

Abstract
Surgery‐induced renal ischemia and reperfusion (I/R) injury and nephrotoxic drugs like cisplatin can cause acute kidney injury (AKI), for which there is no effective therapy. Lipid accumulation is evident following AKI in renal tubules although the mechanisms and pathological effects are unclear. Here, we report that Ehmt2‐encoded histone methyltransferase G9a is upregulated in patients and mouse kidneys after AKI. Renal tubular specific knockout of G9a (Ehmt2 Ksp ) or pharmacological inhibition of G9a alleviates lipid accumulation associated with AKI. Mechanistically, G9a suppresses transcription of the lipolytic enzyme Ces1; moreover, G9a and farnesoid X receptor (FXR) competitively bind to the same promoter regions of Ces1. Ces1 is consistently observed to be downregulated in the kidney of AKI patients. Pharmacological inhibition of Ces1 increases lipid accumulation, exacerbates renal I/R‐injury and eliminates the beneficial effects on AKI observed in Ehmt2 Ksp mice. Furthermore, lipid‐lowering atorvastatin and an FXR agonist alleviate AKI by activating Ces1 and reducing renal lipid accumulation. Together, our results reveal a G9a/FXR‐Ces1 axis that affects the AKI outcome via regulating renal lipid accumulation.
Keywords: acute kidney injury, Ces1, FXR, G9a, lipid toxicity
Subject Categories: Chromatin, Transcription & Genomics; Molecular Biology of Disease
The histone methyltransferase G9a promotes acute kidney injury (AKI) by increasing lipid accumulation via suppressing the lipolytic enzyme Ces1. Inhibition of G9a, activation of Ces1, or lipid‐lowering agents may be considered for AKI therapy.
Introduction
Acute kidney injury (AKI) is a common and serious complication in hospitalized patients. 10–15% of patients admitted to hospitals suffer from AKI, and the prevalence of AKI among the patients in intensive care units is over 50% (Ronco et al, 2019). Due to the difficulty of diagnosis and lack of treatment, AKI causes 1.7 million deaths annually (Rewa & Bagshaw, 2014). Disease, trauma, or surgery induced ischemia and reperfusion (I/R), and the use of nephrotoxic drugs like cisplatin are leading causes of AKI (Basile et al, 2012; Shiva et al, 2020). Major pathological manifestations of AKI include tubular necrosis, cast formation, and denudation of tubular cells, together with upregulation of biomarkers such as kidney injury molecule 1 (KIM1), neutrophil gelatinase‐associated lipocalin (NGAL), blood urea nitrogen (BUN), and serum creatinine (Beker et al, 2018).
Upon injury, renal cells often exhibit energy loss and increased oxidative stress, both of which induce cell death (Emma et al, 2016; Tomsa et al, 2019). Necroptosis, indicated by increased phosphorylation of RIP3 (receptor‐interacting protein kinase 3) and MLKL (mixed lineage kinase domain‐like protein), is the major cell death pathway in AKI (Wang et al, 2016; Jun et al, 2020). Inflammatory cytokines and chemokines released by dead renal cells result in immune cell infiltration, which causes further damage (Bonventre & Zuk, 2004; Bonventre & Yang, 2011). Of all renal cell types, tubular epithelial cells (TECs) are highly sensitive to AKI and are key contributors to pathological development, largely due to their high metabolic rates (Bonventre & Weinberg, 2003; Chevalier, 2016).
Energy is produced in TECs primarily through lipid‐consuming fatty acid beta‐oxidation (FAO; Emma et al, 2016; Sugahara et al, 2019). In diabetic nephropathy, lipid accumulation in TECs is associated with cell death and elevated inflammatory cytokines (Kang et al, 2015; Chen et al, 2019b). Surprisingly, lipids also accumulate in the kidney, predominantly in TECs, in both patients and mice after AKI (Zager et al, 2005; Portilla et al, 2006; Bobulescu, 2010). In contrast to the hyperglycemia or hyperlipidemia that occur in diabetic kidney disease, the regulatory mechanisms for unbalanced lipid metabolism following AKI are largely unknown.
Ehmt2‐encoded G9a is an euchromatin histone methyltransferase that regulates gene expression by manipulating mono‐ and di‐methylation levels on histone H3 lysine 9 (H3K9me1/me2; Shinkai & Tachibana, 2011). We recently reported that G9a plays multiple functions in different organs, including protection from acute liver injury (Zhang et al, 2020b), modulation of hepatic insulin signaling (Xue et al, 2018), and regulation of a muscle‐liver‐fat axis under overnutrition (Zhang et al, 2020a). In the kidney, the level of G9a is positively associated with renal fibrosis in a mouse model of unilateral ureteral obstruction (UUO), while a G9a inhibitor, BIX01294, prevented UUO‐induced fibrosis (Irifuku et al, 2016) and protected against I/R injury (Liu et al, 2021; Sung et al, 2022). Since G9a is expressed in both renal tubules and glomeruli, we wondered whether G9a plays different roles in different types of renal cells. To test this hypothesis, we generated a renal tubular specific G9a knockout mouse to investigate its role in AKI.
In the current study, we report upregulated renal tubular G9a after I/R‐ or cisplatin‐injury in mice and in AKI patients. Using a renal tubular specific G9a knockout (Ehmt2 Ksp ) mouse and a G9a inhibitor, A366, with high specificity and low toxicity, we identified the renal lipolytic enzyme carboxylesterase 1 (Ces1) as a downstream target that is negatively regulated by G9a. Downregulation of CES1 contributes to lipid accumulation and the subsequent pathological lesions in injured kidney. Interestingly, G9a competes with a well‐known lipid metabolism‐related transcription factor, farnesoid X‐activated receptor (FXR), for binding sites on the Ces1 promoter. Two types of lipid‐lowering agents, atorvastatin and an FXR activator, significantly reduce I/R‐induced renal lesions by activating Ces1, thus reducing pathological renal lipid accumulation. Taken together, our findings reveal a renal tubular G9a/FXR‐Ces1 axis in AKI progression and provide new strategies for AKI treatment.
Results
Renal tubular specific G9a deletion alleviated I/R‐induced inflammation and necroptosis
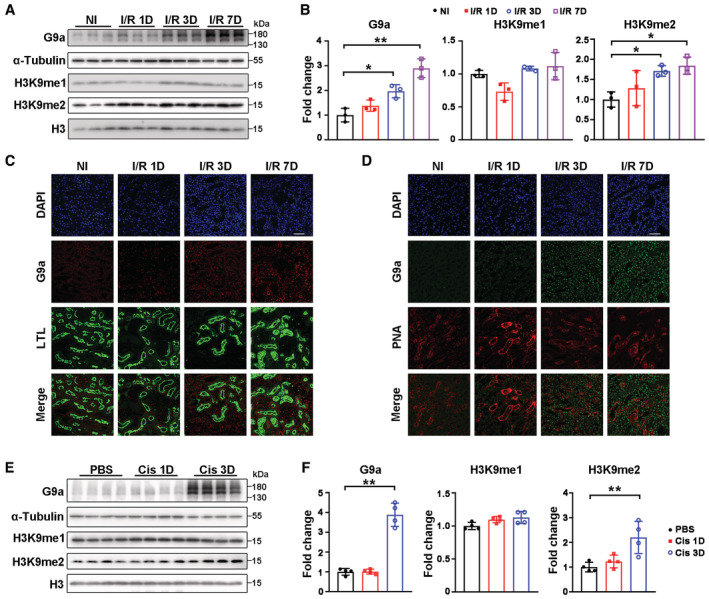
To investigate the potential association of G9a and AKI, the levels of G9a and H3K9me1/2 were determined in mouse kidneys at different time points after I/R injury. G9a and H3K9me2 levels were significantly increased at 3 days after I/R injury (I/R 3D) and remained elevated at 7 days after injury (I/R 7D; Fig EV1A and B). To determine which tubular cell type(s) upregulated expression of G9a after I/R injury, we co‐stained for G9a and LTL (lotus tetragonolobus lectin, detects proximal tubules) or PNA (peanut agglutinin, detects distal tubules and collecting ducts), and found that nuclear staining of G9a was significantly increased in these tubular cells (Fig EV1C and D). Similarly, G9a and H3K9me2 levels were elevated at Day 3 after cisplatin‐induced AKI (Fig EV1E and F). Importantly, G9a was significantly increased in renal tubules of AKI patients compared with the control subjects (Fig 1A). Thus, renal G9a, especially tubular G9a, may play a critical role in AKI.
Figure EV1. Upregulation of G9a and H3K9me2 after renal I/R‐ and cisplatin‐induced AKI.
-
A, BRepresentative immunoblots of G9a and H3K9me1/2 (A) with quantitative results (B) in mouse kidney at indicated time points after renal I/R injury. Three mice per group.
-
C, DRepresentative co‐immunofluorescent staining for G9a (red) with LTL (lotus tetragonolobus lectin, detecting proximal tubules; green) (C) or G9a (green) with PNA (peanut agglutinin, detecting distal tubules and collecting ducts; red) (D) in mouse kidneys at indicated time points after the renal I/R injury. Scale bar = 50 μm.
-
E, FRepresentative immunoblots of G9a and H3K9me1/2 (E) with quantitative results (F) in mouse kidneys at indicated time points after cisplatin injection. Four mice per group.
Data information: NI, non‐injured; Cis, cisplatin; in (B and F), data were presented as means ± SD. Panels B and F (G9a and H3K9me2) were analyzed by 1‐way ANOVA followed by Tukey's test. Panel F (H3K9me1) was analyzed by Mann–Whitney U test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
Figure 1. Renal tubular specific G9a deletion ameliorated renal I/R injury.

-
ARepresentative G9A immunostaining (left) and semi‐quantitative results (right) of human kidney samples. AKI (acute kidney injury), n = 3; Con (control), n = 6. Scale bar = 50 μm.
-
BRepresentative immunoblots of G9a, α‐Tubulin, H3K9me1/2 and H3 in the kidneys of WT and Ehmt2 Ksp mice. C, cortex; M, medulla.
-
CRepresentative renal H&E images (left) with quantitative pathological scores (right) of WT (n = 8) and Ehmt2 Ksp mice (n = 7) at day three after renal I/R injury (I/R 3D). Scale bar = 100 μm.
-
DmRNA levels of renal Kim1 and Ngal of WT (n = 5) and Ehmt2 Ksp (n = 6) mice under I/R 3D injury.
-
E, FRepresentative CD3, Ly6G, and F4/80 immunostaining (E) and quantitative results (F) of WT + NI, Ehmt2 Ksp + NI, WT + I/R 3D and Ehmt2 Ksp + I/R 3D groups (n = 5, 6, 5, 6 of respective groups). Scale bar = 50 μm.
-
G, HRepresentative immunoblots (G) and quantitative results (H) of p‐Rip3 and p‐Mlkl of WT (n = 3) and Ehmt2 Ksp mice (n = 3) under I/R 3D injury.
-
IRepresentative Sirius Red staining (left) with quantitative results (right) on the kidney sections of WT (n = 7) and Ehmt2 Ksp (n = 4) mice at day seven after renal I/R injury (I/R 7D). Scale bar = 50 μm.
-
JmRNA levels of Tgfb1, Fibronectin1 and Collagen1a of WT + NI (n = 4), Ehmt2 Ksp + NI (n = 4), WT + I/R 7D (n = 7) and Ehmt2 Ksp + I/R 7D (n = 4) groups.
-
KRepresentative TUNEL staining (left) with quantitative results (right) of WT (n = 7) and Ehmt2 Ksp mice (n = 4) under I/R 7D injury. Scale bar = 25 μm.
Data information: NI, non‐injured; Rel., relative; in (A, C, D, F, H–K), data were presented as means ± SD. Panels A, H, J and K were analyzed with 2‐tailed, unpaired Student's t test. Panels D (Ngal) and F were analyzed by 1‐way ANOVA followed by Tukey's test. Panels C, D (Kim1), and I were analyzed by Mann–Whitney U test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
To further investigate the impact of renal tubular G9a on AKI, we generated Ehmt2 Ksp mice, which were born at the expected Mendelian frequency, exhibiting normal size and no physical or behavioral abnormalities. Significantly reduced G9a and H3K9me1/me2 levels were observed in the renal cortex and medulla of Ehmt2 Ksp mice (Fig 1B, Appendix Fig S1A–C). Immunostaining showed reduction of G9a in the proximal, distal, and collecting tubules of Ehmt2 Ksp mice after I/R injury (Appendix Fig S1D and E). While no obvious renal morphological difference was observed in Ehmt2 Ksp mice under normal conditions, tubular damage was significantly attenuated in Ehmt2 Ksp mice compared to wildtype (WT) controls at I/R 3D (Fig 1C). The upregulation of renal injury biomarkers Kim1 and Ngal was consistently reduced in injured Ehmt2 Ksp mice compared with those of injured WT mice (Fig 1D). Increased immune cell infiltration is a common manifestation of AKI (Bonventre & Zuk, 2004). Compared to similarly injured WT mice, the number of infiltrating macrophages (F4/80+), neutrophils (Ly6G+), and T cells (CD3+) was significantly lower in the kidneys of Ehmt2 Ksp mice at I/R 3D (Fig 1E and F). Moreover, the necroptosis markers p‐Rip3 and p‐Mlkl were upregulated in injured kidneys of WT mice at I/R 3D; while the increase was reduced in similarly injured Ehmt2 Ksp mice (Fig 1G and H).
Renal tubular specific G9a deletion alleviated renal I/R‐induced fibrosis
Observation of sustained high G9a level at I/R 7D (Fig EV1A–D), the time point when fibrosis usually occurs, encouraged us to investigate its effects on renal fibrosis. Upon injury, Sirius Red staining demonstrated severe renal fibrosis in WT mice, while Ehmt2 Ksp mice showed much less fibrosis and lower transcriptional levels of fibrotic biomarkers including Tgfb1, Fibronectin1, and Collagen1a (Fig 1I and J). Consistently, Ehmt2 Ksp mice also showed significantly less tubular damage and lower Kim1 and Ngal levels at I/R 7D (Fig EV2A and B). Moreover, compared to injured WT mice, significantly fewer infiltrating T cells, macrophages, and TUNEL+ cells, as well as lower p‐Rip3 and p‐Mlkl levels, were observed in Ehmt2 Ksp mice at I/R 7D (Figs 1K and EV2C and D).
Figure EV2. Renal tubular specific G9a deletion ameliorated renal I/R 7D injury, immune cell infiltration and necroptosis.

- Representative H&E staining (left) with pathological scores (right) of WT + NI (n = 7), Ehmt2 Ksp + NI (n = 4), WT + I/R 7D (n = 9) and Ehmt2 Ksp + I/R 7D (n = 4) groups. Scale bar = 100 μm.
- mRNA levels of renal Kim1 and Ngal of WT + I/R 7D (n = 7) and Ehmt2 Ksp + I/R 7D (n = 4).
- Representative CD3 and F4/80 immunostaining (left) and quantitative results (right) of WT + I/R 7D (n = 7) and Ehmt2 Ksp + I/R 7D (n = 4). Scale bar = 50 μm.
- Representative immunoblots (left) and quantitative results (right) of p‐Rip3 and p‐Mlkl of WT + I/R 7D (n = 7) and Ehmt2 Ksp + I/R 7D (n = 4).
Data information: NI, non‐injured; in (A–D), data were presented as means ± SD. Panel A was analyzed by Mann–Whitney U test. Panels B–D were analyzed with 2‐tailed, unpaired Student's t test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
Renal tubular specific G9a deletion reduced I/R injury‐induced lipid accumulation
To identify the pathway(s) affected by renal tubular specific G9a deletion following I/R injury, RNA‐sequencing was performed. Two sets of KEGG pathways were identified based on (i) 6096 differentially expressed genes identified between non‐injured vs. injured kidneys in WT mice and (ii) 4233 differentially expressed genes identified between I/R 3D injured kidneys from Ehmt2 Ksp and WT mice (Fig 2A). Five lipid metabolism‐related pathways were found in both KEGG sets, including thermogenesis, oxidative phosphorylation, peroxisome, fatty acid degradation, and fatty acid metabolism (Fig 2A). The ferroptosis‐related pathway was not enriched; while no significant change in protein levels of Acsl4 and Gpx4, two ferroptosis markers (Hirschhorn & Stockwell, 2019) was found between the kidneys of WT and Ehmt2 Ksp mice, with or without I/R 3D injury (Appendix Fig S2). Therefore, we stained lipids in injured kidneys using Oil Red O and found that I/R‐ and cisplatin‐induced AKI resulted in dramatic renal lipid accumulation (Appendix Fig S3A and B).
Figure 2. Renal tubular specific G9a deletion enhanced lipolysis and fatty acid oxidation after I/R 3D injury.

-
ATop 15 KEGG pathways enriched from significantly downregulated genes between injured kidney vs. non‐injured kidney of WT mice (left), and from significantly upregulated genes between injured kidney of Ehmt2 Ksp mice vs. injured kidney of WT mice (right), at I/R 3D; red bars indicate pathways related to lipid metabolism.
-
B, CHeatmap of RNA‐sequencing data (B) and qPCR validation (C) of indicated genes related to fatty acid metabolism of WT + NI (n = 6), Ehmt2 Ksp + NI (n = 6), WT + I/R 3D (n = 6) and Ehmt2 Ksp + I/R 3D (n = 6) groups.
-
D, EHeatmap of RNA‐sequencing data (D) and qPCR validation (E) of indicated genes related to fatty acid beta‐oxidation of WT + NI (n = 6), Ehmt2 Ksp + NI (n = 6), WT + I/R 3D (n = 6) and Ehmt2 Ksp + I/R 3D (n = 6) groups.
-
FRepresentative Cpt1a immunostaining (up) with quantitative analysis (bottom) in the kidneys of WT + NI (n = 5), Ehmt2 Ksp + NI (n = 5), WT + I/R 3D (n = 5) and Ehmt2 Ksp + I/R 3D (n = 5) groups. Scale bar = 50 μm. IOD, integral optical density.
-
GRepresentative CES1 immunostaining (left) with semi‐quantitative analysis (right) of human kidneys. AKI (acute kidney injury), n = 3; Con (control), n = 6. Scale bar = 50 μm.
-
HRepresentative immunoblots (left) with quantitative results (right) of Ces1 level in the kidneys of WT (n = 3) and Ehmt2 Ksp (n = 3) mice at I/R 3D.
-
I–KCarboxylesterase enzymatic activity (I), representative Oil Red O staining with quantification (J, scale bar = 100 μm), as well as the levels of TG (triglyceride; K, left) and TC (cholesterol; K, right) in the kidney of WT (n = 6) and Ehmt2 Ksp (n = 4) mice at I/R 3D.
Data information: NI, non‐injured; in (C, E–K), data were presented as means ± SD. Panels C (except Ces1f), E (except Acox1 and Pgc1a) and F were analyzed by 1‐way ANOVA followed by Tukey's test. Panels G–J, K(TG) were analyzed with 2‐tailed, unpaired Student's t test. Panels C (Ces1f), E (Acox1 and Pgc1a) and K (TC) were analyzed by Mann–Whitney U test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
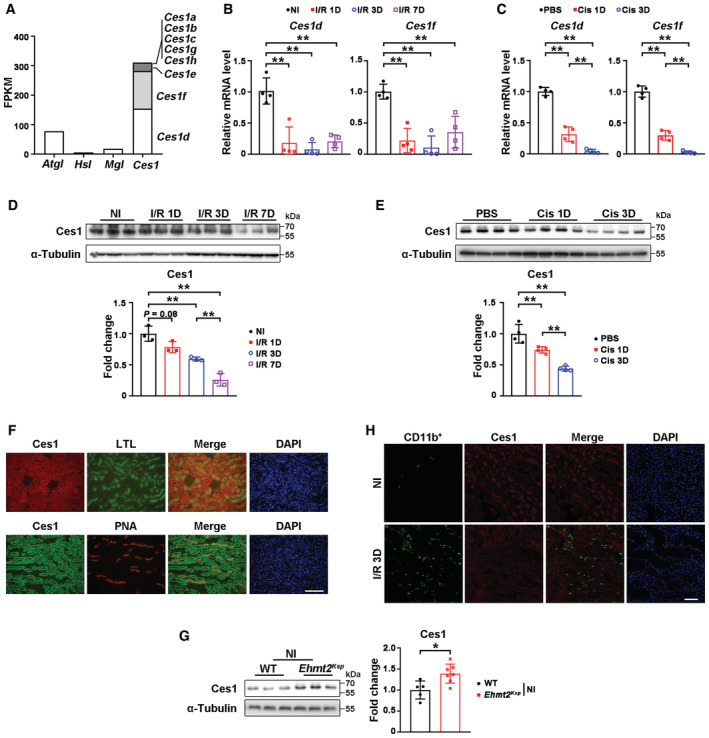
Lipid accumulation may result from an imbalance of fatty acid uptake, lipid synthesis, lipolysis, or fatty acid oxidation (FAO) (Ducasa et al, 2019). Altered transcriptional levels of genes involved in fatty acid uptake (Cd36, Slc27a1, and Slc27a2), lipid synthesis (Dgat1 and Dgat2), neutral TG hydrolases (Atgl, Hs1, and Mgl), lipolysis‐related carboxyl esterase (Ces) family and FAO (Cpt1a, Cpt1b, Acox1, Acadm, Ppara, and Pgc1a) were suggested by RNA‐sequencing, and most of them were further validated using qPCR (Fig 2B–E). The results indicated that downregulation of genes involved in lipolysis (neutral TG hydrolases and Ces family) and FAO may contribute to injury‐induced renal lipid accumulation. Although Atgl, Hs1, and Mgl are generally regarded as the main regulators of intracellular lipolysis (Zimmermann et al, 2009; Zechner, 2015), their abundance in the kidney is relatively low (Fig EV3A). On the other hand, the Ces1 family of triglyceride hydrolases promote lipolysis (Zechner et al, 2012) and consistent with a previous report (Lian et al, 2018), Ces1d and Ces1f are the most abundant members in the kidney (Fig EV3A). Upon I/R injury, significant upregulation of Ces1d/f and most FAO genes, as well as a consistently upregulated level of Cpt1a, a rate‐limiting enzyme for FAO (Schlaepfer & Joshi, 2020), were observed in Ehmt2 Ksp mice compared to WT mice (Fig 2C–F). Moreover, the mRNA levels of Ces1d/f were significantly higher in non‐injured kidneys of Ehmt2 Ksp mice compared to those of WT controls (Fig 2C), suggesting transcriptional regulation of Ces1 by G9a.
Figure EV3. Renal Ces1 location and alteration in mice with or without AKI injury.
-
AAbundance of lipolysis‐related genes in mouse kidney.
-
B, CmRNA levels of Ces1d and Ces1f in mice after I/R injury (B) or cisplatin‐induced AKI (C). Four mice per group.
-
D, ERepresentative immunoblots of Ces1 with quantitative results in the kidney from I/R injury‐induced AKI (n = 3 per group, D) or cisplatin‐induced AKI (n = 4 per group, E).
-
FRepresentative co‐immunofluorescent staining for Ces1 (red) with LTL (green) or Ces1 (green) with PNA (red) in non‐injured kidneys of WT mice. Scale bar = 100 μm.
-
GRepresentative immunoblots with quantitative results of renal Ces1 of WT (n = 5) and Ehmt2 Ksp (n = 7) mice.
-
HRepresentative co‐immunofluorescent staining for CD11b (green) with Ces1 (red) in non‐injured and I/R 3D injured kidneys. Scale bar = 50 μm.
Data information: FPKM, Fragments Per Kilobase of exon model per Million mapped fragments; NI, non‐injured. Cis, cisplatin; in (B–E and G), data were presented as means ± SD. Panels C, E were analyzed by 1‐way ANOVA followed by Tukey's test. Panel G was analyzed with 2‐tailed, unpaired Student's t test. Panels B, D were analyzed by Mann–Whitney U test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
A time‐course study suggested that renal I/R‐ and cisplatin‐induced AKI dramatically downregulated the mRNA levels of Ces1d/f, as well as the total Ces1 protein level in mouse kidneys (Fig EV3B–E). Since there is no commercial antibody for specific Ces1 variants available, for convenience, we used Ces1 to represent the protein level of different isoforms; Ces1d, which has a human homolog (Lian et al, 2018), was used to represent the transcriptional level of the Ces1 family.
Importantly, the renal tubular CES1 protein level was significantly lower in AKI patients compared with control subjects (Fig 2G). In mouse kidney, the majority of Ces1 was expressed in the renal tubular cells under normal conditions (Fig EV3F). Moreover, compared with WT kidneys, the Ces1 protein level was significantly higher in the kidneys of Ehmt2 Ksp mice, with or without I/R 3D injury (Figs 2H and EV3G). According to the Human Protein Atlas (www.proteinatlas.org), a high transcriptional level of CES1 has been reported in immune cells like myeloid dendritic cells and classical monocytes, while increased immune cell infiltration may contribute to the accelerated inflammation and pathological changes after renal I/R injury. However, few CD11b+ cells (a marker of myeloid cells (Rose et al, 2012)) were found under normal conditions and they were not co‐stained with Ces1; although dramatically increased CD11b+ cells were observed following injury, only a few were co‐stained with Ces1 (Fig EV3H), indicating that renal CES1 may exert its major function in the tubules. Moreover, significantly higher carboxylesterase activity, less Oil Red O staining, and lower renal TG level were found in Ehmt2 Ksp mice at I/R 3D (Fig 2I–K), suggesting that the increased carboxylesterase activity may result in reduced lipid accumulation.
Renal tubular specific G9a deletion alleviated cisplatin‐induced injury by preventing lipid accumulation
We next investigated whether Ehmt2 Ksp mice are resistant to nephrotoxic cisplatin induced AKI (Fig 3A). At day three after cisplatin injury, significantly reduced renal G9a and H3K9me2 levels were found in Ehmt2 Ksp mice (Fig 3B). Furthermore, injury‐induced tubular lesions, serum creatinine and BUN levels, and the upregulation of Kim1/Ngal found in injured WT mice, were significantly reduced in injured Ehmt2 Ksp mice (Fig 3C–E). Attenuated Oil Red O staining and renal TG level, accompanied by upregulated FAO‐related genes and Cpt1a protein level were found in injured Ehmt2 Ksp mice (Fig 3F–I). Moreover, the mRNA and protein levels of Ces1, as well as carboxylesterase activity, were significantly higher in cisplatin‐injured Ehmt2 Ksp mice (Fig 3J–L), together with reduced macrophage infiltration and downregulation of p‐Mlk1 (Fig 3M and N). These data support the critical role of G9a in nephrotoxic drug‐induced AKI.
Figure 3. Renal tubular specific G9a deletion alleviated cisplatin‐induced kidney injury by enhancing lipolysis and fatty acid oxidation.

-
AExperimental design for cisplatin‐induced AKI model.
-
BRepresentative immunoblots (left) with quantitative results (right) of G9a and H3K9me1/2 in cisplatin‐injured mouse kidneys.
-
CRepresentative H&E images (left) with quantitative pathological scores (right) of indicated groups. Scale bar = 100 μm.
-
DSerum creatine (Crea) and blood urea nitrogen (BUN) levels of indicated groups.
-
EmRNA levels of Kim1 and Ngal of indicated groups.
-
F, GRepresentative Oil Red O staining with quantification (F, scale bar = 100 μm), and renal TG (triglyceride) and TC (cholesterol) levels (G) of indicated groups.
-
HmRNA levels of fatty acid oxidation related genes of indicated groups.
-
IRepresentative Cpt1a immunostaining (up) with quantitative analysis (bottom) in the kidneys of indicated groups. Scale bar = 50 μm. IOD, integral optical density.
-
J–LmRNA level of Ces1 (J), representative immunoblots with quantitative results of Ces1 (K), and carboxylesterase enzymatic activity (L) of indicated groups.
-
MRepresentative immunostaining for F4/80 and quantitative results of indicated groups. Scale bar = 50 μm.
-
NRepresentative immunoblots (left) with quantitative results (right) of p‐Mlkl of indicated groups.
Data information: Six mice per group. Cis, cisplatin; in (B–N), data were presented as means ± SD. Panels B (G9a), C, D, E (Kim1), F, I, H (except Acadm), J–N were analyzed with 2‐tailed, unpaired Student's t test. Panels B (H3K9me2), E (Ngal), G and H (Acadm) were analyzed by Mann–Whitney U test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
Inhibiting G9a activity attenuated I/R‐ or cisplatin‐induced renal injury by preventing lipid accumulation
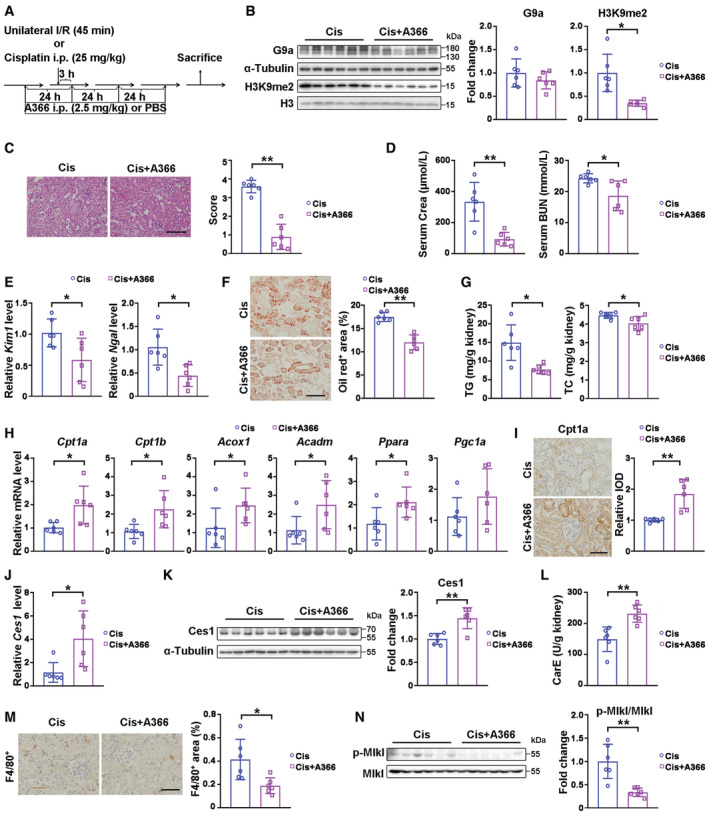
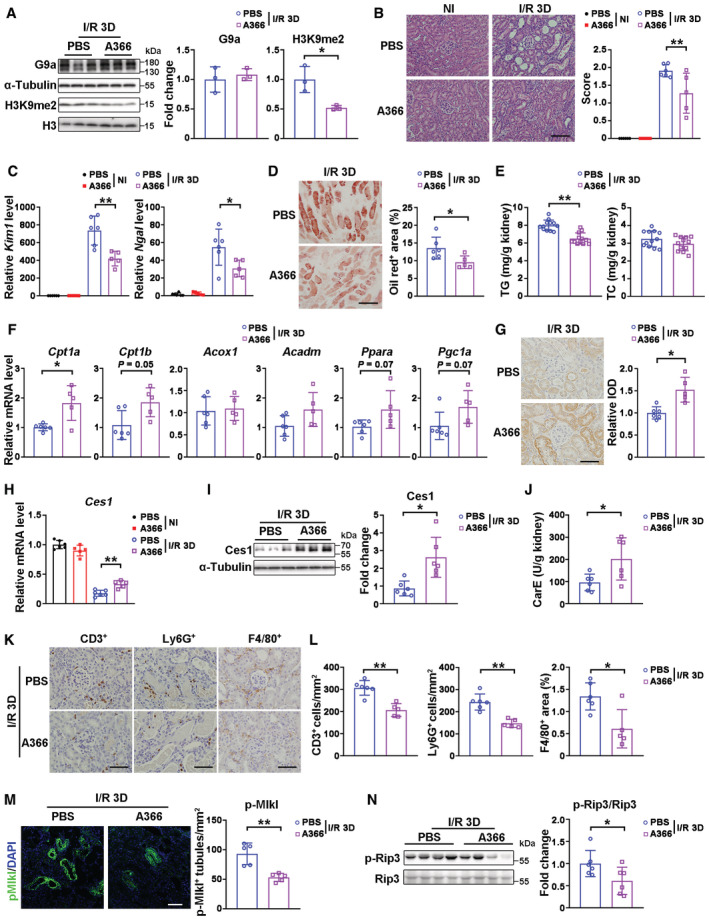
Next, we investigated whether the deleterious effects of G9a in AKI rely on its methyltransferase activity. A G9a methyltransferase inhibitor with high specificity and low toxicity (Sweis et al, 2014; Pappano et al, 2015), A366, was used following I/R (Fig EV4A). A366 treatment significantly reduced the level of H3K9me2 without affecting renal G9a protein level at I/R 3D (Fig 4A). A366 treatment significantly suppressed I/R‐induced tubular lesions, attenuated Kim1/Ngal levels, lipid staining and renal TG level, accompanied by upregulation of FAO‐related genes and Cpt1a protein level (Fig 4B–G). In addition, the mRNA and protein levels of Ces1, and carboxylesterase activity, were significantly increased after A366 treatment at I/R 3D (Fig 4H–J). Consistently, upon I/R injury, A366 treatment reduced the number of F4/80, Ly6G, or CD3 positive cells, and downregulated p‐Mlk1 and p‐Rip3 (Fig 4K–N), suggesting alleviation of immune cell infiltration and necroptosis. Likewise, beneficial effects were also found in A366‐treated mice after cisplatin‐induced AKI (Fig EV4B–N). These data suggest that G9a methyltransferase activity contributes to AKI mediated renal damage.
Figure EV4. A366 ameliorated cisplatin‐induced AKI through reducing lipid accumulation via Ces1 and enhancing fatty acid oxidation.
-
AExperimental design of A366 treatment in I/R‐ or cisplatin‐induced AKI mice model.
-
BRepresentative immunoblots with quantitative results of G9a and H3K9me1/2 in cisplatin‐injured kidneys from mice treated with vehicle or A366.
-
CRepresentative H&E staining with pathological scores of indicated groups. Scale bar = 100 μm.
-
DSerum creatinine (Crea) and blood urea nitrogen (BUN) levels of indicated groups.
-
EmRNA levels of Kim1 and Ngal of indicated groups.
-
F, GRepresentative Oil Red O staining with quantitative results (F, scale bar = 100 μm), and renal TG (triglyceride) and TC (cholesterol) levels (G) of indicated groups.
-
HmRNA levels of fatty acid oxidation related genes of indicated groups.
-
IRepresentative immunostaining for Cpt1a and quantitative results of indicated groups. Scale bar = 50 μm. IOD, integral optical density.
-
J–LmRNA level of Ces1 (J), representative immunoblots with quantitative results of Ces1, and carboxylesterase enzymatic activity (L) of indicated groups.
-
MRepresentative immunostaining for F4/80 and quantitative results of indicated groups. Scale bar = 50 μm.
-
NRepresentative immunoblots with quantitative results of p‐Mlkl for indicated groups.
Data information: Six mice per group. Cis, cisplatin, in (B–N), data were presented as means ± SD. Panels B–G, H (except Acox1 and Acadm), K–M were analyzed with 2‐tailed, unpaired Student's t test. Panels H (Acox1 and Acadm), I, and J were analyzed by Mann–Whitney U test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
Figure 4. A366 ameliorated renal I/R injury through reducing lipid accumulation via Ces1 and enhancing fatty acid oxidation.
-
ARepresentative immunoblots (left) with quantitative results (right) of G9a and H3K9me1/2 in injured kidneys from mice treated with vehicle (n = 3) or A366 (n = 3) at I/R 3D.
-
BRepresentative H&E images (left) with pathological scores (right) of PBS + NI (n = 6), A366 + NI (n = 5), PBS + I/R 3D (n = 6) and A366 + I/R 3D (n = 5) groups. Scale bar = 100 μm.
-
CmRNA levels of Kim1 and Ngal of PBS + NI (n = 6), A366 + NI (n = 5), PBS + I/R 3D (n = 6) and A366 + I/R 3D (n = 5) groups.
-
D, ERepresentative Oil Red O staining with quantification (D, scale bar = 100 μm), and renal TG (triglyceride) and TC (cholesterol) levels (E) of PBS + I/R 3D (n = 12) and A366 + I/R 3D (n = 12) groups.
-
FmRNA levels of fatty acid oxidation related genes of PBS + I/R 3D (n = 6) and A366 + I/R 3D (n = 5) groups.
-
GRepresentative Cpt1a immunostaining with quantitative analysis in the kidneys of PBS + I/R 3D (n = 6) and A366 + I/R 3D (n = 5) groups. Scale bar = 50 μm. IOD, integral optical density.
-
HmRNA level of Ces1 of PBS + NI (n = 6), A366 + NI (n = 5), PBS + I/R 3D (n = 6) and A366 + I/R 3D (n = 5) groups.
-
I, JRepresentative immunoblots (I, left) with quantitative results (I, right) of Ces1, and carboxylesterase enzymatic activity (J) of PBS + I/R 3D (n = 6) and A366 + I/R 3D (n = 6) groups.
-
K, LRepresentative immunostaining for CD3, Ly6G, and F4/80 (K) and quantitative results (L) of PBS + I/R 3D (n = 6) and A366 + I/R 3D (n = 5) groups. Scale bar = 50 μm.
-
MRepresentative immunofluorescence staining for renal p‐Mlkl (left) with score analysis (right) of PBS + I/R 3D (n = 5) and A366 + I/R 3D (n = 5) groups.
-
NRepresentative immunoblots (left) with quantitative results (right) of p‐Rip3 for PBS + I/R 3D (n = 6) and A366 + I/R 3D (n = 6) groups. Scale bar = 50 μm.
Data information: NI, non‐injured; in (A–J and L–N), data were presented as means ± SD. Panels A, D, E (TC), F (except Cpt1b), G, I, J and L–N were analyzed with 2‐tailed, unpaired Student's t test. Panels B, C and H were analyzed by 1‐way ANOVA followed by Tukey's test. Panels E (TG), and F (Cpt1b) were analyzed by Mann–Whitney U test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
G9a affects lipid accumulation via Ces1 transcriptional regulation
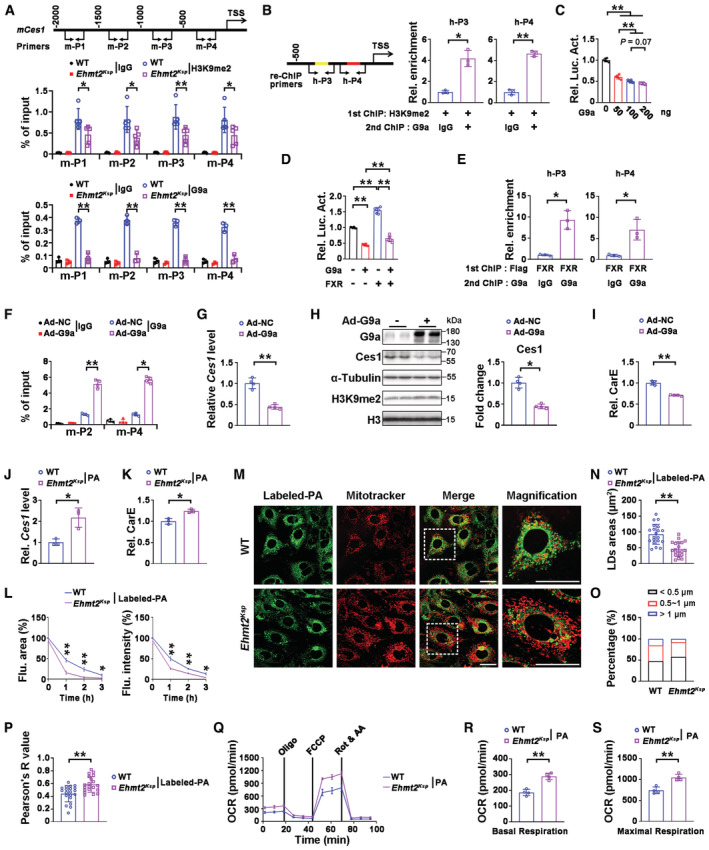
To investigate how G9a regulates Ces1 expression, chromatin immunoprecipitation (ChIP) assays were performed. H3K9me2 and G9a enrichment on the Ces1 promoter were demonstrated in mouse kidney tissue, which were significantly decreased by G9a knockout (Fig 5A). H3K9me2 enrichment on the CES1 promoter was also demonstrated in HK‐2 cells, which was significantly decreased by G9a knockdown or A366 treatment (Appendix Fig S4A–D); similarly, decreased H3K9me2 enrichment on the Ces1 promoter was found in the kidneys of A366 treated mice (Appendix Fig S4E). Furthermore, a ChIP and re‐ChIP assay in HEK293 cells indicated that both H3K9me2 and G9a bound at the same sites (within the −500 to TSS (transcription state site)) on the CES1 promoter (Fig 5B). Taken together, these results indicate that Ces1 expression is transcriptionally regulated by the methyltransferase activity of G9a.
Figure 5. G9a negatively regulated Ces1, affected lipolysis and fatty acid oxidation.
-
AEnrichment of H3K9me2 (middle) or G9a (bottom) at four different regions (m‐P1 to m‐P4, top panel) of the Ces1 promoter in the kidneys from WT (n = 5 for H3K9me2 ChIP, n = 3 for G9a ChIP) and Ehmt2 Ksp (n = 5 for H3K9me2 ChIP, n = 3 for G9a ChIP) mice.
-
BChIP and re‐ChIP assays indicate that G9a and H3K9me2 bind to the same two regions on the Ces1 promoter (h‐P3 and h‐P4 are within the −500 to TSS region, locations shown in the left panel) in HEK293 cells. TSS, transcription start site; n = 3 cultures.
-
CLuciferase assays suggest G9a dosage‐dependently down‐regulates Ces1 promoter in HEK293 cells. n = 3 cultures.
-
DLuciferase assays suggest that G9a blocks the upregulation effects of FXR on the Ces1 promoter in HEK293 cells; n = 3 cultures.
-
EChIP and re‐ChIP assays indicate that G9a and FXR bind to the same two regions on the Ces1 promoter (locations of h‐P3 and h‐P4 are identical to that shown in B; FXR binding sites are indicated as red and yellow within h‐P3 and h‐P4). n = 3 cultures.
-
FEnrichment of G9a at the promoter of Ces1 in Ad‐NC or Ad‐G9a transfected mPTECs (locations of m‐P2 and m‐P4 are identical to that shown in A). n = 4 cultures.
-
G–ImRNA (G) and protein (H) levels of Ces1, as well as the carboxylesterase enzymatic activity (I) in Ad‐NC or Ad‐G9a transfected mPTECs. n = 4 cultures.
-
J, KCes1 mRNA level (J) and carboxylesterase enzymatic activity (K) in mPTECs isolated from WT and Ehmt2 Ksp mice under PA (palmitic acid) treatment. n = 3 cultures.
-
LQuantitative results of fluorescent (Flu.) area (left) and intensity (right) for labeled‐PA treated mPTECs isolated from WT and Ehmt2 Ksp mice at the indicated time. n = 3 cultures.
-
M–PRepresentative images (M) with quantitative results of lipid droplet areas (N) and percentage of different lipid droplet size (O), as well as colocalization of lipid droplet and mitochondria assessed by the Pearson analysis (P), in labeled‐PA treated mPTECs isolated from WT and Ehmt2 Ksp mice at 1 h after treatment. The results were obtained from three biological replicates; a total of 20 cells were imaged in each group for quantification of lipid droplet area (N, O) and colocalization analysis (P). Scale bars = 25 μm.
-
Q–SSeahorse studies (Q) with quantitative results of basal (R) and maximal (S) respiration in PA‐treated mPTECs isolated from WT and Ehmt2 Ksp mice. OCR, oxygen consumption rate; n = 4 cultures.
Data information: Rel., relative. In (A–L and N–S), data were presented as means ± SD. Panels A (G9a ChIP), C, D and F (ChIP on m‐P2 site) were analyzed by 1‐way ANOVA followed by Tukey's test. Panels B, E, G–K, L, N–P, R and S were analyzed with 2‐tailed, unpaired Student's t test. Panels A (H3K9me2 ChIP) and F (ChIP on m‐P4 site) were analyzed by Mann–Whitney U test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
Luciferase assays further demonstrated that G9a inhibited Ces1 transcription in a dose dependent manner in HEK293 cells (Fig 5C). FXR has been reported to transcriptionally activate Ces1 (Xu et al, 2014). Luciferase assays demonstrated that G9a blocked FXR‐mediated transcriptional activation of Ces1 in HEK293 cells (Fig 5D). JASPAR analysis indicated two FXR‐binding sites within the −500 to TSS of the CES1 promoter, and a ChIP and re‐ChIP assay further indicated that both FXR and G9a bound at these two regions (Fig 5E). Overexpression of G9a in mouse primary tubular epithelial cells (mPTECs) consistently increased G9a enrichment on the Ces1 promoter, downregulated Ces1 transcription, and decreased the protein level and enzymatic activity of Ces1 (Fig 5F–I). Consistently, significantly higher Ces1 mRNA level and carboxylesterase activity, along with upregulated FAO genes were found in palmitic acid (PA) treated mPTECs that were isolated from the Ehmt2 Ksp mice (Fig 5J and K, Appendix Fig S5A). To address whether G9a deficiency induced Ces1 upregulation induces functional improvement of FAO, the degradation rate of labeled PA (a fluorescent PA analog) was measured. Faster PA disappearance rates (measured by both fluorescent area and intensity) were found in mPTECs isolated from Ehmt2 Ksp mice (Fig 5L, and Appendix Fig S5B). Furthermore, smaller lipid droplets, as well as increased colocalization between mitochondria and lipid droplets indicating better lipid utilization (Minami et al, 2017), were found in mPTECs isolated from Ehmt2 Ksp mice at early stages after labeled PA treatment (Fig 5M–P). Meanwhile, seahorse studies demonstrated significantly higher basal and maximal respiration in PA‐treated mPTECs isolated from Ehmt2 Ksp mice (Fig 5Q–S), indicating upregulation of cellular bioenergetics as the result of elevated FAO (Miguel et al, 2021).
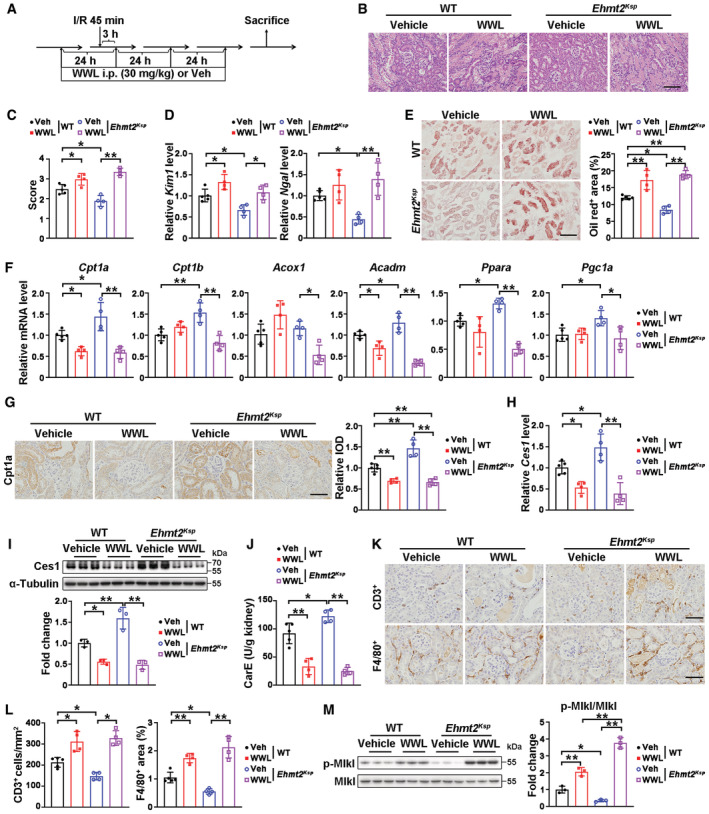
Inhibiting Ces1 activity exacerbated I/R injury in WT mice and blocked the beneficial effects observed in Ehmt2 Ksp KO mice
To confirm that the beneficial effects on AKI mediated renal damage observed in Ehmt2 Ksp mice resulted from Ces1, we treated I/R‐injured WT and Ehmt2 Ksp mice with a Ces1 inhibitor WWL229 (WWL; Fig 6A), which showed dosage‐dependent inhibition of Ces1 activity without affecting its protein level in mPTECs (Appendix Fig S6A and B). WWL significantly exacerbated I/R‐induced tubular lesions and increased Kim1 in WT mice (Fig 6B–D). In I/R‐injured Ehmt2 Ksp mice, the reduced injury indicated by pathological score and tubular injury markers, was prevented by treatment with WWL (Fig 6B–D). WWL treatment also enhanced lipid staining, downregulated mRNA and protein levels of Cpt1a in injured WT mice, while it reversed the reduction in lipid accumulation, prevented transcriptional upregulation of FAO‐related genes and upregulated Cpt1a protein level in injured Ehmt2 Ksp KO mice (Fig 6E–G). Moreover, the mRNA and protein levels of Ces1, and carboxylesterase activity, were significantly decreased in WWL‐treated injured WT mice; meanwhile, WWL treatment abolished the upregulation of CES1 activity in injured Ehmt2 Ksp KO mice (Fig 6H–J). WWL treatment consistently increased the number of F4/80 or CD3 positive cells, and upregulated p‐Mlk1 in injured WT mice; while it prevented the decrease of infiltrating immune cells and necroptosis in injured Ehmt2 Ksp KO mice (Fig 6K–M).
Figure 6. Ces1 inhibitor exacerbated I/R injury in WT mice and blocked the beneficial effects observed in Ehmt2 Ksp KO mice.
-
AExperimental design for WWL229 treatment in a renal I/R‐induced model. All the results shown in this figure were obtained under I/R 3D injury.
-
B, CRepresentative H&E images (B) with pathological scores (C) of WT + Veh (n = 5), WT + WWL (n = 4), Ehmt2 Ksp + Veh (n = 4) and Ehmt2 Ksp + WWL (n = 4) groups. Scale bar = 100 μm.
-
DmRNA levels of Kim1 and Ngal of WT + Veh (n = 5), WT + WWL (n = 4), Ehmt2 Ksp + Veh (n = 4) and Ehmt2 Ksp + WWL (n = 4) groups.
-
ERepresentative Oil Red O staining with quantification of WT + Veh (n = 5), WT + WWL (n = 4), Ehmt2 Ksp + Veh (n = 4) and Ehmt2 Ksp + WWL (n = 4) groups. Scale bar = 100 μm.
-
FmRNA levels of fatty acid oxidation related genes of WT + Veh (n = 5), WT + WWL (n = 4), Ehmt2 Ksp + Veh (n = 4) and Ehmt2 Ksp + WWL (n = 4) groups.
-
GRepresentative Cpt1a immunostaining with quantitative analysis in the kidneys of WT + Veh (n = 5), WT + WWL (n = 4), Ehmt2 Ksp + Veh (n = 4) and Ehmt2 Ksp + WWL (n = 4) groups. Scale bar = 50 μm. IOD, integral optical density.
-
H–JmRNA level of Ces1 of WT + Veh (n = 5), WT + WWL (n = 4), Ehmt2 Ksp + Veh (n = 4) and Ehmt2 Ksp + WWL (n = 4) groups (H); representative immunoblots with quantitative results of Ces1 of WT + Veh (n = 3), WT + WWL (n = 3), Ehmt2 Ksp + Veh (n = 3) and Ehmt2 Ksp + WWL (n = 3) groups (I); and carboxylesterase enzymatic activity (J) of WT + Veh (n = 5), WT + WWL (n = 4), Ehmt2 Ksp + Veh (n = 4) and Ehmt2 Ksp + WWL (n = 4) groups.
-
K, LRepresentative immunostaining for CD3 and F4/80 (K) and quantitative results (L) of WT + Veh (n = 5), WT + WWL (n = 4), Ehmt2 Ksp + Veh (n = 4) and Ehmt2 Ksp + WWL (n = 4) groups. Scale bar = 50 μm.
-
MRepresentative immunoblots (left) with quantitative results (right) of p‐Mlkl for WT + Veh (n = 3), WT + WWL (n = 3), Ehmt2 Ksp + Veh (n = 3) and Ehmt2 Ksp + WWL (n = 3) groups.
Data information: In (C–J, L and M), data were presented as means ± SD. Panels C–J, L (F4/80+) and M were analyzed by 1‐way ANOVA followed by Tukey's test. Pane L (CD3+) was analyzed by Mann–Whitney U test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
FXR agonist GW4064 upregulated Ces1 and alleviated renal I/R injury
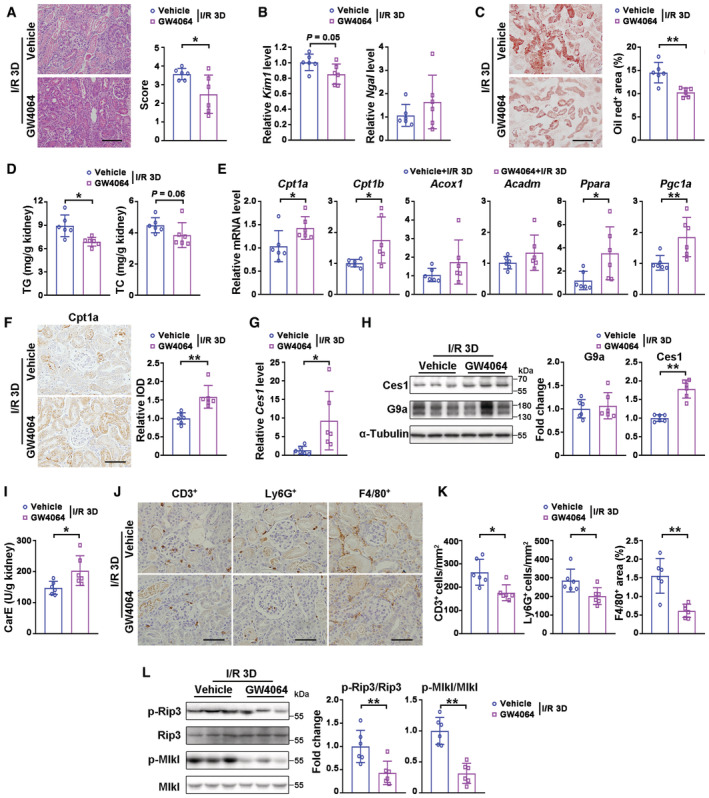
We reasoned that the protective effects against I/R‐injury of Ehmt2 Ksp mice maybe due to increased lipolysis from the higher Ces1 level that prevents lipid accumulation following injury. We explored this possibility by treating cells with an FXR agonist, GW4064 that has been reported to upregulate Ces1 in mPTECs (Sugahara et al, 2019). GW4064 significantly increased Ces1 transcription and attenuated G9a‐mediated inhibition of Ces1 as shown by luciferase assays in HEK293 cells (Appendix Fig S7A). Moreover, at I/R 3D, as compared with the injured controls, GW4064 treatment significantly increased the mRNA and protein levels of FXR, suppressed I/R injury‐induced pathological lesions, downregulated Kim1, as well as suppressed injury‐induced renal lipid accumulation and TG level, without affecting serum TG/TC levels (Fig 7A–D, Appendix Fig S7B–D). Enhanced transcription of FAO‐related genes and increased Cpt1a protein level (Fig 7E and F), as well as increased transcription, protein and activity levels of Ces1 (Fig 7G–I), were observed in injured kidneys treated with GW4064. In addition, GW4064 treatment reduced the number of infiltrating F4/80, Ly6G, or CD3 positive cells, and reduced p‐Rip3 and p‐Mlkl following renal I/R 3D injury, suggesting reduced immune cell infiltration and necroptosis (Fig 7J–L).
Figure 7. FXR agonist GW4064 ameliorated renal I/R injury through reducing lipid accumulation.
-
ARepresentative H&E staining (left) and pathological scores (right) of injured kidney from mice treated with vehicle (n = 6) or GW4064 (n = 6, 30 mg/kg, i.p). Scale bar = 100 μm.
-
BmRNA levels of Kim1 and Ngal of indicated groups.
-
C, DRepresentative Oil Red O staining with quantification (C, scale bar = 100 μm), renal triglyceride and cholesterol levels (D) of indicated groups.
-
EmRNA levels of fatty acid oxidation related genes of indicated groups.
-
FRepresentative Cpt1a immunostaining with quantitative analysis in the kidneys of indicated groups. Scale bar = 50 μm. IOD, integral optical density.
-
G–ImRNA levels of Ces1 (G), representative immunoblots (H, left) with quantitative results (H, right) of Ces1 and G9a, and carboxylesterase enzymatic activity (I) of indicated groups.
-
J, KRepresentative immunostaining for CD3, Ly6G, and F4/80 (J) and quantitative results (K) of indicated groups. Scale bar = 50 μm.
-
LRepresentative immunoblots (left) with quantitative results (right) of p‐Rip3 and p‐Mlkl for indicated groups.
Data information: Six mice per group. In (A–I, K and L), data were presented as means ± SD. Panels A–C, D (TG), E (except Ppara and Pgc1a), G–I, K (F4/80+) and L were analyzed with 2‐tailed, unpaired Student's t test. Panels D (TC), E (Ppara and Pgc1a), F, K (CD3+ and Ly6G+) were analyzed by Mann–Whitney U test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
Atorvastatin treatment reduced renal I/R injury by downregulating renal lipid accumulation and activating Ces1
Hyperlipidemia and hypercholesterolemia have been suggested to be risk factors for AKI (Yang et al, 2012; Akgullu et al, 2015). Inspired by the renal‐protective effects of GW4064 and A366, which decrease lipid accumulation in I/R‐injured kidneys, we hypothesized that clinically effective lipid‐lowering medications such as atorvastatin (ATO) may protect against AKI by affecting post‐injury lipid accumulation. As an orally active inhibitor of HMG‐CoA reductase, ATO has also been reported to reduce renal TG level under high‐fat‐diet stress (Pengrattanachot et al, 2020). ATO pre‐treatment reduced serum TG level, I/R‐induced pathological lesions and Kim1/Ngal levels, as well as significantly suppressing injury‐induced renal lipid accumulation and TG level at I/R 3D (Fig EV5A–F). ATO pre‐treatment consistently promoted the transcription of genes involved in FAO (Fig EV5G). Interestingly, increased mRNA and protein levels of Ces1, as well as carboxylesterase activity, were also found in ATO‐pretreated injured kidneys (Fig EV5H–J). Moreover, ATO pre‐treatment reduced the number of infiltrating F4/80, Ly6G or CD3 positive cells, and decreased p‐Rip3 and p‐Mlkl levels upon I/R 3D injury (Fig EV5K–M).
Figure EV5. Atorvastatin pre‐treatment ameliorated renal I/R injury by reducing lipid accumulation via Ces1 and enhancing fatty acid oxidation.
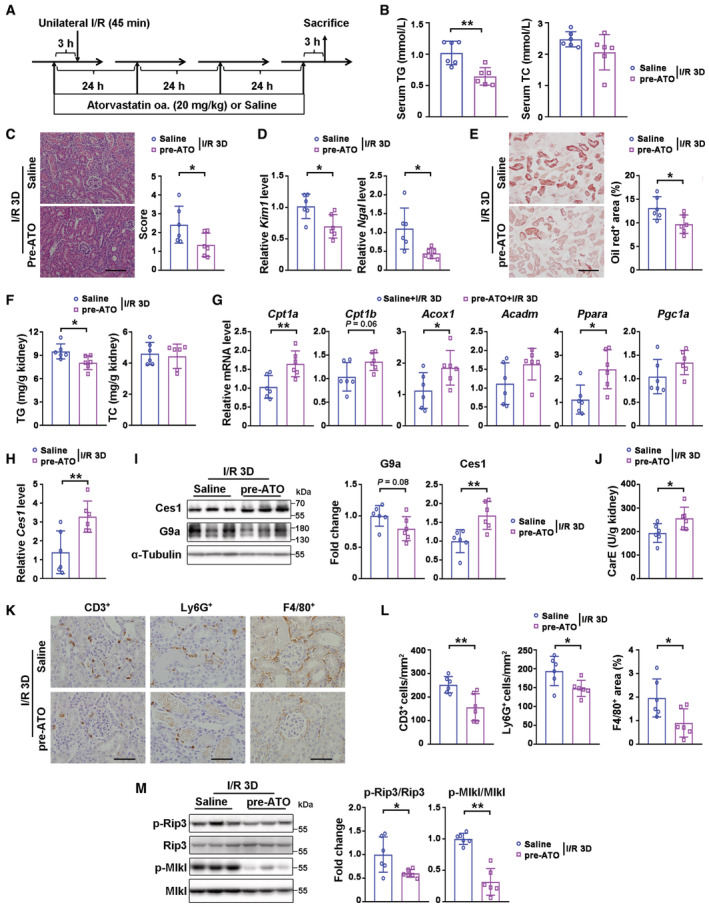
-
AExperimental design for atorvastatin prevention treatment on renal I/R injury.
-
BSerum TG (triglyceride) and TC (cholesterol) levels of indicated groups.
-
CRepresentative H&E staining and pathological scores of I/R‐injured kidneys from mice pre‐treated with vehicle or atorvastatin (pre‐ATO, 20 mg/kg, i.g.). Scale bar = 100 μm.
-
DmRNA levels of Kim1 and Ngal of vehicle and pre‐ATO.
-
E, FRepresentative Oil Red O staining with quantitative results (E, scale bar = 100 μm), renal TG (triglyceride) and TC (cholesterol) levels (F) of indicated groups.
-
GmRNA levels of fatty acid oxidation related genes of indicated groups.
-
H–JmRNA levels of Ces1 (H), representative immunoblots with quantitative results of G9a and Ces1 (I), and carboxylesterase enzymatic activity (J) of indicated groups.
-
K, LRepresentative immunostaining for CD3, Ly6G, and F4/80 (K) and quantitative results (L) of indicated groups. Scale bar = 50 μm.
-
MRepresentative immunoblots with quantitative results of p‐Rip3 and p‐Mlkl of indicated groups.
Data information: Six mice per group. In (B–J, L and M), data were presented as means ± SD. Panels C–E, F (TG), G (except Acadm), H–J, L and M were analyzed with 2‐tailed, unpaired Student's t test. Panels B, F (TC) and G (Acadm) were analyzed by Mann–Whitney U test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
To further investigate whether ATO is an effective intervention against renal I/R, we treated mice with ATO by gavage at 6 or 14 h after renal I/R injury (Appendix Fig S8A and B). Compared to the vehicle control group, ATO intervention started at 6 h (ATO‐6 h) but not 14 h (ATO‐14 h) after the I/R injury significantly reduced AKI‐induced pathological lesions, decreased Kim1/Ngal levels, suppressed injury‐induced renal lipid accumulation and TG level, and reduced serum TG/TC levels (Fig 8A–D, Appendix Fig S8C). Consistent with pre‐treatment, upon renal I/R 3D injury, ATO‐6 h intervention promoted the transcription of FAO‐related genes and increased the protein level of Cpt1a (Fig 8E and F), increased the mRNA and protein levels of Ces1, and carboxylesterase activity (Fig 8G–I), together with reducing the number of infiltrating F4/80, Ly6G or CD3 positive cells, and decreased p‐Rip3 and p‐Mlkl levels (Fig 8J–L). These results support the utility of ATO in AKI prevention/intervention, and revealed a new working mechanism of ATO action associated with facilitating Ces1 related renal lipid metabolism.
Figure 8. Atorvastatin intervention ameliorated renal I/R injury by reducing lipid accumulation via Ces1 and enhancing fatty acid oxidation.
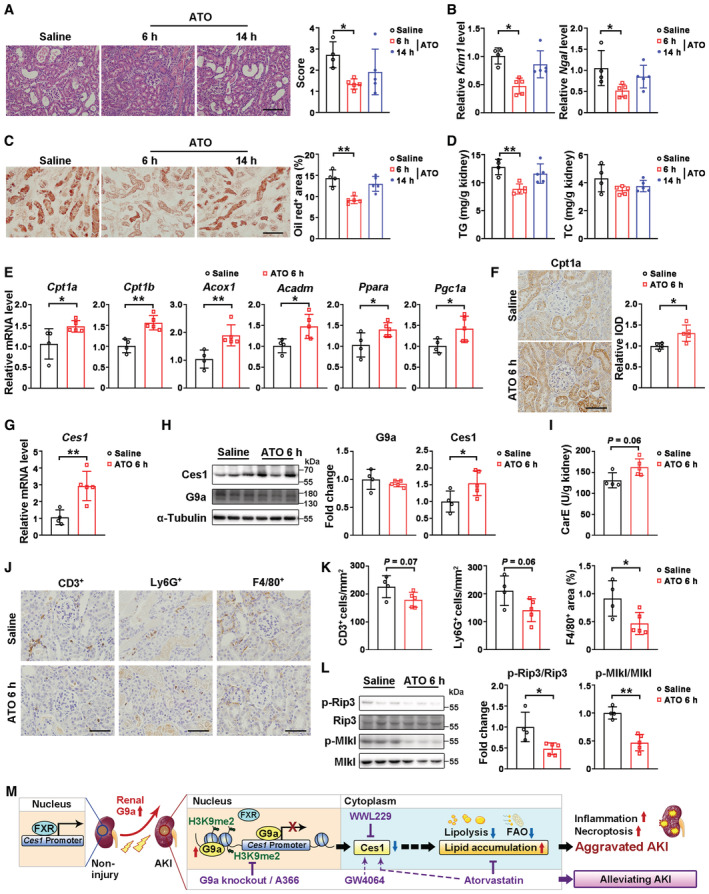
-
ARepresentative H&E staining (left) and pathological scores (right) of injured kidneys from mice received intervention treatment of saline (n = 4) or atorvastatin (20 mg/kg, i.g., first dose at 6‐h after ischemia, n = 5, or 14‐h after ischemia, n = 5). Scale bar = 100 μm.
-
BmRNA levels of Kim1 and Ngal of saline (n = 4), 6 h‐ATO (n = 5) and 14 h‐ATO (n = 5) groups.
-
C, DRepresentative Oil Red O staining with quantification (C, scale bar = 100 μm), renal triglyceride and cholesterol levels (D) of saline (n = 4), 6 h‐ATO (n = 5) and 14 h‐ATO (n = 5) groups.
-
EmRNA levels of fatty acid oxidation related genes of saline (n = 4) and 6 h‐ATO (n = 5).
-
FRepresentative Cpt1a immunostaining with quantitative analysis in the kidneys of saline (n = 4) and 6 h‐ATO (n = 5) groups. Scale bar = 50 μm. IOD, integral optical density.
-
G–ImRNA levels of Ces1 (G), representative immunoblots (H, left) with quantitative results (H, right) of Ces1 and G9a, and carboxylesterase enzymatic activity (I) of saline (n = 4) and 6 h‐ATO (n = 5).
-
J, KRepresentative immunostaining for CD3, Ly6G, and F4/80 (J) and quantitative results (K) of indicated groups. Scale bar = 50 μm.
-
LRepresentative immunoblots (left) with quantitative results (right) of p‐Rip3 and p‐Mlkl for saline (n = 4) and 6 h‐ATO (n = 5).
-
MA schematic representation of the mechanistic model.
Data information: In (A–I, K and L), data were presented as means ± SD. Panels A, B (Ngal), C, and D were analyzed by 1‐way ANOVA followed by Tukey's test. Panels E (except Cpt1a and Acox1), F–H, K and L were analyzed with 2‐tailed, unpaired Student's t test. Panels B (Kim1), E (Cpt1a and Acox1) and I were analyzed by Mann–Whitney U test. *P < 0.05; **P < 0.01.
Source data are available online for this figure.
Discussion
To investigate the role of G9a in TECs associated with AKI, we developed a renal tubular specific G9a knockout mouse model (whole body G9a knockout is embryonic lethal; Tachibana et al, 2002). Our data indicated that significantly increased G9a in renal tubules of injured mice and AKI patients contributes to AKI‐induced necroptosis, inflammation and renal lipid accumulation. The knockout of renal tubular G9a increased lipolysis by upregulating Ces1, a family of primary renal lipolytic enzymes, thus preventing renal lipid toxicity and providing a fuel source for FAO in injured mice. Mechanistically, G9a exerts its function both through H3K9me2 and binding competitively with FXR to the Ces1 promoter to suppress Ces1 transcription (Fig 8M). Together, these results demonstrated that a lipid disorder is regulated by renal G9a/FXR‐Ces1 axis in TECs and is a key contributor to AKI mediated kidney damage.
Previous studies have reported increased G9a protein level in UUO‐injured and at 1 day after I/R‐injury mouse kidneys, as well as in the renal samples of IgA nephropathic patients (Irifuku et al, 2016; Liu et al, 2021). We further demonstrated that the increased renal G9a protein level was not only found during the acute stage of renal I/R (I/R 3D), but also at I/R 7D (the stage at which AKI starts to progress to chronic kideny disease [CKD]; Kellum et al, 2021). This result that in addition to its effects on AKI, G9a may also play a role in CKD, a subject awaits further investigation. Moreover, we also found increased G9a protein level in cisplatin‐injured kidneys. Since cisplatin is a common chemotherapeutic medicine in cancer treatment, the clincal relevance of this finding warrants future study. BIX01294, a G9a inhibitor, has been reported to alleviate renal fibrosis in UUO mouse by regulating klotho (Irifuku et al, 2016), and to attenuate renal I/R injury by reducing the oxidative response through Sirt1 or by reducing inflammation (Liu et al, 2021; Sung et al, 2022). In this study, another G9a inhibitor, A366, with lower IC50, higher specificity and lower toxicity than BIX01294 (Sweis et al, 2014; Pappano et al, 2015), exhibited renal protection effects following both I/R and cisplatin injury (Figs 4 and EV4). To avoid the general issues regarding the specificity and off target effects of inhibitors, we studied and validated our findings in vivo on renal I/R‐ or cisplatin‐induced AKI by using renal tubular specific G9a KO mice. We also noticed that in an in vitro study using a renal tubular epithelial cell model, the inhibition of G9a enhanced ER stress by upregulating ATF4 and XBP1 (Diaz‐Bulnes et al, 2022), while sustained activation of ER stress is associated with aggravated renal damage. Even though the impact of renal G9a on ER stress awaits further in vivo validation, combined with our findings, all of these data indicate that G9a plays complex and systemic physiological and pathological roles.
Moreover, G9a has recently been found to be a critical enhancer of neuronal ferroptosis. G9a activity represses anti‐ferroptotic genes, diminishes intracellular glutathione levels, and triggers an iron‐dependent programmed cell death pathway in an autoimmune encephalomyelitis mouse model (Rothammer et al, 2022). In the present study, no ferroptosis‐related pathway was enriched by knockout of G9a under I/R‐injury conditions using RNA‐sequence analysis. Similar protein levels of ferroptosis markers were found in WT and Ehmt2 Ksp mice, with or without renal I/R injury (Appendix Fig S2). These findings suggested that G9a knockdown/inhibition may involve different regulatory mechanisms in different tissues or models.
The association between lipid imbalance and AKI has been suggested in previous studies (Zager et al, 2005; Portilla et al, 2006; Bobulescu, 2010), however, the underlying mechanism and whether the imbalance of renal lipid metabolism affects AKI progression had not been addressed. Here, we report a novel lipid regulatory mechanism mediated by the G9a/FXR‐Ces1 axis in AKI. More importantly, our data demonstrated that reducing lipid accumulation upon AKI attenuates renal damage. Pharmacological inhibition of G9a, or upregulation of Ces1 by two different lipid‐lowering drugs, all significantly alleviated renal lipid accumulation and the resulting injury (Figs 4, EV4, 6, 7, EV5, 8, and EV5) providing new avenues for AKI treatment. Considering the ubiquitous expression of G9a throughout the body, in contrast to the relatively enriched Ces1 expression in the kidney, further investigation is warranted to determine whether Ces1 is a more suitable therapeutic target than G9a for treatment of AKI. Thus, future studies using tubular‐specific Ces1 overexpression/knockout animals to study the impact of Ces1 on renal lipid accumulation and its association with G9a will facilitate screening for novel Ces1 agonists in AKI setting.
Regarded as a target for cholestatic and metabolic liver diseases, FXR has been reported to be involved in multiple metabolic pathways including bile acid, cholesterol and lipid metabolism, and shows anti‐inflammatory, anti‐fibrotic, and anti‐apoptotic effects (Sun et al, 2021; Panzitt et al, 2022); however, its role in kidney disease remains elusive. Studies using whole body FXR knockout mice have shown both AKI‐promoting and protective effects of FXR (Kim et al, 2021; Xu et al, 2021), which may be due to the different mouse backgrounds used. However, in a renal tubular specific FXR knockout mouse, an AKI‐protective role of FXR has been suggested (Xu et al, 2022). In our studies, we consistently demonstrated that activating FXR with the agonist GW4064 had renal‐protective effects in AKI (Fig 7).
A number of statins are available in the clinic, which exhibit differing profiles, such as potency of lowering total cholesterol, triglycerides, LDL‐cholesterol, and safety in patients with kidney disease (Athyros et al, 2010). ATO is currently the most widely used and cost‐effective statin in clinical practice, and the drug of choice in patients with kidney disease (Athyros et al, 2010). Compared to other statins such as simvastatin, pravastatin, and lovastatin, ATO shows a greater capacity for reducing TG level (Meor Anuar Shuhaili et al, 2017). A clinical study recruiting more than 200,000 patients demonstrated that statins (including ATO) protected against renal complications after major elective surgery and reduced perioperative mortality (Molnar et al, 2011). There are a variety of reports about the different outcomes of ATO use at different dosages in clinic practice for treating AKI. For example, for contrast‐induced AKI, 80 mg/day ATO effectively reduced the risk (Quintavalle et al, 2012), while 40 mg/day of ATO treatment increased the risk (Bei et al, 2017). The difference may be due to the different cohorts and clinical designs in these studies, which await future study with well‐designed cohorts.
Although ATO is well recognized as a lipid lowering drug, its impact on renal diseases in rodent studies have mostly focused on its anti‐oxidative and anti‐inflammatory actions, with limited data regarding its lipid regulatory property (Cusumano et al, 2015; Wang et al, 2017; Hassan et al, 2019). In our study, an ATO prevention/intervention strategy regulated renal lipid metabolism and Ces1 in AKI injured mice, indicating that its capacity to regulate renal lipid profiles may contribute to the renal protection effects. Importantly, the renal‐protective effects of ATO are also associated with Ces1 upregulation, which has not been reported previously.
In conclusion, our findings revealed that the G9a/FXR‐Ces1 axis is an important contributor to AKI mediated kidney damage by regulating renal lipid level opening new therapeutic opportunities. Further clinical evaluation is warranted.
Materials and Methods
Animals
Ehmt2 flox/flox mice on a C57BL/6 background were generated by the Model Animal Research Center of Nanjing University as previously reported (Zhang et al, 2020a,b). Ksp‐Cre mice were a kind gift from Dr. Congyi Wang (Tongji Medical College, China). Ehmt2 flox/flox mice were crossed with Ksp‐Cre mice to generate renal tubular specific G9a knockout mice. Genotyping was performed (primers listed in Appendix Table S1) with Ehmt2 flox/flox ; Cre+ (Ehmt2 Ksp ) as knockout mice, while littermates identified as Ehmt2 flox/flox ; Cre− or Cre+ (Ksp‐Cre) were used as the wildtype controls (WT). For treatments, C57BL/6 mice (8‐week‐old) weighing 25 ± 2 g, were obtained from Hubei Center for Disease Control and Prevention. Males were used in this study, and all mice were randomly grouped without exclusion. Mice were handled according to the Guidelines of the China Animal Welfare Legislation, as approved by the Committee on Ethics in the Care and Use of Laboratory Animals, College of Life Sciences, Wuhan University.
I/R‐ and cisplatin‐induced AKI models and treatments
Ischemic AKI was induced using a renal ischemia–reperfusion (I/R) injury model as previously described (Chen et al, 2017; Wang et al, 2020). Briefly, mice were anesthetized and underwent midline abdominal incisions with the left renal pedicle bluntly clamped for 45 min (unilateral renal occlusion); reperfusion was achieved by removing the clamp. Mice were euthanized at days 1, 3, and 7 to harvest blood and kidneys. The unilateral I/R injury model was used to reduce surgery caused mortality. For cisplatin‐induced AKI, mice were given a single dose of 25 mg/kg cisplatin intraperitoneally (Topscience Biotech., Shanghai, China) or normal saline as described (Ozkok et al, 2016; Yang et al, 2021). Mice were euthanized on Day 1 or 3 to harvest blood and kidneys.
A366 (2.5 mg/kg in PBS; Topscience), GW4064 (30 mg/kg in corn oil; Topscience), and WWL229 (30 mg/kg in PBS containing 1% DMSO, 24% PEG400 and 6% Tween‐80; Topscience) were administrated intraperitoneally, while atorvastatin (20 mg/kg in saline; Topscience) was administrated intragastric. Mice were sacrificed at I/R 3D to harvest blood and kidneys. Depending on experimental purposes, non‐perfused kidneys were divided into two sets. One set of kidneys were transverse cut, with one half fixed with 10% buffered formaldehyde for paraffin sections, and the other half soaked in 30% sucrose in PBS buffer for cryosections. For the other set of kidneys, whole kidney was first minced and then aliquoted for assays including RNA‐sequencing, qPCR, Western blots, triglyceride (TG), total cholesterol (TC), and enzymatic analyses, etc.
Biochemical measurements
Serum creatinine was analyzed with a Siemens ADVIA 2400 automatic biochemistry analyzer using a creatinine reagent kit (Fuxing Chang zheng Medical, Shanghai, China) as previously described (Chen et al, 2017; Wang et al, 2020). Serum BUN was measured using a BUN reagent kit (Jiancheng Bio., Nanjing, China). Renal or serum TG and TC levels were measured with kits per manufacturer's instructions (both from Jiancheng Bio.). Carboxylesterase activity of renal tissue or cultured cells was measured using a micro carboxylesterase (CarE) assay kit (Solarbio Tech., Beijing, China) following the manufacturer's instruction.
Human tissue specimens
Collection and use of human renal biopsy samples, including acute kidney injury samples and the control para‐carcinoma renal tissues, was approved by the Institutional Review Boards of affiliated Hubei Cancer Hospital and Union Hospital of Tongji Medical College (approval numbers LLHBCH2022YN‐015 and (2018)S196, respectively). All subjects included in the study provided written informed consent. The study was performed in accordance with the Declaration of Helsinki.
RNA sequencing and analysis
Total renal RNA was isolated, and RNA sequencing and data analyses were performed by Novogene Bioinformatics (Beijing, China) as previously reported (Liu et al, 2020). Differentially expressed genes were assessed with a threshold of adjusted P‐value < 0.05 and |log2 Fold Change| > 0. A cloud platform (https://magic.novogene.com/) was used for KEGG pathway enrichment, and Graphpad prism (v8.0.1) was used to generate a heatmap.
Quantitative real‐time PCR (qPCR) and Western blots
qPCR and Western blots were performed as previously described (Chen et al, 2022; Wang et al, 2022b). Primers and antibodies are provided in the Appendix Tables S1 and S2.
Cell culture, transfection/infection, and treatments
The human renal tubular cell line HK‐2 (#GDC0152, from CCTCC, China Center for Type Culture Collection) was cultured in DMEM/F12 media (Cytiva, South Logan, UT) plus 15% FBS (Lonsera, Shanghai, China) with less than eight passages used. HEK293 cells (CL‐0005, Procell Biotech, Newport Beach, CA) were cultured in DMEM media (Cytiva) containing 10% FBS. HK‐2 cells and HEK293 were STR authenticated and mycoplasma contamination tested by CCTCC and Procell Biotech. shRNAs targeting human G9a and plasmid pCAGGS‐G9a were constructed to establish stable G9a knockdown HK‐2 cells (Xue et al, 2018). Adeno‐G9a‐3*Flag (Ad‐G9a) and its control (Obio Tech., Shanghai, China) were used to infect cells at an MOI of 50. For A366 treatment, HK‐2 cells were treated with 1 μM A366 or vehicle (DMSO) for 24 h.
Mouse primary tubular epithelial cells (mPTECs) isolation and treatments
Mouse primary tubular epithelial cells were isolated from renal cortex of C57BL/6 mice as previously reported (Wang et al, 2022a). Briefly, minced renal cortex was digested using 1 mg/ml type II collagenase (Sigma‐Aldrich), then sequentially passed through 200‐μm and 70‐μm cell strainers, followed by culturing in RPMI‐1640 medium with 5 mg/l human epidermal growth factor (PeproTech, Rocky Hill, NJ) for further experiments. For treatment, WWL229 was dissolved in DMSO, then added to the medium at final concentration of 0, 25, 50 or 100 μM for 24 h. For PA (Wako, Tokyo, Japan) treatment, PA was dissolved in DMEM media containing 2% bovine serum albumin (Amresco, Solon, OH), then added to the medium at 100 μM for 12 h. For labeled PA treatment, cells were incubated in 25 μM Bodipy FL C16 (a green‐fluorescent PA analog; Invitrogen, Breda, Netherlands) for 6 h.
PA degradation assay
Mouse primary tubular epithelial cells were cultured on coverslips and treated with Bodipy FL C16. Then culture medium was replaced with serum‐limited medium (0.1% FBS) for 0, 1, 2, or 3 h. Cells were fixed and pictures were taken using a TCS SP8 confocal microscope (Leica, Nussloch, Germany). Fluorescence area and intensity of Bodipy FL C16 were quantified using Image J (https://imagej.nih.gov/ij) software with at least 50 cells counted per group and reported as percentage of zero time.
Lipid droplet measurement and analysis
Mouse primary tubular epithelial cells were treated with Bodipy FL C16, followed by serum‐limited medium (0.1% FBS) treatment for 1 h, then 250 nM MitoTracker Red (Thermo Fisher Scientific, Carlsbad, CA) was added for 20 min. Images were taken using a TCS SP8 confocal microscope, and pictures were analyzed using ImageJ software. For lipid droplet quantification, an inter modes threshold method providing best coverage of the lipid droplet (LD) area was applied. The number and diameter of LDs were acquired, and the number of LDs with diameters < 0.5, 0.5–1, and > 1 μm were counted to calculate their proportions in the total population. Pearson's correlation was used to measure the colocalization of LDs and mitochondria.
Oxygen consumption rate (OCR) measurement
Mouse primary tubular epithelial cells were cultured in an XFe24 cell culture microplate (Agilent Technologies, Santa Clara, CA) at a density of 3 × 104 cells per well and treated with 100 μM PA for 12 h. Following incubation, cells were washed with Seahorse XF RPMI medium (Agilent Technologies), followed by incubation in 500 μl Seahorse XF RPMI medium plus 2 mM glutamine, 1 mM pyruvate, and 10 mM glucose for 60 min in a CO2‐free incubator. OCR was analyzed by sequential adding of 1 μM oligomycin, 2 μM carbonyl cyanide 4‐(trifluoromethoxy) phenylhydrazone (FCCP), and 0.5 μM rotenone plus antimycin A (all from XF Cell Mito Stress Test Kit, Agilent Technologies) in a XFe24 extracellular flux analyzer (Seahorse Bioscience, Billerica, MA) following the manufacturer's instruction.
Dual‐luciferase reporter assays
The Ces1 promoter from −500 to TSS (transcription start site) was cloned into a pGL3‐enhancer (Promega). Plasmids pRK‐Flag‐FXR and pRK‐Flag‐G9a were generated by cloning FXR or G9a into a pRK‐5′Flag vector. HEK293 cells were transfected with pGL3‐enhancer‐Ces1, pRL‐TK Renilla luciferase reporter plasmid, and titrated amounts of pRK‐Flag‐G9a or pRK‐Flag‐FXR plasmids. For GW4064 treatment, HEK293 cells were treated with 2 μM GW4064 for 48 h after transfection. Luciferase assays were performed and analyzed as previously described (Zhang et al, 2017; Liu et al, 2020).
Renal histology, and immunohistochemical and immunofluorescent staining
Paraffin embedded sections were used for H&E, Sirius Red, TUNEL assay, or immunohistochemical staining. H&E stained renal sections were assessed double‐blinded to evaluate pathological lesions as previously described (Chen et al, 2015, 2017; Wang et al, 2020). Sirius Red staining was performed to determine the degree of renal fibrosis using a Sirius Red staining kit (SenBeijia Biotech., Nanjing, China). TUNEL‐positive stained cells were detected by an In Situ Cell Death Detection Kit (Roche, Mannheim, Germany) and quantified using Image‐Pro Plus 6.0 software (Media Cybernetics, Bethesda, MD) by manual counting, and reported as number of positively stained cells per mm2 as previously reported (Huang et al, 2022). For immunohistochemical staining, primary antibody for Ly6G, CD3, F4/80, G9a, Ces1 or Cpt1a (information provided in Appendix Table S2) was applied to sections overnight at 4°C. Sections were then incubated with the corresponding biotinylated secondary antibody, sequentially incubated in ABC‐peroxidase solution (Vector laboratories, Burlingame, CA), and finally visualized by 3,3′‐diaminobenzidine (DAB, Cwbiotech, Beijing, China). Quantitative analysis of positively stained cells was performed as previously reported (Sun et al, 2020a,b; Wang et al, 2020).
Renal cryosections were used for Oil Red O staining and immunofluorescent staining. Oil Red O staining was performed as previously described (Liu et al, 2020). p‐Mlkl, G9a or Ces1 antibody (information provided in Appendix Table S2) was applied to the section and sequentially incubated with a corresponding Alexa Fluor labeled secondary antibody (Thermo Fisher Scientific); for renal tubular co‐staining, sections were incubated with 2 μg/ml PNA, or 2 μg/ml LTL (both from Vector Laboratories); for CD11b co‐staining, FITC‐labeled anti‐mouse CD11b (Appendix Table S2) was applied. Finally, sections were covered with DAPI (Sigma‐Aldrich) containing anti‐fading medium (Invitrogen), and images were captured using a TCS SP8 (Leica) confocal microscope.
Chromatin immunoprecipitation (ChIP) and re‐ChIP assay
The ChIP assay was performed as previously described (Chen et al, 2019a; Zhang et al, 2020a). Briefly, cells or minced kidney tissues were crosslinked with 1% formaldehyde, then quenched with glycine. Cross‐linked samples were gently homogenized to disperse cells, and chromatin was digested with micrococcal nuclease (New England Biolabs, Beverly, MA). Chromatin was immunoprecipitated using anti‐H3K9me2, anti‐G9a, rabbit or mouse IgG antibody (Appendix Table S2). Purified DNA was detected by qPCR (primer sequences in Appendix Table S1). Input samples were used as the internal control for comparison between samples. For a ChIP and re‐ChIP assay, Flag‐FXR was first immunoprecipitated with the anti‐Flag antibody from the chromatin fraction of pRK‐Flag‐FXR‐transfected HEK293 cells, or the chromatin was first immunoprecipitated with anti‐H3K9me2 antibody in HEK293 cells. Immunoprecipitated chromatin from the first ChIP was eluted by incubating with 10 mM DTT at 37°C for 1 h, then diluted. The second ChIP was performed using rabbit IgG or anti‐G9a antibody (Appendix Table S2). JASPAR (https://jaspar.genereg.net/), a database for transcription factor‐binding profiles, was used to predict FXR‐binding sites on the CES1 promoter.
Statistical analyses
The data are presented as means ± SD. All statistical analyses were performed using GraphPad Prism. The normal distribution of data was tested by SPSS (version 22). Statistical significance was assessed by an unpaired, 2‐tailed Student's t test or a 1‐way ANOVA with Tukey's multiple‐comparison test when the data was normally distributed; or was assessed by Mann–Whitney U test when the data were not normally distributed; the statistical analyses applied were indicated in the figure legends. Differences were considered statistically significant with P < 0.05.
Author contributions
Dong Yang: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Yu Fan: Formal analysis; validation; investigation. Mingrui Xiong: Formal analysis; validation; investigation; methodology. Yuchen Chen: Data curation; funding acquisition; visualization; writing – original draft; writing – review and editing. Yihao Zhou: Visualization. Xikai Liu: Formal analysis; validation; investigation; visualization; methodology. Yangmian Yuan: Validation; investigation; methodology. Qing Wang: Writing – original draft. Yu Zhang: Methodology. Robert B Petersen: Writing – review and editing. Hua Su: Resources. Junqiu Yue: Resources. Chun Zhang: Resources. Hong Chen: Funding acquisition; project administration. Kun Huang: Conceptualization; resources; data curation; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing. Ling Zheng: Conceptualization; resources; data curation; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
This work is supported by the Natural Science Foundation of China (32271222, 91957114 and 32021003 to LZ; 82273838 and 31971066 to KH; 31500941 to HC), the National Key R&D Program of China (2018YFA0800700 and 2019YFA0802701 to LZ; 2022YFA0806101 to KH), the China Postdoctoral Science Foundation (2021M700050 to YC), and the Natural Science Foundation of Hubei Province (2021CFA004 to KH). The authors appreciate the core facility of College of Life Sciences of Wuhan University, and the Analytical Center of HUST for technical support.
EMBO reports (2023) 24: e56128
Contributor Information
Kun Huang, Email: kunhuang@hust.edu.cn.
Ling Zheng, Email: lzheng@whu.edu.cn.
Data availability
The RNA‐seq data are available at GEO, accession number: GSE183455 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE183455).
References
- Akgullu C, Eryilmaz U, Gungor H, Huyut A, Zencir C, Hekim T (2015) A clinical study about contrast nephropathy: risk factors and the role of beta blockers. Anatol J Cardiol 15: 232–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athyros VG, Tziomalos K, Karagiannis A, Mikhailidis DP (2010) Atorvastatin: safety and tolerability. Expert Opin Drug Saf 9: 667–674 [DOI] [PubMed] [Google Scholar]
- Basile DP, Anderson MD, Sutton TA (2012) Pathophysiology of acute kidney injury. Compr Physiol 2: 1303–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bei WJ, Chen SQ, Li HL, Wu DX, Duan C, Chen PY, Chen JY, Tan N, Xie NJ, Liu Y (2017) Comparing common doses (double‐dose vs usual‐dose) of atorvastatin for preventing contrast‐induced acute kidney injury and mortality after coronary angiography. Medicine (Baltimore) 96: e7501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beker BM, Corleto MG, Fieiras C, Musso CG (2018) Novel acute kidney injury biomarkers: their characteristics, utility and concerns. Int Urol Nephrol 50: 705–713 [DOI] [PubMed] [Google Scholar]
- Bobulescu IA (2010) Renal lipid metabolism and lipotoxicity. Curr Opin Nephrol Hypertens 19: 393–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonventre JV, Weinberg JM (2003) Recent advances in the pathophysiology of ischemic acute renal failure. J Am Soc Nephrol 14: 2199–2210 [DOI] [PubMed] [Google Scholar]
- Bonventre JV, Yang L (2011) Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest 121: 4210–4221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonventre JV, Zuk A (2004) Ischemic acute renal failure: an inflammatory disease? Kidney Int 66: 480–485 [DOI] [PubMed] [Google Scholar]
- Chen H, Wan D, Wang L, Peng A, Xiao H, Petersen RB, Liu C, Zheng L, Huang K (2015) Apelin protects against acute renal injury by inhibiting TGF‐beta1. Biochim Biophys Acta 1852: 1278–1287 [DOI] [PubMed] [Google Scholar]
- Chen H, Wang L, Wang W, Cheng C, Zhang Y, Zhou Y, Wang C, Miao X, Wang J, Wang C et al (2017) ELABELA and an ELABELA fragment protect against AKI. J Am Soc Nephrol 28: 2694–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Huang Y, Zhu X, Liu C, Yuan Y, Su H, Zhang C, Liu C, Xiong M, Qu Y et al (2019a) Histone demethylase UTX is a therapeutic target for diabetic kidney disease. J Physiol 597: 1643–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Han Y, Gao P, Yang M, Xiao L, Xiong X, Zhao H, Tang C, Chen G, Zhu X et al (2019b) Disulfide‐bond a oxidoreductase‐like protein protects against ectopic fat deposition and lipid‐related kidney damage in diabetic nephropathy. Kidney Int 95: 880–895 [DOI] [PubMed] [Google Scholar]
- Chen H, Liu C, Wang Q, Xiong M, Zeng X, Yang D, Xie Y, Su H, Zhang Y, Huang Y et al (2022) Renal UTX‐PHGDH‐serine axis regulates metabolic disorders in the kidney and liver. Nat Commun 13: 3835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier RL (2016) The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Renal Physiol 311: 145–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusumano G, Romagnoli J, Liuzzo G, Ciavarella LP, Severino A, Copponi G, Manchi M, Giubilato S, Zannoni GF, Stigliano E et al (2015) N‐acetylcysteine and high‐dose atorvastatin reduce oxidative stress in an ischemia‐reperfusion model in the rat kidney. Transplant Proc 47: 2757–2762 [DOI] [PubMed] [Google Scholar]
- Diaz‐Bulnes P, Saiz ML, Corte‐Iglesias V, Rodrigues‐Diez RR, Bernardo Florez A, Ruiz Bernet C, Martin Martin C, Ruiz‐Ortega M, Suarez‐Alvarez B, Lopez‐Larrea C (2022) Demethylation of H3K9 and H3K27 contributes to the tubular renal damage triggered by endoplasmic reticulum stress. Antioxidants (Basel) 11: 1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducasa GM, Mitrofanova A, Fornoni A (2019) Crosstalk between lipids and mitochondria in diabetic kidney disease. Curr Diab Rep 19: 144 [DOI] [PubMed] [Google Scholar]
- Emma F, Montini G, Parikh SM, Salviati L (2016) Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol 12: 267–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan SS, Rizk A, Thomann C, Motawie A, Abdelfattah S, Ahmad Z (2019) Preconditioning with atorvastatin against renal ischemia‐reperfusion injury in nondiabetic versus diabetic rats. Can J Physiol Pharmacol 97: 1–14 [DOI] [PubMed] [Google Scholar]
- Hirschhorn T, Stockwell BR (2019) The development of the concept of ferroptosis. Free Radic Biol Med 133: 130–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Xie Y, Yang D, Xiong M, Chen X, Wu D, Wang Q, Chen H, Zheng L, Huang K (2022) Histone demethylase UTX aggravates acetaminophen overdose induced hepatotoxicity through dual mechanisms. Pharmacol Res 175: 106021 [DOI] [PubMed] [Google Scholar]
- Irifuku T, Doi S, Sasaki K, Doi T, Nakashima A, Ueno T, Yamada K, Arihiro K, Kohno N, Masaki T (2016) Inhibition of H3K9 histone methyltransferase G9a attenuates renal fibrosis and retains klotho expression. Kidney Int 89: 147–157 [DOI] [PubMed] [Google Scholar]
- Jun W, Benjanuwattra J, Chattipakorn SC, Chattipakorn N (2020) Necroptosis in renal ischemia/reperfusion injury: a major mode of cell death? Arch Biochem Biophys 689: 108433 [DOI] [PubMed] [Google Scholar]
- Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J et al (2015) Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med 21: 37–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellum JA, Romagnani P, Ashuntantang G, Ronco C, Zarbock A, Anders HJ (2021) Acute kidney injury. Nat Rev Dis Primers 7: 52 [DOI] [PubMed] [Google Scholar]
- Kim DH, Park JS, Choi HI, Kim CS, Bae EH, Ma SK, Kim SW (2021) The critical role of FXR is associated with the regulation of autophagy and apoptosis in the progression of AKI to CKD. Cell Death Dis 12: 320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian J, Nelson R, Lehner R (2018) Carboxylesterases in lipid metabolism: from mouse to human. Protein Cell 9: 178–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Wang J, Wei Y, Zhang W, Geng M, Yuan Y, Chen Y, Sun Y, Chen H, Zhang Y et al (2020) Fat‐specific knockout of Mecp2 upregulates Slpi to reduce obesity by enhancing browning. Diabetes 69: 35–47 [DOI] [PubMed] [Google Scholar]
- Liu H, Wang W, Weng X, Chen H, Chen Z, Du Y, Liu X, Wang L (2021) The H3K9 histone methyltransferase G9a modulates renal ischemia reperfusion injury by targeting Sirt1. Free Radic Biol Med 172: 123–135 [DOI] [PubMed] [Google Scholar]
- Meor Anuar Shuhaili MFR, Samsudin IN, Stanslas J, Hasan S, Thambiah SC (2017) Effects of different types of statins on lipid profile: a perspective on Asians. Int J Endocrinol Metab 15: e43319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miguel V, Tituana J, Herrero JI, Herrero L, Serra D, Cuevas P, Barbas C, Puyol DR, Marquez‐Exposito L, Ruiz‐Ortega M et al (2021) Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J Clin Invest 131: e140695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami S, Yamamoto T, Takabatake Y, Takahashi A, Namba T, Matsuda J, Kimura T, Kaimori JY, Matsui I, Hamano T et al (2017) Lipophagy maintains energy homeostasis in the kidney proximal tubule during prolonged starvation. Autophagy 13: 1629–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnar AO, Coca SG, Devereaux PJ, Jain AK, Kitchlu A, Luo J, Parikh CR, Paterson JM, Siddiqui N, Wald R et al (2011) Statin use associates with a lower incidence of acute kidney injury after major elective surgery. J Am Soc Nephrol 22: 939–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozkok A, Ravichandran K, Wang Q, Ljubanovic D, Edelstein CL (2016) NF‐κB transcriptional inhibition ameliorates cisplatin‐induced acute kidney injury (AKI). Toxicol Lett 240: 105–113 [DOI] [PubMed] [Google Scholar]
- Panzitt K, Zollner G, Marschall HU, Wagner M (2022) Recent advances on FXR‐targeting therapeutics. Mol Cell Endocrinol 552: 111678 [DOI] [PubMed] [Google Scholar]
- Pappano WN, Guo J, He Y, Ferguson D, Jagadeeswaran S, Osterling DJ, Gao W, Spence JK, Pliushchev M, Sweis RF et al (2015) The histone methyltransferase inhibitor A‐366 uncovers a role for G9a/GLP in the epigenetics of leukemia. PLoS One 10: e0131716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengrattanachot N, Cherngwelling R, Jaikumkao K, Pongchaidecha A, Thongnak L, Swe MT, Chatsudthipong V, Lungkaphin A (2020) Atorvastatin attenuates obese‐induced kidney injury and impaired renal organic anion transporter 3 function through inhibition of oxidative stress and inflammation. Biochim Biophys Acta Mol Basis Dis 1866: 165741 [DOI] [PubMed] [Google Scholar]
- Portilla D, Li S, Nagothu KK, Megyesi J, Kaissling B, Schnackenberg L, Safirstein RL, Beger RD (2006) Metabolomic study of cisplatin‐induced nephrotoxicity. Kidney Int 69: 2194–2204 [DOI] [PubMed] [Google Scholar]
- Quintavalle C, Fiore D, De Micco F, Visconti G, Focaccio A, Golia B, Ricciardelli B, Donnarumma E, Bianco A, Zabatta MA et al (2012) Impact of a high loading dose of atorvastatin on contrast‐induced acute kidney injury. Circulation 126: 3008–3016 [DOI] [PubMed] [Google Scholar]
- Rewa O, Bagshaw SM (2014) Acute kidney injury‐epidemiology, outcomes and economics. Nat Rev Nephrol 10: 193–207 [DOI] [PubMed] [Google Scholar]
- Ronco C, Bellomo R, Kellum JA (2019) Acute kidney injury. Lancet 394: 1949–1964 [DOI] [PubMed] [Google Scholar]
- Rose S, Misharin A, Perlman H (2012) A novel Ly6C/Ly6G‐based strategy to analyze the mouse splenic myeloid compartment. Cytometry A 81: 343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothammer N, Woo MS, Bauer S, Binkle‐Ladisch L, Di Liberto G, Egervari K, Wagner I, Haferkamp U, Pless O, Merkler D et al (2022) G9a dictates neuronal vulnerability to inflammatory stress via transcriptional control of ferroptosis. Sci Adv 8: eabm5500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer IR, Joshi M (2020) CPT1A‐mediated fat oxidation, mechanisms, and therapeutic potential. Endocrinology 161: 1–14 [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Tachibana M (2011) H3K9 methyltransferase G9a and the related molecule GLP. Genes Dev 25: 781–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiva N, Sharma N, Kulkarni YA, Mulay SR, Gaikwad AB (2020) Renal ischemia/reperfusion injury: an insight on in vitro and in vivo models. Life Sci 256: 117860 [DOI] [PubMed] [Google Scholar]
- Sugahara S, Kume S, Chin‐Kanasaki M, Tomita I, Yasuda‐Yamahara M, Yamahara K, Takeda N, Osawa N, Yanagita M, Araki SI et al (2019) Protein O‐GlcNAcylation is essential for the maintenance of renal energy homeostasis and function via lipolysis during fasting and diabetes. J Am Soc Nephrol 30: 962–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Geng M, Yuan Y, Guo P, Chen Y, Yang D, Petersen RB, Huang K, Zheng L (2020a) Lmo4‐resistin signaling contributes to adipose tissue‐liver crosstalk upon weight cycling. FASEB J 34: 4732–4748 [DOI] [PubMed] [Google Scholar]
- Sun Y, Wang Q, Zhang Y, Geng M, Wei Y, Liu Y, Liu S, Petersen RB, Yue J, Huang K et al (2020b) Multigenerational maternal obesity increases the incidence of HCC in offspring via miR‐27a‐3p. J Hepatol 73: 603–615 [DOI] [PubMed] [Google Scholar]
- Sun L, Cai J, Gonzalez FJ (2021) The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat Rev Gastroenterol Hepatol 18: 335–347 [DOI] [PubMed] [Google Scholar]
- Sung P, Yang C, Chiang JY, Chen C, Luo C, Yip H (2022) Inhibition of histone methyltransferase G9a effectively protected the kidney against ischemia‐reperfusion injury. Am J Transl Res 14: 3683–3697 [PMC free article] [PubMed] [Google Scholar]
- Sweis RF, Pliushchev M, Brown PJ, Guo J, Li F, Maag D, Petros AM, Soni NB, Tse C, Vedadi M et al (2014) Discovery and development of potent and selective inhibitors of histone methyltransferase g9a. ACS Med Chem Lett 5: 205–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H et al (2002) G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev 16: 1779–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomsa AM, Alexa AL, Junie ML, Rachisan AL, Ciumarnean L (2019) Oxidative stress as a potential target in acute kidney injury. PeerJ 7: e8046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Zhang C, Hu L, Yang C (2016) Necroptosis in acute kidney injury: a shedding light. Cell Death Dis 7: e2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Su YY, Li YQ, Zhang YF, Yang S, Wang JL, Li HY (2017) Atorvastatin alleviates renal ischemia‐reperfusion injury in rats by promoting M1‐M2 transition. Mol Med Rep 15: 798–804 [DOI] [PubMed] [Google Scholar]
- Wang C, Xiong M, Yang C, Yang D, Zheng J, Fan Y, Wang S, Gai Y, Lan X, Chen H et al (2020) PEGylated and acylated Elabela analogues show enhanced receptor binding, prolonged stability, and remedy of acute kidney injury. J Med Chem 63: 16028–16042 [DOI] [PubMed] [Google Scholar]
- Wang J, Xiong MR, Fan Y, Liu CY, Wang Q, Yang D, Yuan YM, Huang YX, Wang S, Zhang Y et al (2022a) Mecp2 protects kidney from ischemia‐reperfusion injury through transcriptional repressing IL‐6/STAT3 signaling. Theranostics 12: 3896–3910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Chen YC, Xie YH, Yang D, Sun YY, Yuan YM, Chen H, Zhang Y, Huang K, Zheng L (2022b) Histone H1.2 promotes hepatocarcinogenesis by regulating signal transducer and activator of transcription 3 signaling. Cancer Sci 113: 1679–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Li Y, Chen WD, Xu Y, Yin L, Ge X, Jadhav K, Adorini L, Zhang Y (2014) Hepatic carboxylesterase 1 is essential for both normal and farnesoid X receptor‐controlled lipid homeostasis. Hepatology 59: 1761–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Li D, Wu J, Zhang M, Shao X, Xu L, Tang L, Zhu M, Ni Z, Zhang M et al (2021) Farnesoid X receptor promotes renal ischaemia‐reperfusion injury by inducing tubular epithelial cell apoptosis. Cell Prolif 54: e13005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Jia P, Fang Y, Jin J, Sun Z, Zhou W, Li J, Zhang Y, Wang X, Ren T et al (2022) Nuclear farnesoid X receptor attenuates acute kidney injury through fatty acid oxidation. Kidney Int 101: 987–1002 [DOI] [PubMed] [Google Scholar]
- Xue W, Huang J, Chen H, Zhang Y, Zhu X, Li J, Zhang W, Yuan Y, Wang Y, Zheng L et al (2018) Histone methyltransferase G9a modulates hepatic insulin signaling via regulating HMGA1. Biochim Biophys Acta Mol Basis Dis 1864: 338–346 [DOI] [PubMed] [Google Scholar]
- Yang D, Lin S, Yang D, Wei L, Shang W (2012) Effects of short‐ and long‐term hypercholesterolemia on contrast‐induced acute kidney injury. Am J Nephrol 35: 80–89 [DOI] [PubMed] [Google Scholar]
- Yang C, Zhang Y, Zeng X, Chen H, Chen Y, Yang D, Shen Z, Wang X, Liu X, Xiong M et al (2021) Kidney injury molecule‐1 is a potential receptor for SARS‐CoV‐2. J Mol Cell Biol 13: 185–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zager RA, Johnson AC, Hanson SY (2005) Renal tubular triglyercide accumulation following endotoxic, toxic, and ischemic injury. Kidney Int 67: 111–121 [DOI] [PubMed] [Google Scholar]
- Zechner R (2015) FAT FLUX: enzymes, regulators, and pathophysiology of intracellular lipolysis. EMBO Mol Med 7: 359–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, Madeo F (2012) FAT SIGNALS‐lipases and lipolysis in lipid metabolism and signaling. Cell Metab 15: 279–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Guo X, Yan W, Chen Y, Ke M, Cheng C, Zhu X, Xue W, Zhou Q, Zheng L et al (2017) ANGPTL8 negatively regulates NF‐kappaB activation by facilitating selective autophagic degradation of IKKgamma. Nat Commun 8: 2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yang D, Yuan Y, Liu C, Chen H, Zhang Y, Wang Q, Petersen RB, Huang K, Zheng L (2020a) Muscular G9a regulates muscle‐liver‐fat axis by Musclin under overnutrition in female mice. Diabetes 69: 2642–2654 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xue W, Zhang W, Yuan Y, Zhu X, Wang Q, Wei Y, Yang D, Yang C, Chen Y et al (2020b) Histone methyltransferase G9a protects against acute liver injury through GSTP1. Cell Death Differ 27: 1243–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann R, Lass A, Haemmerle G, Zechner R (2009) Fate of fat: the role of adipose triglyceride lipase in lipolysis. Biochim Biophys Acta Mol Cell Biol Lipids 1791: 494–500 [DOI] [PubMed] [Google Scholar]