

Abstract
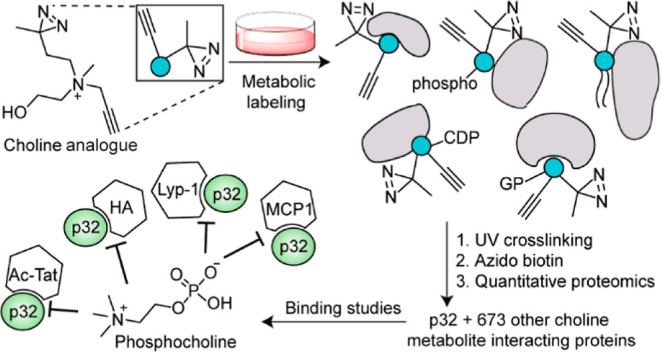
Choline is an essential nutrient for mammalian cells. Our understanding of the cellular functions of choline and its metabolites, independent of their roles as choline lipid metabolism intermediates, remains limited. In addition to fundamental cellular physiology, this knowledge has implications for cancer biology because elevated choline metabolite levels are a hallmark of cancer. Here, we establish a mammalian choline metabolite-interacting proteome by utilizing a photocrosslinkable choline probe. To design this probe, we performed metabolic labeling experiments with structurally diverse choline analogues that resulted in the serendipitous discovery of a choline lipid headgroup remodeling mechanism involving sequential dealkylation and methylation steps. We demonstrate that phosphocholine inhibits the binding of one of the proteins identified, the attractive anticancer target p32, to its endogenous ligands and to the promising p32-targeting anticancer agent, Lyp-1. Our results reveal that choline metabolites play vital roles in cellular physiology by serving as modulators of protein function.
Introduction
Metabolite–protein interactions regulate vital physiological processes including energy production, cellular communication, and protein and nucleic acid biosynthesis.1−3 Therefore, the identification of metabolite-interacting proteins and the delineation of the influence of these interactions on protein function are imperative.
Apart from serving as intermediates in the Kennedy pathway4 (Figure 1a), the physiological roles of choline metabolites are poorly understood. The Kennedy pathway starts with the cellular uptake of choline followed by its sequential conversion into phosphocholine, cytidine 5′-diphospho (CDP) choline, and finally to the phosphatidylcholine (PC) and sphingomyelin (SM) lipids that are catabolized to glycerophosphocholine (GPC). Notably, the cellular levels of these choline metabolites are elevated in a variety of cancers.5 For example, phosphocholine concentrations as high as 9 mM have been reported in the breast cancer cell line MDA-MB-231.6 Non-invasive approaches for the measurement of the cellular levels of choline metabolites are being actively pursued for cancer diagnosis and monitoring cancer progression.5,7 The roles of these metabolites in cancer pathophysiology, however, remain poorly understood. Identification of the choline metabolite-interacting proteome followed by the characterization of the influence of those interactions on protein function is vital for unraveling the extended physiological roles of these metabolites.
Figure 1.

(a) Choline metabolism in mammalian cells, and (b) our strategy of leveraging it to identify the choline metabolite-interacting proteome of mammalian cells. R1, R2, and R3 represent alkyl groups, and CDP, PC, SM, and GP denote cytidine 5′-diphospho, phosphatidylcholine, sphingomyelin, and glycerophospho, respectively.
Herein, we report the discovery of the mammalian choline metabolite-interacting proteome by developing a photoaffinity-labeling8−13 platform that entailed the use of a rationally designed alkynyl diazirine choline analogue capable of metabolically labeling choline metabolites (Figure 1b). The probe design was guided by our metabolic labeling studies focused on interrogating the promiscuity of the Kennedy pathway with respect to accepting choline analogues as precursors. Functional studies on selected protein hits revealed that choline metabolites profoundly modulate protein function. Additionally, metabolic labeling studies resulted in the unexpected discovery of a choline lipid headgroup remodeling mechanism.
Results and Discussion
Choline Lipid Biosynthetic Machinery Is Highly Promiscuous
A critical prerequisite for the success of our approach was the amenability of choline metabolites to undergo metabolic labeling with a choline analogue appended with both the diazirine and the alkynyl/azido groups. Consequently, we evaluated the propensity of structurally diverse choline analogues (compounds 1–9, Figure 2) to metabolically label choline metabolites by characterizing the metabolic labeling of the end products of the pathway, the PC and SM lipids, via lipidomics and cellular imaging. Our lipidomics experiments entailed subjecting lipids extracted from choline analogue-administered HEK293 cells to liquid chromatography with tandem mass spectrometry (LC–MS/MS) in the PIS mode14 (Figure S1, Supporting Information). Our imaging experiments involved treating these cells with azido/alkynyl fluorescent dyes under click chemistry conditions as reported previously with the close structural mimics of choline, propargyl choline15 (1), and azidoethyl choline (2).16,17
Figure 2.

Characterization of the metabolic labeling of mammalian choline lipids with choline analogues. The lipidomics experiments were performed on the lipids extracted from analogue (2 mM)-administered HEK293 cells and subjecting them to LC–MS/MS in the precursor ion scan (PIS) mode. The TICs are depicted on the left along with the m/z values of the daughter ions and the peak retention time values. The extracted ion chromatograms (XICs) with the three most abundant lipids annotated are depicted on the right. LipidView analyses for annotating all the labeled lipids detected are depicted in Figures S2–S10. The imaging experiments involved treating analogue (2 mM)-administered HEK293 cells (left) with either 5-azido fluorescein (for analogues 1, 3–9) or rhodamine alkyne (for analogue 2) under click chemistry conditions. Competition experiments involved the co-administration of native choline and the analogues (2 mM each; right). The scale bar represents 100 μm.
Remarkably, all of the nine analogues successfully labeled cellular choline lipids as demonstrated by the lipidomics analyses depicted in the left panel of Figure 2. Even the analogues that are structurally substantially different from native choline—hexynyl choline (4), the dialkynyl compound 6, benzyl propargyl choline (7), and the trisubstituted derivatives 8 and 9—yielded prominent total ion chromatogram (TIC) peaks for the labeled lipids. Importantly, the labeling efficiency dropped with increasing substituent chain lengths, as demonstrated by a consistent attenuation of TIC peak intensities going from analogue 1 to 4 (Figure 2a–d).
Notably, cells metabolically labeled with our most elaborate analogues (4 and 6–9) did not yield intense fluorescence signals (Figures 2d,f–i, right panel) despite confirmation of metabolic labeling via lipidomics (the corresponding left panels). These results establish the higher sensitivity of our lipidomics workflow as compared to our version of click chemistry-based imaging for characterizing metabolic labeling. In fact, fluorescence imaging as a means to characterize metabolic labeling was compatible with only those analogues (1, 2, 3, and 5) that yielded TIC peak intensities of the order of E8 or above for the labeled lipids.
This survey of choline analogues revealed that extending the chain length of the alkyl substituents leads to reduced metabolic labeling efficiency, motivating us to develop bifunctional choline analogues bearing the diazirine and the alkynyl/azido groups on different alkyl substituents on the choline nitrogen, rather than on the same substituent. The feasibility of this strategy was supported by the successful metabolic labeling of choline lipids by analogues 5, 6, and 7, wherein two N-methyl groups of choline were replaced with longer alkyl groups (Figure 2e–g).
Metabolic Labeling Studies on β-Diazirine Cholines Uncover a Choline Lipid Headgroup Remodeling Mechanism
We began metabolic labeling studies on photocrosslinkable choline analogues with the β-diazirines 10 and 11 and discovered that both of them metabolically label HEK293 choline lipids (Figure 3a,b). Surprisingly however, in-gel fluorescence experiments wherein the lysates of UV-irradiated cells metabolically labeled with 11 were treated with Alexa Fluor 594 azide under click chemistry conditions yielded negligible intensity for fluorescently labeled proteins (Figure 3b, bottom panel). 1H NMR and UV spectroscopy experiments on 11 ruled out lack of UV-reactivity and chemical instability as possible reasons for these poor photocrosslinking yields (Figures S14 and S15).
Figure 3.

Metabolic labeling studies on β-diazirine choline analogues. (a) PIS lipidomics to detect 10-labeled lipids in 10-administered HEK293 cells. (b) Top panel: PIS lipidomics to detect 11-labeled lipids in 11-administered cells. Bottom panel: in-gel fluorescence experiments on cells metabolically labeled with 11. (c) Top panel: imaging experiments on 11 (2 mM)-administered cells (left), and cells administered with both 11 and choline (2 mM each; right). The scale bars represent 100 μm. Middle panel: PIS lipidomics to detect 1-labeled lipids in 11-administered cells. Bottom panel: the Cα–Cβ bond cleavage hypothesis to explain the formation of 1-labeled lipids in 11-administered cells. DAG is an abbreviation for diacylglycerol. (d) Top panel: PIS lipidomics to detect 10-labeled lipids in 11-administered cells. Bottom panel: the Cα–Cβ bond cleavage hypothesis to explain the formation of 10-labeled lipids in 11-administered cells. (e) Expected formation (top) and detection (bottom) of 2-labeled lipids in 12-administered cells. (f) Expected formation (top) and attempted detection (second panel from the top) of 14-labeled lipids in 13-administered cells. Third panel from the top: the detection of 1-labeled lipids in 13-administered cells. Bottom panel: a hypothesis invoking N–C bond cleavage followed by methylation to explain the formation of 1-labeled choline lipids in 13-administered cells. (g) Expected generation (top) and detection (bottom left panel) of D6-labeled (16-labeled) choline lipids due to N–C bond cleavage followed by methylation in D9 choline (15)-administered cells. Bottom right panel: the detection of native choline lipids in cells cultured in standard media devoid of non-natural choline analogues. The LipidView analyses of all XICs are provided in the Supporting Information section. All analogues were administered at 2 mM concentrations unless otherwise stated.
Notably, although 11-labeled lipids yielded a TIC peak intensity similar to that obtained for 7, 8, and 9-labeled lipids (∼E6; Figures 3b and 2g–i), cellular imaging experiments with the former yielded intense fluorescence signals (Figure 3c, top panel) in stark contrast to the latter (Figure 2g–i). In fact, the imaging results for 11 were reminiscent of those obtained with 1 (Figure 2a) engendering the hypothesis that 11-administered cells form 1-labeled choline lipids. Consistent with this hypothesis, lipids isolated from 11-administered cells yielded a high-intensity TIC peak for 1-labeled lipids (∼E8; Figure 3c, middle panel). Cells administered with 11 also formed alkyne-devoid 10-labeled lipids (Figure 3d). A possible explanation for these observations is that 11-labeled choline metabolites form their 1- and 10-labeled counterparts by undergoing Cα–Cβ bond cleavage within cells (the bottom panels of Figure 3c,d).
The TIC peak intensities for 10- and 11-labeled lipids were 2 orders of magnitude lower than those for 1-labeled lipids (Figure 3b–d), establishing that 1-labeled lipids are the dominant metabolically labeled choline lipid species formed in 11-administered cells. HEK293 cells cultured in the presence of 11 for 4, 8, 16, and 24 h revealed that this trend persisted at all these time points (Figure S16). Experiments with 11 on HeLa and MDA-MB-231 cells yielded similar results (Figure S18), demonstrating the generality of this phenomenon across mammalian cell lines. The formation of large amounts of 1-labeled lipids accounts for the high fluorescence intensity observed in the imaging experiments on 11-administered cells (Figure 3c, top panel), whereas the formation of small amounts of 11-labeled choline metabolites explains the low intensity of fluorescently labeled crosslinked proteins obtained in the in-gel fluorescence experiment (Figure 3b, bottom panel). This problem is likely aggravated due to competition for protein-binding sites between the sets of photocrosslinkable choline metabolites labeled with 11 and those labeled with 10.
Two other bifunctional β-diazirine cholines, the azido diazirine 12 (Figures 3e and S21) and S26 (Figure S22), were also processed by mammalian cells in a fashion similar to 11. Interestingly however, metabolic labeling experiments with the branched alkynyl β-diazirine choline 13 did not yield the expected major Cα–Cβ bond cleavage product, that is, ethyl propargyl choline (14)-labeled lipids (top two panels of Figure 3f), and instead, a prominent TIC peak for 1-labeled lipids was obtained (Figure 3f, third panel). The formation of 1-labeled lipids in cells administered with both unbranched and branched β-diazirine cholines supports a mechanism involving the cleavage of the N–C bonds between the choline nitrogen and their N-alkyl substituents followed by methylation (depicted for 13 in Figure 3f, bottom panel), over the Cα–Cβ bond cleavage mechanism proposed above.
Intrigued by the above findings, we investigated whether native choline metabolites are also cellularly processed in a similar fashion by culturing HEK293 cells in the presence of deuterated (D9) choline. PIS analysis yielded dose-dependent signals for D6 choline lipids (removal of the CD3 group due to N–C bond cleavage followed by the incorporation of the −CH3 group) at the same retention time as that for native (H9) choline lipids (Figure 3g), establishing that native choline metabolites are also susceptible to sequential dealkylation and methylation in mammalian cells.
The discovery of this novel choline lipid headgroup remodeling pathway has important implications for disease biology as alterations in cellular PC/PE ratios are associated with the progression of several diseased states, including non-alcoholic fatty liver disease, liver failure, and ulcerative colitis.18 Future studies on characterizing the prevalence of this choline lipid remodeling mechanism in disease-relevant cell lines and animal model systems may provide valuable insights on its relevance to disease pathophysiology. Although the cellular enzymes responsible for the dealkylation and methylation of the choline metabolite derivatives of the analogues reported herein await discovery, enzymes that catalyze the methylation of ethanolamine metabolites and lipids have been previously reported. For example, orthologs of phosphoethanolamine methyltransferase catalyze the conversion of phosphoethanolamine to phosphocholine by sequential N-methylation in plants,19 as well as in multiple species of the Plasmodium family;20 however, these enzymes are believed to be absent in humans. Another example of the N-methylation of ethanolamine metabolites is the formation of choline from ethanolamine, N-methyl ethanolamine, and N,N-dimethyl ethanolamine in rat brain tissue and chicken neuronal cells in culture.21,22 Additionally, the phosphatidylethanolamine N-methyltransferase (PEMT) enzyme is known in N-methylate mammalian PE lipids to yield PC lipids.23 Notably, PEMT has been reported to not be expressed in HEK293 cells,24 the cell line on which we performed the majority of our lipidomics experiments reported herein. In contrast to these previous studies on the N-methylation of ethanolamine metabolites and lipids, reports on the dealkylation of choline metabolites and lipid metabolites are lacking. Clearly, focused efforts on the discovery of the enzymes that orchestrate the dealkylation–methylation of choline metabolites reported herein are desirable.
γ-Diazirine Cholines Are Ideal Metabolic Labeling Probes for Choline Lipids
The γ-diazirine cholines 17 and 18 demonstrated robust metabolic labeling (Figure 4a,b). Notably, the TIC peak intensity for 18-labeled lipids is more than four-fold higher than that obtained for the analogous alkynyl β-diazirine choline compound (11, Figure 3b). Moreover, as expected from an alkynyl choline analogue that yields a TIC peak intensity of <E8, a low fluorescence intensity was obtained upon subjecting cells metabolically labeled with 18 to our imaging conditions (Figure S25a), suggesting that unlike 11, 18 does not form large amounts of 1-labeled lipids. Indeed, lipids isolated from 18-administered cells yielded a small TIC peak (∼E6) for 1-labeled lipids (Figure 4c) as compared to an ∼E8 intensity peak obtained from 11-administered cells (Figure 3c, middle panel). Furthermore, no detectable amount of choline lipids labeled with the alkyne-devoid diazirine (17) was observed (Figure 4d). In fact, the desired 18-labeled choline lipids were the dominant metabolically labeled choline lipids formed in three different 18-administered mammalian cell lines (HEK293, HeLa, and MDA-MB-231; Figure 4e,f). These results unequivocally establish 18 as an ideal probe for metabolically labeling choline metabolites with the diazirine and alkyne groups.
Figure 4.

Metabolic labeling studies on γ-diazirine choline analogues. (a) PIS lipidomics to detect 17-labeled lipids in 17-administered HEK293 cells. (b) PIS lipidomics and LipidView analyses to detect 18-labeled lipids in 18-administered cells. (c) Expected formation (top) and attempted detection (bottom) of 1-labeled lipids in 18-administered cells. (d) Expected formation (left) and detection (right) of 17-labeled lipids in 18-administered cells. (e) Time course plot for TIC peak areas corresponding to 1-, 17-, and 18-labeled lipids in cells cultured in the presence of 18 for 4, 8, 16, and 24 h. (f) TIC peak areas for 1-, 17-, and 18-labeled lipids in HEK293, HeLa, and MDA-MB-231 cells cultured in the presence of 18 for 24 h, respectively. Each data point in the plots depicted in (e,f) is an average of three biological replicates, and the error bars represent the standard error values (representative TICs are depicted in Figures S26−S28). The LipidView analyses of the XICs depicted in (a,c) are provided in the Supporting Information section. All analogues were administered at 2 mM concentrations.
Interestingly, 18 has been previously investigated for metabolically labeling choline lipids in mammalian cells.25 Based on imaging experiments, this previous study concluded that 18 is incapable of labeling choline lipids in HeLa cells. Consistent with this observation, our imaging experiments on HEK293 cells administered with 18 failed to yield fluorescence signals above the background (Figure S25a). However, our lipidomics experiments convincingly demonstrated that 18 metabolically labeled choline lipids in HEK293, HeLa, and MDA-MB-231 cells (Figure 4f). These results establish the superiority of sensitive lipidomics approaches over click chemistry-mediated imaging techniques for characterizing lipidic metabolic labeling.
Discovery of the Mammalian Choline Metabolite-Interacting Proteome
In-gel fluorescence experiments with 18 yielded a multitude of fluorescently labeled proteins that demonstrated a dose-dependent (Figure 5a) and UV exposure time-dependent (Figure 5b) increase in fluorescence intensities. In contrast, neither of the four bifunctional β-diazirine choline analogues, 11, 12, 13, and S26 yielded fluorescently labeled proteins (Figure 5c).
Figure 5.

Identification of the choline metabolite and lipid-interacting proteome of HEK293 cells by using the γ-diazirine choline analogue 18. Panels (a–c) depict in-gel fluorescence data. (a) Photocrosslinking experiments on HEK293 cells metabolically labeled with different concentrations of 18. (b) UV exposure time-dependence on the photocrosslinking efficiency in cells metabolically labeled with 18. (c) Comparison of photocrosslinking efficiency in cells metabolically labeled with 11, 12, 13, 18, and S26. Propargyl choline (1) was used as a negative control. (d) Western blot characterization of the dose-dependence of avidin-enrichment of biotinylated UV-crosslinked proteins obtained from cells metabolically labeled with 18. (e) Plot depicting the results of our six-plex TMT analysis (three +UV and three −UV samples). The averaged enrichment ratio for each of the 902 proteins detected is plotted against their respective p-values, and the 674 high-confidence protein hits (enrichment ratio ≥ 2.00 and p-value of ≤ 0.05) taken forward for further analysis are depicted as either red or blue dots. (f) Western blot validation of the enrichment of the seven proteins marked by blue dots in Figure 5e blotted by using primary antibodies against the individual proteins with their TMT enrichment ratios depicted within parentheses. (g) Subcellular distribution of our protein hits as per the UniProt database. (h) Protein hits classified according to their biochemical functions as per the PANTHER database. (i) Classification of the enzyme protein hits according to the UniProt database. (j) Protein hits genetically linked to various disease classes as per the DAVID database. (k) Pie-chart depicting the classification of the protein hits with respect to their annotation as drug targets according to the DrugBank database. The lists of all the proteins in the above analyses are provided in Table S3.
With optimal metabolic labeling, photocrosslinking, and biotinylation conditions determined, we executed our chemoproteomics workflow depicted in Figure 1b. Treating the cell lysate of UV-irradiated HEK293 cells cultured in the presence of 18 with azido biotin under click chemistry conditions and applying the proteins extracted from this reaction mixture onto an avidin column yielded a dose-dependent enrichment of proteins (Figure 5d). For the proteomics experiments, we performed “+UV” and “–UV” experiments on three 18-administered cell cultures each and subjected the captured proteins to six-plex tandem mass tag (TMT) proteomics. This analysis yielded a total of 674 “high-confidence hits” (enrichment ratio ≥ 2.0 and p-value ≤ 0.05). These proteins are depicted as red and blue dots in Figure 5e, and the complete list is provided in sheet 1 of Table S3. The veracity of these proteomics data was validated by Western blotting-based experiments (Figure 5f) on seven of these proteins (p32, nucleolin, COX 4, ezrin, PGRMC1, VDAC1, and annexin A2), possessing a range of enrichment values (depicted as blue dots in Figure 5e).
Choline Metabolites Interact with a Diverse Range of Cellular Proteins
A survey of the proteins identified revealed that although a majority of these proteins (355 out of 674) are expressed in the cytoplasm, 324 are nuclear and 166 are mitochondrial (Figure 5g and sheet 2 of Table S3). The analysis on the UniProt database revealed that the majority (73%) of them are soluble proteins (sheet 3 of Table S3), consistent with an in-gel fluorescence experiment that yielded a substantially higher fluorescence intensity in the soluble protein fraction as compared to the membrane protein fraction (Figure S29).
Although cellular proteins that interact with water-soluble choline metabolites have not been profiled previously, several proteins have been reported to bind to choline lipids, many of which are also present in our list of protein hits. These proteins include ADP/ATP translocase, the cytochrome c oxidase complex proteins COX 2, 4I1, 5A, 5B, 7A2, 7C, and subunits 1 and 2 of the cytochrome bc1 complex, whose structures depict PC lipids bound to them.26−28 Moreover, 60 of our protein hits were identified previously to bind to PC lipids in HeLa cells (sheet 4 of Table S3).25 Comparison of our protein hits with the PC-interacting yeast mitochondrial proteins identified previously29 revealed that the mammalian homologs of 10 out of the 47 proteins reported in that study are present in our list (sheet 5 of Table S3).
Subjecting our protein hits to the PANTHER classification system resulted in their categorization into 16 functional classes (sheet 6 of Table S3; the 10 most abundant classes are depicted in Figure 5h). Our list contains enzymes belonging to all of the 6 major enzyme subclasses (Figure 5i and sheet 7 of Table S3) and includes 7 of the 10 glycolytic enzymes, 7 of the 8 Krebs cycle enzymes, and proteins belonging to all 5 complexes of the oxidative phosphorylation pathway.
Choline Metabolite-Interacting Proteins Play Important Roles in Diseases and Are Attractive Drug Targets
Analysis on the DAVID resource revealed that 67% of our protein hits were associated with diseases, including 190 with infectious diseases, 109 with cancer, 128 with metabolic disorders, and 99 with neurological disorders (Figure 5j, sheet 8 of Table S3). A total of 370 genes encoding our protein hits are overexpressed in at least 5 different types of cancer according to the ONCOMINE database (sheet 9 of Table S3). Moreover, several of our protein hits are known anticancer drug targets including PARP1, DNA topoisomerase II α, GART, and isocitrate dehydrogenase 2 that are targeted by the drugs olaparib,30 doxorubicin,31 pemetrexed,32 and enasidenib,33 respectively (sheet 10 of Table S3). DrugBank database analysis revealed that the proteins identified in our study are targets for the therapy of several other diseases as well, with 37.1% (a total of 250 proteins) of them known to serve as drug targets (Figure 5k and sheet 10 of Table S3). Interestingly, this percentage is much higher than the percentage of proteins in the entire human proteome pool that are known drug targets (12%).34
Phosphocholine Profoundly Modulates the Binding of p32 with Its Interacting Partners
To evaluate the importance of protein–choline metabolite interactions on protein function, we studied the effect of choline metabolites on the binding of our most enriched protein hit, p32 (Figure 5e and sheet 1 of Table S3), to a variety of its interacting partners. Also known as HABP1, p33, and gC1qR, p32 is a multifunctional protein that plays vital roles in a diverse array of physiological processes and diseased states.35 p32 is expressed at disparate cellular locations and performs diverse functions including facilitating mitochondrial translation,36 orchestrating cell attachment and migration by binding to extracellular matrix components such as hyaluronic acid (HA)37 at the plasma membrane, and contributing to HIV infectivity by binding to the HIV Tat protein within the nucleus.38 Plasma membrane-expressed p32 levels are dramatically enhanced in cancer cells, enabling cell surface p32 to serve both as a diagnostic marker and as a drug target.35 In fact, the promising anticancer and tumor imaging agent, the cyclic peptide Lyp-1, is recruited to cancer cells via its specific binding to cell surface-expressed p32.39 Moreover, small molecule libraries are actively being screened as p32 ligands to identify attractive anticancer lead compounds.40,41
Our studies on p32 began with investigating its binding to individual choline metabolites by performing drug affinity-responsive target stability (DARTS) experiments.42 These experiments involved the treatment of recombinantly produced p32 with the protease cocktail, pronase, in the presence of choline metabolites, followed by SDS-PAGE analysis to evaluate whether they protect p32 from proteolysis. These experiments revealed that although choline, CDP choline, PC, SM, and GPC do not protect p32 from protease digestion (Figure S30d,f–i), pronase-mediated cleavage of p32 was substantially attenuated in the presence of 25 and 50 mM phosphocholine (Figure S30e; left panel). Identical experiments on the myoglobin protein demonstrated negligible phosphocholine-mediated protection against protease cleavage (Figure S30e; right panel), establishing that the protective effect of phosphocholine toward the proteolytic digestion of p32 is due to the specific binding of phosphocholine to p32.
We performed ELISA on the biotinylated recombinant p32 to characterize the effect of choline metabolites on the binding of p32 to its known interacting partners. Considering that phosphocholine is present in concentrations as high as 9 mM in mammalian cells,6 we employed 5 mM of phosphocholine in these experiments. Other choline metabolites were used at 1 mM concentrations as they are present at lower concentrations in mammalian cells, with GPC levels reaching 0.6 mM in MDA-MB-231 breast cancer cells and 1 mM in the PC-3 metastatic prostate cancer cells.6,43
We first focused on the p32-HA complex that is formed on the surface of cancer cells and plays an important role in tumor cell adhesion.35,37 Our ELISA experiments revealed that phosphocholine inhibits p32-HA binding in a competitive fashion, whereas other choline metabolites demonstrated minimal effects (Figure 6a). We next studied another interaction that occurs on cancer cell surfaces, the binding of p32 to the tumor-homing peptide Lyp-1, leading to the peptide’s cellular internalization, enabling it to trigger apoptosis.39 The formation of this complex was profoundly inhibited specifically by phosphocholine (Figure 6b).
Figure 6.

Studies on the modulation of binding of p32 with its interacting partners by choline metabolites. (a–d) ELISA characterization of the effect of choline metabolites on the binding of p32 to HA (a), Lyp-1 (b), acetylated-Tat peptide (c), and MCP1 (d). The metabolite/salt concentrations employed were as follows: 1 mM choline/CDP choline/GPC/NaCl, 5 mM phosphocholine/CaCl2, and 100 μg/mL of PC/SM.
In addition to its important role in cancer, p32 regulates viral infectivity in mammalian cells by binding to the acetylated-Tat protein of HIV-1, resulting in its recruitment to the HIV-1 promoter.38,44 An 18 amino acid-long acetylated peptide spanning residues 36–53 of the Tat protein recapitulates the binding of the acetylated-Tat protein to p32.38 ELISA on this acetylated-Tat peptide revealed that phosphocholine potently inhibits its binding to p32, whereas GPC, CDP choline, choline, PC, and SM demonstrate negligible to moderate inhibitory effects (Figure 6c).
Another p32-interacting mammalian protein is the chemokine, monocyte chemoattractant protein-1 (MCP1). p32 serves as a pro-inflammatory agent by binding to MCP1 and slowing down its cellular degradation, leading to an increase in its cellular levels.45 Our ELISA experiments revealed that phosphocholine inhibits the binding of p32 with MCP1 in a competitive fashion (Figure 6d).
Taken together, our studies on p32 establish that phosphocholine inhibits the binding of p32 to a range of its interacting partners, both endogenous and synthetic.
Choline Metabolites Modulate the Activity of Cellular Enzymes
We also characterized the effects of choline metabolites on the function of five of our enzyme hits: lactate dehydrogenase (LDH), citrate synthase, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), malic enzyme, and glutamic dehydrogenase (sheet 1 of Table S3). LDH is overexpressed in almost all types of cancer, and LDH inhibitors are promising anticancer agents.46 This enzyme catalyzes the conversion of pyruvate to lactate by employing NADH as a coenzyme (Figure S31a). At constant NADH concentrations, we observed that the Km value for pyruvate was reduced ∼five-fold in the presence of 1 mM choline, whereas it was halved in the presence of the same concentrations of CDP choline and GPC (Figure S31b,c). The presence of these three metabolites also caused an attenuation of the Km values of the enzyme in assays wherein the concentration of pyruvate was kept constant, whereas that of NADH was altered, with the largest effect seen with choline (a ∼four-fold reduction, Figure S31b,d). Our experiments on citrate synthase revealed a ∼2.6-fold attenuation of the substrate (oxaloacetate) Km value at constant acetyl CoA concentrations in the presence of PC (Figure S32b,d). Assays on malic enzyme revealed a ∼two-fold attenuation of the Km values of NADP+ in the presence of GPC, phosphocholine and PC, and a similar attenuation of the Km value of malate in the presence of GPC (Figure S34). GAPDH (Figure S33), and glutamate dehydrogenase (Figure S35) was negligibly modulated by choline metabolites.
Conclusions
In summary, our studies have provided novel insights on choline lipid biology and established the importance of choline metabolite–protein interactions in protein function. Our metabolic labeling studies demonstrate that the mammalian choline lipid biosynthetic machinery is remarkably promiscuous, and our choline probes add to the rapidly expanding toolkit of lipidic metabolic labeling probes that include those for labeling both the hydrophobic tails25,47−50 and headgroups50 of lipids, sphingolipids,51−55 inositol lipids,56 and phosphatidic acid.57 Moreover, our discovery that choline lipid headgroups are susceptible to chemical remodeling via sequential dealkylation and methylation in mammalian cells will engender studies focused on elucidating the biochemical basis of this mechanism and its role in lipid homeostasis and disease pathophysiology. Functional studies on six selected protein hits revealed that four of them are modulated by choline metabolites. Our studies on p32 were particularly insightful and revealed that phosphocholine is a potent and general modulator of the binding of this protein with a variety of its interacting partners. These results have important ramifications in cancer biology and also implicate phosphocholine in mechanisms underlying HIV-1 infectivity and the inflammation response. The stage is now set for future studies focused on unraveling the in vivo ramifications of choline metabolite-mediated modulation of the interaction of p32 with its binding partners. For example, experiments involving the extracellular administration of phosphocholine to cancer cells can be performed, leading to the disruption of the binding of cell surface p32 to HA, and the resulting effects on cell migration can be studied. Additionally, to evaluate the effect of phosphocholine-mediated inhibition of the binding of Lyp-1 with cell surface p32, the anticancer activity of Lyp-1 can be evaluated by co-administering Lyp-1 and phosphocholine to cancer cells in culture, followed by assaying for Lyp-1-triggered apoptosis.
Methods
Mass-Spectrometry Details for Lipidomics Experiments
Lipids were extracted from mammalian cells administered with non-natural choline analogues and subjected to LC–MS/MS as per the procedure described in sub-section 5 of the Supporting Information. Lipids eluted from the HILIC column were analyzed and detected on an AB Sciex 4500 QTRAP mass spectrometer equipped with an ESI source heated to 400 °C. The scan speed was maintained at 200 Da/s. The electrospray capillary was held at +5500 V, the curtain gas was set at 35 (arbitrary units), the CAD gas was maintained at a medium level, and the two ion source gases GS1 and GS2 were set at 50 and 60 (arbitrary units), respectively. The PIS mode (schematically described in Figure S1) was used for detecting all the non-natural and native choline lipids to generate the TICs (all TICs are representative of at least three biological replicates). The TIC peaks were processed using the Analyst software to generate the XICs. The compound parameters (declustering potential, entrance potential, collision energy, and collision cell exit potential) were optimized by ramping each of them against the intensity by the direct infusion method. Native choline lipids were detected by employing a PIS of 184.1 Da (m/z for phosphocholine), and the compound parameters were optimized separately. For characterizing metabolic labeling with alkynyl/azido-substituted choline analogues 1–9 and 14, and with the D6 choline analogue 16, the PIS masses of their corresponding phosphoryl derivatives and the optimized compound parameters for the PIS of 184.1 Da (m/z for phosphocholine) were used. For all diazirine analogues, the PIS mass used was 28 Da less than that of the fragment mass of their phosphoryl derivatives to account for the N2 loss that diazirines characteristically undergo during fragmentation. Compound parameters were separately optimized for detecting the β-diazirine 11-labeled choline lipids by using a PIS of 234.1 Da, and the same parameters were used in all PIS experiments for detecting choline lipids labeled with other β-diazirine analogues 10, 12, 13, S26, S35, and S36. Compound parameters were separately optimized for detecting γ-diazirine 18-labeled choline lipids by using a PIS of 248.1 Da, and the same parameters were used to detect 17-labeled choline lipids. Each TIC was background-subtracted with the corresponding blank chromatogram. The optimized compound parameters employed for each analogue are listed in Table S1.
Probing the Binding between p32 and Its Ligands, Lyp-1, HA, Ac-Tat Peptide, and MCP1 Using ELISA
The stock solutions of the ligands were made in PBS at the following concentrations: Ac-Tat peptide (5 mg/mL), MCP1 (11.5 mM), Lyp-1 (2 mg/mL), and HA (10 mg/mL). These solutions were diluted into the sodium carbonate-bicarbonate buffer (50 mM, pH 9.6) to achieve their respective working concentrations (25 μg/mL for Ac-Tat, 100 nM for MCP1, and 50 μg/mL for both Lyp-1 and HA), and 100 μL of each of these solutions was added to 96 well-plates and incubated for 12 h at 4 °C to immobilize the ligands. Subsequently, each well was washed with PBS (300 μL/well × 3) and blocked with 5% (w/v) skimmed milk solution in PBS (300 μL/well) for 2 h at room temperature. The skimmed milk solution was then aspirated off, and the wells were washed three times with 0.05% (v/v) Tween-20 containing PBS (PBST, 300 μL/well) before introducing solutions (100 μL/well) containing different concentrations of biotinylated His-p32 (procedure for producing biotinylated His-p32 is described in sub-section 17C of the Supporting Information) in PBS (as indicated by the data points of the ELISA plots depicted in Figure 6) and choline metabolites (at concentrations mentioned in the legend of Figure 6). Wells administered with 100 μL of PBS instead of biotinylated His-p32 served as a blank. Subsequently, the plates were incubated at room temperature for 2 h, following which, the solutions were aspirated off and the wells were washed three times with PBST (300 μL/well). The bound biotinylated p32 was detected by incubation in a solution containing HRP conjugated to streptavidin in 5% w/v BSA in PBS (100 μL/well, details in Table S2) for 2 h at room temperature. The unbound protein was washed off with PBST (300 μL/well × 3), and the wells were incubated in 100 μL of the substrate solution containing 1.82 mM 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) and 0.03% w/w hydrogen peroxide solution (diluted from a 30% w/w stock) in citrate–phosphate buffer (100 mM, pH 5.0) for 15 min at room temperature under dark. The reaction was stopped by adding sulfuric acid (625 mM, 100 μL/well), and the intensity of the resulting blue-green color was immediately quantified by measuring the absorbance at 405 nm by using an iMark microplate reader (Bio-Rad). The readings obtained from the blank wells containing immobilized Lyp-1/HA/MCP1/Ac-Tat peptide, but not administered with biotinylated p32, were subtracted from those obtained from the experimental samples, and these blank-subtracted values were used to generate a saturation binding curve (Figure 6a–d). Each value is an average of at least three technical replicates.
Acknowledgments
This work was supported by the DBT/Wellcome Trust India Alliance Fellowship [grant number IA/I/14/2/501551] awarded to J.K., and funds from IISER Pune and IISER Bhopal. G.P.J. thanks SERB for the SERB-National Postdoctoral Fellowship [PDF/2016/003750] that he secured under J.K.’s supervision. A.D. and N.T. thank IISER Pune and CSIR, respectively, for their graduate fellowships. We thank S. Shukla and N. Balasubramanian for providing access to the EVOS cell imaging system, A. Gupta for providing access to a UV–visible spectrophotometer, and H. Kumar for providing access to a Zeiss Axio Vert.A1 microscope and a Bio-Rad microtiter plate reader. We thank T. Teesalu for sharing the plasmid encoding the N-terminal histidine-tagged p32. The TMT proteomic experiments were outsourced to the Thermo Fisher Scientific Center for Multiplexed Proteomics at the Harvard Medical School (https://tcmp.hms.harvard.edu/).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00400.
Organic synthetic schemes and procedures, compound characterization data, protocols for metabolic labeling, cellular imaging, lipidomic and LipidView data, photocrosslinking, proteomics, in-gel fluorescence, biotinylation, Western blotting experiments, the recombinant production of p32, production of biotinylated p32, DARTS assay, and enzyme assays. (PDF)
Table S3 depicting the raw proteomics data and lists of protein hits classified with respect to parameters including expression and functional profiles, involvement in diseases, and roles as drug targets. (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- Wang Y.-P.; Lei Q.-Y. Metabolite sensing and signaling in cell metabolism. Signal Transduction Targeted Ther. 2018, 3, 30. 10.1038/s41392-018-0024-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Xu W.; Zhao S. Regulatory roles of metabolites in cell signaling networks. J. Genet. Genom. 2013, 40, 367–374. 10.1016/j.jgg.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Chubukov V.; Gerosa L.; Kochanowski K.; Sauer U. Coordination of microbial metabolism. Nat. Rev. Microbiol. 2014, 12, 327–340. 10.1038/nrmicro3238. [DOI] [PubMed] [Google Scholar]
- Kennedy E. P.; Weiss S. B. The Function of Cytidine Coenzymes in the Biosynthesis of Phospholipides. J. Biol. Chem. 1956, 222, 193–214. 10.1016/s0021-9258(19)50785-2. [DOI] [PubMed] [Google Scholar]
- Glunde K.; Bhujwalla Z. M.; Ronen S. M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. 10.1038/nrc3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori N.; Wildes F.; Takagi T.; Glunde K.; Bhujwalla Z. M. The Tumor Microenvironment Modulates Choline and Lipid Metabolism. Front. Oncol. 2016, 6, 262. 10.3389/fonc.2016.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podo F.; Sardanelli F.; Iorio E.; Canese R.; Carpinelli G.; Fausto A.; Canevari S. Abnormal choline phospholipid metabolism in breast and ovary cancer: Molecular bases for noninvasive imaging approaches. Curr. Med. Imag. 2007, 3, 123–137. 10.2174/157340507780619160. [DOI] [Google Scholar]
- Qin W.; Yang F.; Wang C. Chemoproteomic profiling of protein-metabolite interactions. Curr. Opin. Chem. Biol. 2020, 54, 28–36. 10.1016/j.cbpa.2019.11.003. [DOI] [PubMed] [Google Scholar]
- Šileikytė J.; Sundalam S.; David L. L.; Cohen M. S. Chemical Proteomics Approach for Profiling the NAD Interactome. J. Am. Chem. Soc. 2021, 143, 6787–6791. 10.1021/jacs.1c01302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niphakis M. J.; Lum K. M.; Cognetta A. B. III; Correia B. E.; Ichu T.-A.; Olucha J.; Brown S. J.; Kundu S.; Piscitelli F.; Rosen H.; Cravatt B. F. A Global Map of Lipid-Binding Proteins and Their Ligandability in Cells. Cell 2015, 161, 1668–1680. 10.1016/j.cell.2015.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaxman H. A.; Woo C. M. Mapping the Small Molecule Interactome by Mass Spectrometry. Biochemistry 2018, 57, 186–193. 10.1021/acs.biochem.7b01038. [DOI] [PubMed] [Google Scholar]
- Conway L. P.; Jadhav A. M.; Homan R. A.; Li W.; Rubiano J. S.; Hawkins R.; Lawrence R. M.; Parker C. G. Evaluation of fully-functionalized diazirine tags for chemical proteomic applications. Chem. Sci. 2021, 12, 7839–7847. 10.1039/d1sc01360b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y.; Bond M. R.; Kohler J. J. Photocrosslinkers illuminate interactions in living cells. Mol. Biosyst. 2008, 4, 473–480. 10.1039/b803218a. [DOI] [PubMed] [Google Scholar]
- Brügger B.; Erben G.; Sandhoff R.; Wieland F. T.; Lehmann W. D. Quantitative analysis of biological membrane lipids at the low picomole level by nano-electrospray ionization tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 2339–2344. 10.1073/pnas.94.6.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao C. Y.; Roth M.; Welti R.; Salic A. Metabolic labeling and direct imaging of choline phospholipids in vivo. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 15332–15337. 10.1073/pnas.0907864106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao C. Y.; Roth M.; Welti R.; Salic A. Biosynthetic labeling and two-color imaging of phospholipids in cells. Chembiochem 2015, 16, 472–476. 10.1002/cbic.201402149. [DOI] [PubMed] [Google Scholar]
- Tamura T.; Fujisawa A.; Tsuchiya M.; Shen Y.; Nagao K.; Kawano S.; Tamura Y.; Endo T.; Umeda M.; Hamachi I. Organelle membrane-specific chemical labeling and dynamic imaging in living cells. Nat. Chem. Biol. 2020, 16, 1361–1367. 10.1038/s41589-020-00651-z. [DOI] [PubMed] [Google Scholar]
- van der Veen J. N.; Kennelly J. P.; Wan S.; Vance J. E.; Vance D. E.; Jacobs R. L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572. 10.1016/j.bbamem.2017.04.006. [DOI] [PubMed] [Google Scholar]
- Mou Z.; Wang X.; Fu Z.; Dai Y.; Han C.; Ouyang J.; Bao F.; Hu Y.; Li J. Silencing of phosphoethanolamine N-methyltransferase results in temperature-sensitive male sterility and salt hypersensitivity in Arabidopsis. Plant Cell 2002, 14, 2031–2043. 10.1105/tpc.001701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause R. G. E.; Goldring J. P. D. Phosphoethanolamine-N-methyltransferase is a potential biomarker for the diagnosis of P. knowlesi and P. falciparum malaria. PLoS One 2018, 13, e0193833 10.1371/journal.pone.0193833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kewitz H.; Pleul O. Synthesis of choline from ethanolamine in rat brain. Proc. Natl. Acad. Sci. U.S.A. 1976, 73, 2181–2185. 10.1073/pnas.73.7.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriamampandry C.; Freysz L.; Kanfer J. N.; Dreyfus H.; Massarelli R. Conversion of ethanolamine, monomethylethanolamine and dimethylethanolamine to choline-containing compounds by neurons in culture and by the rat brain. Biochem. J. 1989, 264, 555–562. 10.1042/bj2640555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance D. E. Phospholipid methylation in mammals: from biochemistry to physiological function. Biochim. Biophys. Acta 2014, 1838, 1477–1487. 10.1016/j.bbamem.2013.10.018. [DOI] [PubMed] [Google Scholar]
- Morita S.-y.; Takeuchi A.; Kitagawa S. Functional analysis of two isoforms of phosphatidylethanolamine N-methyltransferase. Biochem. J. 2010, 432, 387–398. 10.1042/bj20100490. [DOI] [PubMed] [Google Scholar]
- Wang D.; Du S.; Cazenave-Gassiot A.; Ge J.; Lee J.-S.; Wenk M. R.; Yao S. Q. Global Mapping of Protein-Lipid Interactions by Using Modified Choline-Containing Phospholipids Metabolically Synthesized in Live Cells. Angew. Chem., Int. Ed. Engl. 2017, 56, 5829–5833. 10.1002/anie.201702509. [DOI] [PubMed] [Google Scholar]
- Pebay-Peyroula E.; Dahout-Gonzalez C.; Kahn R.; Trézéguet V.; Lauquin G. J.-M.; Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- Shinzawa-Itoh K.; Aoyama H.; Muramoto K.; Terada H.; Kurauchi T.; Tadehara Y.; Yamasaki A.; Sugimura T.; Kurono S.; Tsujimoto K.; Mizushima T.; Yamashita E.; Tsukihara T.; Yoshikawa S. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 2007, 26, 1713–1725. 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange C.; Nett J. H.; Trumpower B. L.; Hunte C. Specific roles of protein-phospholipid interactions in the yeast cytochrome bc1 complex structure. EMBO J. 2001, 20, 6591–6600. 10.1093/emboj/20.23.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubbens J.; Ruijter E.; de Fays L. E. V.; de Kruijff B.; Slijper M.; Rijkers D. T. S.; Liskamp R. M. J.; de Kroon A. I. P. M.; de Kroon A. I. Photocrosslinking and click chemistry enable the specific detection of proteins interacting with phospholipids at the membrane interface. Chem. Biol. 2009, 16, 3–14. 10.1016/j.chembiol.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Verhagen C. V. M.; de Haan R.; Hageman F.; Oostendorp T. P. D.; Carli A. L. E.; O’Connor M. J.; Jonkers J.; Verheij M.; van den Brekel M. W.; Vens C. Extent of radiosensitization by the PARP inhibitor olaparib depends on its dose, the radiation dose and the integrity of the homologous recombination pathway of tumor cells. Radiother. Oncol. 2015, 116, 358–365. 10.1016/j.radonc.2015.03.028. [DOI] [PubMed] [Google Scholar]
- Pommier Y.; Leo E.; Zhang H.; Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerger M.; Omlin A.; Cerny T.; Fruh M. The role of pemetrexed in advanced non small-cell lung cancer: special focus on pharmacology and mechanism of action. Curr. Drug Targets 2010, 11, 37–47. 10.2174/138945010790030974. [DOI] [PubMed] [Google Scholar]
- Medeiros B. C.; Fathi A. T.; DiNardo C. D.; Pollyea D. A.; Chan S. M.; Swords R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia 2017, 31, 272–281. 10.1038/leu.2016.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overington J. P.; Al-Lazikani B.; Hopkins A. L. How many drug targets are there?. Nat. Rev. Drug Discov. 2006, 5, 993–996. 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Saha P.; Datta K. Multi-functional, multicompartmental hyaluronan-binding protein 1 (HABP1/p32/gC1qR): implication in cancer progression and metastasis. Oncotarget 2018, 9, 10784–10807. 10.18632/oncotarget.24082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogal V.; Richardson A. D.; Karmali P. P.; Scheffler I. E.; Smith J. W.; Ruoslahti E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell. Biol. 2010, 30, 1303–1318. 10.1128/mcb.01101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deb T. B.; Datta K. Molecular cloning of human fibroblast hyaluronic acid-binding protein confirms its identity with P-32, a protein co-purified with splicing factor SF2. Hyaluronic acid-binding protein as P-32 protein, co-purified with splicing factor SF2. J. Biol. Chem. 1996, 271, 2206–2212. 10.1074/jbc.271.4.2206. [DOI] [PubMed] [Google Scholar]
- Berro R.; Kehn K.; de la Fuente C.; Pumfery A.; Adair R.; Wade J.; Colberg-Poley A. M.; Hiscott J.; Kashanchi F. Acetylated Tat regulates human immunodeficiency virus type 1 splicing through its interaction with the splicing regulator p32. J. Virol. 2006, 80, 3189–3204. 10.1128/jvi.80.7.3189-3204.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song N.; Zhao L.; Zhu M.; Zhao J. Recent progress in LyP-1-based strategies for targeted imaging and therapy. Drug Deliv. 2019, 26, 363–375. 10.1080/10717544.2019.1587047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paasonen L.; Sharma S.; Braun G. B.; Kotamraju V. R.; Chung T. D. Y.; She Z.-G.; Sugahara K. N.; Yliperttula M.; Wu B.; Pellecchia M.; Ruoslahti E.; Teesalu T. New p32/gC1qR Ligands for Targeted Tumor Drug Delivery. Chembiochem 2016, 17, 570–575. 10.1002/cbic.201500564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yenugonda V.; Nomura N.; Kouznetsova V.; Tsigelny I.; Fogal V.; Nurmemmedov E.; Kesari S.; Babic I. A novel small molecule inhibitor of p32 mitochondrial protein overexpressed in glioma. J. Transl. Med. 2017, 15, 210. 10.1186/s12967-017-1312-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomenick B.; Hao R.; Jonai N.; Chin R. M.; Aghajan M.; Warburton S.; Wang J.; Wu R. P.; Gomez F.; Loo J. A.; Wohlschlegel J. A.; Vondriska T. M.; Pelletier J.; Herschman H. R.; Clardy J.; Clarke C. F.; Huang J. Target identification using drug affinity responsive target stability (DARTS). Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 21984–21989. 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerstaff E.; Pflug B. R.; Nelson J. B.; Bhujwalla Z. M. Detection of increased choline compounds with proton nuclear magnetic resonance spectroscopy subsequent to malignant transformation of human prostatic epithelial cells. Cancer Res. 2001, 61, 3599–3603. [PubMed] [Google Scholar]
- Liu Z.; Kato A.; Oyama M.; Kozuka-Hata H.; Arii J.; Kawaguchi Y. Role of Host Cell p32 in Herpes Simplex Virus 1 De-Envelopment during Viral Nuclear Egress. J. Virol. 2015, 89, 8982–8998. 10.1128/jvi.01220-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders E.; Nebel D.; Westman J.; Herwald H.; Nilsson B.-O.; Svensson D. Globular C1q receptor (p33) binds and stabilizes pro-inflammatory MCP-1: a novel mechanism for regulation of MCP-1 production and function. Biochem. J. 2018, 475, 775–786. 10.1042/bcj20170857. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Xiong Y.; Qiao T.; Li X.; Jia L.; Han Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. 10.1002/cam4.1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuerschner L.; Ejsing C. S.; Ekroos K.; Shevchenko A.; Anderson K. I.; Thiele C. Polyene-lipids: a new tool to image lipids. Nat. Methods 2005, 2, 39–45. 10.1038/nmeth728. [DOI] [PubMed] [Google Scholar]
- Thiele C.; Papan C.; Hoelper D.; Kusserow K.; Gaebler A.; Schoene M.; Piotrowitz K.; Lohmann D.; Spandl J.; Stevanovic A.; Shevchenko A.; Kuerschner L. Tracing fatty acid metabolism by click chemistry. ACS Chem. Biol. 2012, 7, 2004–2011. 10.1021/cb300414v. [DOI] [PubMed] [Google Scholar]
- Haberkant P.; Raijmakers R.; Wildwater M.; Sachsenheimer T.; Brügger B.; Maeda K.; Houweling M.; Gavin A.-C.; Schultz C.; van Meer G.; Heck A. J. R.; Holthuis J. C. M. In vivo profiling and visualization of cellular protein-lipid interactions using bifunctional fatty acids. Angew. Chem., Int. Ed. Engl. 2013, 52, 4033–4038. 10.1002/anie.201210178. [DOI] [PubMed] [Google Scholar]
- Ancajas C. F.; Ricks T. J.; Best M. D. Metabolic labeling of glycerophospholipids via clickable analogues derivatized at the lipid headgroup. Chem. Phys. Lipids 2020, 232, 104971. 10.1016/j.chemphyslip.2020.104971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberkant P.; Stein F.; Höglinger D.; Gerl M. J.; Brügger B.; Van Veldhoven P. P.; Krijgsveld J.; Gavin A.-C.; Schultz C. Bifunctional Sphingosine for Cell-Based Analysis of Protein-Sphingolipid Interactions. ACS Chem. Biol. 2016, 11, 222–230. 10.1021/acschembio.5b00810. [DOI] [PubMed] [Google Scholar]
- Höglinger D.; Nadler A.; Haberkant P.; Kirkpatrick J.; Schifferer M.; Stein F.; Hauke S.; Porter F. D.; Schultz C. Trifunctional lipid probes for comprehensive studies of single lipid species in living cells. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 1566–1571. 10.1073/pnas.1611096114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kol M.; Williams B.; Toombs-Ruane H.; Franquelim H. G.; Korneev S.; Schroeer C.; Schwille P.; Trauner D.; Holthuis J. C.; Frank J. A. Optical manipulation of sphingolipid biosynthesis using photoswitchable ceramides. Elife 2019, 8, e43230 10.7554/eLife.43230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Devaraj N. K. Synthetic probes and chemical tools in sphingolipid research. Curr. Opin. Chem. Biol. 2021, 65, 126–135. 10.1016/j.cbpa.2021.08.002. [DOI] [PubMed] [Google Scholar]
- Haberkant P.; Schmitt O.; Contreras F.-X.; Thiele C.; Hanada K.; Sprong H.; Reinhard C.; Wieland F. T.; Brügger B. Protein-sphingolipid interactions within cellular membranes. J. Lipid Res. 2008, 49, 251–262. 10.1194/jlr.d700023-jlr200. [DOI] [PubMed] [Google Scholar]
- Ricks T. J.; Cassilly C. D.; Carr A. J.; Alves D. S.; Alam S.; Tscherch K.; Yokley T. W.; Workman C. E.; Morrell-Falvey J. L.; Barrera F. N.; Reynolds T. B.; Best M. D. Labeling of Phosphatidylinositol Lipid Products in Cells through Metabolic Engineering by Using a Clickable myo-Inositol Probe. ChemBioChem 2019, 20, 172–180. 10.1002/cbic.201800248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bumpus T. W.; Baskin J. M. A Chemoenzymatic Strategy for Imaging Cellular Phosphatidic Acid Synthesis. Angew. Chem., Int. Ed. Engl. 2016, 55, 13155–13158. 10.1002/anie.201607443. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.