

Abstract
Because of their rapid tumor accumulation and normal tissue clearance, ~15-kDa single domain antibody fragments (sdAbs, also known as nanobodies) are an attractive vehicle for developing targeted radiotherapeutics labeled with the 7.2-h α-particle emitting radiohalogen 211At. We have developed a site-specific 211At-labeled prosthetic agent — iso-[211At]AGMB-PODS — that combines a functionality for residualizing radioactivity in tumor cells with a phenyloxadiazolyl methylsulfone (PODS) moiety for linking it to an sdAb. This reagent was evaluated for thiol-selective conjugation to anti-HER2 5F7 sdAb modified to have a C-terminus GGC tail for this purpose. The iso-[211At]AGMB-PODS-5F7GGC conjugate and its 131I-labeled analogue were synthesized in good radiochemical yields with retention of high binding affinity and immunoreactivity. The PODS radioconjugates exhibited high specific binding, internalization and intracellular trapping of radioactivity on HER2-positive BT474 breast carcinoma cells in vitro. Iso-[211At]AGMB-PODS-5F7GGC displayed considerably higher in vitro stability in the presence of cysteine and HSA compared with the maleimide analogue, [211At]MEAGMB-5F7GGC, excellent tumor uptake and high in vivo stability. Superior tumor-to-kidney activity ratios were seen for iso-[211At]AGMB-PODS-5F7GGC and its radioiodinated analogue compared with [177Lu]Lu-DOTA-PODS-5F7GGC. These results suggest that iso-[211At]AGMB-PODS-5F7GGC warrants further evaluation as a targeted α-particle emitting agent for the treatment of HER2-expressing malignancies.
INTRODUCTION
Single domain antibody fragments — sdAbs, also known as VHH molecules and nanobodies — are a promising platform for the selective delivery of radionuclides to cancer cells for nuclear imaging and targeted radionuclide therapy (TRT). Compared with full-length mAbs, sdAbs have many attractive features, some related to their tenfold smaller size (i.e. rapid tumor penetration and normal tissue clearance) and others related to their biological characteristics (i.e. high binding affinity, ease of production chemical, and thermal stability) (1). The radiohalogenation of sdAbs is being pursued with equal or perhaps greater vigor than their radiometallation (2). To wit, the first clinical translation of a radiolabeled sdAb with TRT intent was the recent pilot study evaluating 131I-GMIB-anti-HER2-VHH1 in healthy volunteers and patients with HER2-expressing cancers (3). Work in our laboratory has focused on the development of radiohalogenated conjugates of 5F7 (4) and VHH_1028 (5) - sdAbs that target a trastuzumab-competitive epitope on HER2 extracellular Domain IV and exhibit more extensive internalization and higher HER2 affinity than VHH1. These properties could be advantageous for TRT, and the therapeutic efficacy of iso-[131I]SGMIB-VHH_1028 has recently been demonstrated in two murine models bearing HER2-expressing xenografts (5).
The use of sdAbs for TRT with the α-particle emitting radiohalogen 211At is particularly attractive because of the excellent alignment of the radionuclide’s 7.2-h physical half-life with the pharmacokinetic profile of sdAbs. Moreover, the short tissue range and high potency of α-particles will be of particular value in metastatic disease settings where sdAbs are most likely to have therapeutic impact (1, 2). When labeled with the iso-[211At]SAGMB residualizing prosthetic agent, 5F7 sdAb displayed high tumor uptake, prolonged tumor retention, fast clearance from normal tissues, and excellent in vivo stability against deastatination (6). The in vivo properties of iso-[211At]SAGMB-5F7 compared favorably with those reported for anti-HER2 sdAbs labeled with the α-particle emitting radiometals 213Bi (7) and 225Ac (8). In a recent therapy study, iso-[211At]SAGMB 5F7 and VHH_1028 conjugates showed excellent therapeutic efficacy at a single dose against HER2-positive BT474 breast carcinoma xenografts (9); complete tumor regression was observed in the majority of treated mice with no overt signs of toxicity observed at 200 days post-treatment.
Although the results discussed above are encouraging, one drawback of using radiolabeling prosthetic groups like iso-[211At]SAGMB that contain active esters is that they can react with multiple lysines within the sdAb, producing a mixture of radioconjugates with different properties. The heterogeneity and irreproducibility inherent to random labeling are significant hurdles to the effective deployment of radioimmunoconjugates, particularly at the higher radioactivity levels required for clinical TRT. Indeed, both the site of conjugation and the number of modifications per molecule can have a profound impact on the pharmacokinetic profile, affinity, and therapeutic efficacy of an immunoconjugate (10–13). This is especially true in the context of sdAbs given their small size.
The modification of a single location within a biomolecule via site-specific radiolabeling represents an attractive way to circumvent these issues. The most commonly used approach for site-specific conjugation utilizes the Michael addition reaction between cysteine sulfhydryl groups and maleimide groups. Unfortunately, the succinimidyl thioether linkage that is formed is susceptible to retro-Michael reactions in vivo (14). In the context of radioimmunoconjugates, this can lead to the escape of the labeled prosthetic group and/or the exchange labeling of endogenous thiol-bearing species (15). Yet despite the potential deleterious effects of these processes on normal tissue radioactivity levels, maleimides continue to be used with mixed results, potentially reflecting the influence of the chemical environment on the stability of the maleimide-thiol linkage (14). Of direct relevance to the current study, we recently evaluated the site-specific maleimide-mediated radioiodination of 5F7 at an engineered GGC site at the C-terminus of the sdAb (Figure 1) (16). At early timepoints, the in vivo behavior of the resultant radioimmunoconjugate was comparable to that of native 5F7 that had been randomly radiolabeled using a lysine-based method. However, at 24 h, the maleimide-based radioimmunoconjugate yielded lower tumor-to-tissue ratios for the kidney, spleen, and liver. Moreover, the use of maleimide-based approaches for the 211At-labeling of both 2Rs15d (aka VHH1) (17) and an intact mAb (18) yielded 211At-labeled proteins that were highly susceptible to loss of label in vivo.
Figure 1.
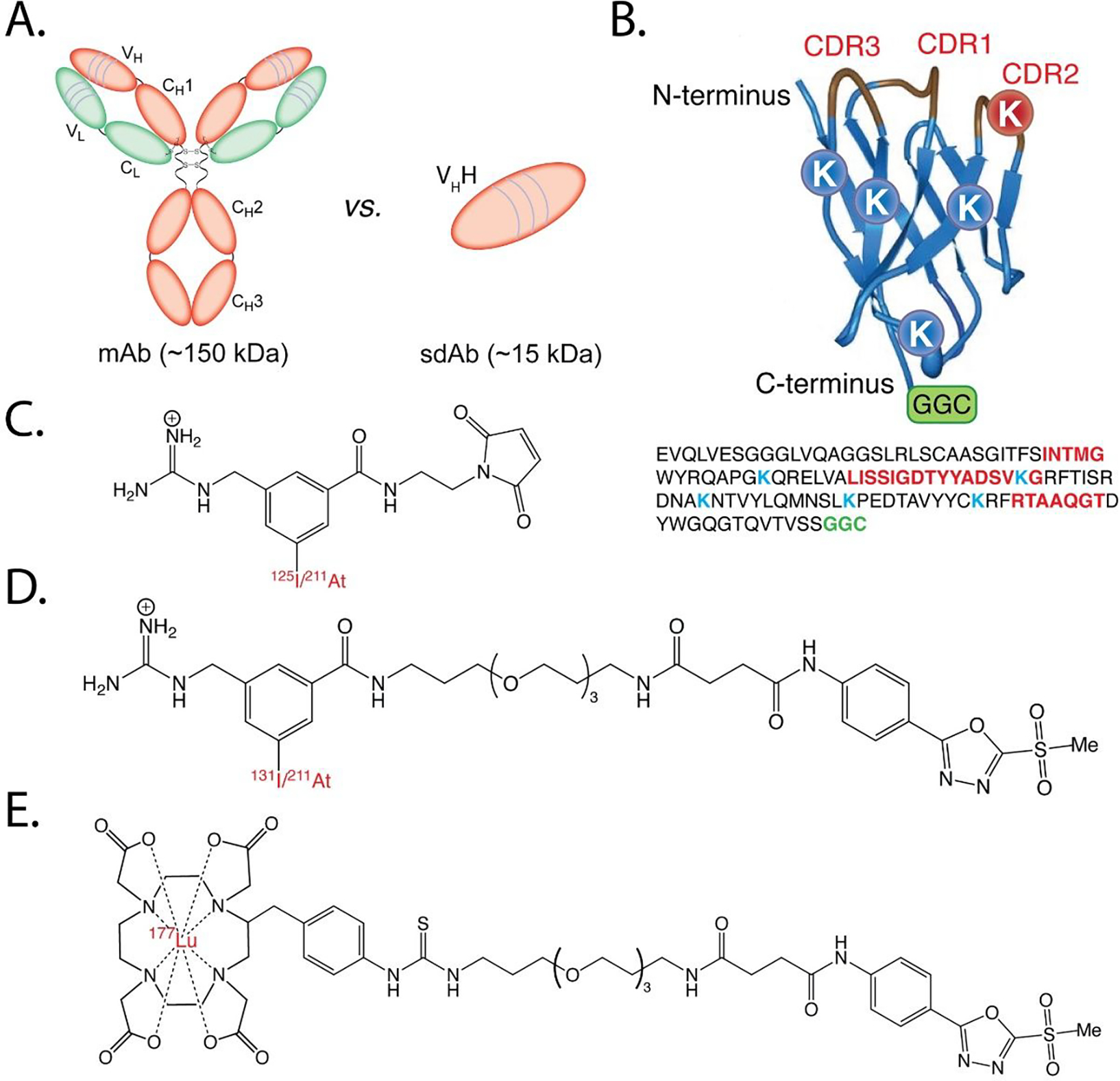
(A) Schematic showing the structures and relative sizes of a monoclonal antibody (mAb) and a single domain antibody (sdAb); (B) ribbon diagram (top) and amino acid sequence (bottom) of 5F7GGC illustrating the positions of the complementarity-determining regions (CDRs), lysine residues (K), and C-terminal GGC conjugation site; (C) structures of [211At]MEAGMB and [125I]MEGMIB; (D) structures of iso-[131l]GMIB-PODS and iso-[211At]AGMB-PODS; (E) structure of [177Lu]Lu-DOTA-PODS
An attractive alternative to maleimide-mediated site-specific conjugation originally described almost a decade ago is predicated on the modification of cysteine residues with bifunctional probes bearing methylsulfonylbenzothiazole groups. This strategy resulted in protein conjugates with higher stability in human plasma than those synthesized via maleimide-thiol chemistry (19), and a subsequent study demonstrated that the methylsulfonylbenzothiazole-thiol linkage provided superior stability even at sites known to be labile after maleimide-thiol conjugations (20). More recently, a phenyloxadiazolyl methylsulfone (PODS) reagent developed for the site-specific cysteine labeling of intact mAbs with radiometals (89Zr, 177Lu) was shown to produce radioimmunoconjugates with markedly superior properties compared with analogs created using maleimide-based probes (21–23). Based on these encouraging results, we have evaluated the possibility of extending the PODS strategy to radiohalogenation and sdAbs, a pair of changes that offer distinct challenges compared with radiometallation and mAbs, respectively. More specifically, we have synthesized PODS-bearing analogues of our residualizing iso-[131ISGMIB and iso-[211At]SAGMB reagents and evaluated their potential utility for labeling a variant of 5F7 sdAb bearing an engineered C-terminus GGC tail (Figure 1). Finally, we have performed a head-to-head comparison between iso-[125I]GMIB-PODS-5F7GGC and 177Lu-labeled PODS 5F7GGC in athymic mice bearing subcutaneous HER2-expressing BT474 human breast carcinoma xenografts.
RESULTS
Chemistry and Radiochemistry.
The radiosyntheses are summarized in Schemes 1–3, while the syntheses of Boc2-iso-SGMIB, Boc2-iso-SGMTB, and PODS have been reported before (16, 21, 22). The synthesis of DOTA-PODS followed a modified method (21) using p-SCN-Bn-DOTA (Macrocyclics, Inc, Texas). The conjugations of Boc2-iso-SGMIB and Boc2-iso-SGMTB to PODS were performed under basic conditions using a common protocol. The products — Boc2-iso-GMIB-PODS and Boc2-iso-GMTB-PODS — were isolated via preparative RP-HPLC in reasonable yields. Iso-GMIB-PODS-5F7GGC, DOTA-PODS-5F7GGC, and Lu-DOTA-PODS-5F7GGC were synthesized in similar yield (~76%). The purities of these immunoconjugates were determined using GP HPLC and all were >95%. Their molecular weights were determined via LC-MS and were 13795.4 (13795.2 calc.), 14046.0 (14046.2 calc.), and 14216.7 (14217.2 calc.), respectively. Non-radioactive iso-SGMIB-PODS was used as a standard for the identification of its radiolabeled analogue, iso-[131I]GMIB-PODS. The radiochemical yield (RCY) and radiochemical purity (RCP) of Boc2-iso-[131I]GMIB-PODS were 70 ± 8% (n = 11) and >99% (RP-HPLC), respectively.
Scheme 1.
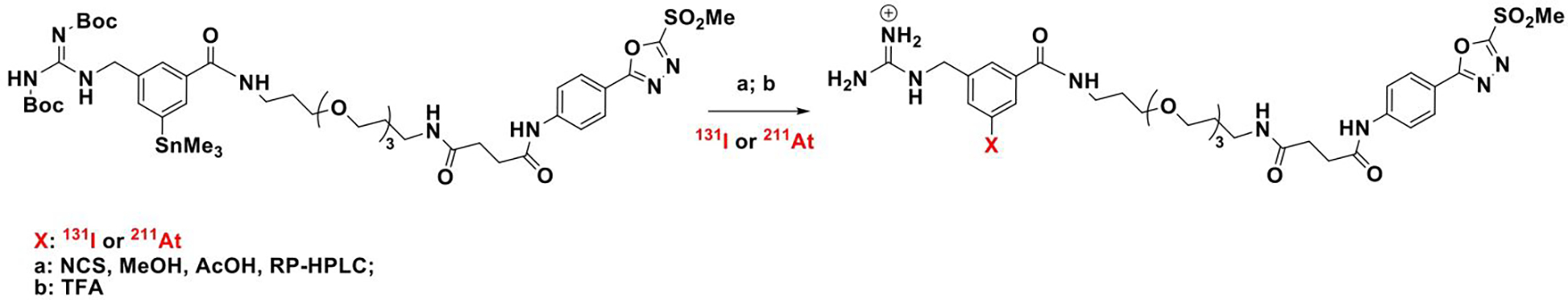
Synthesis of iso-[131l]GMIB-PODS and iso-[211At]AGMB-PODS
Scheme 3.
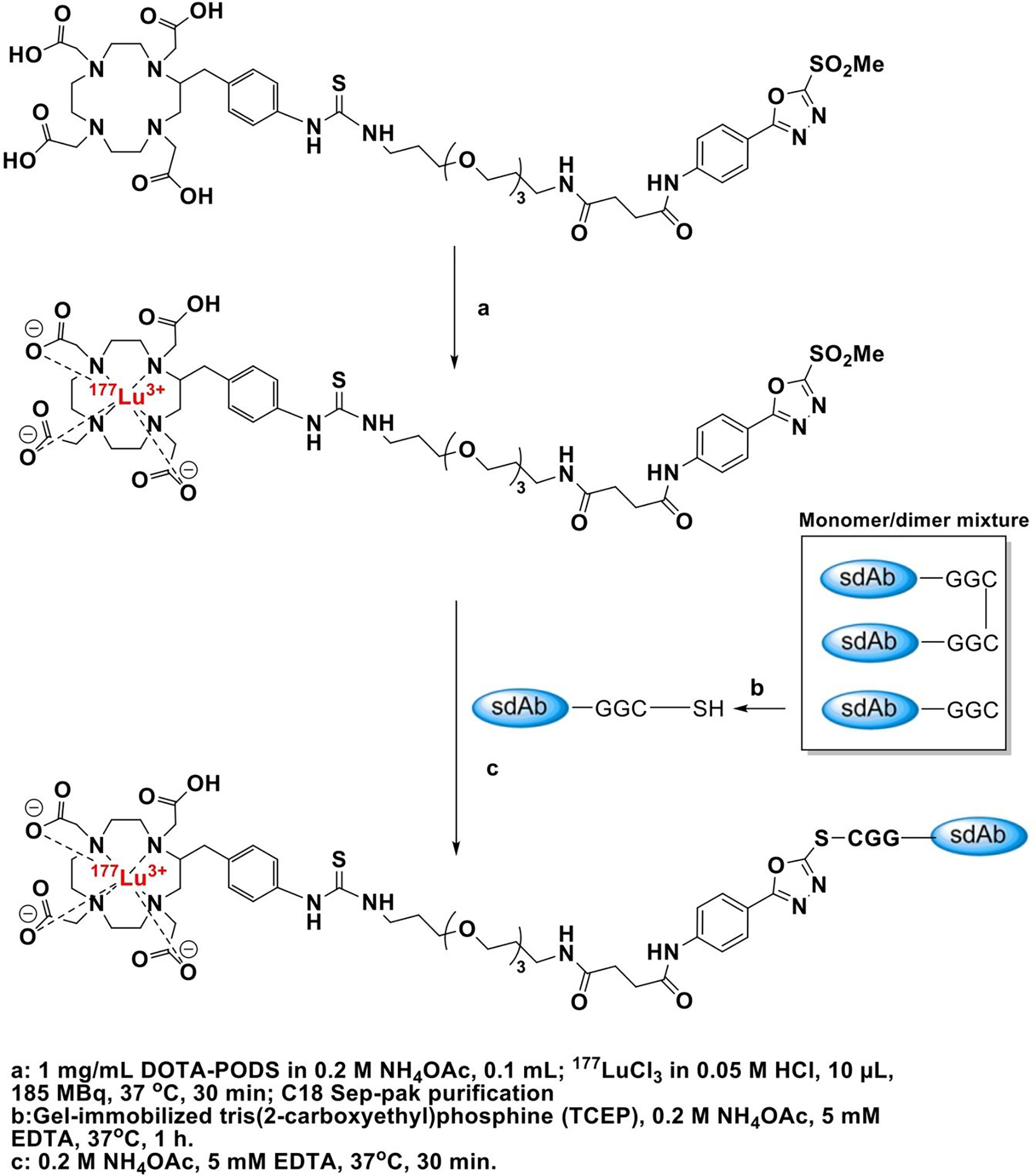
Synthesis of [177Lu]Lu-DOTA-PODS-5F7GGC
The conjugation of iso-[131I]GMIB-PODS to monomeric 5F7GGC was accomplished with a RCY of 58 ± 9% (n=11); the RCP of iso-[131I]GMIB-PODS-5F7GGC determined by SDS-PAGE and GP-HPLC was >99%. The synthesis of iso-[211At]AGMB-PODS was performed with a RCY of 66 ± 5% (n = 6), and the RCP was >99% for each synthesis. The RCY for the conjugation of iso-[211At]AGMB-PODS with 5F7GGC was 64 ± 7% (n = 6), and the RCP of the radiolabeled sdAb determined by SDS-PAGE and GP-HPLC was >99%. [177Lu]Lu-DOTA-PODS was synthesized in almost quantitative yields, and [177Lu]Lu-DOTA-PODS-5F7GGC was synthesized in 35 ± 15% RCY (n = 2) and >99% RCP as determined by SDS-PAGE and GP-HPLC.
Characterization of sdAb Conjugates.
Surface plasmon resonance (SPR) revealed binding constants (Kd) of 0.10, 0.19, and 0.09 nM for iso-GMIB-PODS-5F7GGC, DOTA-PODS-5F7GGC and Lu-DOTA-PODS-5F7GGC, respectively, with recombinant HER2-Fc protein (Figure 2). The immunoreactive fraction (IRF), determined by Lindmo assay (24), was 84.0 ± 1.8% (n = 2) for iso-[131I]GMIB-PODS-5F7GGC, 85.8 ± 2.1% (n = 2) for iso-[211At]AGMB-PODS-5F7GGC, 69.0% (n = 1) for [211At]MEAGMB-5F7GGC, and 71.2% (n = 1) for [177Lu]Lu-DOTA-PODS-5F7GGC. Saturation binding assays using the HER2-expressing SKOV-3 and BT474 cell lines gave Kd values of 3.3 ± 0.5 nM and 5.9 ± 0.8 nM, respectively, for iso-[131I]GMIB-PODS-5F7GGC (Figure 3). Using the BT474 cell line, Kd values of 4.7 ± 0.8, 3.4 ± 0.6, and 5.6 ± 0.9 nM were obtained for iso-[211At]AGMB-PODS-5F7GGC, [211At]MEAGMB-5F7GGC, and [177Lu]Lu-DOTA-PODS-5F7GGC, respectively.
Figure 2.
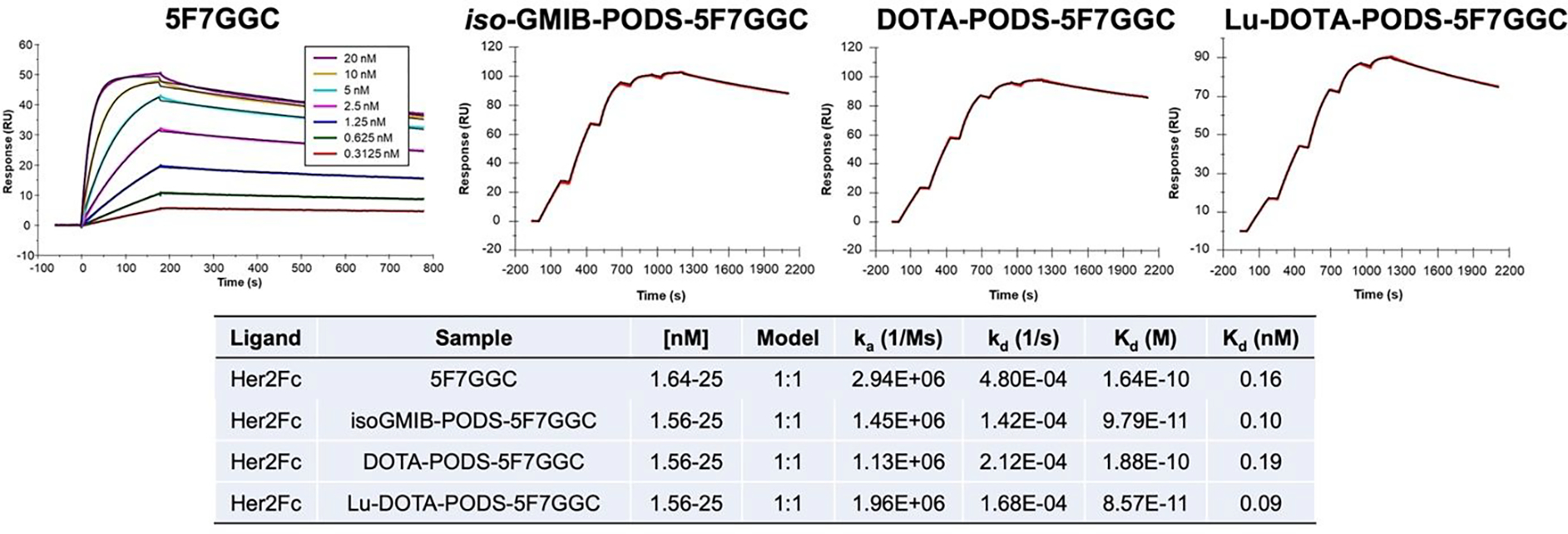
The binding affinities (Kd) of 5F7GGC and 5F7GGC conjugates for HER2 extracellular domain measured by surface plasmon resonance (SPR). The Kd for 5F7GGC was determined using multi-cycle kinetic titration and 5F7GGC conjugates were determined using single-cycle kinetic titration.
Figure 3.
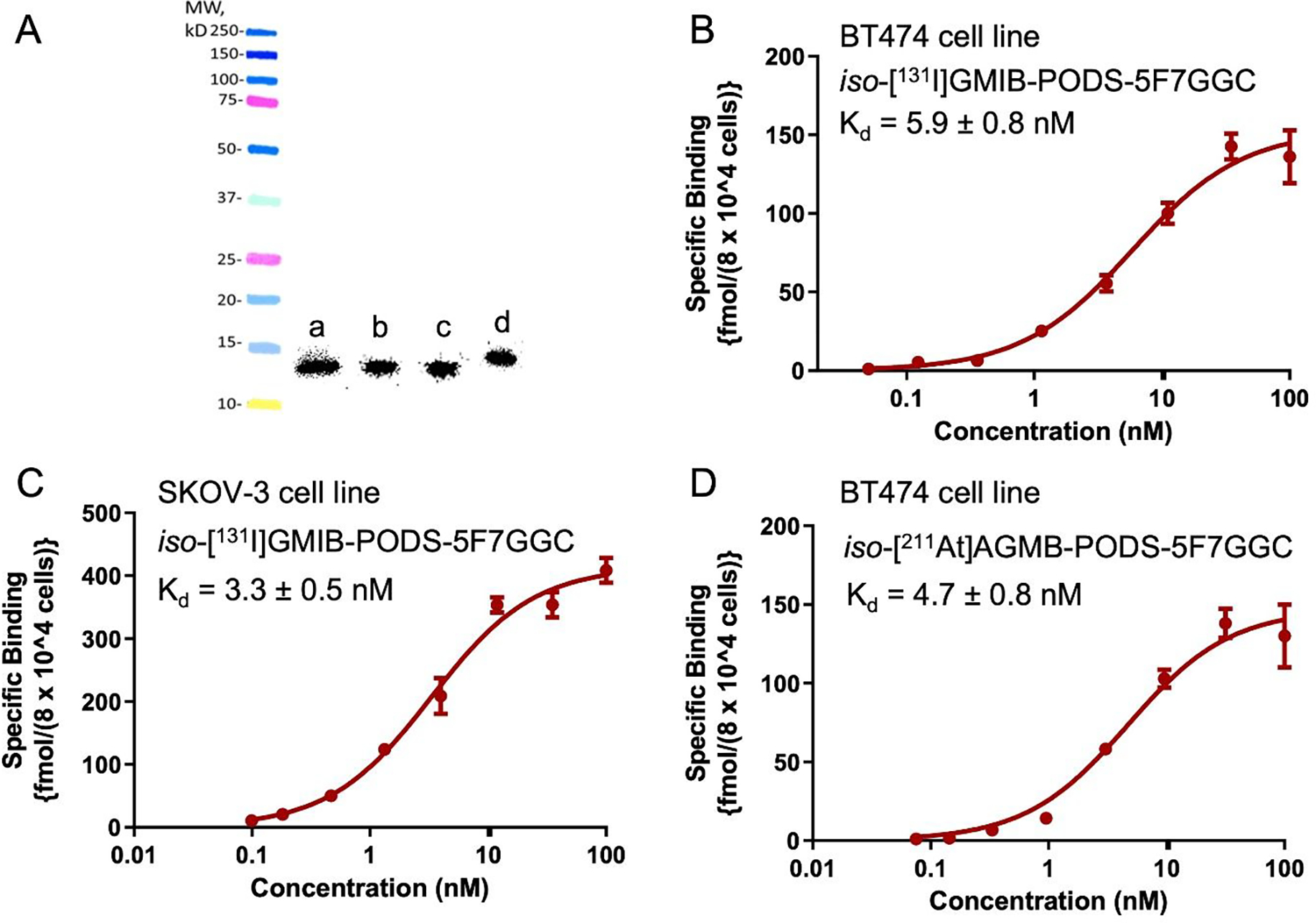
Quality control of the radiolabeled sdAb conjugates. A: SDS-PAGE and phosphor imaging of iso-[131l]GMIB-PODS-5F7GGC (a), [125I]MEGMIB-5F7GGC (b), iso-[211At]AGMB-PODS-5F7GGC (c) and [177Lu]Lu-DOTA-PODS-5F7GGC (d); B: binding affinity of iso-[131l]GMIB-PODS-5F7GGC measured in BT474 cells; C: binding affinity of iso-[131l]GMIB-PODS-5F7GGC measured in SKOV-3 cells; D: binding affinity of/’so-[211At]AGMB-PODS-5F7GGC measured in BT474 cells.
Cellular Retention and Internalization.
In the paired-label internalization assay comparing iso-[131I]GMIB-PODS-5F7GGC and [125I]MEGMIB-5F7GGC, 6.2 ± 0.3% of iso-[131I]GMIB-PODS-5F7GGC was associated with cells after the initial 1 h incubation at 4 °C compared with 5.8 ± 0.4% for [125I]MEGMIB-5F7GGC (difference not statistically significant: P > 0.05). The fraction of this initially bound iso-[131I]GMIB-PODS-5F7GGC that was internalized in the cells after subsequent incubation at 37 °C was 28.4 ± 3.3%, 33.5 ± 1.8%, and 29.0 ± 0.8% at 1, 2, and 4 h, respectively (Figure 4). For [125I]MEGMIB-5F7GGC, these values were 25.0 ± 3.4%, 30.5 ± 2.2%, and 26.7 ± 0.5%. A significantly higher internalized fraction was only observed for iso-[131I]GMIB-PODS-5F7GGC compared to [125I]MEGMIB-5F7GGC at 4 h (P < 0.05). The surface-bound fractions for iso-[131I]GMIB-PODS-5F7GGC and [125I]MEGMIB-5F7GGC were not significantly different at any time point.
Figure 4.
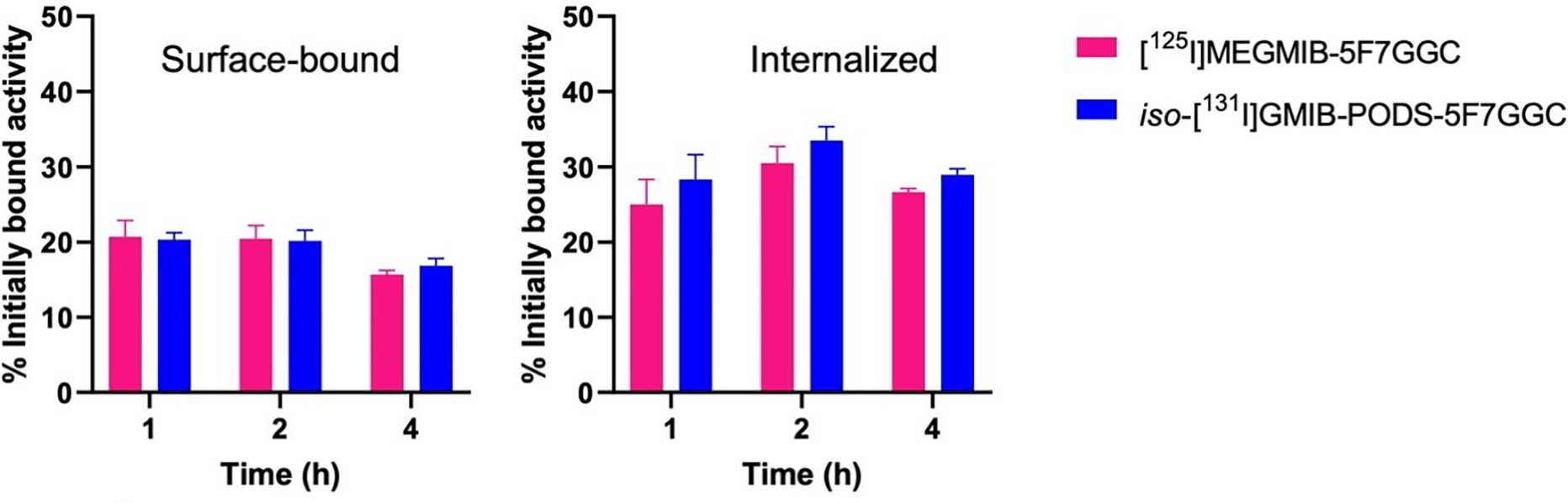
In vitro paired-label internalization assay on HER2-positive BT474 breast carcinoma cells co-incubated with [125I]MEGMIB-5F7GGC and iso-[131l]GMIB-PODS-5F7GGC. The results are presented as surface-bound (left) and internalized (right) fraction of the radioactivity initially bound to the cells after a 1 h incubation at 4°C.
The cellular uptake and internalization of iso-[131I]GMIB-PODS-5F7GG were compared with that of iso-[211At]AGMB-PODS-5F7GGC in a paired-label assay (Figure 5). No significant difference was observed in the uptake after 1 h of incubation at 4 oC, with 7.4 ± 0.8% and 7.5 ± 1.0% of input activity bound to BT474 cells for iso-[131I]GMIB-PODS-5F7GGC and [211At]AGMB-PODS-5F7GGC, respectively. The surface-bound fractions for iso-[131I]GMIB-PODS-5F7GGC ranged from 17.7% to 28.5% of the initially bound activity, and the internalized fractions were between 34.4% and 48.8%. For iso-[211At]AGMB-PODS-5F7GGC, the surface bound fractions were between 15.4% and 29.0%, and the internalized fractions were between 34.1% and 41.8%. The differences between the 131I- and 211At-labeled sdAbs were not statistically significant at any time point.
Figure 5.
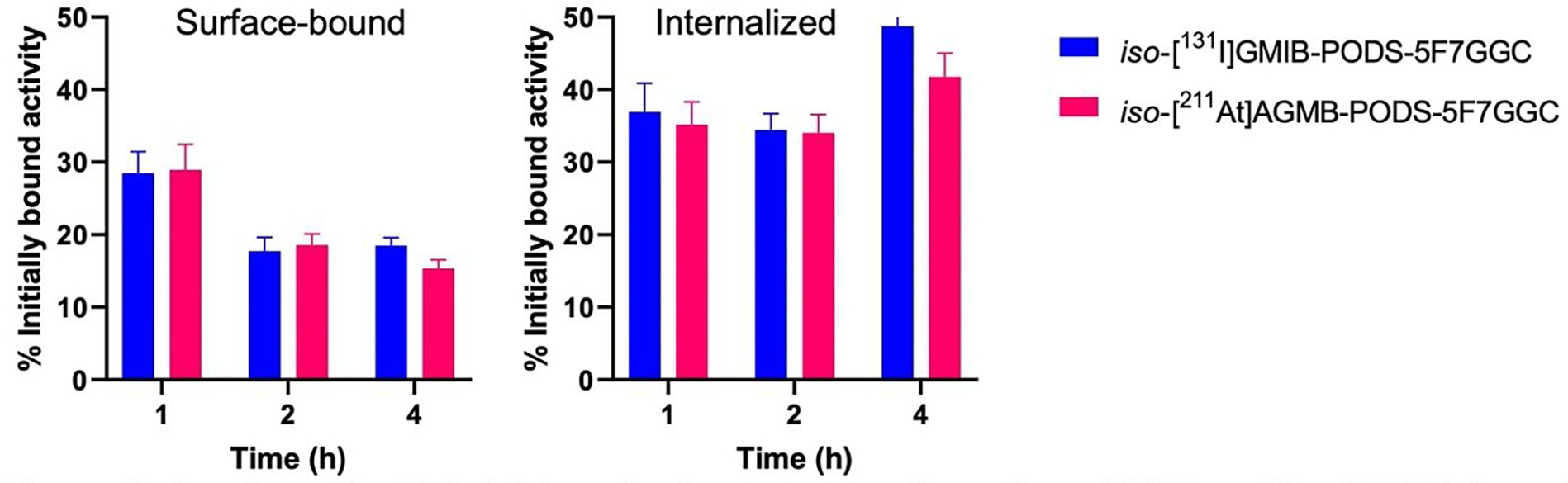
In vitro paired-label internalization assay performed on HER2-positive BT474 breast carcinoma cells co-incubated with iso-[131l]GMIB-PODS-5F7GGC and iso-[211At]AGMB-PODS-5F7GGC. The results are presented as surface-bound (left) and internalized (right) fraction of the radioactivity initially bound to the cells after 1 h incubation at 4°C.
In vitro Stability of [211At]MEAGMB-5F7GGC and iso-[211At]AGMB-PODS-5F7GGC.
The in vitro stability results for the 211At-labeled sdAb are shown in Figure 6. Iso-[211At]AGMB-PODS-5F7GGC showed excellent stability in both PBS and a cysteine solution, with over 95% RCP after 21 h. A decrease in stability was observed in HSA, with 86% and 76% of the 211At remaining associated with the sdAb at 3 and 21 h, respectively. Compared with iso-[211At]AGMB-PODS-5F7GGC, the in vitro stability of [211At]MEAGMB-5F7GGC in cysteine solution and HSA was considerably lower, while no significant difference was observed between the two 211At-labeled sdAbs in PBS. Notably, virtually no radioactivity was associated with intact sdAb at 3 and 21 h for [211At]MEAGMB-5F7GGC in both the cysteine solution and HSA.
Figure 6.
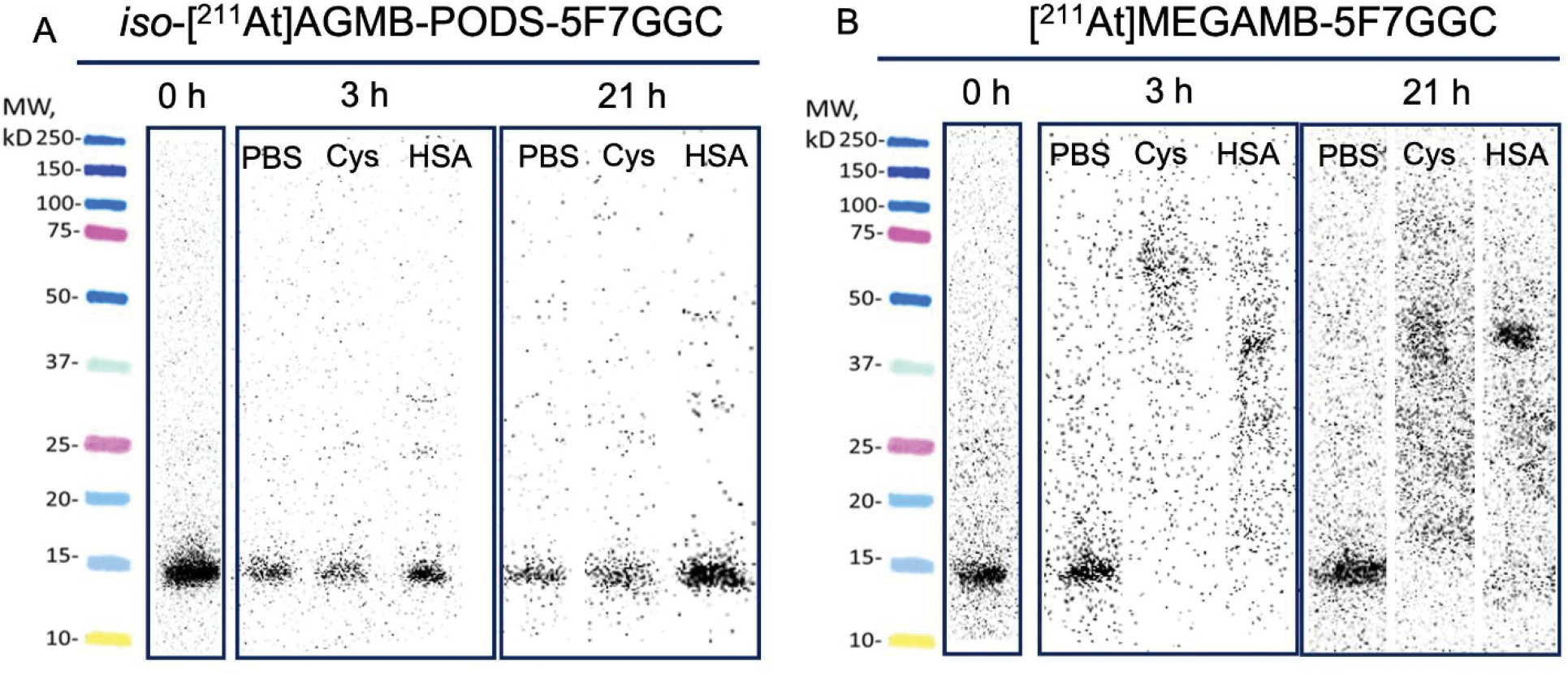
In vitro stability of (A) iso-[211At]AGMB-PODS-5F7GGC and (B) [211At]MEGAMB-5F7GGC in PBS, 50 mM cysteine (Cys) and human serum albumin (HSA). Stability was determined by SDS-PAGE and phosphor imaging.
Biodistribution.
The biodistribution of iso-[131I]GMIB-PODS-5F7GGC and [125I]MEGMIB-5F7GGC were compared in athymic mice both without (Table 1) and with BT474 subcutaneous xenografts (Table 2). In the mice without tumors, the uptake of radioactivity in the kidneys from iso-[131I]GMIB-PODS-5F7GGC at 1 h post-injection (p.i.) (36.3 ± 5.2% ID/g) was significantly higher than that from [125I]MEGMIB-5F7GGC (15.7 ± 1.3% ID/g; P < 0.005). However, at 4 h p.i., the kidney activity concentrations produced by both radioimmunoconjugates were not significantly different (P > 0.05). The uptake of iso-[131I]GMIB-PODS-5F7GGC in the liver at 1 h p.i. was significantly higher (P < 0.01) than that from [125I]MEGMIB-5F7GGC with the trend reversed at 4 h p.i., (P < 0.05). Minimal uptake and retention was observed in other organs. As shown in Table 2, the uptake of iso-[131I]GMIB-PODS-5F7GGC in the BT474 tumors was not significantly different (P > 0.05 for all time points) from that for [125I]MEGMIB-5F7GGC. The differences in the tissue concentrations in normal organs were consistent with those observed in mice without tumors. At 1 h p.i., the tumor-to-kidney activity concentration ratio for [125I]MEGMIB-5F7GGC (0.8 ± 0.1) was significantly higher than that for iso-[131I]GMIB-PODS-5F7GGC (0.3 ± 0.1; P<0.0001); however, the tumor-to-kidney activity concentration ratios of the two conjugates were not significantly different at 4 h p.i. The tumor-to-liver activity concentration ratios for [125I]MEGMIB-5F7GGC were 9.2 ± 1.5, 23.3 ± 6.8, and 63.4 ± 21.2 at 1, 4 and 24 h p.i., respectively, values significantly higher (P < 0.0001) than those for iso-[131I]GMIB-PODS-5F7GGC at each of the timepoints.
Table 1.
Paired-label biodistribution of [125I]MEGMIB-5F7GGC and iso-[131I]GMIB-PODS-5F7GGC in athymic mice without tumors.
| Percent injected dose per grama | ||||||
|---|---|---|---|---|---|---|
|
|
||||||
| 1 h | 4 h | 24 h | ||||
|
|
||||||
| tissue | 125I | 131I | 125I | 131I | 125I | 131I |
| Liver | 3.1 ± 0.4 | 5.2 ± 0.6 | 1.2 ± 0.2 | 0.7 ± 0.3 | 0.4 ± 0.1 | 0.1 ± 0.0 |
| Spleen | 1.4 ± 0.6 | 1.2 ± 0.5 | 0.7 ± 0.1 | 0.4 ± 0.1 | 0.3 ± 0.1 | 0.1 ± 0.0 |
| Lungs | 2.4 ± 0.4 | 3.4 ± 1.2 | 1.2 ± 0.1 | 1.2 ± 0.4 | 0.3 ± 0.2 | 0.4 ± 0.3 |
| Heart | 0.4 ± 0.0 | 0.5 ± 0.1 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Kidneys | 15.7 ± 1.3 | 36.3 ± 5.2 | 1.6 ± 0.2 | 1.9 ± 0.4 | 0.6 ± 0.1 | 0.2 ± 0.0 |
| Stomach | 0.6 ± 0.1 | 0.8 ± 0.3 | 0.3 ± 0.1 | 0.3 ± 0.2 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Sm. Int. | 2.8 ± 0.3 | 4.3 ± 0.7 | 0.8 ± 0.1 | 0.9 ± 0.3 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Lg. Int. | 0.6 ± 0.1 | 0.5 ± 0.1 | 3.9 ± 0.4 | 6.4 ± 0.7 | 0.1 ± 0.0 | 0.1 ± 0.0 |
| Muscle | 0.3 ± 0.1 | 0.3 ± 0.1 | 0.0 ± 0.0 | 0.0 ± 0.00 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Blood | 0.7 ± 0.5 | 0.8 ± 0.4 | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Bone | 0.4 ± 0.1 | 0.5 ± 0.1 | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Brain | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Thyroid | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.1 ± 0.4 | 0.1 ± 0.1 | 0.0 ± 0.0 | 0.2 ± 0.0 |
%ID/g, mean ± SD, n=5
Table 2.
Paired-label biodistribution of iso-[131I]GMIB-PODS-5F7GGC and [125I]MEGMIB-5F7GGC in athymic mice bearing HER2-positive BT474 xenografts.
| Percent injected dose per grama | ||||||
|---|---|---|---|---|---|---|
|
|
||||||
| 1 h | 4 h | 24 h | ||||
|
|
||||||
| tissue | 131I | 125I | 131I | 125I | 131I | 125I |
| Liver | 6.6 ± 1.9 | 1.5 ± 0.4 | 1.4 ± 0.2 | 0.5 ± 0.1 | 0.2 ± 0.1 | 0.1 ± 0.0 |
| Spleen | 0.6 ± 0.2 | 0.4 ± 0.2 | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Lungs | 2.4 ± 1.2 | 1.4 ± 0.3 | 1.0 ± 0.3 | 0.7 ± 0.2 | 0.1 ± 0.1 | 0.2 ± 0.1 |
| Heart | 0.5 ± 0.1 | 0.5 ± 0.2 | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.0 |
| Kidneys | 55.3 ± 14.1 | 16.2 ± 3.3 | 4.8 ± 0.3 | 2.9 ± 0.4 | 0.7 ± 0.2 | 1.2 ± 0.3 |
| Stomach | 0.8 ± 0.1 | 0.7 ± 0.2 | 1.8 ± 3.5 | 1.5 ± 3.0 | 0.1 ± 0.1 | 0.1 ± 0.1 |
| Sm. Int. | 5.6 ± 0.1 | 9.1 ± 1.7 | 3.1 ± 0.7 | 1.6 ± 0.7 | 0.1 ± 0.1 | 0.1 ± 0.0 |
| Lg. Int. | 0.4 ± 0.1 | 1.1 ± 0.4 | 12.8 ± 3.6 | 14.3 ± 2.6 | 0.4 ± 0.2 | 0.2 ± 0.1 |
| Muscle | 0.5 ± 0.4 | 0.7 ± 0.6 | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Blood | 0.8 ± 0.2 | 0.8 ± 0.2 | 0.1 ± 0.0 | 0.3 ± 0.0 | 0.0 ± 0.0 | 0.13 ± 0.0 |
| Bone | 1.2 ± 1.6 | 1.5 ± 1.9 | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Brain | 0.1 ± 0.0 | 0.1 ± 0.1 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Thyroid | 0.1 ± 0.4 | 0.0 ± 0.6 | 0.2 ± 0.1 | 0.1 ± 0.1 | 0.2 ± 0.2 | 0.1 ± 0.1 |
| Tumor | 16.2 ± 4.9 | 13.7 ± 4.5 | 13.7 ± 4.1 | 12.1 ± 3.6 | 4.8 ± 1.5 | 5.7 ± 1.8 |
%ID/g, mean ± SD, n=5
The biodistribution of iso-[131I]GMIB-PODS-5F7GGC and its 211At-labeled analogue — iso-[211At]AGMB-PODS-5F7GGC, —were directly compared in a paired-label experiment using athymic mice bearing BT474 xenografts (Table 3). High tumor uptake and retention was observed for both agents, and differences between the two agents were not statistically significant (P > 0.05 at 1, 4, and 21 h). Kidney activity levels at 1 h p.i. were also similar; however, at 4 and 21 h p.i., the activity concentration of iso-[211At]AGMB-PODS-5F7GGC in the kidneys was significantly higher than that for iso-[131I]GMIB-PODS-5F7GGC (P < 0.05 for 4 and 21 h). The tumor-to-kidney activity concentration ratios for iso-[131I]GMIB-PODS-5F7GGC at 4 and 21 h p.i., respectively, were 2.8 ± 0.5, and 8.0 ± 1.2, significantly higher than those for iso-[211At]AGMB-PODS-5F7GGC: 1.5 ± 0.3 and 2.7 ± 0.4 at (P < 0.01 at 4 h; P < 0.0001 at 21 h; no significant difference was seen at 1 h). A similar trend was seen in the liver. At 1 h p.i., no significant difference was seen between the tumor-to-liver activity concentration ratios of the 211At-labeled (2.5 ± 0.6) and 131I-labeled analogue (2.8 ± 0.8). However, significantly higher tumor-to-liver activity concentration ratios were seen for the 131I-labeled sdAb thereafter: 12.2 ± 3.5 and 27.1 ± 8.9 at 4 and 21 h for 131I, and 7.7 ± 1.8 and 11.6 ± 3.4 for 211At at 4 and 21 h, respectively, (P < 0.05 at 4 h and P < 0.01 at 21 h). The activity concentrations in the stomach for iso-[211At]AGMB-PODS-5F7GGC were significantly higher than those observed for iso-[131I]GMIB-PODS-5F7GGC at 1 h (P < 0.05) and 4 h. Likewise, the uptake of radioactivity in the thyroid for iso-[211At]AGMB-PODS-5F7GGC was significantly higher than observed for iso-[131I]GMIB-PODS-5F7GGC at all time points. Nonetheless, the absolute levels of activity in the stomach and thyroid for both radioimmunoconjugates were consistent with a low level of in vivo dehalogenation for both labeled sdAbs but with the 211At-labeled conjugate showing a higher degree of dehalogenation. When iso-[211At]AGMB-PODS-5F7GGC was evaluated as a single agent in athymic mice bearing BT474 xenografts, tissue activity levels were consistent with those seen in the paired-label study, except for tumor uptake at 4 h and 24 h (Table 4). In the single-label study, the tumor uptake was two-fold higher: 20.5 ± 2.4% ID/g and 8.8 ± 0.8% ID/g at 4 and 24 h, respectively.
Table 3.
Paired-label biodistribution of iso-[131I]GMIB-PODS-5F7GGC and iso-[211At]AGMB-PODS-5F7GGC in athymic mice bearing HER2-positive BT474 xenografts.
| Percent injected dose per grama | ||||||
|---|---|---|---|---|---|---|
|
|
||||||
| 1 h | 4 h | 21 h | ||||
|
|
||||||
| tissue | 131I | 211At | 131I | 211At | 131I | 211At |
| Liver | 4.5 ± 2.2 | 5.0 ± 2.2 | 0.9 ± 0.2 | 1.4 ± 0.2 | 0.2 ± 0.1 | 0.4 ± 0.1 |
| Spleen | 0.4 ± 0.1 | 1.8 ± 0.3 | 0.1 ± 0.0 | 1.3 ± 0.2 | 0.0 ± 0.0 | 0.4 ± 0.2 |
| Lungs | 1.9 ± 0.5 | 4.8 ± 0.2 | 1.4 ± 0.3 | 3.6 ± 0.2 | 0.3 ± 0.2 | 0.8 ± 0.4 |
| Heart | 0.4 ± 0.2 | 0.9 ± 0.3 | 0.0 ± 0.0 | 0.5 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.1 |
| Kidneys | 52.5 ± 18.6 | 53.1 ± 17.5 | 4.0 ± 1.0 | 7.0 ± 1.3 | 0.6 ± 0.2 | 1.5 ± 0.4 |
| Stomach | 0.5 ± 0.3 | 3.2 ± 1.2 | 0.1 ± 0.1 | 4.5 ± 0.9 | 0.1 ± 0.1 | 0.9 ± 0.5 |
| Sm. Int. | 3.3 ± 1.1 | 3.8 ± 1.1 | 0.9 ± 0.2 | 1.4 ± 0.3 | 0.2 ± 0.1 | 0.3 ± 0.1 |
| Lg. Int. | 0.3 ± 0.1 | 0.5 ± 0.1 | 6.2 ± 0.5 | 6.4 ± 0.5 | 0.4 ± 0.2 | 0.4 ± 0.1 |
| Muscle | 0.3 ± 0.1 | 0.4 ± 0.2 | 0.2 ± 0.3 | 0.3 ± 0.3 | 0.1 ± 0.2 | 0.1 ± 0.2 |
| Blood | 0.5 ± 0.2 | 0.8 ± 0.3 | 0.5 ± 1.1 | 0.8 ± 1.1 | 0.4 ± 0.8 | 0.5 ± 0.9 |
| Bone | 0.4 ± 0.3 | 0.6 ± 0.3 | 0.3 ± 0.4 | 0.5 ± 0.4 | 0.0 ± 0.0 | 0.1 ± 0.1 |
| Brain | 0.0 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Thyroid | 0.1 ± 0.2 | 1.1 ± 0.7 | 0.0 ± 0.3 | 1.5 ± 0.4 | 0.2 ± 0.3 | 2.7 ± 1.2 |
| Tumor | 11.9 ± 4.4 | 11.9 ± 4.0 | 10.9 ± 2.4 | 10.6 ± 2.1 | 4.8 ± 1.1 | 4.1 ± 1.0 |
%ID/g, mean ± SD, n=5
Table 4.
Iso-[211At]AGMB-PODS-5F7GGC single-label biodistribution of in athymic mice bearing subcutaneous HER2-positive BT474 xenografts.
| Percent injected dose per grama | |||
|---|---|---|---|
|
|
|||
| tissue | 1 h | 4 h | 24 hb |
|
|
|||
| Liver | 8.4 ± 1.3 | 2.8 ± 0.8 | 0.6 ± 0.1 |
| Spleen | 1.4 ± 0.5 | 1.1 ± 0.4 | 0.3 ± 0.1 |
| Lungs | 2.6 ± 0.7 | 1.7 ± 0.4 | 0.4 ± 0.1 |
| Heart | 0.8 ± 0.2 | 0.4 ± 0.1 | 0.2 ± 0.1 |
| Kidneys | 70.9 ± 12.0 | 11.6 ± 2.4 | 2.5 ± 0.2 |
| Stomach | 3.2 ± 1.2 | 4.9 ± 1.5 | 1.0 ± 0.3 |
| Sm. Int. | 5.1 ± 0.5 | 2.7 ± 1.9 | 0.4 ± 0.1 |
| Lg. Int. | 0.6 ± 0.1 | 12.6 ± 4.5 | 1.0 ± 0.4 |
| Muscle | 0.5 ± 0.3 | 0.3 ± 0.3 | 0.0 ± 0.1 |
| Blood | 0.7 ± 0.3 | 0.3 ± 0.1 | 0.1 ± 0.1 |
| Bone | 0.7 ± 0.3 | 0.5 ± 0.4 | 0.1 ± 0.1 |
| Brain | 0.1 ± 0.1 | 0.1 ± 0.0 | 0.0 ± 0.0 |
| Thyroid | 0.8 ± 0.3 | 1.2 ± 0.4 | 1.1 ± 0.5 |
| Tumor | 15.1 ± 3.3 | 20.5 ± 2.4c | 8.8 ± 0.8 |
%ID/g, mean ± SD, n=5
n=4
n=2
A biodistribution study was performed to directly compare the in vivo performance of iso-[125I]GMIB-PODS-5F7GGC and [177Lu]Lu-DOTA-PODS-5F7GGC in athymic mice bearing BT474 xenografts (Table 5). The tumor uptake values for 125I were 34 ± 8% and 21 ± 3% higher than those for 177Lu at 1 and 4 h, respectively, but 26 ± 8% lower at 24 h. While the radioactivity levels in the kidneys were comparable at 1 h (P > 0.05), they were substantially lower for iso-[125I]GMIB-PODS-5F7GGC (6.3 ± 1.2% ID/g and 1.1 ± 0.2% ID/g) than for [177Lu]Lu-DOTA-PODS-5F7GGC (64.8 ± 13.7% ID/g and 40.8 ± 9.7% ID/g) at 4 and 24 h, respectively (P < 0.0001 at both time points). As a result, the tumor-to-kidney activity concentration ratios were not significantly different between the two conjugates at 1 h (0.3 ± 0.1 for 125I and 0.2 ± 0.1 for 177Lu; P > 0.05). However, at 4 and 24 h, the tumor-to-kidney activity concentration ratios for iso-[125I]GMIB-PODS-5F7GGC were 2.9 ± 0.7 and 5.8 ± 2.0, considerably higher than those for [177Lu]Lu-DOTA-PODS-5F7GGC - 0.2 ± 0.0 and 0.2 ± 0.1 (P < 0.0001 for both time points).
Table 5.
Paired-label biodistribution of iso-[125I]GMIB-PODS-5F7GGC and [177Lu]Lu-DOTA-PODS-5F7GGC in athymic mice bearing subcutaneous HER2-positive BT474 xenografts.
| Percent injected dose per grama | ||||||
|---|---|---|---|---|---|---|
|
|
||||||
| 1 h | 4 h | 24 h | ||||
|
|
||||||
| tissue | 125I | 177Lu | 125I | 177Lu | 125I | 177Lu |
| Liver | 7.8 ± 2.6 | 1.3 ± 0.7 | 1.4 ± 0.4 | 0.6 ± 0.2 | 0.2 ± 0.1 | 0.4 ± 0.0 |
| Spleen | 0.6 ± 0.2 | 0.5 ± 0.2 | 0.1 ± 0.0 | 0.2 ± 0.1 | 0.0 ± 0.0 | 0.2 ± 0.1 |
| Lungs | 4.1 ± 2.6 | 1.7 ± 0.2 | 3.1 ± 1.3 | 1.6 ± 0.8 | 0.8 ± 0.9 | 0.4 ± 0.4 |
| Heart | 0.5 ± 0.2 | 0.6 ± 0.2 | 0.1 ± 0.0 | 0.3 ± 0.1 | 0.0 ± 0.0 | 0.2 ± 0.1 |
| Kidneys | 65.1 ± 20.0 | 73.6 ± 13.9 | 6.3 ± 1.2 | 64.8 ± 13.7 | 1.1 ± 0.2 | 40.8 ± 9.7 |
| Stomach | 1.4 ± 1.5 | 1.2 ± 1.7 | 0.4 ± 0.3 | 0.3 ± 0.2 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Sm. Int. | 5.1 ± 1.2 | 1.5 ± 1.3 | 2.9 ± 1.2 | 0.4 ± 0.2 | 0.1 ± 0.0 | 0.1 ± 0.0 |
| Lg. Int. | 0.5 ± 0.1 | 0.3 ± 0.1 | 11.1 ± 4.0 | 1.0 ± 0.2 | 0.3 ± 0.2 | 0.1 ± 0.0 |
| Muscle | 0.5 ± 0.1 | 0.5 ± 0.2 | 0.1 ± 0.0 | 0.1 ± 0.1 | 0.0 ± 0.0 | 0.1 ± 0.0 |
| Blood | 0.8 ± 0.2 | 1.6 ± 0.3 | 0.1 ± 0.1 | 0.6 ± 0.1 | 0.1 ± 0.1 | 0.3 ± 0.2 |
| Bone | 0.9 ± 0.5 | 0.9 ± 0.6 | 0.1 ± 0.0 | 0.2 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.1 |
| Brain | 0.1 ± 0.0 | 0.1 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Thyroid | 0.2 ± 0.2 | 0.2 ± 0.2 | 0.1 ± 0.0 | 0.1 ± 0.1 | 0.1 ± 0.0 | 0.1 ± 0.1 |
| Tumor | 21.2 ± 8.4 | 15.9 ± 6.2 | 17.4 ± 2.3 | 14.4 ± 1.9 | 6.4 ± 1.7 | 8.5 ± 1.5 |
%ID/g, mean ± SD, n=5
DISCUSSION
Thiol-targeted reagents for protein conjugation originally developed for intact mAbs should be an attractive strategy for the truly site-specific radiolabeling of sdAbs. In most cases, thiols are generated on intact mAbs via the reduction of the multiple disulfide bonds whose number and position vary by IgG class. While this results in a much lower number of potential conjugation sites than are available for lysine-targeted reagents, multiple cysteines can nonetheless still be modified. This can be ameliorated somewhat by genetically inserting capped thiols into the IgG light chain (23); however, in this case, the reaction conditions must be carefully tuned for each mAb to avoid reduction of its native disulfides. sdAbs, in contrast, have an ideal structure for truly site-specific thiol modification, as they generally have only one conserved disulfide linkage that is stable under most reducing conditions (25, 26). Because the C-terminus of sdAbs points away from its CDR regions (27), it represents an excellent position for modification with minimal chance of disrupting antigen binding affinity. This was confirmed in a study demonstrating the superior affinity for three sdAbs modified with IRDye 800CW via different bioconjugation approaches (28).
These factors motivated us to use recombinant methods to add a GGC sequence at the C-terminus of the anti-HER2 sdAb 5F7 to introduce a single cysteine for site-specific labeling (4). Our initial approach utilized an [131I]iodobenzoyl maleimido D-amino acid peptide-bearing prosthetic agent. Unfortunately, however, poor conjugation yields necessitated the modification of the sdAb with 2-iminothiolane so sufficient product for could be obtained for biological evaluation. As a result, the bioconjugation was no longer site specific. More recently, maleimidoethyl 3-(guanidinomethyl)-5-[131I]iodobenzoate (MEGMIB) — a maleimido analogue of the residualizing prosthetic agent iso-SGMIB (29) — was synthesized and evaluated for the site-specific labeling of 5F7GGC (16). Both 5F7 labeled randomly on its lysines using iso-[131I]SGMIB and [131]MEGMIB-5F7GGC exhibited excellent tumor targeting in vivo. However, the maleimido version had higher activity retention in the liver, spleen, and kidneys at 24 h post-injection, suggesting different metabolic patterns for the two radioimmunoconjugates (16). The extension of this labeling strategy to 211At was briefly evaluated via the synthesis of maleimidoethyl 3-[211At]astato-5-(guanidinomethyl)iodobenzoate ([211At]MEAGMB), but the in vivo results were perplexing at best (unpublished observations). As we have determined in the current study and will discuss below, this can be explained by the rapid breakdown of [211At]MEAGMB-5F7GGC in the presence of endogenously abundant thiol-containing species.
Herein, we sought to develop a site-specific labeling strategy for sdAbs with high in vivo stability applicable both to both the highly versatile array of iodine radionuclides and 211At for targeted alpha particle therapy. Although sdAbs labeled randomly on their lysines with reagents like iso-[131I]SGMIB can exhibit high affinity and stability, this has often involved selecting an sdAb without a lysine in its CDR regions (30) or making an analogue in which a CDR lysine is removed (5). To obviate the need for these tactics and take advantage of the benefits of site-specific labeling, we sought to build on the encouraging results we obtained labeling proteins with radiometals using a PODS moiety (21–23, 31). Among the attractive features we hoped to extend to these radiohalogens include efficient and rapid conjugation at biologically relevant pH, and improved in vitro and in vivo stability compared with corresponding conjugates labeled using maleimide chemistry (21, 23). These results encouraged us to combine PODS with our previously validated residualizing prosthetic agents — iso-[131I]SGMIB (29) and iso-[211At]SAGMB (6) — to derive iso-[131I]GMIB-PODS and iso-[211At]AGMB-PODS, respectively. 5F7-GGC labeled with these novel synthons were compared directly with conjugates synthesized using the maleimide-based prosthetic groups [131I]MEGMIB and [211At]MEAGMB. In addition, we directly compared iso-[125I]GMIB-PODS-5F7GGC with 5F7GGC labeled with 177Lu using DOTA-PODS. To the best our knowledge, such paired-label comparison of an sdAb labeled with a radiometal and radiohalogen has not been reported.
The tin precursor Boc2-iso-GMTB-PODS and the iodinated standard iso-GMIB-PODS were synthesized in reasonable yields in two steps. The syntheses of iso-[131I]GMIB-PODS, iso-[211At]AGMB-PODS, and iso-[211At]MEAGMB were performed in 2 steps in a manner similar to reported procedures for iso-[211At]SAGMB (6) and [131I]MEGMIB (16). The conjugation of iso-GMIB-PODS to 5F7GGC gave higher yield than that of MEGMIB to 5F7GGC (16). Similarly, the conjugation of iso-[131I]GMIB-PODS to 5F7GGC had a higher RCY (P=0.0016) than that for [131I]MEGMIB (16). No significant difference in RCY was observed for the conjugation of iso[131I]GMIB-PODS and iso-[211At]AGMB-PODS to 5F7GGC. The construction of [177Lu]Lu-DOTA-PODS-5F7GGC was evaluated in two ways: (1) the synthesis and purification of DOTA-PODS-5F7GGC for direct labeling with [177Lu]LuCl3 and (2) the synthesis and purification of [177Lu]Lu-DOTA-PODS for bioconjugation to 5F7GGC. We found that if DOTA-PODS-5F7GGC was labeled directly with [177Lu]LuCl3, some [177Lu]Lu3+ became loosely bound to DOTA-PODS-5F7GGC, which disassociated when injected into animals (unpublished results). A possible explanation is that the [177Lu]LuCl3 formed colloids at neutral pH and co-eluted with the radioimmunoconjugate from the PD-10 column. [177Lu]Lu-DOTA-PODS-5F7GGC was subsequently performed in two steps to avoid this problem. All non-radioactive versions of these sdAb immunoconjugates exhibited high binding affinity to the extracellular domains of HER2, demonstrating that conjugation with these prosthetic moieties did not diminish HER2 recognition. Moreover, the immunoreactivities, and binding affinities to HER2-expressing cancer cells for all the radioimmunoconjugates were high and consistent with those reported previously for other 5F7 radioconjugates (5, 9, 16).
All of the radioimmunoconjugates in the study exhibited high uptake and internalization into HER2-positive BT474 cells, though some significant differences in behavior were observed. Compared with [125I]MEGMIB-5F7GGC, iso-[131I]GMIB-PODS-5F7GGC showed higher intracellular retention after a 4 h incubation at 37 °C, which likely reflected the higher in vitro stability of iso-[131I]GMIB-PODS-5F7GGC compared with [125I]MEGMIB-5F7GGC. In a prior study, [131I]MEGMIB-5F7GGC was less stable in vitro than iso-[125I]SGMIB-5F7, probably due to hydrolysis and metabolism of the maleimido conjugate (16). Similar differences in behavior were seen when trastuzumab radioiodinated using D-amino acid residualizing peptide conjugates linked both via an active ester and a maleimide moiety were compared (32). It was encouraging that the cellular uptake and intracellular residualization of iso-[211At]AGMB-PODS-5F7GGC was not significantly different than that for iso-[131I]GMIB-PODS-5F7GGC, consistent with a low degree of dehalogenation for both conjugates under in vitro conditions.
The contribution of the PODS moiety to the stability of the radioimmunoconjugates under in vitro conditions was investigated by comparing the behavior of iso-[211At]AGMB-PODS-5F7GGC and iso-[211At]MEAGMB-5F7GGC in three media, two of which contained thiol-bearing substances that are conducive to retro-Michael-mediated degradative processes. Excellent stability was observed for iso-[211At]AGMB-PODS-5F7GGC in PBS, cysteine, and HSA. In contrast, iso-[211At]MEAGMB-5F7GGC quickly degraded within 3 h in both cysteine and HSA. Interestingly, however, significantly higher stability was seen earlier for iso-[131I]MEGMIB-5F7GGC, with 90% of the radioimmunoconjugate intact after a 24-h incubation in human serum (16). The considerably lower stability of iso-[211At]MEAGMB-5F7GGC compared to iso[131I]MEGMIB-5F7GGC could relate to several factors, such as a higher dehalogenation of the [211At]MEAGMB moiety either before or after disassociation from the sdAb conjugate. However, in interpreting these results, it is important to bear in mind that there is a significant difference in the affinity of astatine and iodine for sulfur atoms as well as their susceptibility to dehalogenation. The affinity to sulfur atoms has been an interesting facet of astatine chemistry dating back to the 1980s (33, 34). A more recent publication investigated the interactions of AtO+ species with sulfurbased ligands in aqueous solution, which might influence the in vivo behavior of 211At-labeled agents (35). Because of the transient affinity of astatine for sulfur, care must be taken when translating thiol-based site-specific radioiodination strategies to 211At.
Because of its poor in vitro stability, iso-[211At]MEAGMB-5F7GGC was not evaluated further in biodistribution studies. Except for a confirmatory single-label evaluation of iso-[211At]AGMB-PODS-5F7GGC, biodistribution experiments were done in paired label format. This facilitated the direct comparison of two radioimmunoconjugates while eliminating potentially confounding variables (such as tumor size) that can exist between groups of experimental animals. When iso-[131I]GMIB-PODS-5F7GGC and iso-[125I]MEGMIB-5F7GGC were compared in tumor-bearing mice, there was a trend towards higher tumor uptake for the PODS conjugate at 1 and 4 h, but the differences were not significant. On the other hand, the maleimide-based conjugate exhibited more than two-fold lower kidney activity levels at the 1 h time point in both tumor-bearing and non-tumor bearing mice but not thereafter; by 24 h, the opposite behavior was observed. The low initial kidney uptake of iso-[125I]MEGMIB-5F7GGC is consistent with results published previously (16). As discussed therein, this behavior likely reflects the in vivo lability of the thiosuccinimide linkage generating rapidly excreted labeled catabolites.
Although a different 131I-labeled anti-HER2 sdAb is now being evaluated in patients (3), studies in mice with HER2-expressing xenografts with both randomly labeled 5F7 and the closely related VHH_1028 sdAb have demonstrated considerably greater therapeutic effectiveness for the 211At-labeled compared with the 131I-labeled conjugates (5, 9). For this reason, our primary objective was to develop a site-specific and biologically stable reagent for labeling sdAbs (especially 5F7) with 211At. In order to evaluate the effect of halogen on in vivo behavior, iso-[211At]AGMB-PODS-5F7GGC and iso-[131I]GMIB-PODS-5F7GGC were compared in paired-label format. No significant differences in tumor uptake between 211At and 131I were observed, although 211At levels were significantly higher than those for 131I in spleen, stomach, and thyroid, results that are consistent with a higher degree of dehalogenation for the 211At-labeled sdAb. Similar halogen-dependent trends in tumor and normal tissue uptake were reported for 5F7 randomly labeled using iso-[211At]SAGMB and iso-[131I]SGMIB in SCID mice with BT474 xenografts (6). Unfortunately, the direct comparison of the absolute tissue uptake values for the two paired-label studies is not possible because of differences in biodistribution noted between athymic and SCID mice (9). On the other hand, the biodistribution of iso-[211At]AGMB-VHH_1028 — an sdAb differing from 5F7 by only one amino acid and exhibiting identical in vivo behavior — has been determined in single-label format in athymic mice with BT474 subcutaneous xenografts (9). When iso-[211At]AGMB-PODS-5F7GGC was evaluated in single-label format, its tumor uptake was about twice than that reported for iso-[211At]AGMB-VHH_1028 in the same model. Moreover, levels of 211At in the stomach, thyroid, and spleen were about two times lower for the PODS-based radioimmunoconjugate suggesting it was more stable in vivo than the iso-[211At]SAGMB conjugate.
Finally, the use of PODS for site-specific labeling produces only a single species, providing an opportunity to compare metal and halogen sdAb labeling strategies while excluding heterogeneity as a confounding variable. This is particularly important for sdAbs that bind internalizing targets like HER2 because a labeled sdAb bearing a metal-free chelate could have altered charge and potentially different internalization and/or trapping in tumor cells (36). Significantly higher tumor uptake was observed for iso-[125I]GMIB-PODS-5F7GGC at 1 and 4 h compared with [177Lu]Lu-DOTA-PODS-5F7GGC, with the opposite behavior seen at 24 h. The most notable negative feature of the radioiodinated sdAb is that it showed higher hepatobiliary organ uptake at the earlier time points. On the other hand, the kidney retention of 177Lu was substantially higher than for the radioiodinated agent, with the difference reaching a factor of about 40 at 24 h. The potentially dose-limiting renal activity levels are in line with those reported previously for other sdAbs labeled with 177Lu (37, 38). The results of this head-to-head comparison suggest that for 5F7 (and perhaps other sdAbs), radiohalogenation using our PODS-bearing residualizing agent might be preferable to the use of a radiometal.
CONCLUSION
Herein we report a method for the site-specific radiohalogenation of sdAbs with a prosthetic agent that contains both a residualizing moiety and a thiol-reactive PODS functionality. Excellent preservation of HER2-binding affinity was observed for conjugates derived using this strategy. Compared with iso-[211At]MEAGMB-5F7GGC, superior in vitro stability was seen for iso-[211At]AGMB-PODS-5F7GGC. Excellent tumor targeting was observed for iso-[211At]AGMB-PODS-5F7GGC. These results suggest that iso-[211At]AGMB-PODS-5F7GGC — as well as its 131I-labeled analogue — warrant further evaluation as α- and β-emitting targeted radiotherapeutics for the treatment of HER2-expressing malignancies.
EXPERIMENTAL SECTION
General.
Sodium [125I]iodide (629 GBq/mg) in 0.1 M NaOH was obtained from PerkinElmer (Boston, MA). Sodium [131I]iodide (936 GBq/mg, 236.097 GBq/mL) was obtained from International Isotopes Inc. (Idaho Falls, ID) in a solution of 0.04 M sodium thiosulfate, 0.2 M NaOH and 0.2 M sodium carbonate. All reagents and solvents were purchased from Fisher Scientific unless otherwise stated. Boc2-iso-SGMTB, Boc2-iso-SGMIB, Boc2-MEGMTB, PODS, and PODS-DOTA were synthesized following reported methods (16, 21, 22). Production, purification and characterization details for the anti-HER2 sdAb 5F7GGC have been reported previously (4, 16). Synthesis of [125/131I]MEGMIB-5F7GGC was performed following a reported method (16); its 211At-labeled analogue, [211At]MEAGMB-5F7GGC, was synthesized using an essentially identical procedure. The anti-HER2 antibody, trastuzumab (Roche/Genentech), was obtained from the Duke University Medical Center Pharmacy. All instruments were calibrated and maintained according to standard quality control practices and procedures. Reversed-phase HPLC (RP-HPLC) purification of unlabeled compounds was performed on a Shimadzu HPLC system (Shimadzu Scientific Instruments, Kyoto, Japan). Both the analytical (250 × 2 mm, 5 um, 300 Å) and preparative (250 × 10 mm, 5 um, 300 Å) HPLC columns were purchased from Phenomenex (Jupiter Proteo HPLC columns, Phenomenex, Torrance, CA). HPLC analyses were performed using LabSolutions LC/GC software (Shimadzu Scientific Instruments, Kyoto, Japan). Evaporation of solvents was accomplished with either a vacuum evaporator (Biotage V-10 Touch, V10-2XX, Biotage, Uppsala, Sweden) or a rotary evaporator (Hei-VAP Core, Heidolph, Schwabach, Germany). Gel permeation (GP) HPLC was utilized for purification and identification of the 5F7GGC-prosthetic agent conjugates using an Agilent PL Multisolvent 20 column eluted with pure Milli-Q® water as the mobile phase. Purification of radiolabeled compounds was performed with Agilent 1260 Infinity systems using a reversed-phase HPLC column (Agilent Poroshell 120 C18, 2.7 μm, 4.6 × 50 mm) eluted at a flow rate of 2 mL/min with a gradient consisting of water (solvent A) and acetonitrile (solvent B), both containing 0.1% TFA; the proportion of B was increased linearly from 40% to 80% over 8 min. This system was equipped with a 1260 Infinity Multiple Wavelength Detector (Santa Clara, CA). For monitoring radioactivity, one system was connected to a Dual Scan-RAM flow activity detector/TLC scanner and the other to a Flow-RAM detector (Lablogic, Tampa, FL); both the HPLC and the gamma detectors were controlled by LabLogic Laura software. A CRC-7 dose calibrator (Capintec, Pittsburgh, PA) was used to measure radioactivity at higher levels and for assessing lowe activity levels, either an LKB 1282 (Wallac, Finland) or a Perkin Elmer Wizard II (Shelton, CT) automated gamma counter was used.
Synthesis of Boc2-iso-GMTB-PODS.
N,N-diisopropylethylamine (DIPEA, 8.0 μL, 45.8 μmol, 3.0 eq.) was added to a solution of PODS (24.8 mg, 45. 8 μmol, 3.0 eq.) in 1.0 mL anhydrous dimethylformamide (DMF). A solution of Boc2-iso-SGMTB (10 mg, 15.3 μmol, 1.0 eq.) in 1.0 mL DMF was then added to above and the resultant solution was stirred at room temperature overnight protecting it from light. To determine the progress of the reaction, RP-HPLC was performed using the analytical column (1 mL/min, 5–95% acetonitrile in water (0.1% TFA) over 35 min; tR of product: 25.4 min). Once the reaction was deemed complete, the DMF was evaporated completely with a vacuum evaporator. The resultant crude product was re-constituted in 2 mL acetonitrile, and the solution was filtered with a syringe filter. The product was purified via RP-HPLC using a preparative column (6 mL/min, 5–95% acetonitrile in water (0.1% TFA) in 35 min; tR, product: 29.6 min). The purity of isolated sample was assessed using analytical RP-HPLC (1 mL/min, 5–95% acetonitrile in water with 0.1% TFA over 35 min; tR of product: 25.4 min; >95% purity). The solvents from the pooled HPLC fractions containing the product were removed by lyophilization overnight to yield a white powder (4.9 mg, 30.0% yield): 1H NMR (500 MHz, CDCl3) δ: 9.90 (s, 1H), 9.50 (s, 1H), 7.99 (d, 2H, J = 8.8 Hz), 7.74 (m, 3H), 7.58 (s, 1H), 7.45 (s, 1H), 7.07 (s, 1H), 6.87 (s, 1H), 5.17 (s, 2H), 3.47–3.64 (m, 18H), 3.36 (q, 2H, J = 5.9 Hz), 2.66 (m, 2H), 2.57 (m, 2H), 1.90 (quint, 2H, J = 6.2 Hz), 1.74 (quint, 2H, J = 6.0 Hz), 1.48 (s, 9H), 1.36 (s, 9H), 0.27 (s, 9H). 13C NMR (500 MHz, DMSO-d6) δ: 171.8, 171.5, 167.0, 166.2, 163.2, 162.2, 160.1, 154.5, 144.1, 142.5, 138.5, 137.5, 134.4, 132.9, 129.0, 126.7, 119.6, 116.4, 84.3, 78.3, 70.2, 70.0, 68.7, 68.5, 47.5, 43.4, 37.1, 36.3, 32.2, 30.5, 29.9, 29.8, 28.4, 27.8, 8.8. ESI-MS m/z calculated for C45H69N8O13SSn (M+H)+: 1080.85; found: 1081.0.
Synthesis of Boc2-iso-GMIB-PODS.
DIPEA (6.4 μL, 36.5 μmol, 3.0 eq.) was added to a solution of PODS (19.7 mg, 36.5 μmol, 3.0 eq.) in 1.0 mL DMF. Next, a solution of Boc2-iso-SGMIB (7.5 mg, 12.2 μmol, 1.0 eq.) in 1.0 mL of DMF was added to the PODS mixture. The reaction mixture was stirred at room temperature overnight following the progress of the reaction by RP-HPLC, which was performed using the analytical column (1 mL/min, 5–95% acetonitrile in water with 0.1% TFA in 35 min; tR of product: 23.5 min). When the reaction was deemed to be complete, the DMF was evaporated completely with a vacuum evaporator. The resulting crude product was then reconstituted with 2 mL acetonitrile, and the solution was filtered with a syringe filter. The product was purified via RP-HPLC using a preparative column (6 mL/min, 5–95% acetonitrile in water with 0.1% TFA over 35 min; tR of product: 28.0 min). The purity of isolated sample was assessed using analytical RP-HPLC (1 mL/min, 5–95% acetonitrile in water with 0.1% TFA, 35 min; tR of product: 23.5 min; >95% purity). The pooled HPLC fractions containing the product were lyophilized overnight to yield a white powder (8.5 mg, 67.0% yield): 1H NMR (500 MHz, CDCl3) δ: 9.75 (s, 1H), 9.5 (s, 1H), 8.00 (d, 2H, J = 8.8 Hz), 7.85 (s, 1H), 7.65 (d, 2H, J = 8.5 Hz), 7.60 (s, 1H), 7.56 (s, 1H), 7.30 (s, 1H), 6.87 (s, 1H), 5.13 (s, 2H), 3.52–3.69 (m, 15H), 3.39 (q, 2H, J = 6.0 Hz), 3.51 (s, 3H), 2.70 (m, 2H), 2.59 (m, 2H), 1.90 (quint, 2H, J = 6.1 Hz), 1.74 (quint, 2H, J = 5.9 Hz), 1.48 (s, 9H), 1.36 (s, 9H).13C NMR (500 MHz, DMSO-d6) δ: 171.8, 171.4, 166.2, 165.0, 163.1, 162.2, 159.9, 154.3, 144.2, 141.7, 138.7, 137.0, 134.2, 129.0, 126.0, 119.6, 116.4, 84.4, 78.4, 70.3, 70.0, 68.6, 68.5, 37.2, 36.3, 32.3, 30.5, 29.8, 29.7, 28.4, 27.8. ESI-MS m/z calculated for C42H60N8O13SI (M+H)+: 1043.94; found: 1044.0.
Synthesis of iso-GMIB-PODS.
Trifluoroacetic acid (TFA; 0.7 mL) was added to a solution of Boc2-iso-GMIB-PODS (5.0 mg, 4.8 μmol) in 2.0 mL dichloromethane and the mixture stirred at room temperature for 3 h. The volatiles were evaporated using a rotary evaporator, the crude product re-constituted with 2 mL acetonitrile, and the resultant solution filtered with a syringe filter. The product was purified via RP-HPLC using a preparative column (6 mL/min, 5–95% acetonitrile in water with 0.1% TFA over 35 min; tR of product: 19.3 min). The purity of the isolated sample was assessed using analytical RP-HPLC (1 mL/min, 5–95% acetonitrile in water with 0.1% TFA over 35 min; tR of product: 15.4 min; >95% purity). The pooled HPLC fractions containing the product were lyophilized overnight to yield a white powder (4.0 mg, 87.1% yield; trifluoroacetate salt): 1H NMR (500 MHz, DMSO-d6) δ: 10.42 (s, 1H), 8.57 (t, 1H, J = 5.3 Hz), 8.12 (s, 1H), 8.03 (m, 3H), 7.86 (m, 3H), 7.79 (d, 2H, J = 7.8 Hz), 7.28 (s, 3H), 4.38 (d, 2H, J = 6.0 Hz), 3.69 (s, 3H), 3.25–3.54 (m, 14H), 3.07 (q, 2H, J = 6.3 Hz), 2.61 (t, 2H, J = 6.9 Hz), 2.41 (t, 2H, J = 7.0 Hz), 1.74 (quint, 2H, J = 6.6 Hz), 1.61 (quint, 2H, J = 6.6 Hz). 13C NMR (500 MHz, DMSO-d6) δ: 171.8, 171.5, 166.2, 162.2, 144.1, 129.0, 119.6, 116.4, 70.2, 70.1, 70.0, 68.7, 68.5, 43.4, 40.9, 36.3, 32.2, 30.5, 29.8, 29.1. ESI-MS m/z calculated for C32H46N8O9SI (M+H)+: 844.7; found: 843.6.
Syntheses and Validation of the Anti-HER2 sdAb Conjugates.
The sdAb conjugate iso-GMIB-PODS-5F7GGC was synthesized by a slight modification of a previously reported method for thiol-maleimide conjugation (16). Briefly, the monomeric sdAb 5F7GGC was freshly made by treatment of the sdAb (containing some spontaneously formed dimer) with gel-immobilized TCEP, and iso-GMIB-PODS was added to that solution at a 3:1 iso-GMIB-PODS:5F7GGC molar ratio. The mixture was incubated at 37 oC for 1 h. The iso-GMIB-PODS-5F7GGC conjugate was purified and its molecular weight determined by GP HPLC with an Agilent PL Multisolvent 20 column, connected to an Advion ExpressionL CMS LC-MS System(Ithaca, NY). DOTA-PODS-5F7GGC and Lu-DOTA-PODS-5F7GGC conjugates were synthesized and purified in similar fashion. The binding affinity of the conjugates for HER2 was measured by surface plasmon resonance (Biacore 3000 System, Duke Human Vaccine Institute Shared Resource) using a CM5 sensor chip coated with HER2-Fc protein via NHS activation.
Synthesis of [211At]MEAGMB-5F7GGC sdAb Conjugate.
Synthesis of [211At]MEAGMB and its subsequent conjugation to 5F7GGC was performed by adapting a previously reported method (6, 16). Briefly, 211At, produced as described previously (6), in NCS/methanol (~370 MBq; 200 μL) was added to a vial containing Boc2-MEGMTB (50 μg, 0.07 μmol), and then 2 μL of glacial acetic acid was added. The vial was vortexed for a short time and the reaction was allowed to proceed at 20 °C for 20 min. The volatiles were evaporated under a stream of argon and the residual activity reconstituted in 40% acetonitrile in water (100 μL). This solution was injected onto a reversed-phase HPLC column eluted a flow rate of 2 mL/min with a gradient consisting of 0.1% TFA in water (solvent A) and 0.1% TFA in acetonitrile (solvent B); the proportion of B was linearly increased from 40% to 80% over 8 min. The HPLC fractions containing the product (tR = 4.0 min) were pooled and most of the acetonitrile was removed using a stream of argon. Boc2[211At]MEAGMB was extracted with 2 × 1 mL of ethyl acetate and transferred to a half-dram glass vial. Ethyl acetate was evaporated with argon, TFA (100 μL) was added and the deprotection of Boc2-[211At]MEAGMB was allowed to proceed at 20 °C for 10 min. Subsequently, TFA was removed under a stream of argon, followed by co-evaporation with ethyl acetate (100 μL × 3). Monomeric sdAb 5F7GGC (~100 μg, 60 μL), freshly obtained as described above, was added to the vial containing [211At]MEAGMB and the conjugation was carried out at 37 °C for 45 min. The labeled sdAb was isolated by gel filtration over a PD-10 column using PBS as the mobile phase. Fractions containing [211At]MEAGMB-5F7GGC were pooled for use in the biological experiments described below.
Synthesis of iso-[131I]GMIB-PODS-5F7GGC.
Synthesis of iso-[131I]GMIB-PODS (see below) to monomeric 5F7GGC was performed by adapting a method previously reported for similar compounds (16). Briefly, a methanolic solution (100 μL) of N-chlorosuccinimide (NCS) (0.2 mg/mL), acetic acid (1%;v/v) and sodium [131I]iodide (1–5 μL, 37–185 MBq) was added to a half-dram glass vial containing Boc2-iso-GMTB-PODS (50 μg, 0.05 μmol). The vial was vortexed and the reaction was allowed to proceed at 20 °C for 15 min. The volatiles were evaporated with a stream of argon and the residual activity reconstituted in 40% acetonitrile in water (100 μL). This solution was injected onto a reversed-phase HPLC column eluted at a flow rate of 2 mL/min with a gradient consisting of 0.1% TFA in water (solvent A) and 0.1% TFA in acetonitrile (solvent B); the proportion of B was linearly increased from 40% to 80% over 8 min. The HPLC fractions containing the product (tR = 3.0 min) were pooled and most of the acetonitrile was removed using a stream of argon. The product Boc2-iso-[131I]GMIB-PODS was extracted with 0.5 mL of ethyl acetate and transferred to an half-dram glass vial and the ethyl acetate was evaporated with argon, TFA (100 μL) added and the deprotection of Boc2-iso-[131I]GMIB-PODS was performed at 20 oC for 10 min. Subsequently, TFA was removed with a stream of argon, followed by co-evaporation with ethyl acetate (100 μL × 3). Conjugation of iso-[131I]GMIB-PODS to monomeric 5F7GGC was performed similarly as reported for [131I]MEGMIB (16). Briefly, monomeric sdAb 5F7GGC (~100 μg, 60 μL), freshly obtained as described above, was added to the vial containing iso-[131I]GMIB-PODS and the conjugation was carried out at 37 °C for 45 min. The labeled sdAb was isolated by gel filtration over a PD-10 column using PBS as the mobile phase. Fractions containing the iso-[131I]GMIB-PODS-5F7GGC were pooled for use in the biological experiments described below.
Synthesis of iso-[211At]AGMB-PODS-5F7GGC.
A method previously reported for the synthesis of iso-[211At]SAGMB (6) was adapted for the synthesis of iso-[211At]AGMB-PODS at relatively high 211At activity levels. Astatine-211 in NCS/methanol (~370 MBq; 200 μL) was added to a vial containing Boc2-iso-GMTB-PODS (50 μg, 0.05 μmol), the vial vortexed briefly, and the reaction was allowed to proceed at 20 °C for 15 min. The volatiles were evaporated under a stream of argon and the residual activity reconstituted in 40% acetonitrile in water (100 μL). This solution was injected onto a reversed-phase analytical HPLC column eluted with the same gradient described above. The HPLC fractions containing the product, Boc2-iso-[211At]AGMB-PODS (tR = 3.3 min), were pooled and most of the acetonitrile was removed using a stream of argon. Ethyl acetate (1 mL) was added to extract the Boc2-iso-[211At]AGMB-PODS, the mixture was vortexed for 10 s and the ethyl acetate layer was transferred to a half-dram vial. The ethyl acetate was removed using a stream of argon and TFA was added and incubated at room temperature for 10 min. The TFA was evaporated under a stream of argon, and the remaining TFA was removed by co-evaporating with ethyl acetate (100 uL x 3). Freshly reduced 5F7GGC was added and the mixture was incubated at 37 °C for 45 min. The iso-[211At]AGMB-PODS-5F7GGC was isolated using a PD-10 column as described above.
Synthesis of [177Lu]Lu-DOTA-PODS-5F7GGC.
Lutetium-177, produced at the University of Missouri Research Reactor, was purchased from the National Isotope Development Center, Oak Ridge National Laboratory. Upon receipt, 177Lu (185 MBq, 10 μL) was diluted with 0.1 M HCl (50 μL) and added to a 1 mL Eppendorf tube and DOTA-PODS (150 μL, 1 mg/mL, in 0.2 M NH4OAc, pH=6.3) was added. The reaction mixture was vortexed for 30 s and placed at room temperature for 30 min. After that, the reaction mixture was diluted with 10 mL water and passed through a preconditioned C18 Sep-Pak® cartridge (Waters; 60 mg). The product activity was eluted from the cartridge with ethanol (400 μL) into a half-dram glass vial and the ethanol was evaporated with a gentle stream of argon. Monomeric 5F7GGC sdAb, freshly obtained as described above, was added to the vial containing [177Lu]Lu-DOTA-PODS (180 MBq) and the conjugation was carried out at 37 °C for 45 min. After that, the conjugate was treated with 50 mM EDTA to remove any adventitiously bound 177Lu. The labeled sdAb was isolated by gel filtration over a PD-10 column using PBS as the mobile phase. Fractions containing the [177Lu]Lu-DOTA-PODS-5F7GGC were pooled for use in the biological experiments described below.
Cells and Culture Conditions.
Reagents for cell culture were obtained from Thermo Fisher Scientific (Waltham, MA), except where noted. Cells were cultured at 37 °C in a 5% CO2 humidified incubator. HER2-positive BT474 human breast carcinoma cells were obtained from Duke University Cell Culture Facility and grown in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% FBS, supplemented with 10 mg/ml bovine insulin. SKOV-3 human ovarian carcinoma cells were obtained from the Duke University Cell Culture Facility and grown in McCoy’s 5A medium containing 10% fetal bovine serum and 1% penicillin-streptomycin.
Quality Control of Radiolabeled sdAb Conjugates.
The radiochemical purity of the sdAb conjugates was evaluated by SDS-PAGE/phosphor imaging and GP-HPLC using the Agilent PL Multisolvent 20 column described above. The immunoreactive fraction was determined by the Lindmo method (24) following a reported procedure (39). The HER2 binding affinity of iso-[131I]GMIB-PODS-5F7GGC was determined on SKOV-3 and BT474 cells using a saturation binding assay as described before (40, 41). The binding affinity of iso-[211At]AGMB-PODS-5F7GGC and [211At]MEAGMB-5F7GGC were measured in similar fashion on BT474 cells. Nonspecific binding was determined in parallel assays performed by co-incubating cells with a 100-fold molar excess of trastuzumab.
In vitro Cellular Retention and Internalization Assays.
Cellular retention and intracellular trapping of iso-[131I]GMIB-PODS-5F7GGC in BT474 cells were assessed in single-label format using procedures reported for similar molecules (6, 42). In addition, a paired-label comparison of iso-[131I]GMIB-PODS-5F7GGC and [125I]MEGMIB-5F7GGC was performed on BT474 cells following reported procedures (6, 16). Briefly, cells were incubated with the radiolabeled sdAbs at 4 °C for 30 min, washed with PBS, replenished with fresh medium, and incubated at 37 °C for 1–4 h. Total cell associated and internalized radioactivity was measured at each time point as reported (16). A paired-label comparison of iso-[131I]GMIB-PODS-5F7GGC and iso-[211At]AGMB-PODS-5F7GGC was performed similarly. In these assays, nonspecific uptake was determined in parallel experiments by co-incubating cells with 100-fold molar excess of trastuzumab.
In vitro Stability of [211At]MEAGMB-5F7GGC and iso-[211At]AGMB-PODS-5F7GGC.
The in vitro stability of the 211At-labeled conjugates was evaluated in PBS, 50 mM cysteine solution and human serum albumin (50% in PBS). Briefly, the 211At-labeled sdAb conjugate (3.7 MBq, ~12 μg sdAb) was added to 1 mL of each solution in 10 mL centrifuge tubes. The tubes containing the conjugates were vortexed for 10 s and left at room temperature. The radiochemical purity of each conjugate was determined by SDS-PAGE and phosphor imaging of aliquots of the incubates after 3 h and 21 h.
Biodistribution.
All experiments involving animals were performed under a protocol approved by the Duke University IACUC. Subcutaneous BT474 xenografts were established by inoculating 5-week old female athymic mice (~25 g, obtained from the Duke Cancer Institute) with 20 × 106 BT474 cells in 50% Matrigel (Corning Inc., NY) in the above medium (100 μL), 2 d after implanting an estrogen pellet (0.72 mg 17β-Estradiol; Innovative Research of America, Sarasota, FL) subcutaneously in the back of the neck. Five biodistribution studies were performed: iso-[131I]GMIB-PODS-5F7GGC in tandem with [125I]MEGMIB-5F7GGC in paired-label format in tumor-free athymic mice (1, 4, and 24 h p.i.); iso-[131I]GMIB-PODS-5F7GGC in tandem with [125I]MEGMIB-5F7GGC in paired-label format in athymic mice bearing subcutaneous BT474 xenografts (1, 4, and 24 h p.i.); iso-[131I]GMIB-PODS-5F7GGC in tandem with iso-[211At]AGMB-PODS-5F7GGC in paired-label fashion in athymic mice with subcutaneous BT474 xenografts (1, 4, and 21 h p.i.); iso-[211At]AGMB-PODS-5F7GGC in athymic mice bearing subcutaneous BT474 xenografts (1, 4, and 24 h p.i.); and iso-[125I]GMIB-PODS-5F7GGC in tandem with [177Lu]Lu-DOTA-PODS-5F7GGC in paired-label format in athymic mice bearing BT474 xenografts (1, 4, and 24 h p.i.). In these studies, each mouse received 0.11 –0.22 MBq (1–2 μg) of each labeled sdAb conjugate via the tail vein. At each time point, blood and urine were collected and the mice were killed by an overdose of isofluorane. Tumor and other tissues were harvested, blot-dried, weighed and counted along with injection standards for the administered radionuclides using a dual-channel automated gamma counter. From these counting data, the percentage of the injected dose (%ID) per organ, per gram of tissue (%ID/g) and tumor-to-normal tissue ratios were calculated.
Supplementary Material
Scheme 2.
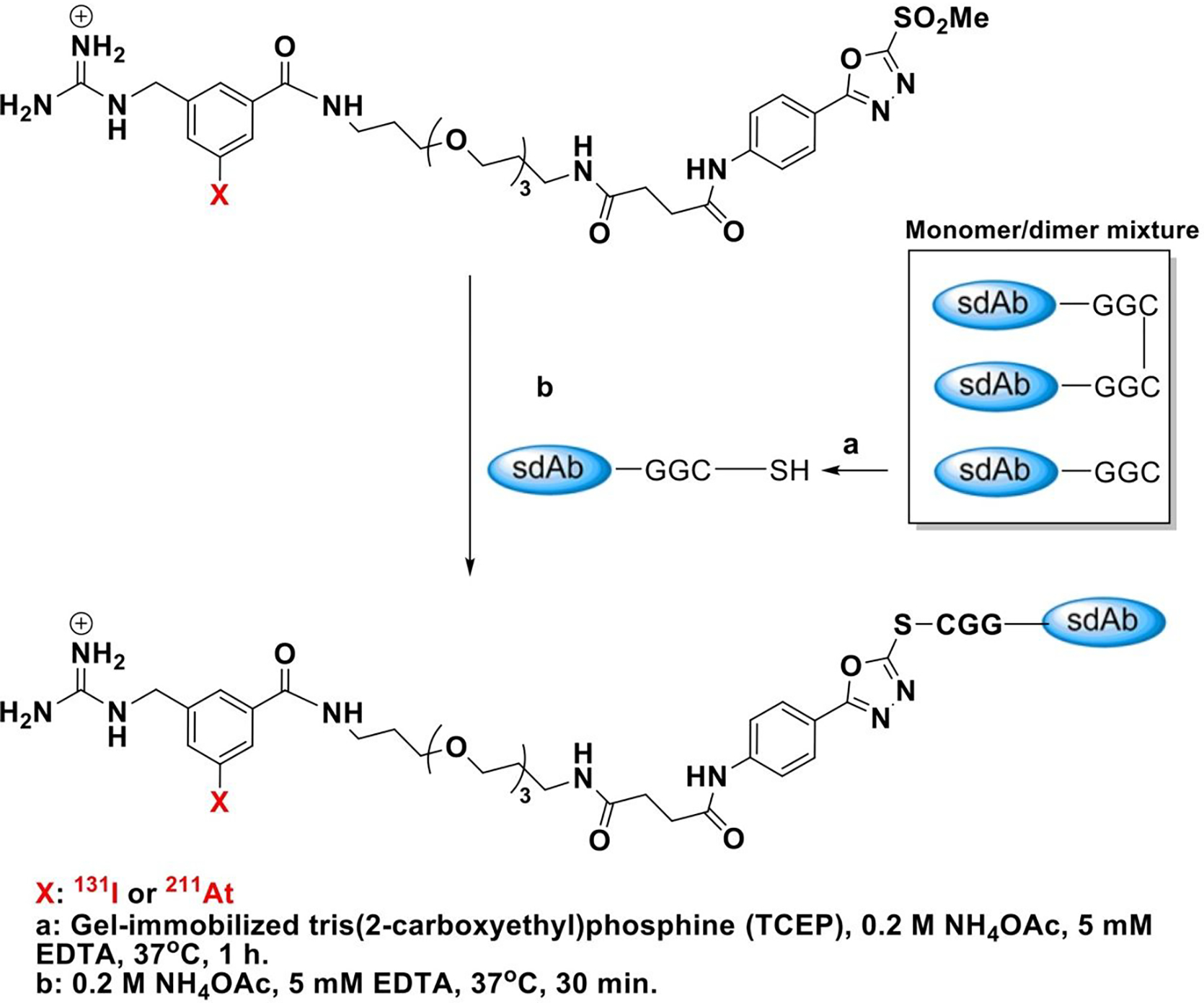
Synthesis of iso-[131l]GMIB-PODS- and iso-[211At]AGMB-PODS-5F7 GGC
Footnotes
The authors declare the following competing financial interest: Y.F., S.M.S., G.V., B.M.Z. and M.R.Z. are co-inventors on a patent application covering this PODS radiohalogenation method and as such may be entitled to a portion of any licensing fees and royalties generated by this technology. That arrangement has been reviewed and approved by Duke University and Hunter College in accordance with their conflict-of-interest policies.
ASSOCIATED CONTENT
Supporting Information
Supplemental Figure 1. Analytical HPLC and ESI-MS analyses for Boc2-iso-GMTB-PODS precursor, Boc2-iso-GMIB-PODS standard and iso-GMIB-PODS standard.
Contributor Information
Yutian Feng, Department of Radiology, Duke University Medical Center, Durham, North Carolina 27710, United States.
Samantha M. Sarrett, Hunter College, City University of New York, New York, New York, 10021 United States Ph.D. Program in Biochemistry, The Graduate Center, City University of New York, New York, New York, 10021.
Rebecca L. Meshaw, Department of Radiology, Duke University Medical Center, Durham, North Carolina 27710, United States
Ganesan Vaidyanathan, Department of Radiology, Duke University Medical Center, Durham, North Carolina 27710, United States.
Mike A. Cornejo, Hunter College, City University of New York, New York, New York 10021, United States Ph.D. Program in Chemistry, The Graduate Center, City University of New York, New York, New York 10021, United States.
Brian M. Zeglis, Hunter College, City University of New York, New York, New York 10021 United States Ph.D. Programs in Biochemistry and Chemistry, The Graduate Center, City University of New York, New York, New York 10021, United States; Department of Radiology, Memorial Sloan Kettering Cancer Center, New York 10065, United States.
Michael R. Zalutsky, Department of Radiology, Duke University Medical Center, Durham, North Carolina 27710, United States
REFERENCES
- 1.Piramoon M, Khodadust F, Hosseinimehr SJ. Radiolabeled nanobodies for tumor targeting: From bioengineering to imaging and therapy. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 2021:188529. [DOI] [PubMed] [Google Scholar]
- 2.Küppers J, Kürpig S, Bundschuh RA, Essler M, & Lütje S (2021). Radiolabeling Strategies of Nanobodies for Imaging Applications. Diagnostics, 2021: 11(9), 1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.D’Huyvetter M, De Vos J, Caveliers V, Vaneycken I, Heemskerk J, Duhoux FP, et al. Phase I trial of 131I-GMIB-Anti-HER2-VHH1, a new promising candidate for HER2-targeted radionuclide therapy in breast cancer patients. J. Nucl. Med. 2021;62(8):1097–105. [DOI] [PubMed] [Google Scholar]
- 4.Pruszynski M, Koumarianou E, Vaidyanathan G, Revets H, Devoogdt N, Lahoutte T, et al. Targeting breast carcinoma with radioiodinated anti-HER2 Nanobody. Nucl. Med. Biol. 2013;40(1):52–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feng Y, Meshaw R, McDougald D, Zhou Z, Zhao X-G, Jannetti SA, et al. Evaluation of an 131I-labeled HER2-specific single domain antibody fragment for the radiopharmaceutical therapy of HER2-expressing cancers. Sci. Rep. 2022;12(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi J, Vaidyanathan G, Koumarianou E, Kang CM, Zalutsky MR. Astatine-211 labeled anti-HER2 5F7 single domain antibody fragment conjugates: radiolabeling and preliminary evaluation. Nucl. Med. Biol. 2018;56:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dekempeneer Y, Caveliers V, Ooms M, Maertens D, Gysemans M, Lahoutte T, et al. Therapeutic efficacy of 213Bi-labeled sdAbs in a preclinical model of ovarian cancer. Mol. Pharm. 2020;17(9):3553–66. [DOI] [PubMed] [Google Scholar]
- 8.Pruszynski M, D’Huyvetter M, Bruchertseifer F, Morgenstern A, Lahoutte T. Evaluation of an anti-HER2 nanobody labeled with 225Ac for targeted α-particle therapy of cancer. Mol. Pharm. 2018;15(4):1457–66. [DOI] [PubMed] [Google Scholar]
- 9.Feng Y, Meshaw R, Zhao X-G, Jannetti SA, Vaidyanathan G, Zalutsky MR. Effective Treatment of Human Breast Carcinoma Xenografts with Single-Dose 211At-Labeled Anti-HER2 Single Domain Antibody Fragment. J. Nucl. Med. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Strop P, Liu S-H, Dorywalska M, Delaria K, Dushin RG, Tran T-T, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem. Biol. 2013;20(2):161–7. [DOI] [PubMed] [Google Scholar]
- 11.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26(8):925–32. [DOI] [PubMed] [Google Scholar]
- 12.Pillow TH, Tien J, Parsons-Reponte KL, Bhakta S, Li H, Staben LR, et al. Site-specific trastuzumab maytansinoid antibody–drug conjugates with improved therapeutic activity through linker and antibody engineering. J. Med. Chem. 2014;57(19):7890–9. [DOI] [PubMed] [Google Scholar]
- 13.Anami Y, Otani Y, Xiong W, Ha SY, Yamaguchi A, Rivera-Caraballo KA, … & Tsuchikama K Homogeneity of antibody-drug conjugates critically impacts the therapeutic efficacy in brain tumors. Cell Reports, 2022: 39(8), 110839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ravasco JM, Faustino H, Trindade A, Gois PM. Bioconjugation with maleimides: a useful tool for chemical biology. Chemistry–A European Journal. 2019;25(1):43–59. [DOI] [PubMed] [Google Scholar]
- 15.Adumeau P, Sharma SK, Brent C, Zeglis BM. Site-specifically labeled immunoconjugates for molecular imaging—part 1: cysteine residues and glycans. Mol. Imaging. Biol. 2016;18(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng Y, Zhou Z, McDougald D, Meshaw RL, Vaidyanathan G, Zalutsky MR. Site-specific radioiodination of an anti-HER2 single domain antibody fragment with a residualizing prosthetic agent. Nucl. Med. Biol. 2021;92:171–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dekempeneer Y, Bäck T, Aneheim E, Jensen H, Puttemans J, Xavier C, et al. Labeling of anti-HER2 nanobodies with astatine-211: Optimization and the effect of different coupling reagents on their in vivo behavior. Mol. Pharm. 2019;16(8):3524–33. [DOI] [PubMed] [Google Scholar]
- 18.Aneheim E, Gustafsson A, Albertsson P, Back T, Jensen H, Palm S, et al. Synthesis and evaluation of astatinated N-[2-(maleimido) ethyl]-3-(trimethylstannyl) benzamide immunoconjugates. Bioconjug. Chem 2016;27(3):688–97. [DOI] [PubMed] [Google Scholar]
- 19.Toda N, Asano S, Barbas CF III. Rapid, Stable, Chemoselective Labeling of Thiols with Julia–Kocieński-like Reagents: A Serum-Stable Alternative to Maleimide-Based Protein Conjugation. Angewandte Chemie International Edition. 2013;52(48):12592–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patterson JT, Asano S, Li X, Rader C, Barbas CF III. Improving the serum stability of site-specific antibody conjugates with sulfone linkers. Bioconjug. Chem 2014;25(8):1402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adumeau P, Davydova M, Zeglis BM. Thiol-reactive bifunctional chelators for the creation of site-selectively modified radioimmunoconjugates with improved stability. Bioconjug. Chem. 2018;29(4):1364–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davydova M, Le Roi GD, Adumeau P, Zeglis BM. Synthesis and bioconjugation of thiol-reactive reagents for the creation of site-selectively modified immunoconjugates. Journal of visualized experiments: JoVE. 2019(145). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharma Sai Kiran, Adumeau Pierre, Keinanen Outi, Sisodiya Vikram, Sarvaiya Hetal, Tchelepi Robert, Korsen Joshua A. et al. Synthesis and Comparative In Vivo Evaluation of Site-Specifically Labeled Radioimmunoconjugates for DLL3-Targeted ImmunoPET. Bioconjug. Chem 32(7), 2021: 1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindmo T, Boven E, Cuttitta F, Fedorko J, Bunn P Jr. Determination of the immunoreactive function of radiolabeled monoclonal antibodies by linear extrapolation to binding at infinite antigen excess. J Immunol. Methods. 1984;72(1):77–89. [DOI] [PubMed] [Google Scholar]
- 25.Audenhove Van, Isabel, and Gettemans Jan. Nanobodies as versatile tools to understand, diagnose, visualize and treat cancer. EBioMedicine 8, 2016: 40–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Debie Pieterjan, Devoogdt Nick, and Hernot Sophie. Targeted nanobody-based molecular tracers for nuclear imaging and image-guided surgery. Antibodies 8(1), 2019: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tasumi Satoshi, Velikovsky C. Alejandro, Xu Gang, Gai S. Annie, Wittrup K. Dane, Flajnik Martin F., Mariuzza Roy A., and Pancer Zeev. High-affinity lamprey VLRA and VLRB monoclonal antibodies. Proceedings of the National Academy of Sciences 106(31), 2009: 12891–12896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kijanka Marta, Warnders Frank-Jan, El Khattabi Mohamed, Hooge Marjolijn Lub-de, van Dam Gooitzen M., Ntziachristos Vasilis, de Vries Liesbeth, Oliveira Sabrina, and van Bergen En Henegouwen Paul MP. Rapid optical imaging of human breast tumour xenografts using anti-HER2 VHHs site-directly conjugated to IRDye 800CW for image-guided surgery. European journal of nuclear medicine and molecular imaging 40(11), 2013: 1718–1729. [DOI] [PubMed] [Google Scholar]
- 29.Choi J, Vaidyanathan G, Koumarianou E, McDougald D, Pruszynski M, Osada T, et al. N-Succinimidyl guanidinomethyl iodobenzoate protein radiohalogenation agents: Influence of isomeric substitution on radiolabeling and target cell residualization. Nucl. Med. Biol. 2014;41(10):802–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’Huyvetter M, De Vos J, Xavier C, Pruszynski M, Sterckx YG, Massa S, et al. 131I-labeled anti-HER2 camelid sdAb as a theranostic tool in cancer treatment. Clin. Cancer. Res. 2017;23(21):6616–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeglis B, Adumeau P, Davydova M. Reagent for site-selective bioconjugation of proteins or antibodies. Google Patents; 2021.
- 32.Pruszynski M, Koumarianou E, Vaidyanathan G, Chitneni S, Zalutsky MR. D-amino acid peptide residualizing agents bearing N-hydroxysuccinimido-and maleimido-functional groups and their application for trastuzumab radioiodination. Nucl. Med. Biol. 2015;42(1):19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dreyer R, Dreyer I, Fischer S, Hartmann H, Rösch F. Synthesis and characterization of cationic astatine compounds with sulphur-containing ligands stable in aqueous solutions. Journal of radioanalytical and nuclear chemistry. 1985;96(3):333–41. [Google Scholar]
- 34.Visser GW, Diemer EL, Kaspersen FM. The nature of the astatine-protein bond. The International Journal of Applied Radiation and Isotopes. 1981;32(12):905–12. [DOI] [PubMed] [Google Scholar]
- 35.Bassal F, Champion J, Pardoue S, Seydou M, Sabatié-Gogova A, Deniaud D, et al. Questioning the Affinity of Electrophilic Astatine for Sulfur-containing Compounds: Unexpected Bindings Revealed. Inorg. Chem. 2020;59(19):13923–32. [DOI] [PubMed] [Google Scholar]
- 36.Vaidyanathan G, White BJ, Affleck DJ, Zhao XG, Welsh PC, McDougald D, Choi J, and Zalutsky MR. SIB-DOTA: A trifunctional prosthetic group potentially amenable for multimodal labeling that enhances tumor uptake of internalizing monoclonal antibodies. Bioorganic & medicinal chemistry 20(24), 2012: 6929–6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.D’huyvetter M, Vincke C, Xavier C, Aerts A, Impens N, Baatout S, et al. Targeted radionuclide therapy with A 177Lu-labeled anti-HER2 nanobody. Theranostics. 2014;4(7):708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’huyvetter M, Aerts A, Xavier C, Vaneycken I, Devoogdt N, Gijs M, et al. Development of 177Lu-nanobodies for radioimmunotherapy of HER2-positive breast cancer: evaluation of different bifunctional chelators. Contrast Media Mol Imaging. 2012;7(2):254–64. [DOI] [PubMed] [Google Scholar]
- 39.Foulon CF, Reist CJ, Bigner DD, Zalutsky MR. Radioiodination via D-amino acid peptide enhances cellular retention and tumor xenograft targeting of an internalizing anti-epidermal growth factor receptor variant III monoclonal antibody. Cancer Res. 2000;60(16):4453–60. [PubMed] [Google Scholar]
- 40.Zhou Z, Chitneni SK, Devoogdt N, Zalutsky MR, Vaidyanathan G. Fluorine-18 labeling of an anti-HER2 VHH using a residualizing prosthetic group via a strain-promoted click reaction: Chemistry and preliminary evaluation. Bioorg. Med. Chem. 2018;26(8):1939–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vaidyanathan G, McDougald D, Choi J, Pruszynski M, Koumarianou E, Zhou Z, et al. N-Succinimidyl 3-((4-(4-[18 F] fluorobutyl)-1 H-1, 2, 3-triazol-1-yl) methyl)-5-(guanidinomethyl) benzoate ([18 F] SFBTMGMB): a residualizing label for 18 F-labeling of internalizing biomolecules. Org. Biomol. Chem. 2016;14(4):1261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Z, Vaidyanathan G, McDougald D, Kang CM, Balyasnikova I, Devoogdt N, et al. Fluorine-18 labeling of the HER2-targeting single-domain antibody 2Rs15d using a residualizing label and preclinical evaluation. Mol. Imaging. Biol. 2017;19(6):867–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.