

Abstract
Primary biliary cholangitis (PBC) is a chronic cholestatic autoimmune liver disease characterized by a destructive, small duct, and lymphocytic cholangitis, and marked by the presence of antimitochondrial antibodies. The incidence and prevalence of PBC vary widely in different regions and time periods, and although disproportionally more common among White non-Hispanic females, contemporary data show a higher prevalence in males and racial minorities than previously described. Outcomes largely depend on early recognition of the disease and prompt institution of treatment, which, in turn, are directly influenced by provider bias and socioeconomic factors. Ursodeoxycholic acid remains the initial treatment of choice for PBC, with obeticholic acid and fibrates (off-label therapy) reserved as add-on therapy for the management of inadequate responders or those with ursodeoxycholic acid intolerance. Novel and repurposed drugs are currently at different stages of clinical development not only for the treatment of PBC but also for its symptomatic management. Here, we summarize the most up-to-date data regarding the epidemiology, prognosis, and treatment of PBC, providing clinically useful information for its holistic management.
INTRODUCTION
Primary biliary cholangitis (PBC) is a chronic inflammatory hepatobiliary disease characterized by a T-cell lymphocyte–mediated destruction of the interlobular bile ducts that can lead to biliary cirrhosis and liver failure if left untreated.1
This autoimmune liver disease results from the combination of environmental triggers, the individual’s genetic susceptibility, and epigenetic factors. Supporting the role of the genetic contribution to disease development, there is a 63% concordance among identical twins,2 a relative risk of 9.13–10.5 in first-degree relatives, and a strong association with HLA and non-HLA allelic variants.3,4 Molecular mimicry between lipoic acids, exogenous antigens, and xenobiotics accounts for the loss of tolerance to biliary epithelial cells and the T-cell cluster differentiation that characterizes the disorder.5–7
The serologic hallmark of PBC is the antimitochondrial antibody (AMA), an autoantibody that targets lipoic acids found in the dehydrogenase complex from the inner mitochondrial membrane, and it is positive in 90%–95% of individuals with PBC.8,9 Given its high disease specificity, a positive AMA serology in patients with cholestasis is sufficient to ascertain the diagnosis of PBC without requiring a liver biopsy.10,11 Approximately 30% of all patients with PBC, and up to 50% of AMA-negative patients, also have detectable titers of PBC-specific antinuclear antibodies (ANA), specifically anti-sp100 and anti-gp210. When present, these antibodies are nearly diagnostic of PBC, thus being particularly helpful in AMA-negative individuals. In addition, anti-gp210 may have prognostic significance as it has been associated with more severe PBC phenotypes.12,13 Other PBC-specific antinuclear antibodies discovered more recently, such as anti-Kelch-like 12 (KLHL12) and anti-hexokinase 1 (HK1), are not yet commercially available, and their clinical utility needs to be validated.14 Regardless, it is notable that AMA-negative and AMA-positive PBC exhibit similar natural history and clinical outcomes, both before and after the onset of cirrhosis.15,16 A proposed algorithm for the diagnosis of PBC in the setting of chronic cholestasis is summarized in Figure 1.
FIGURE 1.
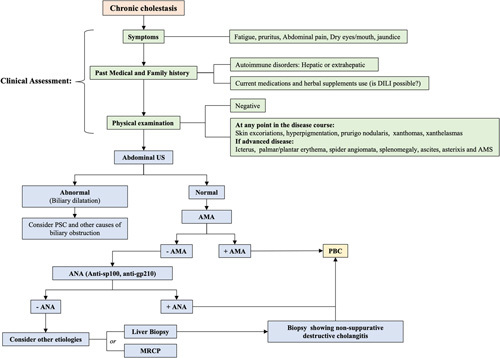
Approach to the diagnosis of PBC. The assessment of patients with cholestasis whether symptomatic or not should include a liver ultrasound and testing for the AMA in serum. A positive AMA in this setting is sufficient to make the diagnosis of PBC. PBC-specific ANAs, ie, anti-sp100 and anti-gp210, can aid in the diagnosis of PBC in individuals with cholestasis and negative AMA serology. Liver biopsy is reserved for those with unclear diagnosis after serologic workup, or when a coexisting process is suspected, including fatty liver disease and autoimmune hepatitis. Abbreviations: AMA, antimitochondrial antibody; AMS, altered mental status; ANAs, antinuclear antibodies; MRCP, magnetic resonance cholangiopancreatography; PBC, primary biliary cholangitis; US, ultrasound.
Most patients are asymptomatic at diagnosis although nearly everyone will develop symptoms within 2 decades. The presence or absence of symptoms at diagnosis or on follow-up does not correlate well with the stage of the disease and has shown no difference in survival outcomes17 (Figure 2). Fatigue is the most common complaint. It is experienced by up to 80% of patients during the disease course and is often rated as the most disabling PBC symptom.18,19 Pruritus closely follows in frequency and may be associated with sleep deprivation, worsening fatigue, depression, social isolation, and self-mutilation, thus leading to significant impairment in quality of life.20 Patients with chronic cholestasis are at risk for developing metabolic bone disease, dyslipidemia, and lipo-soluble vitamin deficiencies (Vitamins A, D, E, and K).21,22
FIGURE 2.
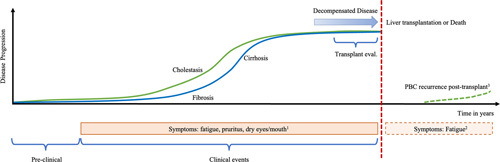
Natural history in primary biliary cholangitis (PBC). Most patients with PBC are diagnosed while asymptomatic. Liver disease progression is inevitable in most untreated individuals, with fibrosis and cirrhosis ensuing as a consequence of the inflammatory and cholestatic processes. The cumulative incidence of major non-neoplastic hepatic complications has been reported as 15% after 15 years of follow-up. Liver transplantation is the only curative treatment option for those with decompensated disease and declining liver function. PBC will recur in 20%–40% of post-transplant patients. Symptoms do not correlate with the degree of cholestasis or fibrosis although patients with more severe disease tend to experience more symptoms in general. Fatigue may not completely resolve post-transplantation even in the absence of recurrent disease. 1Symptoms do not correlate with the disease stage and can occur at any point. 2Fatigue may persist after liver transplant. 3The frequency of post-transplant PBC is highly variable among studies (9%–61%).
In this review, we summarize the most current data regarding the epidemiology, diagnosis, and treatment of PBC focusing on clinically useful information to assist health care providers in the integral management of these complex patients.
EPIDEMIOLOGY
PBC affects people of all sexes, races, and ethnicities. Earlier reports from case-finding studies showed a median female-to-male ratio of 10:1.23 Modern literature has demonstrated that, although PBC remains a female-predominant disease, it is also more prevalent in males than previously thought, with a female-to-male ratio closer to 4–6:1.24,25 PBC in males is often diagnosed at an older age, once the disease is more advanced. PBC in males has also been associated with lower biochemical response to ursodeoxycholic acid (UDCA), greater progression to cirrhosis, higher rates of liver-related death or transplantation, and increased risk for HCC.26,27
While non-Hispanic White patients are disproportionally diagnosed, the incidence and prevalence of PBC in racial minorities are not insignificant, with available US data showing a lower but unrelenting incidence rate and steady increases in prevalence among black and Asian-American patients. Notably, accurate racial epidemiologic estimates are hampered by health care disparities and a lack of disease recognition by providers.24
The incidence and prevalence of PBC increase with age, with a peak range between 60 and 79 years old. PBC is typically identified in middle-aged individuals (40–60 years of age) and is exceptionally rare under 25 years of age.28
The pooled global incidence and prevalence of PBC are estimated to be 1.76 and 14.60 per 100,000 persons, respectively.29 However, these estimates vary significantly according to the search strategy utilized, the size of the study population, and the degree of scrutiny of case finding.30 The incidence of PBC steadily rose until the year 2000 across the globe. Growth rates in North America and Europe have since plateaued, whereas the incidence of PBC in the Asian-Pacific region has continued to increase slowly, likely due to improved case reporting. The prevalence of PBC in all three regions has risen steadily over time, although at a faster pace in North America relative to Europe.29 A 12-year prevalence study from the Fibrotic Liver Disease (FOLD) Consortium found that the PBC prevalence in the United States increased by over 72% among women (33.5–57.8 per 100,000 persons) and over 114% in men (7.7–15.4 per 100,000 persons) during the study period.24 Although this change is likely multifactorial, earlier recognition of the disease and the efficacy of UDCA to prevent morbidity and mortality are likely major contributors.
Contemporary data show that the incidence of PBC is higher in North America (2.75 per 100,000 persons) relative to Europe (1.86 per 100,000 persons) and lowest in the Asian-Pacific region (0.84 per 100,000 persons) (Figure 3A). The pooled prevalence follows a similar trend, being highest in North America (21.81/1000,000), Europe (14.59/100,000), and then in the Asian-Pacific region (9.82/100,000) (Figure 3B).29 However, a recent study suggests that the prevalence of PBC in Asia may be higher than previously reported: 11.9 per 100,000 individuals, with regional differences suggesting a higher prevalence in Japan and China (19.1 cases per 100,000), compared with 9.9 per 100,000 in New Zealand and 3.9 per 100,000 in South Korea and Australia.31 Little is known about the incidence and prevalence of PBC in Africa, Central and South America, and Antarctica given the paucity of population-based studies in those continents.29
FIGURE 3.
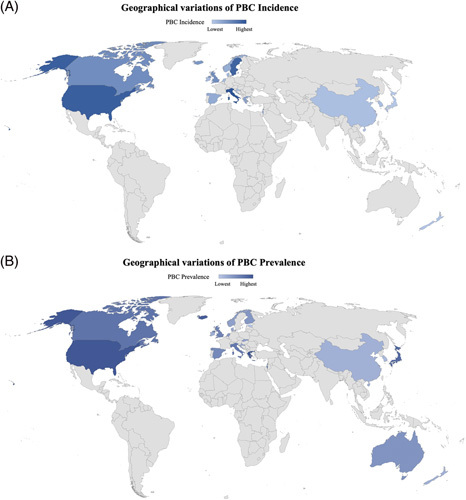
Geographical variations of PBC incidence and prevalence. (A) Global variations in PBC incidence. (B) Global variations in PBC prevalence. Lower to higher rates are qualitatively represented by lighter to darker blue, respectively. Countries with no population-based epidemiologic studies available at the time of this review are represented in gray. Abbreviation: PBC, primary biliary cholangitis.
Continental and country-specific epidemiological differences have also been described within Europe and North America. For example, Eastern European countries have the lowest estimated incidence and prevalence in Europe,29 the northern United Kingdom has a higher incidence of PBC relative to the southern United Kingdom,32 and PBC was significantly more frequent among members of the Aboriginal First Nation’s communities in British Columbia compared with reported rates in Ontario, Canada.33 Genetic susceptibility and environmental factors partially account for these differences, with PBC being more prevalent in industrialized/polluted areas and in patients who smoke cigarettes and use nail polish and hair dye products.34–37
PROGNOSIS
Transplant-free survival
The disease progression and prognosis of PBC have substantially changed in the past few decades. The mean age at PBC diagnosis has steadily increased since the 1970s by 2–3 years per decade and those who have been diagnosed more recently present with milder elevations of liver chemistries and histologic findings, and a better response to UDCA. Although the cause of these variations has not been fully elucidated, it likely reflects a combination of biological factors, changing environmental triggers, longer subclinical periods associated with increased screening in older patients, and overall increased awareness of the disease.38
Racial differences and health care disparities also influence survival in patients with PBC. Black and Hispanic patients have a significantly higher risk of waitlist mortality (HR: 1.26 and HR: 1.41, respectively) and removal from the transplant list due to clinical deterioration compared with Whites.39,40 Similarly, Indigenous Canadian patients with PBC are more commonly diagnosed at advanced disease stages experiencing lower event-free survival and lower transplant-free survival compared with White patients.41
The introduction of UDCA has helped reshape the progression and prognosis of PBC. The transplant-free survival rate for untreated patients has been estimated at 79%, 59%, and 32% at 5, 10, and 15 years, respectively. In contrast, those treated with UDCA experience significantly higher survival rates of 90%, 78%, and 66% at 5, 10, and 15 years, respectively.42 Similar results were obtained after pooling a large cohort of patients from the Global PBC Study Group database in which treated individuals had a 20% higher transplant-free survival rate at 10 years (79.7% among UDCA-treated patients vs. 60.7% among untreated patients) and a statistically significant reduced risk of liver transplant (LT) or death (HR: 0.46) at all histological stages of the disease.43 Treatment with UDCA has additionally been shown to significantly delay the development of esophageal varices (16% in UDCA-treated patients vs. 58% in placebo).44
HCC
Patients with PBC are at a higher risk of developing HCC, but its incidence is certainly lower than that documented for other causes of chronic liver disease (4.17 per 1000 person-years). Male patients with cirrhosis are at the highest risk (9.82 per 1000 patient-years).45,46 A Veterans Affair–based study, including 532 patients with PBC cirrhosis who were predominantly males, demonstrated a higher unadjusted rate of HCC in males (0.9 vs. 0.3, per 100-person-years) but did now show an association on multivariable analysis.47
Reports on the biochemical response to UDCA affecting the development of HCC are conflicting with results markedly influenced by methodologic bias.45,48,49 Despite this, a multicenter study, including 4565 patients from the Global PBC Study Group database, showed that suboptimal response to UDCA is a significant factor predictive of future HCC in patients with early and advanced stage diseases, and remained the most significant factor predictive of future HCC risk (HR: 3.44) on multivariable analysis.48 Periodic screening in patients with cirrhosis is recommended by the American Association for the Study of Liver Diseases (AASLD) and the European Association for the Study of the Liver (EASL)10,11 whether the patient is receiving treatment or not. In addition, the AASLD recommends HCC surveillance in all males with PBC despite the absence of cost-effectiveness analyses.10
Risk stratification
Liver biochemistries and histology
In PBC, serum bilirubin independently and effectively predicts survival.50 However, elevated levels are commonly seen at later stages of the disease, being less useful for initial risk stratification.51 More recent studies indicate a differential prognostic ability of total bilirubin even when within the normal range, with values >0.6× ULN associated with worse outcomes.52
A log-linear association between serum alkaline phosphatase (ALP) and risk of LT and death has been described in patients with PBC, with higher levels translating into decreased transplant-free survival. In a large meta-analysis with nearly 5000 patients with PBC, an ALP level >2× ULN after 1 year of follow-up had the highest predictive ability (C statistic 0.71) although there was no statistically significant difference when compared with other cutoff values such as 1.5× ULN, 1.67× ULN, and 3.0× ULN. The 5-, 10-, and 15-year transplant-free survival rates for patients with ALP levels ≤2.0× ULN were 94%, 84%, and 73%, respectively, and for patients with ALP levels >2.0× ULN, these rates were 81%, 62%, and 50%, respectively (p < 0.0001).42 ALP level is, therefore, considered a reliable marker of treatment response, with lower numbers associated with better prognosis, decreased mortality, and longer transplant-free survival.53 Moreover, combining ALP with either bilirubin42 or gamma-glutamyl transferase (GGT)54 levels in serum further increases the prognostic capability of ALP. Although GGT has been shown to correlate with UDCA nonresponse,54 increased the risk of liver-related death, and LT,55 it should not be used in isolation in the assessment of biochemical response to therapy.
Untreated PBC progresses at an average rate of 1 histological stage every 1.5–2 years, with half of the cases developing cirrhosis at 4 years and only 20% remaining stable over time.56 Histological features that significantly predict the risk of liver fibrosis progression include ductopenia and increasing severity of interface hepatitis.57,58 Treated individuals have a 5-fold reduction in the rate of progression to cirrhosis if diagnosed at an early histologic stage.59,60 However, the presence of advanced fibrosis (stage 3/4) at diagnosis is an independent predictor of poor transplant-free survival (HR: 2.85) even if the biochemical response has been achieved with UDCA.
Response to UDCA treatment
There is currently no consensus regarding the optimal serologic threshold to define response to UDCA. Multiple binary prognostic models have been proposed and utilized in clinical trials (Table 1). Of them, the Paris I criteria are considered the most accurate for transplant-free survival prediction.53 In clinical practice, ALP and total bilirubin normalization or near-normalization are ideal but not always possible.52
TABLE 1.
Assessment of serologic response to ursodeoxycholic acid
| Serologic response to UDCA therapy in PBC | ||
|---|---|---|
| Treatment duration (mo) | Treatment failure if | |
| Rochester | 6 | ALP ≥ 2× ULN or Mayo score ≥4.5 |
| Barcelona | 12 | Decrease in ALP ≤ 40% and ALP ≥ 1× ULN |
| Paris I | 12 | ALP ≥ 3× ULN or AST ≥ 2× ULN or bilirubin > 1 mg/dL |
| Rotterdam | 12 | Bilirubin > 1 mg/dL and/or albumin < 1× ULN |
| Toronto | 24 | ALP ≥ 1.67× ULN |
| Paris-II | 12 | ALP ≥ 1.5× ULN or AST ≥ 1.5× ULN or bilirubin > 1 mg/dL |
| Ehime | 6 | Decrease in GGT ≤ 70% and GGT ≥ 1× ULN |
| Rochester-II | 12 | ALP 2× ULN |
| Global | 12 | ALP 2× ULN |
Abbreviations: ALP, alkaline phosphatase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transferase; ULN, upper limit of normal.
The UK-PBC (https://www.uk-pbc.com/resources/tools/riskcalculator/)61 and the GLOBE (https://www.globalpbc.com/globe)62 scores are derived from the analysis of multicenter PBC cohorts and have been externally validated in large international studies.63 These mathematical models are excellent at estimating outcomes: the UK-PBC score estimates the risk of LT or liver-related death at 5, 10, and 15 years, whereas the GLOBE score estimates the overall survival within 3, 5, 10, and 15 years.61,62
Although the Globe and the UK-PBC scores are considered excellent prognostic tools, they are not exempt from pitfalls. For instance, no specific threshold to classify high-risk or low-risk patients has been defined; therefore, they cannot be used for treatment escalation or de-escalation. Similarly, no validated data regarding their prognostic capability in patients on second-line therapy is currently available.64 An additional limitation is that neither can identify patients who are unlikely to respond to UDCA before treatment initiation. To address this issue, the UK-PBC Research group and the Italian PBC Study Group developed the UDCA Response Score (URS). The URS, which has been externally validated, utilizes pretreatment clinical and serologic variables at the time of PBC diagnosis, ALP at the time of UDCA initiation, patient’s age, and the interval from diagnosis to UDCA initiation in years to identify patients at high risk of failing UDCA monotherapy (http://www.mat.uniroma2.it/~alenardi/URS.html).65,66 Despite having high accuracy, the model has not yet been implemented in routine clinical practice.
Not all patients benefit from UDCA treatment to the same degree, with several studies showing variable responses to the medication according to age, race, and histological stage at diagnosis.67,68 Younger age, advanced fibrosis (stage 3/4) at the time of treatment initiation, and male sex are associated with poor biochemical response to UDCA.59,69 Hispanic patients are more likely to exhibit features of autoimmune hepatitis and to have higher rates of biochemical nonresponse.70 Suboptimal response to UDCA has also been described in those of Indigenous origin.41 Black patients are more likely to respond to UDCA but are less likely to receive treatment.71
Liver stiffness
Liver stiffness measurement (LSM) is widely used as a surrogate marker for fibrosis in chronic liver diseases. In patients with PBC, an LSM below 6.5 kPa or above 11 kPa measured by transient elastography accurately discriminates between the absence or presence of advanced fibrosis with high sensitivity and specificity.72
The utilization of LSM as a prognostic tool in PBC is an evolving field. Earlier data showed that a baseline LSM of >9.6 kPa or an increase in LSM over time of >2.1 kPa/y was associated with 5.1 and 8.4 times increased risks of adverse outcomes, respectively.73 A recent retrospective follow-up study, including almost 4000 patients with PBC, confirmed that elevated LSM assessed by vibration-controlled transient elastography is independently associated with poor clinical outcomes, ie, liver-related complications, liver transplantation, or death. It improves the predictive value of classic biochemical criteria and validated scores. Furthermore, LSM was able to risk-stratify patients into low (LSM < 8 kPa), medium (LSM 8–15 kPa), or high risk (>15 kPa) for poor clinical outcomes with 10-year rates of clinical events among medium- and high-risk patients ranging between 20%–50% and 50%–90%, respectively.74 Re-evaluating liver stiffness after 1 year of therapy with UDCA is likely to provide a more accurate prediction of risk.
Treatment
The management of PBC is centered on staging and treating the disease, its symptoms, and the associated extrahepatic complications (Figures 4 and 5) (Table 2). Although UDCA revolutionized the management of PBC and continues to be cardinal for its treatment, up to 40% of patients do not fully benefit.53 This has switched the focus of clinical research toward developing new or repurposing available medications with the goal of treating inadequate responders. Important therapeutic advances have been made in the last decade, with the US Federal Drug Administration (FDA) granting approval for obeticholic acid (OCA) to be used as second-line therapy in patients without advanced liver disease. Similarly, fibrates are now endorsed as a viable off-label alternative for the management of inadequate responders by medical societies, such as the AASLD, EASL, and the Asian-Pacific Association for the Study of the Liver (APASL),11,75,76 and bezafibrate is a de facto second-line treatment in Japan.77 Unfortunately, these drugs are not devoid of side effects, and their use is limited to a very specific subset of patients. Furthermore, an important paradigm change in the overarching treatment of patients with PBC has been focused on managing symptoms. Multiple new drugs are currently in the pipeline showing promising results in phases II and III clinical trials.
FIGURE 4.
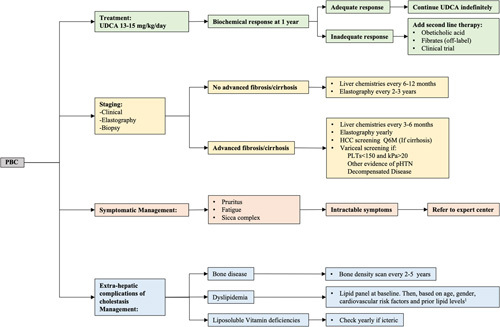
PBC management flow chart. A holistic approach for the management of patients with PBC is recommended and should be based on: (1) PBC treatment; (2) disease staging and monitoring; (3) symptomatic treatment; and (4) extrahepatic complications of cholestasis management. Currently recommended strategy supports assessing response to UDCA after 1 year of treatment although ongoing studies indicate that this may be evaluated at earlier timepoints as well. In addition, while existing guidelines still associate “response to UDCA” with achieving specific cutoff values for ALP and TB, it is likely that the field will evolve in the direction of aiming to normalize liver chemistries. 1Per guidelines published for the general population. AHA/ACC guidelines, ACE guidelines. Abbreviations: PBC, primary biliary cholangitis; pHTN, pulmonary hypertension; PLTs, platelets; UDCA, ursodeoxycholic acid.
FIGURE 5.
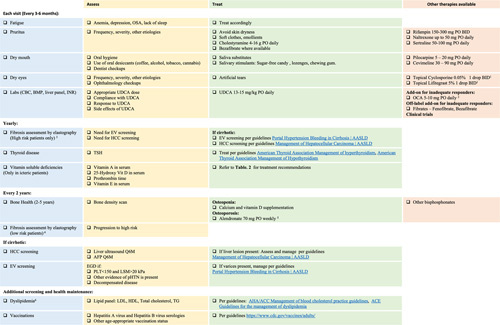
Approach to patients with primary biliary cholangitis (PBC) in the outpatient clinic. PBC is a relatively rare disease, and managing affected patients in the office can be challenging for providers not familiarized with the disorder. Although PBC treatment should be tailored to the patient’s individual needs, we propose a structured approach for its management in the outpatient setting. This should include symptomatic and disease assessment and treatment during each clinic visit, periodic fibrosis staging, and screening for extrahepatic complications of cholestasis when appropriate. For those with advanced disease stages, screening for complications derived from portal hypertension is also necessary. 1Recommend comanagment with ophthalmologist. 2Contraindicated in cirrhosis with portal hypertension and decompensated disease. 3Patients with LSM>10 kPa and/or nonreponders to UDCA. 4Patients with LSM<10 kPa or with appropriate UDCA response. 5Avoid in patients with esophageal varices. 6In the absence of cardiovascular risk factors, hyperlipidemia associated with PBC does not increase the risk of cardiac events. Screening should be performed at baseline and the according to guidelines published for the general population. Abbreviations: AFP, alpha fetoprotein; BMP, basic metabolic panel; CBC, complete blood count; EV, esophageal varices; INR, international normalized ratio; LSM, liver stiffness measurement; pHTN, pulmonary hypertension; PLTs, platelets; TG, triglycerides; TSH, thyroid-stimulating hormone; UDCA, ursodeoxycholic acid.
TABLE 2.
Liposoluble vitamin supplementation
| Vitamin | Serum levels | Formulation names | Repletion dosagea | Maintenance oral dosage (daily) |
|---|---|---|---|---|
| Vitamin Aa | Normal: 30–100 μg/dL | Vitamin A | Absence of symptoms: 10,000–25,000 IU (oral) daily Presence of visual symptoms: 50,000 IU (oral) daily ×1 mo If no response to oral supplementation: 100,000 IU (IM) daily × 3 days followed by 50,000 IU (IM) daily × 2 wk |
15,000 IU × 2 mo |
| Vitamin Da | Normal: > 30 ng/mL | Cholecalciferol (vitamin D3) Ergocalciferol (vitamin D2) |
50,000 IU vitamin D3 (oral) weekly × 8–12 wk | 400–2000 IU vitamin D3 |
| Vitamin Ka | Normal: 0.15–1.0 μg/L | Vitamin K | 2.5–10 mg (oral) twice weekly | 5 mg |
| Vitamin Ea | Normal: 0.5–2.0 mg/dL | Vitamin E (expressed in alpha-tocopherol equivalents) | 200–2000 mg (oral) daily | 15 mg |
Water-miscible formulas are recommended for supplementation in the setting of fat malabsorption.
Abbreviation: IM, intramuscular.
First-line therapy: UDCA
When administered orally, only about 30%–60% of UDCA is absorbed by the bowel (80% small bowel and 20% in the colon), and once inside the hepatocytes, synthetic UDCA requires conjugation with glycine or taurine to be transported across the canalicular domain and into the bile ducts.78 Among other beneficial effects in cholestatic diseases, UDCA increases the biliary pool of hydrophilic bile acids (BA) to 40%–50%,79 stimulates choleresis, increases bile alkalinization through upregulation of anion exchange 2 (AE2) receptor expression, and reduces the hepatocellular and biliary expression of major histocompatibility complex class I and class II proteins, thus decreasing adaptive immune injury. UDCA also stabilizes the mitochondrial membrane preventing the activation of death receptors that ultimately signal apoptosis.1,80
Studies have consistently shown that the use of UDCA improves liver chemistries, delays histological progression, and delays the development of varices.10,11 Although early meta-analyses showed no difference in liver-related mortality, all-cause mortality, and/or transplant-free survival,81–83 a large international collaborative study from the Global PBC Study Group found an improved 10-year cumulative LT-free survival for UDCA-treated individuals (79.7% vs. 60.7%, p < 0.001), confirming the anecdotal experience that UDCA does influence outcomes.43 Furthermore, improved survival with UDCA use has been shown regardless of sex or disease stage and even in those who inadequately respond to the medication.72
Although the use of UDCA has no effect on symptom onset or amelioration, treated patients experience a decreased incidence of non-neoplastic cirrhosis–associated hepatic complications, ie, ascites, variceal bleeding, and HE over time. In a recent retrospective study, the overall cumulative incidence of major non-neoplastic hepatic complications was 9% after 10 years and 15% after 15 years. This incidence sharply decreased to 5.8% after the year 2000, partially due to the gradual introduction of UDCA, which had just been approved to treat PBC a few years earlier.84
All patients with PBC should be started on UDCA at diagnosis, promptly and independently of the degree of cholestasis, fibrosis, or overall clinical status. Where available, the medication should be provided at a therapeutic dose of 13–15 mg/kg/d taken with meals, in a single oral daily dose or divided doses. Higher doses have shown no additional benefits, and dose escalation is not currently recommended.10
UDCA has an excellent safety profile with minimal side effects when administered at the appropriate dose. Some patients do experience weight gain, hair thinning, and gastrointestinal symptoms, such as diarrhea and/or flatulence.85 Limited data have shown UDCA to be safe during pregnancy and breastfeeding with no teratogenic consequences.86
Second-line therapy
Second-line therapy should be considered for all patients with insufficient response to UDCA after 1 year of therapy. Several groups have attempted to identify those patients with an inadequate response even earlier and to allow timely initiation of second-line therapy or clinical trial participation.87–89 However, there is no consensus on how this should be accomplished in practice.
Obeticholic acid
OCA is a synthetically modified hydrophobic BA derived from chenodeoxycholic acid that has a high affinity for Farsenoid X receptors (FXRs). FXRs are transcription factors belonging to the superfamily of nuclear receptors with high expression in the liver and intestines. FXR regulates BA synthesis, conjugation, and transport, as well as various aspects of lipids and glucose metabolism.90 For instance, its activation upregulates the synthesis of short heterodimeric partner, a suppressive protein that inhibits the expression of cholesterol 7∝-hydroxylase. This is a rate-limiting enzyme necessary for the synthesis of BA. FXR also suppresses BA synthesis through FGF-19 and FGF-4 activation, and directly enhances the expression of several BA transporters and uptake proteins.91 The net effect from FXR activation is, therefore, a reduction in the total BA pool by limiting their synthesis and uptake and promoting choleresis.92
OCA is indicated as adjunctive therapy for patients with PBC who inadequately respond to UDCA after 1 year of treatment.10 This medication received conditional FDA approval in 2016, after a phase III randomized controlled trial showed that 47% of the patients receiving OCA 10 mg/d and 46% of those receiving OCA 5–10 mg/d achieved the primary endpoint that is now known as the POISE criteria (serum ALP reduction to <1.67× ULN, with a reduction of at least 15% from baseline and a normal total bilirubin level after 12 mo of treatment).93 This improvement has proven to be sustained in subsequent open-label extension trials.94 OCA can also be used as monotherapy for patients who are intolerant to UDCA.95
Real-world efficacy data support the beneficial effects of OCA in patients with PBC. An Italian cohort including 191 inadequate responders showed a 42.9% response rate according to POISE criteria and 11% by normal range criteria.96 Similarly, a Canadian retrospective cohort study demonstrated a significant reduction in mean ALP and GGT regardless of their initial degree of elevation after 12 months of therapy.97 Both studies encountered a lower response rate for those with cirrhosis, as determined by transient elastography, compared with noncirrhotic counterparts.
A recent study compared the outcomes of death or transplantation among participants in the POISE trial that continued OCA in an open-label extension for up to 6 years to propensity-matched controls from the Global PBC and UK-PBC cohorts. During the 6-year follow-up period, there were 5 composite events in 209 (2.4%) subjects in the POISE study, 135 events in 1381 (10%) patients in the Global PBC cohort, and 281 events in 2135 (13.2%) patients in the UK-PBC participants.98 The use of OCA was associated with 71% and 70% lower hazards of death or liver transplantation compared with participants in the Global PBC and UK-PBC cohorts, respectively.98
The most common side effect of OCA is dose-dependent pruritus, which leads to drug discontinuation in 10%–25% of treated subjects.93,96,97. Therefore, OCA should be started at a low dose and up-titrated slowly to prevent its discontinuation. In addition, FXR activation by OCA causes a negative impact on the lipid panel as it decreases HDL and increases LDL-cholesterol, independently of the dose.
Multicenter case series and data from a VA cohort showing an increased risk of hepatic decompensation and death in individuals with advanced cirrhosis using OCA99,100 led the FDA to issue a label modification in 2021 recommending against its use in patients with cirrhosis and current or prior evidence of decompensation (encephalopathy, ascites, and variceal bleed), or portal hypertension (gastroesophageal varices or persistent thrombocytopenia).75 In summary, OCA is contraindicated in patients with cirrhosis with the Child-Pugh score of B or C, and Child A patients with any evidence of portal hypertension. The drug should be used with caution at a maximum dose of 5 mg/d in those with compensated cirrhosis without portal hypertension.
Off-label therapy: Fibrates
Fibrates are fibric acid derivatives commonly used for the management of atherogenic dyslipidemia, decreasing triglyceride and LDL levels, and increasing HDL levels.101 Although only licensed in the United States as lipid-lowering agents, these drugs have been used off-label for the treatment of cholestatic disorders.
Fibrates are strong peroxisome proliferator–activated receptor (PPAR) agonists. PPAR are ligand-activated transcription factors that belong to the nuclear hormone receptor superfamily. They exist in 3 isoforms: PPAR-α, PPAR-δ, and PPAR-γ, with the former predominantly expressed in hepatocytes. When activated, PPAR-α induces the expression of numerous genes with a number of them involved in lipid pathways that regulate fatty acid oxidation, triglyceride degradation and BA synthesis, metabolism, and transport, reducing BA concentration inside hepatocytes, augmenting phospholipid excretion into the biliary canaliculi, and inhibiting proinflammatory agents within the liver and biliary tree.102
All marketed fibrates, ie, fenofibrate, bezafibrate, pemafibrate, gemfibrozil, and fenofibric acid, are predominantly PPAR-α agonists except for bezafibrate, which also has affinity for PPAR-δ and PPAR-γ. Although bezafibrate is commonly used as second-line therapy in Asia, South America, and Europe, it is not commercially available in the United States.103,104
The phase III randomized, placebo-controlled trial “BEZURSO” assigned 100 inadequate UDCA responders to receive bezafibrate (400-mg PO daily) or placebo in addition to UDCA for 24 months. After 2 years, 31% of patients in the treatment group normalized all liver chemistries compared with none in the placebo arm. Notably, at the end of the study, 67% of the patients in the bezafibrate group had complete ALP normalization and a mean liver stiffness reduction by vibration-controlled transient elastography of 15%. Only 2% of the controls achieved normal ALP levels, and instead, this group experienced a 22% increase in liver stiffness at the end of the study period.105
A recent retrospective nationwide Japanese study collecting data on PBC patients since 1980 showed that bezafibrate add-on therapy decreases all-cause mortality or LT (HR: 0.32) and liver-related mortality or LT (HR: 0.27) with a number-needed-to-treat to prevent 1 additional death or LT of 29, 14, and 8, over 5, 10, and 15 years, respectively.77 Data on outcomes with the use of fenofibrate for inadequate responders are less robust. A few small prospective and retrospective studies have shown improved outcomes, including decompensation-free and transplant-free survival in those receiving combined UDCA and fenofibrate treatment.106–108
Although gastrointestinal and musculoskeletal complaints, such as myalgias and arthralgias, are more common in patients with PBC on dual UDCA-fibrate therapy,109,110 no significant difference in the frequency of serious side effects (including elevations of aminotransferases >5 times the ULN) has been reported when compared with individuals on UDCA monotherapy.109 In the Bezurso trial, 3 out of 50 in the bezafibrate arm, and 1 of 50 in the placebo arm developed elevated transaminases >5 times ULN.105 In addition, serum creatinine levels increased by a median of 5% in the bezafibrate group and decreased by 3% in the placebo group compared with baseline.105 This is a well-described effect of PPAR-α agonists that derive from renal hemodynamic changes and/or an increased creatinine release by muscle but does not reflect renal toxicity.111,112 Liver biochemistries and renal function should be monitored periodically in PBC patients on fibrates, and its use should be avoided in those with decompensated liver disease.
Triple therapy UDCA + OCA + fibrate
Up to half of the patients with PBC and inadequate response to UDCA will fail to achieve an appropriate biochemical response after the addition of second-line therapy. A small real-world study in which OCA or fibrates were used as third-line therapy after dual therapy failure showed an OR of ALP normalization of 3.4 and a gain in ALP reduction of 22% per first year. Fibrates as the third-line therapy had a slightly greater effect on ALP reduction than OCA.113 Triple therapy for inadequate responders also proved to be effective in a retrospective cohort study from Spain that not only demonstrated statistically significant reductions in ALP (p = 0.007), GGT (p = 0.02), and ALT (p = 0.04) but also in the GLOBE score (p = 0.04). Confirmatory prospective clinical trials are needed to validate these results.114
Symptomatic management
Pruritus
Commercially available and investigational drugs used for the management of pruritus generally exert their action through different pathophysiologic targets that: (1) eliminate pruritogens from the enterohepatic circulation or alter their metabolism; (2) modulate the pruritus neural pathway, regulating central and peripheral endogenous opioid, serotonin, or nonspecific nociceptive receptors; or (3) alter the itch perception.115
A systematic stepwise approach is currently recommended by medical societies. Skin moisturizers, emollients, and topical agents, such as camphor or menthol, are often the initial general measures recommended for mildly symptomatic patients due to their low cost and excellent safety profile although their efficacy is unproven.11 Anion-exchange resins are considered first-line therapy for the management of cholestatic itch, with cholestyramine being the only compound FDA-approved for this indication. Cholestyramine is commonly started at a dose of 4-g PO daily and can be increased to a maximum of 16 g per day in divided doses as needed for symptomatic control. Cholestyramine should be taken at least 2–4 hours apart from other drugs—particularly UDCA—since it may prevent their absorption. Additional side effects include abdominal discomfort, bloating, and constipation.116
The use of fibrates to treat cholestatic itch has been proposed based on observational studies and the FITCH trial, in which 55% of bezafibrate-treated patients had more than a 50% reduction in the severity of pruritus as assessed by the visual analog scale.117 Thus, where available, bezafibrate 400 mg/d could be used as a first-line treatment for itching. Evidence for fenofibrate in the treatment of itching is less robust.
Rifampin (150–300-mg PO BID), a pregnane X receptor agonist, enhances the rate of BA metabolism, increases the excretion of pruritogens, and decreases autotoxin levels. Small clinical trials and meta-analyses in children118 and adults have demonstrated its efficacy at decreasing cholestatic itch,119–121 and it is, therefore, considered a second-line treatment option in those who have failed anion-exchange resins. Although rifampin is commonly associated with minor elevation of transaminases, severe hepatitis is rare, and liver biochemistries monitoring is recommended during the first few weeks of treatment and following dose up-titration.122 Patients should also be reminded about the benign orange-red discoloration of bodily fluids that result from the medication and its metabolites.
Endogenous opioids activate inhibitory neurons within the dorsal horn of the spinal cord that modulate the pruritic stimuli. The type of receptor activated is crucial since μ-opioid receptors (MORs) induce itch, whereas stimulation of κ-opioid receptors (KORs) inhibits it.116 Naltrexone (25–50-mg QD) is a MORs blocker that has shown antipruritic properties in small clinical trials123,124 and can be attempted when other treatments have failed. Although naltrexone has shown a significant reduction in cholestatic itch, the magnitude of this effect may be lower than that of rifampicin.125 About 40% of patients on naltrexone experience adverse events with opioid-like withdrawal symptoms being the most common (32%) followed by recurrence of previous pain syndromes (4%).126,127 Naloxone, another MORs blocker, is only available in the intravenous formulation and has been previously used as induction in selected hospitalized patients before transitioning to oral naltrexone.128 Nalfurafine is a KORs agonist only commercially available in Asia.129 Butorphanol is unique given its dual effect of blocking MORs while simultaneously activating KORs. A small case-series showed itch reduction in about two-thirds of the patients on this drug, and although it was safe overall, some patients experienced somnolence and sedation.130
Some selective serotonin receptor inhibitors (SSRI), such as sertraline (50–100-mg QD), modulate afferent itch stimuli, proving to have antipruritic properties in several patients in addition to the therapeutic antidepressant and mood stabilizer properties that characterize this class of medications.131 Like other SSRIs, side effects with the use of sertraline include decreased libido, restlessness, and insomnia.132
Other experimental (nonpharmacologic) treatments, such as plasmapheresis,133 phototherapy,134 nasobiliary drainage,135 and molecular absorbent recirculating system (MARS),136 are available as well, but their use is exclusively reserved for refractory cases.
Fatigue
Although fatigue is extremely common among patients with PBC, its pathophysiology remains largely unknown, and therefore, the development of effective treatments is hindered by the lack of specific biologic targets.137
Medications including antidepressants, such as fluoxetine, and stimulants, such as modafinil, have been shown to be ineffective for the treatment of fatigue in PBC138–141 although modafinil could be considered in patients with associated increased daytime somnolence or other sleep disorders. Clinical outcomes from the BEZURSO trial showed a 28% fatigue reduction with the use of bezafibrate by a self-reported scale.105 This finding requires further validation with prospective placebo-controlled studies utilizing standardized fatigue scales. LT has been shown to improve fatigue to a certain degree, but more than half of the patients continue to report this symptom even after years post-transplantation.142
In clinical practice, the management of fatigue is mainly based on evaluating and treating potential contributing causes, such as anemia, sleep apnea, hypothyroidism, depression, and medications. Since pruritus is most common during the evening and at nighttime, effectively treating it to ensure restful sleep is highly recommended. Maintenance of physical activity and lifestyle changes, such as the development of coping strategies, like introducing rest periods during the day, and avoiding social isolation are also of paramount importance.143
Sicca syndrome
SS is highly prevalent among PBC patients with data showing a variable prevalence of up to 66% depending on the population studied and the methodology utilized. Since Anti-Ro (SS-A) and anti-La (SS-B) antibodies preferentially affect exocrine glands, patients usually report eye and mouth dryness.144 Artificial tears and saliva substitutes are considered the preferred initial treatment given their safety and low cost. Xylitol or sorbitol containing sugar-free mints or gum also increases salivary flow in mild cases.10 For moderate to severe keratoconjunctivitis sicca, comanagement with an ophthalmologist is advised, and medications such as topical cyclosporine or lifitegrast have proven to be beneficial.145 Severe xerostomia cases can be treated with oral pilocarpine or cevimeline, and patients should be reminded about receiving frequent dental checkups and optimal oral hygiene.22
Novel therapies
Several new drugs with diverse pharmacologic targets are currently at different stages of clinical development. These include novel PPAR agonists, nicotinamide-adenine dinucleotide phosphate oxidase (NOX) isoform 1/4 inhibitors, and ileal bile acid transporter (IBAT) inhibitors, among others.
Seladelpar is a PPARδ agonist that showed a significant dose-dependent improvement in biochemical markers of cholestasis persisting during the 52-week study period, with composite response (ALP < 1.67×ULN, ≥ 15% ALP decrease, and normal total bilirubin) rates of 53% and 67%, and ALP normalization rates of 13% and 33% in the seladelpar 5- and 10-mg/d arms, respectively.146 Patients on seladelpar also experienced an improvement in self-reported pruritus, sleep disturbance, and fatigue after 1 year although this was not a placebo-controlled trial.147 Similarly, elafibranor, a dual PPARα/δ agonist, reduced ALP in patients with inadequate response to UDCA by a mean of 48.3% in those treated with 80 mg and by 40.6% in those treated with 120 mg daily. The composite endpoint was achieved by 67% of patients receiving elafibranor 80 mg/d and 79% of patients in the elafibranor 120-mg/d group.148 Saroglitazar, a dual PPAR α/γ agonist, showed a meaningful ALP reduction of 49% and 51% at week 16 in a phase II proof-of-concept trial using 4 and 2 mg of the medication, respectively. Both seladelpar and elafibranor are being evaluated in phase III studies (NCT03301506) (NCT04526665), whereas saroglitazar is undergoing a phase IIb/III trial (NCT05133336).
Setanaxib is a selective NOX isoform 1 and 4 inhibitor. NOX has been implicated in fibrogenesis and cholestasis. PBC patients with inadequate response to UDCA and high-risk disease (LSM > 9.6 kPa at baseline) had liver stiffness reduction (22%) along with improvement of GGT (32.4%) and ALP (24.3%) after 24 weeks of treatment with setanaxib 400 mg twice per day.149,150. A phase II/III trial is currently underway (NCT05014672).
Novel drugs for the management of cholestatic itch have also shown promising results, with IBAT inhibitors such as maralixibat and odevixibat receiving FDA approval in 2021 for the management of Alagille syndrome and progressive familial intrahepatic cholestasis–related pruritus, respectively.151,152 Linerixibat, another IBAT inhibitor, is undergoing a phase III trial (NCT04167358) in patients with PBC after demonstrating significant antipruritic properties in proof-of-concept and phase II studies.153
LIVER TRANSPLANTATION
LT is the only curative treatment for patients with PBC and associated end-stage liver disease.154 Outcomes are excellent with 5-year post-LT mortality, retransplantation, and relisting rates of 8.2%, 14.6%, and 1.9%, respectively, in patients younger than 40 years old and 15.1%, 4.7%, and 1.5%, respectively, in older patients.155 Fortunately, the proportion of waitlisted and transplanted patients has continued to decline steadily due to the early recognition of the disease and prompt initiation of UDCA as discussed.156 Indications for LT in patients with PBC are analogous to LT indications for other chronic liver disorders. Patients with very severe refractory pruritus can be considered for transplantation with the caveat that no MELD exception point is granted for this particular indication due to inadequate evidence of increased risk of pretransplant mortality.
Disease recurrence after transplantation has been well documented. The frequency of post-transplant PBC is highly variable among studies (9%–61%) given their different methodologies, sample sizes, and follow-up times. In addition, no precise diagnostic criteria for recurrent disease have been validated, and clinicians rely mainly on histological findings for diagnosis. AMA remains detectable in all patients with a previous diagnosis of AMA-positive PBC, and therefore, it has no utility in diagnosing post-transplant PBC recurrence.157,158 Prophylactic utilization of UDCA has been shown in retrospective studies to be associated with reduced disease recurrence, all-cause mortality, liver-related mortality, and graft loss.159 The use of UDCA to prevent PBC recurrence after transplant has not been studied prospectively.
Summary and future directions
Although relatively rare, PBC is a well-known cause of chronic liver disease with devastating consequences if left unrecognized. Clinical outcomes not only depend on inherent phenotypic differences, but it is primarily the result of modifiable factors, particularly access to care and the prompt recognition of the disease with early treatment initiation. Contemporary population–based studies have shown a greater incidence and prevalence among males and racial minorities than historically documented, and these groups tend to have a more aggressive disease course than the one classically experienced by White females. Health care providers should be aware of this and appropriately screen patients with chronic cholestasis, whether symptomatic or not.
UDCA continues to be the treatment of choice for PBC. Practitioners should aim for ALP normalization or near-normalization with the understanding that this may not be possible in all cases. Inadequate responders after 1 year of treatment with UDCA should be considered for second-line therapy or clinical trial participation. OCA should be used with caution in patients with cirrhosis and avoided in patients with decompensated disease or evidence of portal hypertension. Patients at high risk for inadequate response to UDCA may also benefit from earlier initiation of adjunctive therapy. A holistic clinical approach to patients with PBC must include the periodic assessment and treatment of symptoms such as fatigue, pruritus, and sicca syndrome, given their association with impaired quality of life, the management of other comorbidities, such as bone disease and vitamin deficiencies, and appropriate health maintenance, including immunizations for HAV and HBV.
Acknowledgments
CONFLICTS OF INTEREST
Cynthia Levy consults and received grants from Calliditas, Cymabay, Genfit, Gilead, GlaxoSmithKline, Intercept, Mirum, and Target RWE. She consults for Ipsen and received grants from Cara, Escient, and Zydus. Binu John advises and is on the speakers’ bureau for GlaxoSmithKline. He advises AstraZeneca and received grants from Gilead, Exact Sciences, and Glycotest. The remaining author has no conflicts to report.
Footnotes
Abbreviations: AASLD, American Association for the Study of Liver Diseases; AE2, anion exchange 2; ALP, alkaline phosphatase; AMA, antimitochondrial antibody; ANA, antinuclear antibody; APSAL, Asian-Pacific Association for the Study of the Liver; AST, aspartate aminotransferase; BA, bile acid; BEC, biliary epithelial cell; EASL, European Association for the Study of the Liver; FDA, US Federal Drug Administration; FXR, Farsenoid X receptor; GGT, gamma-glutamyl transferase; HK1, Hexokinase 1; IBAT, ileal bile acid transporter; IPTW, inverse probability of treatment weighting; INR, international normalized ratio; KLHL12, Kelch-like 12; KORs, κ-opioid receptors; LSM, liver stiffness measurement; LT, liver transplant; MORs, μ-opioid receptors, MRCP, magnetic resonance cholangiopancreatography; NOX, nicotinamide-adenine dinucleotide phosphate oxidase; OCA, obeticholic acid; PBC, primary biliary cholangitis; pHTN, pulmonary hypertension; PPAR, peroxisome proliferator–activated receptors; SS, Sjogren syndrome; SSRI, selective serotonin receptor inhibitor; TE, transient elastography; TG, triglycerides; TSH, thyroid-stimulating hormone; UDCA, ursodeoxycholic acid; ULN, upper limit of normal; URS, ursodeoxycholic acid response score; US, ultrasound; VCTE, vibration-controlled transient elastography.
Contributor Information
Juan Trivella, Email: jtrivell@iu.edu.
Binu V. John, Email: binu.john@va.gov.
Cynthia Levy, Email: clevy@med.miami.edu.
REFERENCES
- 1. Gulamhusein AF, Hirschfield GM. Primary biliary cholangitis: pathogenesis and therapeutic opportunities. Nat Rev Gastroenterol Hepatol. 2020;17:93–110. [DOI] [PubMed] [Google Scholar]
- 2. Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology. 2004;127:485–92. [DOI] [PubMed] [Google Scholar]
- 3. Gulamhusein AF, Juran BD, Lazaridis KN. Genome-wide association studies in primary biliary cirrhosis. Semin Liver Dis. 2015;35:392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lazaridis KN, Juran BD, Boe GM, Slusser JP, de Andrade M, Homburger HA, et al. Increased prevalence of antimitochondrial antibodies in first-degree relatives of patients with primary biliary cirrhosis. Hepatology. 2007;46:785–92. [DOI] [PubMed] [Google Scholar]
- 5. Trivedi PJ, Hirschfield GM. The immunogenetics of autoimmune cholestasis. Clin Liver Dis. 2016;20:15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Juran BD, Lazaridis KN. Environmental factors in primary biliary cirrhosis. Semin Liver Dis. 2014;34:265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Corpechot C, Chretien Y, Chazouilleres O, Poupon R. Demographic, lifestyle, medical and familial factors associated with primary biliary cirrhosis. J Hepatol. 2010;53:162–9. [DOI] [PubMed] [Google Scholar]
- 8. Gershwin ME, Ansari AA, Mackay IR, Nakanuma Y, Nishio A, Rowley MJ, et al. Primary biliary cirrhosis: an orchestrated immune response against epithelial cells. Immunol Rev. 2000;174:210–25. [DOI] [PubMed] [Google Scholar]
- 9. Dahlqvist G, Gaouar F, Carrat F, Meurisse S, Chazouilleres O, Poupon R, et al. Large-scale characterization study of patients with antimitochondrial antibodies but nonestablished primary biliary cholangitis. Hepatology. 2017;65:152–63. [DOI] [PubMed] [Google Scholar]
- 10. Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary biliary cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology. 2019;69:394–419. [DOI] [PubMed] [Google Scholar]
- 11. European Association for the Study of the Liver, Electronic address: easloffice@easloffice.eu; European Association for the Study of the Liver. EASL Clinical Practice Guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67:145–72. [DOI] [PubMed] [Google Scholar]
- 12. Hu SL, Zhao FR, Hu Q, Chen WX. Meta-analysis assessment of GP210 and SP100 for the diagnosis of primary biliary cirrhosis. PLoS One. 2014;9:e101916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakamura M, Kondo H, Mori T, Komori A, Matsuyama M, Ito M, et al. Anti-gp210 and anti-centromere antibodies are different risk factors for the progression of primary biliary cirrhosis. Hepatology. 2007;45:118–27. [DOI] [PubMed] [Google Scholar]
- 14. Leung KK, Hirschfield GM. Autoantibodies in primary biliary cholangitis. Clin Liver Dis. 2022;26:613–27. [DOI] [PubMed] [Google Scholar]
- 15. Cancado GGL, Braga MH, Ferraz MLG, Villela-Nogueira CA, Terrabuio DRB, Cancado ELR, et al. Anti-mitochondrial antibody-negative primary biliary cholangitis is part of the same spectrum of classical primary biliary cholangitis. Dig Dis Sci. 2022;67:3305–12. [DOI] [PubMed] [Google Scholar]
- 16. John BV, Dahman B, Deng Y, Khakoo NS, Taddei TH, Kaplan DE, et al. Rates of decompensation, hepatocellular carcinoma and mortality in AMA-negative primary biliary cholangitis cirrhosis. Liver Int. 2022;42:384–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Prince MI, Chetwynd A, Craig WL, Metcalf JV, James OF. Asymptomatic primary biliary cirrhosis: clinical features, prognosis, and symptom progression in a large population based cohort. Gut. 2004;53:865–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khanna A, Leighton J, Lee Wong L, Jones DE. Symptoms of PBC—pathophysiology and management. Best Pract Res Clin Gastroenterol. 2018;34-35:41–7. [DOI] [PubMed] [Google Scholar]
- 19. Dyson JK, Wilkinson N, Jopson L, Mells G, Bathgate A, Heneghan MA, et al. The inter-relationship of symptom severity and quality of life in 2055 patients with primary biliary cholangitis. Aliment Pharmacol Ther. 2016;44:1039–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mells GF, Pells G, Newton JL, Bathgate AJ, Burroughs AK, Heneghan MA, et al. Impact of primary biliary cirrhosis on perceived quality of life: the UK-PBC national study. Hepatology. 2013;58:273–83. [DOI] [PubMed] [Google Scholar]
- 21. Assis DN. Chronic complications of cholestasis: Evaluation and management. Clin Liver Dis. 2018;22:533–44. [DOI] [PubMed] [Google Scholar]
- 22. Chalifoux SL, Konyn PG, Choi G, Saab S. Extrahepatic manifestations of primary biliary cholangitis. Gut Liver. 2017;11:771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–73. [DOI] [PubMed] [Google Scholar]
- 24. Lu M, Zhou Y, Haller IV, Romanelli RJ, VanWormer JJ, Rodriguez CV, et al. Increasing prevalence of primary biliary cholangitis and reduced mortality with treatment. Clin Gastroenterol Hepatol. 2018;16:1342–50. e1341. [DOI] [PubMed] [Google Scholar]
- 25. Lleo A, Jepsen P, Morenghi E, Carbone M, Moroni L, Battezzati PM, et al. Evolving trends in female to male incidence and male mortality of primary biliary cholangitis. Sci Rep. 2016;6:25906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. John BV, Aitcheson G, Schwartz KB, Khakoo NS, Dahman B, Deng Y, et al. Male sex is associated with higher rates of liver-related mortality in primary biliary cholangitis and cirrhosis. Hepatology. 2021;74:879–91. [DOI] [PubMed] [Google Scholar]
- 27. Shaker M, Mansour N, John BV. Primary biliary cholangitis in males: pathogenesis, clinical presentation, and prognosis. Clin Liver Dis. 2022;26:643–55. [DOI] [PubMed] [Google Scholar]
- 28. Trivedi PJ, Hirschfield GM. Recent advances in clinical practice: epidemiology of autoimmune liver diseases. Gut. 2021;70:1989–2003. [DOI] [PubMed] [Google Scholar]
- 29. Lv T, Chen S, Li M, Zhang D, Kong Y, Jia J. Regional variation and temporal trend of primary biliary cholangitis epidemiology: A systematic review and meta-analysis. J Gastroenterol Hepatol. 2021;36:1423–34. [DOI] [PubMed] [Google Scholar]
- 30. Tanaka A. Current understanding of primary biliary cholangitis. Clin Mol Hepatol. 2021;27:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zeng N, Duan W, Chen S, Wu S, Ma H, Ou X, et al. Epidemiology and clinical course of primary biliary cholangitis in the Asia-Pacific region: a systematic review and meta-analysis. Hepatol Int. 2019;13:788–99. [DOI] [PubMed] [Google Scholar]
- 32. Webb GJ, Ryan RP, Marshall TP, Hirschfield GM. The epidemiology of UK autoimmune liver disease varies with geographic latitude. Clin Gastroenterol Hepatol. 2021;19:2587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yoshida EM, Riley M, Arbour LT. Autoimmune liver disease and the Canadian First Nations Aboriginal Communities of British Columbia’s Pacific Northwest. World J Gastroenterol. 2006;12:3625–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dyson JK, Blain A, Foster Shirley MD, Hudson M, Rushton S, Jeffreys Jones DE. Geo-epidemiology and environmental co-variate mapping of primary biliary cholangitis and primary sclerosing cholangitis. JHEP Rep. 2021;3:100202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gershwin ME, Selmi C, Worman HJ, Gold EB, Watnik M, Utts J, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview-based study of 1032 patients. Hepatology. 2005;42:1194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsumoto K, Ohfuji S, Abe M, Komori A, Takahashi A, Fujii H, et al. Environmental factors, medical and family history, and comorbidities associated with primary biliary cholangitis in Japan: a multicenter case-control study. J Gastroenterol. 2022;57:19–29. [DOI] [PubMed] [Google Scholar]
- 37. Ala A, Stanca CM, Bu-Ghanim M, Ahmado I, Branch AD, Schiano TD, et al. Increased prevalence of primary biliary cirrhosis near Superfund toxic waste sites. Hepatology. 2006;43:525–31. [DOI] [PubMed] [Google Scholar]
- 38. Murillo Perez CF, Goet JC, Lammers WJ, Gulamhusein A, van Buuren HR, Ponsioen CY, et al. Milder disease stage in patients with primary biliary cholangitis over a 44-year period: A changing natural history. Hepatology. 2018;67:1920–30. [DOI] [PubMed] [Google Scholar]
- 39. Cholankeril G, Gonzalez HC, Satapathy SK, Gonzalez SA, Hu M, Khan MA, et al. Increased waitlist mortality and lower rate for liver transplantation in hispanic patients with primary biliary cholangitis. Clin Gastroenterol Hepatol. 2018;16:965–73 e962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Singal AK, Fang X, Kaif M, Hasanin M, McGuire BM, Kuo YF, et al. Primary biliary cirrhosis has high wait-list mortality among patients listed for liver transplantation. Transpl Int. 2017;30:454–62. [DOI] [PubMed] [Google Scholar]
- 41. Roberts SB, Hirschfield GM, Worobetz LJ, Vincent C, Flemming JA, Cheung A, et al. Ethnicity, disease severity, and survival in Canadian patients with primary biliary cholangitis. Hepatology. 2022;76:303–16. [DOI] [PubMed] [Google Scholar]
- 42. Lammers WJ, van Buuren HR, Hirschfield GM, Janssen HL, Invernizzi P, Mason AL, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow-up study. Gastroenterology. 2014;147:1338–49. e1335; quiz e1315. [DOI] [PubMed] [Google Scholar]
- 43. Harms MH, van Buuren HR, Corpechot C, Thorburn D, Janssen HLA, Lindor KD, et al. Ursodeoxycholic acid therapy and liver transplant-free survival in patients with primary biliary cholangitis. J Hepatol. 2019;71:357–65. [DOI] [PubMed] [Google Scholar]
- 44. Lindor KD, Jorgensen RA, Therneau TM, Malinchoc M, Dickson ER. Ursodeoxycholic acid delays the onset of esophageal varices in primary biliary cirrhosis. Mayo Clin Proc. 1997;72:1137–40. [DOI] [PubMed] [Google Scholar]
- 45. Natarajan Y, Tansel A, Patel P, Emologu K, Shukla R, Qureshi Z, et al. Incidence of hepatocellular carcinoma in primary biliary cholangitis: A systematic review and meta-analysis. Dig Dis Sci. 2021;66:2439–51. [DOI] [PubMed] [Google Scholar]
- 46. Rigopoulou EI, Dalekos GN. Current trends and characteristics of hepatocellular carcinoma in patients with autoimmune liver diseases. Cancers (Basel). 2021;13:1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. John BV, Khakoo NS, Schwartz KB, Aitchenson G, Levy C, Dahman B, et al. Ursodeoxycholic acid response is associated with reduced mortality in primary biliary cholangitis with compensated cirrhosis. Am J Gastroenterol. 2021;116:1913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Trivedi PJ, Lammers WJ, van Buuren HR, Pares A, Floreani A, Janssen HL, et al. Stratification of hepatocellular carcinoma risk in primary biliary cirrhosis: a multicentre international study. Gut. 2016;65:321–9. [DOI] [PubMed] [Google Scholar]
- 49. Kuiper EM, Hansen BE, Adang RP, van Nieuwkerk CM, Timmer R, Drenth JP, et al. Relatively high risk for hepatocellular carcinoma in patients with primary biliary cirrhosis not responding to ursodeoxycholic acid. Eur J Gastroenterol Hepatol. 2010;22:1495–502. [DOI] [PubMed] [Google Scholar]
- 50. Dickson ER, Grambsch PM, Fleming TR, Fisher LD, Langworthy A. Prognosis in primary biliary cirrhosis: model for decision making. Hepatology. 1989;10:1–7. [DOI] [PubMed] [Google Scholar]
- 51. Goet JC, Harms MH, Carbone M, Hansen BE. Risk stratification and prognostic modelling in primary biliary cholangitis. Best Pract Res Clin Gastroenterol. 2018;34-35:95–106. [DOI] [PubMed] [Google Scholar]
- 52. Murillo Perez CF, Harms MH, Lindor KD, van Buuren HR, Hirschfield GM, Corpechot C, et al. Goals of treatment for improved survival in primary biliary cholangitis: Treatment target should be bilirubin within the normal range and normalization of alkaline phosphatase. Am J Gastroenterol. 2020;115:1066–74. [DOI] [PubMed] [Google Scholar]
- 53. Corpechot C, Abenavoli L, Rabahi N, Chretien Y, Andreani T, Johanet C, et al. Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis. Hepatology. 2008;48:871–7. [DOI] [PubMed] [Google Scholar]
- 54. Cortez-Pinto H, Liberal R, Lopes S, Machado MV, Carvalho J, Dias T, et al. Predictors for incomplete response to ursodeoxycholic acid in primary biliary cholangitis. Data from a national registry of liver disease. United European Gastroenterol J. 2021;9:699–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gerussi A, Bernasconi DP, O’Donnell SE, Lammers WJ, Van Buuren H, Hirschfield G, et al. Measurement of gamma glutamyl transferase to determine risk of liver transplantation or death in patients with primary biliary cholangitis. Clin Gastroenterol Hepatol. 2021;19:1688–97. e1614. [DOI] [PubMed] [Google Scholar]
- 56. Locke GR, 3rd, Therneau TM, Ludwig J, Dickson ER, Lindor KD. Time course of histological progression in primary biliary cirrhosis. Hepatology. 1996;23:52–56. [DOI] [PubMed] [Google Scholar]
- 57. Corpechot C, Carrat F, Poupon R, Poupon RE. Primary biliary cirrhosis: incidence and predictive factors of cirrhosis development in ursodiol-treated patients. Gastroenterology. 2002;122:652–8. [DOI] [PubMed] [Google Scholar]
- 58. Kumagi T, Guindi M, Fischer SE, Arenovich T, Abdalian R, Coltescu C, et al. Baseline ductopenia and treatment response predict long-term histological progression in primary biliary cirrhosis. Am J Gastroenterol. 2010;105:2186–94. [DOI] [PubMed] [Google Scholar]
- 59. Murillo Perez CF, Hirschfield GM, Corpechot C, Floreani A, Mayo MJ, van der Meer A, et al. Fibrosis stage is an independent predictor of outcome in primary biliary cholangitis despite biochemical treatment response. Aliment Pharmacol Ther. 2019;50:1127–36. [DOI] [PubMed] [Google Scholar]
- 60. Corpechot C, Carrat F, Bonnand AM, Poupon RE, Poupon R. The effect of ursodeoxycholic acid therapy on liver fibrosis progression in primary biliary cirrhosis. Hepatology. 2000;32:1196–9. [DOI] [PubMed] [Google Scholar]
- 61. Carbone M, Sharp SJ, Flack S, Paximadas D, Spiess K, Adgey C, et al. The UK-PBC risk scores: Derivation and validation of a scoring system for long-term prediction of end-stage liver disease in primary biliary cholangitis. Hepatology. 2016;63:930–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lammers WJ, Hirschfield GM, Corpechot C, Nevens F, Lindor KD, Janssen HL, et al. Development and validation of a scoring system to predict outcomes of patients with primary biliary cirrhosis receiving ursodeoxycholic acid therapy. Gastroenterology. 2015;149:1804–12. e1804. [DOI] [PubMed] [Google Scholar]
- 63. Efe C, Tascilar K, Henriksson I, Lytvyak E, Alalkim F, Trivedi H, et al. Validation of risk scoring systems in ursodeoxycholic acid-treated patients with primary biliary cholangitis. Am J Gastroenterol. 2019;114:1101–08. [DOI] [PubMed] [Google Scholar]
- 64. Carbone M, Harms MH, Lammers WJ, Marmon T, Pencek R, MacConell L, et al. Clinical application of the GLOBE and United Kingdom-primary biliary cholangitis risk scores in a trial cohort of patients with primary biliary cholangitis. Hepatol Commun. 2018;2:683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Carbone M, Nardi A, Flack S, Carpino G, Varvaropoulou N, Gavrila C, et al. Pretreatment prediction of response to ursodeoxycholic acid in primary biliary cholangitis: development and validation of the UDCA Response Score. Lancet Gastroenterol Hepatol. 2018;3:626–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yagi M, Matsumoto K, Komori A, Abe M, Hashimoto N, Inao M, et al. A validation study of the Ursodeoxycholic Acid Response Score in Japanese patients with primary biliary cholangitis. Liver Int. 2020;40:1926–33. [DOI] [PubMed] [Google Scholar]
- 67. Carbone M, Mells GF, Pells G, Dawwas MF, Newton JL, Heneghan MA, et al. Sex and age are determinants of the clinical phenotype of primary biliary cirrhosis and response to ursodeoxycholic acid. Gastroenterology. 2013;144:560–9 e567; quiz e513-564. [DOI] [PubMed] [Google Scholar]
- 68. Rabiee A, Polanco NAP, Vara AF, Levy C. Hispanic patients with primary biliary cholangitis have decreased access to care compared to non-Hispanics. J Clin Transl Hepatol. 2020;8:391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cheung AC, Lammers WJ, Murillo Perez CF, van Buuren HR, Gulamhusein A, Trivedi PJ, et al. Effects of age and sex of response to ursodeoxycholic acid and transplant-free survival in patients with primary biliary cholangitis. Clin Gastroenterol Hepatol. 2019;17:2076–84. e2072. [DOI] [PubMed] [Google Scholar]
- 70. Levy C, Naik J, Giordano C, Mandalia A, O’Brien C, Bhamidimarri KR, et al. Hispanics with primary biliary cirrhosis are more likely to have features of autoimmune hepatitis and reduced response to ursodeoxycholic acid than non-Hispanics. Clin Gastroenterol Hepatol. 2014;12:1398–405. [DOI] [PubMed] [Google Scholar]
- 71. Adejumo AC, Akhtar DH, Dennis BB, Cholankeril G, Alayo Q, Ogundipe OA, et al. Gender and racial differences in hospitalizations for primary biliary cholangitis in the USA. Dig Dis Sci. 2021;66:1461–76. [DOI] [PubMed] [Google Scholar]
- 72. Cristoferi L, Calvaruso V, Overi D, Vigano M, Rigamonti C, Degasperi E, et al. Accuracy of transient elastography in assessing fibrosis at diagnosis in naive patients with primary biliary cholangitis: A dual cut-off approach. Hepatology. 2021;74:1496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Corpechot C, Carrat F, Poujol-Robert A, Gaouar F, Wendum D, Chazouilleres O, et al. Noninvasive elastography-based assessment of liver fibrosis progression and prognosis in primary biliary cirrhosis. Hepatology. 2012;56:198–208. [DOI] [PubMed] [Google Scholar]
- 74. Corpechot C, Carrat F, Gaouar F, Chau F, Hirschfield G, Gulamhusein A, et al. Liver stiffness measurement by vibration-controlled transient elastography improves outcome prediction in primary biliary cholangitis. J Hepatol. 2022;77:1545–53. [DOI] [PubMed] [Google Scholar]
- 75. Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary biliary cholangitis: 2021 practice guidance update from the American Association for the Study of Liver Diseases. Hepatology. 2022;75:1012–3. [DOI] [PubMed] [Google Scholar]
- 76. Tanaka A, Ma X, Yokosuka O, Weltman M, You H, Amarapurkar DN, et al. Autoimmune liver diseases in the Asia-Pacific region: Proceedings of APASL symposium on AIH and PBC 2016. Hepatol Int. 2016;10:909–15. [DOI] [PubMed] [Google Scholar]
- 77. Tanaka A, Hirohara J, Nakano T, Matsumoto K, Chazouilleres O, Takikawa H, et al. Association of bezafibrate with transplant-free survival in patients with primary biliary cholangitis. J Hepatol. 2021;75:565–571. [DOI] [PubMed] [Google Scholar]
- 78. Hofmann AF. Pharmacology of ursodeoxycholic acid, an enterohepatic drug. Scand J Gastroenterol Suppl. 1994;204:1–15. [DOI] [PubMed] [Google Scholar]
- 79. Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology. 2002;36:525–31. [DOI] [PubMed] [Google Scholar]
- 80. Arab JP, Cabrera D, Arrese M. Bile acids in cholestasis and its treatment. Ann Hepatol. 2017;16:s53–7. [DOI] [PubMed] [Google Scholar]
- 81. Rudic JS, Poropat G, Krstic MN, Bjelakovic G, Gluud C. Ursodeoxycholic acid for primary biliary cirrhosis. Cochrane Database Syst Rev. 2012;12:CD000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Papatheodoridis GV, Hadziyannis ES, Deutsch M, Hadziyannis SJ. Ursodeoxycholic acid for primary biliary cirrhosis: final results of a 12-year, prospective, randomized, controlled trial. Am J Gastroenterol. 2002;97:2063–70. [DOI] [PubMed] [Google Scholar]
- 83. Goulis J, Leandro G, Burroughs AK. Randomised controlled trials of ursodeoxycholic-acid therapy for primary biliary cirrhosis: a meta-analysis. Lancet. 1999;354:1053–60. [DOI] [PubMed] [Google Scholar]
- 84. Harms MH, Lammers WJ, Thorburn D, Corpechot C, Invernizzi P, Janssen HLA, et al. Major hepatic complications in ursodeoxycholic acid-treated patients with primary biliary cholangitis: Risk factors and time trends in incidence and outcome. Am J Gastroenterol. 2018;113:254–64. [DOI] [PubMed] [Google Scholar]
- 85. Shah RA, Kowdley KV. Current and potential treatments for primary biliary cholangitis. Lancet Gastroenterol Hepatol. 2020;5:306–315. [DOI] [PubMed] [Google Scholar]
- 86. de Vries E, Beuers U. Ursodeoxycholic acid in pregnancy? J Hepatol. 2019;71:1237–45. [DOI] [PubMed] [Google Scholar]
- 87. Cancado GGL, Couto CA, Terrabuio DRB, Cancado ELR, Villela-Nogueira CA, Ferraz MLG, et al. Response to ursodeoxycholic acid may be assessed earlier to allow second-line therapy in patients with unresponsive primary biliary cholangitis. Dig Dis Sci. 2023;68:514–520. [DOI] [PubMed] [Google Scholar]
- 88. Yang C, Guo G, Li B, Zheng L, Sun R, Wang X, et al. Prediction and evaluation of high-risk patients with primary biliary cholangitis receiving ursodeoxycholic acid therapy: an early criterion. Hepatol Int. 2023;17:237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Perez CFM, Ioannou S, Hassanally I, Trivedi P, Corpechot C, Van der Meer AJ, et al. Early identification of insufficient biochemical response to ursodeoxycholic acid in patients with primary biliary cholangitis. Hepatology. 2021;74:759A–60A. [Google Scholar]
- 90. Manne V, Kowdley KV. Obeticholic acid in primary biliary cholangitis: where we stand. Curr Opin Gastroenterol. 2019;35:191–6. [DOI] [PubMed] [Google Scholar]
- 91. Lindor KD. Farnesoid X receptor agonists for primary biliary cirrhosis. Curr Opin Gastroenterol. 2011;27:285–8. [DOI] [PubMed] [Google Scholar]
- 92. Kjaergaard K, Frisch K, Sorensen M, Munk OL, Hofmann AF, Horsager J, et al. Obeticholic acid improves hepatic bile acid excretion in patients with primary biliary cholangitis. J Hepatol. 2021;74:58–65. [DOI] [PubMed] [Google Scholar]
- 93. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375:631–43. [DOI] [PubMed] [Google Scholar]
- 94. Nevens F, Shiffman ML, Drenth JPH, Bowlus CL, Vargas V, Andreone P, et al. Durable response in the markers of cholestasis through 5 years of open-label extension study of obeticholic acid in primary biliary cholangitis. Dig Liver Dis. 2020;52:e30. [Google Scholar]
- 95. Kowdley KV, Luketic V, Chapman R, Hirschfield GM, Poupon R, Schramm C, et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology. 2018;67:1890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. D’Amato D, De Vincentis A, Malinverno F, Vigano M, Alvaro D, Pompili M, et al. Real-world experience with obeticholic acid in patients with primary biliary cholangitis. JHEP Rep. 2021;3:100248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Roberts SB, Ismail M, Kanagalingam G, Mason AL, Swain MG, Vincent C, et al. Real-world effectiveness of obeticholic acid in patients with primary biliary cholangitis. Hepatol Commun. 2020;4:1332–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Murillo Perez CF, Fisher H, Hiu S, Kareithi D, Adekunle F, Mayne T, et al. Greater transplant-free survival in patients receiving obeticholic acid for primary biliary cholangitis in a clinical trial setting compared to real-world external controls. Gastroenterology. 2022;163:1630–42. e1633. [DOI] [PubMed] [Google Scholar]
- 99. Eaton JE, Vuppalanchi R, Reddy R, Sathapathy S, Ali B, Kamath PS. Liver injury in patients with cholestatic liver disease treated with obeticholic acid. Hepatology. 2020;71:1511–4. [DOI] [PubMed] [Google Scholar]
- 100. John BV, Schwartz K, Levy C, Dahman B, Deng Y, Martin P, et al. Impact of obeticholic acid exposure on decompensation and mortality in primary biliary cholangitis and cirrhosis. Hepatol Commun. 2021;5:1426–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Jun M, Foote C, Lv J, Neal B, Patel A, Nicholls SJ, et al. Effects of fibrates on cardiovascular outcomes: a systematic review and meta-analysis. Lancet. 2010;375:1875–84. [DOI] [PubMed] [Google Scholar]
- 102. Ghonem NS, Assis DN, Boyer JL. Fibrates and cholestasis. Hepatology. 2015;62:635–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Jackevicius CA, Tu JV, Ross JS, Ko DT, Carreon D, Krumholz HM. Use of fibrates in the United States and Canada. JAMA. 2011;305:1217–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Honda A, Tanaka A, Kaneko T, Komori A, Abe M, Inao M, et al. Bezafibrate improves GLOBE and UK-PBC Scores and long-term outcomes in patients with primary biliary cholangitis. Hepatology. 2019;70:2035–46. [DOI] [PubMed] [Google Scholar]
- 105. Corpechot C, Chazouilleres O, Rousseau A, Le Gruyer A, Habersetzer F, Mathurin P, et al. A placebo-controlled trial of bezafibrate in primary biliary cholangitis. N Engl J Med. 2018;378:2171–81. [DOI] [PubMed] [Google Scholar]
- 106. Levy C, Peter JA, Nelson DR, Keach J, Petz J, Cabrera R, et al. Pilot study: fenofibrate for patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Aliment Pharmacol Ther. 2011;33:235–42. [DOI] [PubMed] [Google Scholar]
- 107. Hegade VS, Khanna A, Walker LJ, Wong LL, Dyson JK, Jones DEJ. Long-term fenofibrate treatment in primary biliary cholangitis improves biochemistry but not the UK-PBC Risk Score. Dig Dis Sci. 2016;61:3037–44. [DOI] [PubMed] [Google Scholar]
- 108. Cheung AC, Lapointe-Shaw L, Kowgier M, Meza-Cardona J, Hirschfield GM, Janssen HL, et al. Combined ursodeoxycholic acid (UDCA) and fenofibrate in primary biliary cholangitis patients with incomplete UDCA response may improve outcomes. Aliment Pharmacol Ther. 2016;43:283–93. [DOI] [PubMed] [Google Scholar]
- 109. Carrion AF, Lindor KD, Levy C. Safety of fibrates in cholestatic liver diseases. Liver Int. 2021;41:1335–43. [DOI] [PubMed] [Google Scholar]
- 110. Ahmad J, Odin JA, Hayashi PH, Chalasani N, Fontana RJ, Barnhart H, et al. Identification and characterization of fenofibrate-induced liver injury. Dig Dis Sci. 2017;62:3596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Mychaleckyj JC, Craven T, Nayak U, Buse J, Crouse JR, Elam M, et al. Reversibility of fenofibrate therapy-induced renal function impairment in ACCORD type 2 diabetic participants. Diabetes Care. 2012;35:1008–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ting RD, Keech AC, Drury PL, Donoghoe MW, Hedley J, Jenkins AJ, et al. Benefits and safety of long-term fenofibrate therapy in people with type 2 diabetes and renal impairment: the FIELD Study. Diabetes Care. 2012;35:218–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Soret PA, Lam L, Carrat F, Smets L, Berg T, Carbone M, et al. Combination of fibrates with obeticholic acid is able to normalise biochemical liver tests in patients with difficult-to-treat primary biliary cholangitis. Aliment Pharmacol Ther. 2021;53:1138–46. [DOI] [PubMed] [Google Scholar]
- 114. Reig A, Alvarez-Navascues C, Vergara M, Gomez-Dominguez E, Gallego-Moya A, Perez-Medrano IM, et al. Obeticholic acid and fibrates in primary biliary cholangitis: Comparative effects in a multicentric observational study. Am J Gastroenterol. 2021;116:2250–7. [DOI] [PubMed] [Google Scholar]
- 115. Beuers U, Kremer AE, Bolier R, Elferink RP. Pruritus in cholestasis: facts and fiction. Hepatology. 2014;60:399–407. [DOI] [PubMed] [Google Scholar]
- 116. Carrion AF, Rosen JD, Levy C. Understanding and treating pruritus in primary biliary cholangitis. Clin Liver Dis. 2018;22:517–532. [DOI] [PubMed] [Google Scholar]
- 117. de Vries E, Bolier R, Goet J, Pares A, Verbeek J, de Vree M, et al. Fibrates for itch (FITCH) in fibrosing cholangiopathies: a double-blind, randomized, placebo-controlled trial. Gastroenterology. 2021;160:734–43. e736. [DOI] [PubMed] [Google Scholar]
- 118. Yerushalmi B, Sokol RJ, Narkewicz MR, Smith D, Karrer FM. Use of rifampin for severe pruritus in children with chronic cholestasis. J Pediatr Gastroenterol Nutr. 1999;29:442–7. [DOI] [PubMed] [Google Scholar]
- 119. Khurana S, Singh P. Rifampin is safe for treatment of pruritus due to chronic cholestasis: a meta-analysis of prospective randomized-controlled trials. Liver Int. 2006;26:943–8. [DOI] [PubMed] [Google Scholar]
- 120. Podesta A, Lopez P, Terg R, Villamil F, Flores D, Mastai R, et al. Treatment of pruritus of primary biliary cirrhosis with rifampin. Dig Dis Sci. 1991;36:216–20. [DOI] [PubMed] [Google Scholar]
- 121. Bachs L, Pares A, Elena M, Piera C, Rodes J. Effects of long-term rifampicin administration in primary biliary cirrhosis. Gastroenterology. 1992;102:2077–80. [DOI] [PubMed] [Google Scholar]
- 122. Webb GJ, Rahman SR, Levy C, Hirschfield GM. Low risk of hepatotoxicity from rifampicin when used for cholestatic pruritus: a cross-disease cohort study. Aliment Pharmacol Ther. 2018;47:1213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wolfhagen FH, Sternieri E, Hop WC, Vitale G, Bertolotti M, Van Buuren HR. Oral naltrexone treatment for cholestatic pruritus: a double-blind, placebo-controlled study. Gastroenterology. 1997;113:1264–9. [DOI] [PubMed] [Google Scholar]
- 124. Terg R, Coronel E, Sorda J, Munoz AE, Findor J. Efficacy and safety of oral naltrexone treatment for pruritus of cholestasis, a crossover, double blind, placebo-controlled study. J Hepatol. 2002;37:717–22. [DOI] [PubMed] [Google Scholar]
- 125. Tandon P, Rowe BH, Vandermeer B, Bain VG. The efficacy and safety of bile acid binding agents, opioid antagonists, or rifampin in the treatment of cholestasis-associated pruritus. Am J Gastroenterol. 2007;102:1528–36. [DOI] [PubMed] [Google Scholar]
- 126. Mansour-Ghanaei F, Taheri A, Froutan H, Ghofrani H, Nasiri-Toosi M, Bagherzadeh AH, et al. Effect of oral naltrexone on pruritus in cholestatic patients. World J Gastroenterol. 2006;12:1125–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Murray-Brown FL. Naltrexone for cholestatic itch: a systematic review. BMJ Support Palliat Care. 2021;11:217–25. [DOI] [PubMed] [Google Scholar]
- 128. Bergasa NV, Alling DW, Talbot TL, Swain MG, Yurdaydin C, Turner ML, et al. Effects of naloxone infusions in patients with the pruritus of cholestasis. A double-blind, randomized, controlled trial. Ann Intern Med. 1995;123:161–7. [DOI] [PubMed] [Google Scholar]
- 129. Kumada H, Miyakawa H, Muramatsu T, Ando N, Oh T, Takamori K, et al. Efficacy of nalfurafine hydrochloride in patients with chronic liver disease with refractory pruritus: a randomized, double-blind trial. Hepatol Res. 2017;47:972–82. [DOI] [PubMed] [Google Scholar]
- 130. Golpanian RS, Yosipovitch G, Levy C. Use of butorphanol as treatment for cholestatic itch. Dig Dis Sci. 2021;66:1693–9. [DOI] [PubMed] [Google Scholar]
- 131. Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology. 2007;45:666–74. [DOI] [PubMed] [Google Scholar]
- 132. Carvalho AF, Sharma MS, Brunoni AR, Vieta E, Fava GA. The safety, tolerability and risks associated with the use of newer generation antidepressant drugs: a critical review of the literature. Psychother Psychosom. 2016;85:270–88. [DOI] [PubMed] [Google Scholar]
- 133. Krawczyk M, Liebe R, Wasilewicz M, Wunsch E, Raszeja-Wyszomirska J, Milkiewicz P. Plasmapheresis exerts a long-lasting antipruritic effect in severe cholestatic itch. Liver Int. 2017;37:743–7. [DOI] [PubMed] [Google Scholar]
- 134. Decock S, Roelandts R, Steenbergen WV, Laleman W, Cassiman D, Verslype C, et al. Cholestasis-induced pruritus treated with ultraviolet B phototherapy: an observational case series study. J Hepatol. 2012;57:637–41. [DOI] [PubMed] [Google Scholar]
- 135. Appleby VJ, Hutchinson JM, Davies MH. Safety and efficacy of long-term nasobiliary drainage to treat intractable pruritus in cholestatic liver disease. Frontline Gastroenterol. 2015;6:252–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Mullhaupt B, Kullak-Ublick GA, Ambuhl PM, Stocker R, Renner EL. Successful use of the Molecular Adsorbent Recirculating System (MARS) in a patient with primary biliary cirrhosis (PBC) and treatment refractory pruritus. Hepatol Res. 2003;25:442–6. [DOI] [PubMed] [Google Scholar]
- 137. Phaw NA, Leighton J, Dyson JK, Jones DE. Managing cognitive symptoms and fatigue in cholestatic liver disease. Expert Rev Gastroenterol Hepatol. 2021;15:235–41. [DOI] [PubMed] [Google Scholar]
- 138. Lee JY, Danford CJ, Trivedi HD, Tapper EB, Patwardhan VR, Bonder A. Treatment of fatigue in primary biliary cholangitis: a systematic review and meta-analysis. Dig Dis Sci. 2019;64:2338–50. [DOI] [PubMed] [Google Scholar]
- 139. Talwalkar JA, Donlinger JJ, Gossard AA, Keach JC, Jorgensen RA, Petz JC, et al. Fluoxetine for the treatment of fatigue in primary biliary cirrhosis: a randomized, double-blind controlled trial. Dig Dis Sci. 2006;51:1985–91. [DOI] [PubMed] [Google Scholar]
- 140. Silveira MG, Gossard AA, Stahler AC, Jorgensen RA, Petz JL, Ali AH, et al. A Randomized, placebo-controlled clinical trial of efficacy and safety: Modafinil in the treatment of fatigue in patients with primary biliary cirrhosis. Am J Ther. 2017;24:e167–76. [DOI] [PubMed] [Google Scholar]
- 141. Theal JJ, Toosi MN, Girlan L, Heslegrave RJ, Huet PM, Burak KW, et al. A randomized, controlled crossover trial of ondansetron in patients with primary biliary cirrhosis and fatigue. Hepatology. 2005;41:1305–12. [DOI] [PubMed] [Google Scholar]
- 142. Carbone M, Bufton S, Monaco A, Griffiths L, Jones DE, Neuberger JM. The effect of liver transplantation on fatigue in patients with primary biliary cirrhosis: a prospective study. J Hepatol. 2013;59:490–494. [DOI] [PubMed] [Google Scholar]
- 143. Swain MG, Jones DEJ. Fatigue in chronic liver disease: New insights and therapeutic approaches. Liver Int. 2019;39:6–19. [DOI] [PubMed] [Google Scholar]
- 144. Floreani A, Cazzagon N. PBC and related extrahepatic diseases. Best Pract Res Clin Gastroenterol. 2018;34-35:49–54. [DOI] [PubMed] [Google Scholar]
- 145. Sheppard J, Shen Lee B, Periman LM. Dry eye disease: identification and therapeutic strategies for primary care clinicians and clinical specialists. Ann Med. 2023;55:241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Bowlus CL, Galambos MR, Aspinall RJ, Hirschfield GM, Jones DEJ, Dorffel Y, et al. A phase II, randomized, open-label, 52-week study of seladelpar in patients with primary biliary cholangitis. J Hepatol. 2022;77:353–64. [DOI] [PubMed] [Google Scholar]
- 147. Kremer AE, Mayo MJ, Hirschfield G, Levy C, Bowlus CL, Jones DE, et al. Seladelpar improved measures of pruritus, sleep, and fatigue and decreased serum bile acids in patients with primary biliary cholangitis. Liver Int. 2022;42:112–23. [DOI] [PubMed] [Google Scholar]
- 148. Schattenberg JM, Pares A, Kowdley KV, Heneghan MA, Caldwell S, Pratt D, et al. A randomized placebo-controlled trial of elafibranor in patients with primary biliary cholangitis and incomplete response to UDCA. J Hepatol. 2021;74:1344–54. [DOI] [PubMed] [Google Scholar]
- 149. Dalekos G, Invernizzi P, Nevens F, Hans VV, Zigmond E, Andrade RJ, et al. GS-02-Efficacy of GKT831 in patients with primary biliary cholangitis and inadequate response to ursodeoxycholic acid: Interim efficacy results of a phase 2 clinical trial. J Hepatol. 2019;70:e1–e2. [Google Scholar]
- 150. Levy C, Carbone M, Wiesel P, Nevens F, Invernizzi P. Setanaxib reduces cholestasis and fatigue in patients with primary biliary cholangitis and liver stiffness > = 9.6 KPA: Post-hoc analyses from a randomized, controlled, phase 2 trial. Hepatology. 2021;74:782A–3A. [Google Scholar]
- 151. Shirley M. Maralixibat: First approval. Drugs. 2022;82:71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Deeks ED. Odevixibat: First approval. Drugs. 2021;81:1781–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Levy C, Kendrick S, Bowlus CL, Tanaka A, Jones D, Kremer AE, et al. GLIMMER: A randomized phase 2b dose-ranging trial of linerixibat in primary biliary cholangitis patients with pruritus. Clin Gastroenterol Hepatol. 2022. doi: 10.1016/j.cgh.2022.10.032 [DOI] [PubMed] [Google Scholar]
- 154. Aguilar MT, Carey EJ. Current status of liver transplantation for primary biliary cholangitis. Clin Liver Dis. 2018;22:613–24. [DOI] [PubMed] [Google Scholar]
- 155. Singh A, Fritze D, Mansouri M, Lopez R, Poordad F, Lawitz E, et al. Characteristics and outcomes of liver transplantation for primary biliary cholangitis in young patients: Analysis of the United Network for Organ Sharing Database. Transplantation. 2019;103:1191–1198. [DOI] [PubMed] [Google Scholar]
- 156. Harms MH, Janssen QP, Adam R, Duvoux C, Mirza D, Hidalgo E, et al. Trends in liver transplantation for primary biliary cholangitis in Europe over the past three decades. Aliment Pharmacol Ther. 2019;49:285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Silveira MG, Talwalkar JA, Lindor KD, Wiesner RH. Recurrent primary biliary cirrhosis after liver transplantation. Am J Transplant. 2010;10:720–6. [DOI] [PubMed] [Google Scholar]
- 158. Martin EF. Liver transplantation for primary biliary cholangitis. Clin Liver Dis. 2022;26:765–81. [DOI] [PubMed] [Google Scholar]
- 159. Corpechot C, Chazouilleres O, Belnou P, Montano-Loza AJ, Mason A, Ebadi M, et al. Long-term impact of preventive UDCA therapy after transplantation for primary biliary cholangitis. J Hepatol. 2020;73:559–65. [DOI] [PubMed] [Google Scholar]