

Abstract
The Blocks Database WWW (http://blocks.fhcrc.org ) and Email (blocks@blocks.fhcrc.org ) servers provide tools to search DNA and protein queries against the Blocks+ Database of multiple alignments, which represent conserved protein regions. Blocks+ nearly doubles the number of protein families included in the database by adding families from the Pfam-A, ProDom and Domo databases to those from PROSITE and PRINTS. Other new features include improved Block Searcher statistics, searching with NCBI’s IMPALA program and 3D display of blocks on PDB structures.
INTRODUCTION
Blocks are ungapped multiple alignments corresponding to the most conserved regions of proteins. The Blocks Database consists of blocks constructed from documented families of related proteins using the automated PROTOMAT system (1). In addition to searching the Blocks Database for sequence similarities, several enhancements have been introduced for exploiting protein family information implicit in blocks (2). These include blocks-based searching of sequence databanks (3), blocks-versus-blocks searching (4), sequence logo and tree representations of multiple alignments, and PCR primer design using the CODEHOP (COnsensus-DEgenerate Hybrid Oligonucleotide Primer) method (5). During the past year, coverage of the default Blocks Database has increased with the addition of families from several compendiums, and new Blocks Database searching and 3D display options have been implemented.
Blocks+
Previously, lists of protein families for the Blocks Database were obtained from the PROSITE catalog (6) and supplemented with additional families from the PRINTS database (7). Now, additional families are obtained from the Pfam-A (8), ProDom (9) and Domo (10) protein family databases. Blocks for these famililes are computed by extracting SWISS-PROT (11) sequences documented in the source protein family databases and presenting them to the automated PROTOMAT system (1). However, to minimize redundancy, the resulting blocks for a family are added to Blocks+ only if a LAMA blocks-versus-blocks search (4) of them against the current database results in no significant hits. This recursive procedure yields sets of blocks extracted from Pfam-A families not found in either PROSITE or PRINTS, blocks from ProDom not found in the previous three databases and blocks from Domo not found in any of the other databases. The Blocks+ Database (12) represents 9498 blocks from 2129 different protein families as of June 15, 1999 (Fig. 1). Since the multiple alignments in the source family databases are not used, the alignments in Blocks+ may not coincide with them. Therefore, LAMA is used to search each set of blocks in Blocks+ against blocks carved out of these source alignments (2), and WWW links are made when hits are found.
Figure 1.
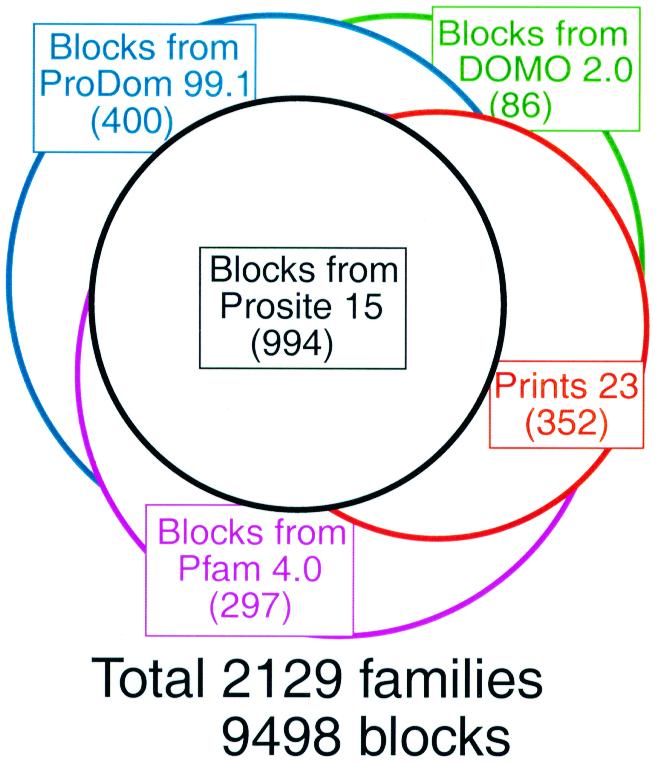
Composition of the Blocks+ Database (as of 15 June 1999).
The Blocks WWW and Email servers provide tools to search DNA and protein queries against Blocks+. As an option to avoid false positive hits, a subset of Blocks+ from which many compositionally biased blocks have been removed can be searched. The Blocks+ Database can also be queried with key words or with blocks or other multiple alignments using the multiple alignment processor and the LAMA search engine. All search results are linked to corresponding entries in the Blocks+ Database which include phylogenetic trees, sequence logos and 3D structures, plus links to other sequence and protein family databases.
IMPROVED Block SEARCHER E-VALUES
The Block Searcher uses the BLIMPS searching program (13) to compare a DNA or protein query sequence with each block in the database of blocks being searched. The results for individual blocks are then analyzed to combine hits for blocks belonging to the same protein family. The original analysis program, BLKSORT, computes E-values for a hit to a family based on ranks (14). A new analysis program, BLKPROB, computes E-values for multiple block hits using methods developed for searches of block queries against sequence databases with the MAST searching tool (15,16). This method requires computing the score distribution for each block, which can be done explicitly when the position-specific scoring matrix (PSSM) derived from a block contains only integers (17). The probability of obtaining the score for the alignment with the query sequence can then simply be looked up in the score distribution. The current implementation computes the complete distributions only for blocks that attain a score greater than the 99.5th percentile score of the distribution; this value is pre-computed and stored with each block. An example of the new output appears in Figure 2a.
Figure 2.


Block Searcher and IMPALA search outputs. A hypothetical Arabidopsis thaliana protein sequence translated from predicted exons in GenBank/EMBL entry U53501 was used to query Blocks+ with a cutoff expected value of 5. Known true positive hits for this query sequence are BL00094 (cytosine DNA methyltransferases) and BL00598 (chromodomains), which are the top two hits for both Block Searcher and IMPALA Searcher. Notice that none of the other hits reported are the same for both methods. Alignments are shown for the top two hits. (a) Block Searcher output. BL00094E and BL00094F were not detected because they are missing from the query as a result of erroneous gene prediction from U53501, confirmed by direct cDNA analysis (21). Each hit consists of one or more blocks from a protein group found in the query sequence. One set of the highest-scoring blocks that are in the correct order and separated by distances comparable to the Blocks Database is selected for analysis. If this set includes multiple blocks the probability that the lower scoring blocks support the highest scoring block is reported. Maps of the database blocks and query sequence are shown: ‘AAA’ represents a block roughly in proportion to its width. ‘:’ represents the minimum distance between blocks in the database. ‘.’ represents the maximum distance between blocks in the database. ‘< >’ indicate the sequence has been truncated to fit the page. The query map is aligned on the highest scoring block. Multiple block hits that are consistent with the highest scoring block are separated by colons. The alignment of the query sequence with the sequence closest to it in the Blocks Database is shown. The distance between detected blocks is listed as (min, max): for the database entry followed by the distance in the query. Upper case in the query indicates at least one occurrence of the residue in that column of the block. (b) IMPALA Searcher output. The IMPALA alignment detects the region corresponding to BL00094A in the query sequence as a separate high scoring segment, which lies 163 aa upstream of BL00094B. The query sequence is aligned with the COBBLER sequence used to make the PSI-BLAST PSSM. In the two alignments shown no gaps have been inserted within the block regions.
The original analysis program with E-values based on ranks is still available as an option and remains the default for the Blocks Email Searcher (to maintain a standardized format for high-volume automatic submissions). However, Email users are encouraged to try the improved analysis program; the required message format is described at http://blocks.fhcrc.org/help/email.html
IMPALA SEARCHER
A new alternative to the Block Searcher for protein queries is the IMPALA Searcher, which has been made available for the Blocks WWW server by the BLAST group at NCBI (18). IMPALA searches a suitably formatted database of PSI-BLAST PSSMs (19). These are constructed for each family in Blocks+ by PSI-BLAST searching with the COBBLER (COnsensus Biasing By Locally Embedding Residues) sequence (3) as query against the SWISS-PROT sequences known to belong to the family. The COBBLER sequence is a representative sequence stretching from 10 aa upstream of the first block to 10 aa downstream of the last block, into which consensus residues deduced from block regions are embedded. PSI-BLAST searching is iterated until convergence, yielding a database of one PSI-BLAST PSSM for each family in Blocks+. Figure 2b shows an example of IMPALA output, which consists of the familiar BLAST output and E-value statistics, and includes links to the Blocks+ families hit. Unlike the Block Searcher, IMPALA may insert gaps in the alignment of the query with the blocks and may also align regions between blocks. Since the Blocks and IMPALA Searchers tend to report the same true positive hits but different false positives (e.g. compare Fig. 2a with b), users who search with both and compare the results may be able to better distinguish true from false hits for challenging queries.
MAPPING Blocks ONTO 3D STRUCTURES
An increasing number of protein families are represented by one or more 3D structures in the PDB database (http://www.rcsb.org/pdb ). To map blocks onto a structure in PDB, MAST (15) is used to search PSSMs against the database of PDB sequences. Segments within corresponding PDB structures are color-coded to indicate the block that they represent. The 3D Blocks representation can be viewed by WWW browsers with helper software that can process Rasmol (20) commands, such as Chime (http://www.mdl.com/chemscape/chime ).
ACCESS
The Blocks WWW server at http://blocks.fhcrc.org implements all of the features described in this article, which should be cited when the Blocks server is used. The Blocks+ Database can also be searched via Email by sending a DNA or protein sequence in FASTA format to blocks@blocks.fhcrc.org
Acknowledgments
ACKNOWLEDGEMENTS
We thank Nick Taylor for implementating 3D Blocks and the NCBI BLAST group, especially Alejandro Schaffer, for making IMPALA software available for searching the Blocks Database. This work is supported by grants from the NIH (GM29009) and the DOE (DE-FG03-97ER62382).
REFERENCES
- 1.Henikoff S. and Henikoff,J.G. (1991) Nucleic Acids Res., 19, 6565–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henikoff J.G., Henikoff,S. and Pietrokovski,S. (1999) Nucleic Acids Res., 27, 226–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henikoff S. and Henikoff,J.G. (1997) Protein Sci., 6, 698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pietrokovski S. (1996) Nucleic Acids Res., 24, 3836–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rose T.M., Schultz,E.R., Henikoff,J.G., Pietrokovski,S., McCallum,C.M. and Henikoff,S. (1998) Nucleic Acids Res., 26, 1628–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hofmann K., Bucher,P., Falquet,L. and Bairoch,A. (1999) Nucleic Acids Res., 27, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Attwood T.K., Flower,D.R., Lewis,A.P., Mabey,J.E., Morgan,S.R., Scordis,P., Selley,J.N. and Wright,W. (1999) Nucleic Acids Res., 27, 220–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bateman A., Birney,E., Durbin,R., Eddy,S.R., Finn,R.D. and Sonnhammer,E.L.L. (1999) Nucleic Acids Res., 27, 260–262. Updated article in this issue: Nucleic Acids Res. (2000), 28, 225–227.9847196 [Google Scholar]
- 9.Corpet F., Gouzy,J. and Kahn,D. (1999) Nucleic Acids Res., 27, 263–267. Updated article in this issue: Nucleic Acids Res. (2000), 28, 267–269.9847197 [Google Scholar]
- 10.Gracy J. and Argos,P. (1998) Bioinformatics, 14, 164–173. [DOI] [PubMed] [Google Scholar]
- 11.Bairoch A. and Boeckmann,B. (1992) Nucleic Acids Res., 20, 2019–2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henikoff S., Henikoff,J.G. and Pietrokovski,S. (1999) Bioinformatics, 15, 471–479. [DOI] [PubMed] [Google Scholar]
- 13.Henikoff S., Henikoff,J.G., Alford,W.J. and Pietrokovski,S. (1995) Gene, 163, GC17–GC26. [DOI] [PubMed] [Google Scholar]
- 14.Henikoff S. and Henikoff,J.G. (1994) Genomics, 19, 97–107. [DOI] [PubMed] [Google Scholar]
- 15.Bailey T.L. and Gribskov,M. (1997) J. Comput. Biol., 4, 45–59. [DOI] [PubMed] [Google Scholar]
- 16.Bailey T.L. and Gribskov,M. (1998) Bioinformatics, 14, 48–54. [DOI] [PubMed] [Google Scholar]
- 17.Tatusov R.L., Altschul,S.F. and Koonin,E.V. (1994) Proc. Natl Acad. Sci. USA, 91, 12091–12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schaffer A.A., Wolf,Y.I., Ponting,C.P., Koonin,E.V., Aravind,L. and Altschul,S.F. (1999) Bioinformatics, in press. [DOI] [PubMed] [Google Scholar]
- 19.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sayle R.A. and Milner-White,E.F. (1995) Trends Biochem. Sci., 20, 374. [DOI] [PubMed] [Google Scholar]
- 21.Henikoff S. and Comai,L. (1998) Genetics, 149, 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]