

Abstract
The discrete steps of transcriptional rewiring have been proposed to occur neutrally to ensure steady gene expression under stabilizing selection. A conflict‐free switch of a regulon between regulators may require an immediate compensatory evolution to minimize deleterious effects. Here, we perform an evolutionary repair experiment on the Lachancea kluyveri yeast sef1Δ mutant using a suppressor development strategy. Complete loss of SEF1 forces cells to initiate a compensatory process for the pleiotropic defects arising from misexpression of TCA cycle genes. Using different selective conditions, we identify two adaptive loss‐of‐function mutations of IRA1 and AZF1. Subsequent analyses show that Azf1 is a weak transcriptional activator regulated by the Ras1‐PKA pathway. Azf1 loss‐of‐function triggers extensive gene expression changes responsible for compensatory, beneficial, and trade‐off phenotypes. The trade‐offs can be alleviated by higher cell density. Our results not only indicate that secondary transcriptional perturbation provides rapid and adaptive mechanisms potentially stabilizing the initial stage of transcriptional rewiring but also suggest how genetic polymorphisms of pleiotropic mutations could be maintained in the population.
Keywords: Azf1, compensatory evolution, Lachancea kluyveri, Sef1, trade‐off
Subject Categories: Chromatin, Transcription & Genomics; Evolution & Ecology; Microbiology, Virology & Host Pathogen Interaction
Evolutionary repair in sef1Δ mutant cells of the yeast Lachancea kluyveri highlights the rapid adaptive strategies to handle growth defects and to compensate for gene misexpression.
Introduction
The diversity of biological systems, including transcription regulatory systems, can be a consequence of adaptive evolution (Tenaillon et al, 2012), either macroscopically in the body plan (Peter Isabelle & Davidson Eric, 2011) or microscopically at the cellular level (Lynch et al, 2014). Altered transcriptional regulation is thought to create phenotypic novelty, thereby playing a crucial role in adaptive evolution and speciation (Carroll, 2000; Wagner & Lynch, 2008; Romero et al, 2012; Mack & Nachman, 2017). One particularly intriguing scenario is “transcriptional rewiring,” which describes changes to a gene regulatory network across species via cis (via polymorphisms in the linked regulatory sequences) or trans (through diffusible products of other loci) mechanisms over evolutionary timescales (Scannell & Wolfe, 2004; Dalal & Johnson, 2017). Complete regulon (a group of co‐regulated and usually functionally correlated target genes) handover from one TF to another (i.e., TF substitution) is an extreme case of transcriptional rewiring (Li & Johnson, 2010). The rewiring process is often constrained due to the deleterious effects of gene misexpression at intermediate stages, rendering the accumulation of regulatory changes problematic. Nevertheless, stabilizing selection in many species is believed to mitigate such constraints upon encountering new mutations by maintaining appropriate gene expression (Tanay et al, 2005; Bedford & Hartl, 2009; Tirosh et al, 2009; Goncalves et al, 2012; Shi et al, 2012; Coolon et al, 2014). In other words, extensive cis‐trans compensation in gene expression underlies transcriptional rewiring (Signor & Nuzhdin, 2018, 2019), potentially enabling “evolutionary tinkering” so that a set of conserved orthologous TFs can be fundamentally repositioned within the regulatory networks of a species (Lavoie et al, 2010). A “redundancy” mechanism has been proposed to explain TF substitution, whereby redundant and/or cooperative machinery of regulation underpins rewiring without giving rise to drastic changes in phenotypic output (Wohlbach et al, 2009; Johnson, 2017). Interestingly, modeling of genomic data from diverse species supports that the evolution of gene expression best fits the “house‐of‐cards” model of stabilizing selection, whereby mutations with large phenotypic effects rather than small effects evolutionarily change current transcriptional networks and allow them to be effectively reshuffled to form new networks (Hodgins‐Davis et al, 2015). However, such large‐effect mutations may be a double‐edged sword due to their deleterious pleiotropic effects (Dittmar et al, 2016). Therefore, we aimed to investigate whether and how compensatory evolution works efficiently to deal with this conflict (trade‐offs from new large‐effect mutations) when the “redundancy” mechanism (redundant and/or cooperative machinery of regulation) is unavailable as new transcriptional networks evolve.
To achieve this goal, we conducted an evolutionary repair experiment. Evolutionary repair is a category of experimental microbial evolution whereby the founder strain contains at least one genetic perturbation (e.g., a single gene deletion; LaBar et al, 2020). A general concern for evolutionary repair experiments is whether such a theoretically rare event can in fact happen in nature because a genetic perturbation would be negatively selected in natural populations and thus kept at a low frequency. Therefore, we chose a target gene whose loss of function causes condition‐dependent deleterious effects, which generally allows the perturbed founders to persist in a changing environment long enough to evolve a compensatory mutation.
Using the yeast L. kluyveri as a model, we first deleted the TF gene SEF1 involved in respiration and growth (Hsu et al, 2021) to create a less‐fit founder strain displaying pleiotropic defects (i.e., misregulation of multiple TCA cycle genes that affect respiration‐related and ‐unrelated traits). The L. kluyveri Sef1 was chosen due to its known condition‐dependent phenotypes, completely characterized direct target genes, simple condition‐responsive regulation, and proper evolutionary divergence from the model baker's yeast (Hsu et al, 2021). All these advantages will help to simplify subsequent investigation after the evolutionary repair experiments. Complete loss of Sef1 excludes the possibility of “stabilizing” evolution via the “redundancy” mechanism. Then, we performed our evolutionary repair experiment through sef1Δ suppressor development to screen for potential large‐effect compensatory mutations. We then identified two adaptive loss‐of‐function mutations of IRA1 and AZF1, respectively, in different selective conditions. We further investigated the pleiotropic effects caused by the azf1 loss‐of‐function mutation. We especially demonstrated that misexpression of multiple TCA cycle genes caused by sef1Δ can be simultaneously compensated by the azf1 loss‐of‐function mutation. Our results demonstrate a process of “quick‐and‐dirty” compensatory evolution in gene expression following a sudden loss of a TF under specific conditions that precluded a smooth transition via the redundancy mechanism. This potentially mimics the extreme case of the incipient stage of transcriptional rewiring wherein the target genes are disconnected from the primary TF without drastic misexpression, allowing the cells to survive and the perturbed transcriptional network to further evolve.
Results
Evolutionary repair rapidly suppressed the fitness defects caused by sef1Δ transcriptional network perturbation
Consistent with our previous work (Hsu et al, 2021), the L. kluyveri sef1Δ mutant is slightly less fit (slower growing) than SEF1 strains maintained at normal temperature (28°C) under fermentative conditions [YPD: yeast extract‐peptone dextrose (Dex)], with incubation under respiratory conditions [YPGly: yeast extract‐peptone glycerol (Gly)] or heat stress (37–39°C) further impairing growth (Fig 1A). The diminished maximal growth rates of the sef1Δ mutant calculated from source growth curves (Appendix Fig S1A and B) under two respiratory conditions (post‐diauxic shift and YPGly, Appendix Fig S1C) are shown in Fig 1B. An RNA‐seq analysis supports these phenotypic outcomes, showing clear downregulation of multiple TCA cycle genes required for respiratory and optimal fermentative growth (Fig 1C, and Datasets EV1 and EV2).
Figure 1. Rapid suppressor development of the sef1Δ mutant.
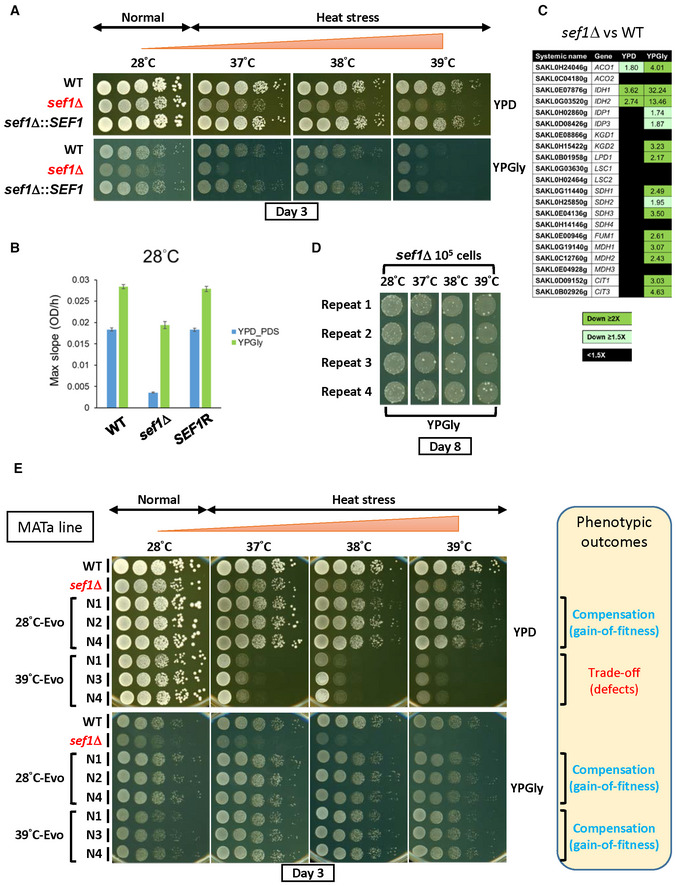
- Growth of the sef1Δ mutant in response to YPD, YPGly, and heat stress. All plates were incubated for 3 days.
- The maximal slope growth rate (OD600/h) of the sef1Δ mutant in liquid cultures. The growth curves were measured at 28°C by using a Tecan plate reader with intermittent shaking. Results are displayed as average max slopes ± standard deviations (SD) from three technical repeats. YPD_PDS: YPD culture grown to post‐diauxic shift phase. “SEF1R” indicates the reconstituted strain sef1Δ::SEF1.
- Differential expression of TCA cycle genes in response to sef1Δ. The RNA expression profiles from L. kluyveri wild‐type and sef1Δ cells grown to early log‐phase under both YPD and YPGly conditions are compared. A total of 21 annotated TCA cycle genes are listed, 18 of which are direct targets of Sef1 and the remaining 3 (IDP1, MDH3, and CIT3) are not (Hsu et al, 2021).
- Rapid formation of spontaneous suppressors of sef1Δ phenotypes. The sef1Δ cells were spotted on YPGly plates at a density of 105 CFU/5 μl spot and incubated at the indicated temperatures for 8 days. Cells of ancestral fitness were grown as a background lawn, while the suppressor clones were grown as bigger colonies.
- The adaptive growth phenotypes of re‐purified sef1Δ suppressor clones (28°C‐ and 39°C‐Evo, MATa line).
Interestingly, we noticed that some colonies displaying greater fitness formed among the sef1Δ population during the growth assay, especially under the more stringent conditions (YPGly + heat stress). Increasing the incubation time accentuated the outcompeting growth of these fitter cells relative to congeners (Fig 1D), suggesting that selection for suppressors of sef1Δ mutant phenotypes may occur rapidly and efficiently. Such adaptive mutations could arise from de novo mutations formed after selection or represent low‐frequency genetic variants that pre‐existed in the founder colony before selection (Teng et al, 2013). In terms of this latter, the hypothetical presence of heterogeneous quasi‐species founder colonies (Appendix Fig S1D) may accelerate the evolution of cells upon losing SEF1, changing their evolutionary trajectories (Appendix Fig S1E). Therefore, we performed a large‐scale sef1Δ suppressor development experiment (in this study, we also consider it as an evolutionary repair experiment) to investigate how cells evolve to deal with the loss of the transcriptional hub SEF1 that affects multiple downstream fitness‐contributing genes.
First, we created the sef1Δ and then the sef1Δchs3Δ (tetrad dissection‐competent) lines from the wild‐type MATa strain, before performing a mating‐type switch to obtain a MATα strain (Appendix Fig S2A). Two independent suppressor development experiments were performed by plating 108 MATa and MATα founder cells on YPGly plates at 28–39°C for 8 days (Appendix Fig S2B, step A). During that process, larger suppressor colonies carrying the correct drug markers were picked (Appendix Fig S2C, steps A, B, and C). Finally, a total of 240 sef1Δ suppressors were isolated, including 144 MATa lines and 96 MATα lines that evolved at different temperatures (Appendix Fig S3A). To characterize the adaptive effects of each clone, we evaluated their fitness by comparing their growth patterns on agar plates against two reference strains (the wild‐type and the corresponding sef1Δ founder), and then assigned a simple fitness score under YPD and YPGly conditions based on fitness categories (Appendix Fig S3B, left and middle columns, with two examples shown in Appendix Fig S3C). The simple fitness scores of all 240 clones are shown in Dataset EV3. To visualize global fitness patterns among the different groups of suppressor clones, we averaged the simple fitness scores and highlighted them with different colors according to the range of mean scores (Appendix Fig S3B, right column, and summarized in Appendix Fig S3D and E). In general, the sef1Δ suppressor selected at 28°C (hereafter, 28°C‐Evo) displayed more improved fitness than the sef1Δ founder under both YPD and YPGly conditions, irrespective of temperature. Hereafter, we describe the 28°C‐Evo clones as “double‐compensation.” In contrast, the sef1Δ suppressors selected under heat‐stressed conditions (hereafter, 37‐, 38‐, or 39°C‐Evo) showed more improved fitness compared to the sef1Δ founder only under YPGly conditions and especially under higher temperatures. Moreover, the 37, 38, and 39°C‐Evo clones showed a severe growth defect relative to the sef1Δ founder when grown under heat‐stressed YPD conditions. We specifically describe these heat‐stressed 37‐, 38‐, and 39°C‐Evo clones as “Dex‐trade‐off and Gly‐compensation” hereafter. Only 30 of the 240 (12.5%) suppressor clones showed inconsistent phenotypic patterns (Appendix Fig S3F), indicating that both the “double‐compensation” and “Dex‐trade‐off and Gly‐compensation” phenotypes are quite representative, implying that at least two major trajectories underlie the rapid evolutionary repair response to SEF1 deletion.
Adaptive phenotypes are monogenically induced by loss‐of‐function azf1 or ira1 mutations
To identify the causal mutations giving rise to the evolved phenotypes, we repurified the suppressor clones from −80°C stocks and confirmed both the “double‐compensation” and “Dex‐trade‐off and Gly‐compensation” phenotypes for two independent MATa and MATα lines (Fig 1E and Appendix Fig S4A). Notably, the randomly picked 37°C‐Evo and 38°C‐Evo clones showed the same pattern of evolved phenotypes as the 39°C‐Evo clones did (Appendix Fig S4B), implying that they may carry the same mutations or mutations eliciting similar suppressive mechanisms. Therefore, only three clones each of 28°C‐Evo MATa, 28°C‐Evo MATα, 39°C‐Evo MATa, and 39°C‐Evo MATα lines were re‐stocked, denoted “N” as indicated in Fig 1E and Appendix Fig S4A, and then subjected to whole‐genome sequencing.
By comparing the resulting genomes with those of their ancestors (i.e., the founder strains), we identified mutated loci in all three clones from the two independent lines (MATa and MATα; Dataset EV4, and Fig 2A and B). Interestingly, only two (IRA1 and FLO1) and four (FLO1, AZF1, SAKL0D15356pg, and AMD2) loci were shared among the respective 28°C‐Evo and 39°C‐Evo lines. The allele frequencies of the FLO1 and AMD2 mutations are < 100% and quite diverse between clones, and SAKL0D15356pg is a pseudogene. Subsequent examination of mutation types revealed that 28°C‐Evo lines carry deletion, missense, or loss‐of‐function (premature stop codon‐gained) mutations in the IRA1 loci (Fig 2C) and 39°C‐Evo lines carry missense and another type of loss‐of‐function mutations (frameshift) in the AZF1 loci (Fig 2D), supporting that ira1 and azf1 loss‐of‐function alleles are the causal mutations in the 28°C‐Evo and 39°C‐Evo suppressors, respectively.
Figure 2. Identification of causal adaptive mutations of sef1Δ phenotypes.
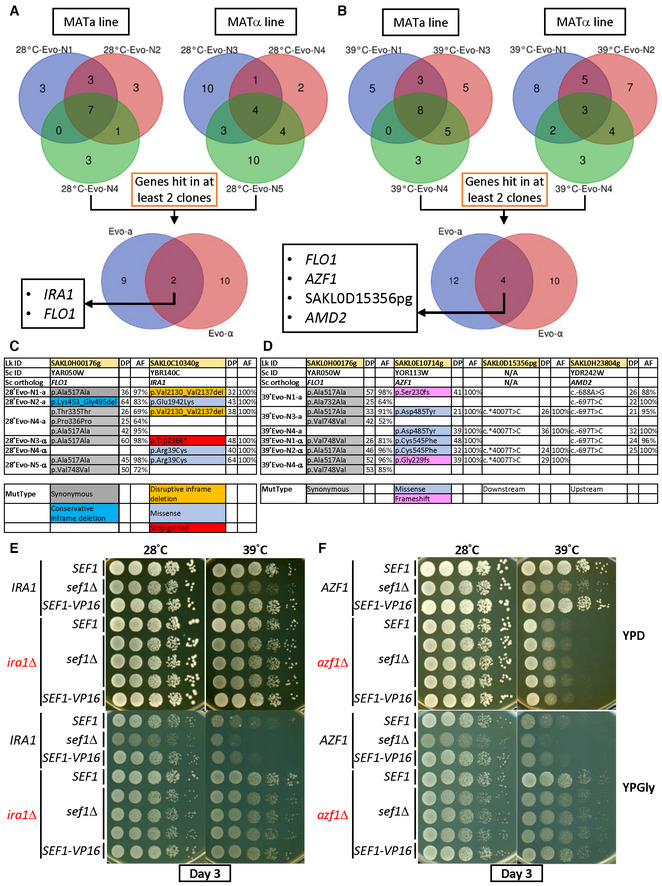
-
AConvergent mutations among 28°C‐Evo sef1Δ suppressors between the MATa and MATα lines.
-
BConvergent mutations among 39°C‐Evo sef1Δ suppressors between the MATa and MATα lines.
-
C, DTypes of candidate causal mutations in (C) 28°C‐Evo sef1Δ suppressors and (D) 39°C‐Evo sef1Δ suppressors. DP, read depth; AF, allele frequency. The mutant types (MutType) are highlighted with different background colors.
-
E, F(E) The adaptive growth phenotypes of re‐constructed sef1Δira1Δ strains. (F) The adaptive growth phenotypes of re‐constructed sef1Δazf1Δ strains. Three independent sef1Δira1Δ and sef1Δazf1Δ clones were tested. SEF1‐VP16 represents the high‐activity SEF1 strain created by fusing the strong viral transcriptional activator VP16 to the C‐terminal of SEF1 as described (Hsu et al, 2021), and one clone for each scenario is shown. SEF1‐VP16 was used to test whether higher Sef1 activity may mask the effect of azf1Δ to examine the epistatic relationship.
To prove that the adaptive phenotypes are monogenic, we performed tetrad dissection analyses by backcrossing the MATa suppressor clones with their MATα founders. We checked three for each of the MATa 28°C‐Evo and MATa 39°C‐Evo clones. After sporulation, one tetrad of each mating pair was dissected. All four spores of each tetrad were phenotyped and the candidate causal mutation loci were sequenced. All tetrads showed a perfect 2‐to‐2 ratio between adaptive versus wild‐type phenotypes, consistent with the 2‐to‐2 genotypes (Appendix Figs S5A–C and S6A–C), indicating a clear monogenic effect of the adaptive mutation. As expected, deletion of IRA1 or AZF1 alone in the parental sef1Δ strains proved sufficient to phenocopy the 28°C‐Evo and 39°C‐Evo suppressors, respectively (Fig 2E and F).
To estimate the overall frequency of azf1 loss‐of‐function mutations and occurrence of the “Dex‐trade‐off and Gly‐compensation” phenotypes, we Sanger sequenced the AZF1 loci of 30 more randomly picked heat stress‐evolved sef1Δ suppressors in both MATa and MATα lines (Fig EV1A and B). In combination with the aforementioned six whole genome‐sequenced clones, about 83.33% (30/36) of them carried azf1 mutations, including 15 premature stop codon‐gained, 11 frame‐shifted, and four missense mutations. The higher proportion of premature stop codon‐gained and frame‐shifted mutations suggests that azf1 loss‐of‐function mutations are the major causal mutations in this batch of suppressors. Moreover, about 85.26% (133/156) of the total heat stress‐evolved sef1Δ suppressors displayed clear “Dex‐trade‐off” phenotypes (Fig EV1C and Appendix Fig S3E), slightly higher than the estimated azf1 frequency (83.33%). This finding indicates the presence of non‐azf1 mutations which also result in the “Dex‐trade‐off and Gly‐compensation” phenotypes (Fig EV1B).
Figure EV1. The estimation of “Dex‐trade‐off” adaptive mutations.
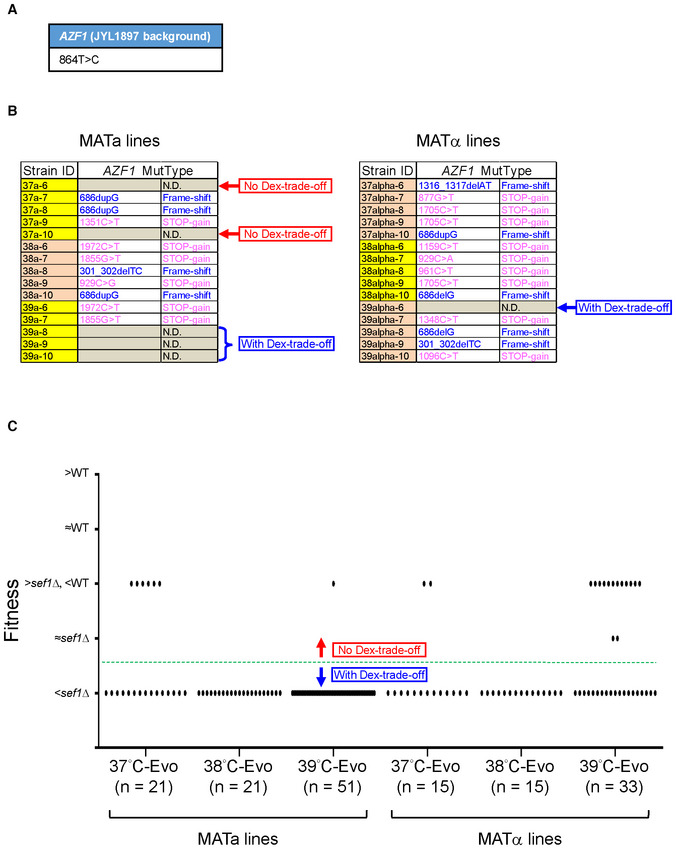
- The parental SNP polymorphism of L. kluyveri AZF1 in the JYL1897 background (the wild‐type of our lab) in comparison with the published reference genome of CBS 3082.
- The AZF1 mutational spectrum of 30 more randomly picked heat stress‐evolved sef1Δ suppressors (five clones each from 37, 38, and 39°C‐Evo sef1Δ suppressors in both MATa and MATα lineages). The mutations were identified by Sanger sequencing. “N.D.” means that there is no mutation detected within the coding region. Whether the clones that carry no azf1 mutation displayed “Dex‐trade‐off” phenotypes or not is indicated on the right side of each table.
- The “Dex‐trade‐off” phenotypic spectrum of 156 heat stress‐evolved sef1Δ suppressors. The “Dex‐trade‐off” phenotypes of each clone are displayed according to the simple fitness scores under the YPD_37°C condition (Dataset EV3). Clones with score 1 (with fitness worse than sef1Δ strain) were defined as “with Dex‐trade‐off” (please also see Appendix Fig S3B for the simple fitness score definition). About 14.74% (23/156) of clones in this batch showed no clear “Dex‐trade‐off.”
Similarly, the overall frequency of ira1 loss‐of‐function mutations was estimated by Sanger sequencing for the IRA1 loci of 10 more randomly picked 28°C‐evolved sef1Δ suppressors in both MATa and MATα lines (Fig EV2A and B). In combination with the aforementioned six whole‐genome‐sequenced clones, about 75% (12/16) of them carried ira1 mutations, including only three premature stop codon‐gained, one frame‐shifted, four in‐frame deletion, and four missense mutations. It is hard to determine the mutation functionalities simply by the mutation types. Therefore, we used qualitative desiccation sensitivity (shown as post‐desiccation viability) to represent the severity of ira1 loss‐of‐function mutations or other mutations generating the same effect as ira1 loss‐of‐function does (Ratnakumar et al, 2011; Welch et al, 2013). As shown in the control assays, the ira1Δ mutations were hypersensitive to desiccation in both wild‐type and sef1Δ backgrounds and the negative control azf1Δ strains were not (Fig EV2C). The loss‐of‐function and hypomorphic ira1 mutations were also distinguishable (Fig EV2D). About 92.86% (78/84) of the total 28°C‐evolved sef1Δ suppressors displayed clear “desiccation hypersensitivity” phenotypes (Fig EV2E), higher than the estimated ira1 frequency (75%). This finding also indicates the presence of non‐ira1 mutations which cause the “desiccation hypersensitivity” phenotypes in this batch of suppressors (Fig EV2B).
Figure EV2. The estimation of “ira1‐related” adaptive mutations.
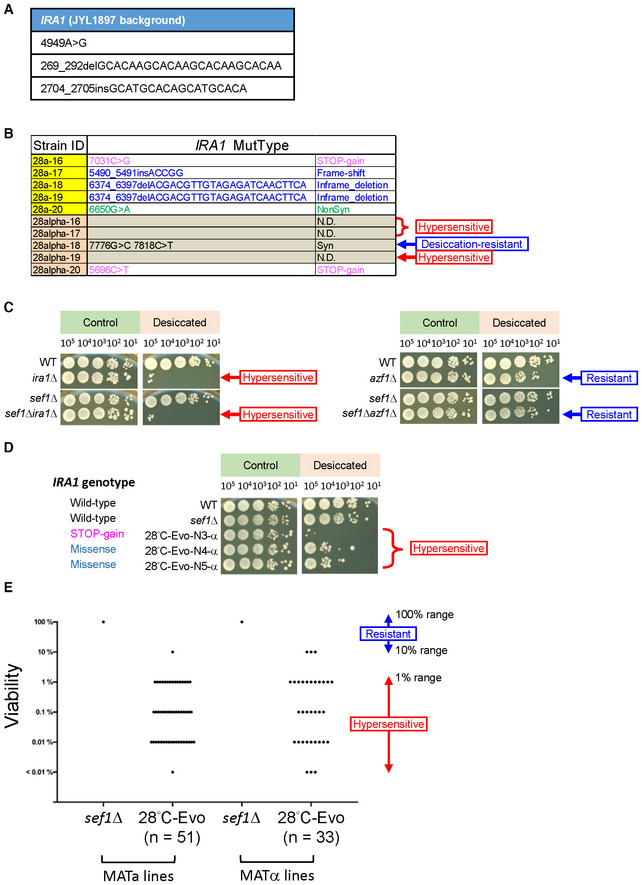
- The parental SNP and indel polymorphisms of L. kluyveri IRA1 in the JYL1897 background (the wild‐type of our lab) in comparison with the published reference genome of CBS 3082.
- The IRA1 mutational spectrum of 10 more randomly picked 28°C‐Evo sef1Δ suppressors (5 clones each in both MATa and MATα lineages). The mutations were identified by Sanger sequencing. “NonSyn” means non‐synonymous mutation. Whether the clones that carry no ira1 mutation displayed “desiccation hypersensitivity” phenotypes or not is indicated on the right side of the table.
- Specific desiccation hypersensitivity of ira1Δ in both wild‐type and sef1Δ backgrounds. The cells grown overnight in YPD were harvested and desiccated by air‐dry at 28°C for 20 h. The post‐desiccation viabilities were determined by spot assays compared to the non‐desiccated controls. The azf1Δ is not sensitive to desiccation.
- The desiccation hypersensitivity of 28°C‐Evo sef1Δ suppressors by taking 28°C‐Evo‐N3, ‐N4, and ‐N5 in the MATα lines as examples. Tested strains carrying different types of ira1 mutations showed different levels of desiccation hypersensitivity.
- The “desiccation hypersensitivity” phenotypic spectrum of 84 28°C‐evolved sef1Δ suppressors. The desiccation sensitivities of each clone are displayed according to the qualitative viabilities (Dataset EV3). Clones with a score ≥ 3 (with post‐desiccation viability ≤ 1% range) were defined as “hypersensitive” to desiccation (please also see Dataset EV3 for the rank definition). About only 7.14% (6/84) of clones in this batch showed no “hypersensitivity.”
Sef1, Ira1, and Azf1 belong to related functional modules
Given general concepts of “modular epistasis” among members in an interaction network (Segrè et al, 2005) and that suppressive interactions (query and modifier mutations) tend to connect functionally related genes (van Leeuwen et al, 2016), we hypothesized that Sef1, Ira1, and Azf1 may be associated with related functional modules. The L. kluyveri IRA1 is homologous to a negative regulator of the Ras signaling pathway in Saccharomyces cerevisiae (Tanaka et al, 1990), which is generally coupled with the cAMP‐PKA pathway that is activated in response to nutrients and the environment and is responsible for regulating metabolism, cell growth, stress resistance, and glucose adaptation (Conrad et al, 2014). L. kluyveri Sef1 is a transcriptional activator (Hsu et al, 2021), whereas Azf1 is a predicted TF based on orthology to S. cerevisiae Azf1 (Stein et al, 1998; Slattery et al, 2006). We speculated that Ras1‐Ira1‐cAMP‐PKA represents the regulatory signaling pathway upstream of Sef1 and Azf1, so we used either chromosome‐integrated or plasmid‐based one‐hybrid assays (Fig 3A) to evaluate the transcriptional activity of Sef1 and Azf1 in response to Ras1‐Ira1‐cAMP‐PKA pathway signaling. We adopted five Ras1‐Ira1‐cAMP‐PKA hyperactive mutants for this experiment, namely ira1Δ (loss of a Ras1‐negative regulator), RAS1 G20V (a hyperactive GTP‐bound Ras1 mutant orthologous to S. cerevisiae RAS2 G19V; Toda et al, 1985), pde2Δ and pde1Δpde2Δ (major loss‐of‐function mutants of high‐affinity cyclic AMP phosphodiesterase), and bcy1Δ (loss of the cAMP‐dependent protein kinase A inhibitor subunit). As shown in Fig 3B and C, hyperactivation of the Ras1‐Ira1‐cAMP‐PKA pathway modulated Sef1 activity by repressing it under the YPD condition and activating it under the YPGly condition. The changes in Sef1 protein abundance in response to ira1Δ were consistent with altered activity (Fig 3D), implying that the underlying regulatory mechanism operates via protein expression or stability.
Figure 3. The Ras1‐Ira1‐PKA pathway regulates Sef1 and Azf1.
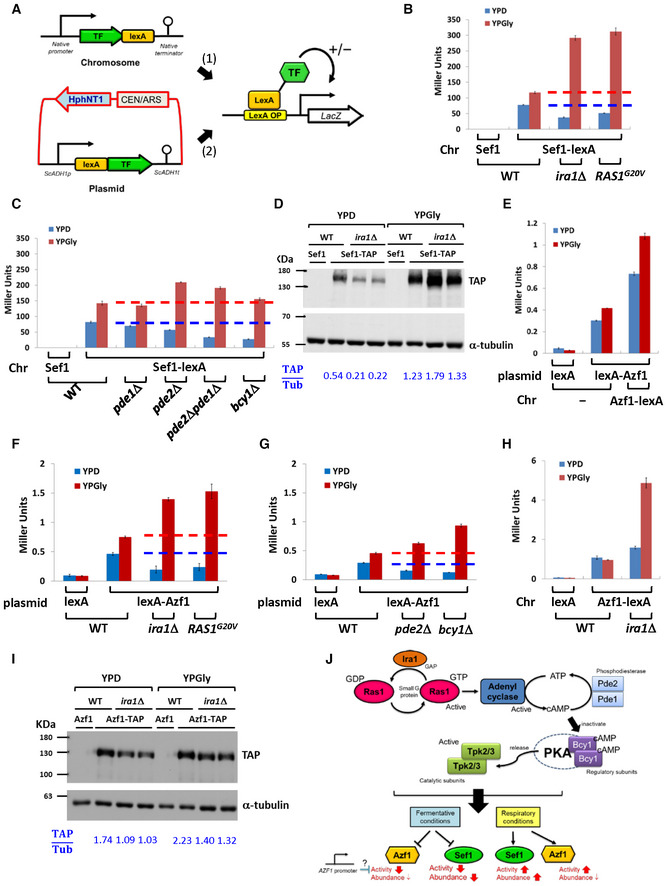
-
ATwo one‐hybrid systems for evaluating transcriptional activities of a TF to the LacZ reporter (Hsu et al, 2021). The first is the chromosome‐integrated system in which the target is C‐terminally fused to the lexA domain and then expressed at the native locus and driven by the native SEF1 promoter. The second is the plasmid‐based system, whereby the target is N‐terminally fused with the lexA domain and is then expressed under the control of a yeast constitutive promoter. Generally, both systems should show consistent results unless the activity of the native promoter is very different from the yeast constitutive promoter.
-
B, CTranscriptional activity of Sef1 in response to hyperactive Ras1‐Ira1 (B) and cAMP‐PKA (C) pathways.
-
DProtein abundance of Sef1 in response to ira1Δ mutation. The normalized band intensities are displayed below the blot as a ratio of TAP‐to‐α‐tubulin signals.
-
ETranscriptional activity of Azf1. Both the plasmid‐based and chromosome systems were used and compared to enhance detection of weak Azf1 activity. The right‐most strain carries two copies of AZF1 (one on the plasmid and the other on the chromosome).
-
F, GTranscriptional activity of Azf1 in response to hyperactive Ras1‐Ira1 (F) and cAMP‐PKA (G) pathways.
-
HTranscriptional activity of Azf1 under the control of a native promoter in response to ira1Δ mutation.
-
IProtein abundance of Azf1 in response to ira1Δ mutation. The normalized band intensities are displayed below the blot as a ratio of TAP‐to‐α‐tubulin signals.
-
JSummarized effects of the hyperactive Ras1‐Ira1‐cAMP‐PKA pathway on Sef1 and Azf1.
Data information: For (B), (C), (E), (F), (G), and (H), LacZ activity was measured using liquid‐galactosidase assay, and results are displayed as average Miller units ± SD from at least three technical repeats. For (B), (C), (F), and (G), the red and blue dashed lines represent the control wild‐type activity in YPD and YPGly, respectively.
Next, we investigated Azf1 according to the same strategy described above, which demonstrated that L. kluyveri Azf1 is a weak transcriptional activator (Fig 3E). Interestingly, hyperactivation of the Ras1‐Ira1‐cAMP‐PKA pathway modulated Azf1 activity in the same way as observed for Sef1 (Fig 3F and G). However, repression of Azf1 activity under the YPD condition was not detected when we used the chromosome‐integrated one‐hybrid constructs (i.e., in which the native promoter was used to express Azf1 fused with C‐terminal lexA; Fig 3H), and altered Azf1 activity could not be simply explained by changes in protein abundance (Fig 3I). These results imply that Azf1 is regulated by a different and more complex Ras1‐Ira1‐cAMP‐PKA pathway‐mediated mechanism than Sef1 (Fig 3J).
Trade‐off effects of the loss‐of‐function azf1 mutation
Mutations of IRA1 and its homologs, as well as for members of the entire Ras1‐PKA pathway, have been identified previously as adaptive hotspots in numerous experimental evolution and suppressor development studies (Kao & Sherlock, 2008; Parts et al, 2011; Wenger et al, 2011; Kvitek & Sherlock, 2013; Lang et al, 2013; van Leeuwen et al, 2016; Venkataram et al, 2016; Huang et al, 2018; Li et al, 2018; Gorter de Vries et al, 2019; Kinsler et al, 2020; Amine et al, 2021; Johnson et al, 2021). Moreover, some arguments suggest that they typically emerge as a response to conditions that involve changes in nutrient abundance, such as leading to uncontrolled cell growth in the absence of glucose (Cazzanelli et al, 2018). Therefore, we chose to focus on the azf1 mutation, the “Dex‐trade‐off and Gly‐compensation” effect, which has not been characterized previously.
To identify the genes regulated by Azf1, first, we confirmed that heat stress did not drastically alter the steady‐state transcriptional activity of Azf1 (Appendix Fig S7A) but only reduced the abundance of Azf1 proteins (Appendix Fig S7B). Accordingly, deletion of AZF1 may trigger responses similar to those observed in cells suffering heat stress. For simplicity, we decided to compare RNA expression profiles directly under the normal temperature (28°C) to reduce profile complexity as much as possible. We undertook five comparisons—sef1Δ/wild‐type, azf1Δ/wild‐type, sef1Δazf1Δ/wild‐type, sef1Δazf1Δ/azf1Δ, and sef1Δazf1Δ/sef1Δ—and differential gene expression for each as summarized in Appendix Fig S7C–G. Detailed profiles and gene ontology (GO) analyses are presented in Datasets EV1, EV2, and [Link], [Link]. In general, we found that the sef1Δ mutation affects fewer genes (Appendix Fig S7C and F) than the azf1Δ mutation does (Appendix Fig S7D, E, and G).
To further link altered gene expression with azf1Δ‐induced phenotypic outcomes, we simplified the enriched GO terms by classifying them as sef1Δ driven (sef1Δ/wild‐type and sef1Δazf1Δ/azf1Δ) or azf1Δ driven (azf1Δ/wild‐type, sef1Δazf1Δ/wild‐type, and sef1Δazf1Δ/sef1Δ). As shown in Fig 4A, under the YPD condition, the azf1Δ‐driven group displays a consistent pattern for enriched GO terms for downregulation of the carbohydrate metabolic process, fatty acid metabolic process, alpha‐amino acid metabolic process, and phosphorus metabolic process. In contrast, the sef1Δ‐driven group only displays consistent enrichment for GO terms relating to downregulation of the isocitrate metabolic process and the TCA cycle. We speculated that the “Dex‐trade‐off” effect of azf1Δ may be attributable to this downregulation. The GO term “carbohydrate metabolic process” of the azf1Δ‐driven group encompasses 80 downregulated genes, including those about core glucose utilization pathways and especially glycolysis (Appendix Fig S8), indicating that glycolysis in the azf1Δ cells may be particularly vulnerable. Indeed, both the azf1Δ and sef1Δazf1Δ mutant lines proved more sensitive to the glycolysis inhibitor, 2‐deoxyglucose (Laussel & Léon, 2020), especially at higher temperatures (Fig 4B and C, and Appendix Fig S9A and B).
Figure 4. Phenotypic response to azf1Δ mutation in the YPD condition.
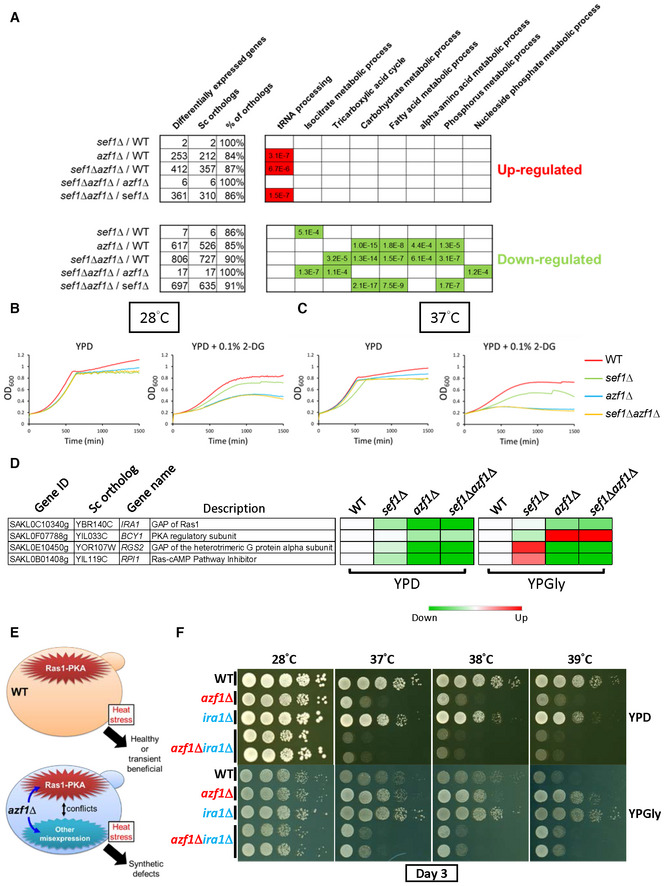
-
ASelected GO terms enriched under the YPD condition. The adjusted P‐values of the hypergeometric test with Bonferroni correction are shown.
-
B, CHypersensitivity of azf1Δ mutants to 2‐deoxyglucose under the YPD condition without heat stress (28°C) (B) or with mild heat stress (37°C) (C).
-
DThe major negative regulators of the Ras1‐PKA pathway are downregulated in response to azf1Δ mutation under the YPD condition. The heatmap was generated from the mean TPM (Transcripts Per Million) ratio of RNA‐seq data relative to wild‐type for each condition. The high‐resolution source table of the heatmap is provided in Dataset EV17.
-
EA proposed model describing how a hyperactive Ras1‐PKA pathway due to downregulation of negative regulators (in the azf1Δ mutant) or ira1Δ mutation (used in experiments) is incompatible with the other gene misexpression induced by azf1Δ.
-
FThe synthetic growth defect of azf1Δira1Δ under the wild‐type background in heat‐stressed conditions.
Moreover, we noticed that several major negative regulators of the Ras1‐PKA pathway, including the aforementioned IRA1 and BCY1, were downregulated under the YPD condition (Fig 4D). Considering that Ras1‐PKA is also a glucose‐responsive signaling pathway (Conrad et al, 2014), misexpression of those negative regulators may contribute to the “Dex‐trade‐off” effect due to conflict between the hyperactive Ras1‐PKA pathway and the phenotypic impacts of other azf1Δ‐driven gene misregulations (Fig 4E). To test that idea, we deleted IRA1 and AZF1 in the wild‐type or sef1Δ background to mimic the extreme downregulation of IRA1. Interestingly, both the azf1Δira1Δ and sef1Δira1Δazf1Δ mutant lines showed a heat stress‐induced synthetic growth defect under both YPD and YPGly conditions (Fig 4F and Appendix Fig S9C). Furthermore, the spores carrying both the mutated ira1 and azf1 alleles from tetrads of mating products between 28°C‐Evo and 39°C‐Evo suppressors also exhibited the synthetic growth defect (Appendix Fig S10A and B). These results not only support the notion that misregulation of the Ras1‐PKA pathway participates in the “Dex‐trade‐off” effect of azf1Δ but also provide corroborating evidence that Ira1 and Azf1 belong to the same functional module.
Similarly, subsequent examination of downregulated genes relating to the GO term “alpha‐amino acid metabolic process” implied that multiple genes involved in Ser, Cys, Asp, Lys, Met, Gln, Glu, Pro, Arg, and Ile metabolism may be defective (Appendix Fig S11A). This scenario is supported by the exacerbated delay in growth (longer lag‐phase) of the azf1Δ and sef1Δazf1Δ mutants when cells were pre‐starved in an amino acid‐depleted medium (Appendix Fig S11B). Notably, we observed delayed growth at normal temperature, which was more severe under heat stress. Thus, taken together, our findings show that under the YPD condition, the azf1Δ mutation places cells in a state of poor core carbon and nitrogen metabolism so that their growth further deteriorates under heat stress, contributing to the “Dex‐trade‐off” effect.
Beneficial effects of the loss‐of‐function azf1 mutation
To connect altered gene expression with the “Gly‐compensation” effect, we investigated the enriched GO term list under the YPGly condition (Fig 5A). The azf1Δ‐driven group displays a consistent pattern of enriched GO terms relating to upregulation of the cell cycle, cytoskeleton organization, DNA repair and replication, response to stress, vesicle‐mediated transport, and chromatin silencing, as well as downregulation of genes related to ribosome/rRNA and tRNA metabolism that are key components in translation. In contrast, the sef1Δ‐driven group only displays consistent enrichment for GO terms pertaining to downregulation of the TCA cycle, as reported in a previous study (Hsu et al, 2021).
Figure 5. Phenotypic response to azf1Δ mutation in the YPGly condition.
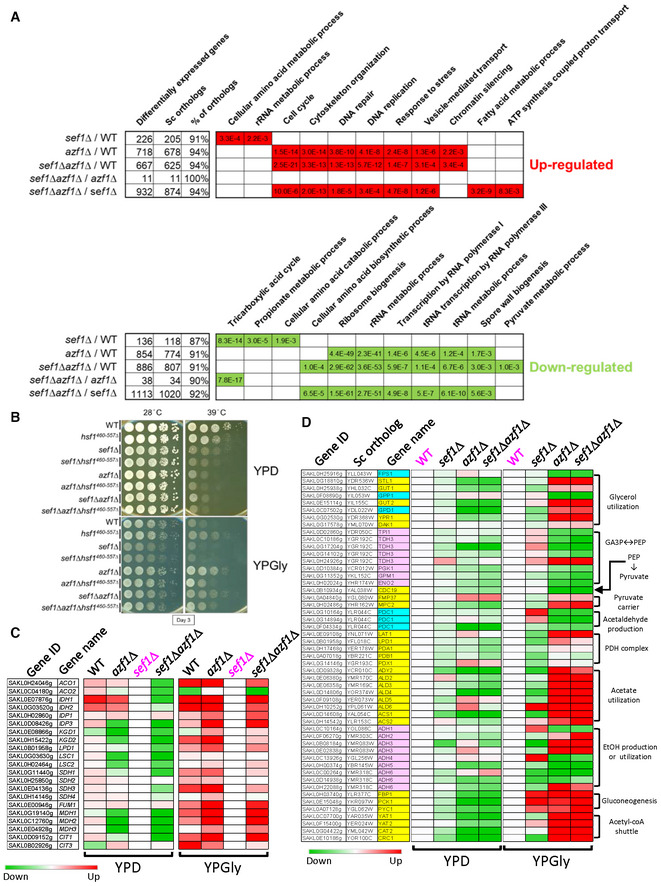
- Selected GO terms enriched under the YPGly condition. The adjusted P‐values of the hypergeometric test with Bonferroni correction are shown.
- The counteracting effect of hypomorphic hsf1 on growth in response to azf1Δ mutation in both wild‐type and sef1Δ backgrounds. HSF1 is an essential gene in yeast (Solís Eric et al, 2016), so a hypomorphic hsf1 mutation with a truncated C‐terminus (460–557 amino acids removed; Sorger, 1990) rather than a null mutant was used in L. kluyveri strains.
- Partial compensation of defective TCA cycle gene expression in sef1Δ mutants by azf1Δ mutation under the YPGly condition. The heatmap was generated using the mean TPM ratio from RNA‐seq data relative to the sef1Δ strain under each condition.
- Transcriptional remodeling of the core glycerol utilization and auxiliary pathways in response to azf1Δ mutation under the YPGly condition. The heatmap was generated using the mean TPM ratio from RNA‐seq data relative to the wild‐type under each condition. The high‐resolution source tables of the heatmaps are provided in Dataset EV17.
The “response to stress” GO term enriched in the azf1Δ‐driven group encompasses 186 upregulated genes, including core heat‐shock response genes and especially heat‐shock proteins (HSPs; Appendix Fig S12A, see “Heat” and “Protein folding” sub‐GO groups), implying a possible link between upregulated HSPs and “Gly‐compensation” under heat‐stressed conditions. HSPs play pivotal roles in protein folding and refolding, as well as in responses to heat and other stresses, and they are globally induced by the transcriptional regulator Hsf1, which is conserved across species (Veri et al, 2018). As expected, we found that a hypomorphic (diminished function) hsf1 mutation partially abrogated the upregulation of representative HSP genes in response to azf1Δ (Appendix Fig S12B), thereby partially limiting the “Gly‐compensation” effect in both azf1Δ and sef1Δazf1Δ mutant lines (Fig 5B). That outcome indicates that the higher basal levels of HSPs induced by Hsf1 contribute to azf1Δ‐induced heat resistance. Moreover, we identified a total of 212 genes related to ribosome/rRNA metabolism and a further 75 genes related to tRNA metabolism that were downregulated in response to azf1Δ (Appendix Fig S13), demonstrating a typical yeast environmental stress response (ESR) in gene expression that involves coupling the upregulation of stress defense genes with downregulation of ribosome biogenesis/protein synthesis genes (Gasch et al, 2000; Ho & Gasch, 2015; Taymaz‐Nikerel et al, 2016).
Furthermore, we noted that the TCA cycle GO term is not enriched among the downregulated genes of the azf1Δ‐driven group, especially for sef1Δazf1Δ/wild‐type, unlike the sef1Δ‐driven group (Fig 5A). Indeed, deletion of AZF1 not only partially restored expression of TCA cycle genes under the YPGly condition (Fig 5C) but it also rescued cellular respiration [as assayed by triphenyl tetrazolium chloride (TTC) reduction assay] (Appendix Fig S14A), thus reinforcing the “Gly‐compensation” effect.
Apart from glycerol, both ethanol and acetate are common non‐fermentable carbon sources preferred by yeast species during respiratory growth, and that trigger a series of coordinated gene expression patterns (Turcotte et al, 2009). Interestingly, in contrast to the pronounced beneficial impact of glycerol, we found that acetate only provided a weak heat stress‐specific benefit to the azf1Δ strains, whereas ethanol did not appear to exert any benefit (Appendix Fig S14B). The same defective growth was observed in the post‐diauxic growth phase of the azf1Δ mutant grown in YPD when ethanol was the major carbon source (Appendix Fig S11B). To link this glycerol‐specific trait to gene expression, we examined more closely the YPGly‐based RNA profiles of the core non‐fermentable carbon metabolic pathways of glycerol utilization (uptake and breakdown), pyruvate production, and pyruvate utilization (more specifically, the PDH‐dependent respiratory pathway and the PDC‐dependent fermentative pathway; Fig 5D). We present in Appendix Fig S14C the remodeled glycerol usage pathways and the predicted metabolic flux in azf1Δ cells (relative to wild‐type) according to the differential gene expression summarized in Fig 5D. In Appendix Fig S14D, we propose a model of glycerol‐driven metabolic remodeling according to Appendix Fig S14C. Our model is based on the notion that azf1Δ cells maintain a high intracellular concentration of glycerol and its derivatives due to increased uptake and restricted pyruvate production (from DHAP to pyruvate), with biased pyruvate flux fueling the TCA cycle due to strong competition for pyruvate between high‐affinity mitochondrial pyruvate carriers plus the PDH complex and the low‐affinity PDC complex (Pronk et al, 1996; Møller et al, 2004).
Moreover, according to the classical definition of “compensation,” a compensatory mutation is a modifier (second) mutation that masks or completely suppresses the defects from the major (first) mutation, and this amelioration should show greater phenotypic gains in the genotypes with the major mutation than the one without (Moore et al, 2000; van Leeuwen et al, 2016). That is, there should be positive epistasis between a compensatory mutation (e.g., loss‐of‐function azf1) and the major mutation (e.g., sef1Δ). To test this idea, we examined the azf1Δ‐induced compensation in gene expression between the sef1Δ and wild‐type backgrounds. As expected, the majority of TCA cycles genes were higher upregulated by azf1Δ under the sef1Δ than the wild‐type background (Fig EV3A), indicating a clear positive epistasis between azf1Δ and sef1Δ genotypes (Fig EV3B). Similar positive‐magnitude epistasis in the differential expression of glycerol utilization and auxiliary pathways as well as the stress‐responsive genes are shown in Fig EV3C–E. Taken together, these results clearly demonstrated that the beneficial azf1Δ causes sef1Δ‐specific compensation in gene expression.
Figure EV3. Positive epistasis in gene expression changes between sef1Δ and azf1Δ genotypes.
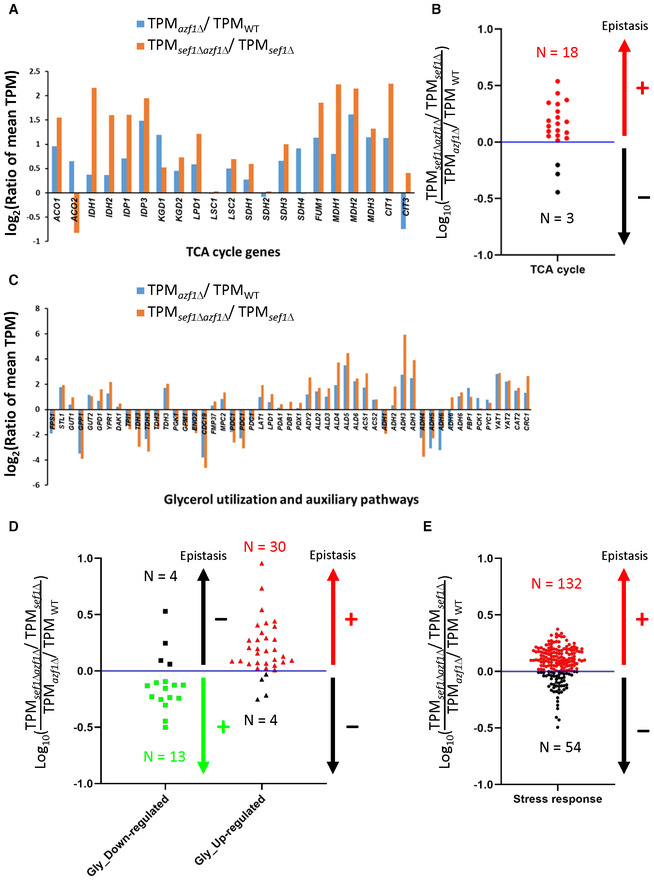
- The compensation of defective TCA cycle gene expression by azf1Δ mutation under the YPGly condition in either wild‐type or sef1Δ background. A total of 21 genes (Fig 5C) were included.
- The epistasis effect of azf1Δ‐induced upregulation of TCA cycle genes in the sef1Δ background compared with the wild‐type.
- The enhanced transcriptional remodeling of glycerol utilization and auxiliary pathways by azf1Δ mutation under the YPGly condition in either wild‐type or sef1Δ background. A total of 51 genes (Fig 5D) were included.
- The epistasis effect of azf1Δ‐induced differential gene expression of glycerol utilization and auxiliary pathways in the sef1Δ background compared with the wild‐type.
- The epistasis effect of azf1Δ‐induced upregulation of stress responsive genes in the sef1Δ background compared with the wild‐type. A total of 186 genes (Appendix Fig S12A) were included.
Data information: For (A) and (C), the gene expression changes are displayed as the log2 values of the mean TPM (from RNA‐seq data) ratios between azf1Δ and AZF1 genotypes under either wild‐type or sef1Δ background. In the Y‐axis, positive values indicate gene upregulation while negative values indicate downregulation. For (B), (D), and (E), the log10 values of the relative mean TPM ratios (the “sef1Δazf1Δ/sef1Δ” group relative to the “azf1Δ/wild type” group) were used to determine the epistasis effects between sef1Δ and azf1Δ genotypes (positive or negative as indicated as “+” or “−”). The dots labeled with red and green indicate that the respective upregulated and downregulated genes show higher azf1Δ‐induced expression changes under the sef1Δ than the wild‐type background (i.e., positive epistasis). The dark blue lines represent the equal relative mean TPM ratio (i.e., there is no detectable epistasis).
Hence, the loss‐of‐function azf1 mutation induces “Gly‐compensation” by: (i) increasing basal transcriptional levels of stress‐defense genes; (ii) restoring TCA cycle gene expression; and (iii) transcriptionally remodeling the metabolic pathways to favor glycerol utilization and more efficient respiration.
azf1Δ cells display cell density‐dependent “Dex‐trade‐off” traits and an evolutionary counteracting strategy
Intriguingly, during growth assays, we noticed unexpectedly that the “Dex‐trade‐off effect” in azf1Δ cells could be alleviated by gradually increasing the initial cell density (Appendix Fig S15). More specifically, the maximal growth rate of azf1Δ cells under “Dex‐trade‐off” conditions was restored to a near wild‐type level when growth was initiated at a higher inoculum size (Fig 6A), hinting that a cell density‐dependent mechanism plays a role in combatting the “Dex‐trade‐off effect.” To prevent extinction, S. cerevisiae cells may display cell density‐dependent cooperative behavior to enable each other and their future generations to survive and replicate at high temperatures (Laman Trip & Youk, 2020). To address if L. kluyveri cells can cooperatively help azf1Δ cells survive, we grew Azf1‐sufficient (denoted AZF1) and azf1Δ strains independently in separate cultures or co‐cultured together and measured the fitness of azf1Δ cells relative to the AZF1 strain by counting viable cell numbers after 20 h of growth under the “Dex‐trade‐off” condition (Appendix Fig S16A). Growth curves (Appendix Fig S16B) revealed that the relative fitness of azf1Δ cells increased under co‐culture conditions relative to the independent culture in both wild‐type and sef1Δ backgrounds (Fig 6B and C). Similar cooperative growth assays on an agar surface (Appendix Fig S16C) generated results consistent with those from the equivalent aforementioned experiments in media (Appendix Fig S16D and E). Taken together, these results indicate enhanced survival of azf1Δ cells under “Dex‐trade‐off” conditions can be facilitated by the presence of surrounding AZF1 cells, mimicking the scenario by which low‐frequency trade‐off mutants can be maintained in a heterogeneous population.
Figure 6. The trade‐off phenotype of azf1Δ mutants is linked to cell wall integrity.
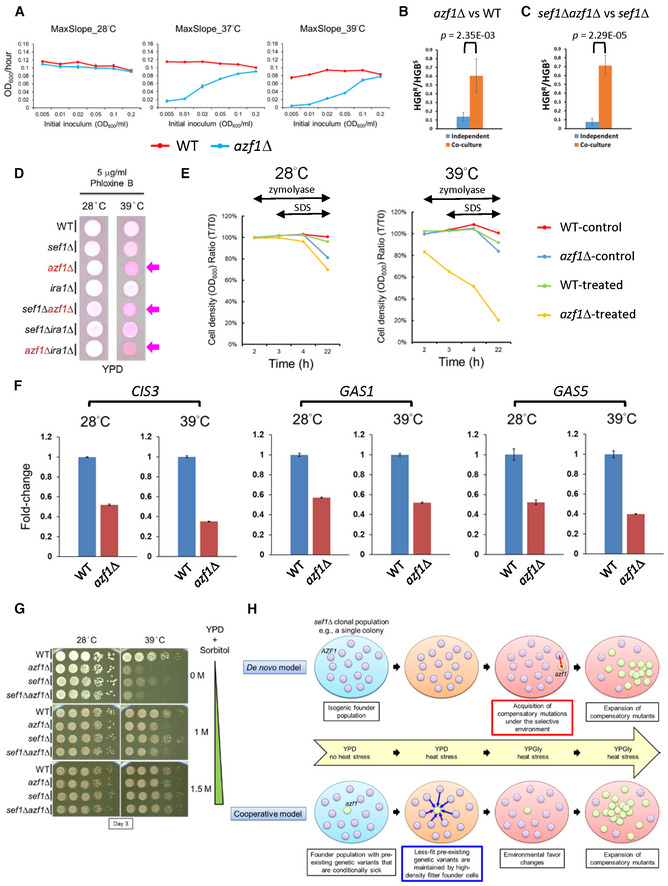
-
AThe effect of inoculum density on the mean maximal growth rate of the azf1Δ population in YPD under heat‐stressed conditions. The growth curves were measured by using a Tecan plate reader with intermittent shaking. Results are displayed as average max slopes ± SD from three technical repeats.
-
B, C(B) The azf1Δ cells are more persistent when co‐grown with wild‐type cells in liquid broth under the “Dex‐trade‐off” condition. (C) The sef1Δazf1Δ cells are more persistent when co‐grown with sef1Δ cells in liquid broth under the “Dex‐trade‐off” condition. For (B) and (C), results are displayed as average HGBR/HGBS ± SD from six technical repeats. HGBR/HGBS represents the ratio of HGB‐resistant and HGB‐sensitive colony numbers. Statistical significance tests were carried out using unpaired Student's t‐tests.
-
DUnder the “Dex‐trade‐off” condition, azf1Δ cells were more severely stained by Phloxine B (pink color). Phloxine B is a negatively charged dye that only stains cells intracellularly when cell membrane intactness has been compromised (Grosfeld et al, 2021). The arrows indicate the azf1Δ‐containing cells that stain heavily by Phloxine B.
-
EThe azf1Δ mutant is more sensitive to mild cell wall disruption. The cells pre‐grown at indicated temperatures in YPD were first treated with 60 units/ml zymolyase at room temperature for 2 h, and then 0.1% SDS was added into the suspension to facilitate cell lysis. Cell intactness (y‐axis) was calculated as the ratio of cell density (OD600/ml) at specific time points relative to T0. Untreated samples were used as the control.
-
FExpression of CIS3 (SAKL0C05676g), GAS1 (SAKL0H00550g), and GAS5 (SAKL0F05456g) in response to azf1Δ mutation under the YPD condition. The relative fold change of each gene is shown as , using CDC34 (SAKL0D02530g) as the endogenous control and the ΔC T value from the wild‐type sample as the corresponding calibration value. Expression levels are displayed as mean fold‐changes ± SD from three technical repeats.
-
GThe addition of sorbitol to the YPD medium to enhance osmolarity suppressed the “Dex‐trade‐off” phenotype of the azf1Δ mutants.
-
HDe novo and cooperative models can explain the rapid evolutionary repair of sef1Δ mutation‐induced phenotypes by AZF1 loss of function. In the de novo model, the azf1 mutant newly forms under the selection brought about by sef1Δ‐suppressive mutations. In the cooperative model, the azf1 mutant pre‐exists as a quasi‐species in the founder population, its persistence being cooperatively supported by its fitter neighboring founder cells under the trade‐off conditions, but it is then selected and undergoes population expansion to be dominant under favorable environmental conditions.
One mechanism by which cell density‐dependent cooperative behavior operates in yeast is secretion of glutathione into the extracellular environment (Laman Trip & Youk, 2020). The level of glutathione secretion required for growth under heat‐stressed conditions depends on the cell population density before extinction. Although it remains unclear how extracellular glutathione helps cells to survive, one study has shown that a mild concentration of surfactants (such as sodium dodecyl sulfate, SDS) to increase cell permeability increased the extracellular concentration of glutathione (Wei et al, 2003). Other studies have indicated that glutathione is required for bacterial and yeast responses to osmotic stress (Smirnova et al, 2001; Jamnik et al, 2006) and that it serves as an osmotic driving force in bile formation (Jamnik et al, 2006). Taken together, these findings prompt the hypothesis that cell density‐dependent cooperative behavior under heat stress may be linked to an osmotic imbalance of cells. Moreover, glutathione might be one but not the only factor contributing to this survival strategy.
Accordingly, we wondered if azf1Δ cells displaying cell density‐dependent cooperative behavior also present an osmotic imbalance. Despite our gene expression data indicating that azf1Δ cells do not preferentially utilize ethanol, that finding is still insufficient to explain the ineffectiveness of ethanol (unlike glycerol) against heat stress. Consequently, we explored if ethanol is also a stressor of cells (Stanley et al, 2010), especially when cell wall integrity (CWI) is compromised (Udom et al, 2019). As expected, compared to wild‐type, all azf1Δ cells not only displayed higher permeability to the dye phloxine B (Fig 6D), but they were also more sensitive to mild cell wall disruption elicited by zymolyase treatment, especially when cells were pre‐grown under heat‐stressed conditions (Fig 6E). Moreover, our finding that gene expression of a vital cell wall glycoprotein (CIS3) and twoβ‐1,3‐glucanosyltransferases (GAS1 and GAS5) required for CWI in yeast (Tomishige et al, 2003; Mazáň et al, 2008) are all downregulated in azf1Δ cells (Fig 6F) supports the notion that the azf1Δ mutants have a weakened CWI. Intuitively, a weakened cell wall (or outer membrane) will trigger hypo‐osmotic stress in cells, leading to cell bursting if the condition is aggravated. Osmotic rebalancing by supplementing the medium with sorbitol can suppress growth defects caused by cell bursting (Lee et al, 1993; Serrano et al, 2006; Beese et al, 2009). We observed complete osmotic rebalancing in azf1Δ mutant cells upon adding 1.5 M of sorbitol into YPD (Fig 6G), implying that the defective CWI of azf1Δ cells alters their optimal osmolarity required for growth and also contributes to their “Dex‐trade‐off” phenotypes. Interestingly, we detected no clear differential gene expression related to glutathione in response to the azf1Δ mutation, indicating that further work is needed to identify the distinct mechanism by which Azf1 in L. kluyveri regulates this cell density‐dependent cooperative behavior.
Discussion
Perturbing the Azf1 transcriptional network can rapidly compensate for disruption of the Sef1 transcriptional network
Our rapid suppressor development strategy allowed us to initiate an evolutionary repair experiment to determine responses to the sef1Δ mutation and to generate a large number of evolved lines carrying early and strongly beneficial mutations with the potential to compensate for the sef1Δ‐caused defects. By performing selection on agar plates, clonal interference (Gerrish & Lenski, 1998) among cells carrying beneficial mutations could be minimized, thereby circumventing a pervasive issue when evolving asexually reproducing organisms in liquid broth (Barrick & Lenski, 2013; Van den Bergh et al, 2018; McDonald, 2019) and maintaining the early adaptive diversity of beneficial mutations (Levy et al, 2015; Blundell et al, 2019). Surprisingly, after phenotyping 240 suppressor clones from two independent lineages, we found that the majority (87.5%) displayed a consistent phenotypic pattern across all tested conditions. Subsequent sequencing and verifications by genetic and phenotypic analyses suggested that they carried a higher proportion of causal mutations hitting IRA1 or AZF1 and other unidentified genes related to Ira1 or Azf1 functions (Figs EV1 and EV2). That finding demonstrates clear evolutionary parallelism and unexpectedly restricted diversity arising from sef1Δ‐adaptive mutations in this batch of the suppressor development experiment.
We consider three possible reasons for that outcome. First, our suppressor colony‐picking strategy could have been biased because we picked the biggest colonies on the agar plates (Appendix Fig S2) and clones of similar fitness most likely carry similar causal mutations, as described above for the beneficial ira1Δ mutation. Second, pre‐existing low‐frequency beneficial mutations in the founder colony may have been selected, with this latter scenario being supported by our identification of the presence of identical mutation types in separate suppressor clones. Indeed, we identified many such cases (Figs 2C and D, and EV1B and EV2B). Identical mutations are less likely to form independently by chance in an asexual population, so we conclude that these mutations, at least in this batch of the experiment, may have pre‐existed in the quasi‐species founder colony that was picked (Appendix Fig S1D and E), acting as rapid adaptive solutions to the changing environment (Teng et al, 2013). Third, the specific type of driving forces may induce specific mutations, as supported by the triple beneficial effects of “restored respiration” (Fig 5C and Appendix Fig S14A), “elevated heat‐shock defense” (Appendix Fig S12), and “augmented glycerol utilization” (Fig 5D) elicited by the azf1 mutation, which together are required for cell survival under our heat‐stressed YPGly condition.
Notably, the azf1 mutations also partially restored the defective TCA cycle gene expression in sef1Δ mutants under the YPGly condition (Fig 5C). This suggests that massive transcriptomic perturbations accompanying fitness restoration might be a general mechanism to facilitate compensatory evolution, as revealed by other studies (Szamecz et al, 2014; McCloskey et al, 2018). However, how azf1 loss‐of‐function assists to stabilize a new transcriptional network without Sef1 is still unclear. We conducted a simple experiment to test whether Azf1 affects IDH2 expression directly or indirectly in L. kluyveri. The IDH2 is a TCA cycle gene directly regulated by Sef1 (Hsu et al, 2021), and its promoter carries one confirmed Sef1‐binding motif and one putative Azf1‐binding motif adjacent to each other (Appendix Fig S17A and B). We deleted the putative Azf1 motif on the IDH2 promoter in the wild‐type and found out this motif loss did not lead to the upregulation of IDH2 the same as azf1Δ did (Appendix Fig S17C). This finding suggests that azf1Δ upregulates TCA cycle genes indirectly, possibly through other down‐regulated transcriptional regulators in response to azf1Δ (Dataset EV15), especially through those transcriptional repressors (Appendix Fig S17D). Elucidating the contributions of these factors to the azf1Δ‐induced gene expression is a potential direction in the future.
The sef1Δ does not provide a strong founder effect to the evolutionary trajectories
Given the potential for founder effects and epistasis (Barrick et al, 2010; Jerison et al, 2017; Wünsche et al, 2017; Rojas Echenique et al, 2019), we wondered whether the sef1Δ founder genotype constrained or shaped the evolution trajectories. We first confirmed that sef1Δ did not result in higher suppression rates than the wild‐type did in our suppressor‐developing conditions by using fluctuation assays (see “mutation rates” in Appendix Fig S18A and B). Then, we demonstrated that ira1Δ and azf1Δ did not cause better fitness improvement (positive epistasis) in the sef1Δ than in the wild‐type backgrounds by using epistasis analyses (Dataset EV13). These two results not only suggest that there is no strong founder effect of sef1Δ but also imply that the wild‐type cells could also evolve in this way. Therefore, we performed a new batch of suppressor development at 28 and 39°C including the wild‐type (Dataset EV16). In the 28°C‐WT‐Evo clones, although at a lower frequency (3 of 20), we still identified evolved clones showing desiccation hypersensitivity (suggesting the occurrence of ira1 loss‐of‐function mutations or other mutations in related pathways; Fig EV4A). Similarly, in the 39°C‐WT‐Evo clones, we also identified 5 of 20 evolved clones showing “Dex‐trade‐offs” (suggesting the occurrence of azf1 loss‐of‐function mutations or other mutations in related pathways). Notably, the new batch MATa and MATα 28°C‐sef1Δ‐Evo clones still showed a high frequency of desiccation hypersensitivity (18/19 and 17/20, respectively) similar to the first batch (Fig EV4A). In contrast, the new batch MATa and MATα 39°C‐sef1Δ‐Evo clones did not have a high frequency of “Dex‐trade‐off” suppressors (2/20 and 3/20, respectively; Fig EV4B). We think the picked quasi‐species founder colonies might still contribute to this discrepancy between two batches of suppressor development experiments.
Figure EV4. Phenotypic surveys for the clones from a new batch of suppressor development including the wild‐type controls.
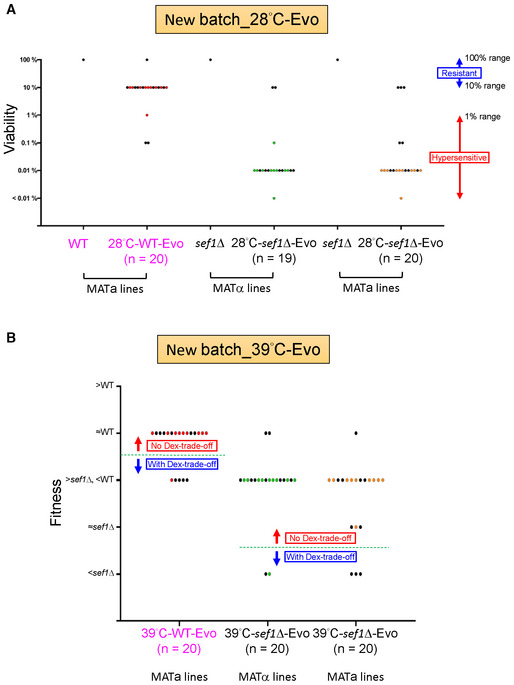
- The “desiccation hypersensitivity” phenotypic spectrum of 20 each 28°C‐evolved new batch clones (one clone of the new batch MATα 28°C‐sef1Δ‐Evo was removed from the collection due to contamination). The desiccation sensitivities of each clone are displayed according to the qualitative viabilities (Dataset EV16) as described above. The sef1Δ background seems more prone to generate adaptive mutations with desiccation hypersensitivity (18/19 in MATα and 17/20 in MATa lines) than the wild‐type background (3/20).
- The “Dex‐trade‐off” phenotypic spectrum of 20 each 39°C‐evolved new batch clones. The “Dex‐trade‐off” phenotypes of each clone are displayed according to the simple fitness scores under the YPD_37°C condition (Dataset EV16) as described above. Clones with score 1 (with fitness worse than sef1Δ strain) were defined as “with Dex‐trade‐off” for the new batch 28°C‐sef1Δ‐Evo while a score ≤ 3 (with fitness worse than the wild‐type strain) for the 28°C‐WT (wild‐type)‐Evo. About 14.74% (23/156) of clones in this batch showed no “Dex‐trade‐off.” Unlike observed in the first batch of suppressor development, there is no clear preference in any genetic background to generate a higher frequency of adaptive mutations with “Dex‐trade‐off” (5/20 in the wild‐type, 2/20 in MATα, and 3/20 in MATa lines).
Data information: For (A) and (B), the dots labeled with colors (red, green, or orange in different lines) stand for clones isolated from the same population in each line while the dots labeled with black from different populations.
Nevertheless, our findings raise a hypothesis that the ira1 and azf1 mutants can form subpopulations in the wild‐type population first and then alleviate the deleterious effects of the following spontaneous sef1 mutations (Fig EV5). This strategy may allow the sef1 mutations to be fixed in the population as long as the sef1 mutants are not less competitive in future environments.
Figure EV5. The historical contingency model for the fates of spontaneous deleterious sef1 loss‐of‐function mutations.
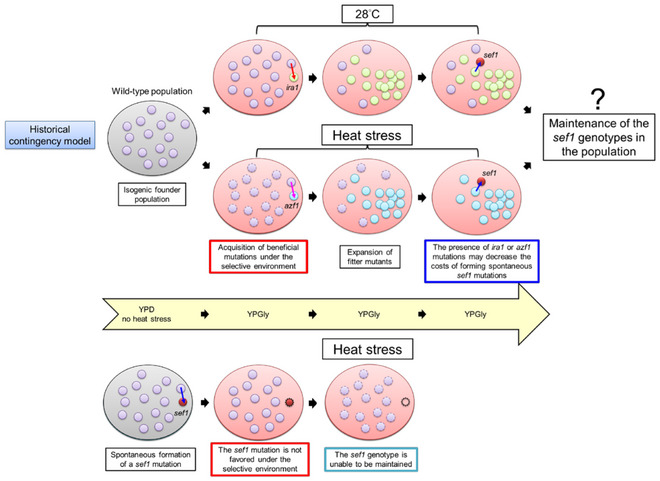
In this hypothetical model, the upper panel shows that the subsequent formation of the sef1 loss‐of‐function mutations under mild (28°C) or harsh (heat stress) selective conditions are preserved by the presence of pre‐existing primary beneficial mutations (e.g., ira1 or azf1) in the founder population (e.g., the wild‐type population). The primary mutations alleviate the deleterious effects of sef1 mutations. The loss‐of‐function sef1 genotypes are unable to be preserved in the population without the primary mutations (bottom panel). However, whether the secondarily formed sef1 subpopulation can compete with the primary population and then expand is not guaranteed, possibly depending on their pleiotropies and the selections by changing environments in the future.
Glucose, Ras1‐PKA signaling, and Azf1
The beneficial effects of the ira1 loss‐of‐function mutation arise from hyperactive Ras‐PKA signaling, leading to uncontrolled cell growth in the absence of the yeast growth‐promoting factor glucose, reminiscent of tumor cell growth (Cazzanelli et al, 2018). That mutation exerted a more general fitness‐improving impact (at least in the conditions used in our study), eliciting a “double‐compensation” effect irrespective of the YPD and YPGly conditions. In contrast, the azf1 loss‐of‐function mutation is pleiotropic in terms of the aforementioned triple beneficial effects and its deleterious impacts relating to mildly defective core carbohydrate metabolism (Fig 4 and Appendix Fig S8), amino acid metabolism (Appendix Fig S11), and CWI (Fig 6). All those defects only arose under the YPD condition and they were exacerbated by heat stress, indicating that they represent life‐history “trade‐offs” (Garland, 2014) and that the azf1 mutant cells are typical specialists (Van den Bergh et al, 2018).
Interestingly, although Azf1 appears functional under both glucose and glycerol conditions, we believe that glucose is the environmental factor sensed by Azf1, that is, Azf1 regulates gene expression in response to the presence or absence of glucose. This speculation is supported by the facts that: (i) Azf1 activity is regulated by the Ras1‐PKA pathway (Fig 3), which is mainly glucose responsive in yeast (Conrad et al, 2014; Cazzanelli et al, 2018); (ii) azf1Δ triggered the opposite expression pattern between stress response and ribosomal genes (Appendix Figs S12A and S13). This opposite expression pattern is also regulated by the PKA pathway in response to the glucose availability (Conrad et al, 2014); and (iii) the presence of glycerol in YPD did not convert “Dex‐trade‐off” to “Gly‐compensation” (Appendix Fig S19), implying that glucose is the trigger. Elucidating the molecular components involved in these processes is an interesting topic for future exploration.
Cooperation maintains trade‐off genotypes
We have revealed that pleiotropic defects (misregulation of multiple TCA genes due to the sef1Δ mutation in this study) can be simultaneously compensated in a short timeframe simply by means of another pleiotropic change (i.e., AZF1 loss of function). Despite potentially being maintained by balancing selection (Mérot et al, 2020), this new pleiotropic change may remain concealed by accompanying trade‐offs within a short evolutionary timescale, especially in unstable environments (Chen & Zhang, 2020). Here, we have demonstrated cell density‐dependent mitigation of such trade‐offs, whereby azf1Δ individuals can persist when densely surrounded by founder cells or until the density of azf1Δ progeny exceeds a threshold (Fig 6A–C and Appendix Fig S16D and E). Although the underlying mechanism remains unknown, this “cooperative persistence” enhances the adaptive potential not only of de novo azf1 mutants when unfavorable environments are encountered but also of pre‐existing azf1 mutants before experiencing selection (Fig 6H), enabling a contingent secondary adaptive step to repair the trade‐offs (Aggeli et al, 2021).
Frequency‐dependent selection (FDS) describes how the fitness of a genotype or phenotype in a population is related to its frequency in the population (Ayala & Campbell, 1974). Positive FDS is a selection regime whereby the fitness of a phenotype increases with its frequency (Ayala & Campbell, 1974). The cell density‐dependent mitigation of trade‐offs in azf1Δ cells represents a simple case of positive FDS. In early models, positive FDS was predicted to reduce diversity due to systematic loss of rare polymorphisms. However, more recent theories predict that positive FDS maintains rather than reduces diversity when populations patchily occupy spatially structured habitats (Molofsky et al, 2001; Molofsky & Bever, 2002; Rendueles et al, 2015). Our evolutionary repair experiments, with characteristics of changing environments (fermentative vs. respiratory; normal vs. heat stressed), local habitats (growth on agar plates), and cell density‐dependent cooperation, is an intuitive example showing that positive FDS maintains microbial diversity and facilitates evolution.
The sef1Δ‐suppressive mutations of AZF1 compensate for perturbed expression of vital TCA cycle genes under selective conditions, reflecting a type of evolutionary tinkering and stabilizing selection (Lavoie et al, 2010; Signor & Nuzhdin, 2018, 2019). The multiple beneficial and trade‐off mutations that accumulate as a consequence of ongoing evolutionary repair processes represent a good source of molecular diversity (LaBar et al, 2020). From the perspective of the macroevolution of transcriptional networks, our study illuminates how compensatory evolution facilitates the stabilization of TCA gene expression under specific conditions. The compensatory evolution maintains the fitness of the mutant cells losing Sef1 and allows a new transcriptional regulatory network without Sef1 to persist, enabling transcriptional rewiring to continue.
Materials and Methods
Genome resources
The genome sequence of Lachancea kluyveri (CBS 3082) was originally from Bioproject accession number PRJNA1445. The sequence and improved annotation can be downloaded from the GRYC (Genome Resources for Yeast Chromosomes; http://gryc.inra.fr) database (Brion et al, 2015; Hsu et al, 2021). The sequences and structures of the L. kluyveri MATa and MATα loci were as per contigs under accession numbers AACE03000005 (Consortium et al, 2009) and AACE01000014 (Butler et al, 2004), respectively.
Strains and plasmid construction
To avoid confusion, we have adopted the term “wild‐type” to describe the parental genotypes used in our study, each of which may carry auxotrophic mutations or specific genetic manipulations, and they acted as reference strains for all comparative experiments unless otherwise stated. The L. kluyveri strains, primers, and plasmids used in this study are listed in Dataset EV14. All DNA fragments used in cloning were PCR amplified using Phusion High‐Fidelity DNA polymerase (F530L, Thermo Fisher Scientific, USA). The PCR products and restriction‐digested products were purified by PCR cleanup or gel extraction using an AccuPrep® PCR/Gel Purification Kit (K‐3037, Bioneer, Korea). DNA cloning into plasmids was achieved by using ligation (T4 DNA ligase, M180A, Promega, USA) or by means of an In‐Fusion® HD Cloning Kit (639650, ClonTech by Takara, Japan). All plasmids were extracted and purified using a Presto™ Mini Plasmid Kit (PDH300, Geneaid, Taiwan). Successful strain constructions were confirmed by genomic DNA extraction, as described previously (Hsu et al, 2011), followed by PCR diagnostics using a homemade Taq DNA polymerase for genotyping.
For gene deletions, the DNA fragments of each deletion module consisting of a selection marker flanked by 5′ and 3′ sequences homologous to the target locus were created by overlap‐extension PCR (Shevchuk et al, 2004). The genes deleted in this study are as follows: SEF1 (SAKL0F12342g), CHS3 (SAKL0C11726g), AZF1 (SAKL0E10714g), IRA1 (SAKL0C10340g), PDE1 (SAKL0F00638g), PDE2 (SAKL0D14344g), and BCY1 (SAKL0F07788g).
For lexA and TAP tagging of Azf1, we used a modified (Hsu et al, 2021) SAT1 flipper method (Reuß et al, 2004). The TAP‐tagging vector pSFS2A‐TAPSacISacIIMut‐28 has a second SacII site near the C‐terminus of the TAP sequence, rendering cloning at the 3′ multiple cloning sites (MCS) problematic. To create a new TAP‐tagging vector, we mutated the second SacII site by amplifying it from pSFS2A‐TAPSacISacIIMut‐28 using the primer pair TAP‐1‐ApaI and TAP‐2‐SacIIMut‐XhoI, and then ligating the product between ApaI‐XhoI sites in the pSFS2A vector to generate pSFS2A‐TAP‐New‐2. The 978‐bp C‐terminal sequence (+1,480 to +2,457 of the ORF without the STOP codon) of AZF1 was cloned into the pSFS2A‐lexA‐5 or pSFS2A‐TAP‐New‐2 vectors between the KpnI‐ApaI sites using In‐Fusion®, and the 411‐bp 3'‐UTR (terminator, +1 to +411 from the STOP codon) sequence of AZF1 was ligated between the SacII‐SacI sites to generate the lexA‐ or TAP‐tagging plasmid for AZF1. The KpnI‐SacI‐digested fragments from both tagging plasmids were used to transform cells. The SklexAOPlacZ‐1 reporter strain and derivatives were transformed to create chromosome‐integrated Azf1 one‐hybrid strains, whereas the JYL1897 wild‐type strain and derivatives were transformed to generate Azf1‐TAP strains. To produce plasmid‐based Azf1 one‐hybrid strains, the entire 2,460‐bp AZF1 ORF with STOP codon was cloned in‐frame into pRS41H‐lexA‐3 between the BamHI‐NotI sites downstream of the lexA sequence, thereby generating pRS41H‐lexA‐SkAzf1‐1, and the plasmid was transformed into the SklexAOPlacZ‐1 strain and derivatives. For Sef1, previously described one‐hybrid and TAP‐tagged strains were used (Hsu et al, 2021).
To create the RAS1 G20V hyperactive one‐hybrid strains, we used the SAT1 flipper method. Gly20 of RAS1 (SAKL0E10252g) was first mutated to Val by overlap‐extension PCR‐based site‐specific mutagenesis (Sambrook, 2001). The 1,395‐bp 5′‐flanking region of the RAS1 G20V fragment (−424 from ATG to STOP) and its 297‐bp 3′ flanking region (+89 to +385 from STOP codon) were then ligated into the 5' and 3' MCSs of the pSFS2A vector between the ApaI‐XhoI sites and the SacII‐SacI sites, respectively. To transform cells, the ApaI‐SacI‐digested fragments from the pSFS2A‐Ras1G20V‐14 plasmid were used during transformation to replace the wild‐type allele.
To create the hsf1 (SAKL0A04576g) hypomorphic strains, we used the DAmP (decreased abundance by mRNA perturbation; Breslow et al, 2008) and decrease‐of‐function hsf1 mutant (Sorger, 1990). In brief, the 475 bp DNA fragment of C‐terminal‐deleted (Δ460–557 aa) hsf1 (SAKL0A04576g, +903 to +1,377 from ATG), a drug marker (KanMX6 or NatMX4), and the 355‐bp 3′ flanking region (+27 to +381 from the STOP codon) were PCR fused and used during transformation to replace the wild‐type allele.
For mating‐type switching from MATa to MATα, we again used the SAT1 flipper method. The 499‐bp 5′ flanking region of the MATα locus near the DIC1 locus, together with the 3,206 bp fragment composed of the 2,587‐bp MATα locus and the 619‐bp 3′ flanking regions of the SLA2 locus, were ligated into pSFS2A vector between the KpnI‐ApaI sites and NotI‐SacII sites, respectively, to create the mating‐type switching plasmid pSFS2A‐5f‐SkMATα‐1. To transform cells, the KpnI‐SacII‐digested fragments from the pSFS2A‐5f‐SkMATα‐1 plasmid were used.
To create the IDH2 (SAKL0G03520g) promoter‐LacZ plasmids, the promoter sequence (−437 to −1 from ATG of IDH2) was PCR amplified and cloned into the pRS41K‐LacZ reporter plasmid between the KpnI‐ApaI sites using by the In‐Fusion method. The putative Azf1‐binding site was deleted by using overlap‐extension PCR‐based mutagenesis. The promoter‐LacZ strains of IDH2 were created by transforming JYL1897 and SkAzf1HA1 with the corresponding plasmids. The transformants were selected in YPD + G418 medium.
All yeast transformations were performed by using electroporation and transformed cells were then selected as described previously (Hsu et al, 2021). Notably, since the Candida albicans MAL2 promoter on the SAT1‐FLIP cassette is leaky in L. kluyveri, the L. kluyveri SAT1‐FLIP transformants could only be selected and propagated in YPD + Nou10 (10 μg/ml Nouseothricin) plates during strain construction procedures. The integrated SAT1‐FLIP cassette does not support efficient growth of L. kluyveri cells in liquid broth with Nouseothricin.
Media, important chemicals, and growth conditions
All media and chemicals used in this study are listed with abbreviations in Dataset EV14. All agar plates contained 2% agar. The basal YP (1% yeast extract and 2% peptone)‐based media were sterilized by autoclaving, except for YPEtOH (YP + 2% ethanol). YPEtOH and all synthetic media were sterilized by filtering through 0.2 μm cup filters (Nalgene™ Rapid‐Flow™ Sterile Single‐Use Bottle Top Filters, 595–4,520; Thermo Fisher Scientific, USA). Some modifications to the recipes for synthetic media are noted in Dataset EV14. All cultures in culture tubes were grown on a drum roller rotating at 65–80 rpm. The normal growth temperature was 28–30°C. Heat stress was applied at 37–39°C, as indicated in the experiments.
Evolutionary repair experiments by suppressor development
The entire procedure is summarized in Appendix Fig S2. In detail, the MATa (SkSef1KChs3HA2) and MATα (SkSef1KChs3Hα1) SEF1‐deleted founder strains (sef1Δ::KanMX6, chs3Δ::HphMX4, ura3 −) were inoculated into a 20‐ml YPD culture and grown at 30°C. After overnight growth, the cells were harvested and resuspended in 2 ml sterile ddH2O at a cell density of ~40 OD600/ml. Approximately 100–120 μl of cell suspensions containing 5 OD600 cells (~108 CFU) were plated onto fresh YPGly plates, with 3–5 repeats per condition. The plates were incubated at 28, 37, 38, or 39°C. After 8 days, outcompeting colonies were re‐streaked on YPGly plates and selected for another 5 days at 28, 37, 38, or 39°C, respectively. Only one large single colony was picked from each re‐streaked population, and then the clones were streaked on YPD + HGB plates and incubated at 28°C for 2–3 days to confirm the original chs3Δ::hphMX4 genotype and to exclude contamination. The HGB‐resistant clones were picked and inoculated into 1‐ml YPD + G418/well cultures in U‐shaped 96‐deep‐well plates (Nunc™ 96‐Well Polypropylene DeepWell™ Storage Plates, 278743, Thermo Fisher Scientific) covered with sterile breathable sealing film (BF‐400‐S, Axygen by Coring, USA). The deep‐well plates were incubated with shaking (Micromixer MX4, FINEPCR, Korea) at 30°C for 24 h to amplify the cells, before confirming the original sef1Δ::KanMX6 genotype. The cells from saturated cultures were spun down and concentrated to ~130 μl, before adding 50 μl of 85% sterile glycerol and transferring them to 96‐well plates (Tissue Culture Testplate 96 wells F‐bottom, 92096, TPP, Switzerland) sealed with adhesive foil (UTI 3101, Smartgene by Ultra Violet, Taiwan) for −80°C storage.
Estimation of suppression rates
Suppression rates were estimated by performing a fluctuation analysis. To set up the primary culture, a single colony of each tested strain, the SEF1‐deleted founder strain (SkSef1KChs3Hα1) and the control wild‐type SEF1 founder strain (SkChs3HA1), was inoculated into a 20 ml YPD culture and grown at 30°C. After 22 h of growth, the cells from the 1 ml culture were harvested and resuspended in sterile ddH2O at a cell density of 1 OD600/ml. The cell suspensions of primary cultures were then used to seed 32 independent secondary cultures (100 μl in 10‐ml YPD, final 0.01 OD600/ml, and 150 μl/well) in U‐shaped 96‐deep‐well plates (Nunc™ 96‐Well Polypropylene DeepWell™ Storage Plates, 278743, Thermo Fisher Scientific) covered with sterile breathable sealing film (BF‐400‐S, Axygen by Coring, USA). The deep‐well plates were incubated with shaking (Micromixer MX4, FINEPCR, Korea) at 30°C to allow accumulation of adaptive mutations. After 20 h of growth, the cultures were harvested and 800‐fold diluted in PBS. The cell densities were then estimated by using the Scepter™ Handheld Automated Cell Counter (PHCC00000, Millipore by Merck). The cells of each secondary culture were diluted in sterile ddH2O at a cell density of 1E+07 CFU/ml. Cells suspensions were track plated (50 μl/track, 8 tracks/plate) onto YPGly plates (Square petri dish; D210‐16, 100 × 100 × 15 mm, Simport Scientific) and incubated at 28 and 39°C for 7 days. Adaptive (out‐competing) colonies were counted and mutation rates were computed using the online Fluctuation AnaLysis CalculatOR “FALCOR” (https://lianglab.brocku.ca/FALCOR/; Hall et al, 2009). The results are displayed in Fig EV2.
The outcompeting clones evolved from the control wild‐type SEF1 founder strain (28 and 39°C‐WT‐Evo) were picked, purified, and stocked as described above with some modifications. Briefly, there were 20 clones picked, and the first 10 clones (clones 01 to 10) were picked from the same evolved population (the same track on the plate) while the other 10 clones (clones 11 to 20) were picked from independent populations (only one clone/track). Generally, we picked the biggest outcompeting colonies among the populations.
Genetic analyses by tetrad dissection
To mate yeast cells, 750 μl (~4 OD600 containing ~1 × 108 CFU) each of MATa and MATα cells from YPD overnight cultures grown at 28°C for at least 20 h were mixed and harvested. The mixed‐cell pellets were washed once with 1 ml sterile ddH2O, resuspended in 10 μl sterile ddH2O, and then spotted onto dry YPD plates. After growing at 28°C for 5–7 h, the cells from the spots were separated by streaking, and candidate zygotes were picked by using a Tetrad Dissection System [Nikon Eclipse 50i Microscope (Nikon, Japan) equipped with a TDM50™ Micromanipulator (Micro Video Instruments, Inc., USA) and a dissection needle (NDL‐010, Singer Instruments, UK)]. The candidate zygotes were incubated on YPD plates at 28°C for 2–3 days and the heterozygous genotypes at the MAT loci were confirmed by gDNA extraction followed by PCR diagnosis for both MATa and MATα alleles.
For sporulation, 1 ml zygotes from YPD overnight culture were pelleted down, washed once with 1 ml sterile ddH2O, and then inoculated into 3 ml Spo medium (2% potassium acetate pH 9, optional: +1% adenine and 1% tryptophan) at 23–25°C. After 5–7 days, the asci from 50 μl sporulation culture were harvested, resuspended in 10 μl of 1 mg/ml zymolyase working solution diluted from 40 mg/ml zymolyase stock solution [Zymolyase® 100 T (07665–55, Nacalai Tesque Inc., Japan), 1× TE (10 mM Tris‐Cl, 1 mM Na2EDTA pH 8), and 5% glucose] in sterile 1 M sorbitol or ddH2O, and digested at room temperature for 5–8 min. The digested asci were streaked on dry YPD plates and the four spores from each tetrad were picked and dissected onto the space of the same YPD plate. The dissected sister spore sets were incubated at 28°C for 2–3 days for further genotyping and phenotyping.
Notably, all strains used for tetrad dissection were in the chs3Δ background, which breaks the interspore bridges between sister spores in asci (Coluccio & Neiman, 2004) and allows separation of sister spores, making L. kluyveri tetrad dissection achievable (Sigwalt et al, 2016).
Sanger sequencing for specific mutations on the AZF1 and IRA1 loci of the spores from tetrad dissection
To sequence the AZF1 locus, the entire AZF1 ORF (−21 from ATG to +17 from STOP) was PCR amplified using Phusion High‐Fidelity DNA polymerase (F530L, Thermo Fisher Scientific) with the primer pair SkAzf1‐UpATG‐1 and SkAzf1‐STOPdown‐2 and an annealing temperature of 52°C. The product was purified by PCR cleanup using an AccuPrep® PCR/Gel Purification Kit (K‐3037, Bioneer), before being subjected to Sanger sequencing with primer SkAzf1‐555‐1‐seq 1 or SkAzf1‐1151‐1‐seq 1. To sequence the IRA1 locus, the second half of the IRA1 ORF (+4,744 from ATG to +40 from STOP) was PCR amplified as detailed above, but with the primer pair SkIra1‐4744‐1‐seq 1 and SkIra1‐STOPdown‐2 and an annealing temperature of 58°C, before undergoing purification and Sanger sequencing with primer SkIra1‐5356‐1‐seq 1 or SkIra1‐5946‐1‐seq 1. All primers used are listed in Dataset EV14. Notably, the AZF1 and IRA1 in JYL1897 (our lab parental strain) background carry.
Sanger sequencing for the whole AZF1 and IRA1 loci of the suppressor collections
The AZF1 locus was sequenced as described above but the entire AZF1 ORF was sequenced using seven sequencing primers. To sequence the IRA1 locus, the first half (−25 to +4,854 from ATG) and the second half (+4,744 from ATG to +40 from STOP) of the IRA1 ORF were PCR amplified as detailed above, with the primer pair SkIra1‐UpATG‐1 and SkIra1‐4854‐2‐seq 1 and primer pair SkIra1‐4744‐1‐seq 1 and SkIra1‐STOPdown‐2, respectively. The product was purified by PCR cleanup using a Zymo Genomic DNA clean & concentration™ kit‐25 (Zymo Research, USA), before undergoing Sanger sequencing with 17 sequencing primers. All primers used are listed in Dataset EV14.
Phenotypic assays
The spot assays, growth curve assays, and TTC reduction assays were performed as described previously (Hsu et al, 2021) with minor modifications. For the spot assays, cells grown in YPD overnight at 28°C were harvested by centrifugation (Eppendorf 5810R centrifuge; A‐4‐62 rotor; 1,500 g; 3 min; 25°C) and serially diluted to the desired cell density with sterile ddH2O. Each dilution (107–103 CFU/ml) was spotted onto agar plates (5 μl/spot, about 105–101 CFU/spot) and incubated at 28°C or other temperatures as indicated. The plates were scanned using a scanner (Epson Perfection V800 Photo) and images were recorded after 3 days or as otherwise indicated.
For the growth curve assays, cells grown overnight in YPD (or other media as indicated) at 28°C were inoculated into 120 μl YPD medium in a 96‐well plate (Tissue Culture Testplate 96 wells F‐bottom, 92096, TPP) at a cell density of 0.2 OD600/ml at indicated temperatures. Cell growth was measured at OD595 in “2 × 2 multiple reads per well” mode every 12 min using a Tecan plate reader (Infinite 200 PRO, Tecan, Switzerland). Tecan software Magellan Version 7.2 was used for data acquisition and analyses. For the Magellan method, we selected the plate definition “[TPP96ft]‐Tecan Plastic Products AG6 Flat Transparent.” Each 12 min cycle comprised 3 min reading, 1 min shaking, 3 min standing, 1 min shaking, 3 min standing, and 1 min shaking. For the glycolysis‐inhibited medium, 0.1% 2‐deoxy‐D‐glucose (D8375, Sigma) was added to the YPD medium. For amino acid pre‐starvation, we used SM + 2X Ura medium (SM supplemented with 80 mg/l uracil) given that we employed ura − strains in this study. Pre‐starvation was performed by inoculating a single colony from a YPD plate into a 5‐ml SM + 2X Ura culture and allowing it to grow for 23 h at 28°C. For cell density‐dependent growth assays, growth curves using different initial cell densities (0.005, 0.01, 0.02, 0.05, 0.1, or 0.2 OD600/ml) in YPD were measured for 24 h at 28, 37, or 39°C. Maximal growth rates were determined by calculating the maximum slope of each growth curve based on 30–40 time points in Magellan software, as illustrated in Appendix Fig S1C.
For the epistasis analysis, the growth curve assays were used as described above with modifications. Briefly, two initial cell densities (0.01 and 0.4 OD600/ml) were used to keep or eliminate the cell density effect of azf1Δ. Maximal growth rates from three technical repeats were determined by calculating the maximum slope of each growth curve based on 10 time points in Magellan software. The relative fitness was calculated by comparing it to the wild‐type and displayed as %. The epistasis status was determined by using the multiplicative model (van Leeuwen et al, 2017), which suggests that the genotype with a pair of loci has fitness values equal to the product of fitness values at the two different loci if there is no epistasis between them.
For the desiccation assays, a single colony of each tested strain was inoculated in 3 ml YPD and grown at 28°C for 20 h. Cells were then harvested and diluted at cell densities of 1 OD600/ml with sterile ddH2O. Two 1 ml aliquots (containing ~107 CFU) were made for each tested strain in 1.5 ml tubes. The desiccated sample was prepared by spinning down cells, removing supernatants, sealing the opened tube with a sterile breathable sealing film (BF‐400‐S, AXYGEN, USA), and air‐drying the cell pellets at 28°C for 20 h. The control samples were prepared by retaining the supernatants and keeping the tubes capped. After 20 h, the cells were rehydrated in 1 ml sterile ddH2O for 1 h. The viability was determined qualitatively by spot assays on YPD plates as described above after 3 days of incubation at 28°C. The desiccation sensitivity was used to represent the severity of ira1 loss‐of‐function mutations or other mutations generating the same effect as ira1 loss‐of‐function does.
For the TTC reduction assays, cells grown overnight in YPD at different temperatures as indicated were harvested and diluted to a cell density of 1 OD600/ml in sterile ddH2O. Then, 5 μl of each cell suspension (~105 CFU) was spotted on YPGly plates. The plates were incubated at 28°C for 2 days, and before being overlaid with 20 ml melted TTC agar (0.1% TTC, 2% agar in sterile PBS [8 g/l NaCl, 0.2 g/l KCl, 1.44 g/l Na2HPO4, and 0.24 g/l KH2PO4 pH 7.4]), it was allowed to solidify for 20 min at room temperature. The plates were incubated at 28°C for red color development. The plates were scanned after 2.5, 8, and 24 h of color development using a scanner (Epson Perfection V800 Photo).
For the Phloxine B permeability assays, cells grown overnight in YPD at 28°C were harvested by centrifugation (Eppendorf 5810R centrifuge; A‐4‐62 rotor; 1,500 g; 3 min; 25°C) and diluted to a cell density of 1 OD600/ml in sterile ddH2O. Then, 5 μl of each cell suspension (~105 CFU) was spotted on YPD plates containing 5 μg/ml Phloxine B (P4030, Sigma). The plates were incubated at 28 or 39°C and scanned after 3 days of pink color development using a scanner (Epson Perfection V800 Photo). To intensify differences in color, the images were processed uniformly by increasing contrast and brightness by 40%.
For the cell wall disruption sensitivity assays, cells grown in YPD for 16 h at 28°C were harvested and re‐inoculated into 5 ml of YPD at a cell density of 0.2 OD600/ml for 24 h at 28 or 39°C. The cells were harvested by centrifugation (Eppendorf 5810R centrifuge; A‐4‐62 rotor; 1,500 g; 3 min; 25°C) and diluted at cell densities of 0.6 OD600/ml with 1 ml of pH 7.6 Tris‐Cl buffer containing 60 units/ml Zymolyase® 100T (07665–55, Nacalai Tesque Inc.) in a disposable 2 ml photometer cuvette (112117, LP Italiana SPA, Italy). The cell walls were digested initially for 2 h in the Zymolase‐hosting buffer, before further digestion using 0.1% SDS at room temperature, with gentle pipetting every 30 min. OD600 at 0, 2, 4, and 22 h was measured using a Beckman DU® 800 UV/Visible spectrophotometer (Beckman Coulter, USA) to evaluate intact cells. Cells with disrupted cell walls lyse in the presence of SDS due to hypoosmolarity, reducing the OD600 value in the cell suspension. The ratio of OD600 at each time point relative to time 0 (0 h) was calculated to represent sensitivity to cell wall disruption, with a lower ratio reflecting higher sensitivity.
For the cooperative growth assays in liquid broths (Appendix Fig S16A), cells grown overnight in YPD (20 h) at 28°C were harvested and diluted to a cell density of 1 OD600/ml in sterile ddH2O. For the “independent cultures,” AZF1 (reference strain, HGB‐sensitive) and azf1Δ (test strain, HGB‐resistant) cell suspensions were diluted independently to a cell density of 0.01 and 0.05 OD600/ml, respectively, in a 1 ml YPD culture. For the “co‐culture,” the AZF1 and azf1Δ cell suspensions were mixed at cell densities of 0.01 and 0.05 OD600/ml, respectively, in the same 1 ml YPD culture. Cell growth was measured at 39°C using a Tecan plate reader (Infinite 200 PRO, Tecan), as described above. After 20 h of growth, 50 μl of each of the “independent cultures” was mixed and resuspended in 900 μl sterile ddH2O, and then 100 μl of the “co‐culture” was resuspended in 900 μl sterile ddH2O. Two suspensions (~0.4 OD600/ml) were serially diluted and plated onto YPD plates at an estimated density of ~102 CFU/plate. The YPD plates were incubated at 28°C for 2 days, before being replicated onto YPD + HGB plates and incubated for 1 day. The ratio of azf1Δ (CFUHGB‐resistant)‐to‐AZF1 (total CFUYPD – CFUHGB‐resistant) cells was calculated from six technical repeats.
For the cooperative growth assays on agar plates (Appendix Fig S16C), cells grown overnight in YPD (20 h) at 28°C were harvested and diluted in sterile ddH2O at cell densities of 1 OD600/ml for the wild‐type and 5 OD600/ml for the azf1Δ mutant (to detect the cooperative growth, more cells of the azf1Δ mutant than the wild‐type needed to be used due to the fitness difference between these two strains under the tested condition). For the “independent cultures,” 5 μl of the wild‐type (reference strain, HGB sensitive, ~105 CFU) and azf1Δ (test strain, HGB resistant, ~5 × 105 CFU) cell suspensions were spotted independently onto a YPD plate. For the “co‐culture,” 5 μl of the mixed wild‐type and azf1Δ cell suspension containing the same CFU as described above were spotted onto the same YPD plate. After incubation at 39°C for 3 days, 7 × 7 mm areas of gel‐hosting cell colonies were sliced out and rehydrated with 1 ml sterile ddH2O at room temperature. After 1 h, the cells were washed down five times by means of mild‐pulse vortexing. Suspensions were transferred to new 2 ml tubes and the wash step was repeated. The new suspensions were combined with the previous ones. Two slices from the “independent cultures” were rehydrated and resuspended together, whereas only one slice from the “co‐culture” was processed. Two suspensions (~3 OD600/ml for the “independent culture” and ~1.6 OD600/ml for the “co‐culture”) were then serially diluted and drop plated. Each dilution (106–103 CFU/ml) was spotted onto the YPD and YPD + HGB plates (5 μl/spot, ~104–101 CFU/spot) and incubated at 28°C for 2–3 days. Spots with < 50 CFU were counted. Notably, instead of using the HGB resistant to HGB‐sensitive ratio, we used the frequency of azf1Δ cells (CFUHGB‐resistant/total CFUYPD) to represent the level of cooperative growth, thereby reducing technical variations caused by the considerable difference in viability between AZF1 and azf1Δ cells on agar plates. The frequencies were calculated from six technical repeats. Due to their lower fitness at 39°C, for cases in the sef1Δ background (sef1Δ, HGB‐sensitive; sef1Δazf1Δ, HGB‐resistant), the cell densities of initial cell suspensions were modified to 2 OD600/ml for the sef1Δ mutant and 4 OD600/ml for the sef1Δazf1Δ mutant.
Beta‐galactosidase assays
LacZ expression levels for the one‐hybrid assays were quantified using liquid β‐galactosidase assays, as described previously but with some modifications (Hsu et al, 2021). In brief, early log‐phase (0.5–1.0 OD600/ml) cells cultured in indicated media were assayed. All subcultures were initiated with 0.2 OD600/ml of inoculum from overnight‐grown cells. One‐hybrid strains from the chromosome‐integrated system were subcultured for 4.5 h, whereas the strains from the plasmid‐based system were subcultured for 5.5 h in the presence of 200 μg/ml HGB. Due to the relatively weak activation of Azf1, more cells (~1 OD600 cells) per reaction and a longer incubation time (90 min) at 37°C were required for colorimetric reactions. LacZ levels are displayed as Miller units.
Genomic DNA extraction for whole‐genome sequencing
Pure genomic DNA was extracted via the phenol–chloroform method, followed by RNase treatment, as described previously but with modifications (Hsu et al, 2011). In brief, ~100 OD600 cells (from a 20 ml YPD overnight culture) per sample were extracted. The cells were resuspended in 200 μl gDNA lysis buffer [2% Triton X‐100, 1% SDS, 100 mM NaCl, 10 mM Tris‐Cl (pH 8), and 1 mM Na2EDTA] and transferred into a 2 ml breaking tube (72.693, Sarstedt) containing 250 μl PCIA (phenol:chloroform:isoamyl alcohol = 25:24:1, 0.1% 8‐quinolinol, pH 8) and a 500 μl volume of glass beads (11079105, BioSpec Products). The cells were lysed by using a FastPrep‐24™ 5G Homogenizer (MP Biomedicals™) at a speed of 6 m/s for 40 s. The aqueous phases were then extracted again using 200 μl PCIA (pH 8). RNase treatment involved adding 10 μl of 10 mg/ml RNase A (R5503, Sigma) into a 400 μl nucleic acid suspension in TE buffer (10 mM Tris‐Cl and 1 mM Na2EDTA, pH 8) and incubating at 37°C for 1 h. The crude gDNA was then precipitated by adding 1 ml of 99% ethanol with 20 μl of 3 M sodium acetate (pH 5.2), followed by mild mixing for 30 min. The nucleic acid pellets were rehydrated in 200 μl ddH2O at 65°C for 10 min and then purified again by using a Zymo Genomic DNA clean & concentration™ kit‐25 (Zymo Research, USA) to remove the small DNA fragments. All procedures followed the manufacturer's instructions, but with the modification of a two‐cycle wash step using 500 μl wash buffer. The final gDNA was eluted with 50 μl of 65°C elution buffer. DNA concentrations were measured using a Qubit™ dsDNA BR Assay Kit (Q32853, Invitrogen by Thermo Fisher Scientific). The quality of the gDNA was checked using the 2200 TapeStation system (G2964AA), Genomic DNA ScreenTape (5,067–5,365), and Genomic DNA Reagents (5,067–5,366) obtained from Agilent Technologies.
DNA‐seq analyses
Isolated genomic DNAs were fragmented using a Covaris M220 Focused‐ultrasonicator (Covaris, USA), and DNA libraries were prepared using a Truseq DNA PCR‐free HT kit (Illumina, USA). Two founders and 12 suppressor clones were sequenced to an estimated average depth of 50‐fold coverage by paired‐end 150 bp read length following the protocol of the NextSeq 500 Mid output v2 (300 cycles) sequencing kit on an Illumina NextSeq 500 System controlled by the NextSeq Control Software (NCS v2.2.0, Illumina). Real‐Time Analysis (RTA v2.4.11, Illumina) software was used to generate the BCL files containing base calls and associated quality scores (Q‐scores). The bcl2fastq Conversion Software (v2.17, Illumina) was used to demultiplex sequencing data and convert the BCL files into FASTQ files, which were then processed by FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc) and summarized in MultiQC (Ewels et al, 2016) to generate a QC report for comparison.
Raw reads were mapped to the reference CBS 3082 genome using the Burrows–Wheeler Aligner (BWA, v0.7.17) and the BWA‐MEM algorithm (Li & Durbin, 2009; preprint: Li, 2013) with default parameters to obtain the BAM files. Mapping rates and sequencing coverages were determined in Samtools v1.9 (Li et al, 2009). Variant calling was performed on the BAM files using GATK v4.1.2.0 (McKenna et al, 2010). According to “hard‐filtering” recommendations for germline short variant (SNP and INDEL) discovery described in the GATK website (https://gatk.broadinstitute.org/hc/en‐us/), we applied the following filtering settings with some modifications for SNP calling: “QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < ‐12.5 || ReadPosRankSum < ‐8.0 || SOR > 4.0,” and for INDEL calling: “QD < 2.0 || FS > 200.0 || ReadPosRankSum < ‐20.0,” to obtain VCF files. Only variants existing in the suppressor clones but not in their founder strain were retained for subsequent analyses. The suppressor‐unique variants were then filtered according to sequencing depth (DP) > 20 and allele frequency (AF) ≥ 0.5 to get the final variant spectra. SnpEff v4.3t (Cingolani et al, 2012) was used to annotate and predict the effects of suppressor‐unique variants on genes. The final unique variants with annotations are listed in Dataset EV4.
RNA extraction and DNase treatment
Approximately 10 OD600 of early log‐phase (0.5–1.0 OD600/ml) cells from subcultures grown from 0.2 OD600/ml of inoculum for 5.5 h under the indicated conditions were harvested. Total RNA was extracted by means of the phenol–chloroform method and treated with the TURBO DNA‐free™ kit (AM1907, Ambion, Invitrogen by Thermo Fisher Scientific), as described (Hsu et al, 2021) but with small modifications. In brief, the cells were resuspended in 500 μl nuclease‐free and ice‐cold RNA lysis buffer and then transferred into a 2 ml breaking tube (72.693, Sarstedt, Germany) containing 250 μl ice‐cold acidic PCIA and a 500 μl volume of glass beads (11079105, BioSpec Products, USA). The cells were lysed using a FastPrep‐24™ 5G Homogenizer (MP Biomedicals™, USA) at a speed of 6 m/s for 40 s at room temperature. The aqueous phases were then extracted by two rounds of 200 μl ice‐cold acidic PCIA treatment, and the nucleic acids were obtained after 2 h of ethanol precipitation. RNA quality was checked using a Bioanalyzer 2100 instrument (Agilent Technologies, USA) with an RNA 6000 Nano LabChip kit (Agilent Technologies). The DNase‐treated RNAs were stored at 80°C for further RT–qPCR or RNA‐seq analyses.
Reverse transcription (RT) and quantitative‐PCR (qPCR) analyses
RT–qPCR was performed as described previously but with some modifications (Hsu et al, 2021). In brief, cDNA synthesis was conducted in the presence of different rRNasin® Ribonuclease Inhibitors (N2511, Promega, USA) at a concentration of 1 μl per 20‐μl reaction. qPCR was performed using the model QuantStudioTM 12 K Flex Real‐Time PCR System (Applied Biosystems by Thermo Fisher Scientific). Primers used for qPCR are listed in Dataset EV14. L. kluyveri CDC34 transcripts (SAKL0D02530g) were used as an endogenous control for qPCR. The average ΔΔC T and standard deviation were determined from at least three technical repeats. The relative fold change of each gene is shown according to the method.
RNA‐seq analyses
For each strain and condition, total RNA was extracted from three biological replicates. RNA‐seq libraries were prepared using a TruSeq Stranded mRNA HT Sample Prep Kit (Illumina) on 2 μg total RNA and sequenced by single‐end 75 bp read length following the protocol of the NextSeq 500 High Output kit v2.5 (75 cycles) sequencing kit on an Illumina NextSeq 500 System. Total read numbers were estimated to be 20 million reads per sample. Raw reads were quality trimmed using Trimmomatic v0.38 (Bolger et al, 2014) with options: “ILLUMINACLIP:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36.” The relative abundance of each gene in units of transcripts per million (TPM) for each sample was quantified using Salmon v1.4.0 (Patro et al, 2017) with options: “‐‐numBootstraps 200 ‐‐validateMappings –gcBias.” Tximport (Soneson et al, 2015) by default was used to convert the estimated transcript abundance files from Salmon to a DESeq2‐compatible dataset. DESeq2 (Love et al, 2014) was used for the analysis of differential gene expression in R v4.0.4 with default settings. In DESeq2, the P‐values attained by the Wald test were corrected for multiple testing using the Benjamini and Hochberg method. Genes with mean TPM < 1 under all conditions were omitted from our analyses. Differences in gene expression with adjusted P‐values < 0.05 and fold‐change of at least two were considered significantly different. All results in the standard DESeq2 output format and the TPM of each gene from three technical repeats are presented in Datasets EV1, EV2, and [Link], [Link].
The heatmaps of selected gene expressions were created using the mean TPM ratio relative to a reference sample, as indicated. The green–white–red color scale ranging from the 5th to 90th percentiles of the selected values was generated in Excel 2016 using the conditional formatting function. The white color represents the median with a value of ~1, indicating no differential expression.
Gene ontology (GO) enrichment analyses and manual simplification
For L. kluyveri GO analyses, gene ontology information borrowed from S. cerevisiae (SGD, http://www.yeastgenome.org; GO Term Finder, version 0.86) was used to analyze the L. kluyveri genes. In brief, the L. kluyveri genes were converted to S. cerevisiae orthologs using a published Lk‐to‐Sc cross‐reference table of orthology (Hsu et al, 2021). L. kluyveri genes in the input list without a clear S. cerevisiae ortholog (~10%) were omitted from our GO enrichment analyses of “biological processes.” The enriched GO terms with adjusted P‐values < 0.01 were considered significant (a more stringent cut‐off for P‐values generally did not affect the results of subsequent analyses). We did not correct the genetic background because L. kluyveri and S. cerevisiae share similar gene numbers in their genomes (~6,000 genes). To browse the GO hierarchy, the enriched GO terms from each set of differentially expressed genes were placed on the QuickGo website (Binns et al, 2009). To “simplify” the enriched GO term list, the resolved GO terms were checked manually and the broader parent terms were removed, while the more specific child terms close to the leaf node of the hierarchy tree were retained to assist in simplifying functional annotations.
Protein extraction and Western blotting
Total protein was extracted by using the NaOH/TCA/HU method with modifications (Lu et al, 2016). In brief, ~3 OD600 cells were resuspended in 1 ml lysis buffer (0.185 M NaOH, 0.75% β‐ME) and lysed on ice for 10 min. Next, we added 150 μl of 55% trichloroacetic acid (TCA, T9159, Sigma, USA) to the lysis suspension at a final percentage of 8% to precipitate proteins. After 15 min on ice, the precipitants were spun down by centrifugation (Eppendorf 5424R centrifuge; FA‐45‐24‐11 rotor; 13 K rpm; 10 min; 4°C) and the supernatant was discarded. The centrifugation step was repeated to remove all residual supernatant. Then, we added 300 μl of high urea (HU) sample buffer (8 M Urea, 5% SDS, 0.2 M Tris–HCl pH 6.5, 1 mM Na2EDTA, and 0.01% bromophenol blue) supplemented with 2 M Tris base in a 50‐to‐3 ratio to the protein pellets, and then incubated them at 65°C for at least 40 min, before subjecting them to vigorous pipetting to sufficiently dissolve the pellets.
Protein extracts were resolved by 8% SDS–PAGE (Sambrook, 2001) under a constant voltage of 80 V for 40 min of stacking followed by 150 V for 2 h of separating, before being blotted onto an Immobilon®‐P PVDF Membrane (IPVH00010, Millipore by Merck) in a transfer buffer (25 mM Tris base, 192 mM glycine, and 20% methanol) under a constant voltage of 30 V for 20 h of transfer at 4°C. Post‐transfer membranes were then blocked with the blocking buffer [1% casein (C5890, Sigma) in PBST (1% Tween® 20 in 1× PBS)] for 1 h. For immunodetection, rabbit polyclonal anti‐TAP antibody (CAB1001, Thermo Fisher Scientific) diluted 1:2,000 in blocking buffer was used to detect TAP‐tagged Sef1 and Azf1. Alpha‐tubulin was used as an internal control and it was detected by means of rabbit monoclonal antibody (ab184970, Abcam, UK) diluted 1:20,000 in blocking buffer. The blots were hybridized with each primary antibody for 2 h at room temperature and washed three times with PBST (5 min each). Finally, the blot was hybridized with Peroxidase‐AffiniPure Donkey Anti‐Rabbit IgG secondary antibody (711‐035‐152, Jackson ImmunoResearch, USA) diluted 1:20,000 in blocking buffer for 1 h at room temperature, and then washed three times with PBST. Chemiluminescence was developed with enhanced ECL reagents (NEL105001EA, PerkinElmer, USA) and visualized by exposure to an X‐ray film. Band intensity was quantified by ImageJ software (https://imagej.nih.gov/ij/index.html; Schneider et al, 2012).
Statistical analysis
Details of statistical analyses are presented in the main text or corresponding figure legends. Statistical significance was established using the unpaired Student's t‐test or the one‐sample t‐test in Excel 2016 or the default functions packaged in each analysis tool.
Author contributions
Po‐Chen Hsu: Conceptualization; data curation; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Yu‐Hsuan Cheng: Data curation; formal analysis; methodology; writing – original draft. Chia‐Wei Liao: Data curation; formal analysis; methodology; writing – original draft. Richard Ron R Litan: Data curation; formal analysis; investigation; visualization. Yu‐Ting Jhou: Formal analysis; investigation; visualization. Florica Jean Ganaden Opoc: Investigation; visualization. Ahmed A A Amine: Investigation. Jun‐Yi Leu: Conceptualization; resources; supervision; funding acquisition; investigation; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Dataset EV11
Dataset EV12
Dataset EV13
Dataset EV14
Dataset EV15
Dataset EV16
Dataset EV17
PDF+
Acknowledgements
We are grateful to Professor Kenneth H. Wolfe (UCD, Ireland) for generously providing the DNA sequences of the L. kluyveri MAT loci used in this study. We are thankful to Dr. Shu‐Yun Tung, the Genomics Core Laboratory (IMB, Academia Sinica, Taiwan) for technical assistance with the RNA‐seq experiments. We thank all the JYL lab members for their helpful discussions and comments on the manuscript. We thank John O'Brien for manuscript editing. This work was supported by Academia Sinica of Taiwan (grant no. AS‐IA‐110‐L01 and AS‐GCS‐110‐01 to J‐YL) and the Taiwan Ministry of Science and Technology (grant no. MOST 110‐2326‐B‐001‐007 to J‐YL). PCH was supported by a MOST post‐doctoral fellowship (MOST 110‐2811‐B‐001‐581). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
EMBO reports (2023) 24: e56019
Data availability
The datasets produced in this study are available in the following databases:
RNA‐seq data: Gene Expression Omnibus database GSE200532 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE200532). The processed results are shown in Datasets EV1, EV2, and [Link], [Link].
DNA‐seq data: NCBI BioProject database PRJNA825358 (https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA825358). The processed results are shown in Dataset EV4.
References
- Aggeli D, Li Y, Sherlock G (2021) Changes in the distribution of fitness effects and adaptive mutational spectra following a single first step towards adaptation. Nat Commun 12: 5193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amine AAA, Liao C‐W, Hsu P‐C, Opoc FJG, Leu J‐Y (2021) Experimental evolution improves mitochondrial genome quality control in Saccharomyces cerevisiae and extends its replicative lifespan. Curr Biol 31: 3663–3670 [DOI] [PubMed] [Google Scholar]
- Ayala FJ, Campbell CA (1974) Frequency‐dependent selection. Annu Rev Ecol Syst 5: 115–138 [Google Scholar]
- Barrick JE, Lenski RE (2013) Genome dynamics during experimental evolution. Nat Rev Genet 14: 827–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrick JE, Kauth MR, Strelioff CC, Lenski RE (2010) Escherichia coli rpoB mutants have increased evolvability in proportion to their fitness defects. Mol Biol Evol 27: 1338–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford T, Hartl DL (2009) Optimization of gene expression by natural selection. Proc Natl Acad Sci USA 106: 1133–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beese SE, Negishi T, Levin DE (2009) Identification of positive regulators of the yeast Fps1 Glycerol Channel. PLoS Genet 5: e1000738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns D, Dimmer E, Huntley R, Barrell D, O'Donovan C, Apweiler R (2009) QuickGO: a web‐based tool for gene ontology searching. Bioinformatics 25: 3045–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell JR, Schwartz K, Francois D, Fisher DS, Sherlock G, Levy SF (2019) The dynamics of adaptive genetic diversity during the early stages of clonal evolution. Nat Ecol Evol 3: 293–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow DK, Cameron DM, Collins SR, Schuldiner M, Stewart‐Ornstein J, Newman HW, Braun S, Madhani HD, Krogan NJ, Weissman JS (2008) A comprehensive strategy enabling high‐resolution functional analysis of the yeast genome. Nat Methods 5: 711–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion C, Pflieger D, Friedrich A, Schacherer J (2015) Evolution of intraspecific transcriptomic landscapes in yeasts. Nucleic Acids Res 43: 4558–4568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler G, Kenny C, Fagan A, Kurischko C, Gaillardin C, Wolfe KH (2004) Evolution of the MAT locus and its Ho endonuclease in yeast species. Proc Natl Acad Sci USA 101: 1632–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SB (2000) Endless forms: the evolution of gene regulation and morphological diversity. Cell 101: 577–580 [DOI] [PubMed] [Google Scholar]
- Cazzanelli G, Pereira F, Alves S, Francisco R, Azevedo L, Dias Carvalho P, Almeida A, Côrte‐Real M, Oliveira MJ, Lucas C et al (2018) The yeast Saccharomyces cerevisiae as a model for understanding RAS proteins and their role in human tumorigenesis. Cell 7: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Zhang J (2020) Antagonistic pleiotropy conceals molecular adaptations in changing environments. Nat Ecol Evol 4: 461–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6: 80–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coluccio A, Neiman AM (2004) Interspore bridges: a new feature of the Saccharomyces cerevisiae spore wall. Microbiology 150: 3189–3196 [DOI] [PubMed] [Google Scholar]
- Conrad M, Schothorst J, Kankipati HN, Van Zeebroeck G, Rubio‐Texeira M, Thevelein JM (2014) Nutrient sensing and signaling in the yeast Saccharomyces cerevisiae . FEMS Microbiol Rev 38: 254–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolon JD, McManus CJ, Stevenson KR, Graveley BR, Wittkopp PJ (2014) Tempo and mode of regulatory evolution in Drosophila . Genome Res 24: 797–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal CK, Johnson AD (2017) How transcription circuits explore alternative architectures while maintaining overall circuit output. Genes Dev 31: 1397–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar EL, Oakley CG, Conner JK, Gould BA, Schemske DW (2016) Factors influencing the effect size distribution of adaptive substitutions. Proc R Soc B Biol Sci 283: 20153065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P, Magnusson M, Lundin S, Käller M (2016) MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32: 3047–3048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland T (2014) Trade‐offs. Curr Biol 24: R60–R61 [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel‐Harel O, Eisen MB, Storz G, Botstein D, Brown PO (2000) Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11: 4241–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Génolevures Consortium , Souciet J‐L, Dujon B, Gaillardin C, Johnston M, Baret PV, Cliften P, Sherman DJ, Weissenbach J, Westhof E et al (2009) Comparative genomics of protoploid Saccharomycetaceae. Genome Res 19: 1696–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrish PJ, Lenski RE (1998) The fate of competing beneficial mutations in an asexual population. Genetica 102–103: 127–144 [PubMed] [Google Scholar]
- Goncalves A, Leigh‐Brown S, Thybert D, Stefflova K, Turro E, Flicek P, Brazma A, Odom DT, Marioni JC (2012) Extensive compensatory cis‐trans regulation in the evolution of mouse gene expression. Genome Res 22: 2376–2384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorter de Vries AR, Voskamp MA, van Aalst ACA, Kristensen LH, Jansen L, van den Broek M, Salazar AN, Brouwers N, Abeel T, Pronk JT et al (2019) Laboratory evolution of a Saccharomyces cerevisiae × S. eubayanus hybrid under simulated lager‐brewing conditions. Front Genet 10: 242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosfeld EV, Bidiuk VA, Mitkevich OV, Ghazy ESMO, Kushnirov VV, Alexandrov AI (2021) A systematic survey of characteristic features of yeast cell death triggered by external factors. J Fungi 7: 886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BM, Ma C‐X, Liang P, Singh KK (2009) Fluctuation AnaLysis CalculatOR: a web tool for the determination of mutation rate using Luria–Delbrück fluctuation analysis. Bioinformatics 25: 1564–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y‐H, Gasch AP (2015) Exploiting the yeast stress‐activated signaling network to inform on stress biology and disease signaling. Curr Genet 61: 503–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgins‐Davis A, Rice DP, Townsend JP (2015) Gene expression evolves under a house‐of‐cards model of stabilizing selection. Mol Biol Evol 32: 2130–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P‐C, Yang C‐Y, Lan C‐Y (2011) Candida albicans Hap43 is a repressor induced under low‐iron conditions and is essential for iron‐responsive transcriptional regulation and virulence. Eukaryot Cell 10: 207–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu P‐C, Lu T‐C, Hung P‐H, Jhou Y‐T, Amine AAA, Liao C‐W, Leu J‐Y (2021) Plastic rewiring of Sef1 transcriptional networks and the potential of nonfunctional transcription factor binding in facilitating adaptive evolution. Mol Biol Evol 38: 4732–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C‐J, Lu M‐Y, Chang Y‐W, Li W‐H (2018) Experimental evolution of yeast for high‐temperature tolerance. Mol Biol Evol 35: 1823–1839 [DOI] [PubMed] [Google Scholar]
- Jamnik P, Medved P, Raspor P (2006) Increased glutathione content in yeast Saccharomyces cerevisiae exposed to NaCl. Ann Microbiol 56: 175–178 [Google Scholar]
- Jerison ER, Kryazhimskiy S, Mitchell JK, Bloom JS, Kruglyak L, Desai MM (2017) Genetic variation in adaptability and pleiotropy in budding yeast. Elife 6: e27167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AD (2017) The rewiring of transcription circuits in evolution. Curr Opin Genet Dev 47: 121–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MS, Gopalakrishnan S, Goyal J, Dillingham ME, Bakerlee CW, Humphrey PT, Jagdish T, Jerison ER, Kosheleva K, Lawrence KR et al (2021) Phenotypic and molecular evolution across 10,000 generations in laboratory budding yeast populations. Elife 10: e63910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao KC, Sherlock G (2008) Molecular characterization of clonal interference during adaptive evolution in asexual populations of Saccharomyces cerevisiae . Nat Genet 40: 1499–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsler G, Geiler‐Samerotte K, Petrov DA (2020) Fitness variation across subtle environmental perturbations reveals local modularity and global pleiotropy of adaptation. Elife 9: e61271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvitek DJ, Sherlock G (2013) Whole genome, whole population sequencing reveals that loss of signaling networks is the major adaptive strategy in a constant environment. PLoS Genet 9: e1003972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBar T, Phoebe Hsieh Y‐Y, Fumasoni M, Murray AW (2020) Evolutionary repair experiments as a window to the molecular diversity of life. Curr Biol 30: R565–R574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laman Trip DS, Youk H (2020) Yeasts collectively extend the limits of habitable temperatures by secreting glutathione. Nat Microbiol 5: 943–954 [DOI] [PubMed] [Google Scholar]
- Lang GI, Rice DP, Hickman MJ, Sodergren E, Weinstock GM, Botstein D, Desai MM (2013) Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature 500: 571–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laussel C, Léon S (2020) Cellular toxicity of the metabolic inhibitor 2‐deoxyglucose and associated resistance mechanisms. Biochem Pharmacol 182: 114213 [DOI] [PubMed] [Google Scholar]
- Lavoie H, Hogues H, Mallick J, Sellam A, Nantel A, Whiteway M (2010) Evolutionary tinkering with conserved components of a transcriptional regulatory network. PLoS Biol 8: e1000329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KS, Irie K, Gotoh Y, Watanabe Y, Araki H, Nishida E, Matsumoto K, Levin DE (1993) A yeast mitogen‐activated protein kinase homolog (Mpk1p) mediates signalling by protein kinase C. Mol Cell Biol 13: 3067–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy SF, Blundell JR, Venkataram S, Petrov DA, Fisher DS, Sherlock G (2015) Quantitative evolutionary dynamics using high‐resolution lineage tracking. Nature 519: 181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv 10.48550/arXiv.1303.3997 [PREPRINT] [DOI] [Google Scholar]
- Li H, Durbin R (2009) Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Johnson AD (2010) Evolution of transcription networks—lessons from yeasts. Curr Biol 20: R746–R753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Venkataram S, Agarwala A, Dunn B, Petrov DA, Sherlock G, Fisher DS (2018) Hidden complexity of yeast adaptation under simple evolutionary conditions. Curr Biol 28: 515–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y‐J, Swamy KBS, Leu J‐Y (2016) Experimental evolution reveals interplay between Sch9 and Polyploid stability in yeast. PLoS Genet 12: e1006409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Field MC, Goodson HV, Malik HS, Pereira‐Leal JB, Roos DS, Turkewitz AP, Sazer S (2014) Evolutionary cell biology: two origins, one objective. Proc Natl Acad Sci USA 111: 16990–16994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack KL, Nachman MW (2017) Gene regulation and speciation. Trends Genet 33: 68–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazáň M, Mazáňová K, Farkaš V (2008) Phenotype analysis of Saccharomyces cerevisiae mutants with deletions in Pir cell wall glycoproteins. Antonie Van Leeuwenhoek 94: 335–342 [DOI] [PubMed] [Google Scholar]
- McCloskey D, Xu S, Sandberg TE, Brunk E, Hefner Y, Szubin R, Feist AM, Palsson BO (2018) Evolution of gene knockout strains of E. coli reveal regulatory architectures governed by metabolism. Nat Commun 9: 3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald MJ (2019) Microbial experimental evolution – a proving ground for evolutionary theory and a tool for discovery. EMBO Rep 20: e46992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 20: 1297–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mérot C, Llaurens V, Normandeau E, Bernatchez L, Wellenreuther M (2020) Balancing selection via life‐history trade‐offs maintains an inversion polymorphism in a seaweed fly. Nat Commun 11: 670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller K, Langkjaer RB, Nielsen J, Piškur J, Olsson L (2004) Pyruvate decarboxylases from the petite‐negative yeast Saccharomyces kluyveri. Mol Genet Genomics 270: 558–568 [DOI] [PubMed] [Google Scholar]
- Molofsky J, Bever JD (2002) A novel theory to explain species diversity in landscapes: positive frequency dependence and habitat suitability. Proc Biol Sci 269: 2389–2393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky J, Bever JD, Antonovics J (2001) Coexistence under positive frequency dependence. Proc Biol Sci 268: 273–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore FB‐G, Rozen DE, Lenski RE (2000) Pervasive compensatory adaptation in Escherichia coli . Proc R Soc Lond B Biol Sci 267: 515–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parts L, Cubillos FA, Warringer J, Jain K, Salinas F, Bumpstead SJ, Molin M, Zia A, Simpson JT, Quail MA et al (2011) Revealing the genetic structure of a trait by sequencing a population under selection. Genome Res 21: 1131–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C (2017) Salmon provides fast and bias‐aware quantification of transcript expression. Nat Methods 14: 417–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter Isabelle S, Davidson Eric H (2011) Evolution of gene regulatory networks controlling body plan development. Cell 144: 970–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk JT, Yde Steensma H, Van Dijken JP (1996) Pyruvate metabolism in Saccharomyces cerevisiae . Yeast 12: 1607–1633 [DOI] [PubMed] [Google Scholar]
- Ratnakumar S, Hesketh A, Gkargkas K, Wilson M, Rash BM, Hayes A, Tunnacliffe A, Oliver SG (2011) Phenomic and transcriptomic analyses reveal that autophagy plays a major role in desiccation tolerance in Saccharomyces cerevisiae . Mol Biosyst 7: 139–149 [DOI] [PubMed] [Google Scholar]
- Rendueles O, Amherd M, Velicer Gregory J (2015) Positively frequency‐dependent interference competition maintains diversity and pervades a natural population of cooperative microbes. Curr Biol 25: 1673–1681 [DOI] [PubMed] [Google Scholar]
- Reuß O, Vik Å, Kolter R, Morschhäuser J (2004) The SAT1 flipper, an optimized tool for gene disruption in Candida albicans . Gene 341: 119–127 [DOI] [PubMed] [Google Scholar]
- Rojas Echenique JI, Kryazhimskiy S, Nguyen Ba AN, Desai MM (2019) Modular epistasis and the compensatory evolution of gene deletion mutants. PLoS Genet 15: e1007958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero IG, Ruvinsky I, Gilad Y (2012) Comparative studies of gene expression and the evolution of gene regulation. Nat Rev Genet 13: 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Scannell DR, Wolfe K (2004) Rewiring the transcriptional regulatory circuits of cells. Genome Biol 5: 206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segrè D, DeLuna A, Church GM, Kishony R (2005) Modular epistasis in yeast metabolism. Nat Genet 37: 77–83 [DOI] [PubMed] [Google Scholar]
- Serrano R, Martín H, Casamayor A, Ariño J (2006) Signaling alkaline pH stress in the yeast Saccharomyces cerevisiae through the Wsc1 cell surface sensor and the Slt2 MAPK pathway. J Biol Chem 281: 39785–39795 [DOI] [PubMed] [Google Scholar]
- Shevchuk NA, Bryksin AV, Nusinovich YA, Cabello FC, Sutherland M, Ladisch S (2004) Construction of long DNA molecules using long PCR‐based fusion of several fragments simultaneously. Nucleic Acids Res 32: e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Ng DWK, Zhang C, Comai L, Ye W, Jeffrey Chen Z (2012) Cis‐ and trans‐regulatory divergence between progenitor species determines gene‐expression novelty in Arabidopsis allopolyploids. Nat Commun 3: 950 [DOI] [PubMed] [Google Scholar]
- Signor SA, Nuzhdin SV (2018) The evolution of gene expression in cis and trans. Trends Genet 34: 532–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signor SA, Nuzhdin SV (2019) Compensatory evolution of gene expression. Trends Genet 35: 890–891 [DOI] [PubMed] [Google Scholar]
- Sigwalt A, Caradec C, Brion C, Hou J, de Montigny J, Jung P, Fischer G, Llorente B, Friedrich A, Schacherer J (2016) Dissection of quantitative traits by bulk segregant mapping in a protoploid yeast species. FEMS Yeast Res 16: fow056 [DOI] [PubMed] [Google Scholar]
- Slattery MG, Liko D, Heideman W (2006) The function and properties of the Azf1 transcriptional regulator change with growth conditions in Saccharomyces cerevisiae . Eukaryot Cell 5: 313–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova GV, Krasnykh TA, Oktyabrsky ON (2001) Role of glutathione in the response of Escherichia coli to osmotic stress. Biochemistry (Moscow) 66: 973–978 [DOI] [PubMed] [Google Scholar]
- Solís Eric J, Pandey Jai P, Zheng X, Jin Dexter X, Gupta Piyush B, Airoldi Edoardo M, Pincus D, Denic V (2016) Defining the essential function of yeast Hsf1 reveals a compact transcriptional program for maintaining eukaryotic Proteostasis. Mol Cell 63: 60–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C, Love MI, Robinson MD (2015) Differential analyses for RNA‐seq: transcript‐level estimates improve gene‐level inferences. F1000Res 4: 1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger PK (1990) Yeast heat shock factor contains separable transient and sustained response transcriptional activators. Cell 62: 793–805 [DOI] [PubMed] [Google Scholar]
- Stanley D, Bandara A, Fraser S, Chambers PJ, Stanley GA (2010) The ethanol stress response and ethanol tolerance of Saccharomyces cerevisiae . J Appl Microbiol 109: 13–24 [DOI] [PubMed] [Google Scholar]
- Stein T, Kricke J, Becher D, Lisowsky T (1998) Azf1p is a nuclear‐localized zinc‐finger protein that is preferentially expressed under non‐fermentative growth conditions in Saccharomyces cerevisiae . Curr Genet 34: 287–296 [DOI] [PubMed] [Google Scholar]
- Szamecz B, Boross G, Kalapis D, Kovács K, Fekete G, Farkas Z, Lázár V, Hrtyan M, Kemmeren P, Groot Koerkamp MJA et al (2014) The genomic landscape of compensatory evolution. PLoS Biol 12: e1001935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Nakafuku M, Satoh T, Marshall MS, Gibbs JB, Matsumoto K, Kaziro Y, Toh‐e A (1990) S. cerevisiae genes IRA1 and IRA2 encode proteins that may be functionally equivalent to mammalian ras GTPase activating protein. Cell 60: 803–807 [DOI] [PubMed] [Google Scholar]
- Tanay A, Regev A, Shamir R (2005) Conservation and evolvability in regulatory networks: the evolution of ribosomal regulation in yeast. Proc Natl Acad Sci USA 102: 7203–7208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taymaz‐Nikerel H, Cankorur‐Cetinkaya A, Kirdar B (2016) Genome‐wide transcriptional response of Saccharomyces cerevisiae to stress‐induced perturbations. Front Bioeng Biotechnol 4: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenaillon O, Rodríguez‐Verdugo A, Gaut RL, McDonald P, Bennett AF, Long AD, Gaut BS (2012) The molecular diversity of adaptive convergence. Science 335: 457–461 [DOI] [PubMed] [Google Scholar]
- Teng X, Dayhoff‐Brannigan M, Cheng W‐C, Gilbert Catherine E, Sing Cierra N, Diny Nicola L, Wheelan Sarah J, Dunham Maitreya J, Boeke Jef D, Pineda Fernando J et al (2013) Genome‐wide consequences of deleting any single gene. Mol Cell 52: 485–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Reikhav S, Levy AA, Barkai N (2009) A yeast hybrid provides insight into the evolution of gene expression regulation. Science 324: 659–662 [DOI] [PubMed] [Google Scholar]
- Toda T, Uno I, Ishikawa T, Powers S, Kataoka T, Broek D, Cameron S, Broach J, Matsumoto K, Wigler M (1985) In yeast, RAS proteins are controlling elements of adenylate cyclase. Cell 40: 27–36 [DOI] [PubMed] [Google Scholar]
- Tomishige N, Noda Y, Adachi H, Shimoi H, Takatsuki A, Yoda K (2003) Mutations that are synthetically lethal with a gas1Δ allele cause defects in the cell wall of Saccharomyces cerevisiae . Mol Genet Genomics 269: 562–573 [DOI] [PubMed] [Google Scholar]
- Turcotte B, Liang XB, Robert F, Soontorngun N (2009) Transcriptional regulation of nonfermentable carbon utilization in budding yeast. FEMS Yeast Res 10: 2–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udom N, Chansongkrow P, Charoensawan V, Auesukaree C, Druzhinina IS (2019) Coordination of the Cell Wall integrity and high‐Osmolarity glycerol pathways in response to ethanol stress in Saccharomyces cerevisiae . Appl Environ Microbiol 85: e00551‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Bergh B, Swings T, Fauvart M, Michiels J (2018) Experimental design, population dynamics, and diversity in microbial experimental evolution. Microbiol Mol Biol Rev 82: e00008‐18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen J, Pons C, Mellor JC, Yamaguchi TN, Friesen H, Koschwanez J, Ušaj MM, Pechlaner M, Takar M, Ušaj M et al (2016) Exploring genetic suppression interactions on a global scale. Science 354: aag0839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen J, Boone C, Andrews BJ (2017) Mapping a diversity of genetic interactions in yeast. Curr Opin Syst Biol 6: 14–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataram S, Dunn B, Li Y, Agarwala A, Chang J, Ebel ER, Geiler‐Samerotte K, Hérissant L, Blundell JR, Levy SF et al (2016) Development of a comprehensive genotype‐to‐fitness map of adaptation‐driving mutations in yeast. Cell 166: 1585–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veri AO, Robbins N, Cowen LE (2018) Regulation of the heat shock transcription factor Hsf1 in fungi: implications for temperature‐dependent virulence traits. FEMS Yeast Res 18: foy041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GP, Lynch VJ (2008) The gene regulatory logic of transcription factor evolution. Trends Ecol Evol 23: 377–385 [DOI] [PubMed] [Google Scholar]
- Wei G, Li Y, Du G, Chen J (2003) Effect of surfactants on extracellular accumulation of glutathione by Saccharomyces cerevisiae . Process Biochem 38: 1133–1138 [Google Scholar]
- Welch AZ, Gibney PA, Botstein D, Koshland DE (2013) TOR and RAS pathways regulate desiccation tolerance in Saccharomyces cerevisiae . Mol Biol Cell 24: 115–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger JW, Piotrowski J, Nagarajan S, Chiotti K, Sherlock G, Rosenzweig F (2011) Hunger artists: yeast adapted to carbon limitation show trade‐offs under carbon sufficiency. PLoS Genet 7: e1002202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlbach DJ, Thompson DA, Gasch AP, Regev A (2009) From elements to modules: regulatory evolution in Ascomycota fungi. Curr Opin Genet Dev 19: 571–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wünsche A, Dinh DM, Satterwhite RS, Arenas CD, Stoebel DM, Cooper TF (2017) Diminishing‐returns epistasis decreases adaptability along an evolutionary trajectory. Nat Ecol Evol 1: 61 [DOI] [PubMed] [Google Scholar]