

Abstract
CRISPR-Cas9-based therapeutic genome editing approaches hold promise to cure a variety of human diseases. Recent findings demonstrate pre-existing immunity for the commonly used Cas orthologs from Streptococcus pyogenes (SpCas9) and Staphylococcus aureus (SaCas9) in humans, which threatens the success of this powerful tool in clinical use. Thus, a comprehensive investigation and potential risk assessment are required to exploit the full potential of the system. Here, we investigated existence of immunity to SpCas9 and SaCas9 in control rhesus macaques (Macaca mulatta) alongside monkeys transplanted with either lentiviral transduced or CRISPR-SpCas9 ribonucleoprotein (RNP)-edited cells. We observed significant levels of Cas9 antibodies in the peripheral blood of all transplanted and non-transplanted control animals. Transplantation of ex vivo transduced or SpCas9-mediated BCL11A enhancer-edited cells did not alter the levels of Cas9 antibodies in rhesus monkeys. Following stimulation of peripheral blood cells with SpCas9 or SaCas9, neither Cas9-specific T cells nor cytokine induction were detected. Robust and durable editing frequencies and expression of high levels of fetal hemoglobin in BCL11A enhancer-edited rhesus monkeys with no evidence of an immune response (>3 years) provide an optimistic outlook for the use of ex vivo CRISPR-SpCas9 (RNP)-edited cells.
Keywords: SaCas9, SpCas9, genome editing, gene therapy, immunity, hematopoietic stem cell transplantation
Graphical abstract
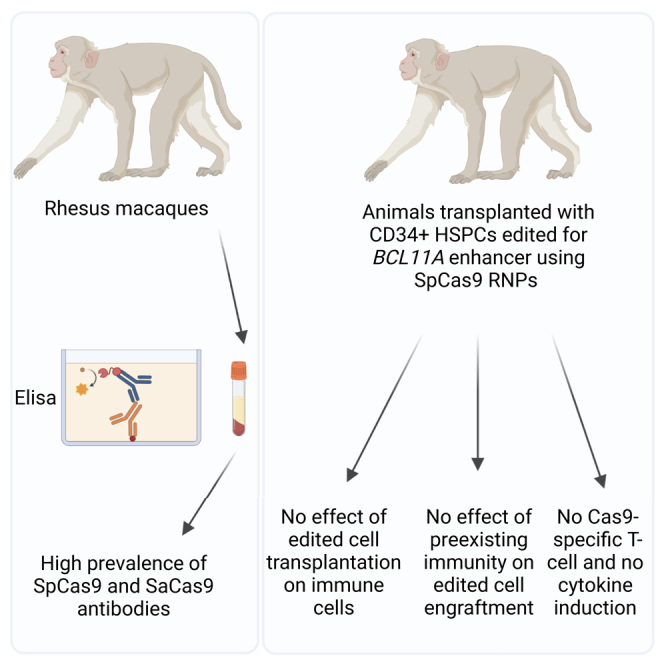
Demirci and colleagues demonstrated that non-transplanted and transplanted rhesus macaques exhibited significant levels of SaCas9 and SpCas9 antibodies, yet no ex vivo cell-mediated immune activity was found. In addition, ex vivo edited CD34+ HSPCs for BCL11A enhancer using SpCas9 RNP efficiently engrafted long term, without evidence of an enhanced immune response.
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated (Cas) protein 9 (Cas9) system, derived from bacteria and archaea, enables precise and highly effective genome manipulation in a wide variety of cells and organisms.1,2 Due to its ease-of-use, low cost, high specificity, and low off-target effects, the method is broadly used as a research tool and has already been incorporated into clinical trials to treat genetic disorders including sickle cell disease (SCD).3,4,5 The editing technology is applied either to knock down gene expression (i.e., BCL11A erythroid enhancer editing) by introducing small insertions/deletions (Indels) mainly through non-homologous end-joining (NHEJ)-mediated repair or to correct underlying disease mutations (i.e., SCD mutation correction) via homology directed repair.6,7,8 Ex vivo progenitor cell editing largely involves delivery of ribonucleoprotein (RNP) complex to cultured cells via electroporation prior to transplantation, whereas in vivo editing depends on transfer of editing toolboxes to target organs/tissues (i.e., using recombinant adeno-associated virus [rAAV] vectors or nanoparticles to target liver).9,10
Given that genome editing is frequently performed using Cas9 orthologs adapted from common bacterial strains that may be pathogenic to human such as Staphylococcus aureus (S. aureus; SaCas9) and Streptococcus pyogenes (S. pyogenes; SpCas9), and pre-existing immunity to these endonucleases has been reported in many healthy individuals,11,12 it is unclear whether this immunity will have an adverse impact on CRISPR-Cas9-edited cells in clinical applications. rAAV are often employed to deliver CRISPR-Cas9 in vivo due to its non-pathogenic nature, ability to infect a wide variety of tissues, and transient expression. Despite these advantages, expected therapeutic benefits were not achieved in some prior clinical work with patients displaying pre-existing adaptive immunity to viral antigens.13,14,15 More specifically, when SpCas9 expressing AAV was introduced into mouse skeletal muscle, both cellular and humoral immune responses accumulated at the target site.16 Similarly, a recent study demonstrated that pre-existing immunity to SaCas9 in mice transduced with AAV encoding SaCas9 could pose a barrier to liver genome editing due to the robust CD8+ cytotoxic lymphocyte (CTL) response that eventually cleared all edited hepatocytes.17
Potential cellular and humoral responses to Cas9 or Cas9-edited cells may vary depending on the editing context, necessitating a case-by-case evaluation of genome editing strategies. Although sustained expression of Cas9 via viral vector delivery has the potential to induce a significant immune response against Cas9/Cas9-edited cells, the effects of transient one-time Cas9 exposure with RNP are yet to be elucidated. Here, we show that rhesus macaques (Macaca mulatta) have acquired immunity to these frequently exploited Cas9 orthologs. Additionally, we have demonstrated that pre-existing immunity to Cas9 orthologs is unaffected in nonhuman primates following SpCas9-edited (RNP) CD34+ hematopoietic stem and progenitor cell (HSPC) transplantation, which may improve prospects for efficient and safe genome editing.
Results
Control rhesus macaques have anti-Cas9 antibodies for both SaCas9 and SpCas9
S. aureus and S. pyogenes are part of the normal microbial flora of humans and nonhuman primates and have the potential to be infectious.18,19,20 Therefore, we investigated whether control non-transplanted rhesus macaques had humoral immunity to SpCas9 and SaCas9. To do so, we profiled the existence of immunoglobulin G (IgG) antibodies against Cas9 proteins in non-transplanted control animals’ serum by an enzyme-linked immunosorbent assay (ELISA). Because pure rhesus albumin was not commercially available, we used human serum albumin (HSA) and bovine serum albumin (BSA) as negative controls. Additionally, since our rhesus colony has been immunized to measles, we used measles antigens as a positive control. Analyzing nine monkey serum samples revealed that all non-transplanted control rhesus monkeys displayed anti-Cas9 antibodies for both SaCas9 and SpCas9 (Figure 1A). Antibody levels for SaCas9 were slightly but not significantly higher than the levels for SpCas9. To confirm reproducibility of the assay, we collected serum samples from the same nine animals 3 months after the first ELISA assay and repeated the analysis. The results demonstrated comparable levels of Cas9 antibodies in the repeat samples, confirming the reproducibility of the assay (Figure 1A).
Figure 1.

Anti-SpCas9 and anti-SaCas9 expressions are present in both non-transplanted and transplanted control animals
(A) Anti-SpCas9 and anti-SaCas9 protein levels were investigated in non-transplanted control animals in two different time points (3 months apart). Each colored, filled circle represents a specific animal, and the same color code is used for both experiments. ns, not significant; ∗p < 0.05 and ∗∗∗p < 0.001. (B) Anti-Cas9 antibody levels were investigated in lentivirally transduced CD34+ HSPC transplanted control animals before and after transplantation. BSA, bovine serum albumin; HSA, human serum albumin. Measles antigens were used as positive control.
Our main question was whether the presence of pre-existing antibodies to Cas9 would trigger an immune response to CRISPR-Cas9-edited cells in our rhesus preclinical model. Before addressing this question, we first evaluated serum samples for two transplanted control animals who received CD34+ HSPCs transduced with a lentiviral vector as a no-CRISPR-Cas9-editing control. Pre- and post-transplantation sample analyses confirmed that the levels for all parameters investigated (anti-SpCas9, anti-SaCas9, and anti-measles) were relatively stable (Figure 1B), and as anticipated, there was no increase in antibody levels observed after transplantation.
Serum anti-Cas9 antibody levels remain stable after infusion of ex vivo edited cells
Next, we evaluated whether serum anti-Cas9 levels change after infusion of CRISPR-Cas9 erythroid-specific BCL11A enhancer- (+58 site) or AAVS1-edited CD34+ HSPCs into total body irradiated animals (Figure 2A). CD34+ HSPCs were edited ex vivo using either 5 μM (for ZM17 and ZM26) or 10 μM (for ZL25 and ZL22) RNP composed of 2× NLS or 3× NLS SpCas9 and erythroid-specific BCL11A enhancer or AAVS1 targeting guide RNA.21 Pre- and post-transplantation blood serum samples were collected and analyzed using the ELISA method. Similar anti-SpCas9 and anti-SaCas9 antibodies levels were detected in the pre- and post-transplantation blood serum samples analyzed by ELISA (Figures 2B–2E). Of note, ZM17 and ZL22 were diagnosed with radiation pneumonitis and euthanized. However, we did not see any increase in the anti-Cas9 antibodies in these animals prior to euthanasia.
Figure 2.

Anti-Cas9 expressions were similar in pre- and post-transplantation periods for BCL11A enhancer-edited animals
(A) Schematic representation of CRISPR-edited CD34+ HSPC transplantation. Note: The schema was made using biorender.com. (B and C) Anti-Cas9 antibody levels were comparable before and after infusion of BCL11A enhancer (+58 site) and AAVS1, (D and E) BCL11A enhancer (+58 site), or (F–H) BCL11A enhancer (+55 and +58 sites) SpCas9-ribonucleaprotein (RNP)-edited CD34+ HSPC transplanted animals. G-CSF, granulocyte colony stimulating factor; RBC, red blood cell; TBI, total body irradiation; BSA, bovine serum albumin; HSA, human serum albumin. Measles antigens were used as positive control.
In addition to targeting only the +58 site in the erythroid-specific BCL11A enhancer, we simultaneously targeted both the +55 and +58 sites using 3× NLS SpCas9 and two guide RNAs (Figure 2A).22 Conditioning in these animals was switched to intravenous busulfan at myeloablative doses to mimic the clinical setting. Of note, busulfan does not provide the degree of immunosuppression we achieve with total body irradiation in this model.23 Additionally, the RNP concentration in this cohort was at 10 μM. Nonetheless, ex vivo CD34+ HSPC editing and infusion did not enhance anti-SpCas9 antibody levels in these busulfan-conditioned animals (Figures 2F–2H). More importantly, editing ratios in the infusion products were 97%–99% and 83%–94% in neutrophils after engraftment with a follow-up of 2.5–3.5 years (Table 1), indicating no adverse effects of anti-SpCas9 on engraftment of edited cells. The presence of anti-Cas9 antibodies in control non-transplanted and transplanted animals along with BCL11A enhancer-edited animals were further confirmed using western blot (Figure 3).
Table 1.
Overview of the transplanted animals used in the study
| Animal ID | Sex | Conditioning | Cas9 | Target | RNP conc. (μM) | Pre-/post-electro-poration culture time (h) | Infusion product (×106 cells/kg) | Infusion product indel (%) or vector copy number (VCN) | Peripheral blood granulocyte indel (%) or VCN | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| ZL52 | F | TBI (5 Gy × 2) | N/A | chromatin loop formation in the β-globin loci | N/A | N/A | 4.72 | VCN; 7.8 | VCN; 0.68 at 12 weeks and 1.29 at 52 weeks post-infusion | Peslak et al.24 |
| ZM31 | F | TBI (5 Gy × 2) | N/A | chromatin loop formation in the β-globin loci | N/A | N/A | 3.22 | VCN; 4.3 | VCN; 0.32 at 12 weeks and 0.49 at 52 weeks post-infusion | |
| 08D247 | F | TBI (5 Gy × 2) | N/A | chromatin loop formation in the β-globin loci | N/A | N/A | 4.03 | VCN; 11.4 | VCN; 0.62 at 12 weeks and 0.27 at 31 weeks post-infusion | Unpublished data |
| ZL25 | M | TBI (5 Gy × 2) | 2× NLS SpCas9 | BCL11A enhancer and AAVS1 | 10 | 0 h/48 h | 1.42 | indel; 73.4% (AAVS1); 40.4% (BCL11A enhancer) | indel; 8.5% at 12 weeks and 6.5% at 52 weeks post-infusion for AAVS1; 3.3% at 12 weeks and 1.5% at 52 weeks post-infusion for BCL11A enhancer | Demirci et al.21 |
| ZL22 | M | TBI (5 Gy × 2) | 2× NLS SpCas9 | BCL11A enhancer and AAVS1 | 10 | 24 h/48 h | 1.23 | indel; 87.8% (AAVS1); 33.2% (BCL11A enhancer) |
indel; 53% at 12 weeks post-infusion for AAVS1; 10% at 12 weeks post-infusion for BCL11A enhancer | |
| ZM26 | M | TBI (5 Gy × 2) | 3× NLS SpCas9 | BCL11A enhancer | 5 | 24 h/48 h | 1.78 | indel; 84.9% | indel; 14.2% at 12 weeks; 11.4% at 52 weeks post-infusion | |
| ZM17 | F | TBI (5 Gy × 2) | 3× NLS SpCas9 | BCL11A enhancer | 5 | 24 h/48 h | 6.06 | indel; 80.9% | indel; 75.8% at 12 weeks; 83.2% at 25 weeks post-infusion | |
| ZL41 | F | busulfan (4 × 5.5 mg/kg) | 3× NLS SpCas9 | BCL11A enhancer | 10 | 24 h/24 h | 4.03 | indel; 97.3% | indel; 73.4% at 12 weeks; 93.9% at 52 weeks post-infusion | Zeng et al.22 |
| 13U018 | F | busulfan (4 × 5.5 mg/kg) | 3× NLS SpCas9 | BCL11A enhancer | 10 | 24 h/24 h | 2.20 | indel; 98.9% | indel; 92.0% at 12 weeks; 84% at 52 weeks post-infusion | |
| RA0013 | F | busulfan (4 × 5.5 mg/kg) | 3× NLS SpCas9 | BCL11A enhancer | 10 | 24 h/24 h | 4.38 | indel; 98.6% | indel; 73.4% at 12 weeks; 93.9% at 52 weeks post-infusion |
Figure 3.

Presence of anti-Cas9 antibodies in rhesus serum was confirmed using western blot
Both anti-SpCas9 and anti-SaCas9 antibodies were detected in all rhesus animals and time points tested. Control transplanted animals were transplanted with C34+ HSPCs transduced with lentiviruses.
Cellular immune responses to SpCas9 and SaCas9 were below the limit of detection
To investigate the presence of Cas9-specific T cells in rhesus peripheral blood mononuclear cells (PBMCs), we investigated the frequency of cytokine (IFN-γ, IL-2, and TNF-α) positive CD3+ T cells and CD154 and CD137 expression among CD3+ T cells post-stimulation (Figure S1). Although significant levels of induction were detected in positive control groups (PMA + ionomycin; PMA/I), no statistical differences were noted for stimulated and non-stimulated SpCas9 and SaCas9 groups (Figures 4A and 4B). Furthermore, IFN-γ enzyme-linked immunospot (ELISpot) assay demonstrated no difference between Cas9 antigen stimulated PBMCs versus unstimulated controls (Figure 4C). To exclude that limited processing of full-length SpCas9 and SaCas9 protein contributed to our inability to detect Cas9-specific T cells, we performed IFN-γ ELISpot assays with SpCas9 and SaCas9 derived pepmixes, which consist of 15-amino-acid-long peptides with an 11 amino acid overlap. These are routinely used to detect or reactivate antigen-specific T cells with high sensitivity.25,26,27 We performed IFN-γ ELISpot assays with pepmixes on unstimulated PBMCs and on PBMCs that were activated for 9 days with SpCas9 or SaCas9 pepmixes in the presence of IL7/IL15 to increase their frequency (Figures 5A and S2A). PBMCs stimulated with CMV/AdV pepmixes, DMSO (negative), and PMA/I (positive) served as controls. No SpCas9- or SaCas9-specific T cells could be detected at baseline or on day 9 post reactivation, while CMV/Adv-specific T cells were detected in three out of nine samples post activation (Figures 5B–5D and S2B–S2D). Thus, using two orthogonal approaches, we were not able to detect SpCas9- or SaCas9-specific T cells in rhesus PBMCs.
Figure 4.

No cytokine or cell surface marker induction was observed after either SpCas9 or SaCas9 stimulation
Peripheral blood mononuclear cells (PBMCs) were collected from both transplanted and non-transplanted animals and stimulated with SpCas9 or SaCas9 (10 μg/mL) and analyzed for (A) interferon-gamma (IFN-γ), interleukin-2 (IL-2), or tumor necrosis factor alpha (TNF-α) or (B) CD154 or CD137 positive CD3+ T cell frequencies. PMA/I (phorbol-12-myristate-13-acetate/ionomycin) was used as a positive control. No Stim, no stimulation. (C) PBMCs from two human and eight rhesus donors were left unstimulated (NC) or treated with phytohemagglutinin (PHA, 5 μg/mL) as a positive control or Cas9 antigens (20 μg/mL) and tested by IFN-γ ELISpot. ns, not significant, and ∗∗∗p < 0.001.
Figure 5.

SpCas9-specific T cells are not detected by IFN-γ ELISpot assay
(A) Outline of experiment performed on nine rhesus macaque PBMCs. (B) IFN-γ ELISpot performed on day 0. (C and D) IFN-γ ELISpot assay results performed on day 9 following stimulation with (C) SpCas9 or (D) rhCMV/HuAdV pepmixes. Saturated: complete confluency of spot-forming cells (SFC).
Discussion
Although the prevalence of anti-Cas9 antibodies differs from study to study, most likely due to the differences in the study population, the detection technique, and its sensitivity and specificity, pre-existing immunity to Cas9 has indeed been reported in animals and humans.12,28,29 The pre-existing immunity to bacterial-derived Cas9 represents a significant potential hurdle for CRISPR-based therapies. A comprehensive understanding of the immune response of the host toward CRISPR-Cas9-edited cells/Cas proteins is essential to design long-lasting therapeutic strategies with the least adverse effects on the host/edited cells. When CRISPR cargos were delivered using AAV vectors, apoptosis induction in target tissues, loss of recombinant AAV genomes, and complete elimination of genome-edited cells have been observed in mice17 and canine30 models due to pre-existing immunity to SaCas9 and SpCas9, respectively. Additional genome editing studies in mice have shown similar results, with inefficient genome editing outcomes due to prevalent immunity against AAV-delivered Cas9, acting as a barrier to long-lasting therapy and repeated dosing.16,31 Similarly, Pten targeted CRISPR cargo delivered using Adenovirus vectors (AdV)-induced SpCas9-specific immune response in mice.32 In contrast, delivery of Cas9 by lipid nanoparticles to human hepatocytes appears to be well tolerated with sustained editing inferred by the absence of target gene product.33 In this manner, AAV- and AdV-based Cas9 delivery providing long-term protein expression could be the reason for robust immune system activation. Additionally, little is known about ex vivo Cas9 RNP-mediated gene editing immune response. As Cas9 protein is mostly cleared within 24 h,34,35 common genome editing protocols for ex vivo Cas9 RNP editing (1–2 days resting after electroporation) would theoretically be safe. Nonetheless, since there is a major concern for Cas-specific immunity and several ongoing and future clinical trials are designed with ex vivo Cas9 RNP-edited cells, there remains an open question to be answered. Here, we assessed the consequences of pre-existing immunity for both SpCas9 and SaCas9 along with Cas9-specific immune response in rhesus monkeys transplanted with ex vivo SpCas9 RNP-edited CD34+ HSPCs.
As S. aureus and S. pyogenes naturally exist in normal flora of rhesus monkeys,18,19,20 we first evaluated potential pre-existing anti-Cas9 antibodies in the rhesus serum samples using an established ELISA method. Stable and significant levels for both anti-Cas9 antibodies were detected in the control animals at two different time points 3 months apart, as well as in lentiviral vector transduced CD34+ HSPC transplanted animals at pre- and post-transplant periods (Figure 1). As expected, because no Cas9 was used during lentiviral transduction, no increase in the anti-Cas9 antibodies was noted for lentiviral vector transduced CD34+ HSPC transplanted animals. The presence of anti-Cas9 antibodies in serum was further confirmed using western blot (Figure 3). Further evaluations on ex vivo SpCas9 RNP-edited CD34+ HSPC transplanted animals that were targeted at the BCL11A enhancer +58 site or both the +58 and +55 sites were conducted (Figure 2). Interestingly, the results revealed stable antibody levels before and after transplantation indicating no obvious effects of transplanted edited cells on the animal’s immunity against SpCas9. Although the cells were edited using SpCas9, we also examined levels of anti-SaCas9 in the transplanted animals as a control group. As anticipated, no differences in antibody levels for SaCas9 were observed before and after transplantation.
We also investigated cellular immunity by measuring IFN-γ, IL-2, and TNF-α producing CD3+ cells or surface marker expression (CD154 and CD137) upon stimulation of rhesus PBMCs with SaCas9 or SpCas9 protein (Figure 4). In addition, we performed IFN-γ ELISpot assays using whole protein (Figure 4) or SaCas9- or SpCas9-derived pepmixes (Figure 5) to detect Cas9-specific T cells. We could not detect Cas9-specific cytokine production or cell surface marker induction in any of our performed assays, indicating that rhesus macaques have either no or a very low frequency (<1 than in 100,000 cells) of Cas9-specific T cells in their peripheral blood, based on the sensitivity of ELISpot assays.36 One plausible explanation is that the SpCas9 antibodies detected in the study were pre-existing, likely induced a long time prior to our investigation. These antibodies are generated by long-lived plasma cells, which play a pivotal role in humoral immunity and do not necessitate T cell assistance. Additionally, our evaluation of Cas9-specific T cells was limited to the peripheral blood. However, it should be noted that only approximately 2% of lymphocytes are present in peripheral blood, and a higher frequency of Cas9-specific T cells may exist in other lymphoid organs that were not assessed in this study.37 It is also worth to mention that Wagner et al.28 previously reported a CD8+ T cell response was triggered by SpCas9 stimulation, while Simhadri et al.38 demonstrated CD4+ T cell response prompted by SaCas9 stimulation. Wagner et al. also reported an increased frequency of regulatory T cells (Treg) specific to SpCas9 that could possibly be masking the effect of CD8+ T cells and may potentially reduce the severity of an immune response. Elevated levels of Tregs could dampen the cellular immune response and could potentially be the reason why we did not see any cellular response. In order to assess such a possible scenario, further experiments are warranted to investigate separate CD4+/CD8+ and Treg fractions in these animals.
Previous studies have shown that the transfer of Cas9 using viral systems can result in longer expression, leading to a stronger immune response.17,32 However, clinical trials that use HSPCs exposed to RNP complexes ex vivo39,40 or lipid nanoparticle-mediated RNP transfer33 do not appear to cause significant immune system activation. While it remains unclear whether resting post-electroporation is necessary to prevent an immune cell response, our research and clinical trials investigating ex vivo edited cells suggest that incubation for 24–48 h post-electroporation is safe. For applications that require longer Cas9 expression, additional precautions such as immunogenic Cas9 masking, using Cas9 orthologs from non-pathogenic microorganisms, or immune tolerance induction should be considered. Looking forward, further research is needed to fully understand the mechanisms of immune response to different gene editing approaches and to develop innovative strategies for minimizing potential immune reactions.
While nonhuman primates are valuable tools in research and developmental stages, they are not perfect models, similar to other preclinical models. Recent studies have suggested that there are major differences in immune cell regulation and adaptive response between human and nonhuman primates.41,42,43 For instance, T cell proliferative response following T cell receptor activation is stronger in human compared with nonhuman primates.44 Although our results display an optimistic outlook for ex vivo SpCas9-edited cell transplantation approaches in clinics, such differences in the immune cell regulation and activation could limit the definitive use of certain nonhuman primates in anti-Cas9 investigations before clinical trials. Therefore, it is imperative that clinical trials evaluating ex vivo or in vivo Cas9-edited cell transplantation also incorporate comprehensive assessments of serum anti-Cas9 levels and immune response to edited cells to pave the way for more effective and safe clinical approaches.
Conclusions
Immunogenicity against Cas proteins has been a subject of debate in recent years due to its potential adverse effects on long-lasting therapeutic CRISPR genome editing strategies. Understanding of the phenomenon is critical in order to design better genome editing modalities for patients. Our current study provides an optimistic outlook for the use of SpCas9 RNPs for ex vivo BCL11A enhancer editing of CD34+ HSPCs to reactivate fetal hemoglobin as no deleterious effects were noted in rhesus monkeys with pre-existing Cas9 immunity. Durable and robust engraftment of the edited cells in these animals was observed with no evidence of an enhanced immune response. Still, no cell-mediated immune activity was able to be detected in rhesus PBMCs induced by SaCas9 or SpCas9. However, this study only covers the RNP modality of SpCas9 editing, and it still needs to be determined if other forms of editing having longer expressions, such as mRNA, plasmid, or AAV, would be safe or not. These results are promising yet need further comprehensive understanding regarding the mechanisms involved in induction of these immune responses in order to be used in clinical decision-making.
Materials and methods
Animals
Animal samples were used following a National Heart, Lung, and Blood Institute (NHLBI) Animal Care and Use Committee approved protocol (protocol no: H-0136R4). Rhesus CD34+ HSPCs were mobilized using Granulocyte Colony Stimulating Factor (G-CSF, Thousand Oaks, CA) and AMD3100 (Plerixafor, Sigma-Aldrich) regimen and enriched as described previously.21 For BCL11A enhancer-editing animals, cells were either directly electroporated (Animal ID: ZL25) or pre-stimulated in XVIVO-10 (Lonza) media containing human stem cell factor (SCF, R&D Systems), human thrombopoietin (TPO, R&D Systems), and recombinant human Flt3-ligand (Flt3-L, R&D Systems) (100 ng/mL each) for 24 h. Rhesus HSPCs were electroporated with RNP complexes consisting of either 2×NLS-Cas9 (Animal IDs: ZL25 and ZL22) or 3×NLS-Cas9 (all other editing animals), and AAVS1 and BCL11A enhancer (+58 site) targeting guide RNAs (Animal IDs: ZL25 and ZL22), single BCL11A enhancer (+58 site) targeting guide RNA (Animal IDs: ZM26 and ZM17), or two BCL11A enhancers (+55 and +58 sites) targeting guide RNAs (Animal IDs: ZL41, 13U018, and RA0013). After electroporation, cells were rested in the same media for either 48 h (Animal IDs: ZL25, ZL22, ZM26, and ZM17) or 24 h (Animal IDs: ZL41, 13U018, and RA0013). Rested cells either freshly infused (Animal IDs: ZL25 and ZL22) or cryopreserved before infusion (all other editing animals). The lentiviral control animals (ZM31, ZL52, and 08D247) were transplanted with CD34+ HSPCs transduced with lentiviruses carrying artificial zinc-finger constructs targeting chromatin loop formation in the β-globin loci. Cells were transduced at high cell density conditions as described earlier.24 Detailed information on the editing/transduction animals of the study can be found in Table 1 and our previously published reports.21,22,24
Enzyme-linked immunosorbent assay
An ELISA protocol was adapted from a previously published work.11 Briefly, 96-well Maxisorp plates (Thermo Fisher Scientific) were coated with each antigen (SpCas9 [Wolfe laboratory, University of Massachusetts Medical School, Worcester, MA, USA],45 SaCas9 [Aldevron], native measles virus [Bio-rad], BSA [MilliporeSigma], or human albumin [MilliporeSigma]) at a concentration of 1 μg/ml overnight at 4°C in a 1× bicarbonate buffer solution (MilliporeSigma). Plates were then washed three times for 5 min using a one-percent wash buffer (TBST, pH 8.0; Sigma-Aldrich). Plates were subsequently blocked for 2 h at room temperature with 5% non-fat milk blocking solution. Next, 1:20 diluted serum samples were added and incubated for 2 h at room temperature and then washed three times. Goat anti-Rhesus Fc antibody (SouthernBiotech) was diluted 1:10,000 times in 5% non-fat milk blocking solution and applied before being incubated for 1 h at room temperature. Following the addition of the 3,3′,5,5′-tetramethylbenzidine substrate solution (Thermo Fisher Scientific), the reaction was stopped by adding 1 N sulfuric acid (Thermo Fisher Scientific) and the absorbance at 450 nm was measured using a microplate reader (microplate reader Fluostar Omega; BMG Labtech).
Intracellular cytokine staining
Staining and analysis for intracellular cytokines was conducted as previously described.11 In short, cells were thawed and diluted 1:10 in RPMI 1640 medium (Corning) with 5% Rhesus AB serum, 1,000 U/ml of penicillin, and 1,000 ng/mL of streptomycin, and 25 U/ml of benzonase nuclease (Santa Cruz Biotechnology). The medium was aspirated after the cells had been spun at 400 g for 10 min. The cells were then resuspended in 1 mL of medium containing 25 U/ml of benzonase and passed through a 70-μm filter. Finally, 9 mL of the medium containing 25 U/ml of benzonase was added to the cells, and they were spun at 400 g for 10 min. After removing the media and resuspending the cells in benzonase-free medium, 5 × 105 PBMCs were plated in a 12-well plate (Corning), and they were rested overnight. Afterward, 10 μg/mL of each antigen was administered to the cells. After 2 h, brefeldin A (Abcam) was added to the cells, and cells were incubated for an additional 4 h. After washing twice with the staining buffer (1% Rhesus serum albumin [Bioworld] and 0.02% sodium azide in phosphate buffer solution-PBS, pH 7), cells were stained for CD3 (APC-nonhuman primate anti-CD3, clone 10D12, Miltenyi Biotec), CD4 (V450-human anti-CD4, clone L200, BD Biosciences), and CD8a (Brilliant violet 650 - human anti-CD8a, # 301042, BioLegend). Then, using a Cytofix/Cytoperm kit (BD Biosciences), cells were fixed/permeabilized and then stained for intracellular cytokines, IFN-γ (PE-human anti-IFN-γ, Clone B27, BD Biosciences), and IL-2 (BV510 - human anti-IL-2, Clone MQ1-17H12, BD Biosciences). Cells were then washed twice with permeabilization buffer (BD Biosciences), resuspended in staining buffer, and analyzed with a BD FACSymphony Flow Cytometer.
CD137/CD154 analysis
As described above, cells were thawed, plated, and allowed to rest overnight. Cells were treated with each antigen at a concentration of 10 μg/mL for 16 h after an overnight rest. Then, cells were stained with CD3, CD4 (V450-human anti-CD4, Clone L200, BD Biosciences), CD8a, and CD137 (BV510-human anti-CD137, Clone 4B4-1), and CD154 (APC-Cy7-human anti-CD154, BD Biosciences). Cells were washed twice in staining buffer and assessed using the Symphony flow cytometer.
Western blotting
SpCas9 and SaCas9 proteins (1 μg) were resolved in NuPAGE LDS sample buffer (NP0007, Thermo Fisher Scientific) and NuPAGE sample reducing agent (NP0004, Thermo Fisher Scientific). Samples were then loaded into NuPAGE Bis-Tris 4%–12% gradient gels (NP0335BOX, Thermo Fisher Scientific) and ran with NuPAGE MOPS SDS running buffer (NP0001, Thermo Fisher Scientific) combined with NuPAGE antioxidant (NP0005, Thermo Fisher Scientific). The proteins were then electro-transferred to polyvinylidene fluoride nitrocellulose membranes (PB5210, Thermo Fisher Scientific) using iBlot 2 Dry Blotting System (IB21001, Invitrogen). Membranes were blocked for 1 h at room temperature in 5% milk (Sc-2325, Santa Cruz Biotechnology) in PBS (25- 507B, GenClone) with Tween (PBST) (28352, Thermo Fisher Scientific) on a shaker followed by overnight incubation at 4°C with 1:10 dilution of monkey serums in PBST with 0.5% non-fat milk. Membranes were washed five times for 5 min with PBST and then incubated for 1–2 h at room temperature with anti-rhesus antibody horseradish peroxide-conjugated secondary antibody (Abcam) at a dilution of 1:5,000. Membranes were then washed five times for 5 min with PBST and developed using ECL western blot reagent, Super Signal West Dura Extended Duration Substrate (34076, Thermo Fisher Scientific). Membranes were analyzed using chemiluminescence via Fuji Amersham Imager 680 blot and imager (Cytvia).
PBMC stimulation with pepmixes
PBMCs from rhesus macaques were stimulated for 90 min at 37°C with 5% CO2 with SaCas9 (22.6 pmol), SpCas9 (30 pmol), or a combination of rhCMV/HuAdV (30 pmol) pepmixes per 1×106 cells. All pepmixes are described in the ELISpot assay section. Cells were then cultured for 9 days in A-CTL media. A-CTL media composition is as follows: 55% Advanced RPMI-1640 (1X) (Gibco, #12633-012), 45% Click’s Medium (EHAA) (Irvine Scientific, #9195), 5% Human AB Serum Negative for HBsAG, HIV-1/2, HCV, (Corning, #35-060-CI), and 1% GlutaMAX supplement (Thermo Fisher Scientific, # 35050061) supplemented with IL-7 (10 ng/mL) (Recombinant Human IL-7; Peprotech, #200-07) and IL-15 (100 ng/mL) (Recombinant Human IL-15; Peprotech, #200-15).
ELISpot assay
IFN-γ ELISpot assays were performed as previously described.46 Briefly, cells were plated in triplicates at a seeding density of 0.5–1 ×105 cells/well, and antigen-specific T cell reactivity was measured after direct stimulation with whole SpCas9 or Sacas9 (10–20 μg/mL) (SpCas9 [Wolfe laboratory], Sacas9 [Aldevron]) or peptide libraries, dissolved in DMSO consisting of 15-amino-acid-long peptides with an 11-amino-acid overlap that covered the coding regions of the following proteins: rhesus cytomegalovirus rhCMV (Rh112) (#PM-rhCMV-Rh112); adenovirus (AdV) serotype 5 penton protein (#PM-PM-HAdV5) and AdV serotype 3 hexon protein (#PMHAdV3); and SpCas9 (two subpools) (#PM-STRP1-Cas9), all obtained from JPT Peptide Technology (Acton, MA). In the absence of a commercially available library for SaCas9, individual peptides were synthesized by Genemed Synthesys (San Antonio, TX), and the library was assembled in the laboratory. Media (no peptide, DMSO only) served as a negative control. PHA 5 μg/mL (Thermo Fisher Scientific, #50-1129-264) or PMA/I (Thermo Fisher Scientific, #00-4970-03) was used as positive control. After 18–20 h of incubation, plates were developed, dried at room temperature, and sent to Zellnet Consulting (New York, NY) for quantification. The frequency of T cells specific to each antigen was corrected for confluency and then normalized as spot-forming cells per 1×105 cells.
Statistical analysis
The data were statistically analyzed with one-way analysis of variance followed by the Tukey post hoc test using GraphPad Prism 9.0.2 software (GraphPad Software, CA). Data were represented as mean ± SD and considered significantly different at ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Data availability
Data are available upon request.
Acknowledgments
This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute (NHLBI) at the National Institutes of Health (NIH), NIH grant 8 U01AI157189-03 (S.G.). D.E.B was supported by the National Heart, Lung, and Blood Institute (OT2HL154984, P01HL053749) and the St. Jude Children's Research Hospital Collaborative Research Consortium. We thank animal facility staffs and veterinarians at the NIH Animal Center for their support. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author contributions
K.E., W.H., M.B.N.K., R.M., J.Z., R.C., N.U., U.G., J.M., and S.D. performed experiments and analyzed results. S.D. made figures. A.C.B., A.E.K., N.D.L., and R.E.D. performed animal care, transplantation, and sample derivation. S.A.M. and S.A.W. produced 3× NLS SpCas9 protein. S.D., G.A.B., S.G., D.E.B., L.S.K, R.E.D, and J.F.T. designed the research. K.E., W.H., M.B.N.K., J.M., S.D., and. J.F.T. wrote the paper. All authors reviewed, edited, and commented on the manuscript.
Declaration of interests
S.G. has patent applications in the fields of cell and/or gene therapy for cancer. S.G. is a consultant of TESSA Therapeutics, a DSMB member of Immatics, and has received honoraria from Tidal, Catamaran Bio, Sanofi, and Novartis within the last 2 years.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.04.004.
Contributor Information
John F. Tisdale, Email: johntis@nhlbi.nih.gov.
Selami Demirci, Email: selami.demirci@nih.gov.
Supplemental information
References
- 1.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cox D.B.T., Platt R.J., Zhang F. Therapeutic genome editing: prospects and challenges. Nat. Med. 2015;21:121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Demirci S., Leonard A., Haro-Mora J.J., Uchida N., Tisdale J.F. CRISPR/Cas9 for sickle cell disease: applications, future possibilities, and challenges. Adv. Exp. Med. Biol. 2019;1144:37–52. doi: 10.1007/5584_2018_331. [DOI] [PubMed] [Google Scholar]
- 5.Demirci S., Leonard A., Tisdale J.F. Genome editing strategies for fetal hemoglobin induction in beta-hemoglobinopathies. Hum. Mol. Genet. 2020;29:R100–R106. doi: 10.1093/hmg/ddaa088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S., et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martyn G.E., Wienert B., Yang L., Shah M., Norton L.J., Burdach J., Kurita R., Nakamura Y., Pearson R., Funnell A.P., et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat. Genet. 2018;50:498–503. doi: 10.1038/s41588-018-0085-0. [DOI] [PubMed] [Google Scholar]
- 8.Uchida N., Li L., Nassehi T., Drysdale C.M., Yapundich M., Gamer J., Haro-Mora J.J., Demirci S., Leonard A., Bonifacino A.C., et al. Preclinical evaluation for engraftment of CD34+ cells gene-edited at the sickle cell disease locus in xenograft mouse and non-human primate models. Cell Rep. Med. 2021;2 doi: 10.1016/j.xcrm.2021.100247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finn J.D., Smith A.R., Patel M.C., Shaw L., Youniss M.R., van Heteren J., Dirstine T., Ciullo C., Lescarbeau R., Seitzer J., et al. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 2018;22:2227–2235. doi: 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 10.Zhao H., Li Y., He L., Pu W., Yu W., Li Y., Wu Y.-T., Xu C., Wei Y., Ding Q., et al. In vivo AAV-CRISPR/Cas9–mediated gene editing ameliorates atherosclerosis in familial hypercholesterolemia. Circulation. 2020;141:67–79. doi: 10.1161/CIRCULATIONAHA.119.042476. [DOI] [PubMed] [Google Scholar]
- 11.Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L., Bode N.M., et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simhadri V.L., McGill J., McMahon S., Wang J., Jiang H., Sauna Z.E. Prevalence of pre-existing antibodies to CRISPR-associated nuclease Cas9 in the USA population. Mol. Ther. Methods Clin. Dev. 2018;10:105–112. doi: 10.1016/j.omtm.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Ozelo M.C., Hoots K., Blatt P., Konkle B., et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 14.Mingozzi F., High K.A. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122:23–36. doi: 10.1182/blood-2013-01-306647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mingozzi F., Maus M.V., Hui D.J., Sabatino D.E., Murphy S.L., Rasko J.E.J., Ragni M.V., Manno C.S., Sommer J., Jiang H., et al. CD8+ T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- 16.Chew W.L., Tabebordbar M., Cheng J.K., Mali P., Wu E.Y., Ng A.H., Zhu K., Wagers A.J., Church G.M. A multifunctional AAV–CRISPR–Cas9 and its host response. Nat. Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li A., Tanner M.R., Lee C.M., Hurley A.E., De Giorgi M., Jarrett K.E., Davis T.H., Doerfler A.M., Bao G., Beeton C., Lagor W.R. AAV-CRISPR gene editing is negated by pre-existing immunity to Cas9. Mol. Ther. 2020;28:1432–1441. doi: 10.1016/j.ymthe.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrier C., Elliott T., Ledney G. Resident bacteria in a mixed population of rhesus macaque (Macaca mulatta) monkeys: a prevalence study. J. Med. Primatol. 2009;38:397–403. doi: 10.1111/j.1600-0684.2009.00366.x. [DOI] [PubMed] [Google Scholar]
- 19.Davis K.L., Gonzalez O., Kumar S., Dick E.J., Jr. Pathology associated with Streptococcus spp. infection in baboons (Papio spp.) Vet. Pathol. 2020;57:714–722. doi: 10.1177/0300985820941496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fremont J.J., Marini R.P., Fox J.G., Rogers A.B. Acute respiratory distress syndrome in two rhesus macaques (Macaca mulatta) J. Am. Assoc. Lab. Anim. Sci. 2008;47:61–66. PMC2691537. [PMC free article] [PubMed] [Google Scholar]
- 21.Demirci S., Zeng J., Wu Y., Uchida N., Shen A.H., Pellin D., Gamer J., Yapundich M., Drysdale C., Bonanno J., et al. BCL11A enhancer–edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J. Clin. Invest. 2020;130:6677–6687. doi: 10.1172/JCI140189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeng J., Demirci S., Nguyen M.A., Lin L.Y., Maitland S.A., Mintzer E., Wu Y., Pellin D., Tangprasittipap A., Vong C., et al. Combined+ 58 and+ 55 BCL11A enhancer editing Yields exceptional efficiency, specificity and HbF induction in human and NHP preclinical models. Blood. 2021;138:1852. doi: 10.1182/blood-2021-152634. [DOI] [Google Scholar]
- 23.Uchida N., Nassehi T., Drysdale C.M., Gamer J., Yapundich M., Bonifacino A.C., Krouse A.E., Linde N., Hsieh M.M., Donahue R.E., et al. Busulfan combined with immunosuppression allows efficient engraftment of gene-modified cells in a rhesus macaque model. Mol. Ther. 2019;27:1586–1596. doi: 10.1016/j.ymthe.2019.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peslak S.A., Demirci S., Chandra V., Ryu B., Bhardwaj S.K., Jiang J., Rupon J.W., Throm R.E., Uchida N., Leonard A., et al. Forced enhancer-promoter rewiring to alter gene expression in animal models. Mol. Ther. Nucleic Acids. 2023;31:452–465. doi: 10.1016/j.omtn.2023.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hont A.B., Powell A.B., Sohai D.K., Valdez I.K., Stanojevic M., Geiger A.E., Chaudhary K., Dowlati E., Bollard C.M., Cruz C.R.Y. The generation and application of antigen-specific T cell therapies for cancer and viral-associated disease. Mol. Ther. 2022;30:2130–2152. doi: 10.1016/j.ymthe.2022.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leen A.M., Bollard C.M., Mendizabal A.M., Shpall E.J., Szabolcs P., Antin J.H., Kapoor N., Pai S.-Y., Rowley S.D., Kebriaei P., et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. 2013;121:5113–5123. doi: 10.1182/blood-2013-02-486324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vasileiou S., Turney A.M., Kuvalekar M., Mukhi S.S., Watanabe A., Lulla P., Ramos C.A., Naik S., Vera J.F., Tzannou I., Leen A.M. Rapid generation of multivirus-specific T lymphocytes for the prevention and treatment of respiratory viral infections. Haematologica. 2020;105:235–243. doi: 10.3324/haematol.2018.206896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagner D.L., Amini L., Wendering D.J., Burkhardt L.-M., Akyüz L., Reinke P., Volk H.-D., Schmueck-Henneresse M. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2019;25:242–248. doi: 10.1038/s41591-018-0204-6. [DOI] [PubMed] [Google Scholar]
- 29.Ferdosi S.R., Ewaisha R., Moghadam F., Krishna S., Park J.G., Ebrahimkhani M.R., Kiani S., Anderson K.S. Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat. Commun. 2019;10:1842. doi: 10.1038/s41467-019-09693-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hakim C.H., Kumar S.R.P., Pérez-López D.O., Wasala N.B., Zhang D., Yue Y., Teixeira J., Pan X., Zhang K., Million E.D., et al. Cas9-specific immune responses compromise local and systemic AAV CRISPR therapy in multiple dystrophic canine models. Nat. Commun. 2021;12:6769. doi: 10.1038/s41467-021-26830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moreno A.M., Palmer N., Alemán F., Chen G., Pla A., Jiang N., Leong Chew W., Law M., Mali P. Immune-orthogonal orthologues of AAV capsids and of Cas9 circumvent the immune response to the administration of gene therapy. Nat. Biomed. Eng. 2019;3:806–816. doi: 10.1038/s41551-019-0431-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D., Mou H., Li S., Li Y., Hough S., Tran K., Li J., Yin H., Anderson D.G., Sontheimer E.J., et al. Adenovirus-mediated somatic genome editing of Pten by CRISPR/Cas9 in mouse liver in spite of Cas9-specific immune responses. Hum. Gene Ther. 2015;26:432–442. doi: 10.1089/hum.2015.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gillmore J.D., Gane E., Taubel J., Kao J., Fontana M., Maitland M.L., Seitzer J., O’Connell D., Walsh K.R., Wood K., et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 2021;385:493–502. doi: 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- 34.DeWitt M.A., Corn J.E., Carroll D. Genome editing via delivery of Cas9 ribonucleoprotein. Methods. 2017;121-122:9–15. doi: 10.1016/j.ymeth.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim S., Kim D., Cho S.W., Kim J., Kim J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith J.G., Liu X., Kaufhold R.M., Clair J., Caulfield M.J. Development and validation of a gamma interferon ELISPOT assay for quantitation of cellular immune responses to varicella-zoster virus. Clin. Diagn. Lab. Immunol. 2001;8:871–879. doi: 10.1128/CDLI.8.5.871-879.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blum K.S., Pabst R. Lymphocyte numbers and subsets in the human blood: do they mirror the situation in all organs? Immunol. Lett. 2007;108:45–51. doi: 10.1016/j.imlet.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 38.Simhadri V.L., Hopkins L., McGill J.R., Duke B.R., Mukherjee S., Zhang K., Sauna Z.E. Cas9-derived peptides presented by MHC Class II that elicit proliferation of CD4+ T-cells. Nat. Commun. 2021;12:5090. doi: 10.1038/s41467-021-25414-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.-S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 40.Fu B., Liao J., Chen S., Li W., Wang Q., Hu J., Yang F., Hsiao S., Jiang Y., Wang L., et al. CRISPR–Cas9-mediated gene editing of the BCL11A enhancer for pediatric β0/β0 transfusion-dependent β-thalassemia. Nat. Med. 2022;28:1573–1580. doi: 10.1038/s41591-022-01906-z. [DOI] [PubMed] [Google Scholar]
- 41.Bjornson-Hooper Z.B., Fragiadakis G.K., Spitzer M.H., Chen H., Madhireddy D., Hu K., Lundsten K., McIlwain D.R., Nolan G.P. A comprehensive atlas of immunological differences between humans, mice, and non-human primates. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.867015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu S., Zhao Y., Yu W., Yang Y., Gao J., Wang J., Kuang D., Yang M., Yang J., Ma C., et al. Comparison of nonhuman primates identified the suitable model for COVID-19. Signal Transduct. Target. Ther. 2020;5:157. doi: 10.1038/s41392-020-00269-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magalhaes I., Vudattu N.K., Ahmed R.K., Kühlmann-Berenzon S., Ngo Y., Sizemore D.R., Wehlin L., Weichold F., Andersson J., Skeiky Y.A.W., et al. High content cellular immune profiling reveals differences between rhesus monkeys and men. Immunology. 2010;131:128–140. doi: 10.1111/j.1365-2567.2010.03284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soto P.C., Stein L.L., Hurtado-Ziola N., Hedrick S.M., Varki A. Relative over-reactivity of human versus chimpanzee lymphocytes: implications for the human diseases associated with immune activation. J. Immunol. 2010;184:4185–4195. doi: 10.4049/jimmunol.0903420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu Y., Zeng J., Roscoe B.P., Liu P., Yao Q., Lazzarotto C.R., Clement K., Cole M.A., Luk K., Baricordi C., et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019;25:776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Louis C.U., Straathof K., Bollard C.M., Gerken C., Huls M.H., Gresik M.V., Wu M.-F., Weiss H.L., Gee A.P., Brenner M.K., et al. Enhancing the in vivo expansion of adoptively transferred EBV-specific CTL with lymphodepleting CD45 monoclonal antibodies in NPC patients. Blood. 2009;113:2442–2450. doi: 10.1182/blood-2008-05-157222. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon request.