

Abstract
Voltage-gated sodium (Nav) channels are targeted by a number of widely used and investigational drugs for the treatment of epilepsy, arrhythmia, pain, and other disorders. Despite recent advances in structural elucidation of Nav channels, the binding mode of most Nav-targeting drugs remains unknown. Here we report high-resolution cryo-EM structures of human Nav1.7 treated with drugs and lead compounds with representative chemical backbones at resolutions of 2.6-3.2 Å. A binding site beneath the intracellular gate (site BIG) accommodates carbamazepine, bupivacaine, and lacosamide. Unexpectedly, a second molecule of lacosamide plugs into the selectivity filter from the central cavity. Fenestrations are popular sites for various state-dependent drugs. We show that vinpocetine, a synthetic derivative of a vinca alkaloid, and hardwickiic acid, a natural product with antinociceptive effect, bind to the III-IV fenestration, while vixotrigine, an analgesic candidate, penetrates the IV-I fenestration of the pore domain. Our results permit building a 3D structural map for known drug-binding sites on Nav channels summarized from the present and previous structures.
Subject terms: Cryoelectron microscopy, Pharmaceutics, Permeation and transport, Sodium channels
Voltage-gated sodium (Nav) channels are targeted by various clinically applied and investigational drugs. Here cryo-EM structures of Nav1.7 bound to 7 compounds with diverse chemical skeletons reveal the structural basis of action of these drugs and provide a 3D structural map for drug binding sites on Nav channels.
Introduction
Voltage-gated sodium (Nav) channels govern membrane excitability in neurons and muscles1,2. Nine subtypes of Nav channels, Nav1.1-Nav1.9, are responsible for firing electrical signals with tissue specificity3. Consistent with their fundamental physiological significance, aberrant activities of Nav channels are linked to a wide range of pathogenic conditions. Nav channels are directly targeted by a number of FDA-approved anti-epilepsy drugs (AEDs), painkillers, and antiarrhythmic agents4–8.
High-resolution (cryo-EM) structures of eukaryotic Nav channels reported in recent years offered an advanced understanding of the mode of action (MOA) of some of the drugs at atomic level. For instance, the antiarrhythmic drugs, flecainide, quinidine, and propafenone are all accommodated in the central cavity of the pore domain (PD) of Nav1.5, but with non-overlapping binding poses9–11. Bulleyaconitine A (BLA), an active ingredient isolated from Aconitum plant for pain management and for the treatment of rheumatoid arthritis in China, cuts off the ion permeation by sitting right below the selectivity filter (SF) in the structure of Nav1.312. Nonetheless, the accurate binding mode of most of the Nav-targeting drugs is yet to be unveiled. Such information will not only reveal the chemical basis for the MOA of the clinically applied drugs, but also lay an important foundation for future drug development13.
We sought to identify the druggable sites on Nav channels by carrying out a systematic structural investigation using human Nav1.7-β1-β2 as the scaffold14,15. Nav1.7, encoded by SCN9A and primarily localized to the dorsal root ganglion neurons, has been explored as a prominent target for pain management16. Loss of function of Nav1.7 leads to indifference to pain17–19, whereas mutations that enhance channel activities are found in patients with primary erythromelalgia, paroxysmal extreme pain disorder and small fiber neuropathy20–23. Despite extensive efforts from a number of preeminent pharmaceutical companies, most of the Nav1.7-targeting drug candidates failed to meet the endpoint(s) of phase II trials8,24–26. We reckoned that structural details of Nav1.7 in complex with clinically applied drugs and lead compounds may afford a molecular insight that will benefit rational drug design or/and optimization.
In the structures of eukaryotic Nav channels, the core α subunit, which generally consists of 1500-2200 residues, is well resolved for the transmembrane (TM) and extracellular regions12,15,27–34. The four 6-TM (S1-S6) containing repeats exhibit a pseudo symmetry33,35,36. The S1-S4 segments in each repeat constitute the flanking voltage-sensing domain (VSD), and the S5-S6 segments enclose the PD that is responsible for selective and gated ion permeation28,33,34,37. Above the PD, several heavily glycosylated extracellular loops (ECL) are stabilized by multiple disulfide bonds32,38.
To explore potential druggable sites, we selected representative drugs and lead compounds with diverse chemical backbones (Supplementary Table 5). Our efforts were unfortunately limited by the availability of the chemicals. Here we present cryo-EM structural analysis of Nav1.7 treated with the following drugs: a local anesthetic drug bupivacaine (Marcaine) and a chemically related AED lacosamide (Vimpat), an anticonvulsant drug carbamazepine (Tegretol) that is also used for the treatment of trigeminal neuralgia and epilepsy39–41, a synthetic vincamine derivative vinpocetine (Cavinton) that can target Nav channels for the treatment of cerebrovascular disorders, a natural product hardwickiic acid isolated from plant Croton californicus with antinociceptive effect, and vixotrigine (also known as raxatrigine), which is a state- and use-dependent Nav1.7 blocker under clinical investigation for ameliorating trigeminal pain and neuropathic pain42–44. Our present structural studies, along with previous reports, reveal that the PD is a versatile receptor for ligands with distinct chemical formulas and structures.
Results
Cryo-EM analysis of Nav1.7 with different drugs
We started with more drugs than the abovementioned ones, including a local anesthetic lidocaine, an analgesic under development funapide (XEN402)45, a pain killer candidate DSP-22308, and a selective Nav1.7 inhibitor PF-05089771 that targets VSDIV46,47. To validate the function of some purchased antagonists, we performed preliminary whole-cell patch-clamp electrophysiological recordings in HEK293T cells (Supplementary Figs. 1–2 and Supplementary Tables 1–2). Based on these characterizations, the antagonists were incubated with purified Nav1.7-β1-β2 complex for 1 h at a final concentration of at least 10-fold higher than their measured IC50 values before cryo-sample preparation.
Following our reported cryo-EM data acquisition and processing protocols14,33, we obtained ten 3D EM reconstructions for the Nav1.7-β1-β2 complex treated with different small molecules at overall resolutions of 2.6−3.2 Å (Supplementary Figs. 3–4, Supplementary Figs. 8–10 and Supplementary Table 3). Cross comparison of 3D EM maps immediately reveals unambiguously resolved densities for bupivacaine (BPV), lacosamide (LCM), carbamazepine (CBZ), vinpocetine (VPC), hardwickiic acid (HDA), vixotrigine (VXT), and PF-05089771. However, no density is found for lidocaine, and the local resolution is insufficient to unambiguously assign funapide or DSP-2230, even when these compounds were added throughout the entire purification procedure during repeated attempt to resolve the ligands (Supplementary Table 5).
The lack of density for lidocaine and limited resolutions for funapide and DSP-2230 may result from various reasons. Detergents, which were applied at high concentrations, may interfere with the drug binding. In fact, we sought to solve the structure of human Nav1.5 bound to lidocaine with multiple attempts, but never observed any ligand density. It is thus not surprising that no lidocaine is found in the EM map for Nav1.7 either. Structures of both Nav1.5 and Nav1.7 display similar inactivated state. It is also possible that some ligands may prefer different conformations that are yet to be resolved. In the following, we will discuss the well resolved ligands only.
Recognition of PF-05089771 by Nav1.7-VSDIV was previously examined on a NavAb chimera where the extracellular half of the VSD is replaced by the counterpart in Nav1.7-VSDIV47. Our incentive to solve the structure of PF-05089771 bound to wild-type (WT) human Nav1.7 was to verify if the chimeric VSD can faithfully recapitulate all binding details. The binding mode of PF-05089771 in the intact Nav1.7-VSDIV is remarkably similar to the chimeric one (Supplementary Fig. 5). As coordination details and the molecular basis for the subtype-specificity have been thoroughly discussed by Ahuja and co-workers, we will not elaborate on this one. In the following, we will focus on drug binding to the PD.
A common site for BPV, LCM, and CBZ
In the 3D EM map of all WT human Nav channels, the intracellular gate is penetrated by a linear density that likely belongs to the detergent molecule glyco-diosgenin (GDN) or digitonin. In the maps of Nav1.7 treated with BPV, LCM, and CBZ, the linear density is replaced by shorter ones with distinct features that respectively match the three drugs (Fig. 1a). Instead of wedging through the gate, these densities block it from the intracellular side. Therefore, BPV, LCM, and CBZ occupy the same site beneath the intracellular gate, which we name as site BIG. Unexpectedly, LCM has a second binding pose that directly plugs into the selectivity filter from the central cavity (Fig. 1b, c). We will discuss the second site in a later session. For illustration simplicity, we will name LCM as LCM-1 and LCM-2 in site BIG and the central cavity, respectively.
Fig. 1. BPV, LCM, and CBZ bind to the same site beneath the intracellular gate (site BIG).

a Distinct densities for the three drugs at site BIG. From left to right, the chemical structures (ball and sticks) and corresponding densities (semi-transparent cloud) are shown for GDN (Nav1.7-apo, PDB code: 7W9K), bupivacaine (BPV, brown), lacosamide (LCM-1, grey), and carbamazepine (CBZ, purple). The densities for the small molecules are presented at similar levels in Chimera, 5 σ for GDN, 4 σ for BPV, 4 σ for LCM-1, and 4.5 σ for CBZ. For visual clarity, only part of the S6I and S6III segments are shown. b The three drugs overlay at Site BIG. A bottom view (left) and a side view (right) of the superimposed pore domain (PD, domain colored) with bound drugs (BPV, LCM, and CBZ) are shown. The same color scheme for the four repeats is applied throughout the manuscript. c Drugs at site BIG directly block the intracellular gate. Shown here is a cross-section view of ligand-bound Nav1.7 PD in an electrostatic surface representation. Two distinct binding poses are observed for LCM (grey spheres), one at site BIG (LCM-1) and the other below the SF (LCM-2). d Further contracted intracellular gate in the presence of the site BIG-binding drugs. Left: Structural comparison of Nav1.7-BPV and Nav1.7-apo (silver). Middle: The pore radii of Nav1.7 bound to different ligands are calculated in HOLE and tabulated. Right: The α→ π transition of S6IV and rearrangement of the gating residues, such as Leu398, Leu964, Ile1457 and Ile1756 (shown as ball and sticks), in the presence of BPV lead to gate contraction. LCM and CBZ-bound structures display nearly identical conformations to that of Nav1.7-BPV.
Analysis of the ion permeation path using HOLE48 shows that the intracellular gate is substantially narrowed (Fig. 1d). The radius of the constriction site falls to ~1 Å, which is more than 1 Å shorter than that in the apo channel (PDB code: 7W9K)14. Gate contraction results from an α→π transition in the middle of the S6IV segment, in addition to minor inward movements of S6II and S6III (Fig. 1d). Similar conformational changes have been observed for Nav1.7 upon binding to the peptide toxins Protoxin-II (ProTx-II) or Huwentoxin-IV (HWTX-IV)14. ProTx-II binds to the extracellular side of VSDII and VSDIV, which are respectively known as Site 4 and Site 3 for neurotoxin binding to Nav channels; HWTX-IV recognizes Site 315, 49,50. It is interesting that peptide toxins and small molecule drugs that bind to unrelated sites lead to similar gate rearrangement.
In apo-Nav1.7, the intracellular gate is of an oval contour constituted by Leu398, Leu960, Phe963, Ile1453, Val1752, and Tyr1755 from the four S6 segments. In the presence of the drugs BPV, LCM, or CBZ, the three S6 segments other than S6I all slightly move toward the center in addition to the α→π transition of S6IV. Consequently, four gating residues, Leu398, Leu964, Ile1457, and Ile1756, move closer to tighten the gate (Fig. 1d). It is noted that Leu964, Ile1457, and Ile1756 are all engaged in the coordination of the three drugs (Fig. 2). Such conformational shifts are thus coupled to site BIG formation.
Fig. 2. Drug coordination at the BIG site.

Binding details of (a) BPV, (b) LCM-1, and (c) CBZ at site BIG are presented in identical side (top) and intracellular views (bottom). The drugs are shown as ball and sticks, and surrounding residues are shown as sticks. The potential hydrogen bonds are indicated with red dashed lines.
Site BIG
Site BIG is constituted by the cytosolic residues on the S6 tetrahelical bundle that are adjacent to the gating residues. Although the three drugs completely overlap in site BIG, the coordinating details vary. BPV is primarily surrounded by hydrophobic residues, including Leu964, Leu967, Leu968, and Phe971 from S6II, Ile1457 from S6III, and Ile1756 and Leu1760 from S6IV (Fig. 2a). The 3,5-dimethylphenyl and the piperidine rings of BPV, linked by an amide group, align on a similar plane against the BIG site, leaving the butyl tail pointing to the cytoplasmic side.
The purchased BPV is a racemate of dextro-(R)- and levo-(S)-bupivacaine. To investigate if BPV binding has racemic specificity, we performed in-silico docking simulations. The aromatic ring and the piperidine basic nitrogen of these two isomers largely overlap in site BIG. The primary difference in the binding poses of the isomers is the opposite orientations of the butyl tail (Supplementary Fig. 6). Therefore, both racemic isomers of BPV can be recognized at site BIG.
LCM-1 and CBZ occupy the same cavity of site BIG as BPV, but form additional hydrogen bonds (H-bonds) (Fig. 2b, c). The amide nitrogen from the central amide group of LCM-1 is H-bonded with the carboxyl group of Glu406 on S6I (Fig. 2b), and the carbonyl oxygen of CBZ receives an H-bond from Asn1461 on S6III (Fig. 2c). The three clinical drugs share a conserved binding mode, wherein the aromatic rings project into the center of the pore, leaving the flexible tail to the intracellular side. Drugs with the same chemical skeleton may share a similar binding pose.
LCM-2 directly blocks the SF
The central cavity of voltage-gated ion channels (VGICs) is a versatile platform for various chemicals. Eukaryotic Nav and Cav channels comprise four non-identical repeats, thereby offering more docking sites than homo-tetrameric VGICs.
Beneath the ceiling of the central cavity, the Y-shaped LCM-2 is positioned off the central axis, closer to repeats I and IV (Fig. 3a, b). The benzyl ring is embedded in a pocket enclosed by residues Ser1697, Ile1744, Phe1748, Val1751, and Val1752. The two amides are H-bonded to the backbone carbonyl group of Thr1696 on the P1 loop of repeat IV, forming a stable anchor for LCM-2. The carbonyl oxygen from the acetylamino moiety interacts with Lys1406, which is one constituent of the DEKA motif on repeat III, and the amide nitrogen engages in an H-bond with Gln360, which is stabilized by the side chain hydroxyl group of Ser390 (Fig. 3c). The extensive H-bond network may account for the stronger density of LCM-2 than that of LCM-1 (Figs. 1a, 3a).
Fig. 3. Lacosamide binds to two distinct sites.
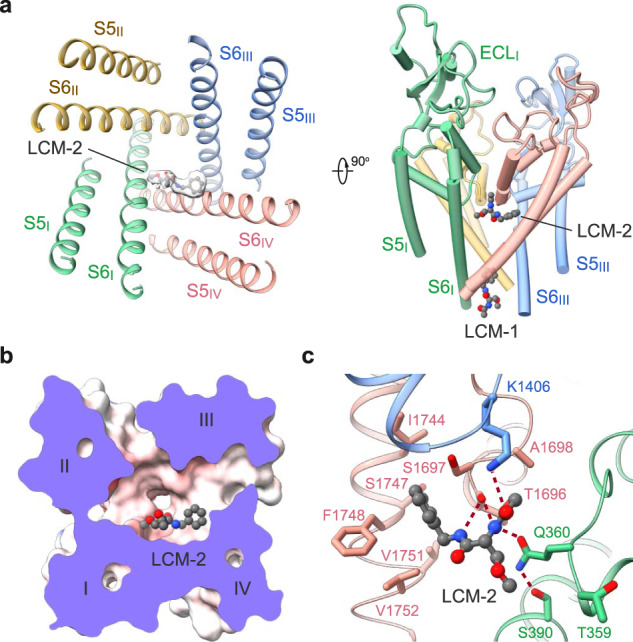
a Accommodation of lacosamide at the second binding site in the central cavity. The contour level for LCM-2 in the left panel is 5 σ. b A cut-open electrostatic surface of Nav1.7-LCM-2 viewed from the extracellular side. c Coordination of LCM-2 in the central cavity. Surrounding residues are shown as sticks. The potential hydrogen bonds are indicated with red dashed lines.
Vinpocetine and hardwickiic acid bind to the III–IV fenestration
The III–IV fenestration in the L-type Cav channels is a well-known site for dihydropyridine compounds13, 51,52. The corresponding site in Nav channels has been suggested to be targeted by local anesthetics53–57. Although no density for lidocaine is found around this site, densities for VPC and HDA were resolved40,58. A battery of hydrophobic residues on S6III and S6IV are engaged in accommodating VPC or HDA, including Thr1404 on P1III, Trp1332 on S5III, Thr1448, Leu1449, and Phe1452 on S6III, and Ser1697, Ile1744 and Phe1748 in repeat IV (Fig. 4a–c). Notably, the binding pose for VPC, which extends deeper into the central cavity, only partially overlaps with that of HDA in the III–IV fenestration (Fig. 4a).
Fig. 4. Versatile fenestration binding sites on the PD.

a VPC and HDA are accommodated at the III-IV fenestration, and the elongated linear VXT projects into the central cavity from the IV-I fenestration. Two perpendicular views of the PD are shown. b Different binding poses at the fenestration sites. Shown here are cut-open electrostatic surfaces viewed from the extracellular side. The drugs are shown as spheres. c Details of VPC, HDA, and VXT coordination. d Different states of the IV-I fenestration in the presence of distinct pore blockers. The fenestration, which exists in the apo or VXT, HDA and VPC-bound channel, is gone in the presence of BPV, CBZ, or LCM accompanying the α→π transition of S6IV.
Vixotrigine penetrates the IV–I fenestration
A stretch of elongated density is observed traversing the IV-I fenestration in VXT-treated Nav1.7. Linear density with similar pose is often found to belong to a lipid tail in apo-Nav channels. Fortunately, the bulges that are characteristic of the three ring moieties facilitated model building of VXT (Fig. 4a, b). VXT is held by a number of hydrophobic residues on S6I and S6IV, including Phe1692, Thr1695 and Thr1696 on P1IV, Ile386, Phe387 and Phe391 on S6I, and Val1751 and Tyr1755 on S6IV. The amido group from pyrrolidine moiety and carbonyl oxygen are H-bonded to the phenolic hydroxyl group of Tyr1755 on S6IV, forming a stable anchor for VXT (Fig. 4c).
Unlike VPC, the majority of which is within the cavity (Fig. 4b), VXT tunnels through the IV-I fenestration, with its acylamino moiety below the SF and its benzyl ring contacting the lipid bilayer (Fig. 4a, b). It is noted that the IV-I fenestration is sensitive to the α→π transition of S6IV (Fig. 4d). In the π form, the fenestration is gone. Such binding pose thus immediately provides a molecular interpretation for the dependence on channel states for VXT binding.
Discussion
A structural map for drug binding sites on Nav channels
Nav channels are well-known targets for natural toxins and clinically applied drugs. Seven sites (Site 1 – Site 7) have been mapped to the primary sequence of Nav channels based on decades of rigorous functional characterizations4 (Fig. 5a). With the growing number of high-resolution structures of Nav channels in the presence of toxins and drugs, the conventional definition of binding sites may no longer be able to reflect the diversity and complexity of the drug binding sites, particularly those on the PD. Hereby we map the known ligand binding sites to the resolved structures of Nav channels and propose a letter-coded nomenclature system to depict these druggable sites in the 3D space.
Fig. 5. Structural mapping of binding sites for Nav antagonists.

a Conventional mapping of drug and toxin-binding sites on the primary sequence of Nav channels. b Structural mapping of VSD sites for accommodating peptidic and small molecule ligands. Based on the new nomenclature system, these sites are named as VnEC/M/P. V: VSD; n: the repeat number; E: extracellular; C: cavity. M: membrane; P: pore domain. c The PD hosts multiple drug-binding sites. Left: Representative Nav inhibitors are mapped to the structural model of the PD. The peptide toxin μ-conotoxin KIIIA is shown as a pale pink surface that is encaged by the extracellular loops (Site E). TTX is shown as orange spheres at the entrance to the SF (Site S). Quinidine, propafenone and cannabidiol are shown as blue, purple and green spheres that bind to the cavity (Site C), the intracellular gate (Site G) and the inactivation site (Site I)65, respectively. A diagram of the druggable sites on the PD are shown on the right. Site F refers to the fenestration sites.
The conventional Site 3 and Site 4 refer to the extracellular segments of VSDIV and VSDII, respectively (Fig. 5a). Whereas small molecules like PF-05089771 resides in an extracellular cavity of VSDIV, peptide toxins like ProTx-II and HWTX-IV bind to the extracellular periphery of the VSD, partly immersed in the lipid bilayer14, 15,59–61. Some other peptide toxins, such as Dc1a and AaH2, employ the adjacent pore segments as scaffold to secure the interaction with the VSD62–64. To distinguish these different binding poses on VSDs, we propose to name the binding sites as following: VnEC for the extracellular cavity (n = 1, 2, 3, 4, referring to repeats I, II, III and IV, respectively), VnEM and VnEP for the extracellular site involving the membrane and the pore domain, respectively. Per this definition, PF-05089771 binds to site V4EC, HWTX-IV and LqhIII to V4EM, and Dc1a to site V2EP (Fig. 5b). There is no known ligand that binds to the intracellular side of any VSD. If such binder is discovered or engineered in the future, the corresponding accommodation site can be named similarly as VnIC/M/P, where I stand for intracellular.
The pore domain hosts remarkably diverse ligand binding sites, in part owing to the asymmetry of the four repeats. The dome enclosed by the disulfide bond-stabilized ECLs represents an important receptor site for peptide toxins like μ-conotoxin KIIIA28. To acknowledge the contribution of ECLs from all repeats instead of the previously defined Site 1b, which refers to the residues on ECLII, we suggest naming this region site E (Fig. 5c).
Below the ECLs, the outer mouth to the SF vestibule is notoriously known as Site 1 or Site 1a that is targeted by the prototypical neurotoxins tetrodotoxin and saxitoxin15,33,62. We propose to name it Site S, standing for selectivity filter. Along the permeation path, the binding sites between the SF and site BIG may be named as Site C for the cavity and Site G for the gate (Fig. 5c). As the central cavity is rather spacious for a small molecule, Site C can be further specified as UC, CC, and LC for upper cavity, central cavity, and lower cavity, respectively. Based on this classification, LCM-2 binds to site UC. It is noted that the bulk of LCM-2 is off the central axis, residing closer to repeats I and IV. The specific site can be referred to as UC14 when the asymmetry needs to be highlighted. We also propose to use letter F to refer to the fenestration sites, such as Site F1 for the I-II fenestration and F4 for IV-I. Thus, bulleyaconitine A, vinpocetine and vixotrigine bind to Sites F1, F3 and F4, respectively. In the accompanying paper, we describe the action of cannabidiol (CBD) on Nav1.7. Among the two binding poses of CBD, one involves the cavity where the fast inactivation motif is accommodated, and thus named site I65 (Fig. 5c).
Implications on drug discovery
The nine subtypes of Nav channels, owing to their tissue-specific distributions and unique biophysical properties, are preferentially associated with different disorders. Nevertheless, these channels share a high degree of sequence similarity, posing a major challenge for the development of subtype-specific drugs. Sites C, G, BIG, and F all involve the S6 segments, which are highly conserved (Supplementary Fig. 7). It is therefore unsurprising that many drugs that bind to these sites are non-selective Nav blockers. Nevertheless, there are subtype-specific inhibitors for Site C. For example, A-803467, which binds to Site CC4, is a Nav1.8-selective blocker66. The subtype specificity is conferred by remote allosteric loci, adding another tier of complications to drug design31.
The S5 and S6 segments below the SF exhibit marked structural shifts associated with their functional states. For instance, the fenestrations may exist only in the activated and inactivated states, but not the resting state. The volume of the central cavity fluctuates accordingly67. Furthermore, the axial rotation of the S6 segments accompanying the transition between the α and π helical turns results in reposition of S6 residues, reshaping the interior of the cavity. In addition, fenestrations and intracellular gate may be related to the entrance and exit pathways of small molecules. Consequently, many drugs that bind to sites C, F, G, and BIG are state-dependent. Such complexity necessitates structural resolution of the channels in different functional states.
Site V4C has been extensively explored for subtype-specific antagonists, such as PF-05089771 for Nav1.7 and ICA121431 for Nav1.3. An overlooked region is site E, which exhibits lower sequence conservation among Nav channels. Supporting this notion, KIIIA shows a strong subtype-selective sensitivity28,68. Site E may deserve more attention for the development of biologics that are of therapeutic potentials.
The development of non-addictive analgesics represents an unmet healthcare need. For a long time, rational design targeting Nav channels was impeded by the lack of accurate structures. Despite recent structure boom of Nav channels, AI-facilitated drug discovery is still hindered by the limitation of the training database wherein the number of ligand-bound structures remains scarce. Moreover, the conformational dynamics is beyond the current capacity of structure prediction. Therefore, experimental structures of target proteins bound to ligands are indispensable for drug discovery. Structural analyses reported here provide an important foundation to aid rational design of drugs targeting Nav channels.
Methods
Transient expression and purification of the human Nav1.7-β1-β2 channel complex
The plasmids for human Nav1.7, β1 and β2 are the same as reported previously15. HEK293F cells (Invitrogen) were cultured in SMM 293T-II medium (Sino Biological Inc.) under 5% CO2 in a Multitron-Pro shaker (Infors, 130 r.p.m.) at 37 °C, and transfected with plasmids when the cell density reached 2.0 × 106 cells per ml. For 1 liter cell culture, approximately 2.5 mg plasmids (1.5 mg for Nav1.7 plus 0.5 mg for β1 and 0.5 mg for β2) were pre-incubated with 4 mg 25-kDa linear polyethylenimines (PEIs, Polysciences) in 25 mL fresh medium for 20 min before adding into cell culture. Transfected cells were cultured for 48 h before harvesting.
Protein purification was carried out following our optimized protocol15. 20 L cells co-transfected with Nav1.7-β1-β2 were used for one batch of sample preparation. They were harvested by centrifugation at 800 g and resuspended in the lysis buffer containing 25 mM Tris-HCl (pH 7.5) and 150 mM NaCl. The suspension was supplemented with 1% (w/v) n-dodecyl-β-D-maltopyranoside (DDM, Anatrace), 0.1% (w/v) cholesteryl hemisuccinate Tris salt (CHS, Anatrace), and protease inhibitor cocktail containing 2 mM phenylmethylsulfonyl fluoride (PMSF), aprotinin (6.5 μg/mL), pepstatin (3.5 μg/mL), and leupeptin (25 μg/mL). For channels treated with carbamazepine or lacosamide,1 mM drug molecule was supplemented throughout the purification procedure, while for channels treated with hardwickiic acid, 10 μM drug molecule was added throughout the whole purification process. After incubation at 4 °C for 2 h, the cell lysate was ultra-centrifuged at 20,000 g for 45 min, and the supernatant was applied to anti-Flag M2 affinity gel (Sigma) by gravity at 4 °C. The resin was rinsed four times with the wash buffer (buffer W) containing 25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.05% glycol-diosgenin (GDN, Anatrace), and the protease inhibitor cocktail. Proteins were eluted with buffer W plus 200 μg/mL FLAG peptide (Sigma). The eluent was then applied to Strep-Tactin Sepharose (IBA) and the purification protocol was similar to the previous steps except that the elution buffer was buffer W plus 2.5 mM D-Desthiobiotin (IBA). It was then concentrated using a 100-kDa cut-off Centricon (Millipore) and further purified through Superose-6 column (GE Healthcare) in buffer W. The peak fractions were pooled and concentrated to ~1 mg/mL, and further incubated with the small molecule drugs for 1 h before cryo-EM sample preparation. Additional carbamezapine (2 mM) and lacosamide (3 mM) were supplemented, making the final concentrations for small molecule drugs to 50 μM for vixotrigine and vinpocetine, 230 μM for bupivacaine, 3 mM for carbamazepine, 4 mM for lacosamide, 10 μM for hardwickiic acid, and 50 μM for PF-05089771. Lacosamide was purchased from Cayman Chemical, and other drugs were purchased from MedChemExpress.
Whole cell electrophysiology
For whole-cell patch-clamp recordings, HEK293T cells (Invitrogen) were cultured in Dulbecco’s Modified Eagle Medium (DMEM, BI) supplemented with 4.5 mg/mL glucose and 10% (v/v) fetal bovine serum (FBS, BI). Cells for recordings were plated onto glass coverslips and transiently co-transfected with the expression plasmid for Nav1.7 and an eGFP-encoding plasmid using lipofectamine 2000 (Invitrogen). Cells with green fluorescence were selected for patch-clamp recording 18–36 h after transfection. All experiments were performed at room temperature. No further authentication was performed for the commercially available cell line. Mycoplasma contamination was not tested.
The whole-cell Na+ currents were recorded using an EPC10-USB amplifier with Patchmaster software v2*90.2 (HEKA Elektronic), filtered at 3 kHz (low-pass Bessel filter) and sampled at 50 kHz. The borosilicate pipettes (Sutter Instrument) had a resistance of 2–4 MΩ and the electrodes were filled with the internal solution composed of (in mM) 105 CsF, 40 CsCl, 10 NaCl, 10 EGTA, 10 Hepes, pH 7.4 with CsOH. The bath solutions contained (in mM): 140 NaCl, 4 KCl, 10 Hepes, 10 D-glucose, 1 MgCl2, 1.5 CaCl2, pH 7.4 with NaOH. Data were analyzed using Origin (OriginLab) and GraphPad Prism (GraphPad Software).
To determine inhibition of the inactivated state by PF-05089771, cells were clamped at −120 mV for 200 ms before stepping to −70 mV (for Nav1.7) or −80 mV (for Nav1.5) for 5 s in order to get about 50% channel inactivated. This was followed by a 5 ms return to −120 mV preceding a 50 ms test pulse to 0 mV. To investigate the resting state blockage of Nav1.7 by the small molecules, cells were held at −120 mV and stepped to +0 mV for 50 ms. Drugs were dissolved in dimethyl sulfoxide (DMSO, final concentration in the perfusion buffer is less than 0.1%). Solutions with indicated concentrations were freshly prepared and perfused to the recording cell for up to 10 mins to get to the maximal block using a multichannel perfusion system (VM8, ALA). Prior to drugs perfusion, cells were recorded for 5 min to establish stable peak current. Concentration-response curve was fitted with: Y = Bottom + (Top − Bottom) / (1 + w10^((LogIC50-X) * Hill Slope)), where IC50 is the concentration of small drugs that blocks 50% of the current and X is log of indicated concentration, and Hill Slope is slope factor. All data points are presented as mean ± SEM and n is the number of experimental cells from which recordings were obtained. Statistical significance was assessed using one-way ANOVA analysis and extra sum-of-squares F test.
Cryo-EM data acquisition
Aliquots of 3.5 μL freshly purified Nav1.7 with small drugs were placed on glow-discharged holey carbon grids (Quantifoil Au 400 mesh, R1.2/1.3). Grids were blotted for 3.0 s and plunge frozen in liquid ethane cooled by liquid nitrogen with Vitrobot Mark IV (Thermo Fisher). For vixotrigine/vinpocetine complex, electron micrographs were acquired on a Titan Krios electron microscope (Thermo Fisher) operating at 300 kV and equipped with a Gatan K3 Summit detector and GIF Quantum energy filter. A total of 5280/4534 movie stacks for Nav1.7 bound to vixotrigine/vinpocetine were automatically collected using AutoEMation69 with a slit width of 20 eV on the energy filter and a preset defocus ranging from −1.8 µm to −1.5 µm in super-resolution mode at a nominal magnification of 81,000. Each stack was exposed for 2.56 s with 0.08 s per frame, resulting in 32 frames per stack. For hardwickiic acid complex, electron micrographs were acquired on a Titan Krios electron microscope (Thermo Fisher) operating at 300 kV and equipped with a Gatan K3 Summit detector and GIF Quantum energy filter. A total of 6389 movie stacks for Nav1.7 bound to hardwickiic acid were automatically collected using AutoEMation69 with a slit width of 20 eV on the energy filter and a preset defocus ranging from −1.8 µm to −1.5 µm in super-resolution mode at a nominal magnification of 64,000. Each stack was exposed for 2.56 s with 0.08 s per frame, resulting in 32 frames per stack. For the carbamezapine/lacosamide complex, electron micrographs were acquired on a Titan Krios electron microscope (Thermo Fisher) operating at 300 kV and equipped with a Cs corrector, Gatan K2 Summit detector and GIF Quantum energy filter. A total of 4254/4268 movie stacks for Nav1.7 bound to carbamezapine/lacosamide were automatically collected using SerialEM70 with a slit width of 20 eV on the energy filter and a preset defocus ranging from −1.6 µm to −1.2 µm in super-resolution mode at a nominal magnification of 105,000. Each stack was exposed for 5.6 s with 0.175 s per frame, resulting in 32 frames per stack. For the PF-05089771/bupivacaine complex, electron micrographs were acquired on a Titan Krios electron microscope (Thermo Fisher) operating at 300 kV and equipped with Gatan K3 Summit detector and GIF Quantum energy filter. 6106/7361 movie stacks of Nav1.7 bound to PF-05089771/bupivacaine were automatically collected using EPU (Thermo Fisher) with a slit width of 20 eV on the energy filter and a preset defocus ranging from −1.8 µm to −1.5 µm in super-resolution mode at a nominal magnification of 81,000. Each stack was exposed for 2.56 s with 0.08 s per frame, resulting in 32 frames per stack. The total dose was 50 e−/Å2 for all the datasets. The stacks were motion-corrected with MotionCor271 or Warp72 and binned 2-fold, resulting in 1.0825 Å/pixel for vinpocetine and vixotrigine, resulting in 1.0979 Å/pixel for hardwickiic acid, 1.114 Å/pixel for carbamezapine/lacosamide and 0.865 Å/pixel for PF-05089771/bupivacaine. Dose weighting was performed as described73. Defocus values were estimated using Gctf74 or cryoSPARC75.
Image processing
For simplicity, the data processing of PF-05089771 was selected as representative with a diagram showed in Supplementary Fig. 3. A total of 7,991,055 particles were automatically picked using template picking in cryoSPARC. After 2D classification, a total of 551,861 good particles were selected, these particles were subject to one round of non-uniform refinement in cryoSPARC76, resulting in 3D reconstructions at 2.8 Å. The resulting particles were subject to one additional round of Heterogeneous Refinement in cryoSPARC, with the number of classes set to 6. A total of 492,421 particles were selected and applied to another round of Non-uniform Refinement and CTF refinement, resulting in final reconstructions at overall resolutions of 2.7 Å (Supplementary Fig. 4 and Supplementary Table 3). Other datasets were processed with the same workflow.
Model building and structure refinement
Model building was performed based on the map acquired in the data processing. The starting model of Nav1.7 (PDB 7W9K), was fitted into the EM map by Chimera77. All Nav1.7 residues, lipids, and sugar moieties were manually checked in COOT78. The chemical properties of amino acids were taken into consideration during model building.
Structural refinement was performed using phenix.real_space_refine application in PHENIX79 with secondary structure and geometry restraints. Over-fitting of the overall model was monitored by refining the model in one of the two independent maps from the gold-standard refinement approach and testing the refined model against the other map80. Statistics of the map reconstruction and model refinement can be found in Supplementary Table 3.
In silico molecular docking
After removing BPV from the Nav1.7-BPV complex, both dextro-(R)- and levo-(S)-bupivacaine were docked against Nav1.7 using Schrödinger Suite 2018-1 (Schrödinger, Inc.) to investigate the racemic specificity of BPV on Nav1.7. The initial small molecule structures were generated and optimized using LigPrep program81 with the OPLS3 force field82, while the protein structure was processed using the default setting within Protein Preparation Wizard with the coordinates of the Nav1.7-BPV complex as input. Molecular docking was performed using the Glide program with the extra-precision docking method (Glide XP).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We thank Xiaomin Li, Fan Yang, and Jianlin Lei for technical support during EM image acquisition. We thank the Tsinghua University Branch of China National Center for Protein Sciences (Beijing) for providing the cryo-EM facility support. We thank the computational facility support from Tsinghua University Branch of China National Center for Protein Sciences (Beijing). We also thank the cryo-EM facility at Princeton Imaging and Analysis Centre. This work was funded by National Natural Science Foundation of China (32271252). X.F. has been supported by the HFSP long-term fellowship (LT000754) from the International Human Frontier Science Program Organization (HFSPO). N.Y. had been supported by the Shirley M. Tilghman endowed professorship from Princeton University in 2017–2022.
Source data
Author contributions
Q.W., H.J., X.F., K.W. and J.L. prepared the sample. Q.W., J.H. and X.F. collected the EM data. Q.W., X.F. and G.H. analyzed the EM data and calculated the EM map. X.F., G.H. and X.P. built and refined the atomic model. X.J. designed and performed the electrophysiological experiments. Q.W., H.J., X.F., K.W., X.J., G.H. discussed and analyzed the results. Q.W., H.J., X.F., X.P and N.Y.analyzed the structure and wrote the paper.
Peer review
Peer review information
Nature Communications thanks Shangyu Dang and Vladimir Yarov-Yarovoy for their contribution to the peer review of this work. A peer review file is available.
Data availability
The data that support this study are available from the corresponding authors upon request. The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-35193 (Nav1.7-BPV), EMD-40238 (Nav1.7-LCM), EMD-40239 (Nav1.7-CBZ), EMD-35197 (Nav1.7-VPC), EMD-35975 (Nav1.7-HDA), EMD-35198 (Nav1.7-VXT), and EMD-35194 (Nav1.7-PF-05089771). Coordinates have been deposited in the Protein Data Bank under accession codes 8I5B (Nav1.7-BPV), 8S9B (Nav1.7-LCM), 8S9C (Nav1.7-CBZ), 8I5X (Nav1.7-VPC), 8J4F (Nav1.7-HDA), 8I5Y (Nav1.7-VXT), and 8I5G (Nav1.7-PF-05089771). The sequences of human Nav1.7, β1 and β2 are available in the following links: Nav1.7 (UniProtID:Q15858); β1 (UniProtID:Q07699); β2 (UniProtID:O60939).
Previously solved structures mentioned in this study are under the accession codes in PDB: 7W9K; 6J8J; 6J8I; 6J8E; 7W9M; 7W9T; 7W9P; 5EK0; 7XMF; 7XMG; 7XM9; 6LQA; 7FBS; 8G1A.
The source data underlying Fig. 1d, Supplementary Fig. 2a–b and 4 are provided as a Source Data file. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Qiurong Wu, Jian Huang, Xiao Fan, Kan Wang, Xueqin Jin.
Contributor Information
Jian Huang, Email: jh6493@princeton.edu.
Xiao Fan, Email: xiaof@princeton.edu.
Xiaojing Pan, Email: panxj@tsinghua.edu.cn.
Nieng Yan, Email: nyan@tsinghua.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-023-38942-3.
References
- 1.Hille, B. Ion channels of excitable membranes. 3rd edn, (Sinauer, 2001).
- 2.Hodgkin AL, Huxley AF. Resting and action potentials in single nerve fibres. J. Physiol. 1945;104:176–195. doi: 10.1113/jphysiol.1945.sp004114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharm. Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 4.Chahine, M. in Handbook of Experimental Pharmacology, 1 online resource (X, 450 pages 480 illustrations, 455 illustrations in color (Springer International Publishing: Imprint: Springer, Cham, 2018).
- 5.McKerrall SJ, Sutherlin DP. Na(v)1.7 inhibitors for the treatment of chronic pain. Bioorg. Med. Chem. Lett. 2018;28:3141–3149. doi: 10.1016/j.bmcl.2018.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Santos R, et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017;16:19–34. doi: 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson MR, Kaminski RM. A systems-level framework for anti-epilepsy drug discovery. Neuropharmacology. 2020;170:107868. doi: 10.1016/j.neuropharm.2019.107868. [DOI] [PubMed] [Google Scholar]
- 8.Alsaloum M, Higerd GP, Effraim PR, Waxman SG. Status of peripheral sodium channel blockers for non-addictive pain treatment. Nat. Rev. Neurol. 2020;16:689–705. doi: 10.1038/s41582-020-00415-2. [DOI] [PubMed] [Google Scholar]
- 9.Jiang D, et al. Structure of the cardiac sodium channel. Cell. 2020;180:122–134 e110. doi: 10.1016/j.cell.2019.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang, D. et al. Structure of the cardiac sodium channel. Cell, 10.1016/j.cell.2019.11.041 (2019).
- 11.Li Z, et al. Structural basis for pore blockade of the human cardiac sodium channel Na(v) 1.5 by the antiarrhythmic drug quinidine*. Angew. Chem. Int. Ed. Engl. 2021;60:11474–11480. doi: 10.1002/anie.202102196. [DOI] [PubMed] [Google Scholar]
- 12.Li X, et al. Structural basis for modulation of human Na(V)1.3 by clinical drug and selective antagonist. Nat. Commun. 2022;13:1286. doi: 10.1038/s41467-022-28808-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao X, et al. Structural basis for the severe adverse interaction of sofosbuvir and amiodarone on L-type Ca(v) channels. Cell. 2022;185:4801–4810 e4813. doi: 10.1016/j.cell.2022.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang G, et al. High-resolution structures of human Na(v)1.7 reveal gating modulation through alpha-pi helical transition of S6(IV) Cell Rep. 2022;39:110735. doi: 10.1016/j.celrep.2022.110735. [DOI] [PubMed] [Google Scholar]
- 15.Shen H, Liu D, Wu K, Lei J, Yan N. Structures of human Na(v)1.7 channel in complex with auxiliary subunits and animal toxins. Science. 2019;363:1303–1308. doi: 10.1126/science.aaw2493. [DOI] [PubMed] [Google Scholar]
- 16.Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The Na(V)1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 2013;14:49–62. doi: 10.1038/nrn3404. [DOI] [PubMed] [Google Scholar]
- 17.Cox JJ, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–898. doi: 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldberg YP, et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin. Genet. 2007;71:311–319. doi: 10.1111/j.1399-0004.2007.00790.x. [DOI] [PubMed] [Google Scholar]
- 19.Ahmad S, et al. A stop codon mutation in SCN9A causes lack of pain sensation. Hum. Mol. Genet. 2007;16:2114–2121. doi: 10.1093/hmg/ddm160. [DOI] [PubMed] [Google Scholar]
- 20.Fertleman CR, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52:767–774. doi: 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Yang Y, et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 2004;41:171–174. doi: 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cummins TR, Dib-Hajj SD, Waxman SG. Electrophysiological properties of mutant Nav1.7 sodium channels in a painful inherited neuropathy. J. Neurosci. 2004;24:8232–8236. doi: 10.1523/JNEUROSCI.2695-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faber CG, et al. Gain of function Nanu1.7 mutations in idiopathic small fiber neuropathy. Ann. Neurol. 2012;71:26–39. doi: 10.1002/ana.22485. [DOI] [PubMed] [Google Scholar]
- 24.Chernov-Rogan T, et al. Mechanism-specific assay design facilitates the discovery of Nav1.7-selective inhibitors. Proc. Natl Acad. Sci. USA. 2018;115:E792–E801. doi: 10.1073/pnas.1713701115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Theile JW, Fuller MD, Chapman ML. The selective Nav1.7 inhibitor, PF-05089771, interacts equivalently with fast and slow inactivated Nav1.7 channels. Mol. Pharm. 2016;90:540–548. doi: 10.1124/mol.116.105437. [DOI] [PubMed] [Google Scholar]
- 26.Cao L, et al. Pharmacological reversal of a pain phenotype in iPSC-derived sensory neurons and patients with inherited erythromelalgia. Sci. Transl. Med. 2016;8:335ra356. doi: 10.1126/scitranslmed.aad7653. [DOI] [PubMed] [Google Scholar]
- 27.Pan, X. et al. Comparative structural analysis of human Na(v)1.1 and Na(v)1.5 reveals mutational hotspots for sodium channelopathies. Proc. Natl Acad. Sci. USA118, 10.1073/pnas.2100066118 (2021). [DOI] [PMC free article] [PubMed]
- 28.Pan X, et al. Molecular basis for pore blockade of human Na(+) channel Na(v)1.2 by the mu-conotoxin KIIIA. Science. 2019;363:1309–1313. doi: 10.1126/science.aaw2999. [DOI] [PubMed] [Google Scholar]
- 29.Pan, X. et al. Structure of the human voltage-gated sodium channel Na(v)1.4 in complex with beta1. Science362, 10.1126/science.aau2486 (2018). [DOI] [PubMed]
- 30.Li, Z. et al. Structure of human Na(v)1.5 reveals the fast inactivation-related segments as a mutational hotspot for the long QT syndrome. Proc. Natl Acad. Sci. USA118, 10.1073/pnas.2100069118 (2021). [DOI] [PMC free article] [PubMed]
- 31.Huang X, et al. Structural basis for high-voltage activation and subtype-specific inhibition of human Na(v)1.8. Proc. Natl Acad. Sci. USA. 2022;119:e2208211119. doi: 10.1073/pnas.2208211119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan X, Huang J, Jin X, Yan N. Cryo-EM structure of human voltage-gated sodium channel Nav1.6. Proc. Natl Acad. Sci. USA. 2023;120:e2220578120. doi: 10.1073/pnas.2220578120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen, H. Z. et al. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science355, 10.1126/science.aal4326 (2017). [DOI] [PubMed]
- 34.Yan Z, et al. Structure of the Na(v)1.4-beta1 complex from electric eel. Cell. 2017;170:470–482 e411. doi: 10.1016/j.cell.2017.06.039. [DOI] [PubMed] [Google Scholar]
- 35.Muroi Y, Arcisio-Miranda M, Chowdhury S, Chanda B. Molecular determinants of coupling between the domain III voltage sensor and pore of a sodium channel. Nat. Struct. Mol. Biol. 2010;17:230–U214. doi: 10.1038/nsmb.1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, et al. Crystal structure of an orthologue of the NaChBac voltage-gated sodium channel. Nature. 2012;486:130–134. doi: 10.1038/nature11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noda M, et al. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature. 1984;312:121–127. doi: 10.1038/312121a0. [DOI] [PubMed] [Google Scholar]
- 38.Patino GA, Isom LL. Electrophysiology and beyond: multiple roles of Na+ channel beta subunits in development and disease. Neurosci. Lett. 2010;486:53–59. doi: 10.1016/j.neulet.2010.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fischer TZ, et al. A novel Nav1.7 mutation producing carbamazepine-responsive erythromelalgia. Ann. Neurol. 2009;65:733–741. doi: 10.1002/ana.21678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang YC, Huang CS, Kuo CC. Lidocaine, carbamazepine, and imipramine have partially overlapping binding sites and additive inhibitory effect on neuronal Na+ channels. Anesthesiology. 2010;113:160–174. doi: 10.1097/ALN.0b013e3181dc1dd6. [DOI] [PubMed] [Google Scholar]
- 41.Blair HA. Bupivacaine/Meloxicam prolonged release: a review in postoperative pain. Drugs. 2021;81:1203–1211. doi: 10.1007/s40265-021-01551-9. [DOI] [PubMed] [Google Scholar]
- 42.Al-Kuraishy HM, Al-Gareeb AI, Naji MT, Al-Mamorry F. Role of vinpocetine in ischemic stroke and poststroke outcomes: a critical review. Brain Circ. 2020;6:1–10. doi: 10.4103/bc.bc_46_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jensen TS. Selective sodium channel blockers in trigeminal neuralgia. Lancet Neurol. 2017;16:255–256. doi: 10.1016/S1474-4422(17)30017-0. [DOI] [PubMed] [Google Scholar]
- 44.Cai, S. et al. Hardwickiic acid and hautriwaic acid induce antinociception via blockade of tetrodotoxin-sensitive voltage-dependent sodium channels. ACS Chem. Neurosci.10, 1716–1728 (2019). [DOI] [PubMed]
- 45.Kocot-Kepska, M. et al. Topical treatments and their molecular/cellular mechanisms in patients with peripheral neuropathic pain-narrative review. Pharmaceutics13, 10.3390/pharmaceutics13040450 (2021). [DOI] [PMC free article] [PubMed]
- 46.McCormack K, et al. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc. Natl Acad. Sci. USA. 2013;110:E2724–E2732. doi: 10.1073/pnas.1220844110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahuja S, et al. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science. 2015;350:aac5464. doi: 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- 48.Smart OS, Neduvelil JG, Wang X, Wallace BA, Sansom MS. HOLE: a program for the analysis of the pore dimensions of ion channel structural models. J. Mol. Graph. 1996;14:354–360, 376. doi: 10.1016/s0263-7855(97)00009-x. [DOI] [PubMed] [Google Scholar]
- 49.Priest BT, Blumenthal KM, Smith JJ, Warren VA, Smith MM. ProTx-I and ProTx-II: gating modifiers of voltage-gated sodium channels. Toxicon. 2007;49:194–201. doi: 10.1016/j.toxicon.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 50.Xiao Y, et al. Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain ii voltage sensor in the closed configuration. J. Biol. Chem. 2008;283:27300–27313. doi: 10.1074/jbc.M708447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao Y, et al. Molecular basis for ligand modulation of a mammalian voltage-gated Ca(2+) channel. Cell. 2019;177:1495–1506 e1412. doi: 10.1016/j.cell.2019.04.043. [DOI] [PubMed] [Google Scholar]
- 52.Gao S, Yan N. Structural basis of the modulation of the voltage-gated calcium ion channel Ca(v) 1.1 by dihydropyridine compounds*. Angew. Chem. Int Ed. Engl. 2021;60:3131–3137. doi: 10.1002/anie.202011793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahern CA, Eastwood AL, Dougherty DA, Horn R. Electrostatic contributions of aromatic residues in the local anesthetic receptor of voltage-gated sodium channels. Circ. Res. 2008;102:86–94. doi: 10.1161/CIRCRESAHA.107.160663. [DOI] [PubMed] [Google Scholar]
- 54.Ragsdale DS, McPhee JC, Scheuer T, Catterall WA. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science. 1994;265:1724–1728. doi: 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- 55.Yarov-Yarovoy V, et al. Molecular determinants of voltage-dependent gating and binding of pore-blocking drugs in transmembrane segment IIIS6 of the Na(+) channel alpha subunit. J. Biol. Chem. 2001;276:20–27. doi: 10.1074/jbc.M006992200. [DOI] [PubMed] [Google Scholar]
- 56.Yarov-Yarovoy V, et al. Role of amino acid residues in transmembrane segments IS6 and IIS6 of the Na+ channel alpha subunit in voltage-dependent gating and drug block. J. Biol. Chem. 2002;277:35393–35401. doi: 10.1074/jbc.M206126200. [DOI] [PubMed] [Google Scholar]
- 57.Nguyen PT, DeMarco KR, Vorobyov I, Clancy CE, Yarov-Yarovoy V. Structural basis for antiarrhythmic drug interactions with the human cardiac sodium channel. Proc. Natl Acad. Sci. USA. 2019;116:2945–2954. doi: 10.1073/pnas.1817446116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jo S, Bean BP. Lidocaine binding enhances inhibition of Nav1.7 channels by the sulfonamide PF-05089771. Mol. Pharm. 2020;97:377–383. doi: 10.1124/mol.119.118380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Legros C, et al. Expression of the standard scorpion alpha-toxin AaH II and AaH II mutants leading to the identification of some key bioactive elements. Biochim Biophys. Acta. 2005;1723:91–99. doi: 10.1016/j.bbagen.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 60.Gao S, et al. Employing NaChBac for cryo-EM analysis of toxin action on voltage-gated Na(+) channels in nanodisc. Proc. Natl Acad. Sci. USA. 2020;117:14187–14193. doi: 10.1073/pnas.1922903117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wisedchaisri G, et al. Structural basis for high-affinity trapping of the Na(V)1.7 channel in its resting state by tarantula toxin. Mol. Cell. 2021;81:38–48 e34. doi: 10.1016/j.molcel.2020.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen, H. et al. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science362, 10.1126/science.aau2596 (2018). [DOI] [PubMed]
- 63.Clairfeuille, T. et al. Structural basis of alpha-scorpion toxin action on Na(v) channels. Science363, 10.1126/science.aav8573 (2019). [DOI] [PubMed]
- 64.Bende NS, et al. A distinct sodium channel voltage-sensor locus determines insect selectivity of the spider toxin Dc1a. Nat. Commun. 2014;5:4350. doi: 10.1038/ncomms5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang., J. et al. Cannabidiol inhibits Nav channels through two distinct binding sites. Nat Commun. (2023). [DOI] [PMC free article] [PubMed]
- 66.Jarvis MF, et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc. Natl Acad. Sci. USA. 2007;104:8520–8525. doi: 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang G, et al. Unwinding and spiral sliding of S4 and domain rotation of VSD during the electromechanical coupling in Na(v)1.7. Proc. Natl Acad. Sci. USA. 2022;119:e2209164119. doi: 10.1073/pnas.2209164119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang MM, et al. Structure/function characterization of micro-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. J. Biol. Chem. 2007;282:30699–30706. doi: 10.1074/jbc.M704616200. [DOI] [PubMed] [Google Scholar]
- 69.Lei J, Frank J. Automated acquisition of cryo-electron micrographs for single particle reconstruction on an FEI Tecnai electron microscope. J. Struct. Biol. 2005;150:69–80. doi: 10.1016/j.jsb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 70.Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 71.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tegunov D, Cramer P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods. 2019;16:1146–1152. doi: 10.1038/s41592-019-0580-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Grant, T. & Grigorieff, N. Measuring the optimal exposure for single particle cryo-EM using a 2.6 angstrom reconstruction of rotavirus VP6. Elife4, 10.7554/eLife.06980 (2015). [DOI] [PMC free article] [PubMed]
- 74.Zhang K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016;193:1–12. doi: 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife7, 10.7554/eLife.42166 (2018). [DOI] [PMC free article] [PubMed]
- 76.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 77.Pettersen EF, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 78.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D. Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Amunts A, et al. Structure of the yeast mitochondrial large ribosomal subunit. Science. 2014;343:1485–1489. doi: 10.1126/science.1249410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 82.Harder E, et al. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016;12:281–296. doi: 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support this study are available from the corresponding authors upon request. The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-35193 (Nav1.7-BPV), EMD-40238 (Nav1.7-LCM), EMD-40239 (Nav1.7-CBZ), EMD-35197 (Nav1.7-VPC), EMD-35975 (Nav1.7-HDA), EMD-35198 (Nav1.7-VXT), and EMD-35194 (Nav1.7-PF-05089771). Coordinates have been deposited in the Protein Data Bank under accession codes 8I5B (Nav1.7-BPV), 8S9B (Nav1.7-LCM), 8S9C (Nav1.7-CBZ), 8I5X (Nav1.7-VPC), 8J4F (Nav1.7-HDA), 8I5Y (Nav1.7-VXT), and 8I5G (Nav1.7-PF-05089771). The sequences of human Nav1.7, β1 and β2 are available in the following links: Nav1.7 (UniProtID:Q15858); β1 (UniProtID:Q07699); β2 (UniProtID:O60939).
Previously solved structures mentioned in this study are under the accession codes in PDB: 7W9K; 6J8J; 6J8I; 6J8E; 7W9M; 7W9T; 7W9P; 5EK0; 7XMF; 7XMG; 7XM9; 6LQA; 7FBS; 8G1A.
The source data underlying Fig. 1d, Supplementary Fig. 2a–b and 4 are provided as a Source Data file. Source data are provided with this paper.