

Summary
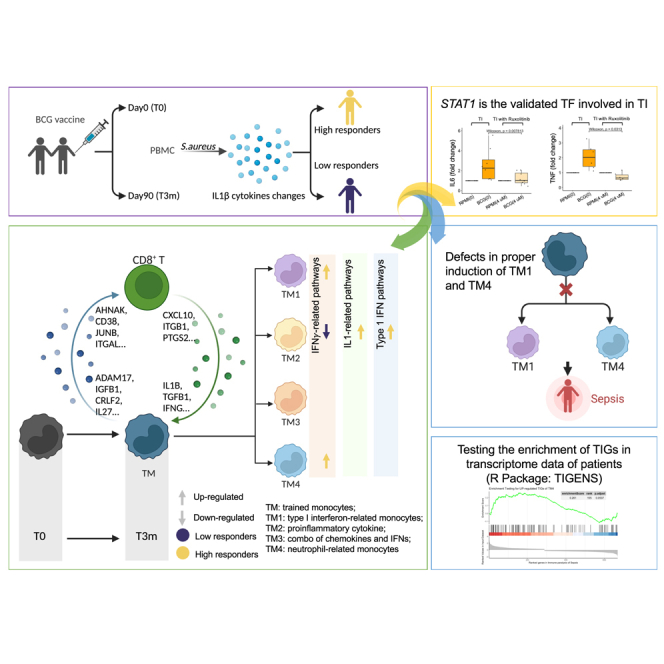
Bacillus Calmette-Guérin (BCG) vaccination is a prototype model for the study of trained immunity (TI) in humans, and results in a more effective response of innate immune cells upon stimulation with heterologous stimuli. Here, we investigate the heterogeneity of TI induction by single-cell RNA sequencing of immune cells collected from 156 samples. We observe that both monocytes and CD8+ T cells show heterologous transcriptional responses to lipopolysaccharide, with an active crosstalk between these two cell types. Furthermore, the interferon-γ pathway is crucial in BCG-induced TI, and it is upregulated in functional high responders. Data-driven analyses and functional experiments reveal STAT1 to be one of the important transcription factors for TI shared in all identified monocyte subpopulations. Finally, we report the role of type I interferon-related and neutrophil-related TI transcriptional programs in patients with sepsis. These findings provide comprehensive insights into the importance of monocyte heterogeneity during TI in humans.
Keywords: trained immunity, BCG, monocyte heterogeneity, IFN-γ, STAT1, single-cell RNA-seq, in vivo training
Graphical abstract
Highlights
-
•
IFN-γ plays an important role in amplifying trained immunity response
-
•
Monocytes show heterogeneous trained immunity capacity after in vivo BCG vaccination
-
•
Trained monocytes are regulated by different transcription factors including STAT1
-
•
A developed tool for user to test trained immunity signatures in transcriptome data
Li et al. show that BCG vaccination induces an enhanced antimicrobial response upon secondary stimulation, and this effect is heterogeneous at single-cell level. These findings provide comprehensive insights into the molecular mechanism of trained immunity and its role in immune-mediated diseases.
Introduction
In 1921, the live-attenuated Bacillus Calmette-Guérin (BCG) vaccine was introduced against tuberculosis. Interestingly, epidemiological studies have shown that BCG may also provide protection against all-cause mortality in children. These effects were due to an increased resistance against heterologous infections, including neonatal sepsis and respiratory tract infections.1 These non-specific protective effects of BCG have been later validated in many independent studies, showing non-specific protection by BCG in experimental models of bacterial, viral, fungal, and parasitic infections.2 For instance, a recent murine model study reported that intravenous BCG delivery causes enhanced production of cytokines such as interleukin-6 (IL-6), IL-1β, and tumor necrosis factor (TNF) by both splenocytes and peritoneal macrophages upon ex vivo restimulation with numerous heterologous pathogens.3 The capacity of BCG (and other vaccines or microbes) to increase non-specific resistance to heterologous pathogens is at least partly due to epigenetic and metabolic reprogramming leading to increased antimicrobial properties of innate immune cells, a process called trained immunity (TI).4
BCG vaccination is also known to induce TI in human innate immune cell populations such as myeloid cells. For weeks and even months after BCG vaccination, monocytes increase their production of proinflammatory cytokines upon reinfection with the same or different pathogen,5,6 while neutrophils display enhanced antimicrobial killing capacities.7 In addition, metabolic remodeling of monocytes by a first stimulation (such as a vaccination) can lead to a faster and stronger activation of gene transcription upon reinfection.8,9 To determine whether monocytes respond heterogeneously after being trained, recently our group performed an in vitro study with an experimental model of TI and observed three distinct TI-associated monocyte subpopulations: one population that did not respond better after BCG vaccination (non-trainable), one of monocytes that produced more chemokines (MC, which produces CXCL9/10/11), and one that improved its capacity to produce both chemokines and proinflammatory cytokines (MCI, which induces also more TNF, IL-1β, and IL-6).10 While this study sheds light on the monocyte heterogeneity in relation to transcriptional programs underlying TI, it remains elusive how the monocytes function and interact as well as how the genes are regulated in an in vivo environment, which is crucial for understanding clinical conditions.
In the present study, we studied in vivo BCG-induced TI and generated single-cell RNA sequencing (scRNA-seq) data before and 3 months after BCG vaccination from peripheral blood mononuclear cells (PBMCs) of 39 healthy individuals having either a strong (high responders, 19 individuals) or poor (low responders, 20 individuals) TI response to BCG. At both time points, cells were stimulated with lipopolysaccharide (LPS) to mimic a secondary infection or left unstimulated as a control. We identified monocytes and CD8+ T cells to have the largest sets of trained immunity response genes (TIGs), and we reported on their dense crosstalk. Four distinct monocyte subpopulations were found with distinct functions in relation to their post-BCG TI responses. Notably, interferon-γ (IFN-γ)-related pathways were upregulated in high responders, especially in the monocyte subpopulation with gene transcriptional responses enriched for IFN-related marker genes CALHM6, HES4, CXCL10, TNF, CD69, and IFIT2. The opposite result was shown in low responders. Furthermore, to reach a better understanding of each monocyte subpopulation, a method combining data-driven results and a reference database was employed to find the enriched transcription factors (TFs) for the various TIGs. Interestingly, STAT1 overlapped across all subpopulations, and functional experiments validated its function in BCG-induced TI.
Results
Single-cell transcriptome profiling of PBMCs trained by in vivo BCG vaccination
A total of 325 healthy volunteers were included in the 300BCG vaccination cohort.11 PBMCs were isolated at day 0 (T0, before BCG vaccination) and 3 months after BCG vaccination (T3m), after which the cells were stimulated with or without LPS. Since TI is characterized by enhanced responses of proinflammatory cytokines upon restimulation as compared with untrained cells, the IL-1β concentration after stimulation with Staphylococcus aureus of samples at T0 and T3m was also measured. The fold change (FC) of IL-1β between T3m and T0 was used as a measure for BCG-induced TI. Subsequently, we selected 39 individuals at the extremes of this TI spectrum: high (FC ≥ 2 of S. aureus-induced IL-1β production; N = 19) and low (FC < 2; N = 20) responders (Figures 1A and S1A). To validate whether the differences in monocyte cell counts at baseline would influence the definition of high and low responders, we calculated the correlation between TI response markers, IL-1β production capacity, and monocyte cell counts measured by fluorescence-activated cell sorting. Our results did not reveal a significant correlation between these parameters (p = 0.1). PBMCs from these selected 39 individuals were processed for scRNA-seq using the 10x Genomics protocol. In total, we profiled ∼200,000 PBMCs after filtering doublets, followed by quality control. To investigate the transcriptome profiles of trained cells, 97,022 cells from LPS stimulation before BCG (T0_LPS) and LPS stimulation 3 months after BCG (T3m_LPS) were considered and analyzed. Based on known marker genes and reference database, the uniform manifold approximation and projection (UMAP) plot revealed eight major immune cell types, namely monocytes, CD4+ T cells, CD8+ T cells, natural killer (NK) cells, B cells, platelets, plasmacytoid dendritic cells, and myeloid dendritic cells (Figure 1B). The expression of top marker genes of each identified cell type are displayed in Figure 1C. All cell clusters were uniformly distributed among individuals, batches, and sexes, which in turn suggested minimal effect of technical batch or donors (Figures S1B–S1D).
Figure 1.

Overview of scRNA-seq dataset
(A) Experimental design of scRNA-seq. Peripheral blood mononuclear cells (PBMCs) were collected before (T0) and 3 months after (T3m) BCG vaccination. At each time point, cells were stimulated with LPS (or RPMI as a control). Since IL-1β enhanced response is one of the major features to characterize TI, we measured the production of IL-1β and calculated the FC between T3m and T0, stimulated by S. aureus after 24 h. Thirty-nine individuals were selected with diverse responses to TI and divided into high (FC ≥ 2; 19 individuals) and low (FC < 2; 20 individuals) responders. See also Figure S1A.
(B) UMAP visualization of cell types in PBMCs based on known marker genes and reference database. Among them, eight major cell types were identified: monocytes, CD4+ T cells, CD8+ T cells, natural killer (NK) cells, B cells, platelets, plasmacytoid dendritic cells (pDC), and myeloid dendritic cells (mDC). See also Figures S1B–S1D.
(C) Expression level of markers identified in main PBMC types.
(D) Cell proportion comparison between T0 and T3m in each cell type. Analysis was conducted using all 39 individuals, high responders, and low responders, respectively.
(E) The number of up- and downregulated TI response genes (TIGs) using all 39 individuals(left) and the same analysis were followed by dividing the individuals into high and low responders (right). Red bars represent the number of genes upregulated after being trained and blue bars represent the number of genes downregulated. Bars with a yellow icon below the x axis correspond to high responders, and bars with a dark-blue icon correspond to low responders. See also Figures S2A and S2B.
We then systematically compared the cell proportion of each major cell type between T0 (before training by BCG) and T3m (after training by BCG) using Dirichlet regression analysis12 and observed no significant differences (Figure 1D). After stratifying individuals into high and low responders, no significant cell proportion changes were observed in either of them. However, an interesting pattern was observed in high responders after being trained (T3m_LPS), i.e., the proportion of monocytes increased, while the opposite was observed in low responders (Figure 1D). To detect whether the changes in monocyte proportion between high and low responders was significant, we performed Fisher’s exact test and observed a significantly increased (two-sided, p <2.2 × 10−16) monocyte proportion after being trained in high responders. This supports the notion that monocytes are one of the key factors for individual response variation to TI.
Further, to gain an overview of transcriptome characteristics of the in vivo induced TI effect, we calculated differentially expressed genes (DEGs) by comparing T0_LPS and T3m_LPS, which were defined as trained immunity response genes (TIGs) (Table S1). In line with the role of monocytes in heterologous BCG effects, we observed a large number of TIGs in monocytes (Figure 1E). Interestingly, we also identified abundant TIGs in CD8+ T cells. Subsequently, a stratified analysis in high and low responders separately revealed that in monocytes, although the number of downregulated TIGs in high (n = 79) and low responders (n = 76) was approximately equal, more TIGs were upregulated in high responders (n = 128) than in low responders (n = 11). Thus, more upregulated TIGs in monocytes of high responders indicated that the difference in monocytes’ response to TI might be one of the key reasons to explain the differences between high and low responders. In contrast, in CD8+ T cells, we detected an almost similar number of upregulated TIGs in high (n = 54) and low (n = 74) responders, and more downregulated TIGs in low responders (n = 112) than in high responders (n = 20).
In parallel, we noticed that the number of DEGs responding to BCG vaccination, which was calculated by comparing cells between T3m_RPMI and T0_RPMI (Figures S2A and S2B) in monocytes and CD8+ T cells, separately, was not significantly different between high and low responders. This contrasted with the number of TIGs in high and low responders (two-sided Fisher’s exact test, p < 0.01). This suggests that transcriptomic responses to BCG vaccination show a modest association with TI response.
Interferon-γ-related pathways contribute to heterogeneity of TI response
Subsequently, pathway analysis was performed to investigate the biological functions of TIGs associated with BCG-induced TI in monocytes. In total, 225 and 87 TIGs were identified within the monocytes of the high and low responders, respectively. The type I IFN signaling pathway was significantly upregulated in high responders but not in low responders (Figure 2A). Notably, type I IFNs activate the immune response after infection.13 Similarly, the type II IFN pathway was upregulated in high responders while being downregulated in low responders (Figures 2A and S2C–S2J). The opposite direction of type II IFN pathway regulation after BCG vaccination in low and high responders suggests an important role of IFN-γ in BCG-induced TI, as supported by several recent studies.14,15
Figure 2.

scRNA-seq analysis of TI characteristics
(A) Pathway enrichment analysis using up- and downregulated TIGs in high and low responders in monocytes separately. Pink area means pathways were upregulated, while the blue area means pathways were downregulated. Different patterns were shown between high and low responders. Specifically, IFN-γ pathways were upregulated in high responders but downregulated in low responders. See also Figures S2C–S2J.
(B) Effect size (log2FC) comparison of TIGs identified in monocytes. Values on the x axis were log2FC in high responders, while values on the y axis were from low responders. Yellow dots represent those TIGs only significant in high responders while dark-blue ones were only significant in low responders. Gray dots represent TIGs that were significant in both groups.
(C) Enriched pathways using TIGs that were significantly upregulated in high responders but downregulated in low responders. See also Figure S3A.
(D and E) Cell-cell interaction between monocytes and CD8+ T cells. Ligands were highly variable genes in senders, and targets were TIGs in receivers. In (D), monocytes were set as sender, while in (E) CD8+ T cells were set as sender. See also Figures S3B–S3E.
A comparison of effect size of TIGs between high and low responders (Figure 2B) showed that 179 out of 274 TIGs (65.32%) had the same direction (both either upregulated or downregulated in high and low responders), demonstrating a shared response pattern between high and low responders. However, there were 194 and 56 TIGs (Bonferroni corrected p value <0.05) that were specifically significant in either high or low responders, respectively. Among 31 TIGs that showed significant differential expression in both groups, 14 genes (45.16%), i.e., GBP5, LAP3, GBP1, WARS, ACOD1, PSMB9, IRF1, CREM, APOL2, CDC42SE2, LRRC75A, APOL1, HAPLN3, and FBXO6, were upregulated in high responders but downregulated in low responders. These genes were enriched in IFN-γ pathways (Benjamini-Hochberg [BH]) corrected p value <0.05) (Figure 2C). Additionally, there were 70 significant TIGs in either high or low responders but with opposite effect (i.e., upregulated in high responders and downregulated in low responders, fourth quadrant of Figure 2B). Those genes also showed an enrichment in IFN-γ-related pathways (BH corrected p value <0.05, Figure S3A). These findings are consistent with the hypothesis that IFN-γ pathways play a key role during induction of TI.
Crosstalk between monocyte and CD8+ T cells in trained immunity
Since a large number of TIGs were also detected in CD8+ T cells (Figure 1E), we further investigated the biological functions associated with upregulated (n = 135) and downregulated (n = 79) TIGs in CD8+ T cells. Many of the enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways among the upregulated TIGs in CD8+ T cells were the same as in upregulated TIGs of monocytes, including NK cell-mediated cytotoxicity, suggesting a possible interaction between monocytes and CD8+ T cells in TI (Figures S3B–S3E). To investigate the putative crosstalk between monocytes and CD8+ T cells during TI, NicheNet16 was used to estimate the ligands-targets pairs based on gene expression and reference database. When monocytes were set as senders, we found that monocytes likely interact directly with CD8+ T cells via several ligands, including IL27 and CRLF2 (Figure 2D). Previous studies have shown that IL27, together with IL12, is regulated in human macrophages that interact with Mycobacterium tuberculosis, with an important role in T cell activation.17 In addition, the CRLF2 gene interacts with IL7R and subsequently thymic stromal lymphopoietin to activate downstream lymphopoiesis pathways.18 Although we detected a large number of ligands-targets connections while setting CD8+ T cells as senders, the lymphocyte products IFNG19 and TGFB1 seemed to play key roles during interaction with monocytes (Figure 2E). Our data suggested that monocytes and CD8+ T cells may interact with each other in intensive interactions during induction of TI. The TIGs from monocytes influenced the reaction of CD8+ T cells, and the changes in CD8+ T cells may assist monocytes to respond to BCG-induced training. Therefore, CD8+ T cells is likely to be an important source of IFN-γ that amplifies the TI responses after BCG vaccination. These data suggest that CD8+ T cells may be an important source of IFN-γ, amplifying the TI responses after BCG vaccination.
Four heterogeneous monocyte subpopulations are identified upon in vivo BCG training
To systematically study the heterogeneity of in vivo BCG-induced TI effects in monocytes in an unbiased manner, we analyzed 12,703 TI-associated monocytes from T3m_LPS and T0_LPS from all the individuals in our data. The workflow of this analysis was as follows: (1) we performed unsupervised clustering based on the changes of most variable genes between T3m_LPS and T0_LPS; (2) we calculated the TIGs (specifically between T3m_LPS and T0_LPS) for each identified subpopulation from step 1; (3) we performed enrichment analysis based on the identified TIGs from step 2 to detect further biological heterogeneity. In detail, initially we selected the top 50 most variable genes from the monocytes and performed unsupervised clustering based on the expression difference between T3m_LPS and T0_LPS: this methodology identified four subpopulations (Figure 3A). Subsequently, for visualizing the distribution of four identified monocyte subpopulations, we extracted monocytes at T3m_LPS and mapped corresponding cells in each subpopulation to UMAP directly, demonstrating the transcriptional heterogeneity of the TI effect induced by BCG vaccination (Figure 3B). Additionally, the robustness of clustering results was confirmed by the same analysis using either the top 100 or top 300 (Figures S4A and S4B) most variable genes.
Figure 3.

Subclustering of monocytes based on TI responses, which are changes between T3m_LPS and T0_LPS
(A) Unsupervised clustering using the top 50 most variable genes in monocytes. For each gene in each cell, the value was calculated by the difference in T3m_LPS and the mean value of the corresponding gene in the corresponding individual in T0_LPS. Spearman correlation was calculated between cells, and k-means was used to cluster cells. In total, four distinct monocytes were identified. See also Figures S4A and S4B.
(B) UMAP visualization of identified monocyte subpopulations. Monocytes were extracted from the UMAP of PBMC in T3m_LPS, and cells in each subpopulation were mapped to UMAP directly. Four subpopulations were independently distributed, which may demonstrate their heterogeneous functions in TI.
(C) Expression level of markers in four defined monocyte subpopulations. Based on the markers, four subpopulations were defined: type I IFN-related monocytes (TM1); proinflammatory cytokine monocytes (mainly IL-1 pathway) (TM2); proinflammatory monocytes (combination of chemokines and IFNs) (TM3); and neutrophil-related monocytes (TM4).
(D) Comparative analysis of four monocyte subpopulations and previously reported monocyte subpopulations (i.e., MCI and MC) using an in vitro dataset. Markers of MCI and MC were selected from Zhang et al.10 The expression pattern indicated that TM1–TM3 were subtypes of MCI while TM4 tended to have characteristics similar to those of MC.
To characterize the four monocyte subpopulations, we assessed the main genes changing transcriptionally in each subpopulation. Based on these characteristics we could loosely define subpopulations of trained monocytes as: TM1 (characterized by CALHM6, HES4, CXCL10, TNF, CD69, and IFIT2) enriched in type I IFN-related pathways; TM2 (characterized by CXCL1-CXCL3, THBS1, SLC7A11, CCL20, F3, and CSF3) as mainly enriched in proinflammatory cytokine genes (mainly IL-1 pathway); TM3 (characterized by CCL2, IFI6, IFI44L, and CCL7) as enriched in a combination of chemokines and IFNs; and TM4 (characterized by ZEB2, ACTG1, PFN1, LYZ, S100A9, OLR1, S100A8, and ZFAS1) as enriched in neutrophil-related genes (Figure 3C). We used the known markers of classical monocytes (CD14), non-classical monocytes (FCGR3A), M1 (CD80, CD86), and M2 (CD163, CD68) to check whether the four identified heterogeneous subpopulations were overlapped with one of these classically divided monocyte subsets. First, we did not observe the expression of CD14 and FCGR3A in our dataset. Second, we assessed CD80, CD86, CD163, and CD68 expression and found that TM1 showed overlap with M1 while TM2–TM4 overlapped with subsets of M2 (Figures S4C and S4D).
Next, we compared the four identified monocyte subpopulations with the three previously reported trained monocyte subpopulations after in vitro TI induction, i.e., MCI, MC, and non-trained subsets.10 We observed that MC signature genes were highly expressed in TM4, whereas MCI signatures were highly expressed in TM1, TM2, and TM3. For example, IL1B, TNF, and IL6, which were markers of MCI, were expressed higher in TM1–TM3 than in TM4. This result suggested that TM4 was comparable with MC whereas MCI can be further subclassified into TM1–TM3 (Figure 3D). The TM3 population has the lowest number of DEGs after BCG vaccination, and is therefore most closely related to the in vitro non-trained monocyte subset.
The trained immunity monocyte subpopulations have distinct functions
To understand the functions of these four distinct monocyte subpopulations, we identified TIGs in each monocyte subpopulation (Figure 4A) followed by pathway enrichment analysis. The number of TIGs in TM1 was the largest compared with the other three subpopulations. TM3 showed the least number of TIGs. A different pattern was observed in TM2, in which 89% TIGs were downregulated. In TM4, most of the TIGs (90%) were upregulated.
Figure 4.

TI characteristics of four monocyte subpopulations
(A and B) The number of up- and downregulated TIGs in each subpopulation. The results of pathway enrichment analysis using up- and downregulated TIGs identified in (A), separately, are summarized in (B). See also Figures S4C–S4F. Pathways in the pink area were upregulated and in the blue area downregulated. IFN-γ pathways were upregulated in TM1 but downregulated in TM2.
(C and D) Comparisons of the number of shared and unique TIGs in high and low responders in each population. The results of enrichment analysis (C) using specific up- and downregulated TIGs in high and low responders, separately, are summarized in (D). Pathways in the pink area were upregulated and in the blue area downregulated. Interestingly, IFN-γ pathways were upregulated in TM1 in high responders but were downregulated in TM2 in low responders. This again indicated the importance of IFN-γ pathways in TI and the role of these pathways in explaining individual diversity in responding to TI. See also Figures S5C–S5H and S6A–S6D.
(E) Further investigation comparing monocytes and CD8+ T cells explored communications between four monocyte subpopulations and CD8+ T cells. The expression level of the top 20 most active ligands identified in four subpopulations are shown as a dot plot. Different ligands were interacting with CD8+ T cells in each subpopulation, and TM1 was the most active cell type. Heatmap shows the potential regulatory relationship between ligands in monocyte subpopulations and TIGs in CD8+ T cells. See also Figures S6E–S6G.
Pathway enrichment analysis revealed that upregulated TIGs in TM1 and TM4 were enriched in IFN-γ-related pathways. On the contrary, IFN-γ and type I IFN-related pathways were downregulated in TM2 (Figure 4B). Gene ontology enrichment analysis revealed that the upregulated TIGs in the monocyte subpopulations were associated with several immune-related pathways, e.g., the nuclear factor (NF)-κB signaling pathway in TM1 and TM4, the IL-1 receptor binding pathway in TM2, and the response to IL-1 in TM3 (Figure S4E). Downregulated TIGs were enriched for IL-12-related pathways in TM1 and type I IFN in TM2, while IL-1-related pathways were lowered by BCG vaccination in TM4 (Figure S4F). In the KEGG enrichment analysis, although the number of TIGs in these four subpopulations was different, some pathways were shared between them (Figures S4G and S4H). For example, for upregulated TIGs Toll-like receptor signaling pathway, NOD-like receptor signaling pathway, RIG-1-like receptor signaling pathway, and TNF signaling pathway were upregulated in TM1, TM2, and TM3 but downregulated in TM4 (Figure 4B). This was also consistent with what we have observed above (Figure 3D), i.e., the expression level of TNF is lower in TM4 compared with other subpopulations. The IL-17 signaling pathway was enriched for upregulated genes in TM2 and TM3 but was downregulated in TM1 and TM4. These findings suggest that monocyte responses after BCG vaccination are heterogeneous, with four major types of transcriptional response.
To validate the robustness of the identified four BCG-induced TI monocyte subpopulations, we assessed single-cell transcriptome profiles from three additional, independent individuals from the same cohort (300BCG cohort) using the same technology (10x Genomics). Unsupervised clustering analysis here again revealed four TI-induced monocyte subpopulations (Figures S5A and S5B). We also checked the enriched pathways for upregulated TIGs and downregulated TIGs separately. Twelve out of 33 enriched pathways of upregulated TIGs, including NOD-like receptor signaling pathway, RIG-1-like receptor signaling pathway, NF-κB signaling pathway, cytokine-cytokine receptor interaction, and IL-17 signaling pathways, were replicated in the corresponding subpopulations. While looking into the TIGs of each enriched pathway, we found that on average 55% of them can be replicated. For downregulated TIGs, 13 out of 30 enriched pathways, including Toll-like receptor signaling pathway, legionellosis, pertussis, AGE-RAGE signaling pathway in diabetic complications, and graft-versus-host disease, can be replicated (Figures S5C and S5D). This high level of replication, despite the lower power of the second study, strongly suggests the robustness of the pathways identified in the four TI monocyte subpopulations.
To further understand how the functions of these four distinct monocyte subpopulations differ depending on the individual functional TI response after BCG vaccination, we stratified individuals into high and low responders and then compared the number of TIGs between them in each monocyte subpopulation (Figure 4C). In TM1, 63.6% of the TIGs were shared between high and low responders, whereas TIGs of TM2–TM4 were mostly specific to either high or low responders. TM3 again showed the least number of TIGs compared with the other three subpopulations, thereby suggesting that those cells were less trainable than the others. IFN-γ-related pathways were upregulated in TM1 for TIGs specifically in high responders but downregulated in TM2 for TIGs specific in low responders. Similar to the findings above (Figure 2A), this also suggests that IFN-γ played an important role in differing high and low responders (Figures 4D, S5E–S5H, and S6A–S6D).
Crosstalk between TI monocyte subpopulations and other immune cells
Considering the heterogeneous monocyte response after BCG-induced TI and the large number of TIGs in CD8+ T cells, we performed ligands-targets interaction between each monocyte subpopulation (senders) and CD8+ T cells (receivers) in T3m_LPS (after being trained) separately. Most of the active ligands, including AMAD17, PTPRC, IL18, CLCF1, TNF, IL15, and EBI3, were detected in TM1. EBI3 in TM1 and CRLF2 in TM4 were the actively communicating ligands with large numbers of TIGs in CD8+ T cells (Figure 4E). We did not identify unique ligands in TM3 that interact with CD8+ T cells, which may again illustrate that TM3 monocytes are not as well trained as the other subpopulations.
As discussed above, we found that upregulated TIGs in TM1 and TM4 were also enriched in pathways related to T cells (Figure S4E). Subsequently, we further explored whether the four different monocyte subpopulations interacted among themselves or with other immune cells, especially the communication between TM1/TM4 and T cells. Analysis was performed between each monocyte subpopulation (set as receiver) and all cell types (including TM1–TM4, CD8+ T cells, CD4+ T cells, NK cells, and B cells, set as the sender) separately. We observed that SEMA4D in CD8+ T cells communicated with BCL3, CDKN1A, GADD45B, PLEC, PTMA, UBC, and ELBO in TM4 (Figures S6E–S6G). CD4+ T cells interacted with TM1 via PTPRC (CD45). We also observed that TM1 was the most active sender during communication and interacted with all four monocyte subpopulations. Specifically, IFNG expressed in NK cells, TNF expressed in TM1, and IL1B and IL1A expressed in TM1, TM2, and TM3 showed interactions with TIGs detected in either TM1/TM2/TM3 but not in TM4. Interestingly, some ligands, such as TNF and ADM expressed in TM1 and CCL2 expressed in TM3, were also interacting with the other two subpopulations, i.e., either TM2/TM3 or TM1/TM2, respectively, but not with TM4. We observed that these ligands were also TIGs (Table S1), thereby suggesting interactions among TM1, TM2, and TM3 during TI. However, the interaction between TM4 and other cell types showed very different patterns. For example, unlike in TM1–TM3, an interaction between TM4 and NK cells was missing. These results suggest a similarity in interactions mediated by TM1, TM2, and TM3 with other immune cells. Based on these data and the expression level of previously identified MCI signatures (Figure 3D), the notion that the TM1–TM3 trained monocytes identified here are subpopulations of MCI cells (while TM3 is most closely related also to non-trainable cells) was further consolidated.
Trained monocyte subpopulations are regulated by different transcription factors including STAT1
We next aimed to identify the active transcription regulators and reconstruct the gene regulatory network important for the induction of the distinct TI programs in the four monocyte subpopulations. First, the RcisTarget package in SCENIC20 was used to identify TFs that may regulate the TIGs upon BCG vaccination. GENIE321 was then used to reconstruct the gene regulatory network based on the identified TFs and TIGs in each subpopulation separately. TF-targets links were randomly permuted 500 times, and the area under the receiver-operating characteristic curve (AUC) and the area under the precision-recall curve (AUPR) and their corresponding p values were calculated (Figure 5A). In total, 41 TFs were found to be significantly enriched in TIGs from the four monocyte subpopulations (AUC p < 0.05 and AUPR p < 0.05). Among the detected TFs, 65% were shared among two or more subpopulations. TM1, TM3, and TM4 had six (XBP1, HIVEP2, GTF2B, JUND, FOSL1, and MEF2A), five (ETS2, ATF4, NFAT5, PRDM1, and STAT6) and one (KLF6) specific TFs, respectively (Table S2). Specifically, STAT1 was one of the master TFs shared among all four subpopulations, together with BACH1, IRF7, NFKB1, and CEBPB (Figure 5B). Among the TFs shared by all four monocyte subpopulations, pathways in which STAT1 functioned as a crucial TF regulate the IFN-γ effects.22 To validate the regulatory role of STAT1 in TI, monocytes were stimulated with RPMI or BCG in the absence or presence of the STAT1 inhibitor ruxolitinib (4 μM). Ruxolitinib significantly inhibited TI induced by BCG (Wilcoxon test, p = 0.0078 and p = 0.031, respectively) (Figure 5C), demonstrating the role of STAT1 for the induction of TI.
Figure 5.

TF analysis of monocyte subpopulations
(A) Workflow of TF identification. First, RcisTarget was performed to search the regulons from a human database using TIGs in each subpopulation separately. GENIE3 was used to reconstruct gene regulatory networks between identified TFs and TIGs using our scRNA-seq dataset. TF-target links were ranked on the basis of importance score. Then for each identified TF, we labeled the link with this specific TF as 1 with the others labeled as 0 and shuffled the rank 500 times to calculate AUC p value and AUPR p value. Those TFs with both AUC p < 0.05 and AUPR p < 0.05 were considered significant.
(B) Unique and overlapping identified TFs among four monocyte subpopulations. Potential TFs in each subpopulation are colored dark green. See also Figure S6H.
(C) Lab validation of STAT1. Monocytes were stimulated with RPMI or BCG. Both simulations were performed separately with a carrier control (DMSO, 0) or with 4 μM ruxolitinib. Comparison of FC of IL-6 and TNF (both are TI markers) between after being trained and being trained with ruxolitinib, separately, revealed that, after being trained with ruxolitinib, the TI effect decreases significantly (Wilcoxon test, p = 0.007813 and p = 0.0313, respectively).
Subsequently, transcriptomic data in T3m_LPS were used to separately predict the targets of STAT1 in high and low responders, employing GENIE3. It is interesting that the targets of STAT1 in TM1 were completely unique between high and low responders, while some were shared between the two groups in other subpopulations (Figure S6H). We detected that IFN-γ-related pathways were upregulated in TM1, especially in high responders (Figures 4B and 4D), which may again indicate that IFN-γ-related pathways acting through STAT1 play an important role in BCG-induced TI.
Involvement of TM1 and TM4 trained immunity monocyte subpopulations in diseases
To explore whether and how our identified TI-induced monocyte subpopulations were also related to diseases, we tested the enrichment of expression for genes responding to M. tuberculosis infections (the BCG targeted disease23) in the four monocyte subpopulations. M. tuberculosis-responding genes were enriched in the upregulated TIGs in TM1 but downregulated TIGs in TM2, and they were mainly involved in IFN-γ-related pathways (BH corrected p value <1 × 10−25).
To further understand the function of TIGs in immune diseases, as well as the specific monocyte subpopulation involved, we used up- and downregulated DEGs between sepsis patients and healthy individuals. Our goal was to test the potential overlap and enrichment of TIGs with sepsis DEGs as an illustrative example of an important immune-mediated disease. Based on the circulating ferritin concentrations and data from the previous study,24 we compared three groups of sepsis patients with either immune paralysis, macrophage activation-like syndrome (MALS), or an unclassified immune profile. Sepsis-responding genes were obtained by comparing each group of patients and healthy controls separately. First, we calculated the DEGs between each subgroup of patients and healthy controls in sepsis scRNA-seq data. Next, we ranked our TIGs based on the log2 fold change (log2FC) in decreasing order. Thereafter, we mapped the DEGs to the ranked TIGs and calculated the enrichment score. We found that upregulated TIGs in TM1 were enriched in the gene dataset that was downregulated in the MALS and immune paralysis sepsis patients (Figure 6A). These genes were related to the IFN-γ and inflammatory response pathway (BH corrected p value <0.05) (Figure 6B and Table S3). This suggests a defective capacity of monocytes from sepsis patients to mount an effective TM1 transcriptional program. Similarly, we observed that down- and upregulated TIGs in TM1 and TM4 (Figures 6C and 6D), respectively, were enriched in the genes from immune paralysis and MALS patients. Among important pathways, they were all enriched for genes of the complement pathway (Figure 6E and Table S3). An opposite pattern, i.e., downregulated TIGs in TM1 but upregulated in patients, may contribute to susceptibility to sepsis, while upregulation of TIGs in both TM4 and patients might provide protection against sepsis. A similar pattern was detected in ICU-SEPSIS patients of the second sepsis dataset25 (Figure 6C).
Figure 6.

TIG association with sepsis disease
(A and B) Upregulated genes in TM1 were all enriched in downregulated genes of macrophage activation-like syndrome (MALS) and immune paralysis patients compared with healthy controls. Enrichment analysis using enriched genes identified from MALS and immune paralysis (A), which were upregulated in TM1 but downregulated in patients, separately, are summarized in (B). IFN-γ responses and inflammatory responses were significantly enriched in both patients.
(C) Downregulated genes in TM1 were enriched in upregulated genes in patients in immune paralysis and MALS patients compared with healthy controls. The same result was also detected in ICU-SEPSIS patients from the second cohort.
(D) In two independent sepsis cohorts, we observed that upregulated genes in TM4 are enriched in upregulated genes in both immune paralysis and ICU-SEPSIS patients compared with healthy controls.
(E) Pathway enrichment analysis using enriched genes from (C) and (D) showed genes that were upregulated in TM1 and downregulated in TM4, but upregulated in patients (compared with healthy controls), were all enriched in the complement pathway. These analyses may indicate the balance between TM1 and TM4.
Altogether, these results suggest that TI monocyte subpopulations (TM1 and TM4) are involved in immune response in infections, and defects in proper induction of TM1 and TM4 TI transcriptional programs can lead to an increased severity of sepsis or a higher risk of disease progression (toward MALS or immune paralysis).
To facilitate the understanding of TI in diseases, we have implemented the above analysis into an R tool, TIGENS, for testing the enrichment of gene signatures from different TI monocyte subpopulations in transcriptome data of patients. TIGENS is freely available at GitHub of CiiM (https://github.com/CiiM-Bioinformatics-group/TIGENS).
Genes associated with TI responses show higher expression in TM4 subpopulation
To further validate the function of the identified TI monocyte subpopulations, we tested whether the expression of genes associated with TI is enriched in these cells. First, we performed trained immunity quantitative trait locus (TI QTL) mapping to calculate the correlation between the ratio (between T3m_LPS and T0_LPS) of cytokine response (IL-1β) and SNP genetic variants in 289 samples of the 300BCG cohort. Subsequently, we extracted genes located around SNPs having a TI QTL p value of <1 × 10−3, within a 250 kb window both upstream and downstream, and tested whether they were enriched in the above identified TIGs in each monocyte subpopulation, respectively. Among these four subpopulations, the neutrophil-related monocyte subpopulation (TM4) showed the highest number of upregulated TIGs (38) that also contained QTLs (Table 1). Considering TM4 also had a greater number of upregulated TIGs compared with TM2 and TM3, we set the TIGs of the other three subpopulations as the background to test the significance of enrichment. Fisher’s exact test showed that the enrichment was significant (p < 0.05), which suggested that TI QTL proximity SNPs were more likely to influence TI response in TM4. To check the robustness of our results, we selected a p value of <1 × 10−4 as another threshold and found that the number of TIGs in TM4 (neutrophil-related monocyte subpopulation) was consistently larger than in other subpopulations (Table S4).
Table 1.
Analysis of association between monocyte subpopulations and TI combining genetics dataset
| TM1 | TM2 | TM3 | TM4 | |
|---|---|---|---|---|
| No. of TIGs located within IL-1β QTL areas | 29 | 1 | 0 | 38 |
| No. of TIGs | 350 | 19 | 35 | 309 |
We calculated the number of TIGs located within IL-1β QTL areas. Fisher’s exact test showed that TI QTL proximity SNPs were more likely to influence TI response in TM4 (p < 0.05).
Discussion
BCG vaccination not only protects against tuberculosis but also against unrelated infections and all-cause mortality.3 This is thought to be at least in part mediated by the induction of TI, which results in a more effective activation of innate host defense mechanisms in an antigen-independent manner.4,26 However, there is a large variation in the TI response to BCG between individuals, and the molecular mechanisms behind this variation are not known. The 300BCG cohort has been designed to comprehensively investigate the immunological effects of BCG vaccination.11 One very important aspect that is basically unknown is the heterogeneity of the TI response at a cellular level: are there different subpopulations of trained innate immune cells, and what are their characteristics and roles in disease? In this study, we presented single-cell transcriptome profiles of immune cells isolated from 39 healthy individuals, stimulated ex vivo with LPS before and 3 months after BCG vaccination. We identified four different populations of TI monocytes characterized by specific activation of immune pathways after BCG vaccination. We also reported the role of STAT1 and type II IFN pathway for the induction of TI, and the defects in proper induction of these programs in patients with severe sepsis.
One of the interesting findings was to identify the effects of BCG-induced TI at the level of different immune cell populations. When assessed per immune cell type, we found a large number of TIGs, mainly in monocytes and CD8+ T cells. Subsequently, we also observed cell-cell communication networks between monocytes and CD8+ T cells during TI, and these intense interactions suggested that although monocytes represent the main substrate of TI, their function was strongly influenced by CD8+ T cells, while CD8+ T cells were also influenced by monocytes after BCG vaccination. The importance of interaction between monocytes and T cells for induction of TI is supported by a recent study on TI in a malaria model.14 Importantly, the data on the enrichment of the type II IFN pathway in the induction of the TI programs may suggest a role of IFN-γ produced by CD8+ cells in these effects. This remains to be formally demonstrated, but IFN-γ has been shown to be crucial for the induction of TI by BCG also in animal models.15
Another interesting finding of our study was that we observed individual variations in TI while measuring cytokine production capacity before and after vaccination: high responders (N = 19) and low responders (N = 20) were separated based on the FC of one of the key TI response markers, i.e., a change in IL-1β production capacity following BCG vaccination. To explore the reasons and mechanisms leading to individual diversity, enrichment analysis using TIGs was performed which, interestingly, showed that IFN-γ-related pathways were upregulated in monocytes from high responders but downregulated in monocytes from low responders. Finally, an important argument for the role of the type II IFN pathway for the induction of TI was provided by the complementary data on the enrichment of STAT1 motifs in TI genes and the functional experiment showing inhibitory effects of the JAK/STAT pathway inhibitor ruxolitinib on the induction of TI.
Previous studies have shown that monocytes and macrophages mediate protective effects upon multiple stimulations from various pathogens.6,27 However, the heterogeneous response of monocytes in TI after vaccination was not studied. An in vitro study has recently attempted to unveil monocyte heterogeneity after being trained, and reported three populations of trained monocytes—MCI (monocytes producing higher chemokines and inflammatory cytokines), MC (monocytes producing higher chemokines), and non-trained monocytes (monocytes that do not increase their function after training)—based on the corresponding TI response profiles.10 We wanted to explore whether similar heterogeneity exists after in vivo vaccination with BCG, a canonical inducer of TI. We therefore compared single-cell transcriptional profiles before and after BCG vaccination using an unsupervised clustering method. This enabled us to identify four monocyte subpopulations (TM1, TM2, TM3, and TM4) with distinct TI characteristics, immune functions, and regulated TFs. We observed that TM1–TM3 expressed the earlier described MCI signatures, while TM4 expressed MC signatures. The presence of several subclasses of trained MCI monocytes in vivo was suggestive of subtle signals that were masked in the in vitro model. Interestingly, IFN-γ-related pathways were upregulated in high responders in both TM1 and TM4 but downregulated in low responders in TM2, which argued for specific pathways of induction for these distinctive TI programs.
TFs are cell-type or condition specific and have a very important role in determining the cellular function. Therefore, we used TIGs to detect subpopulation-specific TFs, which in turn helped us to interpret the distinguishing characteristic of each trained monocyte subpopulation. Interestingly, we identified both unique TFs corresponding to each subpopulation and shared ones across all subpopulations. Among them, STAT1, which was a crucial TF in regulating IFN-γ effects, seemed to act as a master TF across all four monocyte subpopulations. The effect of STAT1 in regulating TI was experimentally validated by pharmacological inhibition in vitro.
While very interesting from an immunological point of view, a crucial question regards the importance of the TM1–TM4 TI programs for human diseases, which would in turn provide insights into immunotherapy targets. As innate immune responses are crucial for host defense, we investigated these transcriptional programs in sepsis, which comprises different endotypes based on severity or specific immune responses.24 Importantly, we observed that the trained monocyte programs had dysregulated expression in patients with severe sepsis. In detail, the genes that were downregulated TIGs in TM1 were significantly upregulated in patients with severe forms of sepsis. In contrast, the upregulated TIGs had different behavior whether they belonged to the TM1 or TM4 programs: upregulated TM4 TIGs were also higher in patients, whereas the upregulated TIGs in TM1 were downregulated in patients. These data strongly suggest that it is especially the TM1 TI program that is defective in patients with severe sepsis, opening the possibility to identify specific immune-based approaches to sepsis in the future that could target the pathways belonging to TM1 TI. The response to type II IFNs is an important component of the TM1 TI program, and IFN-γ is currently in clinical trials for patients with sepsis (https://clinicaltrials.gov/ct2/show/NCT04990232).
Limitations of the study
This study also has limitations. First, we have focused on monocytes, which are probably the most important cell type in TI, but earlier studies have also reported that BCG affects several other innate immune cell types including NK cells, neutrophils, dendritic cells, and even adaptive immune cell types, e.g., T cells.4,28 Future studies should also focus on how these cells respond to BCG vaccination-induced TI. For example, it would be interesting to explore whether there are any subpopulations of other innate immune cells after being trained and how they interact and cooperate with each other upon secondary stimuli. Second, our study investigated post-BCG changes up to 3 months after vaccination, but future studies should investigate the effects of BCG beyond 3 months. Third, in this study we demonstrate the importance of the crosstalk between monocytes and CD8+ T cells for induction of TI, and this finding is based solely on the data generated by the BCG vaccination. The important role of T cell/monocyte interaction for an efficient induction of TI has been also recently demonstrated in the case of malaria.14 However, whether this interaction is important for other stimuli that induce TI remains to be investigated by future studies. Fourth, the definition of high- and low-TI responders was limited to the FCs of ex vivo IL-1β production. In the future, an unbiased and generalized model should be established to explore individual response differences upon stimulations or vaccinations in TI studies. Finally, ligands-targets communication could only provide cell-cell interaction between two groups. However, we found that upregulated TIGs in both monocytes and CD8+ T cells were all enriched in the NK-cell-mediated cytotoxicity pathway, which may imply a more complicated network between monocyte CD8+ T cells and NK cells during TI. In addition, since multi-omics datasets such as epigenomics, metabolomics, and proteomics are available, integrating different layers of data could be powerful and may uncover deeper biological information.
Taken together, using an in vivo BCG-induced TI model, independent data-driven cross-check, and functional experimental validation, we have identified four distinctive TI transcriptional programs that characterize monocyte subpopulations in vivo. We unraveled and demonstrated the importance of IFN-γ-related pathways and STAT1 regulation for these identified trained monocyte subpopulations, and we reported defective induction of these programs in patients with severe sepsis. We believe that our findings will provide a crucial starting point for studying cell-type heterogeneity during TI and allow for a better understanding across different cell types and diseases.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Staphylococcus aureus | Clinical isolate | N/A |
| Biological samples | ||
| PBMC from BCG vaccinated subjects | Radboud University Medical Center | 300BCG |
| Chemicals, peptides, and recombinant proteins | ||
| Bacille Calmette-Guérin Vaccine | Intervax | Bulgaria strain |
| Lipopolysaccharide | Sigma-Aldrich | From E. coliserotype 055:B5, L2880 |
| Roswell Park Memorial Institute medium (RPMI) | Invitrogen | Cat# 22406031 |
| Ficoll-Paque | GE Healthcare | Cat# 17-1440-03 |
| Critical commercial assays | ||
| Chromium Next GEM Single Cell 3′ Library & Gel Bead Kit v3.1 | 10X Genomics | Cat# 1000121 |
| Chromium Next GEM Chip G Single Cell Kit | 10X Genomics | Cat# 1000120 |
| NovaSeq 6000 S4 Reagent Kit v1 (200 cycles) | Illumina | Cat# 20027466 |
| Buffy coats | Sanquin | E2824R00/B2825R00 |
| Pan monocyte isolation kit human | Miltenyi Biotec | Cat# 130-096-537 |
| Ruxolitinib | Invivogen | Cat# 941678-49-5 |
| Human IL-1β ELISA | R&D systems | Cat# DY201 |
| Deposited data | ||
| Single-cell RNA-seq sequencing | This paper | EGAS00001006990 |
| TIGEN | This paper | https://github.com/CiiM-Bioinformatics-group/TIGENS |
| Experimental models: Organisms/strains | ||
| 300BCG cohort (Human Functional Genomics project) | N/A | http://www.humanfunctionalgenomics.org |
| Software and algorithms | ||
| bcf2fastq | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html |
| CellRanger (v3.1.0) | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest/ |
| souporcell (v2.0) | Heaton et al.29 | https://github.com/wheaton5/souporcell |
| Seurat (v4.0.0) | Butler et al.30 | https://satijalab.org/seurat/index.html |
| SingleR (v1.4.1) | Aran et al.31 | https://github.com/dviraran/SingleR |
| ComplexHeatmap (v2.7.7) | Gu et al.32 | https://jokergoo.github.io/ComplexHeatmap-reference/book/ |
| ClusterProfiler (v3.18.1) | Yu et al.33 | https://github.com/YuLab-SMU/clusterProfiler |
| FUMA | Watanabe et al.34 | https://fuma.ctglab.nl |
| NicheNet (v1.0.0) | Browaeys et al.16 | https://github.com/saeyslab/nichenetr |
| Circlize (v0.4.12) | Gu et al.35 | https://jokergoo.github.io/circlize_book/book/ |
| SCENIC (v1.2.4) | Aibar et al.20 | https://github.com/aertslab/SCENIC |
| GENIE3 (v1.12.0) | Huynh-Thu et al.21 | https://github.com/aertslab/SCENIC |
| Cytoscape (v3.7.1) | Shannon et al.36 | https://cytoscape.org |
| MatrixEQTL (v2.3) | Shabalin et al.37 | http://www.bios.unc.edu/research/genomic_software/Matrix_eQTL/ |
| ggplot2 (v3.3.6) | Wickham et al.38 | https://ggplot2.tidyverse.org |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yang Li (yang.li@helmholtz-hzi.de).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
300BCG study design
To study the immunological effects of BCG vaccination, 325 healthy (44% male and 56% female) adult volunteers of Western European ancestry were included in the 300BCG cohort between April 2017 and June 2018 in the Radboud University Medical Center. Healthy volunteers were recruited using local advertisements and flyers in Nijmegen (the Netherlands) and were compensated for participation. After written informed consent was obtained, EDTA blood was collected, followed by administration of a standard dose of 0.1 mL BCG (BCG-Bulgaria, InterVax) intradermally in the left upper arm by a medical doctor. Vaccination of study participants was organized in batches of 6–16 subjects per day. Three months after BCG vaccination, additional blood samples were collected. The exclusion criteria comprised the use of systemic medication, except for oral contraceptives or acetaminophen, and antibiotics within three months before inclusion, previous BCG vaccination, history of tuberculosis, any febrile illness four weeks before and during participation, any vaccination within three months before participation, and a medical history of immunodeficiency. The fold change (FC) of IL-1β between T3m and T0 was used as a measure for BCG-induced TI. Subsequently, we selected 39 individuals at the extremes of this TI spectrum: high (FC ≥ 2 of S. aureus-induced IL-1β production; N = 19) and low (FC < 2; N = 20) responders.
Study approval
The 300BCG (NL58553.091.16) study was approved by the Arnhem-Nijmegen Medical Ethical Committee. Written informed consent was obtained before any research procedure was initiated. The study was performed in accordance with the declaration of Helsinki.
Method details
PBMC isolation and stimulation
Peripheral blood mononuclear cell (PBMC) isolation was performed by density centrifugation over Ficoll-Paque (GE healthcare, UK). Cells were washed twice in PBS and suspended in RPMI culture medium (Roswell Park Memorial Institute medium, Invitrogen, CA, USA) supplemented with 50 mg/mL gentamicin (Centrafarm), 2 mM glutamax (GIBCO), and 1mM pyruvate (GIBCO). In a total volume of 200 μL/well, 5×105 PBMCs were cultured in round bottom 96-well plates (Greiner) and stimulated ex vivo with 5 × 106 CFU/mL heat-killed S. aureus before vaccination and 3 months after vaccination. IL-1β production was measured after 24 h incubation (37°C, 5% CO2) in supernatants using commercial ELISA kits, in accordance with the manufacturer’s instructions, and the fold change in cytokine production (after vaccination compared to baseline) was used as a measurement of the magnitude of the trained immunity response and was used to identify low vs. high responders. In addition, isolated PBMCs were stimulated for 4 h with either RPMI (control) or LPS 10 ng/mL (serotype 055: B5; Sigma).
Single-cell library preparation and RNA-sequencing
In total, 156 samples were mixed into 32 pooled libraries. In each pool, an equal number of cells from 3 or 5 different donors were pooled together. Single cell gene expression libraries were generated on the 10x Genomics Chromium platform using the Chromium Next GEM Single Cell 3′ Library & Gel Bead Kit v3.1 and Chromium Next GEM Chip G Single Cell Kit (10x Genomics) according to the manufacturer’s protocol. Libraries were sequenced on a NovaSeq 6000 S4 flow cell using v1 chemistry (Illumina) with 28bp R1 and 90bp R2 run settings. These pools were sequenced in 3 batches in which conditions and timepoints were mixed to minimize potential batch effects.
Pilot validation dataset
Individuals from the 300BCG cohort were vaccinated in the morning with 0.1 mL of BCG (BCG vaccine strain Bulgaria; Intervax). PBMCs isolated from 3 healthy donors (19, 24, and 25 years of age, all men) before vaccination and 90 days after vaccination were stimulated ex vivo with RPMI medium (control) or 10 ng/mL LPS. In total, 12 samples (3 donors × 2 vaccination-status × 2 restimulation-status) were applied to 10× Genomics scRNA-seq in 1 batch. CellRanger v3.1.0 was used to process scRNA-seq of the in vivo study. To generate a digital gene expression matrix for each sample, we mapped their reads to the GRCh38 human reference genome and recorded the number of UMIs. UMI count matrices were then imported to R/Seurat package for downstream analyses.
Quantification and statistical analysis
Data pre-processing and demultiplexing
In each library, bcf2fastq Conversion Software (Illumina) was used to convert BCL files to FASTQ files, along with sample sheet including 10x barcodes. The proprietary 10x Genomics STAR in CellRanger pipeline (v3.1.0) was used to align read data to GRCh38/b38 (downloaded from 10X Genomics). We set the parameter of setting of expected cells to 2000. Finally, a gene expression matrix was generated which recorded UMIs count of each gene in each cell.
In each library, duplicates were removed using pre-mapped bam files, and cells were assigned to their individuals of origin using souporcell (v2.0)29 and, 91.98% cells were retrieved for downstream analysis. Since T0 data of one individual contained few cells at T3m, we discarded it from downstream analysis. Subsequently, the souporcell tool was employed to cluster cells based on allele counts using hierarchical clustering strategy and assign individuals to clusters. Further, the genotype dataset of each individual was used to cross-check the consistency between the assigned individuals via souporcell and individual phenotypes id.
Data quality control
Cells with: 1) MT-genes percentage more than 25% and 2) number of detected genes less than 100 or more than 5,000 were removed. Also, only genes expressed in at least 5 cells (leaving ∼200k cells and 21,975 genes) were considered for the downstream analysis.
Data integration and clustering
In total, cells were collected from 3 batches in 10, 10, 12 pools, respectively. In each pool, 5 samples from different individuals at different timepoints with different stimulations were pooled randomly. Seurat (v4.0.0)30 package of R (v4.0.2) was used to integrate and analyze all data together. In brief, at first, for each independent dataset from each pool, UMI counts were normalized (log(10000x + 1)) and the top 2000 variable features were selected using NormalizeData and FindVariableFeatures function with default parameters, respectively. Later, repeated features were identified across all independent datasets, and utilized for scaling and PCA analysis on each dataset. Instead of canonical correlation analysis, in order to speed up calculation for integration and avoid overcorrection,30 reciprocal PCA was used via the SelectIntegrationFeatures function to detect integration anchors. Followed by IntegrateData, the integrated dataset was scaled and clustered using default parameters. Cell clusters were further annotated combining the results from SingleR31 package of R (HumanPrimaryCellAtlasData, BlueprintEncodeData, MonacoImmuneData, DatabaseImmuneCellExpressionData and NovershternHematopoieticData were selected as reference) and the expression level of known cell markers (CD4+ T cells: IL7R, CD3D; CD8+ T cells: CD8A, CD8B; Monocytes: CD14, IL1B; NK cells: GNLY, NKG7; B cells: CD79A; mDC: HLA-DPA1, HLA-DPB1; pDC: CTSC, TSPAN13; Platelet: PPBP). We also found a group of T cells with a high expression of HSPA1A, HSPA1B and labeled as HSP(T). After removing undefined cells, the remaining cells (CD4+ T cells = 65170, CD8+ T cells = 31926, Monocytes = 31601, NK cells = 28796, B cells = 21356, mDC = 2211, pDC = 1223) across all individuals and conditions were used for downstream analysis.
Unsupervised clustering for trained monocytes
FindVariableFeatures function of the Seurat was employed to detect the top variable genes in monocytes and perform unsupervised clustering. Initially, we calculated the mean value of each gene at T0 in each individual. Then the expression value of each gene in each monocyte of each individual at T3m was subtracted by the corresponding mean value of each gene of monocytes of each individual at T0 and define the difference as "TI changes". Later, based on TI changes, unsupervised clustering employing Spearman correlation in ComplexHeatmap package (v2.7.7)32 was performed to estimate the similarity between cells and, subsequently, monocytes were clustered into 4 groups using k-means. Then we extracted monocytes from PBMC and mapped the clustered sub-populations to extracted monocytes for visualization.
Differentially expressed genes tests and enrichment analysis
Differential gene expression analysis was performed using FindMarkers/FindAllMarkers function with Wilcoxon Rank-Sum test in Seurat. Genes expressed in at 10% cells and having p_val_adj <0.05 (Bonferroni correction) were considered significant. In each identified main cell type, the BCG response genes were compared between T3m_RPIM and T0_RPIM. For each main cell type and monocyte sub-population, TIGs were defined based on the changes between T3m_LPS and T0_LPS. Additionally, the same analyses were also done for high- and low-responders separately.
Enrichment analysis
For the enrichment test, significant gene sets were subjected to enrichGO and enrichKEGG function (pAdjustMethod = "BH", qvalueCutoff = 0.05) of ClusterProfiler (v3.18.1)33 package of R, separately. In order to focus on the immune-related pathways, enrichment of genes that were upregulated in high-responders but downregulated in low-responders in monocytes and disease association analysis, GENE2FUNC function in FUMA34 was used and MsigDB H (hallmark gene sets) database39 was set as background. Pathways with BH correction <0.05 were regarded as significant.
Cell-cell interaction analysis
NicheNet (v1.0.0)16 was used to infer cell-cell interaction in the T3m_LPS based on aggregated prior information of ligand-receptor, signaling and gene regulatory data. In brief, initially, communications analysis was performed between two main cell types having the largest numbers of TIGs, i.e., monocytes and CD8+ T cells, and one-time monocytes and CD8+ T cells were set as ''receiver', and ''sender', respectively and other time vice-versa. For the communications between monocyte sub-populations and all the cell types, we set each sub-population as ''sender', separately. In each analysis, TIGs were regarded as sets of interest (targets) and ligands were identified from senders. The genes expressed in at least 10% cells from senders were considered as background genes. Top 20 ligands were selected based on the Pearson correlation coefficients with TIGs. Cell-cell interaction was visualized using circlize (v0.4.12).35
Transcription factors of each sub-population and gene regulatory network reconstruction
For each monocyte sub-population, SCENIC (v1.2.4)20 was used to find activated regulons of TIGs separately. Specifically, RcisTarget (v1.10.0) was applied to identify cis-regulatory motifs of TIGs using hg38_refseq-r80_10kb_up_and_down_tss.mc9nr.feather database. GENIE3 (v1.12.0)21 was used to reconstruct the gene regulatory network while targets were TIGs. Further, to select the top TFs based on ranked TF-target links obtained from GENIE3, for each TF, we labeled each link with the specific TF as 1 and others as 0 and calculated AUC and AUPR value. Then we performed random shuffles for 500 times and TFs with AUC-pvalue <0.05 and AUPR-pvalue <0.05 were considered as potential TFs. Cytoscape36 was used for gene regulatory (between identified TFs and TIGs) network visualization.
Gene set enrichment analyses for disease association study
To understand the association between TIGs and immune diseases, gene set enrichment analysis of TIGs was performed using the GSEA function in ClusterProfiler package33 of R' 'TERM2GENE' was the ranked TIGs based on their avg_log2fc (calculated by Seurat) in each sub-population and '''geneList' was the differentially expressed genes between patients and healthy control in each disease group.
Genotype data pre-processing
DNA samples of individuals were genotyped using the commercially available SNP chip, Infinium Global Screening Array MD v1.0 from Illumina. optiCall 0.7.040 with default settings was used for genotype calling. Genetic variants with a call rate ≤0.01 were excluded, as were variants with Hardy-Weinberg equilibrium (HWE) ≤ 0.0001, and minor allele frequency (MAF) ≤ 0.001. The strands and variants identifiers were aligned to the 1000 Genomes reference panel using Genotype Harmonizer.41 One sample was excluded from the pre-imputed dataset due to high relatedness. We then imputed the samples on the Michigan imputation server using the human reference consortium (HRC r1.1 2016)42 as a reference panel. Data were phased using Eagle v2.3, and we filtered out genetic variants with an R2 < 0.3 for imputation quality. We identified and excluded 17 genetic outliers, and selected 4,296,841 SNPs with MAF 5% for follow-up QTL mapping.
Association with cytokine QTL
TI cytokine QTL analysis was performed using MatrixEQTL37 package of R. After filtering outliers, 278 individuals were left. The changes of TI associated cytokines, i.e. IL-1β, between T3m and T0 with S. aureus stimulation after 24 h, were only considered. Prior to analysis, cytokine measurements were log2-transformed using the Shapiro-Wilk normality test for normal distribution check and SNPs with MAF >0.05 were removed. Age, sex and cell counts were considered as covariates in the linear model. TIGs within ± 250kb of TI cytokine-QTLs were selected as being regulated. Fisher’s exact test (two-sided, p value <0.05) was used to identify which monocyte sub-population is significantly correlated with TI.
Independent cohort validation of sub-populations identification
Cells with: 1) MT-genes percentage more than 25% and 2) number of detected genes less than 100 or more than 5,000 were removed. UMI counts were normalized (log(10000x + 1)) and the top 2000 variable features were selected using NormalizeData and FindVariableFeatures function with default parameters, respectively. PCA was performed based on the 2,000 most variable features identified in Seurat. The cells were then clustered using the Louvain algorithm based on the first 20 PCA dimensions with a resolution of 0.7. For 2D data visualization, we performed UMAP also based on the first 20 PCA dimensions. TI markers of each identified sub-population were used as features. Each feature in each cell in T3m_LPS was subtracted by the mean value of the corresponding gene in the corresponding individual in T0_LPS. Spearman correlation was performed to calculate the similarity between cells in T3m_LPS and k-means was used to cluster cells.
STAT1 validation experiment
For the STAT1 validation experiment, we used a previously optimized in vitro TI model. PBMCs were isolated as described above, but from buffy coats (Sanquin) instead of fresh blood. Untouched monocytes were isolated by negative magnetic separation (Pan monocyte isolation kit human, Miltenyi Biotec). All monocyte culture was performed in RPMI (described above) further supplemented with 10% human pooled serum. Briefly, the monocytes were stimulated for 24 h with RPMI or BCG for 24 h, in the presence of 4 μM Ruxolitinib or a carrier control (DMSO). The cells were washed and incubated for 5 days with an intermittent medium refresh on day 3. On day 6, monocytes were re-stimulated with LPS for 24 h. Subsequently, cytokine production was assessed using ELISA as described above.
Data visualization
In general, the R package Seurat and ggplot2 (v3.3.6)38 were used to plot figures.
Additional resources
Clinical trial registry (NCT04990232) for the human sepsis trial data described in the manuscript can be found here (https://clinicaltrials.gov/ct2/show/NCT04990232).
Acknowledgments
We thank the volunteers of the 300BCG cohort for their participation in this study. This work was supported with funds provided by ERC starting grant 948207, NWO ASPASIA, and Radboud University Medical Center Hypatia grant to Y.L., ERC advanced grant 833247 and Spinoza grant of the Netherlands Organization for Scientific research to M.G.N., Helmholtz Initiative and Networking Fund to C.J.X. (1800167), China Scholarship Council, a PhD scholarship to W.L., and Singh-Chhatwal-Postdoctoral Fellowship Program to Z.Z.
Author contributions
Conceptualization, Y.L. and M.G.N.; methodology, S.J.C.F.M.M.; formal analysis, W.L.; investigation, S.J.C.F.M.M., V.A.C.M.K., R.J.R., L.C.J.d.B., V.P.M., and K.E.v.M.; resources, B.Z., J.F., Z.Z., A.A., L.Z., R.v.C., C.-J.X., and L.A.B.J.; validation, W.L. and R.J.R.; supervision, Y.L. and M.G.N.; writing – original draft, W.L., S.J.C.F.M.M., Y.L., and M.G.N.; writing– review & editing, all authors.
Declaration of interests
M.G.N. and L.A.B.J. are scientific founders of TTxD and Lemba Therapeutics. M.G.N. is a founder of BioTRIP.
Published: May 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112487.
Supplemental information
Data and code availability
-
•
Single-cell RNA-seq data has been deposited at European Genome-phenome Archive (EGA), which is hosted by the EBI and the CRG, under accession number EGAS00001006990 (https://ega-archive.org/studies/EGAS00001006990).
-
•
All original code has been deposited at https://github.com/CiiM-Bioinformatics-group/TIGENS and is publicly available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Benn C.S., Netea M.G., Selin L.K., Aaby P. A small jab - a big effect: nonspecific immunomodulation by vaccines. Trends Immunol. 2013;34:431–439. doi: 10.1016/j.it.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 2.Netea M.G., Joosten L.A.B., Latz E., Mills K.H.G., Natoli G., Stunnenberg H.G., O'Neill L.A.J., Xavier R.J. Trained immunity: a program of innate immune memory in health and disease. Science. 2016;352:aaf1098. doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Neill L.A.J., Netea M.G. BCG-induced trained immunity: can it offer protection against COVID-19? Nat. Rev. Immunol. 2020;20:335–337. doi: 10.1038/s41577-020-0337-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Netea M.G., Domínguez-Andrés J., Barreiro L.B., Chavakis T., Divangahi M., Fuchs E., Joosten L.A.B., van der Meer J.W.M., Mhlanga M.M., Mulder W.J.M., et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020;20:375–388. doi: 10.1038/s41577-020-0285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quintin J., Cheng S.C., van der Meer J.W.M., Netea M.G. Innate immune memory: towards a better understanding of host defense mechanisms. Curr. Opin. Immunol. 2014;29:1–7. doi: 10.1016/j.coi.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 6.Kleinnijenhuis J., Quintin J., Preijers F., Joosten L.A.B., Ifrim D.C., Saeed S., Jacobs C., van Loenhout J., de Jong D., Stunnenberg H.G., et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. USA. 2012;109:17537–17542. doi: 10.1073/pnas.1202870109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moorlag S.J.C.F.M., Rodriguez-Rosales Y.A., Gillard J., Fanucchi S., Theunissen K., Novakovic B., de Bont C.M., Negishi Y., Fok E.T., Kalafati L., et al. BCG vaccination induces long-term functional reprogramming of human neutrophils. Cell Rep. 2020;33:108387. doi: 10.1016/j.celrep.2020.108387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng S.C., Quintin J., Cramer R.A., Shepardson K.M., Saeed S., Kumar V., Giamarellos-Bourboulis E.J., Martens J.H.A., Rao N.A., Aghajanirefah A., et al. mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arts R.J.W., Carvalho A., La Rocca C., Palma C., Rodrigues F., Silvestre R., Kleinnijenhuis J., Lachmandas E., Gonçalves L.G., Belinha A., et al. Immunometabolic pathways in BCG-induced trained immunity. Cell Rep. 2016;17:2562–2571. doi: 10.1016/j.celrep.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B., Moorlag S.J., Dominguez-Andres J., Bulut Ö., Kilic G., Liu Z., van Crevel R., Xu C.J., Joosten L.A., Netea M.G., Li Y. Single-cell RNA sequencing reveals induction of distinct trained-immunity programs in human monocytes. J. Clin. Invest. 2022;132:e147719. doi: 10.1172/JCI147719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koeken V.A., de Bree L.C.J., Mourits V.P., Moorlag S.J., Walk J., Cirovic B., Arts R.J., Jaeger M., Dijkstra H., Lemmers H., et al. BCG vaccination in humans inhibits systemic inflammation in a sex-dependent manner. J. Clin. Invest. 2020;130:5591–5602. doi: 10.1172/JCI133935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marco J.M. WU Vienna University of Economics and Business; 2014. DirichletReg: Dirichlet Regression for Compositional Data in R, Research Report Series. [Google Scholar]
- 13.Ivashkiv L.B., Donlin L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014;14:36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crabtree J.N., Caffrey D.R., de Souza Silva L., Kurt-Jones E.A., Dobbs K., Dent A., Fitzgerald K.A., Golenbock D.T. Lymphocyte crosstalk is required for monocyte-intrinsic trained immunity to Plasmodium falciparum. J. Clin. Invest. 2022;132:e139298. doi: 10.1172/JCI139298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaufmann E., Sanz J., Dunn J.L., Khan N., Mendonça L.E., Pacis A., Tzelepis F., Pernet E., Dumaine A., Grenier J.C., et al. BCG educates hematopoietic stem cells to generate protective innate immunity against Tuberculosis. Cell. 2018;172:176–190.e19. doi: 10.1016/j.cell.2017.12.031. [DOI] [PubMed] [Google Scholar]
- 16.Browaeys R., Saelens W., Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods. 2020;17:159–162. doi: 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- 17.Robinson C.M., Nau G.J. Interleukin-12 and interleukin-27 regulate macrophage control of Mycobacterium tuberculosis. J. Infect. Dis. 2008;198:359–366. doi: 10.1086/589774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fielding A.K. JAK-ing up treatment for CRLF2-R ALL. Blood. 2022;139:645–646. doi: 10.1182/blood.2021014196. [DOI] [PubMed] [Google Scholar]
- 19.Bhat P., Leggatt G., Waterhouse N., Frazer I.H. Interferon-gamma derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis. 2017;8:e2836. doi: 10.1038/cddis.2017.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aibar S., González-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., Rambow F., Marine J.C., Geurts P., Aerts J., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14:1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huynh-Thu V.A., Irrthum A., Wehenkel L., Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS One. 2010;5:e12776. doi: 10.1371/journal.pone.0012776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qing Y., Stark G.R. Alternative activation of STAT1 and STAT3 in response to interferon-gamma. J. Biol. Chem. 2004;279:41679–41685. doi: 10.1074/jbc.M406413200. [DOI] [PubMed] [Google Scholar]
- 23.Berry M.P.R., Graham C.M., McNab F.W., Xu Z., Bloch S.A.A., Oni T., Wilkinson K.A., Banchereau R., Skinner J., Wilkinson R.J., et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Inge G., Valerie A.C.M.K., Athanasios K., Wenchao L., Nikolaos A., Bowen Z., Georgia D., Chengjian X., Evangelos J., Giamarellos -B., et al. Single-cell transcriptomics differentiates hyperinflammation from immune paralysis in sepsis patients. medRxiv. 2023 https://medrxiv.org/cgi/content/short/2023.03.17.23287390v1 Preprint at. [Google Scholar]
- 25.Reyes M., Filbin M.R., Bhattacharyya R.P., Billman K., Eisenhaure T., Hung D.T., Levy B.D., Baron R.M., Blainey P.C., Goldberg M.B., Hacohen N. An immune-cell signature of bacterial sepsis. Nat. Med. 2020;26:333–340. doi: 10.1038/s41591-020-0752-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quintin J., Saeed S., Martens J.H.A., Giamarellos-Bourboulis E.J., Ifrim D.C., Logie C., Jacobs L., Jansen T., Kullberg B.J., Wijmenga C., et al. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host Microbe. 2012;12:223–232. doi: 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ball G.F., Faris P.L., Hartman B.K., Wingfield J.C. Immunohistochemical localization of neuropeptides in the vocal control regions of two songbird species. J. Comp. Neurol. 1988;268:171–180. doi: 10.1002/cne.902680204. [DOI] [PubMed] [Google Scholar]
- 28.de Bree L.C.J., Mourits V.P., Koeken V.A., Moorlag S.J., Janssen R., Folkman L., Barreca D., Krausgruber T., Fife-Gernedl V., Novakovic B., et al. Circadian rhythm influences induction of trained immunity by BCG vaccination. J. Clin. Invest. 2020;130:5603–5617. doi: 10.1172/JCI133934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heaton H., Talman A.M., Knights A., Imaz M., Gaffney D.J., Durbin R., Hemberg M., Lawniczak M.K.N. Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes. Nat. Methods. 2020;17:615–620. doi: 10.1038/s41592-020-0820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aran D., Looney A.P., Liu L., Wu E., Fong V., Hsu A., Chak S., Naikawadi R.P., Wolters P.J., Abate A.R., et al. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 33.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Watanabe K., Taskesen E., van Bochoven A., Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017;8:1826. doi: 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gu Z., Gu L., Eils R., Schlesner M., Brors B. Circlize Implements and enhances circular visualization in R. Bioinformatics. 2014;30:2811–2812. doi: 10.1093/bioinformatics/btu393. [DOI] [PubMed] [Google Scholar]
- 36.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shabalin A.A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wickham H. Springer-Verlag; New York: 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- 39.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shah T.S., Liu J.Z., Floyd J.A.B., Morris J.A., Wirth N., Barrett J.C., Anderson C.A. optiCall: a robust genotype-calling algorithm for rare, low-frequency and common variants. Bioinformatics. 2012;28:1598–1603. doi: 10.1093/bioinformatics/bts180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deelen P., Bonder M.J., van der Velde K.J., Westra H.J., Winder E., Hendriksen D., Franke L., Swertz M.A. Genotype harmonizer: automatic strand alignment and format conversion for genotype data integration. BMC Res. Notes. 2014;7:901. doi: 10.1186/1756-0500-7-901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCarthy S., Das S., Kretzschmar W., Delaneau O., Wood A.R., Teumer A., Kang H.M., Fuchsberger C., Danecek P., Sharp K., et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016;48:1279–1283. doi: 10.1038/ng.3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Single-cell RNA-seq data has been deposited at European Genome-phenome Archive (EGA), which is hosted by the EBI and the CRG, under accession number EGAS00001006990 (https://ega-archive.org/studies/EGAS00001006990).
-
•
All original code has been deposited at https://github.com/CiiM-Bioinformatics-group/TIGENS and is publicly available as of the date of publication.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.