of cellular tension. Not only did this prevent cells from initiating a catabolic program, overexpression of Yap/Taz preserved accessibility at specific targeted chromatin loci following loss of tension. Taken together, these data support that cellular tension, acting through Yap/Taz/Tead, promotes an anabolic state in tendon fibroblasts, and thus may serve as a novel mechanosignaling mechanism regulating the initiation and pathogenesis of tendinopathy.
Results
Cellular Tension Negatively Regulates Extracellular Matrix Degradative Pathways.
To investigate the role of tensile cues in directing tendon-resident fibroblast function, we developed an in vivo loss-of-tension model in which we resected a distal segment of the flexor digitorum longus tendon (FDL) in the mouse forefoot, resulting in retraction and loss-of-tension in the proximal region of the tendon. Injuring the tendon distal to the pulley at the ankle allowed for the proximal portion of the tendon to be analyzed while minimizing effects from the early stages of healing (e.g., clot formation, granulation tissue) at the injury site (Fig. 1A and SI Appendix, Fig. S1). To examine the early response of the cell nucleus to the loss of tension, we quantified the nuclear aspect ratio and the dispersion (i.e., higher dispersion indicates cells/nuclei are more disorganized) of DAPI-stained tissue sections at 24 h postresection. Interestingly, acute loss of tension reduced nuclear aspect ratio and increased nuclear dispersion (Fig. 1 B and C) in resected tendons compared to uninjured contralateral controls. These data demonstrate that in vivo tensile cues are central to the maintenance of overall nuclear shape and positioning.
Fig. 1.
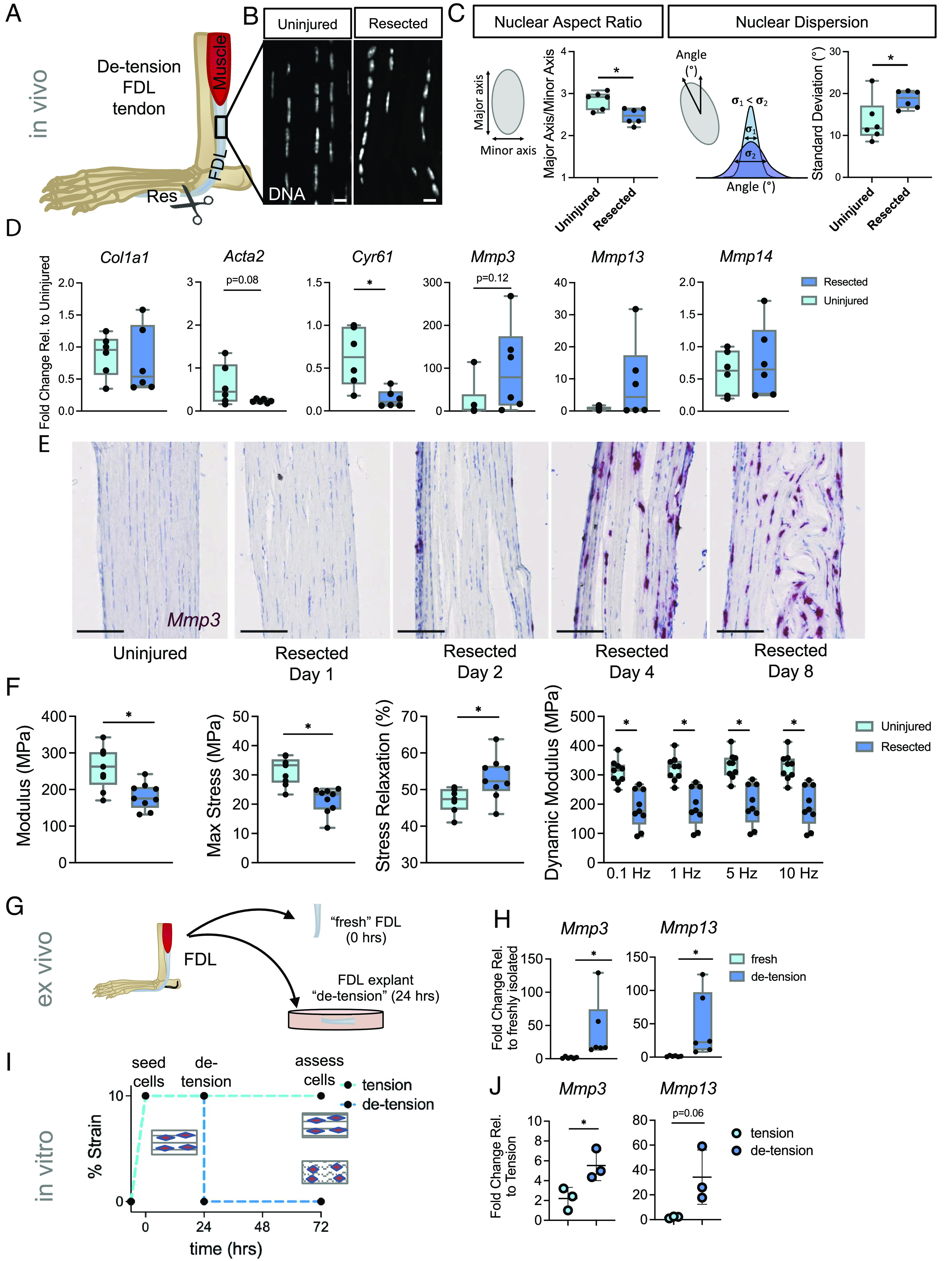
Pathologic acute mechanoresponses of tendon fibroblasts to de-tensioning. (A) Schematic showing approach to de-tensioning the FDL tendon in vivo. (B) DAPI staining depicting nuclear morphology following loss of tension (Scale bar, 10 μm.) (C) Schematic and quantification of nuclear aspect ratio and nuclear dispersion angle of tendon fibroblasts 24 h following loss of tension in the FDL (n = 6 biological replicates). (D) qRT-PCR analysis of FDL tendons 24 h following resection (n = 6 biological replicates). (E) RNAscope analysis for Mmp3 over time following resection of the FDL tendon (Scale bar, 100 μm.) (F) Mechanical properties of resected FDL tendons and their contralateral uninjured controls on day 8 post-resection (n = 9). (G) Schematic showing FDL explant de-tensioning experiment. (H) qRT-PCR analysis of FDL explants (n = 6/group). (I) Schematic showing scaffold de-tensioning experiment (using aligned electrospun scaffolds). (J) qRT-PCR analysis of cells from scaffolds that were either left in tension or de-tensioned for 48 h (n = 3/group). *P < 0.05 evaluated by paired t test. Error bars represent SD.
To study how these early changes in nuclear shape might affect downstream gene expression, we performed qRT-PCR on tendons collected 24 h after resection. While analysis of transcripts from the de-tensioned tendons showed no change in matrix anabolic genes (i.e., Col1a1), we observed an increase in the expression of matrix catabolism genes [i.e., matrix metalloproteinases (MMPs)], demonstrating that the acute early response to a loss of tensile cues is an upregulation of matrix degradative gene programs (Fig. 1D). Interestingly, we also noted reduced expression of Acta2 and Cyr61, established mechanoresponsive genes (Fig. 1D), suggesting that tendon fibroblasts are responsive to the loss of tensile mechanical cues in vivo. Using RNAscope, we performed a time course experiment to better understand the kinetics of Mmp3 expression following resection. Interestingly, we observed a progressive increase in Mmp3 within the resident tendon cells over time (Fig. 1E) suggesting that the early acute response to a loss of tension leads to persistent changes in matrix catabolic gene expression. To confirm that elevation of Mmp3 was not from other myeloid cells known to produce MMPs, we performed RNAscope for Mmp3 and Csf1r, a common myeloid marker(27). Microscopy images revealed little overlap of Mmp3+ cells and Csf1r+ cells at 8 d after resection (SI Appendix, Fig. S2). Collectively, these data demonstrate that de-tensioning results in acute, and long-term, changes in expression of matrix catabolic genes in resident tendon fibroblasts, potentially leading to subsequent degenerative tendon tissue.
To assess the long-term functional consequence of loss-of-tension on the tendon tissue, we performed mechanical testing on FDL tendons 8 d following loss of tension. We observed a decrease in elastic modulus and maximum stress in the resected tendons compared to the contralateral controls (Fig. 1F). Additionally, the dynamic modulus decreased while stress relaxation increased in the resected tendons (Fig. 1F). Notably, the change in mechanical properties was driven by a substantial change in tissue structure, with the cross-sectional area increasing in the resected tendons, with no change in stiffness and maximum force (SI Appendix, Fig. S4). These findings of increased tissue swelling and reduced material properties are consistent with previous in vivo stress-shielding studies using an external fixator on the rabbit patellar tendon (28, 29). Taken together, these data show that loss of tension to the FDL following resection injury leads to changes in nuclear shape/positioning and early activation of a catabolic program that progressively worsens leading to structural remodeling and mechanical weakening of the tendon.
To confirm that the increase in catabolic gene expression we saw after de-tensioning in vivo is indeed caused by loss of tension, we utilized both ex vivo and in vitro assays where applied tension was removed. First, we cultured FDL tendon explants under load-deprived free-floating conditions for 24 h. We found that under this de-tensioned state the expression of both Mmp3 and Mmp13 was upregulated compared to freshly isolated FDL tendons (Fig. 1 G and H). Next, we cultured primary mouse tendon fibroblasts on aligned, pretensioned polycaprolactone scaffolds (4). After seeding and attachment, the tension was released to simulate a loss of tension and cells were collected for analysis 48 h after loss of tension (Fig. 1I). In accordance with our in vivo and ex vivo data, we similarly observed an upregulation of genes involved in matrix catabolism (Mmp3, Mmp13) (Fig. 1J). Collectively, these experiments demonstrate that the acute response to loss of applied tension to tendons is the induction of MMP gene expression, which ultimately leads to changes in tissue mechanical properties.
Mechanical Regulation of Epigenomic and Transcriptomic State of Tendon Fibroblasts.
Given the changes in cell morphology and rapid induction of MMP gene expression following loss of applied tension, we next sought to elucidate the short-term cellular mechanoresponse which regulates this transcriptional program. To do so, we de-tensioned tendon fibroblasts in vitro using two small-molecule inhibitors of proteins involved in mechanosignaling, blebbistatin inhibiting myosin contraction (biomechanical intervention) and Y27632 inhibiting Rho kinase (biochemical intervention). Using freshly isolated tendon fibroblasts (Fig. 2A), we administered blebbistatin (10 µM) and separately Y27632 (30 µM) and analyzed a broad set of matrix catabolic genes by qRT-PCR. Similar to de-tensioning in vivo, we found marked upregulation of matrix catabolic genes, particularly following inhibition of myosin contractility, with less dramatic changes with Rho kinase inhibition (Fig. 2B), demonstrating that cellular tension plays a key role in regulating expression of these pathways. Taken together, these data suggest that an early response to loss of tensile cues could be altered expression of matrix degradation pathways, a potentially critical step in the progression to chronic tendinopathy.
Fig. 2.

Cellular tension maintains epigenetic and transcriptional homeostasis in tendon fibroblasts. (A) Schematic showing approach for isolation and culture/experimental conditions for tendon fibroblasts in vitro. (B) qRT-PCR analysis of tendon fibroblasts 6 h following addition of blebbistatin (10 µM) or Y27632 (30 µM) (n = 3 biological replicates). *P < 0.05 evaluated by unpaired one-way ANOVA with Tukey’s correction for multiple comparison test. Error bars represent SD. (C) Volcano plot from RNAseq analysis showing differentially expressed genes (adjusted P value ≤ 0.05 and a fold-change of ≤−1 or ≥ 1) in blue (n = 3 biological replicates). (D) gene ontology analysis of gene sets identified in (C). (E) Volcano plot from ATACseq analysis showing differentially accessible genomic loci (adjusted P value ≤ 0.05 and a fold-change of ≤−1 or ≥ 1) in blue (n = 3 biological replicates). (F) De novo transcription factor motif enrichment analysis from the genomic loci identified in (E). Transcription factors are ranked by statistical enrichment (P value). (G) Representative changes in chromatin accessibility following blebbistatin treatment cross-referenced with publicly available Yap ChIPseq (GSE83863). Genomic distance represents distance from gene transcriptional start site (TSS). Scale bar represents 1 kb. (H) Representative gene expression changes of collagen catabolism genes and Yap/Taz target genes following blebbistatin treatment. The color scale shown is normalized read counts, with the normalization carried out relative to the highest read count in that row (for a given gene).
To investigate the transcriptional and epigenetic mechanisms through which intracellular tension mediates this extracellular matrix homeostasis, we administered blebbistatin (10 µM) for 6 h and performed RNAseq and ATACseq. Following blebbistatin treatment, RNAseq analysis revealed increased transcripts from 570 genes and decreased transcripts from 462 genes (Fig. 2C). Gene ontology (GO) analysis of those gene lists identified that inhibition of cytoskeletal tension did indeed increase collagen catabolism pathways, confirming a role for cellular tension in regulating genes involved in extracellular matrix homeostasis (Fig. 2D). A complete list of GO terms from this analysis can be found in Datasets S1 and S2. Interestingly, ATACseq revealed that blebbistatin reduced accessibility at 954 chromatin loci and increased accessibility at only 17 chromatin loci. These results indicate that active cytoskeletal tension, on the whole, acts to positively regulate chromatin accessibility (Fig. 2E).
To identify potential regulators driving these epigenetic and transcriptional changes, we performed de novo transcription factor motif enrichment analysis within the chromatin loci that were either increased or decreased in accessibility following blebbistatin treatment (Fig. 2E). This unbiased motif analysis identified Tead as the leading transcription factor (Fig. 2F). The Tead family of proteins function as transcriptional co-activators for Yap and Taz, which are major mechanoresponsive transcription factors involved in cellular mechanosensing (15). This suggests that tensile cues could mediate extracellular matrix homeostasis through a Yap/Taz/Tead axis. Further supporting this idea, co-occupancy analysis of publicly available Yap ChIPseq data (GSE83863) (30) demonstrated a robust overlap of Yap occupancy with differentially accessible chromatin loci following blebbistatin treatment (Fig. 2G). Analysis of Yap/Taz target genes within our RNAseq data confirmed a reduction in transcripts of Yap/Taz target genes as well as an increase in transcripts of a broad group of MMPs. Collectively, these data demonstrate that loss of cellular tension disrupts chromatin state, modulating accessibility at Yap/Taz/Tead target loci, resulting in an increase in transcriptional activity of genes in matrix degradation pathways.
Yap/Taz/Tead Represses Matrix Degradation Pathways.
Given that the ATACseq analysis pointed to Yap and/or Taz as potential key mediators of the response to loss of cytoskeletal tension in tendon fibroblasts, we next used a B6.C-Tg(CMV-cre)1Cgn/J;TetO-YapS127A; Gt(ROSA)26Sortm1(rtTA, EGFP)Nagy/J mouse (herein referred as YAP-constitutively active or YAP-CA) which overexpresses a mutated constitutively-active human YAP1 gene following doxycycline administration to further assess the role of Yap/Taz in these cells (31, 32). We isolated primary fibroblasts from tail tendons from adult YAP-CA mice and found robust YAP overexpression following 48 h of doxycycline administration (10 ng/mL) (Fig. 3 A and B). To better understand the role of Yap/Taz in regulating chromatin state, we administered doxycycline for 48 h and then performed ATACseq. ATACseq analysis revealed that overexpression of YAP resulted in an increase of accessibility at 7,297 chromatin loci and only decreased accessibility at 316 chromatin loci (Fig. 3C). These data are consistent with YAP maintaining chromatin “openness,” potentially acting as a pioneer factor directly regulating chromatin accessibility. As expected, de novo motif enrichment analysis within the chromatin loci that changed accessibility following YAP overexpression revealed Tead as the leading candidate, confirming a Yap/Taz/Tead axis as directly regulating chromatin state (Fig. 3D). Investigation of specific gene loci that changed in accessibility also confirmed an increase of accessibility of Yap/Taz/Tead target genes (e.g., Cyr61, Nuak2, and Amotl1) (Fig. 3E). Interestingly, we also observed a marked reduction of accessibility for genes involved in matrix degradation (e.g., Mmp2 and Mmp3) (Fig. 3E) demonstrating a potential role of Yap/Taz in modulating matrix degradation pathways.
Fig. 3.
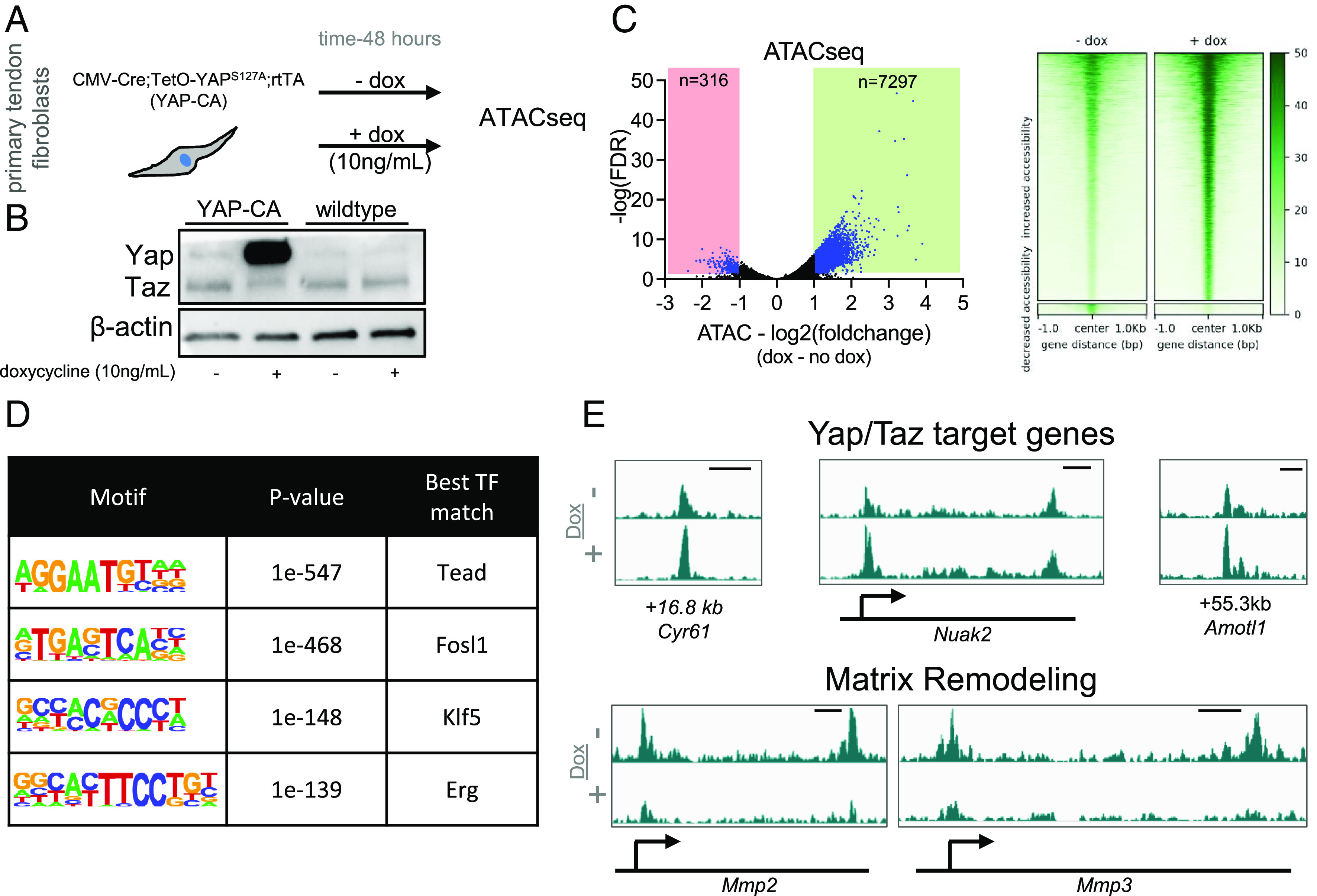
Yap overexpression maintains chromatin accessibility and reduces accessibility near matrix degradation genes. (A) Schematic showing approach to examine the role of Yap in maintenance of chromatin accessibility. (B) Western blot showing robust overexpression of Yap following 48 h of doxycycline treatment (10 ng/mL). (C) Volcano plot from ATACseq analysis showing differentially accessible genomic loci (adjusted P value ≤ 0.05 and a fold-change of ≤−1 or ≥ 1) in blue (n = 3 biological replicates). Heatmaps of chromatin accessibility changes following blebbistatin treatment are also shown. (D) De novo transcription factor motif enrichment analysis from the genomic loci identified in (C). (E) Representative chromatin accessibility tracks showing an increase of accessibility of Yap/Taz target genes (Cyr61, Nuak2, and Amotl1) and a reduction of accessibility of Mmp2 and Mmp3 following Yap overexpression. Genomic distance represents distance from gene TSS. Scale bar represents 1 kb.
To test whether Yap/Taz directly regulates expression of genes involved in matrix degradation, we performed gain of Yap function assays using fibroblasts isolated from YAP-CA mice and administered doxycycline for 48 h. We found that overexpression of YAP robustly repressed Mmp2, Mmp3, Mmp13, and Mmp14 (Fig. 4A), consistent with our chromatin accessibility findings (Fig. 3E). We further administered siRNA targeting Yap1 and Wwtr1 (Taz) in wildtype tendon fibroblasts for 3 d and then analyzed gene expression by qRT-PCR. Consistent with our gain-of-function experiment, we found that depletion of Yap/Taz resulted in strong upregulation of Mmp2, Mmp3, Mmp13, and Mmp14 (Fig. 4B). To test whether YAP/TAZ also regulates MMP expression in human tendon fibroblasts, we administered siRNA targeting both YAP1 and WWTR1 (TAZ) for 3 d and then analyzed gene expression by qRT-PCR. Analysis revealed significant upregulation of MMP3 and MMP13, with mild upregulation of MMP2 and MMP14 (Fig. 4C) in these human cells, consistent with our prior results in mouse cells (Fig. 4B).
Fig. 4.

Yap/Taz regulates MMP gene expression and nascent protein deposition and remodeling. (A) qRT-PCR analysis of mouse tendon fibroblasts following gain-of-function of Yap/Taz (n = 3 biological replicates). (B) qRT-PCR analysis of mouse tendon fibroblasts following loss-of-function of Yap/Taz (n = 3 biological replicates). (C) qRT-PCR analysis of human tendon fibroblasts following loss-of-function of Yap/Taz (n = 3 biological replicates). (D) Schematic showing approach to examine the role of Yap/Taz and blebbistatin in regulating nascent protein deposition and remodeling. (E) Integrated intensity of nascent protein production of tendon fibroblasts after treatment. Each data point represents nascent protein deposition from a single cell. Data from two biological replicates are grouped together. (F) Representative images from data generated in (E). Scale bar represents 50 µm. *P < 0.05 evaluated by unpaired t test. Error bars represent SD.
To test whether these pathways functionally modulate fibroblast matrix remodeling in a three-dimensional fibrous setting, we performed functional noncanonical amino acid tagging (FUNCAT) to enable labeling of nascent extracellular matrix protein deposition at a single cell resolution(4, 33, 34). Fibroblasts were treated with siRNA targeting Yap1 and Wwtr1 (Taz) for 48 h on plastic, after which they were seeded onto fibrous scaffolds for 2 d for FUNCAT analysis (Fig. 4D). Depletion of Yap1 and Wwtr1 (Taz) resulted in a mild reduction of nascent protein deposition (Fig. 4E). To test whether Yap/Taz gain of function increased nascent protein deposition, we similarly depleted Lats1 and Lats2, the primary upstream kinases in the Hippo pathway which directly phosphorylate Yap/Taz, and thereby block their function(15). Depletion of Lats1/2 (gain of Yap/Taz function) resulted in an increase, albeit not statistically significant, in nascent protein deposition (Fig. 4E). To examine whether blebbistatin regulates matrix remodeling function in tendon fibroblasts, we seeded tendon fibroblasts on fibrous scaffolds for 2 d, after which blebbistatin was added for 24 h (Fig. 4D). Compared to DMSO (dimethylsulfoxide, vehicle) control, blebbistatin potently reduced nascent protein deposition (Fig. 4 E and F). Taken together, these data demonstrate that Yap/Taz are repressors of matrix degradation gene programs and that they act through direct epigenetic regulation of their respective genomic loci. Furthermore, Yap/Taz, and blebbistatin, modulate tendon fibroblast nascent protein production.
Overexpression of YAP Protects Chromatin Mechanohomeostasis Following Loss of Cytoskeletal Tension.
Our data demonstrated that loss of cytoskeletal tension disrupted chromatin homeostasis (Fig. 2E), resulting in increased expression of matrix degradative gene programs (Fig. 2 D and H), potentially through a loss of Yap/Taz signaling (Fig. 2F), and that Yap/Taz are regulators of expression of matrix degradative gene programs (Fig. 4 A–C). This raised the interesting possibility that overactivation of Yap/Taz might serve as a counterbalance to the change in chromatin and transcriptional state that occurs with the loss of tensional homeostasis in cells. To test whether gain of Yap function could protect nuclear state following loss of tension, we overexpressed YAP in tendon fibroblasts from YAP-CA mice, administered blebbistatin for 6 h, and then collected cells for RNA and ATAC analysis (Fig. 5A). Using qRT-PCR, we found that overexpression of YAP prevented the blebbistatin-induced downregulation of known Yap/Taz target genes (e.g., Cyr61) while also preventing the upregulation of matrix degradative gene programs (e.g., Mmp2, Mmp3, Mmp10, Mmp13, and Mmp14) (Fig. 5B). These data support that activating Yap signaling could protect the transcriptional state of the cell following de-tensioning. Interestingly, ATACseq analysis comparing Yap overexpressing cells with or without blebbistatin treatment revealed that nonmuscle myosin inhibition still reduced accessibility even with Yap overexpression (Fig. 5 C and D). While these data are consistent with our prior blebbistatin ATACseq data from wildtype fibroblasts (Fig. 2E), we intriguingly no longer found Tead as the leading candidate in the motif enrichment (Fig. 5E), indicating that alternative Yap/Taz independent pathways could be contributing to the loss of chromatin accessibility under these conditions.
Fig. 5.
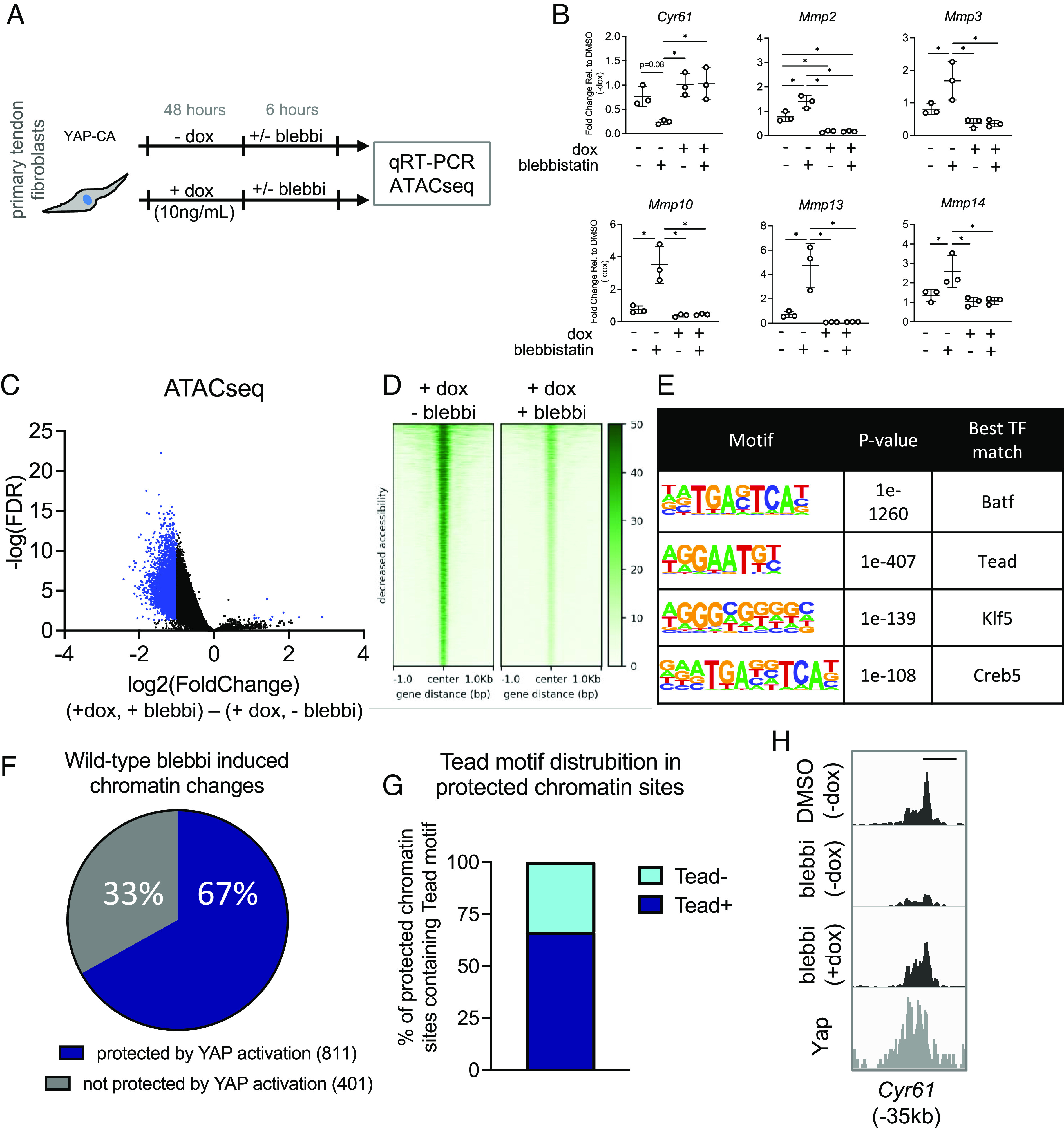
Activation of Yap signaling protects against catabolic changes from loss of cellular tension by maintaining chromatin accessibility at mechanoresponsive loci. (A) Approach to examine whether Yap gain-of-function could attenuate transcriptional and epigenetic changes following loss of tension. (B) qRT-PCR analysis of tendon fibroblasts following Yap overexpression and blebbistatin treatment (n = 3 biological replicates). (C) Volcano plot from ATACseq analysis showing differentially accessible genomic loci (adjusted P value ≤ 0.05 and a fold-change of ≤ −1 or ≥ 1) in blue (n = 3 biological replicates). (D) Heatmaps of chromatin accessibility changes following blebbistatin treatment. (E) De novo transcription factor motif enrichment analysis from the genomic loci identified in (C). (F) Percentage of chromatin accessibility changes in wildtype tendon fibroblasts following blebbistatin treatment that no longer changed in accessibility following Yap overexpression. (G) Distribution of the Tead motif in the protected chromatin sites identified in (F). (H) Representative ATAC data showing protection of chromatin accessibility changes in Yap overexpressing cells following blebbistatin treatment. Genomic distance represents distance from gene TSS. Scale bar represents 1 kb. *P < 0.05 evaluated by unpaired t test. Error bars represent SE.
To assess whether Yap overexpression in the context of blebbistatin treatment protected the accessibility of chromatin binding sites that were inhibited in wildtype cells (Fig. 2E), we specifically examined the accessibility profiles of those chromatin loci in Yap overexpressing cells treated with blebbistatin compared with cells only overexpressing Yap. Interestingly, we found that 67% (n = 811) of the chromatin loci that changed accessibility in wildtype cells with blebbistatin no longer changed in chromatin accessibility in the Yap overexpressing cells following blebbistatin treatment (Fig. 5F). This demonstrates that activation of Yap protects the chromatin state following cellular de-tensioning. We next examined these 811 chromatin loci for presence of the Tead DNA binding motif and found motif enrichment in 2/3rd (66.5%) of these chromatin loci (Fig. 5G). Finally, we cross-examined the protected ATAC loci with publicly available Yap ChIPseq data and found strong overlap of Yap occupancy at the sites of protected chromatin loci following loss of tension (Fig. 5H). Collectively, these data reveal that overexpression of Yap protects both chromatin accessibility and transcriptional state following loss of tensile cues in fibroblasts.
Discussion
This study demonstrates an epigenetic role for Yap/Taz in regulating the initial mechanoresponse to loss of tension and the subsequent activation of a catabolic program in tendon fibroblasts (Fig. 6). Upon disruption of tensional homeostasis in vivo, nuclei round up and become disorganized, and matrix degradation pathways are activated resulting in a weakening of the tendon tissue. Similarly, loss of cell tension in vitro leads to an overall reduction in chromatin accessibility, resulting in the activation of matrix degradation pathways. Using paired ATAC/RNAseq we identify Yap/Taz/Tead as central epigenetic and transcriptional regulators of the nuclear response to loss of tension. Depletion of Yap and Taz elevates gene expression of MMPs, similar to that which occurs with cytoskeletal de-tensioning, while overexpression of Yap represses MMP gene expression, even when the cytoskeleton is de-tensioned. These results suggest a direct epigenetic role of Yap and Taz proteins, based on the tensional state of the cell, in the repression of genes involved in matrix degradation. Finally, we show that overexpression of Yap preserves chromatin accessibility at distinct mechanoresponsive genomic loci following cellular de-tensioning.
Fig. 6.

Summary schematic displaying loss of tension results in a Yap/Taz-mediated reduction of chromatin accessibility, increasing matrix catabolism in tendon fibroblasts. Created with BioRender.com.
Disruption of tensional homeostasis is an important factor in the progression of a wide variety of chronic diseases, including cancer, cardiovascular disease, and tendinopathies (35–37). Our finding that inhibition of tensional homeostasis activates matrix degradation through an epigenetic and transcriptional Yap and Taz axis provides a novel mechanism by which disruption of tensile cues leads directly to persistent matrix degradation and/or remodeling. Interestingly, we find that loss of tensile cues reduced chromatin accessibility, which is consistent with recent work showing that reducing matrix stiffness, a different mechanical input to the cell, also reduced chromatin accessibility (23, 24). While these previous studies did not primarily focus on Yap/Taz, a distinct Yap/Taz signature was observed in their datasets, suggesting that Yap/Taz could be principal mechanoepigenetic regulators of chromatin state. This is also consistent with prior work highlighting the role of Yap and Taz in chromatin remodeling (38). Furthermore, consistent with our observations, previous work demonstrated that activation of signaling pathways which block Yap/Taz signaling (e.g. cAMP, statins) initiate matrix degradation in tendons and other tissues (39–41). These studies, taken together with our findings, demonstrate that the nucleus is exquisitely sensitive to mechanical cues, with increased mechanosignaling (i.e., tension, matrix rigidity) promoting global chromatin accessibility. However, additional studies are needed to determine whether the changes in chromatin state are due to transcription factor abundance in the nucleus (i.e., Yap/Taz) or to direct mechanical control of chromatin accessibility and transcription via nuclear (and so chromatin) deformation (16). Such insights into these mechanisms may very well inform critical epigenetic programs that regulate disease progression.
Tendinopathy, particularly from overuse, is thought to arise from tissue microdamage. This microdamage, in turn, impairs transmission of tensile mechanical signals through the tissue, resulting in underloading of endogenous tendon cells (7, 42). The disease often leads to aberrant cell and matrix responses, such as chondroid metaplasia of the tendon, further attenuating ECM-to-cell strain transfer (8, 43). At the level of chromatin organization (STORM imaging of H2B clusters), this results in aberrant packing and inability to reorganize with changing mechanical inputs in cells from end-stage disease (44). Interestingly, tendinopathic specimens also contain myofibroblasts and appear to undergo chronic remodeling, which may be an attempt by the resident tendon fibroblasts to re-tension their matrix to re-establish tensional homeostasis(35). Similar myofibroblast-like changes, matrix remodeling, and engagement of the cytoskeleton are seen in de-tensioned tendon explants ex vivo (45, 46). Ongoing failure to restore the proper tensional set point is likely a contributing factor in the progression of the disease. Our finding that acute de-tensioning results in a marked induction of a catabolic transcriptional program and that the Yap/Taz/Tead axis protects the initial chromatin state following loss of tension, preventing this catabolic response, could inform mechanisms that regulate the initiation and early progression of tendinopathy. Beyond the catabolic response, acute de-tensioning also elicits cell death which can be protected through inhibition of cell contractility (4, 5). Therefore, it is probable that control of cellular tension is required to maintain cell fate and tissue homeostasis, a process that is likely controlled through Yap and Taz (47). Importantly, our findings offer new insight towards the development of novel mechanotherapeutic targets that could directly regulate this balance in cellular tension to attenuate disease progression.
The models that we employed, FDL resection and blebbistatin treatment, were extreme versions of de-tensioning. There are several other in vivo [e.g., overuse activities (48), fatigue loading (10), stress shielding with external fixators (49), casting (50), botulinum toxin injections (5)] and ex vivo [e.g., free floating or fatigue loaded tendon explants (46, 51)] preclinical model systems that can be used to elicit partial or full de-tensioning, which yield a catabolic response at varying degrees. We chose the more extreme of these models to investigate the early mechanoresponse to acute changes in tension. While more chronic loading regimens may better model the activities and duration seen in overuse tendinopathies, they require days or weeks of stimulation to elicit the catabolic response, reducing our ability to pinpoint this early response to de-tensioning. As such, the timing of the response and the cofactors beyond tension that influence it (e.g., inflammation) are likely different between models and should be considered when making comparisons. This is also important to consider clinically, as disuse conditions such as muscle weakness poststroke (52) or prolonged period of immobilization from casting (53, 54) can have detrimental effects on tendon integrity but likely have different timing and covariables, compared to overuse tendinopathy.
While we found that both cellular de-tensioning and Yap/Taz regulate expression of MMPs, we only found changes in chromatin accessibility at MMP genomic loci when over-expressing a mutated and constitutively active Yap (Fig. 3E). While the relationship between ATAC signal and gene expression is thought to be linear (i.e., increase in accessibility is associated with increased expression), temporal analysis of these assays during dynamic changes, such as single factor perturbations, is less clear (55). We performed ATAC/RNAseq at 6 h after blebbistatin and Y27632 treatment, and so it is possible this short duration was too early for observable and measurable ATAC differences at MMP loci to occur. Alternatively, it is possible that additional transcriptional machinery downstream of Yap/Taz could also be regulating MMP expression, further convoluting the mechanism by which cellular tension regulates MMP expression. Delineating the precise contribution of ATAC signal and MMP gene expression will require future temporal analysis of these genomic loci in response to prolonged and altered mechanical signaling.
Collectively, our findings provide new insights into the mechanisms by which applied tension leads to mechanosignaling that directs cell behavior at the transcriptional and chromatin accessibility level. This epigenetic program, particularly chromatin accessibility and suppression of matrix catabolism, is mediated by Yap/Taz. These findings open new avenues for further investigation of these understudied biological processes in the context of tissue formation, homeostasis, and disease progression.
Methods
Animals.
All animal procedures were approved by the University of Pennsylvania’s Institutional Animal Care and Use Committee. The following mice were used: Tg(CMV-cre)1Cgn/J (Jackson Laboratory, strain #006054) (32), TetO-YapS127A (gift from Carmago and Calloway labs, Harvard University), Gt(ROSA)26Sortm1(rtTA, EGFP)Nagy/J (Jackson Laboratory, strain #005670) (56), Scx-GFP (57), Col1a1-CFP (58), and wildtype CD-1 IGS (Charles-River, strain #022).
FDL Resection Surgeries.
The FDL tendon consists of a proximal region that attaches to the FDL muscle, a wrap-around region that glides along the distal-medial edge of the tibia and is surrounded by a synovial sheath, and the distal region in the forefoot that attaches to the phalanges of digits 2 to 5. In the FDL resection procedure, first mice (2 to 3 mo old) were anesthetized with isoflurane (1 to 3%), given pre-surgical analgesia, and sterilely prepped. The distal FDL was exposed and a 3 mm segment proximal to the bifurcation point was resected in order to de-tension the proximal region of the tendon (SI Appendix, Fig. S1). The skin was closed with 4-0 nylon suture and the animal was returned to unrestricted cage activity. This model was used to induce acute de-tensioning on the tendon while also assessing the proximal region that is less effected by inflammation and granulation tissue of the healing tissue at the resected site in the forefoot.
Tissue Sectioning and RNA In Situ Hybridization.
Following euthanasia, hindlimbs were harvested, fixed in RNase-free 4% paraformaldehyde (PFA) for 24 h with the ankle at 90° of flexion, decalcified in ethylenediaminetetraacetic acid containing 2% PFA for 7 d, and embedded in optimal cutting temperature compound. Tape-stabilized (Cryofilm 2C, Section-Lab Co. Ltd.) sagittal sections were made and then stained for Mmp3 (Advanced Cell Diagnostics, Cat No. 480961) using the RNAscope 2.5 HD Assay (Advanced Cell Diagnostics, Inc.) for Mmp3 and Csf1r (Advanced Cell Diagnostics, Cat No. 428191) using the RNAscope 2.5 HD Duplex Assay with the custom pretreatment reagent (Cat No. 300040) according to the manufacturer’s protocol. Brightfield tiled scans were acquired using a Plan-Apochromat 10×/0.45 objective on the Zeiss Axio Scan.Z1 digital slide scanner.
Nuclear Aspect Ratio and Dispersion Measurements.
Hindlimbs were harvested, fixed, decalcified, and sectioned using cryofilm as above. Sections were counterstained with DAPI and imaged using a Plan-Apochromat 10×/0.45 objective on the Zeiss Axio Scan.Z1 digital slide scanner. DAPI images were thresholded to create a binary image and then the “analyze particles” function in Fiji was employed to calculate aspect ratio and orientation angle. The orientation angle for each nucleus was normalized to the long axis of the tendon and the SD of this unimodal distribution was calculated for each image. Three different sections were analyzed and averaged to create the dispersion measurement for each tendon.
Mechanical Testing of FDL Tendons.
FDL tendons were isolated and tendon cross-sectional areas (CSA) were measured using a custom laser device (59). Sandpaper was applied to the proximal and distal ends of the tendon using cyanoacrylate to create a gauge length of 3 mm. Tendons were mounted within a mechanical testing system (Instron 5848) and underwent a viscoelastic mechanical testing protocol. The protocol included preconditioning (10 cycles of 0.5% strain amplitude oscillations at 1 Hz centered at 1% strain), a viscoelastic stress relaxation (3% strain rapidly applied and maintained for 5 min) and frequency sweep (10 cycles of 0.0125% strain amplitude oscillations centered at 3% strain at 0.1, 1, 5 and 10 Hz), and a quasistatic ramp to failure (0.1% strain per second). Structural and material properties were calculated from CSA measurements and load–displacement data using a custom MATLAB script (60).
FDL Tendon Explant.
FDL tendons from 3-mo–old wildtype CD1 mice (n = 6) were isolated. The FDL from the right limb was digested directly in TRIzol LS whereas the FDL from the left limb was cultured under load-deprived free-floating conditions for 24 h in cell culture medium (Dulbecco's Modified Eagle Medium (DMEM) supplemented with 5% fetal bovine serum (FBS) and antibiotics-antimycotics) at 37 °C. The cultured tendon was then digested in TRIzol LS for qRT-PCR (see details below).
Primary Tendon Fibroblast Isolation and Cell Culture.
Tails from 6 to 8-wk–old male and female CD1 wildtype mice were cut into 3-cm–long segments. Tendon fascicles were pulled from the tail and digested with an enzymatic solution (DMEM, 0.2 mg/mL Liberase DL, and 100 U/mL DNase I, Thermo-Fisher Scientific) for 60 min at 37 °C. Digestive solution was inactivated with DMEM containing 10% FBS and antibiotics–antimycotics. Cell and tissue suspensions were passed through a 70 mM filter and centrifuged at 300 g at 4 °C for 10 min. Cells were then resuspended in cell culture medium (DMEM supplemented with 10% FBS and antibiotics-antimycotics) and then seeded on collagen I coated tissue-culture plastic.
To confirm that our tendon digestions and cultures yield primarily tissue resident tendon fibroblasts, we performed tendon digestions on tendons isolated from double reporter mice containing Scx-GFP and Col1a1-CFP. Cultures of tendon fibroblasts from these mice showed that all cells were double positive (GFP+ and CFP+) confirming selective isolation and culture of tendon fibroblasts (SI Appendix, Fig. S3).
Human tendon fibroblasts were purchased from Zen-Bio and maintained in the same media as above.
All cells were used between passage 1 to 3. Blebbistatin (Caymen Chemicals) was used at 10 uM in DMEM supplemented with 0.1% FBS with antibiotics-antimycotics. Y27632 (Caymen Chemicals) was used at 30 uM in DMEM supplemented with 0.1% FBS with antibiotics–antimycotics. Concentrations of blebbistatin and Y27632 were used in accordance with previous literature reporting the use of these compounds on fibroblasts (12, 61). Working concentration of DMSO was used at 0.1% (v/v). We confirmed that DMSO does not influence transcript abundance of genes studied in this manuscript when used at this concentration (SI Appendix, Fig. S4B).
Electrospun Scaffold Preparation and Cell Seeding.
Electrospun PCL scaffolds were prepared as previously described (4, 62). Briefly, a 14% weight/volume solution of PCL in 1:1 tetrahydrofuran and dimethylformamide was loaded into a 10 mL syringe and extruded through a spinneret charged to 14 kV (1 kVcm−1 effective gradient to ground) and collected on a rotating, grounded mandrel. Spinning was conducted at room temperature in a humidity-controlled room (35% relative humidity). Aligned scaffolds were prepared by tuning the mandrel surface velocity (14 m/s). These electrospun mats were cut into 10 mm × 40 mm strips with the long axis corresponding to the prevailing fiber direction. Scaffolds for cell seeding were sterilized and rehydrated in 30-min steps of progressively weaker ethanol (100%, 70%, 50%, and 30%) followed by 2 × 30 min washes in sterile PBS and overnight coating in fibronectin (20 mg/mL).
Scaffold De-Tensioning.
Aligned PCL scaffolds were generated as above. Prior to cell seeding, fibronectin-coated scaffolds were pretensioned using a custom clamp device to a set 10% strain (pre-tensioned scaffolds). Primary mouse tendon fibroblasts (n=3 biological replicates) were seeded at 250,000 cells/scaffold on the top surface of the pre-tensioned scaffold (area for cell attachment: 1.5 cm2). The scaffolds were divided into two groups: 1) tension and 2) de-tension. After 24 h, scaffolds were either left in tension or were de-tensioned by cutting the scaffolds at the clamp using a scalpel. Subsequently, both tensioned and de-tensioned scaffolds were cultured for another 2 d and then cells were collected for RNA analysis. Briefly, scaffolds were washed in ice-cold PBS, then placed in RNA lysis buffer supplemented with beta-mercaptoethanol. RNA was then isolated as described below using the Qiagen RNeasy Mini Plus kit.
FUNCAT of Nascent Matrix.
Nascent matrix deposition of tendon fibroblast cells on PCL scaffolds was assessed through FUNCAT staining (33). Briefly, cell culture media (DMEM) without l-methionine was supplemented with the methionine analogue AHA at 100 µM. Scaffolds were seeded with 5,000 cells per scaffold and cultured for 48 h. Following culture, scaffolds were stained with the click chemistry fluorophore AFDye 488 DBCO (1:167 in PBS with 1% BSA) that labels azides in a copper-free reaction. Following 40 min of staining at 37 °C, scaffolds were fixed for 18 min in 4% PFA before staining with AlexaFluor 546 phalloidin (1:1,000) and Hoechst (1:10,000). Imaging was conducted on a confocal microscope as described above. Image analysis was conducted in ImageJ. Briefly, a sum slices projection was analyzed for local fiber organization and cell area from the blue and red channels, respectively. The nascent matrix integrated intensity was calculated from thresholded images.
RNAi Knockdown.
RNAi was carried out with SMARTpool: ON-TARGETplus siRNA’s specific for mouse Yap1 and Wwtr1 (Dharmacon) or scramble nontargeting siRNA as a control. Primary mouse tendon fibroblasts were transfected with Lipofectamine RNAiMAX (Thermo Fisher Scientific) in DMEM supplemented with 10% FBS and antibiotics-antimycotics for 72 h, after which they were reseeded on tissue-culture plastic at lower density (7,500 cells/cm2). To confirm on target efficacy of the pooled siRNA oligos, we tested two different groups of RNA oligos (one targeting Yap1 and one targeting Wwtr1) (SI Appendix, Fig. S4A).
Protein Lysis and Western Blotting.
For protein analysis, cells were lysed with RIPA buffer (Thermo Fisher Scientific) supplemented with Halt protease and phosphatase inhibitor (Thermo Fisher Scientific). Protein concentration was quantified with the Pierce BCA Protein Assay kit (Thermo Fisher Scientific). A total of 5 µg of protein was loaded per sample. Samples were loaded onto 4 to 15% gradient SDS-PAGE gels, then transferred to 0.2 µm pore-size PVDF membranes (Bio-Rad). Blots were blocked in blocking buffer (5% milk, 0.1% Tween-20 in 1× TBS) and then treated with primary antibodies (YAP/TAZ, anti-rabbit, Cell Signaling, 8418 and beta-Actin, antimouse, Cell Signaling, 3700) overnight at 4 °C. Next, blots were washed 3× in 1× TBS with 0.1% Tween-20. Blots were then incubated in species specific secondary HRP-conjugated antibody (anti-rabbit, Promega, W4011 and anti-mouse, Promega, W4021) for 1 h at room temperature. Blots were then washed 3× in 1× TBS with 0.1% Tween-20. Protein bands were visualized using Super Signal West Pico Plus (Thermo Fisher Scientific), and images were acquired using a ChemiDoc XRS Imaging System (BioRad).
RNA Isolation, cDNA Synthesis, and qRT-PCR.
In the FDL resection experiments, the skin was removed from the hindlimbs and the FDL was isolated such that the distal ~1 mm of tissue, closest to the injury site was removed and the remainder of the midsubstance up to the myotendinous junction was homogenized with a mortar and pestle in TRIzol LS. In the cell culture experiments, the cells were lysed directly in RNA lysis buffer supplied in RNeasy Mini Kit (Qiagen). mRNA isolation was carried out using the Qiagen mRNeasy Plus Mini Kit (Qiagen). RNA concentration was quantified using a NanoDrop spectrophotometer. cDNA was synthesized using the SuperScript VILO IV kit (Thermo Fisher Scientific). Quantitative real-time PCR (qRT-PCR) was completed using Fast SYBR reagents (Thermo Fisher Scientific) and analyzed using a QuantStudio 6 Pro (Applied Biosystems). All qRT-PCR experiments are analyzed with the delta delta Ct method. Results were further normalized to a random control sample within that experiment. Primer sequences are provided in SI Appendix, Table S1.
RNA Sequencing and Analysis.
RNA quality was evaluated using a BioAnalyzer. RNA libraries were prepared using Illumina truSeq polyA high-throughput kit and single-end reads were sequenced to 100 base pairs using an Illumina NovaSeq. Reads were aligned to the mm10 genome using Hisat2. Picard was used to generate bam files and sorted by chromosomal coordinates. FeatureCounts was used to generate count files (63). Deseq2 was used for differential expression analysis between groups (n = 3 biological replicates/group) (64). Gene ontology analysis was completed using DAVID (65, 66).
ChIPseq Analysis.
Publicly available Yap ChIPseq data was downloaded from GSE83863 (30). Sequenced reads were aligned to the mm10 build of the mouse genome using Bowtie2 with default settings (67). SAM files were converted to BAM and were sorted by chromosomal coordinates using Picard SortSam. Duplicates were removed using Picard Mark Duplicates. BAM files were then used to call peaks using MACS2, with default settings with a q threshold of 0.05 (68). To generate BigWig files, deepTools BamCoverage was used with default settings and a bin size of 10 bp (69). Integrative Genome Viewer was used to visualize BigWig files on the mm10 genome track.
ATACseq and Analysis.
Fifty thousand cells were subjected to Assay for Transposase Accessible sequencing (ATACseq) following the published protocol (70). DNA fragment size was analyzed using a Fragment Analyzer. Libraries were sequenced to 50 base pairs from both ends using an Illumina NovaSeq. Raw reads were aligned to the mm10 genome using Bowtie2. Picard and Samtools were used to generate bam files and to filter out duplicates and mitochondrial reads. Peaks were called using MACS2 using the following options: “--keep-dup all -q 0.01 -no model.” Diffbind was used to identify differentially accessibility chromatin loci between groups (n = 3 biological replicates/group) (70). Motif analysis was completed using the findMotifsGenome.pl command within Homer (71).
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (TXT)
Dataset S02 (TXT)
Acknowledgments
We would like to acknowledge the following funding sources: NIH (R00AR067283, R01AR075418, P50AR080581, and P30 AR069619) and the Department of Veterans Affairs (IK6 RX003416). Additional support was provided by a Gilliam Fellowship (GT13516) from the Howard Hughes Medical Institute. We would like to thank the Penn Genomic Sequencing Core for their assistance with RNA/ATACseq and the Penn Microscopy Core. We would also like to thank Drs. Fernando Carmago and Jenna Galloway for supplying the YAP-CA mice. We would finally like to thank all members of the Mauck and Dyment labs for thoughtful discussion and feedback during the development of this project.
Author contributions
D.L.J., G.F.H., X.J., R.C.L., M.K.E., E.D.B., A.S., T.P.L., M.P.N., J.D.B., R.L.M., and N.A.D. designed research; D.L.J., G.F.H., X.J., R.C.L., M.K.E., E.D.B., A.S., T.P.L., and M.P.N. performed research; D.L.J., G.F.H., X.J., R.C.L., M.K.E., E.D.B., A.S., T.P.L., and M.P.N. contributed new reagents/analytic tools; D.L.J., G.F.H., X.J., R.C.L., M.K.E., E.D.B., A.S., T.P.L., M.P.N., R.L.M., and N.A.D. analyzed data; and D.L.J., G.F.H., X.J., R.C.L., M.K.E., E.D.B., A.S., M.P.N., J.D.B., R.L.M., and N.A.D. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. A.J.E. is a guest editor invited by the Editorial Board.
Contributor Information
Robert L. Mauck, Email: lemauck@pennmedicine.upenn.edu.
Nathaniel A. Dyment, Email: dyment@pennmedicine.upenn.edu.
Data, Materials, and Software Availability
Raw and analyzed ATACseq and RNAseq data generated in this study are available through the Gene Expression Omnibus under the GEO accession GSE207896 (72).
Supporting Information
References
- 1.Szczesny S. E., Lee C. S., Soslowsky L. J., Remodeling and repair of orthopedic tissue: Role of mechanical loading and biologics: Part I: Tendon and ligament; meniscus. Am. J. Orthop. Belle Mead. NJ 39, 525 (2010). [PMC free article] [PubMed] [Google Scholar]
- 2.Saint-Maurice P. F., et al. , Association of daily step count and step intensity with mortality among US adults. JAMA 323, 1151–1160 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Korvick D. L., et al. , The use of an implantable force transducer to measure patellar tendon forces in goats. J. Biomech. 29, 557–561 (1996). [DOI] [PubMed] [Google Scholar]
- 4.Bonnevie E. D., et al. , Aberrant mechanosensing in injured intervertebral discs as a result of boundary-constraint disruption and residual-strain loss. Nat. Biomed. Eng. 3, 998–1008 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maeda T., et al. , Conversion of mechanical force into TGF-β-mediated biochemical signals. Curr. Biol. CB 21, 933–941 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riley G., Tendinopathy–from basic science to treatment. Nat. Clin. Pract. Rheumatol. 4, 82–89 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Arnoczky S. P., Lavagnino M., Egerbacher M., The mechanobiological aetiopathogenesis of tendinopathy: Is it the over-stimulation or the under-stimulation of tendon cells? Int. J. Exp. Pathol. 88, 217–226 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Millar N. L., et al. , Tendinopathy. Nat. Rev. Dis. Primer 7, 1 (2021). [DOI] [PubMed] [Google Scholar]
- 9.Lake S. P., Ansorge H. L., Soslowsky L. J., Animal models of tendinopathy. Disabil. Rehabil. 30, 1530–1541 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Sun H. B., et al. , Coordinate regulation of IL-1beta and MMP-13 in rat tendons following subrupture fatigue damage. Clin. Orthop. 466, 1555–1561 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.D’Urso M., Kurniawan N. A., Mechanical and physical regulation of fibroblast-myofibroblast transition: From cellular mechanoresponse to tissue pathology. Front. Bioeng. Biotechnol. 8, 609653 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu F., et al. , Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J. Cell Biol. 190, 693–706 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marinković A., Liu F., Tschumperlin D. J., Matrices of physiologic stiffness potently inactivate idiopathic pulmonary fibrosis fibroblasts. Am. J. Respir. Cell Mol. Biol. 48, 422–430 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang X., et al. , Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am. J. Respir. Cell Mol. Biol. 47, 340–348 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pocaterra A., Romani P., Dupont S., YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 133 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Tajik A., et al. , Transcription upregulation via force-induced direct stretching of chromatin. Nat. Mater. 15, 1287–1296 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J., Lewis A. H., Grandl J., Touch, tension, and transduction – the function and regulation of piezo ion channels. Trends Biochem. Sci. 42, 57–71 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Andreu I., et al. , Mechanical force application to the nucleus regulates nucleocytoplasmic transport. Nat. Cell Biol. 24, 896–905 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elosegui-Artola A., et al. , Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 171, 1397–1410.e14 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Dupont S., Wickström S. A., Mechanical regulation of chromatin and transcription. Nat. Rev. Genet. 2022, 1–20 (2022). [DOI] [PubMed] [Google Scholar]
- 21.Passini F. S., et al. , Shear-stress sensing by PIEZO1 regulates tendon stiffness in rodents and influences jumping performance in humans. Nat. Biomed. Eng. 5, 1457–1471 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker C. J., et al. , Nuclear mechanosensing drives chromatin remodelling in persistently activated fibroblasts. Nat. Biomed. Eng. 5, 1485–1499 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stowers R. S., et al. , Matrix stiffness induces a tumorigenic phenotype in mammary epithelium through changes in chromatin accessibility. Nat. Biomed. Eng. 3, 1009–1019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones D. L., et al. , ZNF416 is a pivotal transcriptional regulator of fibroblast mechanoactivation. J.Cell Biol. 220, e202007152 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nava M. M., et al. , Heterochromatin-driven nuclear softening protects the genome against mechanical stress-induced damage. Cell 181, 800–817.e22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le H. Q., et al. , Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 18, 864–875 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Sasmono R. T., et al. , A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood 101, 1155–1163 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto E., Hayashi K., Yamamoto N., Mechanical properties of collagen fascicles from stress-shielded patellar tendons in the rabbit. Clin. Biomech. Bristol Avon 14, 418–425 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Majima T., et al. , Biomechanical effects of stress shielding of the rabbit patellar tendon depend on the degree of stress reduction. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 14, 377–383 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Croci O., et al. , Transcriptional integration of mitogenic and mechanical signals by Myc and YAP. Genes Dev. 31, 2017–2022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Camargo F. D., et al. , YAP1 increases organ size and expands undifferentiated progenitor cells. Curr. Biol. 17, 2054–2060 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Schwenk F., Baron U., Rajewsky K., A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23, 5080–5081 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McLeod C. M., Mauck R. L., High fidelity visualization of cell-to-cell variation and temporal dynamics in nascent extracellular matrix formation. Sci. Rep. 6, 38852 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loebel C., Mauck R. L., Burdick J. A., Local nascent protein deposition and remodelling guide mesenchymal stromal cell mechanosensing and fate in three-dimensional hydrogels. Nat. Mater. 18, 883–891 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lavagnino M., Arnoczky S. P., In vitro alterations in cytoskeletal tensional homeostasis control gene expression in tendon cells. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 23, 1211–1218 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Paszek M. J., et al. , Tensional homeostasis and the malignant phenotype. Cancer Cell 8, 241–254 (2005). [DOI] [PubMed] [Google Scholar]
- 37.DuFort C. C., Paszek M. J., Weaver V. M., Balancing forces: Architectural control of mechanotransduction. Nat. Publ. Group 12, 308–319 (2011), 10.1038/nrm3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hillmer R. E., Link B. A., The roles of hippo signaling transducers Yap and Taz in chromatin remodeling. Cells 8, 502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haak A. J., et al. , Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci. Transl. Med. 11, eaau6296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Diaz Espinosa A. M., et al. , Dopamine D1 receptor stimulates cathepsin K-dependent degradation and resorption of collagen I in lung fibroblasts. J. Cell Sci. 133, jcs248278 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eliasson P., et al. , Simvastatin and atorvastatin reduce the mechanical properties of tendon constructs in vitro and introduce catabolic changes in the gene expression pattern. PloS One 12, e0172797 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szczesny S. E., et al. , Crimped nanofibrous biomaterials mimic microstructure and mechanics of native tissue and alter strain transfer to cells. ACS Biomater. Sci. Eng. 3, 2869–2876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Han W. M., et al. , Microstructural heterogeneity directs micromechanics and mechanobiology in native and engineered fibrocartilage. Nat. Mater. 15, 477–484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heo S.-J., et al. , Aberrant chromatin reorganization in cells from diseased fibrous connective tissue in response to altered chemomechanical cues. Nat. Biomed. Eng. 7, 177–191 (2022), 10.1038/s41551-022-00910-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gardner K., Lavagnino M., Egerbacher M., Arnoczky S. P., Re-establishment of cytoskeletal tensional homeostasis in lax tendons occurs through an actin-mediated cellular contraction of the extracellular matrix. J. Orthop. Res. 30, 1695–1701 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Arnoczky S. P., Lavagnino M., Egerbacher M., Caballero O., Gardner K., Matrix metalloproteinase inhibitors prevent a decrease in the mechanical properties of stress-deprived tendons: An in vitro experimental study. Am. J. Sports Med. 35, 763–769 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Nardone G., et al. , YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun. 8, 15321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carpenter J. E., Flanagan C. L., Thomopoulos S., Yian E. H., Soslowsky L. J., The effects of overuse combined with intrinsic or extrinsic alterations in an animal model of rotator cuff tendinosis. Am. J. Sports Med. 26, 801–807 (1998). [DOI] [PubMed] [Google Scholar]
- 49.Yamamoto N., et al. , Effects of stress shielding on the mechanical properties of rabbit patellar tendon. J. Biomech. Eng. 115, 23–28 (1993). [DOI] [PubMed] [Google Scholar]
- 50.Matsumoto F., Trudel G., Uhthoff H. K., Backman D. S., Mechanical effects of immobilization on the Achilles’ tendon. Arch. Phys. Med. Rehabil. 84, 662–667 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Blache U., et al. , Inhibition of ERK 1/2 kinases prevents tendon matrix breakdown. Sci. Rep. 11, 6838 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao H., Ren Y., Wu Y.-N., Liu S. Q., Zhang L.-Q., Ultrasonic evaluations of Achilles tendon mechanical properties poststroke. J. Appl. Physiol. Bethesda Md 1985, 843–849 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dideriksen K., et al. , Tendon collagen synthesis declines with immobilization in elderly humans: No effect of anti-inflammatory medication. J. Appl. Physiol. Bethesda Md 1985, 273–282 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Couppé C., et al. , The effects of immobilization on the mechanical properties of the patellar tendon in younger and older men. Clin. Biomech. Bristol Avon 27, 949–954 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Kiani K., Sanford E. M., Goyal Y., Raj A., Changes in chromatin accessibility are not concordant with transcriptional changes for single-factor perturbations. bioRxiv [Preprint] (2022). 10.1101/2022.02.03.478981 (Accessed 3 February 2022). [DOI] [PMC free article] [PubMed]
- 56.Belteki G., et al. , Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res. 33, e51 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pryce B. A., Brent A. E., Murchison N. D., Tabin C. J., Schweitzer R., Generation of transgenic tendon reporters, ScxGFP and ScxAP, using regulatory elements of the scleraxis gene. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 236, 1677–1682 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Bilic-Curcic I., et al. , Visualizing levels of osteoblast differentiation by a two-color promoter-GFP strategy: Type I collagen-GFPcyan and osteocalcin-GFPtpz. Genes. N. Y. N 2000, 87–98 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Peltz C. D., Perry S. M., Getz C. L., Soslowsky L. J., Mechanical properties of the long-head of the biceps tendon are altered in the presence of rotator cuff tears in a rat model. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 27, 416–420 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dourte L. M., et al. , Mechanical, compositional, and structural properties of the mouse patellar tendon with changes in biglycan gene expression. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 31, 1430–1437 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mih J. D., Marinkovic A., Liu F., Sharif A. S., Tschumperlin D. J., Matrix stiffness reverses the effect of actomyosin tension on cell proliferation. J. Cell Sci. 125, 5974–5983 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Driscoll T. P., Cosgrove B. D., Heo S.-J., Shurden Z. E., Mauck R. L., Cytoskeletal to Nuclear Strain Transfer Regulates YAP Signaling in Mesenchymal Stem Cells. Biophys. J. 108, 2783–2793 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang D. W., Sherman B. T., Lempicki R. A., Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 66.Huang da W., Sherman B. T., Lempicki R. A., Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012), 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y., et al. , Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ramírez F., et al. , deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016), 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Corces M. R., et al. , An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962 (2017), 10.1038/nmeth.4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heinz S., et al. , Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jones D. L., Mauck R. L., Dyment N. A., Mechano-epigenetic regulation of extracellular matrix homeostasis via Yap and Taz. NCBI Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE207896. Deposited 11 July 2022. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (TXT)
Dataset S02 (TXT)
Data Availability Statement
Raw and analyzed ATACseq and RNAseq data generated in this study are available through the Gene Expression Omnibus under the GEO accession GSE207896 (72).