

Abstract
Beta-glucocerebrosidase is a lysosomal hydrolase, encoded by GBA1 that represents the most common risk gene associated with Parkinson’s disease (PD) and Lewy Body Dementia. Glucocerebrosidase dysfunction has been also observed in the absence of GBA1 mutations across different genetic and sporadic forms of PD and related disorders, suggesting a broader role of glucocerebrosidase in neurodegeneration. In this review, we highlight recent advances in mechanistic characterization of glucocerebrosidase function as the foundation for development of novel therapeutics targeting glucocerebrosidase in PD and related disorders.
Keywords: Glucocerebrosidase, GBA1, Parkinson’s disease, neurodegeneration, lysosomes
Graphical Abstract
Introduction
Parkinson’s Disease (PD) is a debilitating neurodegenerative movement disorder that presents with a diverse array of symptomatology. The disease signature most notably associated with motor symptoms (bradykinesia, rigidity, postural instability, and tremor) is the selective degeneration of dopaminergic neurons in the substantia nigra [1]. However, PD is a progressive disorder that also affects other neuronal subpopulations leading to non-motor symptoms such as cognitive decline, behavioral and mood disorders, and autonomic dysfunction [2,3]. PD is pathologically characterized by eosinophilic inclusions known as Lewy bodies or Lewy neurites consisting of the aggregated protein alpha-synuclein (αSyn) [4].
Although predominantly known as an idiopathic disorder, approximately 15% of PD cases are considered familial with up to 10% inherited in Mendelian pattern [5]. Several genetic risk factors identified since the late 1990s have been studied to better understand convergent mechanisms potentially applicable to sporadic PD [6,7]. Interestingly, more than half of PD risk genes identified in GWAS studies are associated with putative variants linked to lysosomal storage disorders [8]. GBA1, the gene encoding for beta-glucocerebrosidase (GCase), is the most common genetic risk factor for PD that has been involved in PD pathogenesis [9,10]. Glucocerebrosidase is a member of the coordinated lysosomal expression and regulation (CLEAR) network that functions in glycosphingolipid processing and ceramide metabolism [11,12]. Given the established genetic link between GBA1 and PD, several studies have aimed to understand GBA1-based mechanisms that contribute to PD-related neurodegeneration.
Several excellent reviews have comprehensively described the link of GCase deficiency and dysfunction to PD. In this review, we provide an update on mechanistic studies exploring the role of GCase in PD, including how non-cell autonomous GCase dysfunction may contribute to PD pathogenesis, and highlight important considerations to better understand GCase-pathophysiology and effective targeting for therapeutic development.
The Association of Gaucher Disease and GBA1-PD
Glucocerebrosidase is a 497-amino acid protein which functions within the acidic lumen of lysosomes to hydrolyze glycolipids and sphingolipids. Synthesized GCase is transported from the endoplasmic reticulum (ER) to the lysosome by the lysosomal integral membrane protein-2 (LIMP2) encoded by SCARB2 [13]. Lysosomal GCase functions independently from homologous cytosolic glucosidases, glucocerebrosidase-2 and -3 (GBA2/GBA3), which do not have genetic associations with PD, but GBA2 has been linked to hereditary spastic paraplegia [14]. Biallelic mutations in the GBA1 gene are known to cause Gaucher Disease (GD), a rare, pan-ethnic lysosomal storage disorder that ranges in a broad spectrum of clinical presentations and ages of onset [15]. Although a majority of GD cases present as a disease of the peripheral organs (known as Type 1 GD), a small fraction of GD cases manifest in neuronopathic disease (Types 2 and 3) which features focal neurodegeneration and brainstem dysfunction [15,16]. A vast majority of GD associated mutations cause loss-of GCase activity which leads to substrate accumulation in lysosomes, most commonly observed in the form of engorged macrophages (termed “Gaucher Cells”) clustered in the spleen, liver, lungs, and bone marrow [16]. The severity and rate of progression of disease is variable amongst GD patients and associated to particular risk variants. For example, the most common missense mutation, p.N370S (now commonly referred to as p.N409S due to an updated annotation featuring an additional 39-residue leader sequence) typically is characterized by milder phenotypes associated with Type 1 GD [17,18]. The other common variant, p.L444P (i.e. p.L483P), is observed across all three subtypes of GD and is considered a more severe mutation. To date, almost 400 mutations in the GBA1 locus have been identified throughout coding and non-coding sequences, possibly contributing to heterogeneity in disease course and progression [19].
In 1996, Neudorfer described six cases of GD-associated Parkinsonism with cardinal features including tremor, rigidity, bradykinesia, and speech impairment [20]. These findings were later validated in larger studies showing a higher propensity of GD patients to develop PD [18,21]. Interestingly, the higher incidence of Parkinsonism in GD patients was also observed in first- and second-degree family members of GD patients, indicating that GCase function, even in heterozygous carriers, may be an important factor in PD pathogenesis [22,23]. Sidransky et. al. confirmed the link between GBA1 heterozygosity and PD in an international, multicenter study that compared 5691 PD and 4898 control subjects and found an odds ratio of 5.43 for GBA mutation carriers to develop PD [9]. Since then, multiple studies have reproduced these findings with the incidence of PD in GBA-mutant carriers ranging from 5–20% depending on the populations of interest. Several studies have provided excellent summaries of the distribution mutations across ancestries [19,24,25]. Recent additional studies of carrier frequency were conducted in India, the Netherlands, Ireland, and New Zealand [26–29]. However, as exome-sequencing and whole genome sequencing for GBA1 can be problematic due to a highly analogous pseudogene on chr.1 (GBAP1) [30], utilizing refined methods to analyze comprehensive mutant status from populations is necessary to understanding the scope of GBA1-PD populations [31].
The distribution of mutations has been well reviewed in previous work [19,32,33]. However, new PD-associated mutations continue to be uncovered, most recently including the p.N227S mutation found in GD patients in Chinese population studies [34]. Although a majority of PD-associated variants overlap with causative GD mutations, several PD-selective variants have also been identified which are not considered pathogenic for GD (e.g. p.D443N, p.E326K, p.K7E, and p.T369M) [33,35–37]. The data suggest some risk variants may contribute specifically to the development of PD pathologies without inducing GD pathophysiological sequalae.
Interestingly, the calculated relative risk of developing PD is similar between GD patients and heterozygous GBA1 mutation carriers (RR of 21.4 to 30, respectively) [38,39]. It is relevant to note that although GBA carriers have a substantially higher odds ratio of developing a synucleinopathy than the general population, mutations are poorly penetrant, and a vast majority of carriers do not manifest with disease [40]. Penetrance is hypothesized to be linked to a combination of genetic, epigenetic, and environmental modifiers that modulate GCase-linked pathologies.
Clinical Manifestations of GBA1-PD
GBA1-PD is marginally distinguishable from the classical PD. Disease onset in GBA1-carriers is accelerated by approximately 2–6 years, depending on variant and population [9,33,41]. The acceleration of motor dysfunction coincides with a shorter, but more prevalent, prodromal phase of disease that characteristically features anosmia, autonomic dysfunction, neuropsychiatric and behavioral disorders, and early motor dysfunction [42]. Honeycutt et. al. reported no change in the severity of motor prodrome in GBA-carriers, but indicated a more rapid conversion to PD or cognitive impairment from prodromal indications [43]. Data also suggest an association between the severity of variant and the acceleration of disease onset [44]. GBA1-PD patients are reported to experience faster motor symptom progression and a more rapid conversion to Hoehn and Yahr Stage 3 (onset to postural instability) [45,46]. A long-term, UK-based study of mutant carriers conducted by Stoker et. al. validated higher rates of dementia in mutant carriers and also indicated earlier mortality in patients carrying pathogenic, GD-associated mutations [47].
In addition to canonical PD symptomatology, GBA1-PD patients commonly develop non-motor symptoms at significantly higher rates than non-mutant carriers, including neuropsychiatric sequalae and cognitive deficit. Several studies have demonstrated a higher prevalence of cognitive decline and dementia in GBA1-PD compared to non-mutant carriers [33,46,48,49]. This disease progression is observed with higher frequencies of neocortical and limbic neuropathology. A recent study analyzed CSF GCase activity of PD patients (with and without mutations) and control subjects and found GCase activity to be lower at the time of diagnosis in patients who develop dementia within 10 years compared to cognitively normal patients [50]. These data suggest CSF GCase activity may be an effective prognostic differentiator for newly diagnosed patients. Although the difference in PD prevalence is marginal between GD patients and GBA1 heterozygotes, two studies in Ashkenazi Jewish populations indicate GD-PD patients demonstrate earlier ages-of-onset, and may develop more pronounced motor and non-motor deficits than GBA1-PD patients, suggesting a potential dose effect of GCase in the development of PD symptomatology [51,52].
The prevalence of non-motor symptomatology in GBA1-PD, most notably cognitive decline, raises question of the association of GBA mutations with other synucleinopathies or neurodegenerative illnesses that feature non-motor pathologies. GWAS studies have suggested an even stronger association between GBA1 variants and Dementia with Lewy Bodies (DLB) than with PD, with an adjusted odds ratio observed to be 8.28 [53]. Similarly, GBA1 was identified amongst a subtype of Alzheimer’s disease patients with concomitant DLB pathology (LBD-AD), providing further evidence that cortical and hippocampal neurons are susceptible to GCase pathologies [54]. One clinical study investigated the GBA1-PD specific variants p.E365K and p.T408M and highlighted lower cognitive performance and neuroimaging signs of more advanced disease in variant carriers vs wild-type PD patients [55]. Interestingly, it has been suggested that mutant status may exacerbate cognitive deficits in patients undergoing deep brain stimulation in the subthalamic nucleus (STN-DBS) [56].
A recent GWAS analysis also identified GBA1 as a significant risk allele for REM sleep and behavioral disorder (RBD), the most predictive prodromal syndrome of conversion to synucleinopathy with >80% of diagnosed patients developing PD, DLB or multiple system atrophy (MSA) [57,58]. Interestingly, the study also identified GWAS hits in the loci of SNCA, TMEM175, and SCARB2, all of which directly associate with GCase or mediate GCase function within the lysosome [57]. Collectively, these data suggest a potent association with pathways involved in lysosomal GCase function and the development of neuronal synucleinopathies. Mutations have also been linked to the oligodendroglial synucleinopathy, MSA, [59], although data on the association is conflicting with some studies suggesting no genetic association between variants and MSA [60,61].
Although a majority of mutant carriers will not go on to develop PD, there are lines of evidence that suggest non-manifesting GBA mutant carriers experience subtle clinical changes that may indicate preliminary PD conversion. A study from the Parkinson’s Progression Markers Initiative (PPMI) investigated a longitudinal cohort of GBA non-manifesting carriers and found higher scores in the Movement Disorders Society-Unified Parksinson’s Disease Rating Scale (MDS-UPDRS) in carriers (9.5) vs control subjects (4.6) indicating a subtle clinical dysfunction that may precede DAT deficits [62]. Neuroimaging studies of non-manifesting carriers, however, have shown highly-variable and conflicting evidence of a prodromal PD signature [63]. Similarly, studies in discordant siblings have shown no development of clinical Parkinsonism in non-manifesting carriers [64,65]. These data caution against over-interpreting non-penetrant GBA1-mutation carriers as eventual converters to PD, and suggest the need for more comprehensive longitudinal studies to better evaluate the penetrance of GBA1-PD.
Glucocerebrosidase and Alpha-synuclein
Neuropathological studies and concordant clinical presentation of non-motor symptoms have suggested a strong association with GCase and alpha-synuclein (αSyn) pathology. Unlike other common PD risk variants such as LRRK2 or PRKN, Lewy pathology (LP) is commonly seen in GBA1-PD. LP has also been observed in the brainstem, cortex, and hippocampal regions of GD patients that develop DLB-like phenotypic dysfunction and Parkinsonism, suggesting a prominent association between GCase and αSyn [16]. αSyn is an intrinsically disordered 140-amino acid protein most prevalently found in synaptic compartments of neurons. Although the precise function and requirement of αSyn remains unclear, studies suggest αSyn plays a role in synaptic vesicular dynamics and transmission [66,67]. αSyn belongs to a class of amyloidogenic proteins which have a propensity to aggregate and induce proteopathic templating of naïve protein under pathologic conditions.
Since the near parallel discoveries of αSyn as a causative gene for PD [68] and its predominance as a protein constituent in LP [69], a substantial body of work has characterized mechanisms by which αSyn may cause cellular toxicities and neurodegeneration [4]. Several mutations [70–74] and multiplications [75,76] of the SNCA locus further confirmed the genetic association of αSyn and PD. Gunder et. al. demonstrated increased αSyn levels in the substantia nigra of post-mortem patients with GBA mutations [77]. These findings coincide with data illustrating decreased GCase activity in the substantia nigra of PD and DLB patients [78]. GWAS analysis of modifiers identified variants near the SNCA locus amongst two candidate loci that may have a significant role in GBA1-PD penetrance, indicating a potential genetic interaction in addition to protein interactions [79].
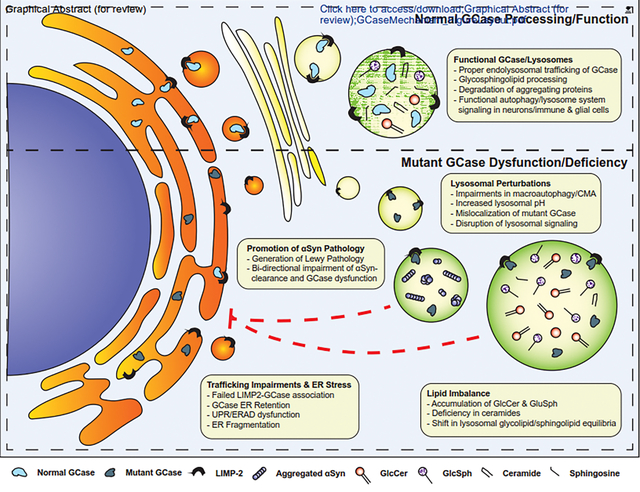
Our group has demonstrated a bi-directional association between GCase and αSyn, whereby GCase impairment leads to accumulation of αSyn in iPSC-derived dopaminergic neurons [10]. Furthermore, accumulated GlcCer from GCase deficiency can stabilize intermediate aggregate structures to drive the generation of high-molecular weight αSyn species [10]. Conversely, aggregated αSyn was observed to decrease GCase activity in iPSC-derived neurons and post-mortem brains of patients with idiopathic forms of disease, illustrating a positive feedback mechanism of αSyn-GCase toxicity [10]. These data suggest that this feedback loop would lead to decreased activity of wild-type or mutant GCase in any cell that accumulates αSyn. However, this mechanism does not explain preferential vulnerability of midbrain dopaminergic neurons in PD. Our subsequent work suggested that the activity of wild-type or mutant GCase can be decreased by accumulation of oxidized dopamine in dopaminergic neurons [80]. While the effects of αSyn on trafficking of GCase can affect GCase activity in dopaminergic and non-dopaminergic cells, the effect of oxidized dopamine would be seen only in dopaminergic populations. Mitochondrial oxidant stress and dysfunctional synaptic vesicle endocytosis contribute to increased oxidized dopamine in PD patient dopaminergic neurons [81]. Since oxidized dopamine and neuromelanin were detected in human but not mouse dopaminergic neurons, our data highlight the importance of human models for studying dysfunction of nigral dopaminergic neurons in PD.
We have also observed decreased GCase activity and concomitant PD pathology in neurons derived from patients with alternative familial PD mutations, including LRRK2, PRKN, and DJ-1, implicating convergent GCase-αSyn pathology across PD subtypes [82]. Collectively, these results suggest that direct targeting of wild-type glucocerebrosidase may improve pathogenic phenotypes across synucleinopathies. To this end, we identified allosteric GCase modulators increase wild-type GCase activity in dopaminergic neurons from patients with various forms of PD [82]. These modulators improved lysosomal dysfunction, lowered oxidized dopamine, αSyn, and glucosylceramide in patient neurons. Activation of wild-type GCase may serve as a potential therapeutic target for multiple synucleinopathies that exhibit decreased GCase activity.
The direct interaction between GCase and αSyn is still relatively unknown. A study from Yap et. al. identified C-terminal interactions between αSyn and GCase at sub-cytosolic pH, which was tempered by p.N370S mutant GCase [83]. Thus, αSyn may feature a direct binding motif that facilitates processing. However, it is uncertain how aggregated forms of αSyn modify binding capacity with or without the presence of GCase mutations. Kuo et. al. observed that misfolded mutant GCase is aberrantly bound to the lysosomal membrane in post-mortem brains of PD patients [84]. This mislocalization leads to interference and disruption in chaperone-mediated autophagy (CMA) and consequently leads to αSyn aggregation and induced DA neurodegeneration, thus providing an indirect mechanism for the GCase-αSyn pathological cascade [84]. A recent study of GBA1-PD fibroblasts used a shotgun lipidomic method to differentiate p.L444P-patients from control subjects and sporadic PD cases [85]. Lipid extracts from the p.L444P fibroblasts rapidly accelerated αSyn aggregation upon co-incubation, indicating a permissive lipid profile for αSyn pathology that may be promoted through impaired GCase activity [85].
Several studies have utilized animal models to understand how mutations may affect αSyn seeding, propagation, and toxicity. One study in a drosophila model of αSyn neurodegeneration confirmed several loss-of-function enhancers of αSyn toxicity, including SCARB2, SMPD1, CTSD, all of which are associated with lysosomal function, or more directly, GCase function [86]. In the p.D409V transgenic mouse model, heterozygous animals showed no histopathological aggravation of αSyn pathology or behavioral insults compared to wild-type littermates after unilateral injection of αSyn pre-formed fibrils (PFFs) into the olfactory bulb [87]. However, two independent studies injecting PFFs into the striata of p.L444P heterozygous mice show enhanced formation and spread of αSyn inclusions compared to control subjects [88,89]. Mahoney-Crane et. al. reported pathological exacerbation specifically in the hippocampus, whereas the rate of nigrostriatal and cortical pathologies was unaffected [89]. Previous reports have demonstrated the diversity of αSyn pathology profiles are contingent to the site of PFF injection and the corresponding neural networks associated with the target brain region [90–92]. However, these studies in GBA1-mutant model systems also suggest GCase modification of αSyn aggregation kinetics may be dependent on the particular pathogenic variant. In studies in both primary murine neuronal cultures and mouse models, Henderson et. al. show GCase inhibition does not induce αSyn aggregation, but is permissive to already initiated pathological processes in which pathological αSyn attenuates GCase activity [93]. Intriguingly, the indirect association between αSyn seeding and GCase activity has also been tested in peripheral tissues. In a study modeling gut-to-brain pathological αSyn spread, delivery of a peripheral-targeting AAV carrying GBA1 was efficient in reducing enteric nervous system αSyn pathology and highlighted potential therapeutic benefit of restoring active GCase in peripheral tissues [94]. It is hypothesized that a fraction of PD pathologies may initiate from peripheral induction points with CNS contacts such as the gut [95]. Studies exploring these axes of pathological initiation and transfer are useful in understanding what role peripheral GCase deficiencies may play in CNS disease.
The imbalance of lipid pathways upon glucocerebrosidase deficiency
The primary known function of GCase is the hydrolysis of glucosyl residues from glucosylceramide (GlcCer) and glucosylsphingosine (GlcSph), although several other glycosphingolipid moieties may also be substrates specific to lysosomal GCase. Mutation-induced impairment in GCase activity thus shifts the stoichiometry of glycosphingolipid processing causing an abundance of unprocessed lysosomal substrates and changes in ceramide levels. However, the specific role of how substrate-product imbalance contributes to GBA1-PD pathophysiology has been conflicting and difficult to resolve.
GlcCer has been demonstrated to directly mediate αSyn aggregation dynamics [10]. However, post-mortem assessment of GlcCer levels in the brains of synucleionpathy patients show conflicting data, with one study suggesting age-dependent accumulation in PD patients [96] and others showing no changes compared to control subjects [97,98]. Some evidence suggests GlcSph levels may also have an association with PD pathology. Taguchi et. al. showed in vitro GlcSph specifically induces seed-competent αSyn oligomerization that can template naïve αSyn in neurons [99]. They further show GlcSph accumulation to precede GlcCer accumulation in a PD mouse model generated from GD mice crossed with αSyn transgenic mice [99]. Recent data quantifying lipid content in plasma from p.N370S carriers in PD and non-PD populations showed increases in GlcSph in mutant carriers compared to controls, but were unable to differentiate the PD from non-PD cohort [100].
Methods to detect lipid accumulation in post-mortem brains have several potential confounding variables that may generate lower signal-to-noise ratios. For example, different cell types have large distributions of GCase expression and activity. Isolating neuronal glycosphingolipid content from glial fractions is technically challenging and may not present the most relevant lipid profiles for neurodegeneration. Also, the mass spectra signal of GlcCer and GlcSph may be contaminated by enantiomeric glycosphingolipids such as galactosylceramides which are prevalent in CNS tissue. Additionally, lipid content is subject to variability due to post-mortem intervals and tissue processing methods. Other studies have investigated substrate accumulation profiles from CSF, but have similarly found conflicting or negative results from GBA1-PD patients [98,101]. Thus, it is problematic to establish conclusions of substrate accumulation in GBA1-PD from the current literature, and studies to validate GBA-substrate/product ratios will require larger cohorts and more consistent methodology than have previously been utilized [102].
Although the evidence of GCase substrate accumulation from PD patient data has been conflicting, evidence from animal models of substrate accumulation led to the development of Venglustat, a small molecule inhibitor of glucosylceramide synthase, for the treatment of GBA1-PD [103]. Phase II clinical data (ClinicalTrials.gov Identifier: NCT02906020) suggested effective target engagement and lowering of CSF GlcCer at 4 weeks (a decrease of 72% from baseline in Japanese patients and 74.3% from baseline in non-Japanese patients at highest treatment dose) [104]. However, treated patients showed no signs of improvement in UPDRS part II or III [104]. Although the phase II study was not powered to detect meaningful clinical changes, further development of Venglustat for treatment of GBA1-PD was suspended. These findings suggest GlcCer accumulation may not be an ideal pharmacological target for effective therapy, or may indicate substrate reduction therapy to be ineffective in combating GBA1-PD.
GCase impairment may also contribute to impaired ceramide processing, which may play a significant role in cellular health and function. Ceramides are important constituents in lipid membrane stabilization and signaling [105]. In a recent study, our group showed lysosomal ceramides activate Cathepsin B which, in turn, promotes cleavage of prosaposin to saposin C, the coactivator of lysosomal GCase [106]. In PRKN-mutant models of PD, deficient ceramide levels correlated with impaired GCase activity [106]. Conversely, treatment with an inhibitor of acid ceramidase to upregulate ceramide rescued Cathepsin B activation [106]. However, clinical data of ceramide levels in GBA1-PD are conflicting. One study comparing brain ceramide levels in patients with Lewy Body Disease (LBD) vs age-matched controls showed elevations of ceramide in LBD regardless of variant status [107]. Indeed, these data collectively suggest altered sphingolipid processing in patients with Lewy Body Disease, but does not show clean directionality on ceramide levels.
It is also possible that GCase mediated lipid dysregulation may be challenged beyond ratios of specific GCase substrates, suggesting greater lipid imbalances that may influence pathologies. Interestingly, several other proteins involved in glycosphingolipid enzymatic processing have also been implicated as PD risk genes, specifically functioning in the ceramide metabolism pathway (e.g. GALC, GLA, SMPD1, ASAH1) [6,8,108]. These findings suggest a collective dysregulatory network which may lead to lipid imbalances and cellular dysfunction. Recent data suggests that plasma multiple glycosphingolipid levels may be abnormal in PD patients with or without select GBA mutations, nominating lipid dyshomeostasis as a convergent phenomenon across PD subtypes [109]. Studies using unbiased lipidomic analyses across glycosphingolipid processing with respect to GCase activity may provide insight into functional requirement and makeup of lipid profiles with respect to disease progression and tissue type.
Glucocerebrosidase and the Autophagic Lysosomal System
Lysosomal network genes and enzymes function in a carefully regulated and coordinated manner as part of cellular autophagy-lysosome system. As such, many hypotheses of GCase-related cellular dysfunction connect GCase-induced impairments to global lysosomal/autophagic dysfunction. Several studies have explored and documented global impairments in the autophagy lysosomal system as a product of GCase deficiency [110–112]. GCase deficiency has also been associated with disruption to chaperone-mediated autophagic programs [84].
However, autophagic responses to GCase damage may differ depending on model system. One study investigating the role of proteasomal turnover and autophagic regulation in mutant flies with gba1b deficiency (the drosophila ortholog of GBA1) showed no GCase-associated perturbation in global autophagy or other protein regulation systems [113]. The study did find higher levels of extracellular vesicle synthesis and release in gba1b mutants, indicating a potential pathologic role in vesicle cycling and protein aggregation [113]. One study investigating post-mortem brain tissue assessed sphingolipid hydrolase activity to determine whether network sphingolipid dysregulation contributed to PD decline [96]. The study identified GCase impairment to be accompanied by a network of dysfunctional hydrolase activities, leading to impairments in complex ganglioside concentrations [96]. Importantly, sphingolipid processing impairments were correlated with aging in control subjects, but were more pronounced in PD subjects. These findings suggest the concept of lysosome enzymatic fatigue as a product of aging, which may provide important context in the malignancy and penetrance of GBA mutations.
Deficiencies in other lysosomal-associated proteins have been shown to induce GCase pathologies. We and others have found that patients with progranulin mutations (GRN) that develop frontal temporal dementia (FTD) show lower levels of GCase activity [114,115]. Using iPSC-derived cortical neurons, we showed GRN-mutations fail to convert prosaposin into saposin-C, a critical activator of functional GCase [114]. GRN-deficits in GCase activity have also been reported to be a product of incompletely glycosylated GCase protein [115]. These findings were replicated in GRN KO mice, with evidence that GCase activity deficits in neurons can be corrected through administration of AAV-progranulin [115]. It has been suggested that progranulin regulates GCase activity through a number of different mechanisms. Progranulin has been shown to directly bind to GCase and regulate lysosomal compartmentalization of GCase [116,117]. In additional to failed GCase activation through saposin C, progranulin deficiency also causes dysregulation of bis(monoacylglycerol)phosphate (BMP), an anionic phospholipid that has been associated with GCase regulation [118].
GCase function has also been intriguingly linked with another common PD risk gene, LRRK2 (which encodes for leucine-rich repeat kinase-2). Similar to GBA1, LRRK2 dysregulation is linked to both genetic and sporadic forms of PD. Our group has shown that LRRK2-mutant iPSC-derived dopaminergic neurons show lower GCase activity that can be rescued through LRRK2 inhibition, primarily through Rab10-mediated regulation of lysosomal GCase [119]. A study investigating p.D409V murine astrocytes also showed rescue of lysosomal pathologies through inhibition of LRRK2 [120]. Studies in transgenic mice have shown a significant depletion of GCase protein in LRRK2 KO mouse brains [121]. Clinical studies have recently highlighted an interaction in compound heterozygous and LRRK2 mutant carriers indicating a potential role for LRRK2 to modify dysfunction. A study monitoring patient performance on the Montreal Cognitive Assessment (MoCA) indicated LRRK2/GBA1-mutant carriers had slower rates of decline than GBA1-mutant carriers [122]. Yahalom et. al. described similar data from a smaller cohort that showed lower incidence of RBD, dementia and psychosis in the dual mutant LRRK2/GBA1 cohort [123]. The interaction between LRRK2 modulation of GBA1 requires further study to understand mechanistic links between the two proteins in relevant cell types.
Impairment of Glucocerebrosidase Trafficking and ER Stress
Over the last decade, studies have described the role of ER stress in PD pathophysiology. One prominent hypothesis is that mutant misfolded GCase, due primarily to the prevalence of nonsynonymous missense mutations, fails to traffic to the lysosomal compartment and induces proteostatic stress signaling and ER-associated degradation (ERAD) causing ER stress. Bendikov-Bar et. al. showed over 50% of p.L444P mutant GCase in GD-patient derived fibroblasts was retained in the ER and polyubiquitinated for proteasomal degradation [124]. Similarly, using p.N370S patient-derived fibroblasts, Thomas et. al. showed GBA haploinsufficiency to be accompanied by lower LIMP2 expression levels, thus decreasing efficiency of GCase trafficking to the lysosome [125]. Another study used heterozygous p.N370S patient-derived iPSCs differentiated into dopaminergic neurons to show upregulated unfolded protein response (UPR) and ER-stress markers compared to control DA neurons [126]. The study also highlighted a retention of high-molecular weight GCase isoforms, most likely attributed to improper GCase glycosylation processing in the golgi due to ER retention [126].
Other studies have also shown human cellular models of GCase inhibition and dysfunction to lead to ER stress, including several that link αSyn dysregulation and aggregation as a cause and consequence of ER-mediated GCase impairment and failure to reach lysosomes. Smith et. al. showed ER-GCase retention and ER stress was specific to the p.L444P variant compared to the p.E326K mutation in patient fibroblasts [127]. Certain mutant variants of GCase may induce improper folds or negatively impact LIMP2 binding which may promote ER retention and stress. Correcting GCase misfolding has been an attractive target for therapeutic intervention, as multiple studies have investigated the efficacy of molecular chaperones to rescue GCase pathologies. The repurposed chaperone molecule Ambroxol was previously shown to enhance GCase levels in mutant fibroblasts from GD and GBA1-carrier PD patients and healthy controls [128]. Subsequent in vivo validation studies confirmed Ambroxol increased GCase activity in the brains of rodents and non-human primates [128,129]. Most recently, after the successful completion of both phase I [130] and phase II clinical trials (ClinicalTrials.gov Identifier: NCT05287503), a large-scale, multi-center phase III clinical trial was confirmed, indicating evidence of potential clinical utility for chaperone-based pharmacological agents. Several other small molecule chaperones have also progressed through various stages of clinical development for the treatment of both GD and GBA1-PD [131].
However, it is still unclear whether ER stress is driven primarily through direct GCase interaction or through secondary GCase mechanisms such as αSyn aggregation and lysosomal dysfunction. Stojkovska et. al. demonstrated that aggregated αSyn induced ER fragmentation and disrupted proper protein folding in midbrain dopaminergic cultures [132]. These pathologies could be rescued through the use of small-molecule drugs that promote ER proteostasis and trafficking [132]. These findings also add to the notion that many PD pathophysiologies are driven through impaired proteostatic machinery, both in the autophagy-lysosomal and the ubiquitin-proteasomal systems, suggesting a class of mechanistic targets that may be relevant to multiple PD subtypes.
Glucocerebrosidase Deficiency in Immune Cells
Mechanistic studies exploring the link of GCase deficiency and PD pathogenesis have largely centered on cell-autonomous neuronal dysfunction. However, recent work has highlighted how GCase abnormalities and impaired lysosomal function in immune cells and glia may contribute to neurodegenerative processes. Indeed, the prominence of dyslipidemic Gaucher Cells in GD suggests a particular vulnerability in myeloid cells to GCase impairment [133]. Lysosomes are critical sensors in scavenging/antigen-presenting cell populations such as myeloid cells and lymphocytes. Lysosomes drive cellular uptake programs like phagocytosis and modulate gene expression to mediate local microenvironments and induce appropriate cytokine/chemokine signaling. Thus, dysfunctional GCase, or associated enzymes like LRRK2 and progranulin with high expression in immune cells, may have significant impact on disease.
Neuroinflammation is a universal signature in the pathophysiology of synucleinopathies. PD neuropathology in accompanied by recruitment of both reactive microglia and astrocytes to primary sites of lesions [134]. Neuroimaging PET tracer studies also validate the localization of activated microglia to the substantia nigra of idiopathic PD patients [135,136]. Temporal post-mortem analysis of PD brains that received therapeutic transplants of fetal stem cells have also suggested that the temporal development of naïve LP is preceded by focal recruitment of CD45+ microglia, indicating a role in microglial signaling and reactivity in the development in pathology [137]. Recent studies in animal models of αSyn seeding have also demonstrated inflammatory glial processes to modify the kinetics of αSyn pathology [138,139]. Aside from CNS glial populations, both peripheral macrophages and T-cells have been implicated in PD pathogenesis [140,141]. T-cells from PD patients have been shown to bind to αSyn antigenic epitopes and may play a role in directly interacting with resident glial populations or dopaminergic neurons presenting MHC Class I [141].
There are several linking factors which implicate the role of GCase impairment in immune and inflammatory cell modification of neuronal pathologies. Microglial activation and cytokine release have been prominently associated with multiple animal models of GCase deficiency [142–144]. Studies utilizing the nestin-CRE floxed mouse modeling GCase impairment in neurons demonstrate engagement of lipid-engorged mac-2+ microglia in regions preceding neuronal loss and behavioral deficits [145,146]. Mutant mice as well as mice treated with CBE show microglial reactivity [144,147,148]. Soria et. al. generated a mouse with selective KO in dopaminergic neurons and observed prominent microglial activation without overt neurodegeneration or αSyn aggregation [149]. These data suggest a contribution in glial-specific GCase impairment in the degenerative thresholding of dopaminergic neurons. Astrocytic pathologies have also been observed in GBA1-PD models. Primary murine astrocyte cultures with p.D409V mutations show decreased lysosome counts and higher lysosomal pH than control astrocytes [120].
Peripheral monocytes and lymphocytes are also potent reservoirs of lysosomal hydrolase activity, including prominent GCase expression and activity [150,151]. Impaired glycosphingolipid processing can activate peripheral myeloid cells and induce cytokine production and release through GlcCer accumulation [152]. Studies have shown peripheral monocytes collected from both idiopathic PD and GBA1-PD patients have significantly dampened GCase activity, potentially highlighting a robust source for biomarker development and, ultimately, target engagement. Recently, Wallings et. al. described a multiplexed flow-cytometry based readout for GCase and LRRK2 activity from PD patient PBMCs, suggesting their utility as a reliable tandem biomarker for immune-related deficiencies associated with PD pathophysiology [153]. Longitudinal studies monitoring peripheral GCase enzyme activity correlated with disease course are ultimately needed to determine whether peripheral immune cells can serve as a surrogate measure for brain GCase activity.
Stemming from evidence in GD of differential secretion patterns of cytokines and chemokines, several groups have investigated cytokine release as a function of GCase activity. One study investigating GBA1- and LRRK2-mutant carriers showed no discernable differences in CSF or peripheral cytokine levels between groups [154]. These findings are in contrast to other studies that have found differential cytokine release profiles in GBA1-PD patients [155,156]. Assays to measure cytokine levels have markedly variable sensitivities and may generate conflicting results. Larger scale studies with consistent methodologies are necessary to resolve the pattern of cytokine/chemokine release related to GCase pathology.
Although there are convincing pathologies relevant to immune/inflammatory involvement in GBA1-PD, the relative contributions of GCase impairment in these cells, or their mechanisms associated with neuronal dysfunction are enigmatic. Furthermore, studies have shown clear disparities in immune cell signatures and function in humans vs animal model systems [157,158]. Thus, studies exploring human-cell based interactions of these cell types, perhaps through co-culture methods or organotypic/organoid modeling systems, will be important to highlight how compounded GCase pathologies in multiple cell types may cause neurodegenerative disease.
Challenges in Resolving Glucocerebrosidase Function in Disease
Several challenges exist to better understand how GCase dysregulation and impairment may contribute to the development of PD pathophysiology. A primary barrier to functional genomic understanding of GCase has been species disparities in GCase regulation, function, and mutational output depending on model system. For example, the p.N370S mutation most commonly found in PD patients causes embryonic lethality in mice [159]. Similarly, the frequently used transgenic animal model for GCase therapeutic targeting, p.D409V, has not been associated with the development of PD (although the p.D409H mutation associated with PD has also been used to monitor pathological effects of mutant GCase) [160,161]. Further development of GCase mutant transgenic lines and mutation-specific pathologies may illuminate key gaps in functional understanding and associations to genotype-specific pathophysiology. Studies employing both animal model systems in conjunction with human-cell based paradigms may be advantageous in clarifying GCase biology.
Another critical roadblock is poor penetrance of mutations and the development of disease. Recent efforts have utilized patient derived iPSCs to determine genetic modifiers of GCase penetrance. A large-cohort GWAS study investigating genetic risk loci for GBA-risk and age-of-onset identified variants near the SNCA and CTSB loci to be the most significant modifiers of GCase, although without very prominent effects [79]. One study on genetic modification of the GBA1 locus interrogated regulatory interactions and suggests expression in the CNS (SN and cortex) is mediated through trans-regulatory action from other chromosomes, whereas peripheral GCase expression is mediated through cis-regulatory elements [162]. Another study screened 305 PD patients vs 207 controls to identify GBA variant modifiers and found the strongest interactors to be an alternate variant in the GBA1 locus and variants in genes that cause mucopolysaccharidoses [163]. Better understanding of specific SNPs or loci that modify CNS GCase expression will be necessary to understand differential expression in tissue/cell types as well as identify genetic targets for intervention. Recent advances in pooled and arrayed CRISPR screening methods should be employed to determine how modified or shut-down expression across the genome may impact GCase activity and function. However, a significant challenge with current methods is accounting for epigenetic and environmental triggers and facilitators that contribute to modified penetrance. As iPSCs largely lose epigenetic regulatory signals during the reprogramming phase, recent efforts have turned to direct fibroblast-neuron differentiation programs to better understand genetic regulatory tags in patient-derived material [164,165].
Recent efforts to enhance the probing of patient GCase function have highlighted the importance of higher resolution, more selective assays in clinical characterizations of GBA1-PD. GCase activity assays feature non-specific noise from extra-lysosomal glucocerebrosidases (GBA2) as well as activity from other glucosidase family enzymes. The development of flow-cytometry based probes specific to lysosomal compartmentalization have improved the target signal of GCase activity detection, but can still be noisy. Methods for optimizing readouts of GCase activity have been discussed in previous reviews [166]. A recent study from Deen et. al. describes the development of lysotropic GCase fluorophores (LysoFQ-GBA) to be used for enhanced and targeted GCase activity measurements specifically in lysosomes of patient-derived tissues [167]. Similarly, multiplexed fluorescent probe systems to monitor GCase activity in tandem with other PD-associated enzymatic activity (LRRK2) may provide more appropriate context in the relative role of lysosomal GCase activity with other convergent pathologies identified in PD patients [153].
Lastly, integrating GCase dysfunction across relevant cell types will be critical moving forward, particularly with respect to therapeutic targeting. For example, it is still unclear what role GCase activity in CNS glia or peripheral monocyte and lymphocyte populations plays in facilitating neurodegenerative pathologies. Furthermore, new studies, particularly including scRNA and snRNA datasets, continue to confirm the level of heterogeneity found in these cell types and their potential roles in mediating CNS microenvironments. Current efforts for GCase replacement, either through enzyme replacement therapy or gene therapy, may be limited by ineffective comprehensive targeting of the appropriate cell types. For example, AAV serotypes currently in clinical use for the treatment of PD and other neurological disorders feature poor microglial tropism which may be necessary for effective target engagement. Establishing better GCase and lysosomal functional profiles across these cell types, as well as their potential interactions with neurons, will be important to know to guide targeting strategies for improved GCase function.
Conclusions
Our knowledge of GBA1-PD has increased significantly over the course of the past two decades. Although the genetic link of GBA and PD has been well established, understanding how GCase plays a role in PD and related disorders has been limited by the lack of adequate model systems and the tools to accurately measure the activity of lysosomal GCase. Despite these barriers, important recent work has provided better insight into GCase function in different cell types across both the central nervous system and the periphery. This expanded picture of GCase dysfunction provides a platform to evaluate GCase-associated mechanisms in the context of other pathogenic pathways that have been implicated in PD. Hopefully, this will lead to improved translational studies for the development of effective therapeutic strategies for PD and other related neurodegenerative diseases.
Research Highlights.
Mutations in GBA1 are the most prevalent genetic risk factor for Parkinson’s disease.
Lysosomal GCase dysfunction is a conserved mechanisms across genetic and idiopathic forms of disease.
Enhancing GCase activity and function, in both normal and mutant protein, may be a powerful therapeutic avenue.
Methods to analyze lysosomal GCase activity and function require standardization and higher signal-to-noise ratios for proper assessment of pathology.
Identifying modifiers of GBA-PD penetrance will be critical to understand GCase dysfunction in disease.
Non-neuronal glucocerebrosidase dysfunction, particularly in immune and glial cells, may contribute to neurodegeneration and requires future assessment.
Acknowledgements
This work was supported by the National Institute of Neurological Disorders and Stroke (D.K., 5R35NS122257). D.C. is supported by a training grant from the National Institute of Aging (T32AG020506).
Footnotes
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
D.K. is the founder of Vanqua Bio, Lysosomal Therapeutics, Inc., and serves on the Scientific Advisory Board of Intellia Therapeutics, AcureX Therapeutics, Leal Therapeutics, The Silverstein Foundation and serves as a Venture Partner at OrbiMed.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Savitt JM, Dawson VL, Dawson TM, Diagnosis and treatment of Parkinson disease: molecules to medicine, J Clin Invest. 116 (2006) 1744–1754. 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Lang AE, Obeso JA, Time to move beyond nigrostriatal dopamine deficiency in Parkinson’s disease, Annals of Neurology. 55 (2004) 761–765. 10.1002/ana.20102. [DOI] [PubMed] [Google Scholar]
- [3].Obeso JA, Stamelou M, Goetz CG, Poewe W, Lang AE, Weintraub D, Burn D, Halliday GM, Bezard E, Przedborski S, Lehericy S, Brooks DJ, Rothwell JC, Hallett M, DeLong MR, Marras C, Tanner CM, Ross GW, Langston JW, Klein C, Bonifati V, Jankovic J, Lozano AM, Deuschl G, Bergman H, Tolosa E, Rodriguez-Violante M, Fahn S, Postuma RB, Berg D, Marek K, Standaert DG, Surmeier DJ, Olanow CW, Kordower JH, Calabresi P, Schapira AHV, Stoessl AJ, Past, Present, and Future of Parkinson’s Disease: A Special Essay on the 200th Anniversary of the Shaking Palsy, Mov Disord. 32 (2017) 1264–1310. 10.1002/mds.27115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wong YC, Krainc D, α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies, Nat Med. 23 (2017) 1–13. 10.1038/nm.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lesage S, Brice A, Parkinson’s disease: from monogenic forms to genetic susceptibility factors, Human Molecular Genetics. 18 (2009) R48–R59. 10.1093/hmg/ddp012. [DOI] [PubMed] [Google Scholar]
- [6].Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, Bras J, Young E, von Coelln R, Simón-Sánchez J, Schulte C, Sharma M, Krohn L, Pihlstrøm L, Siitonen A, Iwaki H, Leonard H, Faghri F, Gibbs JR, Hernandez DG, Scholz SW, Botia JA, Martinez M, Corvol J-C, Lesage S, Jankovic J, Shulman LM, Sutherland M, Tienari P, Majamaa K, Toft M, Andreassen OA, Bangale T, Brice A, Yang J, Gan-Or Z, Gasser T, Heutink P, Shulman JM, Wood NW, Hinds DA, Hardy JA, Morris HR, Gratten J, Visscher PM, Graham RR, Singleton AB, Adarmes-Gómez AD, Aguilar M, Aitkulova A, Akhmetzhanov V, Alcalay RN, Alvarez I, Alvarez V, Bandres-Ciga S, Barrero FJ, Yarza JAB, Bernal-Bernal I, Billingsley K, Blauwendraat C, Blazquez M, Bonilla-Toribio M, Botía JA, Boungiorno MT, Bras J, Brice A, Brockmann K, Bubb V, Buiza-Rueda D, Cámara A, Carrillo F, Carrión-Claro M, Cerdan D, Chelban V, Clarimón J, Clarke C, Compta Y, Cookson MR, Corvol J-C, Craig DW, Danjou F, Diez-Fairen M, Dols-Icardo O, Duarte J, Duran R, Escamilla-Sevilla F, Escott-Price V, Ezquerra M, Faghri F, Feliz C, Fernández M, Fernández-Santiago R, Finkbeiner S, Foltynie T, Gan-Or Z, Garcia C, García-Ruiz P, Gasser T, Gibbs JR, Heredia MJG, Gómez-Garre P, González MM, Gonzalez-Aramburu I, Guelfi S, Guerreiro R, Hardy J, Hassin-Baer S, Hernandez DG, Heutink P, Hoenicka J, Holmans P, Houlden H, Infante J, Iwaki H, Jesús S, Jimenez-Escrig A, Kaishybayeva G, Kaiyrzhanov R, Karimova A, Kia DA, Kinghorn KJ, Koks S, Krohn L, Kulisevsky J, Labrador-Espinosa MA, Leonard HL, Lesage S, Lewis P, Lopez-Sendon JL, Lovering R, Lubbe S, Lungu C, Macias D, Majamaa K, Manzoni C, Marín J, Marinus J, Marti MJ, Martinez M, Torres IM, Martínez-Castrillo JC, Mata M, Mencacci NE, Méndez-del-Barrio C, Middlehurst B, Mínguez A, Mir P, Mok KY, Morris HR, Muñoz E, Nalls MA, Narendra D, Noyce AJ, Ojo OO, Okubadejo NU, Pagola AG, Pastor P, Errazquin FP, Periñán-Tocino T, Pihlstrom L, Plun-Favreau H, Quinn J, R’Bibo L, Reed X, Rezola EM, Rizig M, Rizzu P, Robak L, Rodriguez AS, Rouleau GA, Ruiz-Martínez J, Ruz C, Ryten M, Sadykova D, Scholz SW, Schreglmann S, Schulte C, Sharma M, Shashkin C, Shulman JM, Sierra M, Siitonen A, Simón-Sánchez J, Singleton AB, Suarez-Sanmartin E, Taba P, Tabernero C, Tan MX, Tartari JP, Tejera-Parrado C, Toft M, Tolosa E, Trabzuni D, Valldeoriola F, van Hilten JJ, Keuren-Jensen KV, Vargas-González L, Vela L, Vives F, Williams N, Wood NW, Zharkinbekova N, Zharmukhanov Z, Zholdybayeva E, Zimprich A, Ylikotila P, Shulman LM, von Coelln R, Reich S, Savitt J, Agee M, Alipanahi B, Auton A, Bell RK, Bryc K, Elson SL, Fontanillas P, Furlotte NA, Huber KE, Hicks B, Jewett EM, Jiang Y, Kleinman A, Lin K-H, Litterman NK, McCreight JC, McIntyre MH, McManus KF, Mountain JL, Noblin ES, Northover CAM, Pitts SJ, Poznik GD, Sathirapongsasuti JF, Shelton JF, Shringarpure S, Tian C, Tung J, Vacic V, Wang X, Wilson CH, Anderson T, Bentley S, Dalrymple-Alford J, Fowdar J, Gratten J, Halliday G, Henders AK, Hickie I, Kassam I, Kennedy M, Kwok J, Lewis S, Mellick G, Montgomery G, Pearson J, Pitcher T, Sidorenko J, Silburn PA, Vallerga CL, Visscher PM, Wallace L, Wray NR, Xue A, Yang J, Zhang F, Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies, The Lancet Neurology. 18 (2019) 1091–1102. 10.1016/S1474-4422(19)30320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, Graham RR, A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci, Nat Genet. 49 (2017) 1511–1516. 10.1038/ng.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Robak LA, Jansen IE, van Rooij J, Uitterlinden AG, Kraaij R, Jankovic J, Heutink P, Shulman JM, International Parkinson’s Disease Genomics Consortium (IPDGC), Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease, Brain. 140 (2017) 3191–3203. 10.1093/brain/awx285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen C-M, Clark LN, Condroyer C, De Marco EV, Dürr A, Eblan MJ, Fahn S, Farrer MJ, Fung H-C, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen G-J, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan E-K, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu Y-R, Zabetian CP, Zhao Y, Ziegler SG, Multicenter Analysis of Glucocerebrosidase Mutations in Parkinson’s Disease, New England Journal of Medicine. 361 (2009) 1651–1661. 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mazzulli JR, Xu Y-H, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D, Gaucher Disease Glucocerebrosidase and α-Synuclein Form a Bidirectional Pathogenic Loop in Synucleinopathies, Cell. 146 (2011) 37–52. 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A, Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways, Human Molecular Genetics. 20 (2011) 3852–3866. 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- [12].Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A, A Gene Network Regulating Lysosomal Biogenesis and Function, Science. 325 (2009) 473–477. 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- [13].Fujita H, Ezaki J, Noguchi Y, Kono A, Himeno M, Kato K, Isolation and sequencing of a cDNA clone encoding 85kDa sialoglycoprotein in rat liver lysosomal membranes, Biochemical and Biophysical Research Communications. 178 (1991) 444–452. 10.1016/0006-291X(91)90127-S. [DOI] [PubMed] [Google Scholar]
- [14].Woeste MA, Wachten D, The Enigmatic Role of GBA2 in Controlling Locomotor Function, Frontiers in Molecular Neuroscience. 10 (2017). https://www.frontiersin.org/articles/10.3389/fnmol.2017.00386 (accessed December 12, 2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, Levade T, Astudillo L, Serratrice J, Brassier A, Rose C, Billette de Villemeur T, Berger MG, A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments, Int J Mol Sci. 18 (2017) 441. 10.3390/ijms18020441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Furderer ML, Hertz E, Lopez GJ, Sidransky E, Neuropathological Features of Gaucher Disease and Gaucher Disease with Parkinsonism, Int J Mol Sci. 23 (2022) 5842. 10.3390/ijms23105842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hruska KS, LaMarca ME, Scott CR, Sidransky E, Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA), Human Mutation. 29 (2008) 567–583. 10.1002/humu.20676. [DOI] [PubMed] [Google Scholar]
- [18].Tayebi N, Walker J, Stubblefield B, Orvisky E, LaMarca ME, Wong K, Rosenbaum H, Schiffmann R, Bembi B, Sidransky E, Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism?, Molecular Genetics and Metabolism. 79 (2003) 104–109. 10.1016/S1096-7192(03)00071-4. [DOI] [PubMed] [Google Scholar]
- [19].Parlar SC, Grenn FP, Kim JJ, Blauwendraat C, Gan-Or Z, Classification of GBA1 variants in Parkinson’s disease; the GBA1-PD browser, (2022) 2022.09.27.22280421. 10.1101/2022.09.27.22280421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Neudorfer O, Giladi N, Elstein D, Abrahamov A, Turezkite T, Aghai E, Reches A, Bembi B, Zimran A, Occurrence of Parkinson’s syndrome in type 1 Gaucher disease, QJM: An International Journal of Medicine. 89 (1996) 691–694. 10.1093/qjmed/89.9.691. [DOI] [PubMed] [Google Scholar]
- [21].Machaczka M, Rucinska M, Skotnicki AB, Jurczak W, Parkinson’s syndrome preceding clinical manifestation of Gaucher’s disease, American Journal of Hematology. 61 (1999) 216–217. . [DOI] [PubMed] [Google Scholar]
- [22].Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E, Parkinsonism among Gaucher disease carriers, Journal of Medical Genetics. 41 (2004) 937–940. 10.1136/jmg.2004.024455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lwin A, Orvisky E, Goker-Alpan O, LaMarca ME, Sidransky E, Glucocerebrosidase mutations in subjects with parkinsonism, Molecular Genetics and Metabolism. 81 (2004) 70–73. 10.1016/j.ymgme.2003.11.004. [DOI] [PubMed] [Google Scholar]
- [24].Lim JL, Lohmann K, Tan AH, Tay YW, Ibrahim KA, Abdul Aziz Z, Mawardi AS, Puvanarajah SD, Lim TT, Looi I, Ooi JCE, Chia YK, Muthusamy KA, Bauer P, Rolfs A, Klein C, Ahmad-Annuar A, Lim S-Y, Glucocerebrosidase (GBA) gene variants in a multiethnic Asian cohort with Parkinson’s disease: mutational spectrum and clinical features, J Neural Transm. 129 (2022) 37–48. 10.1007/s00702-021-02421-0. [DOI] [PubMed] [Google Scholar]
- [25].Migdalska-Richards A, Schapira AHV, The relationship between glucocerebrosidase mutations and Parkinson disease, Journal of Neurochemistry. 139 (2016) 77–90. 10.1111/jnc.13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Biswas A, Sadhukhan D, Biswas A, Das SK, Banerjee TK, Bal PS, Pal S, Ghosh A, Ray K, Ray J, Identification of GBA mutations among neurodegenerative disease patients from eastern India, Neuroscience Letters. 751 (2021) 135816. 10.1016/j.neulet.2021.135816. [DOI] [PubMed] [Google Scholar]
- [27].Olszewska DA, McCarthy A, Soto-Beasley AI, Walton RL, Magennis B, McLaughlin RL, Hardiman O, Ross OA, Lynch T, Association Between Glucocerebrosidase Mutations and Parkinson’s Disease in Ireland, Frontiers in Neurology. 11 (2020). https://www.frontiersin.org/articles/10.3389/fneur.2020.00527 (accessed November 8, 2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].den Heijer JM, Cullen VC, Quadri M, Schmitz A, Hilt DC, Lansbury P, Berendse HW, van de Berg WDJ, de Bie RMA, Boertien JM, Boon AJW, Contarino MF, van Hilten JJ, Hoff JI, van Mierlo T, Munts AG, van der Plas AA, Ponsen MM, Baas F, Majoor-Krakauer D, Bonifati V, van Laar T, Groeneveld GJ, A Large-Scale Full GBA1 Gene Screening in Parkinson’s Disease in the Netherlands, Movement Disorders. 35 (2020) 1667–1674. 10.1002/mds.28112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Graham OEE, Pitcher TL, Liau Y, Miller AL, Dalrymple-Alford JC, Anderson TJ, Kennedy MA, Nanopore sequencing of the glucocerebrosidase (GBA) gene in a New Zealand Parkinson’s disease cohort, (2019) 748335. 10.1101/748335. [DOI] [PubMed]
- [30].Gustavsson EK, Sethi S, Gao Y, Brenton J, García-Ruiz S, Zhang D, Garza R, Reynolds RH, Evans JR, Chen Z, Grant-Peters M, Macpherson H, Montgomery K, Dore R, Wernick AI, Arber C, Wray S, Gandhi S, Esselborn J, Blauwendraat C, Douse CH, Adami A, Atacho DAM, Kouli A, Quaegebeur A, Barker RA, Englund E, Platt F, Jakobsson J, Wood NW, Houlden H, Saini H, Bento CF, Hardy J, Ryten M, Pseudogenes limit the identification of novel common transcripts generated by their parent genes, (2022) 2022.10.21.513169. 10.1101/2022.10.21.513169. [DOI]
- [31].Toffoli M, Chen X, Sedlazeck FJ, Lee C-Y, Mullin S, Higgins A, Koletsi S, Garcia-Segura ME, Sammler E, Scholz SW, Schapira AHV, Eberle MA, Proukakis C, Comprehensive short and long read sequencing analysis for the Gaucher and Parkinson’s disease-associated GBA gene, Commun Biol. 5 (2022) 1–10. 10.1038/s42003-022-03610-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu G, Boot B, Locascio JJ, Jansen IE, Winder-Rhodes S, Eberly S, Elbaz A, Brice A, Ravina B, van Hilten JJ, Cormier-Dequaire F, Corvol J-C, Barker RA, Heutink P, Marinus J, Williams-Gray CH, Scherzer CR, for the I.G. of P.D.P. (IGPP) Consortium, Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s, Annals of Neurology. 80 (2016) 674–685. 10.1002/ana.24781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Neumann J, Bras J, Deas E, O’Sullivan SS, Parkkinen L, Lachmann RH, Li A, Holton J, Guerreiro R, Paudel R, Segarane B, Singleton A, Lees A, Hardy J, Houlden H, Revesz T, Wood NW, Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease, Brain. 132 (2009) 1783–1794. 10.1093/brain/awp044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lee N, Moon H-J, Park S-H, Moon J-Y, Park K-K, Kim J-H, Lee J-H, Generation of Parkinson’s disease patient-derived human induced pluripotent stem cells line (PNUSCRi001-A) carrying a N227S mutation in GBA gene, Stem Cell Research. 65 (2022) 102959. 10.1016/j.scr.2022.102959. [DOI] [PubMed] [Google Scholar]
- [35].Asselta R, Rimoldi V, Siri C, Cilia R, Guella I, Tesei S, Soldà G, Pezzoli G, Duga S, Goldwurm S, Glucocerebrosidase mutations in primary parkinsonism, Parkinsonism Relat Disord. 20 (2014) 1215–1220. 10.1016/j.parkreldis.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Davis MY, Johnson CO, Leverenz JB, Weintraub D, Trojanowski JQ, Chen-Plotkin A, Van Deerlin VM, Quinn JF, Chung KA, Peterson-Hiller AL, Rosenthal LS, Dawson TM, Albert MS, Goldman JG, Stebbins GT, Bernard B, Wszolek ZK, Ross OA, Dickson DW, Eidelberg D, Mattis PJ, Niethammer M, Yearout D, Hu S-C, Cholerton BA, Smith M, Mata IF, Montine TJ, Edwards KL, Zabetian CP, Association of GBA Mutations and the E326K Polymorphism With Motor and Cognitive Progression in Parkinson Disease, JAMA Neurology. 73 (2016) 1217–1224. 10.1001/jamaneurol.2016.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mallett V, Ross JP, Alcalay RN, Ambalavanan A, Sidransky E, Dion PA, Rouleau GA, Gan-Or Z, GBA p.T369M substitution in Parkinson disease: Polymorphism or association? A meta-analysis, Neurology Genetics. 2 (2016). 10.1212/NXG.0000000000000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bultron G, Kacena K, Pearson D, Boxer M, Yang R, Sathe S, Pastores G, Mistry PK, The risk of Parkinson’s disease in type 1 Gaucher disease, J Inherit Metab Dis. 33 (2010) 167–173. 10.1007/s10545-010-9055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McNeill A, Duran R, Hughes DA, Mehta A, Schapira AHV, A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers, J Neurol Neurosurg Psychiatry. 83 (2012) 853–854. 10.1136/jnnp-2012-302402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Anheim M, Elbaz A, Lesage S, Durr A, Condroyer C, Viallet F, Pollak P, Bonaïti B, Bonaïti-Pellié C, Brice A, Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers, Neurology. 78 (2012) 417–420. 10.1212/WNL.0b013e318245f476. [DOI] [PubMed] [Google Scholar]
- [41].Gan-Or Z, Giladi N, Rozovski U, Shifrin C, Rosner S, Gurevich T, Bar-Shira A, Orr-Urtreger A, Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset, Neurology. 70 (2008) 2277–2283. 10.1212/01.wnl.0000304039.11891.29. [DOI] [PubMed] [Google Scholar]
- [42].Lopez GJ, Lichtenberg J, Tayebi N, Ryan E, Lecker AL, Sidransky E, Longitudinal evaluation of olfactory function in individuals with Gaucher disease and GBA1 mutation carriers with and without Parkinson’s disease, Frontiers in Neurology. 13 (2022). https://www.frontiersin.org/articles/10.3389/fneur.2022.1039214 (accessed November 8, 2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Honeycutt L, Montplaisir JY, Gagnon J-F, Ruskey J, Pelletier A, Gan-Or Z, Postuma RB, Glucocerebrosidase mutations and phenoconversion of REM sleep behavior disorder to parkinsonism and dementia, Parkinsonism & Related Disorders. 65 (2019) 230–233. 10.1016/j.parkreldis.2019.04.016. [DOI] [PubMed] [Google Scholar]
- [44].Malek N, Weil RS, Bresner C, Lawton MA, Grosset KA, Tan M, Bajaj N, Barker RA, Burn DJ, Foltynie T, Hardy J, Wood NW, Ben-Shlomo Y, Williams NW, Grosset DG, Morris HR, Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study, J Neurol Neurosurg Psychiatry. 89 (2018) 702–709. 10.1136/jnnp-2017-317348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Winder-Rhodes SE, Evans JR, Ban M, Mason SL, Williams-Gray CH, Foltynie T, Duran R, Mencacci NE, Sawcer SJ, Barker RA, Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort, Brain. 136 (2013) 392–399. 10.1093/brain/aws318. [DOI] [PubMed] [Google Scholar]
- [46].Brockmann K, Srulijes K, Hauser A-K, Schulte C, Csoti I, Gasser T, Berg D, GBA-associated PD presents with nonmotor characteristics, Neurology. 77 (2011) 276–280. 10.1212/WNL.0b013e318225ab77. [DOI] [PubMed] [Google Scholar]
- [47].Stoker TB, Camacho M, Winder-Rhodes S, Liu G, Scherzer CR, Foltynie T, Evans J, Breen DP, Barker RA, Williams-Gray CH, Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson’s disease, J Neurol Neurosurg Psychiatry. 91 (2020) 695–702. 10.1136/jnnp-2020-322857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jiang Z, Huang Y, Zhang P, Han C, Lu Y, Mo Z, Zhang Z, Li X, Zhao S, Cai F, Huang L, Chen C, Shi Z, Zhang Y, Ling F, Characterization of a pathogenic variant in GBA for Parkinson’s disease with mild cognitive impairment patients, Mol Brain. 13 (2020) 102. 10.1186/s13041-020-00637-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].McNeill A, Duran R, Proukakis C, Bras J, Hughes D, Mehta A, Hardy J, Wood NW, Schapira AHV, Hyposmia and Cognitive Impairment in Gaucher Disease Patients and Carriers, Mov Disord. 27 (2012) 526–532. 10.1002/mds.24945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Oftedal L, Maple-Grødem J, Dalen I, Tysnes O-B, Pedersen KF, Alves G, Lange J, Association of CSF Glucocerebrosidase Activity With the Risk of Incident Dementia in Patients With Parkinson Disease, Neurology. (2022). 10.1212/WNL.0000000000201418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Thaler A, Gurevich T, Bar Shira A, Gana Weisz M, Ash E, Shiner T, Orr-Urtreger A, Giladi N, Mirelman A, A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype, Parkinsonism & Related Disorders. 36 (2017) 47–51. 10.1016/j.parkreldis.2016.12.014. [DOI] [PubMed] [Google Scholar]
- [52].Alcalay RN, Dinur T, Quinn T, Sakanaka K, Levy O, Waters C, Fahn S, Dorovski T, Chung WK, Pauciulo M, Nichols W, Rana HQ, Balwani M, Bier L, Elstein D, Zimran A, Comparison of Parkinson risk in Ashkenazi Jewish Gaucher patients and GBA heterozygotes, JAMA Neurol. 71 (2014) 752–757. 10.1001/jamaneurol.2014.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nalls MA, Duran R, Lopez G, Kurzawa-Akanbi M, McKeith IG, Chinnery PF, Morris CM, Theuns J, Crosiers D, Cras P, Engelborghs S, De Deyn PP, Van Broeckhoven C, Mann DMA, Snowden J, Pickering-Brown S, Halliwell N, Davidson Y, Gibbons L, Harris J, Sheerin U-M, Bras J, Hardy J, Clark L, Marder K, Honig LS, Berg D, Maetzler W, Brockmann K, Gasser T, Novellino F, Quattrone A, Annesi G, De Marco EV, Rogaeva E, Masellis M, Black SE, Bilbao JM, Foroud T, Ghetti B, Nichols WC, Pankratz N, Halliday G, Lesage S, Klebe S, Durr A, Duyckaerts C, Brice A, Giasson BI, Trojanowski JQ, Hurtig HI, Tayebi N, Landazabal C, Knight MA, Keller M, Singleton AB, Wolfsberg TG, Sidransky E, A Multicenter Study of Glucocerebrosidase Mutations in Dementia With Lewy Bodies, JAMA Neurology. 70 (2013) 727–735. 10.1001/jamaneurol.2013.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, Buchman AS, Larson EB, Crane PK, Kaye JA, Kramer P, Woltjer R, Kukull W, Nelson PT, Jicha GA, Neltner JH, Galasko D, Masliah E, Trojanowski JQ, Schellenberg GD, Yearout D, Huston H, Fritts-Penniman A, Mata IF, Wan JY, Edwards KL, Montine TJ, Zabetian CP, GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology, Neurology. 79 (2012) 1944–1950. 10.1212/WNL.0b013e3182735e9a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Greuel A, Trezzi J-P, Glaab E, Ruppert MC, Maier F, Jäger C, Hodak Z, Lohmann K, Ma Y, Eidelberg D, Timmermann L, Hiller K, Tittgemeyer M, Drzezga A, Diederich N, Eggers C, GBA Variants in Parkinson’s Disease: Clinical, Metabolomic, and Multimodal Neuroimaging Phenotypes, Movement Disorders. 35 (2020) 2201–2210. 10.1002/mds.28225. [DOI] [PubMed] [Google Scholar]
- [56].Pal G, Mangone G, Hill EJ, Ouyang B, Liu Y, Lythe V, Ehrlich D, Saunders-Pullman R, Shanker V, Bressman S, Alcalay RN, Garcia P, Marder KS, Aasly J, Mouradian MM, Link S, Rosenbaum M, Anderson S, Bernard B, Wilson R, Stebbins G, Nichols WC, Welter M-L, Sani S, Afshari M, Verhagen L, de Bie RMA, Foltynie T, Hall D, Corvol J-C, Goetz CG, Parkinson Disease and Subthalamic Nucleus Deep Brain Stimulation: Cognitive Effects in GBA Mutation Carriers, Annals of Neurology. 91 (2022) 424–435. 10.1002/ana.26302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Krohn L, Heilbron K, Blauwendraat C, Reynolds RH, Yu E, Senkevich K, Rudakou U, Estiar MA, Gustavsson EK, Brolin K, Ruskey JA, Freeman K, Asayesh F, Chia R, Arnulf I, Hu MTM, Montplaisir JY, Gagnon J-F, Desautels A, Dauvilliers Y, Gigli GL, Valente M, Janes F, Bernardini A, Högl B, Stefani A, Ibrahim A, Šonka K, Kemlink D, Oertel W, Janzen A, Plazzi G, Biscarini F, Antelmi E, Figorilli M, Puligheddu M, Mollenhauer B, Trenkwalder C, Sixel-Döring F, Cochen De Cock V, Monaca CC, Heidbreder A, Ferini-Strambi L, Dijkstra F, Viaene M, Abril B, Boeve BF, Scholz SW, Ryten M, Bandres-Ciga S, Noyce A, Cannon P, Pihlstrøm L, Nalls MA, Singleton AB, Rouleau GA, Postuma RB, Gan-Or Z, Genome-wide association study of REM sleep behavior disorder identifies polygenic risk and brain expression effects, Nat Commun. 13 (2022) 7496. 10.1038/s41467-022-34732-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Berg D, Borghammer P, Fereshtehnejad S-M, Heinzel S, Horsager J, Schaeffer E, Postuma RB, Prodromal Parkinson disease subtypes — key to understanding heterogeneity, Nat Rev Neurol. 17 (2021) 349–361. 10.1038/s41582-021-00486-9. [DOI] [PubMed] [Google Scholar]
- [59].Mitsui J, Matsukawa T, Sasaki H, Yabe I, Matsushima M, Dürr A, Brice A, Takashima H, Kikuchi A, Aoki M, Ishiura H, Yasuda T, Date H, Ahsan B, Iwata A, Goto J, Ichikawa Y, Nakahara Y, Momose Y, Takahashi Y, Hara K, Kakita A, Yamada M, Takahashi H, Onodera O, Nishizawa M, Watanabe H, Ito M, Sobue G, Ishikawa K, Mizusawa H, Kanai K, Hattori T, Kuwabara S, Arai K, Koyano S, Kuroiwa Y, Hasegawa K, Yuasa T, Yasui K, Nakashima K, Ito H, Izumi Y, Kaji R, Kato T, Kusunoki S, Osaki Y, Horiuchi M, Kondo T, Murayama S, Hattori N, Yamamoto M, Murata M, Satake W, Toda T, Filla A, Klockgether T, Wüllner U, Nicholson G, Gilman S, Tanner CM, Kukull WA, Stern MB, Lee VM-Y, Trojanowski JQ, Masliah E, Low PA, Sandroni P, Ozelius LJ, Foroud T, Tsuji S, Variants associated with Gaucher disease in multiple system atrophy, Ann Clin Transl Neurol. 2 (2015) 417–426. 10.1002/acn3.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Srulijes K, Hauser A-K, Guella I, Asselta R, Brockmann K, Schulte C, Soldà G, Cilia R, Maetzler W, Schols L, Wenning GK, Poewe W, Barone P, Wüllner U, Oertel W, Berg D, Goldwurm S, Gasser T, No association of GBA mutations and multiple system atrophy, European Journal of Neurology. 20 (2013) e61–e62. 10.1111/ene.12086. [DOI] [PubMed] [Google Scholar]
- [61].Sailer A, Scholz SW, Nalls MA, Schulte C, Federoff M, Price TR, Lees A, Ross OA, Dickson DW, Mok K, Mencacci NE, Schottlaender L, Chelban V, Ling H, O’Sullivan SS, Wood NW, Traynor BJ, Ferrucci L, Federoff HJ, Mhyre TR, Morris HR, Deuschl G, Quinn N, Widner H, Albanese A, Infante J, Bhatia KP, Poewe W, Oertel W, Höglinger GU, Wüllner U, Goldwurm S, Pellecchia MT, Ferreira J, Tolosa E, Bloem BR, Rascol O, Meissner WG, Hardy JA, Revesz T, Holton JL, Gasser T, Wenning GK, Singleton AB, Houlden H, A genome-wide association study in multiple system atrophy, Neurology. 87 (2016) 1591–1598. 10.1212/WNL.0000000000003221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Simuni T, Uribe L, Cho HR, Caspell-Garcia C, Coffey CS, Siderowf A, Trojanowski JQ, Shaw LM, Seibyl J, Singleton A, Toga AW, Galasko D, Foroud T, Tosun D, Poston K, Weintraub D, Mollenhauer B, Tanner CM, Kieburtz K, Chahine LM, Reimer A, Hutten SJ, Bressman S, Marek K, Arnedo V, Clark A, Fraiser M, Kopil C, Chowdhury S, Sherer T, Daegele N, Casaceli C, Dorsey R, Wilson R, Mahes S, Salerno C, Crawford K, Casalin P, Malferrari G, Weisz MG, Orr-Urtreger A, Montine T, Baglieri C, Christini A, Russell D, Dahodwala N, Giladi N, Factor S, Hogarth P, Standaert D, Hauser R, Jankovic J, Saint-Hilaire M, Richard I, Shprecher D, Fernandez H, Brockmann K, Rosenthal L, Barone P, Espay A, Rowe D, Marder K, Santiago A, Hu S-C, Isaacson S, Corvol J-C, Martinez JR, Tolosa E, Tai Y, Politis M, Smejdir D, Rees L, Williams K, Kausar F, Williams K, Richardson W, Willeke D, Peacock S, Sommerfeld B, Freed A, Wakeman K, Blair C, Guthrie S, Harrell L, Hunter C, Thomas C-A, James R, Zimmerman G, Brown V, Mule J, Hilt E, Ribb K, Ainscough S, Wethington M, Ranola M, Santana HM, Moreno J, Raymond D, Speketer K, Carvajal L, Carvalo S, Croitoru I, Garrido A, Payne LM, Viswanth V, Severt L, Facheris M, Soares H, Mintun MA, Cedarbaum J, Taylor P, Biglan K, Vandenbroucke E, Sheikh ZH, Bingol B, Fischer T, Sardi P, Forrat R, Reith A, Egebjerg J, Hillert GA, Saba B, Min C, Umek R, Mather J, Santi SD, Post A, Boess F, Taylor K, Grachev I, Avbersek A, Muglia P, Merchant K, Tauscher J, Clinical and dopamine transporter imaging characteristics of non-manifest LRRK2 and GBA mutation carriers in the Parkinson’s Progression Markers Initiative (PPMI): a cross-sectional study, The Lancet Neurology. 19 (2020) 71–80. 10.1016/S1474-4422(19)30319-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Filippi M, Balestrino R, Basaia S, Agosta F, Neuroimaging in Glucocerebrosidase-Associated Parkinsonism: A Systematic Review, Movement Disorders. 37 (2022) 1375–1393. 10.1002/mds.29047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Biegstraaten M, van Schaik IN, Aerts JMFG, Langeveld M, Mannens MMAM, Bour LJ, Sidransky E, Tayebi N, Fitzgibbon E, Hollak CEM, A monozygotic twin pair with highly discordant Gaucher phenotypes, Blood Cells Mol Dis. 46 (2011) 39–41. 10.1016/j.bcmd.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lopez G, Steward A, Ryan E, Groden C, Wiggs E, Segalà L, Monestime GM, Tayebi N, Sidransky E, Clinical Evaluation of Sibling Pairs With Gaucher Disease Discordant for Parkinsonism, Movement Disorders. 35 (2020) 359–365. 10.1002/mds.27916. [DOI] [PubMed] [Google Scholar]
- [66].Man WK, Tahirbegi B, Vrettas MD, Preet S, Ying L, Vendruscolo M, De Simone A, Fusco G, The docking of synaptic vesicles on the presynaptic membrane induced by α-synuclein is modulated by lipid composition, Nat Commun. 12 (2021) 927. 10.1038/s41467-021-21027-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Burré J, The Synaptic Function of α-Synuclein, J Parkinsons Dis 5 (n.d.) 699–713. 10.3233/JPD-150642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL, Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease, Science. 276 (1997) 2045–2047. 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- [69].Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M, Alpha-synuclein in Lewy bodies, Nature. 388 (1997) 839–840. 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- [70].Conway KA, Harper JD, Lansbury PT, Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease, Nat Med. 4 (1998) 1318–1320. 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- [71].Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Tortosa EG, del Ser T, Muñoz DG, de Yebenes JG, The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia, Annals of Neurology. 55 (2004) 164–173. 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]
- [72].Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, Aasly JO, Rajput A, Rajput AH, Jon Stoessl A, Farrer MJ, Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease, Movement Disorders. 28 (2013) 811–813. 10.1002/mds.25421. [DOI] [PubMed] [Google Scholar]
- [73].Lesage S, Anheim M, Letournel F, Bousset L, Honoré A, Rozas N, Pieri L, Madiona K, Dürr A, Melki R, Verny C, Brice A, for the FPDGS Group, G51D α-synuclein mutation causes a novel Parkinsonian–pyramidal syndrome, Annals of Neurology. 73 (2013) 459–471. 10.1002/ana.23894. [DOI] [PubMed] [Google Scholar]
- [74].Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Pöyhönen M, Paetau A, A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology, Neurobiology of Aging. 35 (2014) 2180.e1–2180.e5. 10.1016/j.neurobiolaging.2014.03.024. [DOI] [PubMed] [Google Scholar]
- [75].Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K, α-Synuclein Locus Triplication Causes Parkinson’s Disease, Science. 302 (2003) 841–841. 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- [76].Chartier-Harlin M-C, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A, α-synuclein locus duplication as a cause of familial Parkinson’s disease, The Lancet. 364 (2004) 1167–1169. 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- [77].Gündner AL, Duran-Pacheco G, Zimmermann S, Ruf I, Moors T, Baumann K, Jagasia R, van de Berg WDJ, Kremer T, Path mediation analysis reveals GBA impacts Lewy body disease status by increasing α-synuclein levels, Neurobiology of Disease. 121 (2019) 205–213. 10.1016/j.nbd.2018.09.015. [DOI] [PubMed] [Google Scholar]
- [78].Moors TE, Paciotti S, Ingrassia A, Quadri M, Breedveld G, Tasegian A, Chiasserini D, Eusebi P, Duran-Pacheco G, Kremer T, Calabresi P, Bonifati V, Parnetti L, Beccari T, van de Berg WDJ, Characterization of Brain Lysosomal Activities in GBA-Related and Sporadic Parkinson’s Disease and Dementia with Lewy Bodies, Mol Neurobiol. 56 (2019) 1344–1355. 10.1007/s12035-018-1090-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Blauwendraat C, Reed X, Krohn L, Heilbron K, Bandres-Ciga S, Tan M, Gibbs JR, Hernandez DG, Kumaran R, Langston R, Bonet-Ponce L, Alcalay RN, Hassin-Baer S, Greenbaum L, Iwaki H, Leonard HL, Grenn FP, Ruskey JA, Sabir M, Ahmed S, Makarious MB, Pihlstrøm L, Toft M, van Hilten JJ, Marinus J, Schulte C, Brockmann K, Sharma M, Siitonen A, Majamaa K, Eerola-Rautio J, Tienari PJ, The 23andMe Research Team, Pantelyat A, Hillis AE, Dawson TM, Rosenthal LS, Albert MS, Resnick SM, Ferrucci L, Morris CM, Pletnikova O, Troncoso J, Grosset D, Lesage S, Corvol J-C, Brice A, Noyce AJ, Masliah E, Wood N, Hardy J, Shulman LM, Jankovic J, Shulman JM, Heutink P, Gasser T, Cannon P, Scholz SW, Morris H, Cookson MR, Nalls MA, Gan-Or Z, Singleton AB, on behalf of the International Parkinson’s Disease Genomics Consortium (IPDGC), Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia, Brain. 143 (2020) 234–248. 10.1093/brain/awz350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obermaier CD, Strojny C, Savas JN, Kiskinis E, Zhuang X, Krüger R, Surmeier DJ, Krainc D, Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease, Science. 357 (2017) 1255–1261. 10.1126/science.aam9080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Nguyen M, Krainc D, LRRK2 phosphorylation of auxilin mediates synaptic defects in dopaminergic neurons from patients with Parkinson’s disease, Proceedings of the National Academy of Sciences. 115 (2018) 5576–5581. 10.1073/pnas.1717590115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Burbulla LF, Jeon S, Zheng J, Song P, Silverman RB, Krainc D, A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease, Science Translational Medicine. 11 (2019) eaau6870. 10.1126/scitranslmed.aau6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Yap TL, Gruschus JM, Velayati A, Westbroek W, Goldin E, Moaven N, Sidransky E, Lee JC, α-Synuclein Interacts with Glucocerebrosidase Providing a Molecular Link between Parkinson and Gaucher Diseases, J Biol Chem. 286 (2011) 28080–28088. 10.1074/jbc.M111.237859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kuo S-H, Tasset I, Cheng MM, Diaz A, Pan M-K, Lieberman OJ, Hutten SJ, Alcalay RN, Kim S, Ximénez-Embún P, Fan L, Kim D, Ko HS, Yacoubian T, Kanter E, Liu L, Tang G, Muñoz J, Sardi SP, Li A, Gan L, Cuervo AM, Sulzer D, Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy, Science Advances. 8 (2022) eabm6393. 10.1126/sciadv.abm6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Galvagnion C, Marlet FR, Cerri S, Schapira AHV, Blandini F, Di Monte DA, Sphingolipid changes in Parkinson L444P GBA mutation fibroblasts promote α-synuclein aggregation, Brain. 145 (2022) 1038–1051. 10.1093/brain/awab371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yu M, Ye H, De-Paula RB, Mangleburg CG, Wu T, Li Y, Duong D, Allen GI, Seyfried NT, Al-Ramahi I, Botas J, Shulman JM, Functional screening of lysosomal storage disorder genes identifies modifiers of alpha-synuclein mediated neurodegeneration, (2022) 2022.07.23.501240. 10.1101/2022.07.23.501240. [DOI] [PMC free article] [PubMed]
- [87].Johnson ME, Bergkvist L, Stetzik L, Steiner JA, Meyerdirk L, Schulz E, Wolfrum E, Luk KC, Wesson DW, Krainc D, Brundin P, Heterozygous GBA D409V and ATP13a2 mutations do not exacerbate pathological α-synuclein spread in the prodromal preformed fibrils model in young mice, Neurobiology of Disease. 159 (2021) 105513. 10.1016/j.nbd.2021.105513. [DOI] [PubMed] [Google Scholar]
- [88].Migdalska-Richards A, Wegrzynowicz M, Harrison IF, Verona G, Bellotti V, Spillantini MG, Schapira AHV, L444P Gba1 mutation increases formation and spread of α-synuclein deposits in mice injected with mouse α-synuclein pre-formed fibrils, PLOS ONE. 15 (2020) e0238075. 10.1371/journal.pone.0238075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Mahoney-Crane CL, Viswanathan M, Russell D, Curtiss RAC, Freire J, Bobba SS, Coyle SD, Kandebo M, Yao L, Wan B-L, Hatcher NG, Smith SM, Marcus JN, Volpicelli-Daley LA, Neuronopathic GBA1L444P mutation accelerates Glucosylsphingosine levels and formation of hippocampal alpha-synuclein inclusions, J. Neurosci. (2022). 10.1523/JNEUROSCI.0680-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Henderson MX, Cornblath EJ, Darwich A, Zhang B, Brown H, Gathagan RJ, Sandler RM, Bassett DS, Trojanowski JQ, Lee VMY, Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis, Nat Neurosci. 22 (2019) 1248–1257. 10.1038/s41593-019-0457-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Rey NL, George S, Steiner JA, Madaj Z, Luk KC, Trojanowski JQ, Lee VM-Y, Brundin P, Spread of aggregates after olfactory bulb injection of α-synuclein fibrils is associated with early neuronal loss and is reduced long term, Acta Neuropathol. 135 (2018) 65–83. 10.1007/s00401-017-1792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Chatterjee D, Sanchez DS, Quansah E, Rey NL, George S, Becker K, Madaj Z, Steiner JA, Ma J, Escobar Galvis ML, Kordower JH, Brundin P, Loss of One Engrailed1 Allele Enhances Induced α-Synucleinopathy, Journal of Parkinson’s Disease. 9 (2019) 315–326. 10.3233/JPD-191590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Henderson MX, Sedor S, McGeary I, Cornblath EJ, Peng C, Riddle DM, Li HL, Zhang B, Brown HJ, Olufemi MF, Bassett DS, Trojanowski JQ, Lee VMY, Glucocerebrosidase activity modulates neuronal susceptibility to pathological α-synuclein insult, Neuron. 105 (2020) 822–836.e7. 10.1016/j.neuron.2019.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, Mazmanian SK, Volpicelli-Daley LA, Gradinaru V, Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice, Nat Neurosci. 23 (2020) 327–336. 10.1038/s41593-020-0589-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Horsager J, Andersen KB, Knudsen K, Skjærbæk C, Fedorova TD, Okkels N, Schaeffer E, Bonkat SK, Geday J, Otto M, Sommerauer M, Danielsen EH, Bech E, Kraft J, Munk OL, Hansen SD, Pavese N, Göder R, Brooks DJ, Berg D, Borghammer P, Brain-first versus body-first Parkinson’s disease: a multimodal imaging case-control study, Brain. 143 (2020) 3077–3088. 10.1093/brain/awaa238. [DOI] [PubMed] [Google Scholar]
- [96].Huebecker M, Moloney EB, van der Spoel AC, Priestman DA, Isacson O, Hallett PJ, Platt FM, Reduced sphingolipid hydrolase activities, substrate accumulation and ganglioside decline in Parkinson’s disease, Mol Neurodegeneration. 14 (2019) 40. 10.1186/s13024-019-0339-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Gegg ME, Sweet L, Wang BH, Shihabuddin LS, Sardi SP, Schapira AHV, No evidence for substrate accumulation in Parkinson brains with GBA mutations, Movement Disorders. 30 (2015) 1085–1089. 10.1002/mds.26278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Boutin M, Sun Y, Shacka JJ, Auray-Blais C, Tandem Mass Spectrometry Multiplex Analysis of Glucosylceramide and Galactosylceramide Isoforms in Brain Tissues at Different Stages of Parkinson Disease, Anal. Chem. 88 (2016) 1856–1863. 10.1021/acs.analchem.5b04227. [DOI] [PubMed] [Google Scholar]
- [99].Taguchi YV, Liu J, Ruan J, Pacheco J, Zhang X, Abbasi J, Keutzer J, Mistry PK, Chandra SS, Glucosylsphingosine Promotes α-Synuclein Pathology in Mutant GBA-Associated Parkinson’s Disease, J Neurosci. 37 (2017) 9617–9631. 10.1523/JNEUROSCI.1525-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Surface M, Balwani M, Waters C, Haimovich A, Gan-Or Z, Marder KS, Hsieh T, Song L, Padmanabhan S, Hsieh F, Merchant KM, Alcalay RN, Plasma Glucosylsphingosine in GBA1 Mutation Carriers with and without Parkinson’s Disease, Movement Disorders. 37 (2022) 416–421. 10.1002/mds.28846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Lerche S, Schulte C, Wurster I, Machetanz G, Roeben B, Zimmermann M, Deuschle C, Hauser A-K, Böhringer J, Krägeloh-Mann I, Waniek K, Lachmann I, Petterson X-MT, Chiang R, Park H, Wang B, Liepelt-Scarfone I, Maetzler W, Galasko D, Scherzer CR, Gasser T, Mielke MM, Hutten SJ, Mollenhauer B, Sardi SP, Berg D, Brockmann K, The Mutation Matters: CSF Profiles of GCase, Sphingolipids, α-Synuclein in PDGBA, Movement Disorders. 36 (2021) 1216–1228. 10.1002/mds.28472. [DOI] [PubMed] [Google Scholar]
- [102].Milenkovic I, Blumenreich S, Futerman AH, GBA mutations, glucosylceramide and Parkinson’s disease, Current Opinion in Neurobiology. 72 (2022) 148–154. 10.1016/j.conb.2021.11.004. [DOI] [PubMed] [Google Scholar]
- [103].Sardi SP, Viel C, Clarke J, Treleaven CM, Richards AM, Park H, Olszewski MA, Dodge JC, Marshall J, Makino E, Wang B, Sidman RL, Cheng SH, Shihabuddin LS, Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models, Proc Natl Acad Sci U S A. 114 (2017) 2699–2704. 10.1073/pnas.1616152114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Peterschmitt MJ, Saiki H, Hatano T, Gasser T, Isaacson SH, Gaemers SJM, Minini P, Saubadu S, Sharma J, Walbillic S, Alcalay RN, Cutter G, Hattori N, Höglinger GU, Marek K, Schapira AHV, Scherzer CR, Simuni T, Giladi N, Sardi SP, Fischer TZ, Safety, Pharmacokinetics, and Pharmacodynamics of Oral Venglustat in Patients with Parkinson’s Disease and a GBA Mutation: Results from Part 1 of the Randomized, Double-Blinded, Placebo-Controlled MOVES-PD Trial, J Parkinsons Dis. 12 (n.d.) 557–570. 10.3233/JPD-212714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Plotegher N, Bubacco L, Greggio E, Civiero L, Ceramides in Parkinson’s Disease: From Recent Evidence to New Hypotheses, Frontiers in Neuroscience. 13 (2019). https://www.frontiersin.org/articles/10.3389/fnins.2019.00330 (accessed December 12, 2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Kim MJ, Jeong H, Krainc D, Lysosomal ceramides regulate cathepsin B-mediated processing of saposin C and glucocerebrosidase activity, Human Molecular Genetics. 31 (2022) 2424–2437. 10.1093/hmg/ddac047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Kurzawa-Akanbi M, Tammireddy S, Fabrik I, Gliaudelytė L, Doherty MK, Heap R, Matečko-Burmann I, Burmann BM, Trost M, Lucocq JM, Gherman AV, Fairfoul G, Singh P, Burté F, Green A, McKeith IG, Härtlova A, Whitfield PD, Morris CM, Altered ceramide metabolism is a feature in the extracellular vesicle-mediated spread of alpha-synuclein in Lewy body disorders, Acta Neuropathol. 142 (2021) 961–984. 10.1007/s00401-021-02367-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Wise AH, Yang A, Naik H, Stauffer C, Zeid N, Liong C, Balwani M, Desnick RJ, Alcalay RN, Parkinson’s disease prevalence in Fabry disease: A survey study, Mol Genet Metab Rep. 14 (2017) 27–30. 10.1016/j.ymgmr.2017.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].te Vruchte D, Sturchio A, Priestman DA, Tsitsi P, Hertz E, Andréasson M, Markaki I, Wallom K-L, Platt F, Svenningsson P, Glycosphingolipid Changes in Plasma in Parkinson’s Disease Independent of Glucosylceramide Levels, Movement Disorders. 37 (2022) 2129–2134. 10.1002/mds.29163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Bae E-J, Yang NY, Lee C, Lee H-J, Kim S, Sardi SP, Lee S-J, Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and α-synuclein aggregation, Exp Mol Med. 47 (2015) e153. 10.1038/emm.2014.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Schöndorf DC, Aureli M, McAllister FE, Hindley CJ, Mayer F, Schmid B, Sardi SP, Valsecchi M, Hoffmann S, Schwarz LK, Hedrich U, Berg D, Shihabuddin LS, Hu J, Pruszak J, Gygi SP, Sonnino S, Gasser T, Deleidi M, iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis, Nat Commun. 5 (2014) 4028. 10.1038/ncomms5028. [DOI] [PubMed] [Google Scholar]
- [112].Awad O, Sarkar C, Panicker LM, Miller D, Zeng X, Sgambato JA, Lipinski MM, Feldman RA, Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells, Human Molecular Genetics. 24 (2015) 5775–5788. 10.1093/hmg/ddv297. [DOI] [PubMed] [Google Scholar]
- [113].Thomas RE, Vincow ES, Merrihew GE, MacCoss MJ, Davis MY, Pallanck LJ, Glucocerebrosidase deficiency promotes protein aggregation through dysregulation of extracellular vesicles, PLOS Genetics. 14 (2018) e1007694. 10.1371/journal.pgen.1007694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Valdez C, Ysselstein D, Young TJ, Zheng J, Krainc D, Progranulin mutations result in impaired processing of prosaposin and reduced glucocerebrosidase activity, Hum Mol Genet. 29 (2020) 716–726. 10.1093/hmg/ddz229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Arrant AE, Roth JR, Boyle NR, Kashyap SN, Hoffmann MQ, Murchison CF, Ramos EM, Nana AL, Spina S, Grinberg LT, Miller BL, Seeley WW, Roberson ED, Impaired β-glucocerebrosidase activity and processing in frontotemporal dementia due to progranulin mutations, Acta Neuropathologica Communications. 7 (2019). 10.1186/s40478-019-0872-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Jian J, Zhao S, Tian Q-Y, Liu H, Zhao Y, Chen W-C, Grunig G, Torres PA, Wang BC, Zeng B, Pastores G, Tang W, Sun Y, Grabowski GA, Kong MX, Wang G, Chen Y, Liang F, Overkleeft HS, Saunders-Pullman R, Chan GL, Liu C, Association Between Progranulin and Gaucher Disease, EBioMedicine. 11 (2016) 127–137. 10.1016/j.ebiom.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Zhou X, Paushter DH, Pagan MD, Kim D, Santos MN, Lieberman RL, Overkleeft HS, Sun Y, Smolka MB, Hu F, Progranulin deficiency leads to reduced glucocerebrosidase activity, PLOS ONE. 14 (2019) e0212382. 10.1371/journal.pone.0212382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Logan T, Simon MJ, Rana A, Cherf GM, Srivastava A, Davis SS, Low RLY, Chiu C-L, Fang M, Huang F, Bhalla A, Llapashtica C, Prorok R, Pizzo ME, Calvert MEK, Sun EW, Hsiao-Nakamoto J, Rajendra Y, Lexa KW, Srivastava DB, van Lengerich B, Wang J, Robles-Colmenares Y, Kim DJ, Duque J, Lenser M, Earr TK, Nguyen H, Chau R, Tsogtbaatar B, Ravi R, Skuja LL, Solanoy H, Rosen HJ, Boeve BF, Boxer AL, Heuer HW, Dennis MS, Kariolis MS, Monroe KM, Przybyla L, Sanchez PE, Meisner R, Diaz D, Henne KR, Watts RJ, Henry AG, Gunasekaran K, Astarita G, Suh JH, Lewcock JW, DeVos SL, Di Paolo G, Rescue of a lysosomal storage disorder caused by Grn loss of function with a brain penetrant progranulin biologic, Cell. 184 (2021) 4651–4668.e25. 10.1016/j.cell.2021.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Ysselstein D, Nguyen M, Young TJ, Severino A, Schwake M, Merchant K, Krainc D, LRRK2 kinase activity regulates lysosomal glucocerebrosidase in neurons derived from Parkinson’s disease patients, Nat Commun. 10 (2019) 5570. 10.1038/s41467-019-13413-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Sanyal A, DeAndrade MP, Novis HS, Lin S, Chang J, Lengacher N, Tomlinson JJ, Tansey MG, LaVoie MJ, Lysosome and Inflammatory Defects in GBA1-Mutant Astrocytes Are Normalized by LRRK2 Inhibition, Movement Disorders. 35 (2020) 760–773. 10.1002/mds.27994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Ferrazza R, Cogo S, Melrose H, Bubacco L, Greggio E, Guella G, Civiero L, Plotegher N, LRRK2 deficiency impacts ceramide metabolism in brain, Biochemical and Biophysical Research Communications. 478 (2016) 1141–1146. 10.1016/j.bbrc.2016.08.082. [DOI] [PubMed] [Google Scholar]
- [122].Ortega RA, Wang C, Raymond D, Bryant N, Scherzer CR, Thaler A, Alcalay RN, West AB, Mirelman A, Kuras Y, Marder KS, Giladi N, Ozelius LJ, Bressman SB, Saunders-Pullman R, Association of Dual LRRK2 G2019S and GBA Variations With Parkinson Disease Progression, JAMA Network Open. 4 (2021) e215845. 10.1001/jamanetworkopen.2021.5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Yahalom G, Greenbaum L, Israeli-Korn S, Fay-Karmon T, Livneh V, Ruskey JA, Roncière L, Alam A, Gan-Or Z, Hassin-Baer S, Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson’s disease: Risk estimates and genotype-phenotype correlations, Parkinsonism & Related Disorders. 62 (2019) 179–184. 10.1016/j.parkreldis.2018.12.014. [DOI] [PubMed] [Google Scholar]
- [124].Bendikov-Bar I, Ron I, Filocamo M, Horowitz M, Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant, Blood Cells, Molecules, and Diseases. 46 (2011) 4–10. 10.1016/j.bcmd.2010.10.012. [DOI] [PubMed] [Google Scholar]
- [125].Thomas R, Moloney EB, Macbain ZK, Hallett PJ, Isacson O, Fibroblasts from idiopathic Parkinson’s disease exhibit deficiency of lysosomal glucocerebrosidase activity associated with reduced levels of the trafficking receptor LIMP2, Molecular Brain. 14 (2021) 16. 10.1186/s13041-020-00712-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Fernandes HJR, Hartfield EM, Christian HC, Emmanoulidou E, Zheng Y, Booth H, Bogetofte H, Lang C, Ryan BJ, Sardi SP, Badger J, Vowles J, Evetts S, Tofaris GK, Vekrellis K, Talbot K, Hu MT, James W, Cowley SA, Wade-Martins R, ER Stress and Autophagic Perturbations Lead to Elevated Extracellular α-Synuclein in GBA-N370S Parkinson’s iPSC-Derived Dopamine Neurons, Stem Cell Reports. 6 (2016) 342–356. 10.1016/j.stemcr.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Smith LJ, Bolsinger MM, Chau K-Y, Gegg ME, Schapira AHV, The GBA variant E326K is associated with alpha-synuclein aggregation and lipid droplet accumulation in human cell lines, (2022) 2022.06.01.494130. 10.1101/2022.06.01.494130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].McNeill A, Magalhaes J, Shen C, Chau K-Y, Hughes D, Mehta A, Foltynie T, Cooper JM, Abramov AY, Gegg M, Schapira AHV, Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells, Brain. 137 (2014) 1481–1495. 10.1093/brain/awu020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Migdalska-Richards A, Ko WKD, Li Q, Bezard E, Schapira AHV, Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate, Synapse. 71 (2017) e21967. 10.1002/syn.21967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Mullin S, Smith L, Lee K, D’Souza G, Woodgate P, Elflein J, Hällqvist J, Toffoli M, Streeter A, Hosking J, Heywood WE, Khengar R, Campbell P, Hehir J, Cable S, Mills K, Zetterberg H, Limousin P, Libri V, Foltynie T, Schapira AHV, Ambroxol for the Treatment of Patients With Parkinson Disease With and Without Glucocerebrosidase Gene Mutations: A Nonrandomized, Noncontrolled Trial, JAMA Neurology. 77 (2020) 427–434. 10.1001/jamaneurol.2019.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Han T-U, Sam R, Sidransky E, Small Molecule Chaperones for the Treatment of Gaucher Disease and GBA1-Associated Parkinson Disease, Frontiers in Cell and Developmental Biology. 8 (2020). https://www.frontiersin.org/articles/10.3389/fcell.2020.00271 (accessed February 9, 2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Stojkovska I, Wani WY, Zunke F, Belur NR, Pavlenko EA, Mwenda N, Sharma K, Francelle L, Mazzulli JR, Rescue of α-synuclein aggregation in Parkinson’s patient neurons by synergistic enhancement of ER proteostasis and protein trafficking, Neuron. 110 (2022) 436–451.e11. 10.1016/j.neuron.2021.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Boddupalli CS, Nair S, Belinsky G, Gans J, Teeple E, Nguyen T-H, Mehta S, Guo L, Kramer ML, Ruan J, Wang H, Davison M, Kumar D, Vidyadhara D, Zhang B, Klinger K, Mistry PK, Neuroinflammation in neuronopathic Gaucher disease: Role of microglia and NK cells, biomarkers, and response to substrate reduction therapy, ELife. 11 (2022) e79830. 10.7554/eLife.79830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Kam T-I, Hinkle JT, Dawson TM, Dawson VL, Microglia and Astrocyte Dysfunction in Parkinson’s Disease, Neurobiol Dis. 144 (2020) 105028. 10.1016/j.nbd.2020.105028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Liu S-Y, Qiao H-W, Song T-B, Liu X-L, Yao Y-X, Zhao C-S, Barret O, Xu S-L, Cai Y-N, Tamagnan GD, Sossi V, Lu J, Chan P, Brain microglia activation and peripheral adaptive immunity in Parkinson’s disease: a multimodal PET study, Journal of Neuroinflammation. 19 (2022) 209. 10.1186/s12974-022-02574-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Ouchi Y, Imaging neuroinflammation to monitor α-synucleinopathy, The Lancet Neurology. 16 (2017) 763–764. 10.1016/S1474-4422(17)30244-2. [DOI] [PubMed] [Google Scholar]
- [137].Olanow CW, Savolainen M, Chu Y, Halliday GM, Kordower JH, Temporal evolution of microglia and α-synuclein accumulation following foetal grafting in Parkinson’s disease, Brain. 142 (2019) 1690–1700. 10.1093/brain/awz104. [DOI] [PubMed] [Google Scholar]
- [138].Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, Robertson AAB, Butler MS, Rowe DB, O’Neill LA, Kanthasamy AG, Schroder K, Cooper MA, Woodruff TM, Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice, Science Translational Medicine. 10 (2018) eaah4066. 10.1126/scitranslmed.aah4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Kim T-K, Bae E-J, Jung BC, Choi M, Shin SJ, Park SJ, Kim JT, Jung MK, Ulusoy A, Song M-Y, Lee JS, Lee H-J, Di Monte DA, Lee S-J, Inflammation promotes synucleinopathy propagation, Exp Mol Med. (2022) 1–14. 10.1038/s12276-022-00895-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Lindestam Arlehamn CS, Dhanwani R, Pham J, Kuan R, Frazier A, Rezende Dutra J, Phillips E, Mallal S, Roederer M, Marder KS, Amara AW, Standaert DG, Goldman JG, Litvan I, Peters B, Sulzer D, Sette A, α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease, Nat Commun. 11 (2020) 1875. 10.1038/s41467-020-15626-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes JP, Liong C, McMurtrey C, Hildebrand WH, Mao X, Dawson VL, Dawson TM, Oseroff C, Pham J, Sidney J, Dillon MB, Carpenter C, Weiskopf D, Phillips E, Mallal S, Peters B, Frazier A, Lindestam Arlehamn CS, Sette A, T cells of Parkinson’s disease patients recognize α–synuclein peptides, Nature. 546 (2017) 656–661. 10.1038/nature22815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Brunialti E, Villa A, Mekhaeil M, Mornata F, Vegeto E, Maggi A, Di Monte DA, Ciana P, Inhibition of microglial β-glucocerebrosidase hampers the microglia-mediated antioxidant and protective response in neurons, Journal of Neuroinflammation. 18 (2021) 220. 10.1186/s12974-021-02272-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Zhang Z, Wang X, Lin Y, Pan D, A multifaceted evaluation of microgliosis and differential cellular dysregulation of mammalian target of rapamycin signaling in neuronopathic Gaucher disease, Frontiers in Molecular Neuroscience. 15 (2022). https://www.frontiersin.org/articles/10.3389/fnmol.2022.944883 (accessed December 12, 2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Rocha EM, Smith GA, Park E, Cao H, Graham A-R, Brown E, McLean JR, Hayes MA, Beagan J, Izen SC, Perez-Torres E, Hallett PJ, Isacson O, Sustained Systemic Glucocerebrosidase Inhibition Induces Brain α-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice, Antioxid Redox Signal. 23 (2015) 550–564. 10.1089/ars.2015.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Enquist IB, Bianco CL, Ooka A, Nilsson E, Månsson J-E, Ehinger M, Richter J, Brady RO, Kirik D, Karlsson S, Murine models of acute neuronopathic Gaucher disease, Proceedings of the National Academy of Sciences. 104 (2007) 17483–17488. 10.1073/pnas.0708086104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Farfel-Becker T, Vitner EB, Kelly SL, Bame JR, Duan J, Shinder V, Merrill AH Jr, Dobrenis K, Futerman AH, Neuronal accumulation of glucosylceramide in a mouse model of neuronopathic Gaucher disease leads to neurodegeneration, Human Molecular Genetics. 23 (2014) 843–854. 10.1093/hmg/ddt468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Ginns EI, Mak SK-K, Ko N, Karlgren J, Akbarian S, Chou VP, Guo Y, Lim A, Samuelsson S, LaMarca ML, Vazquez-DeRose J, Manning-Boğ AB, Neuroinflammation and α-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction, Molecular Genetics and Metabolism. 111 (2014) 152–162. 10.1016/j.ymgme.2013.12.003. [DOI] [PubMed] [Google Scholar]
- [148].Mus L, Siani F, Giuliano C, Ghezzi C, Cerri S, Blandini F, Development and biochemical characterization of a mouse model of Parkinson’s disease bearing defective glucocerebrosidase activity, Neurobiol Dis. 124 (2019) 289–296. 10.1016/j.nbd.2018.12.001. [DOI] [PubMed] [Google Scholar]
- [149].Soria FN, Engeln M, Martinez-Vicente M, Glangetas C, López-González MJ, Dovero S, Dehay B, Normand E, Vila M, Favereaux A, Georges F, Lo Bianco C, Bezard E, Fernagut P-O, Glucocerebrosidase deficiency in dopaminergic neurons induces microglial activation without neurodegeneration, Human Molecular Genetics. 26 (2017) 2603–2615. 10.1093/hmg/ddx120. [DOI] [PubMed] [Google Scholar]
- [150].Harms AS, Ferreira SA, Romero-Ramos M, Periphery and brain, innate and adaptive immunity in Parkinson’s disease, Acta Neuropathol. 141 (2021) 527–545. 10.1007/s00401-021-02268-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Avenali M, Cerri S, Ongari G, Ghezzi C, Pacchetti C, Tassorelli C, Valente EM, Blandini F, Profiling the Biochemical Signature of GBA-Related Parkinson’s Disease in Peripheral Blood Mononuclear Cells, Movement Disorders. 36 (2021) 1267–1272. 10.1002/mds.28496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Nagata M, Izumi Y, Ishikawa E, Kiyotake R, Doi R, Iwai S, Omahdi Z, Yamaji T, Miyamoto T, Bamba T, Yamasaki S, Intracellular metabolite β-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity, Proc Natl Acad Sci U S A. 114 (2017) E3285–E3294. 10.1073/pnas.1618133114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Wallings RL, Hughes LP, Staley HA, Simon ZD, McFarland NR, Alcalay RN, Garrido A, Martí MJ, Sarró ET, Dzamko N, Tansey MG, WHOPPA Enables Parallel Assessment of Leucine-Rich Repeat Kinase 2 and Glucocerebrosidase Enzymatic Activity in Parkinson’s Disease Monocytes, Front Cell Neurosci. 16 (2022) 892899. 10.3389/fncel.2022.892899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Thaler A, Omer N, Giladi N, Gurevich T, Bar-Shira A, Gana-Weisz M, Goldstein O, Kestenbaum M, Shirvan JC, Cedarbaum JM, Orr-Urtreger A, Regev K, Shenhar-Tsarfaty S, Mirelman A, Mutations in GBA and LRRK2 Are Not Associated with Increased Inflammatory Markers, JPD. 11 (2021) 1285–1296. 10.3233/JPD-212624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Galper J, Balwani M, Fahn S, Waters C, Krohn L, Gan-Or Z, Dzamko N, Alcalay RN, Cytokines and Gaucher Biomarkers in Glucocerebrosidase Carriers with and Without Parkinson Disease, Mov Disord. 36 (2021) 1451–1455. 10.1002/mds.28525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Miliukhina IV, Usenko TS, Senkevich KA, Nikolaev MA, Timofeeva AA, Agapova EA, Semenov AV, Lubimova NE, Totolyan AA, Pchelina SN, Plasma Cytokines Profile in Patients with Parkinson’s Disease Associated with Mutations in GBA Gene, Bull Exp Biol Med. 168 (2020) 423–426. 10.1007/s10517-020-04723-x. [DOI] [PubMed] [Google Scholar]
- [157].Mancuso R, Van Den Daele J, Fattorelli N, Wolfs L, Balusu S, Burton O, Liston A, Sierksma A, Fourne Y, Poovathingal S, Arranz-Mendiguren A, Sala Frigerio C, Claes C, Serneels L, Theys T, Perry VH, Verfaillie C, Fiers M, De Strooper B, Stem-cell-derived human microglia transplanted in mouse brain to study human disease, Nat Neurosci. 22 (2019) 2111–2116. 10.1038/s41593-019-0525-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Fattorelli N, Martinez-Muriana A, Wolfs L, Geric I, De Strooper B, Mancuso R, Stem-cell-derived human microglia transplanted into mouse brain to study human disease, Nat Protoc. 16 (2021) 1013–1033. 10.1038/s41596-020-00447-4. [DOI] [PubMed] [Google Scholar]
- [159].Xu Y-H, Quinn B, Witte D, Grabowski GA, Viable Mouse Models of Acid β-Glucosidase Deficiency: The Defect in Gaucher Disease, The American Journal of Pathology. 163 (2003) 2093–2101. 10.1016/S0002-9440(10)63566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Latham T. e., Theophilus B. d. m., Grabowski G. a., Smith F. i., Heterogeneity of Mutations in the Acid β-Glucosidase Gene of Gaucher Disease Patients, DNA and Cell Biology. 10 (1991) 15–21. 10.1089/dna.1991.10.15. [DOI] [PubMed] [Google Scholar]
- [161].Kim D, Hwang H, Choi S, Kwon SH, Lee S, Park JH, Kim S, Ko HS, D409H GBA1 mutation accelerates the progression of pathology in A53T α-synuclein transgenic mouse model, Acta Neuropathol Commun. 6 (2018) 32. 10.1186/s40478-018-0538-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Schierding W, Farrow S, Fadason T, Graham OEE, Pitcher TL, Qubisi S, Davidson AJ, Perry JK, Anderson TJ, Kennedy MA, Cooper A, O’Sullivan JM, Common Variants Coregulate Expression of GBA and Modifier Genes to Delay Parkinson’s Disease Onset, Movement Disorders. 35 (2020) 1346–1356. 10.1002/mds.28144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Straniero L, Rimoldi V, Monfrini E, Bonvegna S, Melistaccio G, Lake J, Soldà G, Aureli M, Shankaracharya P. Keagle, Foroud T, Landers JE, Blauwendraat C, Zecchinelli A, Cilia R, Di Fonzo A, Pezzoli G, Duga S, Asselta R, Role of Lysosomal Gene Variants in Modulating GBA-Associated Parkinson’s Disease Risk, Movement Disorders. 37 (2022) 1202–1210. 10.1002/mds.28987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Drouin-Ouellet J, Legault EM, Nilsson F, Pircs K, Bouquety J, Petit F, Shrigley S, Birtele M, Pereira M, Storm P, Sharma Y, Bruzelius A, Vuono R, Kele M, Stoker TB, Ottosson DR, Falk A, Jakobsson J, Barker RA, Parmar M, Age-related pathological impairments in directly reprogrammed dopaminergic neurons derived from patients with idiopathic Parkinson’s disease, Stem Cell Reports. 17 (2022) 2203–2219. 10.1016/j.stemcr.2022.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Qin H, Zhao A-D, Sun M-L, Ma K, Fu X-B, Direct conversion of human fibroblasts into dopaminergic neuron-like cells using small molecules and protein factors, Military Medical Research. 7 (2020) 52. 10.1186/s40779-020-00284-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Ysselstein D, Young TJ, Nguyen M, Padmanabhan S, Hirst WD, Dzamko N, Krainc D, Evaluation of Strategies for Measuring Lysosomal Glucocerebrosidase Activity, Movement Disorders. 36 (2021) 2719–2730. 10.1002/mds.28815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Deen MC, Zhu Y, Gros C, Na N, Gilormini P-A, Shen DL, Bhosale S, Anastasi N, Wang R, Shan X, Harde E, Jagasia R, Lynn FC, Vocadlo DJ, A versatile fluorescence-quenched substrate for quantitative measurement of glucocerebrosidase activity within live cells, Proceedings of the National Academy of Sciences. 119 (2022) e2200553119. 10.1073/pnas.2200553119. [DOI] [PMC free article] [PubMed] [Google Scholar]