

Abstract
Adaptor protein 2-associated kinase 1 (AAK1) is a member of the Ark1/Prk1 family of serine/threonine kinases and plays a role in modulating receptor endocytosis. AAK1 was identified as a potential therapeutic target for the treatment of neuropathic pain when it was shown that AAK1 knock out (KO) mice had a normal response to the acute pain phase of the mouse formalin model, but a reduced response to the persistent pain phase. Herein we report our early work investigating a series of pyrrolo[2,1-f][1,2,4]triazines as part of our efforts to recapitulate this KO phenotype with a potent, small molecule inhibitor of AAK1. The synthesis, structure–activity relationships (SAR), and in vivo evaluation of these AAK1 inhibitors is described.
Graphical Abstract
Keywords: Adaptor protein 2-associated kinase 1, AAK1, Neuropathic pain, Kinase inhibitor
Introduction
Adaptor associated kinase 1 (AAK1) is a member of the Ark1/Prk1 family of serine/threonine kinases involved in modulating clathrin coated endocytosis by binding clathrin and regulating the function of the adaptor protein 2 (AP2) complex [1, 2]. AAK1 is also able to bind and phosphorylate the endocytic protein Numb [3], and has been shown to be a positive regulator of Notch signaling, in part through phosphorylation of Numb [4]. AAK1 has been implicated in a broad range of diseases including a number of central nervous system (CNS) indications such as: schizophrenia [5], Parkinson’s disease [6], amyotrophic lateral sclerosis [7], and Alzheimer’s disease [8]. Inhibitors of AAK1 also have antiviral activity by blocking receptor-mediated endocytosis, which is a mechanism for virus entry into cells. AAK1 has been postulated as a target for the treatment of hepatitis C [9, 10], dengue and Ebola viruses [11], rabies virus [12], and more recently for SARS-CoV-2 [13–18].
Our interest in AAK1 as a therapeutic target arose from the finding that AAK1 knock-out (KO) mice show reduced pain response in the persistent pain phase of the mouse formalin model, while still having a normal response in the acute pain phase [19]. We recently published our work exploring aryl amide-based AAK1 inhibitors [20], as well as quinazoline- and quinoline-based inhibitors [21]. Additional recent publications describe our structure–activity relationship (SAR) and in vitro/in vivo optimization studies that ultimately led to the discovery of a clinical compound, LX9211 [22, 23]. Herein we describe our early work on the identification of pyrrolo[2,1-f][1,2,4]triazine-based inhibitors of AAK1 that could recapitulate the KO phenotype and helped establish properties needed in an AAK1 inhibitor.
Results and discussion
In the initial stages of our AAKI program our objective was to identify small molecule, selective AAK1 inhibitors with properties suitable for evaluation to determine if the KO pain phenotype could be recapitulated in vivo. At the time of this work, the only reported AAK1 inhibitor in the literature was (N-[5-(4-cyanophenyl)-1H-pyrrolo[2,3-b]pyridin-3-yl]pyridine-3-carboxamide) (1), a dual inhibitor of AAK1 and liver kinase B1 (LKB1) [24]. To find novel leads, we conducted a screen of our in-house compound collection of kinase inhibitors and identified several active compounds, including pyrrolo[2,1-f][1,2,4]triazine 2, Fig. 1.
Fig. 1.
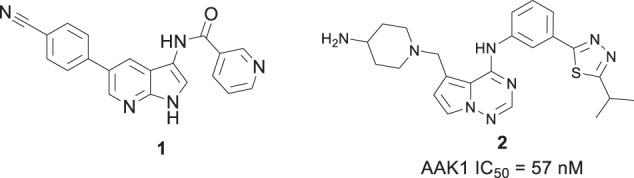
Structure of LKB1/AAK1 dual inhibitor (1) and pyrrolo[2,1-f][1,2,4]triazine 2
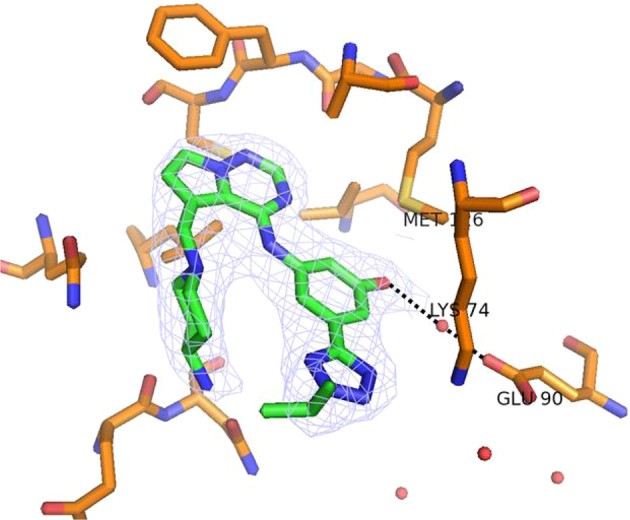
An x-ray crystal structure of 2 bound to AAK1 was obtained (PDB ID 8GMC) (Fig. 2) to aid in the SAR development of this series. The molecule binds in the ATP pocket of the enzyme as shown with the following interactions: (a) The pyrrolotriazine has the typical donor-acceptor-donor (D-A-D) hydrogen-bonding interactions with the Asp127 (carbonyl) and Cys129 (carbonyl and N-H) in the hinge region; (b) the isopropyl group extends into a pocket comprised of the p-loop and engages in hydrophobic interactions with the lipophilic residues lining the pocket; (c) the amino-piperidine moiety binds in the sugar pocket. In addition, the piperidine nitrogen forms an intramolecular hydrogen bonding interaction with the anilinic N-H (d), which holds the molecule in a conformation that allows for efficient binding in the ATP pocket. Additional features of note in Fig. 2 include: engagement of a nitrogen of the thiadiazole in a hydrogen bonding interaction with Lys74 (e) and the presence of a water molecule bound to Glu90 (f), which could potentially be reached by substitution off of the phenyl ring.
Fig. 2.
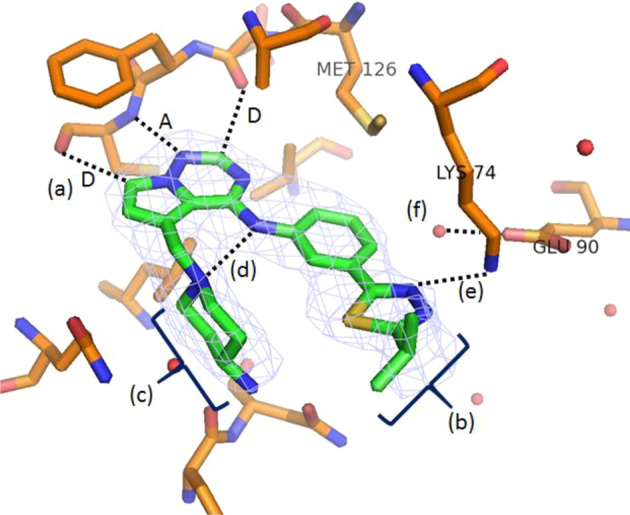
X-ray co-crystal structure of 2 bound to AAK1 (PDB ID 8GMC). a Donor-acceptor-donor (D-A-D) interaction with hinge region residues; b hydrophobic interactions with p-loop; c binding of amino piperidine in sugar pocket; d H-bond between the piperidine nitrogen and the anilinic N-H; e H-bonding between thiadiazole nitrogen and Lys74; and f water molecule bound to Glu90. Water molecules are shown as red spheres
The synthesis of the biaryl side chain of 2 is shown in Scheme 1. Commercially available 3-nitrobenzoyl chloride (3) was treated with isobutyrohydrazide to form intermediate 4. The resultant hydrazide was cyclized in the presence of Lawesson’s reagent to form thiadiazole 5. Reduction of the nitro group with Zn and ammonium chloride provided aniline 6. N-((4-chloropyrrolo[2,1-f][1,2,4]triazin-5-yl)methyl)-N,N-diethylethanaminium bromide (7) [25] was treated with 6 to displace the chloride, and then with 4-(N-Boc-amino)-piperidine to displace the triethylammonium group to provide 8. The Boc group was removed with trifluoroacetic acid to afford 2. The modular nature of the synthesis allowed for rapid SAR development of the series.
Scheme 1.

Reagents and conditions: a isobutyrohydrazide, DIEA, DCM, 23 °C, 30 min, 94% yield; b Lawesson’s Reagent, toluene, 100 °C, 12 h, 96% yield; c zinc powder (100 mesh), NH4Cl, EtOH, 0 °C – reflux, 6 h, 93% yield; d 6, MeCN, 75 °C, 30 min, then 4-(N-Boc amino)-piperidine, DIEA, 75 °C, 30 min, 33% yield; e TFA, ClCH2CH2Cl, room temperature, 3 h, 81% yield
With a promising lead in 2 and a crystal structure for guidance, we set out to optimize the potency of this series. The thiazole-phenyl side chain was varied by modifying the heteroaryl ring and substituents on this ring (R) as shown in Table 1. The isopropyl group of 2 was modified to determine the effect of branching and chain length. Conversion of the isopropyl to an n-propyl had no effect on potency (9), whereas cyclization to the cyclopropyl led to a 2-fold decrease in potency (10). Shortening the chain from n-propyl (9) to methyl (11) led to a 4-fold decrease in potency. Switching the heteroaryl group to a 1,2,4-oxadiazole (12) led to a modest improvement in potency relative to the 1,3,4-thiadiazole analog (2). Increasing the number of carbons on R to t-butyl (13), neopentyl (14), or 2-methyl-2-propanol (15) had negligible effect on potency. Cyclization to either a cyclopentyl (16) or cyclohexyl (17) group was also well tolerated. Replacement of the heteroaryl group with a tetrazole as in 18 gave a 7-fold boost in potency relative to the 1,3,4-thiadiazole, 2. As seen in the crystal structure in Fig. 2, the heteroaryl side chain picks up a hydrogen-bonding interaction with Lys74. The increased electron density of the tetrazole relative to the thiadiazole should strengthen this hydrogen bond, which may account for the improved potency. Finally, the installation of an unsubstituted oxazole (19), led to a significant decrease in potency. In general, heterocycles with a side chain of three or more carbons were well tolerated in this region of the molecule.
Table 1.
SAR studies: effect of varying the heterocyclic side chain
 | ||
|---|---|---|
| Compound | Aryl-R | IC50a nM (±SD) |
| 2 | 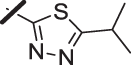 |
57 (±9) |
| 9 | 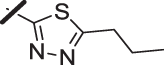 |
54 (±38) |
| 10 | 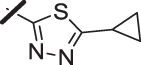 |
100 (±71) |
| 11 | 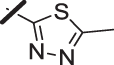 |
210 (±26) |
| 12 | 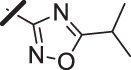 |
21 (±11) |
| 13 | 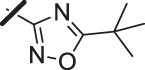 |
10 (±7.1) |
| 14 | 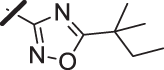 |
22 (±13) |
| 15 | 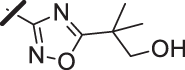 |
15 (±7.2) |
| 16 | 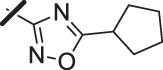 |
16 (±3.6) |
| 17 | 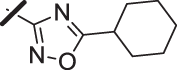 |
11 (±6.2) |
| 18 | 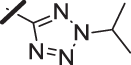 |
7.9 (±5.8) |
| 19 | 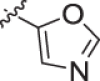 |
770 (±100) |
aAAK1 IC50 values are means of two or more experiments, standard deviation is given in parentheses
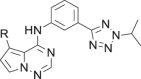
The amino piperidine in 18 was also varied as shown in Table 2. Connecting the piperidine through an amide linkage (20) led to a 200-fold decrease in potency relative to the carbon-linked analog (18). This finding is consistent with the crystal structure of 2, Fig. 2. The nitrogen of the amide linked piperidine is no longer able to form an intramolecular hydrogen bond to the anilinic N-H, and as a result the piperidine of 20 is likely twisted away from the biaryl moiety leading to poor binding to the enzyme. The terminal 4-amino group (21) could be substituted with a methyl group while maintaining binding. Both piperazine (22) and morpholine (23) were tolerated as piperidine replacements with only a 4-5-fold decrease in potency. Switching from the 4-amino piperidine (18) to the shorter primary amine (24) led to a 10-fold decrease in potency. Surprisingly, the potency could be regained by removal of the NH2 to afford the simple methyl analog (25). Further removal of the methyl to give the unsubstituted analog 26 again led to a 10-fold decrease in potency. Within this series, both 18 and 25 show comparable potency, however, the 4-aminopiperidine analog 18 had significantly greater aqueous solubility (>7.96 mg/mL solubility at pH 6.5 vs. <0.001 mg/mL for 25).
Table 2.
SAR studies: effect of varying the amino side chain
 | ||
|---|---|---|
| Compound | R | IC50a nM (±SD) |
| 18 | 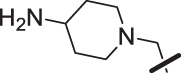 |
21 (±3.1) |
| 20 | 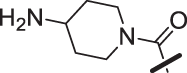 |
4200 (±280) |
| 21 | 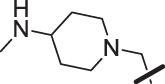 |
12 (±9.8) |
| 22 | 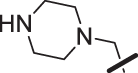 |
90 (±49) |
| 23 | 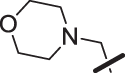 |
99 (±31) |
| 24 | 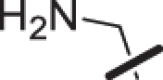 |
220 (±93) |
| 25 | 23 (±1.4) | |
| 26 | 220 (±110) | |
aAAK1 IC50 values are means of two or more experiments, standard deviation is given in parentheses
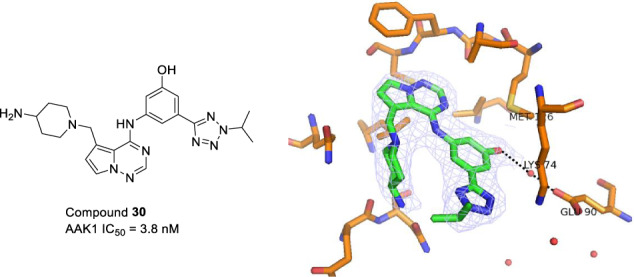
In the crystal structure of 2 bound to AAK1 a water molecule bound to Glu90 was observed (Fig. 2). We hypothesized that an ortho- or para-substituent on the phenyl group could either displace the water and engage in an additional hydrogen bonding interaction with Glu90, or engage in a water-bridged hydrogen bond to the residue leading to a boost in potency. This was explored with analogs shown in Table 3. Relative to the unsubstituted phenyl analog 18, the para-fluorophenyl analog 27 showed a 5-fold decrease in potency, the meta-fluoro analog 28 was equipotent, while the ortho-fluoro analog 29 showed a 100-fold decrease in potency. Switching to an OH in the preferred meta-position 30 led to a 2-fold improvement in potency relative to 18. Incorporation of a larger group at the meta-position, such as CHF2 (31) or OiPr (32) led to a 25-fold and 100-fold decrease in potency, respectively. A crystal structure of 30 bound to AAK1 (PDB ID 8GMD), Fig. 3, shows that the phenolic OH picks up a water-bridged hydrogen bond to Glu90 in the back pocket of the kinase, which helps explain the superior potency of 30 relative to 31 and 32.
Table 3.
SAR studies: effect of varying phenyl substitution
 | ||||
|---|---|---|---|---|
| Compound | o | m | p | IC50,a nM (±SD) |
| 18 | H | H | H | 7.9 (±5.8) |
| 27 | H | H | F | 21 (±3.1) |
| 28 | H | F | H | 9.3 (±3.1) |
| 29 | F | H | H | 830 (±70) |
| 30 | H | OH | H | 3.8 (±1.1) |
| 31 | H | CHF2 | H | 97 (±13) |
| 32 | H | OiPr | H | 400 (±130) |
aAAK1 IC50 values are means of two or more experiments, standard deviation is given in parentheses
Fig. 3.
X-ray co-crystal structure of 30 PDB ID 8GMD). The phenol OH picks up a water-bridging hydrogen bond to Glu90. Water molecules are shown as red spheres
Compound 30 was selected for in vivo proof of concept studies based on its potency. Compound 19 was included to allow for comparison with a structurally similar, but much less potent inhibitor. AAK1 KO mice were previously shown to have a reduced persistent pain response in the formalin model of acute and persistent pain [19] as measured by a reduction in flinching. To determine if we could recapitulate this phenotype with a small molecule AAK1 inhibitor, we administered 30 and the less potent analog 19 in the formalin pain model [26] (Fig. 4). The compounds were dosed either subcutaneously (s.c.) or intraperitoneally (i.p.) to maximize the exposure. Compound 30 was dosed at 10, 30, and 60 mg/kg, s.c. Gabapentin was included as a positive control, dosed at 200 mg/kg, i.p. The number of flinches was measured over the course of 65 min and plasma and brain exposures were obtained from these animals at the end of the study (~90 min post-dose). Efficacy was seen with both gabapentin (200 mg/kg) and compound 30 at the 60 mg/kg dose. The plasma exposure for compound 19 was 11.9 μM (estimated free plasma exposure based on plasma protein binding of 99.5% = 60 nM) (Table 4). AAK1 is expressed both in the CNS and in the periphery [1] and it was unknown at the time of this early work if CNS penetration was necessary to see efficacy in pain models. While compound 30 showed efficacy in the formalin model of persistent pain, despite its negligible CNS penetration (B/P ~ 0.02), it should be noted that it was later shown that penetration into the CNS was necessary to see robust efficacy with AAK1 inhibitors in models of neuropathic pain (for example, spinal nerve ligation (SNL) model) [19]. Compound 19 did not show efficacy in the formalin model when dosed up to 30 mg/kg, i.p. (Fig. 4b) even though a similar level of plasma exposure was achieved relative to compound 30 at the 60 mg/kg dose. The total plasma exposure for 19 at a dose of 30 mg/kg was 8.4 μM, 90-min post dose (118 nM free plasma exposure based on 98.6% plasma protein binding) (Table 4). Compound 19 also showed improved, albeit modest, total brain exposure (1.8 μM) relative to compound 30. Based on the structural similarity as well as the similar kinase selectivity (see Supplementary Information) of the two analogs, the results seen in the formalin model can likely be attributed to the difference in AAK1 potency. Compound 30 was also examined in a mouse rotarod assay [27, 28] to rule out confounding side effects that could masquerade as efficacy. Compound 30 showed no side effects up to the top doses of 60 mg/kg (Supplementary Information). This data is consistent with the anti-nociceptive effect of 30 being driven by AAK1 inhibition. It is noteworthy that at a dose of 200 mg/kg, gabapentin shows behavioral side effects (i.e., sedation) in this model.
Fig. 4.

a Dose response of 30 in the mouse formalin model; b Dose response of 19 in the mouse formalin model
Table 4.
Plasma and brain exposures of Compounds 19 and 30 from mouse formalin model taken 90 min post-dose
| Compound 19 and 30 - Mouse Formalin Exposure Data (90 min post-dose) | |||||
|---|---|---|---|---|---|
| Compound | Dose (mg/kg), route | Total Plasma (μM) | Free Plasma (nM) | Total Brain (μM) | B/P |
| 19 | 30, i.p. | 8.4 ± 1.5 | 118 | 1.84 ± 1.84 | 0.22 |
| 30 | 10, s.c. | 2.5 ± 2.1 | 13 | 0.04 ± 0.03 | 0.03 |
| 30, s.c. | 5.4 ± 1.1 | 27 | 0.06 ± 0.02 | 0.01 | |
| 60, s.c. | 11.9 ± 1.2 | 60 | 0.15 ± 0.03 | 0.01 | |
The broad kinase selectivity of compounds 19 and 30 was assessed in a panel consisting of 351 kinases (Ambit/DiscoverX/Eurofins) [29]. Compounds 2 and 18, which also showed efficacy in the mouse formalin model (data not shown) were also tested in the kinase panel. The four analogs all displayed modest selectivity, inhibiting 6–11% of the kinases tested (complete panel shown in Supplemental Information). Of these hits, seven kinases other than AAK1 (BIKE, GPRK4, MSSK1, PIP5K2B, PKCD, RIOK1, and RIOK3) were inhibited by the three analogs that showed efficacy in the pain model (compounds 2, 18, and 30), but not for the inactive compound 19. BIKE, also known as BMP2K, is the most closely related kinase to AAK1 in this family with 74% sequence identity to AAK1 within their respective kinase domains [2]. While there is no reported connection of these seven off-target kinase hits with pain, contribution to efficacy from inhibition these other kinases cannot definitively be ruled out. It is noteworthy, however, that potent AAK1 inhibitors from a structurally diverse set of chemotypes, with differentiated kinase selectivity, have shown activity in the mouse formalin model [19–33] suggesting that the efficacy of analogs 2, 18, and 30 is largely due to inhibiting AAK1.
Conclusions
A series of pyrrolo[2,1-f][1,2,4]triazine analogs was prepared to assess their potential as inhibitors of AAK1. Starting from an analog with an IC50 value of ~60 nM, identified through screening of our in house kinase compound collection, we were able to improve potency >10x to find analogs with IC50 values in the single digit nM range. Enhanced potency could be achieved by picking up an additional water-bridging H-bonding interaction with the Glu90 residue in the back pocket of the kinase. Compound 30, a potent AAK1 inhibitor, showed efficacy in the formalin model of acute and persistent pain, while the structurally similar compound 19 with poor AAK1 potency did not show efficacy in this model. These data, along with the AAK KO mouse data, helped demonstrate that small molecule inhibitors of AAK1 could be useful for the treatment of pain. This concept is currently being tested in the clinic with a more advanced brain-penetrant compound [23].
Compliance with ethical standards
Conflict of interest
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Conner SD, Schmid SL. Identification of an adaptor-associated kinase, AAK1, as a regulator of clathrin-mediated endocytosis. J Cell Biol. 2002;156:921–9. doi: 10.1083/jcb.200108123.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sorrell FJ, Szklarz M, Abdul Azeez KR, Elkins JM, Knapp S. Family-wide structural analysis of human Numb-associated protein kinases. Structure. 2016;24:401–11. doi: 10.1016/j.str.2015.12.015.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sorensen EB, Conner SD. AAK1 regulates Numb function at an early step in clathrin-mediated endocytosis. Traffic. 2008;9:1791–800. doi: 10.1111/j.1600-0854.2008.00790.x.. [DOI] [PubMed] [Google Scholar]
- 4.Gupta-Rossi N, Ortica S, Meas-Yedid V, Heuss S, Moretti J, Olivo-Marin JC, et al. The adaptor-associated kinase 1, AAK1, is a positive regulator of the Notch pathway. J Biol Chem. 2011;286:18720–30. doi: 10.1074/jbc.M110.190769.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abdel-Magid AF. Inhibitors of Adaptor-Associated Kinase 1 (AAK1) May Treat Neuropathic Pain, Schizophrenia, Parkinson’s Disease, and Other Disorders. ACS Med Chem Lett. 2017;8:595–7. doi: 10.1021/acsmedchemlett.7b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Latourelle JC, Pankratz N, Dumitriu A, Dumitriu A, Wilk JB, Goldwurm S, et al. Genomewide association study for onset age in Parkinson disease. BMC Med Genet. 2009;10:98. doi: 10.1186/1471-2350-10-98.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi B, Conner SD, Liu J. Dysfunction of endocytic kinase AAK1 in ALS. Int J Mol Sci. 2014;15:22918–32. doi: 10.3390/ijms151222918.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu X, Ke M, Yu W, Wang X, Xiao Q, Gu M, et al. Periodic variation of AAK1 in an Aβ1–42-induced mouse model of Alzheimer’s disease. J Mol Neurosci. 2018;65:179–89. doi: 10.1007/s12031-018-1085-3.. [DOI] [PubMed] [Google Scholar]
- 9.Neveu G, Barouch-Bentov R, Ziv-Av A, Gerber D, Jacob Y, Einav S. Identification and targeting of an interaction between a tyrosine motif within hepatitis C virus core protein and AP2M1 essential for viral assembly. PLoS Pathog. 2012;8:e1002845. doi: 10.1371/journal.ppat.1002845.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neveu G, Ziv-Av A, Barouch-Bentov R, Berkerman E, Mulholland J, Einav S. AP-2-associated protein kinase 1 and cyclin G-associated kinase regulate hepatitis C virus entry and are potential drug targets. J Virol. 2015;89:4387–404. doi: 10.1128/JVI.02705-14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verdonck S, Pu S-Y, Sorrell FJ, Elkins JM, Froeyen M, Gao L-J, et al. Synthesis and structure–activity relationships of 3,5 disubstitutedpyrrolo[2,3-b]pyridines as inhibitors of adaptor-associated kinase 1 with antiviral activity. J Med Chem. 2019;62:5810–31. doi: 10.1021/acs.jmedchem.9b00136.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang C, Wang J, Shuai L, Ma X, Zhang H, Liu R, et al. The serine/threonine kinase AP2-associated kinase plays an important role in rabies virus entry. Viruses. 2020;12:45–60. doi: 10.3390/v12010045.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zuo W. Single-cell RNA expression profiling of ACE2, the putative receptor of SARS-CoV-2. Am J Respir Crit Care Med. 2020;202:756–559. doi: 10.1164/rccm.202001-0179LE.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395:565–74. doi: 10.1016/S0140-6736(20)30251-8.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pottoo FH, Abu-Izneid T, Ibrahim AM, Javed MN, AlHajri N, Hamrouni AM. Immune system response during viral infections: immunomodulators, cytokine storm (CS) and immunotherapeutics in COVID-19. Saudi Pharm J. 2021;29:173–87. doi: 10.1016/j.jsps.2020.12.018.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X, Zhang Y, Qiao W, Zhang J, Qi Z. Baricitinib, a drug with potential effect to prevent SARS-COV-2 from entering target cells and control cytokine storm induced by COVID-19. Int Immunopharmacol. 2020;86:106749. doi: 10.1016/j.intimp.2020.106749.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184:5298–07. doi: 10.4049/jimmunol.0902819.. [DOI] [PubMed] [Google Scholar]
- 18.Stebbing J, Krishnan V, de Bono S, Ottaviani S, Casalini G, Richardson PJ, et al. Mechanism of baricitinib supports artificial intelligence-predicted testing in COVID-19 patients. EMBO Mol Med. 2020;12:e12697. doi: 10.15252/emmm.202012697.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kostich W, Hamman BD, Li Y-W, Naidu S, Dandapani K, Feng J, et al. Inhibition of AAK1 kinase as a novel therapeutic approach to treat neuropathic pain. J Pharmacol Exp Ther. 2016;358:371–86. doi: 10.1124/jpet.116.235333.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hartz RA, Ahuja VT, Nara SJ, Kumar CMV, Brown JM, Bristow LJ, et al. Discovery, Structure-activity relationships, and in vivo evaluation of novel aryl amides as brain penetrant adaptor protein 2-associated kinase 1 (AAK1) inhibitors for the treatment of neuropathic pain. J Med Chem. 2021;64:11090–28. doi: 10.1021/acs.jmedchem.1c00472. [DOI] [PubMed] [Google Scholar]
- 21.Hartz RA, Ahuja VT, Nara SJ, Kumar CMV, Manepalli RKVLP, Sarvasiddhi SK, et al. Bicyclic heterocyclic replacement of an aryl amide leading to potent and kinase-selective adaptor protein 2-associated kinase 1 inhibitors. J Med Chem. 2022;65:4121–55. doi: 10.1021/acs.jmedchem.1c01966. [DOI] [PubMed] [Google Scholar]
- 22.Luo G, Chen L, Kostich WA, Hamman B, Allen J, Easton A, et al. Discovery and optimization of biaryl alkyl ethers as a novel class of highly selective, CNS-penetrable, and orally active adaptor protein-2-associated kinase 1 (AAK1) inhibitors for the potential treatment of neuropathic pain. J Med Chem. 2022;65:4534–64. doi: 10.1021/acs.jmedchem.1c02132. [DOI] [PubMed] [Google Scholar]
- 23.Luo G, Chen L, Kostich WA, Hamman B, Allen J, Easton A, et al. Discovery of (S)-1-((2',6-Bis(difluoromethyl)-[2,4'-bipyridin]-5-yl)oxy)-2,4-dimethylpentan-2-amine (BMS-986176/LX-9211): A highly selective, CNS penetrable, and orally active adaptor protein-2 associated kinase 1 inhibitor in clinical trials for the treatment of neuropathic pain. J Med Chem. 2022;65:4457–80. doi: 10.1021/acs.jmedchem.1c02131. [DOI] [PubMed] [Google Scholar]
- 24.Bamborough P, Drewry D, Harper G, Smith GK, Schneider K. Assessment of chemical coverage of kinome space and its implications for kinase drug discovery. J Med Chem. 2008;51:7898–14. doi: 10.1021/jm8011036.. [DOI] [PubMed] [Google Scholar]
- 25.Fink BE, Norris D, Mastalerz H, Chen P, Goyal B, Zhao Y, et al. Novel pyrrolo[2,1-f][1,2,4]triazin-4-amines: Dual inhibitors of EGFR and HER2 protein tyrosine kinases. Bioorg Med Chem Lett. 2011;21:781–85. doi: 10.1016/j.bmcl.2010.11.100.. [DOI] [PubMed] [Google Scholar]
- 26.Dubuisson D, Dennis SG. The formalin test: a quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats. Pain. 1977;4:161–74. doi: 10.1016/0304-3959(77)90130-0.. [DOI] [PubMed] [Google Scholar]
- 27.Dunham NW, Miya TS. A note on a simple apparatus for detecting neurological deficit in rats and mice. J Am Pharm Assoc Am Pharm Assoc. 1957;46:208–9. doi: 10.1002/jps.3030460322.. [DOI] [PubMed] [Google Scholar]
- 28.Watzman N, Barry H, III, Buckley JP, Kinnard WJ., Jr Semiautomatic system for timing rotarod performance. J Pharm Sci. 1964;53:1429–30. doi: 10.1002/jps.2600531142.. [DOI] [PubMed] [Google Scholar]
- 29.Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–32. doi: 10.1038/nbt1358.. [DOI] [PubMed] [Google Scholar]