

SUMMARY
Harmful algal blooms of the toxic haptophyte Prymnesium parvum are a recurrent problem in many inland and estuarine waters around the world. Strains of P. parvum vary in the toxins they produce and in other physiological traits associated with harmful algal blooms, but the genetic basis for this variation is unknown. To investigate genome diversity in this morphospecies, we generated genome assemblies for fifteen phylogenetically and geographically diverse strains of P. parvum including Hi-C guided, near-chromosome level assemblies for two strains. Comparative analysis revealed considerable DNA content variation between strains, ranging from 115 Mbp to 845 Mbp. Strains included haploids, diploids, and polyploids, but not all differences in DNA content were due to variation in genome copy number. Haploid genome size between strains of different chemotypes differed by as much as 243 Mbp. Syntenic and phylogenetic analyses indicate that UTEX 2797, a common laboratory strain from Texas, is a hybrid that retains two phylogenetically distinct haplotypes. Investigation of gene families variably present across the strains identified several functional categories associated with metabolic and genome size variation in P. parvum including genes for the biosynthesis of toxic metabolites and proliferation of transposable elements. Together, our results indicate that P. parvum is comprised of multiple cryptic species. These genomes provide a robust phylogenetic and genomic framework for investigations into the eco-physiological consequences of the intra- and inter-specific genetic variation present in P. parvum and demonstrate the need for similar resources for other harmful algal bloom-forming morphospecies.
eTOC Blurb
Wisecaver et al. presents genomes for fifteen strains of Prymnesium parvum, a cause of harmful algal blooms around the world. They uncover unprecedented sequence-level, gene family, and genome architecture evolution and provide evidence for both cryptic speciation and hybridization in this protist morphospecies.
Graphical Abstract
INTRODUCTION
Environmentally disruptive harmful algal blooms are increasing in frequency and severity in many marine, brackish, and freshwater ecosystems1–3. Knowledge of genetic variation within harmful algal bloom populations can aid efforts to predict when blooms will occur and model bloom impacts in a changing climate. However, genome-level data are currently limited for harmful algal bloom-forming species4. One such species, Prymnesium parvum, is a unicellular microalga belonging to the haptophyte clade of eukaryotes5. Globally, harmful algal blooms of P. parvum have considerable ecologic and economic impacts. In the southwestern United States, particularly in Texas, blooms are correlated with several troubling environmental trends including increased use of glyphosate herbicides, increased concentrations of atmospheric CO2, and decreased wetland cover6,7.
P. parvum’s success as a harmful algal bloom-forming species likely results from several interrelated aspects of its biology. The species is extremely eurythermal and euryhaline, tolerating temperatures ranging from 2 to 32 °C and salinities ranging from 0.5 to 125% of typical seawater8. Although P. parvum is photosynthetic and maintains permanent chloroplasts, it is also a mixotroph and able to ingest organic material via phagocytosis9,10. Despite its small size, P. parvum is a ferocious facultative predator capable of swarming and killing various prey including bacteria, other microbial eukaryotes, aquatic invertebrates, and even fish11–14. The species produces toxins that are thought to assist in this predatory behavior and/or inhibit the activity of competitors and grazers5. Heterotrophy and toxicity can increase in P. parvum cultures grown in sub-optimal conditions, e.g., when nutrients are limiting or water temperature/salinity is at the edge of what a strain can tolerate8,10,15.
P. parvum produces a variety of toxic metabolites16. Most conspicuous are the prymnesins, massive ladder-frame polyether compounds that are unique to P. parvum but structurally similar to the phycotoxins of many harmful algal bloom-forming dinoflagellates17. More than 50 different prymnesins have been identified and are grouped into types based on the number of carbons in their aglycone backbone. The backbone of A-type prymnesins contain 91 carbons, while B-type and C-type prymnesins contain 85 and 83 carbons, respectively18,19. To date, all strains of P. parvum produce just one type of prymnesin, and strains that produce the same prymnesin type (i.e., chemotype) group together in phylogenies of rDNA sequences19,20. The strong agreement between phylogeny and chemotype —despite strains of different chemotypes often existing in the same geographical location—suggests that there is limited gene flow between chemotypes of P. parvum19.
Strains of P. parvum may appear morphologically indistinguishable, but the species harbors considerable phenotypic diversity. In addition to the previously discussed diversity in prymnesin chemotypes, the amount of prymnesin produced varies between strains20 as does overall toxicity20–23. Predatory behavior also varies between P. parvum strains, even between strains taken from the same bloom24. Moreover, several abiotic factors including temperature, salinity, pH, and irradiance have distinct effects on the relative growth rate and toxicity of different strains23,25–27, demonstrating remarkable genotype by environment interactions and adaptive plasticity in P. parvum.
Extensive physiological and biochemical variation between strains of P. parvum indicates either the existence of multiple cryptic species19 or extreme standing genetic variation in this morphospecies. However, the genetic differences between strains have not yet been quantified. Here, we generated fifteen sequenced genomes of P. parvum strains and report considerable genomic variation, both at the nucleotide level and in terms of gene family presence/absence. Our comparative phylogenetic analysis supports monophyletic origins of A-, B-, and C-type prymnesins, with C-type strains having more within-clade sequence diversity compared to A- and B-types. Moreover, we found that the three prymnesin chemotypes of P. parvum have dramatically different haploid genome sizes. In addition to haploid strains identified in all three chemotypes, we also identified A- and C-type diploid strains, as well as A- and B-type 4n tetraploids. Lastly, we present evidence that UTEX 2797, a common laboratory strain from Texas, is a hybrid that retains two phylogenetically distinct haplotypes.
RESULTS
Prymnesium parvum genome assemblies.
We obtained Hi-C scaffolded, highly contiguous genome assemblies of two P. parvum strains from Texas: UTEX 2797 and 12B1. UTEX 2797 was selected for sequencing due to its use in numerous toxicology and physiology experiments of P. parvum19,20,22,27–32. However, preliminary analysis of k-mer frequencies revealed that UTEX 2797 displays high sequence-level heterozygosity (Figure 1A), which can complicate genome assembly. Therefore, we also selected strain 12B121, which had no observable heterozygosity to obtain a homozygous reference assembly (Figure 1A). UTEX 2797 produces A-type prymnesins18,19, and our chemotyping analysis revealed strain 12B1 produces A-type prymnesins as well (Figure S1).
Figure 1. Genome metrics for P. parvum Hi-C scaffolded long-read assemblies.
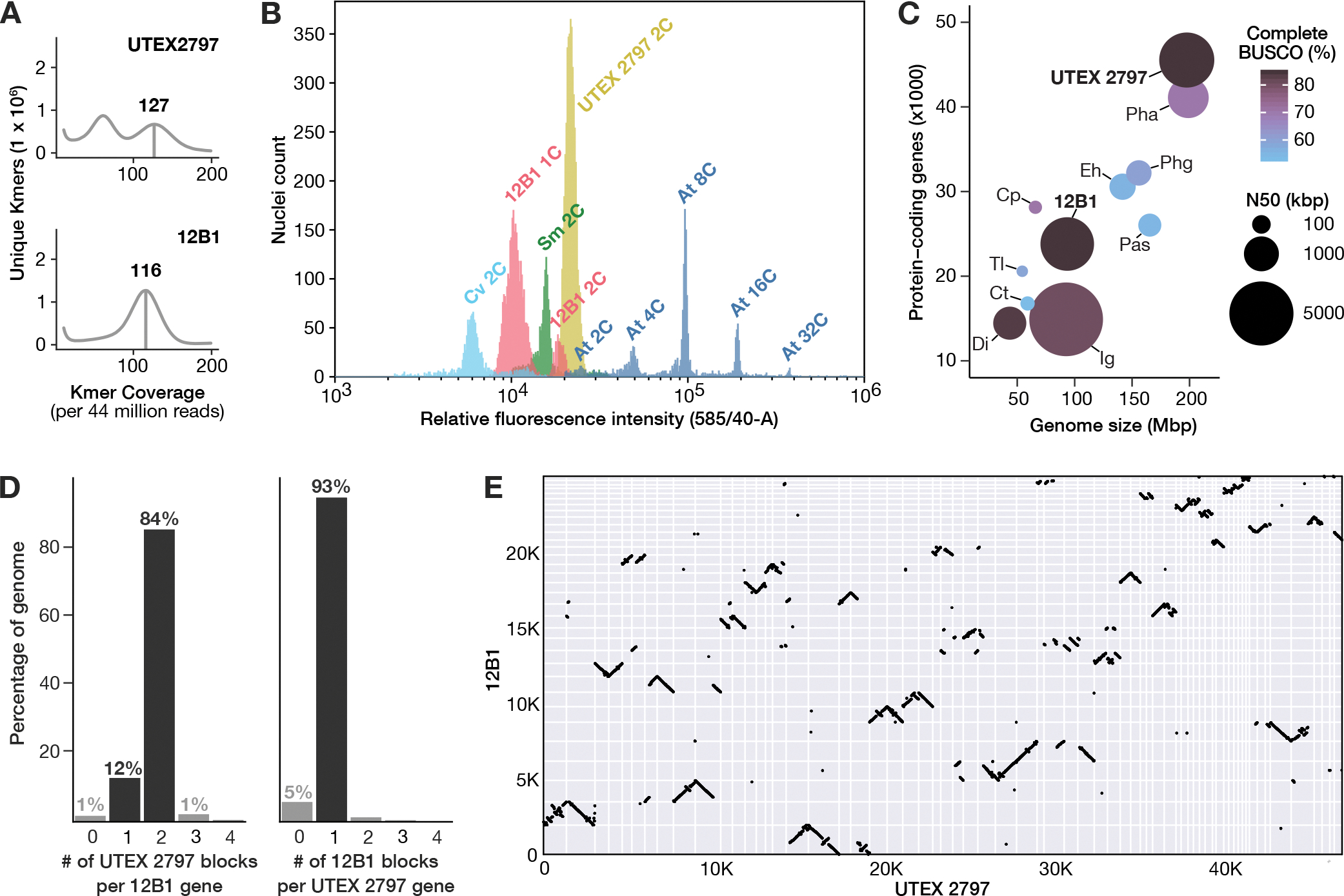
A) K-mer frequency plots showing estimated heterozygosity for strains UTEX 2797 and 12B1. The homozygous k-mer peaks are indicated by vertical bars, with the number above the homozygous peaks indicating the coverage of maximal unique k-mers (CMUKs). UTEX 2797 has high heterozygosity as indicated by the large second peak at half the k-mer coverage of the homozygous peak. B) Histogram indicating the relative fluorescent intensity of propidium iodide-stained nuclei for strains 12B1 (pink) and UTEX 2797 (yellow) relative to genome standards: Cv, Chlorella vulgaris (light blue); Sm, Selaginella moellendorfii (green); At, Arabidopsis thaliana Col-0 (dark blue). C) Comparison of assembly completeness and contiguity across eleven haptophyte genome assemblies. Cp, Chrysochromulina parva; Ct, Chrysochromulina tobinii; Dl, Diacronema lutheri; Eh, Emiliania huxleyi; Ig, Isochrysis galbana; Pas, Pavlovales sp.; Pha, Phaeocystis antarctica; Phg, Phaeocystis globosa; Tl, Tisochrysis lutea. D) Ratio of syntenic block counts between P. parvum genome assemblies for strains 12B1 and UTEX 2797. Syntenic blocks of UTEX 2797 per 12B1 gene (left) and syntenic blocks of 12B1 per UTEX 2797 gene (right) are shown, indicating a clear 1:2 pattern of 12B1 to UTEX 2797. E) Macrosynteny of the 12B1 and UTEX 2797 genomes. Syntenic gene pairs are denoted by black points and positionally oriented by scaffold (grid squares). See also Table S1, Table S2, Table S3.
The nuclear DNA content of both strains was estimated using flow cytometry (Figure 1B). The majority of 12B1 nuclei contained an average of 0.12 pg DNA, which corresponds to approximately 115.6 million base pairs (Mbp). The 12B1 flow cytometry histogram also contained a second, substantially smaller peak corresponding to approximately 0.23 pg DNA (228 Mbp), indicating that some cells may have been in the process of dividing (i.e., the G2 phase of the cell cycle) (Figure 1B). The UTEX 2797 flow cytometry histogram contained a single dominant peak corresponding to 0.28 pg DNA, or 274.4 Mbp (Figure 1B). A cryptic sexual lifecycle for P. parvum has been proposed that alternates between haploid and diploid forms, which are only distinguishable by transmission electron microscopy of the cells’ outer scales33. However, neither syngamy nor meiosis have been observed in P. parvum, and its lifecycle remains unknown. Therefore, we must be cautious when inferring the haploid genome size and relative ploidy states of different strains, and we tentatively labeled the dominant peaks in 12B1 and UTEX 2797 as 1C and 2C, respectively (Figure 1B).
The Hi-C scaffolded genome assembly for 12B1 consisted of 34 scaffolds spanning 93.6 Mbp with an N50 of 3.2 Mbp (Table S1). The UTEX 2797 assembly was over twice the length of the 12B1 assembly at 197.6 Mbp and consisted of 66 scaffolds with an N50 of 3.4 Mbp (Table S1). Compared to nine other haptophyte assemblies, the UTEX 2797 and 12B1 assemblies are the second and third most contiguous (Figure 1C). We predicted and annotated 23,802 and 45,535 protein-coding genes in the genomes of 12B1 and UTEX 2797, respectively (Table S1). The recovery of BUSCO conserved single-copy genes in both strains is the greatest of any currently available haptophyte genome assembly (Figure 1C). While only 5.1% of BUSCOs were duplicated in 12B1, the majority of BUSCOs (75.5%) were duplicated in UTEX 2797 (Table S3).
Synteny and collinearity analyses further highlight the duplicated state of the UTEX 2797 genome assembly. A strong 2:1 synteny pattern is observed between the genes of UTEX 2797 and 12B1; 93% of UTEX 2797 genes are syntenic to a single region in 12B1, whereas 84% of 12B1 genes are syntenic to two regions in the UTEX 2797 genome (Figure 1D). Most 12B1 scaffolds are collinear with two syntenic regions in the UTEX 2797 assembly, but an abundance of large structural variants (e.g., inversions, indels, translocations) are apparent both between the two reference strains as well as between syntenic scaffolds of UTEX 2797 (Figure 1E). Together, these data indicate that the assembly of UTEX 2797 consists of two largely non-collapsed haplotypes, which is consistent with the strain being a highly heterozygous diploid. Due to the extreme heterozygosity present in UTEX 2797, we suspected that the strain may have arisen via hybridization of two divergent parents, as has been observed in other eukaryotic lineages including diatoms34, fungi35,36, and plants37. However, further investigation required data from additional P. parvum strains.
To enable phylogenetic analysis of P. parvum and investigate the origin of the divergent haplotypes in UTEX 2797, we selected thirteen additional P. parvum strains from different geographical locations and prymnesin chemotypes (Table S1). In addition to 12B1 and UTEX 2797, we included four other strains that produce A-type prymnesins: 12A1, CCMP2941, CCMP3037, and RCC3703 (Figure S1)19. Three strains in the analysis are already known to produce B-type prymnesins (K-0081, K-0374, and KAC-39), and four to produce C-type prymnesins (K-0252, RCC191, RCC1433, and RCC1436)18,19. We chemotyped two additional strains (RCC3426 and UTEX 995) for this analysis and determined that RCC3426 produces B-type prymnesins and UTEX 995 produces C-types (Figure S1). We assembled and annotated the genomes of these thirteen additional strains using Illumina short reads (Table S1). Genome assembly lengths ranged from 77.0 Mbp (CCMP3707) to 92.9 Mbp (RCC1436). As expected of short-read assemblies, genome contiguity was low, with contig N50s that ranged from 3414 bp (K-0081) to 8872 bp (RCC1433). Nevertheless, coding gene space was well represented, and the number of protein coding genes ranged from 26,458 (CCMP3037) to 32,339 (UTEX 995) (Table S1). BUSCO scores ranged from 65% (UTEX 995) to 81% (RCC1433), comparable to BUSCO scores observed in other Illumina-only haptophyte genome assemblies (Table S3). However, it is notable that 10 unique BUSCOs were not recovered in any of the 24 haptophyte genomes investigated (Table S3). This suggests that some BUSCOs may be absent in haptophytes or too divergent to be recovered using the current BUSCO sequence models.
High sequence divergence within a unicellular morphospecies.
To determine the relatedness among strains of different chemotypes, we used 2699 single-copy orthogroups (SCOGs) present in all 15 strains for concatenation- and coalescent-based species tree analyses. Support for the species tree topology was generally high across the tree but low within A-type and B-type clades (Figure 2A, Figure S2). Low support values corresponded to areas of disagreement between the concatenation and coalescent based phylogenies (Figure S2).
Figure 2. Phylogenomic and breadth of coverage analyses in P. parvum.
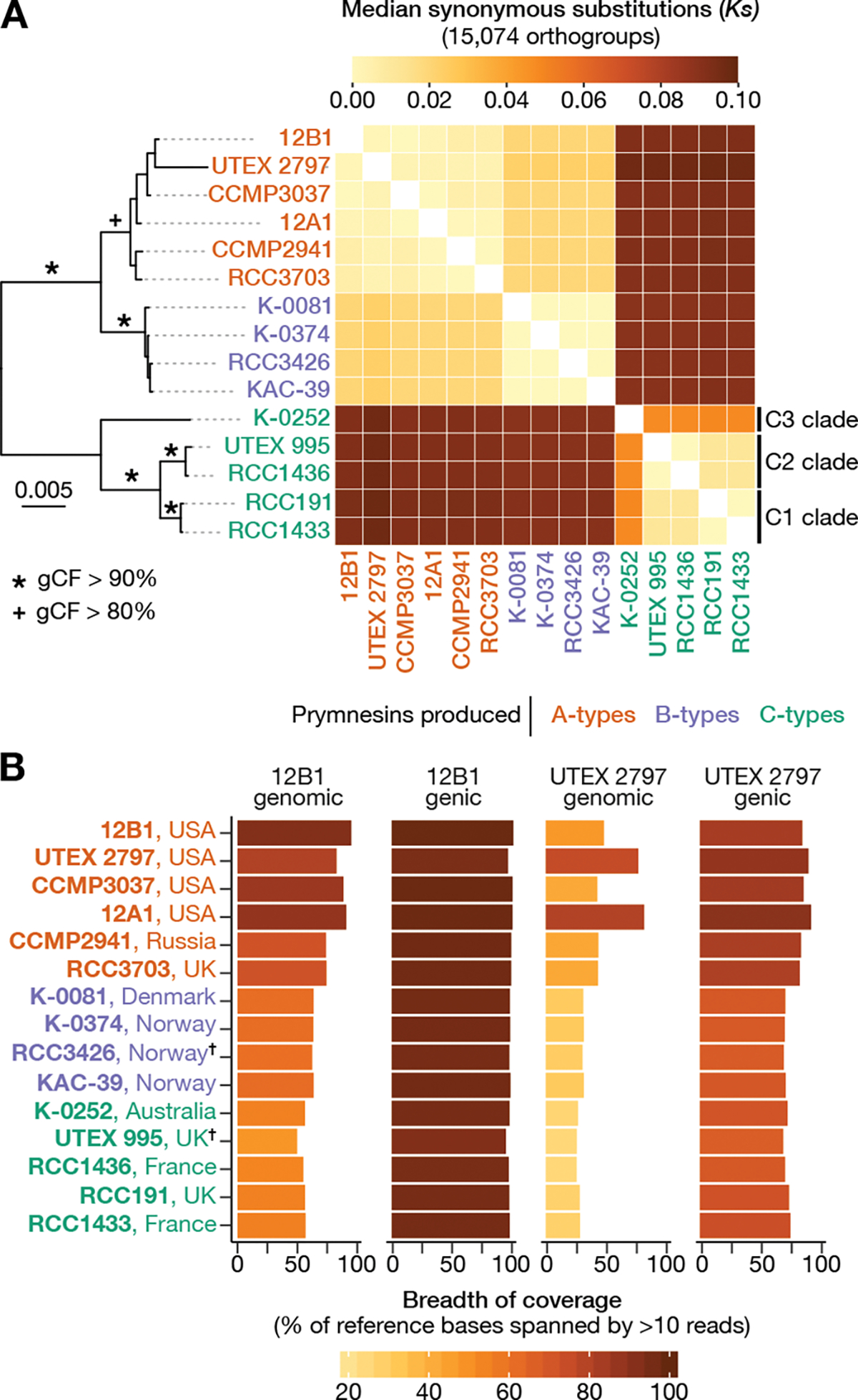
A) Concatenation-based ML species phylogeny. The heatmap shows the median synonymous substitutions per synonymous site (Ks) between all strains. B) Breadth of coverage (BOC) bar plots showing the percent of 12B1 and UTEX 2797 reference bases spanned by >10 Illumina reads from each strain. BOC is provided for all genomic positions and genic space as separate statistics. See also Figure S1, Figure S2.
Divergence among strains was evaluated was estimated based on the average number of substitutions per synonymous site (Ks) between gene pairs in 15,074 orthogroups. These orthogroups were selected based on their presence in 10 or more strains and robust nucleotide alignments. Average Ks within A- and B-type clades was extremely low at 0.003 and 0.000, respectively (Figure 2A). The C-type strains formed three distinct groups based on Ks: clade C1 (RCC191 and RCC1433), clade C2 (RCC1436, UTEX 995), and clade C3 (K-0252). Median Ks was elevated when C-type strains were compared to each other (Ks = 0.0241) with the C3 strain, K-0252, from Australia acting as a significant outlier when compared to other C-types (Ks = 0.048) (Figure 2A). The largest Ks values occurred when C-type strains were compared to A- and B-types (Ks = 0.093). These results suggest that the best root location for the P. parvum species tree is along the branch separating C-type strains from A- and B-types, supporting the supported the monophyletic origin of A-, B-, and C-type prymnesins (Figure 2A).
A second approach was used to explore strain relatedness that was complimentary Ks and based on breadth of coverage (BOC) of each strain’s Illumina reads to the 12B1 reference genome. Average BOC varied dramatically between chemotypes, ranging from 91.7% in A-type strains, 72.7% in B-types, and 63.4% in C-types (Figure 2B). A-type strains from the U.S.A had a higher average BOC (95.4%) compared to the two A-type strains from Europe (84.2%). All strains maintained high sequence coverage across 12B1’s genic space, ranging from 97.5% in other A-type strains to 91.2% in strain UTEX 995 (Figure 2B). This pattern is similar to that seen in interspecific comparisons of Arabidopsis spp. (Streptophyta), in which protein-coding genes are conserved while intergenic sequences show significant divergence38.
Phylogenetically divergent haplotypes within a single strain.
Average breadth of coverage was strikingly low for most strains when aligned to the UTEX 2797 assembly (Figure 2B). The two exceptions were UTEX 2797 (i.e., aligned to itself, BOC = 86%) and another strain from Texas, 12A1 (BOC = 91.7%). This pattern of low coverage across the UTEX 2797 assembly is consistent with UTEX 2797 having two divergent haplotypes. Strain 12A1 likely shares these or closely related haplotypes as its reads map well across the UTEX 2797 assembly while the reads from other A-type strains primarily map to one UTEX 2797 haplotype or the other (Figure 2B).
If the two haplotypes of UTEX 2797 have divergent evolutionary histories, as suggested by the BOC analysis, this could lead to high levels of incongruence between the input gene trees (in which UTEX 2797 may group in two locations reflecting the evolutionary histories of its two haplotypes) and the inferred species tree (which forces each strain to appear only once). Therefore, we performed a gene tree-species tree reconciliation analysis using GRAMPA39 to test whether a multi-labeled (MUL) species tree in which UTEX 2797 appeared twice was a more parsimonious representation of the input gene trees compared to the original single-labeled species tree. Seven of 29 possible MUL phylogenies had a smaller parsimony score, i.e., required fewer total gene duplication and loss events, compared to the single-labeled species tree (Table S4). In the most parsimonious MUL species tree, UTEX 2797 grouped in two locations: with other A-type strains from the U.S.A (hereafter clade A1) and with CCMP2941 and RCC3703 from Europe (hereafter clade A2) (Figure 3). Plotting the genomic distribution of UTEX 2797 genes that group with either clade A1 or A2 revealed a striking pattern. Most scaffolds formed syntenic pairs in the which one scaffold had a high proportion of genes that grouped with subclade A1 and the second scaffold contained genes that grouped with subclade A2 (Figure 3). To quantify the divergence between the UTEX 2797 haplotypes, we compared the distribution of synonymous substitutions of UTEX 2797 genes that grouped with either the A1 or A2 clades. This analysis revealed a bimodal distribution of Ks values in which A1 genes in UTEX 2797 were essentially identical to A1 strains 12B1 and CCMP3037 (Ks = 0) and more divergent to A2 strains RCC3703 and CCMP2941 (Ks = 0.009) (Figure S3). This pattern was reversed in A2 genes (Figure S3). From these results we can infer that UTEX 2797 was formed via the hybridization of an A1-like and an A2-like parent.
Figure 3. Phylogenetically distinct haplotypes of P. parvum strain UTEX 2797.
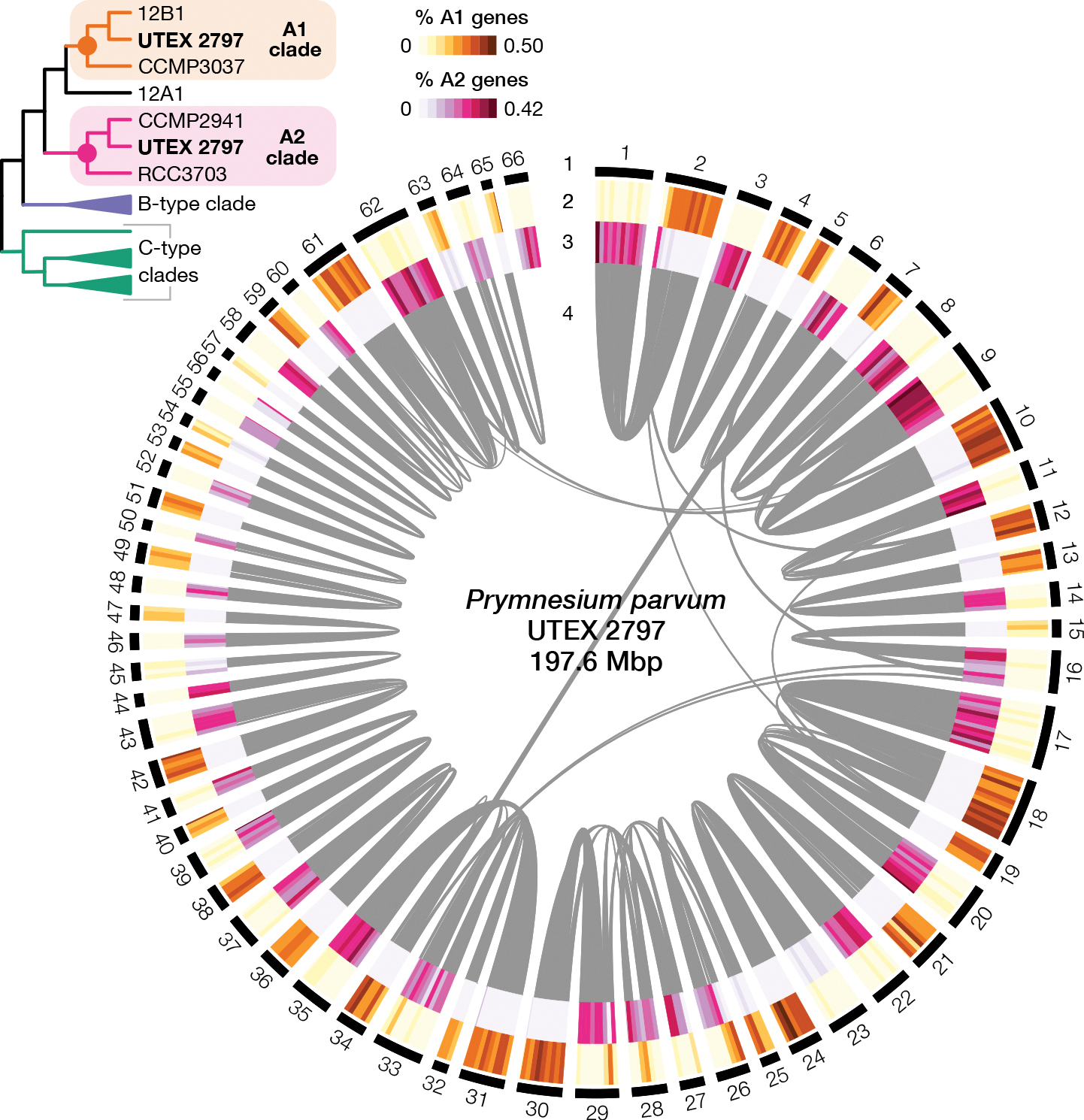
Circos plot showing the 66 scaffolds of UTEX 2797 with four tracks (1) outer black track indicates scaffolds, (2) orange heatmap illustrates the percentage of genes in 50 kbp windows that group within the A1 clade, (3) pink heatmap indicates the percentage of genes in the same 50 kbp windows that group within the A2 clade, and (4) syntenic blocks (≥ 15 syntenic genes per block) are designated as gray bands. Multi-labeled (MUL) species tree (top left) shows the topology of the A1 and A2 clades as determined by the GRAMPA gene tree-species tree reconciliation analysis. See also Figure S3, Table S4.
Inter-strain variation in DNA content and haploid genome size.
Flow cytometry revealed that DNA content varied dramatically among P. parvum strains (Figure 4A). Strain 12B1 had the smallest amount of nuclear DNA at 115.6 Mbp, while K-0081 (a B-type strain) had the largest at 845.6 Mbp. Large variation in DNA content between strains of P. parvum has previously been interpreted as differences in ploidy33. However, in our analysis, the relative DNA content between strains did not discretely cluster around integer fold changes (Figure 4A), as would be expected if all variation in nuclear DNA content was due to differences in genome copy number (i.e., ploidy). Given the sequence-level divergence between strains in our analysis, it is possible that differences between strains could extend to the size of their haploid genome as well. Moreover, due to the limited understanding of the P. parvum lifecycle, the ploidy of different strains was not immediately clear. This creates a circular puzzle, as we cannot infer the haploid genome size of a strain from flow cytometry without a priori knowing the strain’s ploidy.
Figure 4. Summary of ploidy, heterozygosity, and genome size diversity in P. parvum.
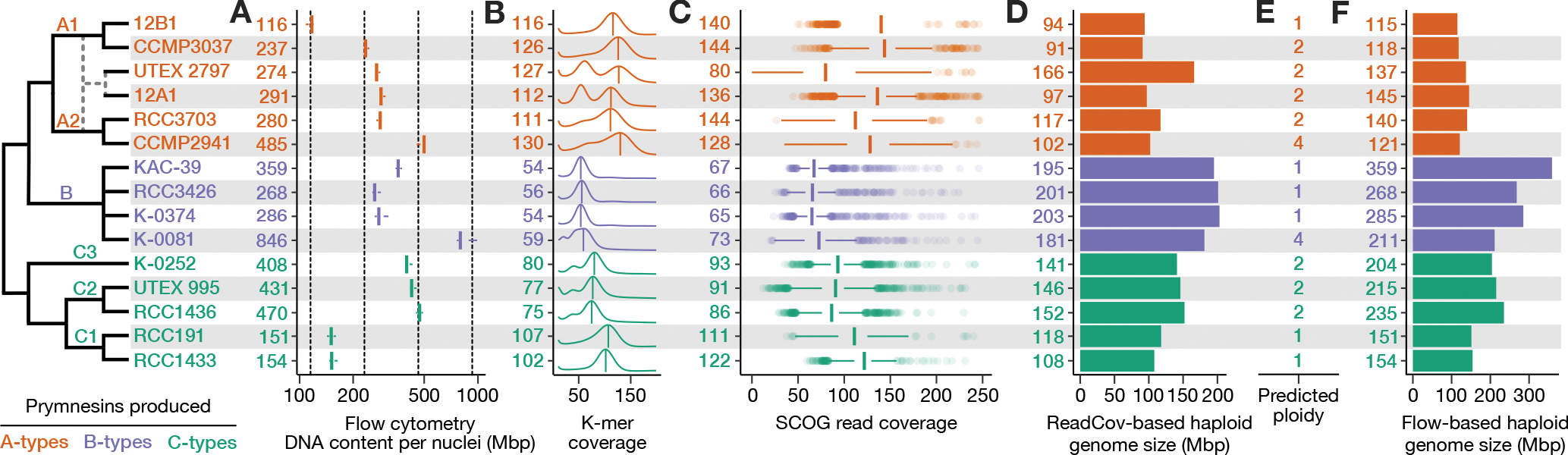
Evolutionary model (left) depicts strain relationships, including a predicted hybridization (dashed lines) giving rise to strains UTEX 2797 and 12A1. A) Boxplots depicting total DNA content per nuclei (Mbp) based on flow cytometry. Numbers (left) indicate mean Mbp per strain. Vertical dashed lines indicate 1x, 2x, 4x, 8x Mbp relative to 12B1. B) K-mer coverage plots depict estimated heterozygosity; numbers indicate CMUKs, i.e., the homozygous k-mer peaks labeled by the vertical bars. Heterozygosity can be qualitatively assessed by the presence and relative height of a second peak at half the k-mer coverage of the homozygous peak. C) Boxplots depicting the distribution of read coverage for 2699 SCOGs; numbers indicate median read coverage. D) Haploid genome size was estimated using read coverage (total read length divided by median Illumina read coverage of SCOGs). E) Predicted ploidy was determined by cross-referencing DNA content (A) with sequencing-based estimated of genome size (B-D). F) Haploid genome size was estimated using flow cytometry (total DNA content divided by proposed ploidy). See also Table S1.
To address this limitation, we used two sequence-based approaches to estimate the relative haploid genome size between strains. First, k-mer frequency analyses were performed using the Illumina reads from each strain. The coverage of maximal unique k-mers (CMUK) of the homozygous peak in k-mer frequency plots was used as a proxy for haploid genome size, with larger CMUKs indicative of smaller haploid genomes and vice versa. CMUK varied dramatically between strains with different prymnesin chemotypes, ranging from an average of 55.8 in B-type producers to 120.3 in A-types (Figure 4B, Table S1). Average CMUK also varied between C-type strains with strains in the C1 clade having an average CMUK of 104.5 compared to strains in C2 and C3 clades, which had an average CMUK of 77.3 (Figure 4B, Table S1). This pattern indicates considerable differences in haploid genome size between clades with the A-type clade having the smallest haploid genome size and the B-type clade having the largest.
The amount of heterozygosity also varied considerably between strains (Figure 4B). In addition to 12B1, six strains (A1 strain CCMP3037; B strains K-0374, KAC-39, RCC3426; and C1 strains RCC191, RCC1433) showed little to no evidence of heterozygosity, which could indicate that, like 12B1, these strains are haploid. However, in the case of A1 strain CCMP3037, its flow cytometry estimated DNA content was twice that of 12B1 despite the two strains having similar CMUKs peaks (Figure 4A,B). This indicates that CCMP3037 is a diploid strain with low levels of heterozygosity. For all other low heterozygosity strains in B and C1 clades, decreased CMUK relative to 12B1 corresponds with increased DNA content, suggesting that these strains are haploids. All other strains displayed comparatively moderate to high levels of heterozygosity, indicating that they are either diploids (2n) or polyploid (3n or greater). Notably, strain 12A1 had a pronounced heterozygous peak indicating extremely high levels of heterozygosity, like that observed in UTEX 2797 (Figure 4B). This, coupled with 12A1’s high BOC against the UTEX 2797 diploid assembly, indicates that 12A1 is also a hybrid strain.
Average read coverage across 2699 single-copy orthogroups (SCOGs) was used as a second sequence-based estimate of haploid genome size (Figure 4C, Table S1). SCOG and CMUK coverage estimates were in agreement for most strains, except for UTEX 2797, which had significantly lower average SCOG coverage compared to the location of its CMUK peak (Figure 4B,C). This discrepancy is due to the nature of its hybrid genome and Hi-C scaffolded assembly with resolved haplotypes. To be assigned to a SCOG, genes in UTEX 2797 have likely returned to single-copy post hybridization, resulting in lower coverage compared to genes that have been retained in duplicate. Using the Lander-Waterman equation40,41, the sequence-based estimate of haploid genome size ranged from 91 Mbp in A-type strain CCMP3031 to 203 Mbp in B-type strain K-0374 (Figure 4D).
Cross-referencing total DNA content with the sequence-based haploid genome size, we were able to assign a predicted ploidy level for each strain (Figure 4E, Table S1). Six strains were determined to be haploids, and seven (including hybrids 12A1 and UTEX 2797) were diploids. Two strains, A-type CCMP2941 and B-type K-0081 appear tetraploid. Using this predicted ploidy, we also determined flow cytometry-based estimates of haploid genome size (Figure 4F, Table S1). The A clade has the smallest genome size (average = 130 Mbp), followed by the C1 clade (153 Mbp). The C3 strain (K-0252) had an estimated genome size of 204 Mbp, and the C2 clade had an average genome size of 225 Mbp. Lastly, B clade strains had the largest genomes (average = 281 Mbp), while also having the largest amount of intra-clade variation in genome size (Figure 4F).
Accessory gene families and horizontal gene transfer.
To investigate the impact of genome variation on gene family evolution in P. parvum, we performed a pangenome analysis orthogroups (i.e., gene families) and identified a total of 47,043 orthogroups including 16,453 core orthogroups present in all strains; 20,738 accessory orthogroups present in 2–14 strains; and 9852 singleton orthogroups unique to a single strain. The number of shared orthogroups generally clustered by prymnesin type, with some exceptions (Figure 5A). Strains 12B1 and UTEX 2797 did not cluster with other A-types, likely in part due to the differences between their Hi-C scaffolded assemblies and the other Illumina-only assemblies. The C-types also clustered into two groups based on orthogroup membership, with C2 strains (RCC1436 and UTEX 995) clustering separate from C1 and C3 strains (Figure 5A).
Figure 5. Pan genome analysis of P. parvum.
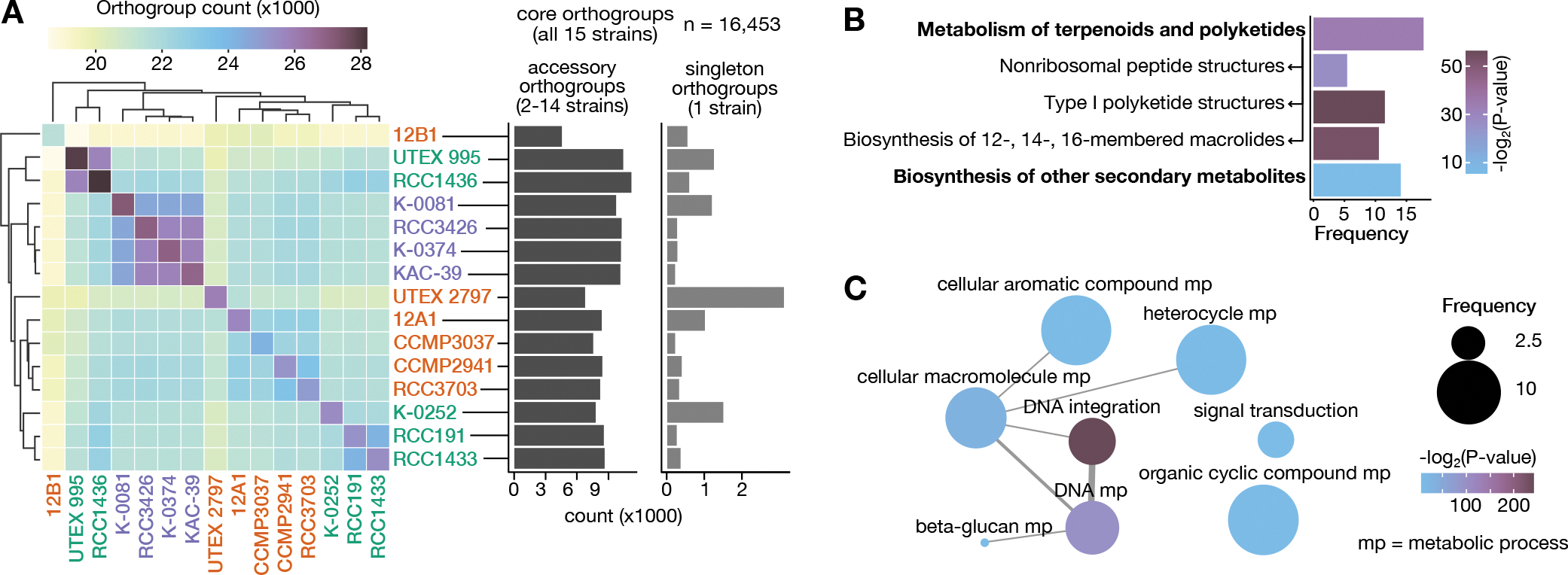
A) Hierarchically clustered heatmap showing the number of orthogroups shared by each strain pair. Strains are colored based on the prymnesin type produced (as in Figure 2). Center diagonal indicates the total number of orthogroups, including singletons, present in each strain. Bar charts indicate the number of accessory orthogroups and singleton orthogroups in each strain. B) Significantly enriched KEGG pathways (unbolded) and pathway categories (bolded); arrows point to KEGG pathway parent category. Bar height indicates frequency of the annotation in accessory orthogroups with one or more KEGG annotations. C) Significantly enriched GO categories in accessory orthogroups. Width of network edges indicate the degree of similarity between GO terms as calculated by REVIGO59. Bubble size indicates frequency of the annotation in accessory orthogroups with one or more GO annotations. See also Table S5.
A significant percentage of all orthogroups (44.1%) were variably present in 2–14 strains. Many of these orthogroups (n = 16,633; 80.2%) could not be assigned any functional annotation. Among the accessory orthogroups that could be annotated (n = 4105; 19.8%), several belong to enriched functional categories that could be associated with metabolic and genome size variation in P. parvum. KEGG specialized metabolic pathways, including those for the biosynthesis of type I polyketides, macrolides, and nonribosomal peptides, were enriched in accessory orthogroups compared to core and singleton orthogroups (BH adjusted p-value < 0.01; Figure 5B, Table S5). Moreover, 31 Gene Ontology (GO) categories were enriched in the accessory orthogroups, the most significantly enriched of which was GO:0015074, DNA integration (BH adjusted p-value = 3.68E-73; Figure 5C, Table S5). Most orthogroups assigned to this GO category were annotated as integrase-like enzymes, common components of lysogenic viruses and transposable elements. We investigated the 60 genes annotated with the DNA integration GO term (GO:0015074) in 12B1 and UTEX 2797 and found that all genes fell within 400 bp of a predicted repeat. Many of these genes were found within Ngaro LTRs, which intersected 31.3% of integrase-like genes in 12B1. This pattern reveals that enrichment of the DNA integration GO term is driven by transposable element integrase genes that have been incorporated into the gene model predictions. We investigated the phylogenetic distribution of this pattern by performing functional enrichment on gene families uniquely present in A-, B-, and C-type strains. We found that the DNA integration GO term was enriched in the B- and C-type specific orthogroups (BH adjusted p-values = 3.94e−15 and 1.72e−4, respectively) but was not significantly enriched in A-type specific orthogroups (Table S5), suggesting that an expansion of transposon copy number may contribute to the greater haploid genome sizes of the B- and C-type clades.
One potential source of accessory genes in a pangenome is horizontal gene transfer (HGT)42. To investigate the role of HGT in the genome evolution of P. parvum, we performed a combined Alien Index and phylogenetic analysis43. We identified 11 HGT events into Prymnesium after the genus diverged from other haptophytes. Phylogenetic trees of these HGTs showed clear donor lineages with strong node support (Figure S4, Table S6). Nine of the 11 HGTs had expression support (maximum length-scaled TPMs > 30; Table S6). Two HGTs were likely acquired from eukaryotic donors: HGT01, a bicarbonate transporter of diatom origin, and HGT02, a gene of unknown function that grouped with pelagophytes. Eight HGTs (HGT03 - HGT10) were likely acquired from bacteria. All the bacterially derived HGTs were enzyme-coding with diverse metabolic activities (Table S6). For example, HGT09 grouped phylogenetically with sequences from marine bacteria (Figure 6A), including bmp5, a decarboxylating flavin-dependent halogenase involved in the biosynthesis of polybrominated natural products in Pseudoalteromonas spp.44. Lastly, HGT11 was a Clp protease that grouped with large dsDNA megaviruses that infect eukaryotic algae (Figure 6B).
Figure 6. Horizontal gene transfer in P. parvum.
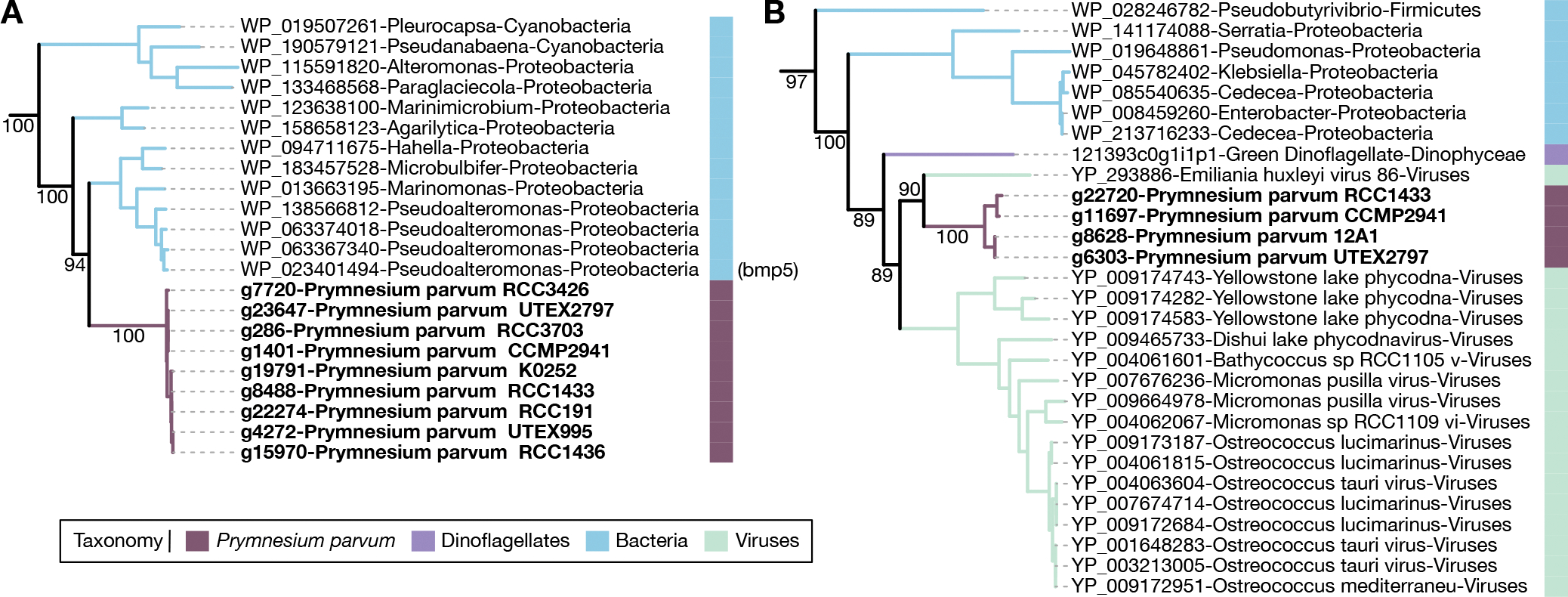
A) Flavin-dependent halogenase phylogeny showing HGT from marine bacteria. The bmp5 gene functionally characterized in Pseudoalteromonas is labeled. B) Clp protease phylogeny showing HGT from viruses. For both phylogenies, numbers along select branches indicate ultrafast bootstrap support values for the descendant nodes. See also Figure S4, Figure S5, Figure S6, Table S6.
The genomic neighborhood of HGT11 in UTEX 2797 contains six additional genes with a top BLAST hit to EhV-86, a dsDNA megavirus that infects the haptophyte Emiliania huxleyi45 (Figure S5). These additional genes were not recovered in our primary screen for HGT due to a limited number of hits to proteins in the NCBI RefSeq database, making these genes insufficient for phylogenetic analysis. However, their sequence similarities to EhV-86 suggests that this 50 kbp region of viral-like genes was likely acquired as a single block (Figure S5). Only one gene in this region (UTEX2797g6289, a major capsid protein) had a significant hit to the dsDNA megavirus PpDNAV, which was recently isolated from P. parvum46 (Figure S5). EhV-86 and PpDNAV are both members of the Phycodnaviridae, a clade of large DNA viruses that infect eukaryotic algae. The genomes of sequenced Phycodnaviridae range in size from 160 to 560 kb and contain hundreds of genes47. The Phycodnaviridae are typically lytic in nature; however, one virus in this group, EsV-1 can integrate into its host genome47. It is unclear whether the HGT11 viral region in UTEX 2797 is the result of viral integration or was acquired though an alternative mechanism. None of the genes in this region were expressed under any of the growth conditions surveyed, and whether these genes are functional in UTEX 2797 is unknown.
Several horizontally transferred genes were part of the accessory genome and variably present across strains (Table S6). Some of this variation is likely due to ancestral HGTs being lost in some clades; e.g., HGT07 and HGT10 both appear to have been gained in an ancestor of Prymnesium and subsequently lost in the B-type clade (Figure S4). HGT11, the Clp protease gene of viral origin, was notable due to its presence in only four disparate strains: three A-type strains (UTEX 2797, 12A1, and CCMP2941) and one C-type (RCC1433) (Figure 6B). If this gene was acquired in a shared ancestor of all four strains, at least six independent loss events would be required to explain this presence/absence pattern. An alternative explanation could be that multiple independent acquisitions of a viral Clp protease have occurred in P. parvum. We checked for shared synteny between UTEX 2797 and the three additional strains that contain HGT11. In 12A1, HGT11 is located on a 20-gene scaffold (Scaf652979) which shares nearly perfect synteny with UTEX 2797, including five genes within the proposed viral fragment (Figure S6), which suggests that UTEX 2797 and 12A1 share the same HGT event. In contrast, the scaffolds that contain HGT11 in CCMP2941 and RCC1433 are syntenic with each other, but neither are syntenic with UTEX 2797 (Figure S6). This suggests that the HGT that gave rise to the Clp protease in CCMP2941 and RCC1433 may be independent from that of UTEX 2797 and 12A1. Long-read assemblies of additional strains including 12A1, CCMP2941, and RCC1433 are needed to confirm this pattern.
DISCUSSION
The harmful algal bloom-forming eukaryote, Prymnesium parvum, possesses considerable genomic diversity. Combining data from synonymous substitutions, reference genome coverage, phylogenetics, and genome size, the fifteen strains in our analysis can be subdivided into six distinct clades consisting of at least three cryptic species (Figure 4). Evidence for these cryptic species comes from the fact that prymnesin chemotype is phylogenetically segregated, which indicates that the P. parvum A-, B-, and C-type clades are reproductively isolated. Here, we provide further support for these clades being separate cryptic species based on the extreme variation in their genome size, as excessive chromosome-level differences will likely inhibit proper chromosome pairing during meiosis. Another haptophyte with population-level genomic data is the coccolithophore, E. huxleyi. And like P. parvum, E. huxleyi forms a species complex with a pan genome that shows significant variation in terms of gene content and reference genome coverage48. If this pattern extends to additional haptophyte morphospecies, it suggests that species diversity may be severely underestimated in this ecologically important lineage of algae.
The most dramatic difference in P. parvum genome size occurred between the sister clades of A- and B- type strains (Figure 4F). These clades show limited substitutions at synonymous sites (Ks = 0.02; Figure 2A), which suggests a relatively recent divergence in genome size. Possible mechanisms for genome expansion in B-type strains includes whole-genome duplication (WGD) and/or proliferation of transposable elements. Our analysis of Ks distributions showed no evidence of recent WGD in the last common ancestor of the B-clade. Instead, gene functions associated with transposable elements were enriched in gene families unique to B-type strains (Table S5), suggesting that the increase in their genome size could be due to an increase in transposition activity of these elements. A high-quality reference genome of a B-type strain would enable further investigation of repeat diversity and expansion across these two clades and clarify the mechanism(s) of genome size variation in P. parvum.
Although P. parvum is currently considered a single morphospecies, earlier taxonomic descriptions split the species in two (P. parvum and P. patelliferum) based on differences in the morphology of the organic scales that cover their cell surface49. When the rDNA ITS1 region was found to be identical in strains of P. parvum and P. patelliferum isolated from the same geographic location, the two species were merged into one50. Later, two strains originally identified as P. parvum (K-0081 and strain RL10parv93, not included in our analysis) were found to have higher amounts of DNA compared to three strains labeled P. patelliferum (K-0252, RCC191, and a third strain not assessed here, RLpat93)33. This observation led Larsen and Edvardsen (1998) to propose a cryptic lifecycle for P. parvum that alternates between flagellated haploid and diploid forms. Our analysis identified strains of different ploidy states, supporting the existence of a cryptic sexual lifecycle. Moreover, the existence of a hybrid strain (UTEX 2797) with two phylogenetically divergent parental genotypes indicates that syngamy can occur in P. parvum. Lastly, P. parvum retains the full complement of conserved meiosis and recombination genes present in other haptophytes51. The combined evidence suggests that P. parvum is capable of sexual reproduction. Our assessment of ploidy across these strains, which includes flow cytometry and read-based estimates of haploid genome size, indicates that K-0081 is a tetraploid, not a diploid as previously thought, and that K-0252 is a diploid and not a haploid (Figure 4). Given the inclusion of these strains in the hypothesis about scale morphology and ploidy, our new results suggest that scale morphology is not diagnostic of ploidy state. Analysis of the fifteen strains reported here places variation in ploidy and genome size in a phylogenetic framework, facilitating future investigation into cellular morphology and sexual reproduction in P. parvum.
Our phylogenomic analysis also reveals that UTEX 2797, a Texas P. parvum strain frequently used in growth and toxicity experiments, is a hybrid of A1 and A2 parents (Figure 4A). To our knowledge, this is only the second genome level analysis of hybridization in a protist or eukaryotic alga34. Regarding the evolutionary history of UTEX 2797, it is intriguing that the A1 strains in our analysis were isolated from the U.S.A while the A2 strains were isolated from Europe, and it is tempting to speculate that hybridization was a result of a recent introduction of the A2 lineage in Texas. However, further investigation into the origin of UTEX 2797 requires additional taxon sampling of A-type strains in Texas and around the world. Our analyses suggest that the high heterozygosity strain 12A1 is a hybrid as well, but a long-read based assembly for 12A1 is required to determine if it arose from the same hybridization event as UTEX 2797. The evolutionary outcome of hybridization in P. parvum is also unknown. Are these hybrid strains capable of sexual reproduction? Genome divergence between A1 and A2 clades may be such that homologous chromosomes are unable to pair correctly during meiosis. If so, these hybrids may be effectively trapped as diploids and only able to reproduce asexually. Alternatively, whole-genome duplication following hybridization (allopolyploidy) has been shown to restore fertility in hybrid yeast36, which could make these P. parvum hybrids reproductively haploids. Characterizing the sexual lifecycle of P. parvum would allow researchers to address several outstanding questions regarding the ecological and evolutionary consequences of extreme genetic variation in these toxic bloom forming eukaryotes. Knowledge of the P. parvum lifecycle would enable the design of mating tests to determine the reproductive status of hybrid strains. Mating tests could also be used to assess reproductive isolation between divergent populations, as has been done in other morphologically indistinguishable cryptic species complexes52–54.
Previous work indicates that harmful algal blooms of P. parvum are comprised of multiple genotypes21,24,55, and our results reveal that the genome-level differences between these genotypes can be dramatic. For example, strains 12A1 and 12B1 were isolated from the same Texas bloom in 2010 and consistently show different cell-level behaviors involved in toxicity and predation of microalgal prey21,24. Here, we show that characteristic differences between 12A1 and 12B1 extend to large differences in their genomes as well, with 12A1 being a hybrid diploid and 12B1 having a streamlined haploid genome. Moreover, significant gene-level differences exist between these two strains, with 7% (n = 1485) of 12B1 orthogroups absent in 12A1. An even larger percentage, 22% (n = 5709), of 12A1 orthogroups are absent in 12B1, including one that was horizontally acquired (HGT11; Figure 6B). Gene families that are variably present across strains in our analysis include those for the biosynthesis of type I polyketides, (Figure 5B), the class of specialized metabolites that includes prymnesins17. This is unsurprising given the structural diversity in prymnesins that have already been characterized in P. parvum18,19. However, it is notable that 12A1 and 12B1, both A-type strains, show variable representation of eighteen orthogroups assigned to the type I polyketide KEGG pathway (map01052; Table S5). If any of these orthogroups are involved in toxin biosynthesis, phenotypic differences between these two strains could extend to their toxin profiles as well. The coding capacity of P. parvum has also been expanded by HGT. Prymnesins are halogenated metabolites, and though we have not functionally characterized it, it is tantalizing to note that a halogenase is among the HGT candidates in the P. parvum genomes (HGT09; Figure 6A). HGT is often a source of metabolic innovation43,56–58 and elucidating the prymnesin pathway will determine if this gene is relevant to the production of these metabolites. More work is needed to identify the genetic factors associated with toxin production and the phenotypic differences between these diverse strains of P. parvum. Additionally, work is needed to identify the selective advantage of different phenotypes and whether such advantages fluctuate between bloom and non-bloom conditions. The genome assemblies and phylogenomic analysis reported here are thus essential resources that will enable future investigation into the eco-physiological consequences of hidden genetic diversity in this harmful algal bloom-forming morphospecies.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Jennifer Wisecaver (jwisecav@purdue.edu).
Materials availability
P. parvum strains 12B1 and 12A1 have been deposited in the UTEX Culture Collection of Algae at UT-Austin under UTEX accessions UTEX LB ZZ1299 and UTEX LB ZZ1300.
Data and code availability
All genome assemblies, predicted CDS and protein sequences, multiple sequence alignments, tree files, and other related data files are available through FigShare (https://doi.org/10.6084/m9.figshare.21376500). Scripts for chromatogram production are available on Figshare https://doi.org/10.6084/m9.figshare.22267066.v1. All other scripts are available through GitHub (https://github.com/WisecaverLab/Pparvum_genome_diversity). Raw sequencing reads have been deposited in the Sequence Read Archive database under accession number PRJNA807128. LC-MS data from chemotyping is available in the EBI MetaboLights database under accession MTBLS5893.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Culturing methods.
Strains and their respective media types are summarized in Table S1. Cultures were kept at 20 °C using a 12:12 light dark cycle and irradiance of 40–200 μmol photos m−2 s−1.
METHOD DETAILS
Flow cytometry.
Nuclei for genome size estimation were isolated using LB01 buffer (15 mM Tris, 2 mM Na2-EDTA, 0.5 mM spermine tetrahydrochloride, 80 mM KCl, 20 mM NaCl, 0.1% (v/v) Triton X-100, pH=8.0)60,61. For P. parvum, 1 mL of exponentially growing culture was centrifuged at 1000 × g for 10 minutes. The supernatant was decanted, and the cell pellet was flash-frozen in liquid nitrogen. The pellet was resuspended in 1 mL of ice-cold LB01 buffer. For the Chlorella vulgaris genome standard, 1 mL of exponentially growing liquid culture (Carolina Biological) was centrifuged at 1000 × g for 10 minutes. The cell pellet was resuspended in 1 mL ice-cold LB01 buffer and transferred to a 2 mL ZR BashingBead Lysis Tube (Zymo Research Cat #S6003–50). The tube was agitated using a Disruptor Genie (Scientific Industries Model #SI-D238) for 5 minutes. For the Selaginella moellendorfii genome standard, leaf and stem tissue (provided by Dr. Scott McAdam, Purdue University) were chopped using a razor blade with 1.5 mL ice-cold LB01 buffer added per 100 mg tissue for 3 minutes. For the Arabidopsis thaliana genome standard, rosette tissue from the Col-0 reference accession (provided by Dr. Clint Chapple, Purdue University) was chopped using a razor blade with 1.5 mL ice-cold LB01 buffer added per 100 mg tissue for 3 minutes. All nuclei suspensions were filtered through a 40 μm cell strainer and kept at 4 °C until use.
RNaseA and propidium iodide were added to nuclei suspensions at a final concentration of 0.4 mg/mL and 0.05 mg/mL respectively. After briefly vortexing, the nuclei were incubated at 4 °C in the dark for 3 hours. Samples were analyzed using a BD Accuri C6 Plus flow cytometer. All samples were gathered using a flow rate of 14 μL/min and a core size of 10 μm. Fluorescence values were gathered using a 488-nm laser using a 585/40 nm band pass filter and a 670 nm long pass filter. Data from at least 300 nuclei were collected per sample. Biological replicates were performed on three subsequent afternoons at approximately the same time. Samples were processed using the FlowCal Python library62. The endoreduplicative nature of the A. thaliana tissue enabled identification of 2C, 4C, 8C, 16C, and 32C nuclei in this species63. A line of best fit through the genome standards was calculated using ordinary least squares linear regression. The linear model was then used to estimate the nuclear DNA content in picograms for P. parvum strains, which was converted to estimated base pairs assuming a conversion factor of 1 pg = 0.98 × 109 base pairs64.
Prymnesin chemotyping.
P. parvum strains were cultured in media consisting of L1 (Guillard and Morton, 2003) in GF/F glass fiber filter (Whatman, USA) filtered seawater (Scripps Institution of Oceanography seawater system) diluted to 25% salinity with 18 MOhm water with 1.5 mM added NaHCO3 to adjust for the lack of carbon in 18 MOhm water65. Cultures were grown in 100 mL of media in 500 mL glass Erlenmeyer flasks without external aeration or shaking. Exponential phase culture (>= 100e3 cells/mL, <= 600e3 cells/mL; late log phase) was harvested via filtration onto 47 mm glass fiber filters (GF/B of GF/F, Whatman) using laboratory vacuum. The resulting filters were stored in 50 mL polypropylene (PP) centrifuge tubes at −80 °C until extraction.
PP tubes with the collected biomass filters were shaken with 25 mL of ethyl acetate at 37 °C (220 RPM) for 20 minutes, and the resulting yellow-green extract was discarded. We chose ethyl acetate rather than the standard cold acetone for pre-extraction based on reports that the cold-acetone lipid-solubilization step can lead to PRYM losses due to variable quantities of water on the filter and resulting extraction65. The ethyl acetate pre-extraction step was repeated 2–3 times until the resulting extract was without coloration. The tube was shaken with 25 mL of methanol (MeOH) at 37 °C for 30 minutes. After centrifugation at 4000 × g for 10 minutes at 20 °C, the MeOH extract was decanted into a 25 mL pear-shaped flask (P/N 9477–06, Ace Glass) and rotary evaporated to dryness at 30 °C under reduced pressure (50 mbar, 150 RPM). The resulting residue was redissolved in 500 μL MeOH and filtered through a 0.2 μm PFTE filter (P/N: CIPT-02, American Chromatography Supplies) into a glass HPLC vial. For each sample, 20 μL was injected onto an Agilent 1260 Infinity HPLC system coupled to an Agilent 6530 quadrupole time-of-flight (QToF) mass spectrometer.
Compounds were separated via gradient HPLC on an Agilent 1260 Infinity system, via C18 reversed phase chromatography (Phenomenex Kinetex C18 150 × 4.6 mm, 5 μm, 100 Å, with C18 guard column), at 0.7 mL/min with the following solvents and gradients: Solvent A: H2O + 0.1% v/v formic acid, Solvent B: acetonitrile + 0.1% v/v formic acid. Compounds were eluted with a gradient of 10% to 100% B over 30 minutes. To wash and equilibrate the column for subsequent injections, the gradient was then held at 100% B until 36 minutes, decreased to 50% over 3 minutes, returned to 10% over 3 minutes, and equilibrated at 10% B for 3 minutes for a total run time of 45 minutes. Under these conditions PRYM-1 and PRYM-2 eluted at around 55–57% B (15.19 to 15.68 min).
The Agilent 6530 QToF MS was configured in either the 3200 m/z, HiRes (4GHz) instrument state or the high dynamic range (2GHz) instrument state. In either case the instrument was quick tuned and mass calibrated just before use. The instrument was set to positive ionization mode. The source parameters were 300 °C gas temp, 11 L/min drying gas, nebulizer at 45 psig, source voltage at 4000V, fragmentor at 100V, Skimmer at 65V, OCT 1 Rf Vpp at 750 V. N2 gas was supplied by a Parker NitroFlowLab N2 generator at ~>90% purity. The reference mass infusion and locking were disabled, due to an overlap with the PRYM aglycone [M+2H]2+ isotopic peaks with the vendor supplied lock mass. The LC flow was diverted to waste from 0 to 6 minutes and MS acquisition was not performed. At 6 minutes, the LC flow was switched to the MS source & the instrument began acquiring MS1 data from 125–3200 m/z, at a rate of 4 spectra/second, in the auto MS2 mode with a 250 ms dwell time per subsequent MS2 scan. Between 1–3 of the most abundant precursors per MS1 cycle were chosen for fragmentation, with dynamic exclusion of precursors after their selection. A fixed collision energy of 20 was used. All data was acquired in profile mode. MS acquisition was stopped after 43 minutes.
The resulting LC-MS data was transformed from Agilent MassHunter .d format to .mzML format using Proteowizard v 3.0.2030366, with parameters ‘--zlib’. Data in .mzML format were analyzed using with MZmine2 v2.5367, while .d format data were analyzed with Agilent MassHunter (B.05.00). PRYM-1 and PRYM-2 were identified by comparison of their characteristic multi-chlorinated MS1 isotopologue ionization intensity pattern to in silico calculated MS1 isotopologue intensity patterns using the enviPat Web 2.4 tool68. Coeluting [M+H]+, [M+2H]2+, and presumed aglycone in-source fragments, each showing the characteristic MS1 pattern, were detected. Chromatograms were produced using pymzML69, matplotlib70, and svgutils71, and production scripts are available on Figshare https://doi.org/10.6084/m9.figshare.22267066.v1. Raw data is available on Metabolights at study ID MTBLS5893.
Genome sequencing and assembly.
Genomic DNA for Illumina sequencing was extracted from P. parvum cell pellets using the CTAB method according to the following protocol https://dx.doi.org/10.17504/protocols.io.b5qhq5t672. Extracted DNA was purified using a Genomic DNA Clean and Concentrator kit (Zymo Research). Sequencing libraries were constructed and sequenced to produce 150 bp paired-end reads using one of two approaches: 1) libraries were prepared using a TruSeq DNA PCR-Free library prep kit (Illumina, San Diego, CA), and sequenced using an Illumina NovaSeq 6000 at the Purdue Genomics Center 2) libraries were prepared using an NEBNext DNA library prep kit (New England Biolabs Inc.) and sequenced using an Illumina NovaSeq 6000 by Novogene Corporation Inc. (Sacramento, CA). Illumina gDNA read quality was assessed by FastQC v0.10.073. Short-read only genome assemblies were performed by Abyss v2.2.474 using a k-mer size of 96. Contigs less than 500 bp in length and those flagged as bacterial contamination (see below) were discarded.
Strains were grown in xenic conditions; therefore, bacterial contamination in the Illumina assemblies was identified using BlobTools v1.1.175. For each strain, Illumina gDNA reads were aligned to the Abyss assembly using BWA-MEM v0.7.1576 to generate a coverage BAM file. Contigs were queried against the NCBI nucleotide (nt) database (accessed September 11, 2021) using blastn v2.11.077. DIAMOND v2.0.8.14678 was used to query contigs against a custom protein databases that consisted of NCBI RefSeq (release 207)79 sequences supplemented with additional predicted protein sequences from MMETSP80 and the 1000 Plants transcriptome sequencing project (1KP)81. The custom protein database used in the BlobTools analysis is available from the authors as well as through the following link: https://www.datadepot.rcac.purdue.edu/jwisecav/custom-refseq/2021-08-02/. The BlobTools taxrule ‘bestsumorder’ determined the taxonomic assignment of each contig, prioritizing information from protein hits first. Contigs denoted as non-eukaryotic in origin were removed to produce the final filtered assembly. Lastly, BBSplit v38.8782 was used to retain only Illumina reads that mapped to the BlobTools filtered assembly (hereafter referred to as filtered Illumina reads). This filtering step removed 14% of paired reads on average for each strain (Table S1). UTEX 995 was an outlier with 52% of its reads removed due to a large amount of contamination from Proteobacteria and Bacteroidetes. All BlobTools results, including blobplot figures, are available for download from the project’s FigShare data repository (see Data Availability).
For long-read sequencing with Oxford Nanopore Technologies (ONT), high molecular weight DNA was extracted from isolated P. parvum nuclei using the following protocol https://dx.doi.org/10.17504/protocols.io.7b7hirn83. At least 1.5 μg of gDNA was used as input for an Oxford Nanopore LSK-109 library ligation kit and sequenced on R9 MinION flow cells. Base calling was performed with Guppy v2.3.584. Reads less than 3 kbp long or with quality scores less than 7 were discarded. Different assembly approaches were selected to optimize for either assembly contiguity (in the case of low heterozygosity 12B1) or the amount of resolved haplotypes (in the case of high heterozygosity UTEX 2797). The 12B1 long-read assembly was created using both Nanopore and Illumina gDNA data via MaSuRCA v3.3.185 with the following parameters: LHE_COVERAGE=60, CA_PARAMETERS=cgwErrorRate=0.15, KMER_COUNT_THRESHOLD=2, CLOSE_GAPS=1, JF_SIZE=5000000000. The UTEX 2797 long-read assembly was created using ONT reads only via Canu v2.1.186 with an expected genome size of 200 Mbp. Both assembly types were error corrected via five rounds of polishing with Illumina gDNA reads that were first aligned to the assembly with using BWA-MEM v0.7.15 76 and polished with Pilon v1.23 using default settings87.
To identify mitochondria- and plastid-derived contigs, predicted proteins from the Emiliania huxleyi mitochondrial (Accn: NC_005332.1) and plastid (Accn: NC_007288.1) genomes were queried using tblastn v2.11.077 against both long-read genome assemblies. A single contig corresponding to the plastid genome was identified in both assemblies. One contig was flagged as mitochondrial in origin in UTEX 2797, while no mitochondria-derived contig was identified in 12B1. All three organelle-derived contigs were excluded from the nuclear genome assemblies and are available for download from the project’s FigShare data repository (see Data Availability).
Chromatin conformation capture data was generated using a Phase Genomics (Seattle, WA) Proximo Hi-C 2.0 Kit, which is a commercially available version of the Hi-C protocol88. Following the manufacturer’s instructions for the kit, intact cells were crosslinked using a formaldehyde solution, digested using the DPNII restriction enzyme, end repaired with biotinylated nucleotides, and proximity ligated to create chimeric molecules composed of fragments from different regions of the genome that were physically proximal in vivo. Molecules were pulled down with streptavidin beads, processed into an Illumina-compatible sequencing library, and sequenced on the Illumina HiSeq platform as 2×150 bp reads. Illumina reads were aligned to the long-read assemblies (Canu assembly for UTEX 2797 and MaSuRCA assembly for 12B1) using BWA-MEM v0.7.1576 with the -5SP options specified, and all other options default. SAMBLASTER89 was used to flag PCR duplicates, which were then excluded. Alignments were filtered with SAMtools90 using the -F 2304 filtering flag to remove non-primary and secondary alignments. Putative misjoined contigs were broken using Juicebox91,92 based on Hi-C alignments. Kraken v293 identified eukaryotic contigs, which were selected for scaffolding, and prokaryotic contaminants, which were discarded. The same alignment procedure was repeated from the beginning on this corrected assembly. Phase Genomics’ Proximo Hi-C genome scaffolding platform was used to create chromosome-scale scaffolds from the corrected assembly as previously described94. As in the LACHESIS method95, this process computes a contact frequency matrix from the aligned Hi-C read pairs, normalized by the number of DPNII restriction sites (GATC) on each contig, and constructs scaffolds that optimize expected contact frequency and other statistical patterns in Hi-C data. Approximately 60,000 separate Proximo runs were performed to optimize the number of scaffolds and scaffold construction to make the scaffolds as concordant with the oybserved Hi-C data as possible.
During the Alien Index analysis (see Horizontal Gene Transfer methods section below), we flagged two 12B1 MaSuRCA contigs (scf7180000001543 on 12B1-Scaf8 and scf7180000001202 on 12B1-Scaf32) as bacterial contamination in the scaffolded 12B1 assembly. Both contigs were located at the ends of scaffolds, all genes on the contigs were of bacterial origin, and the percent identity to the database sequences was high (> 80% for all genes), indicating that these bacterially derived contigs were incorporated into the Hi-C scaffolded assembly in error rather than true horizontal gene transfer. Therefore, these contig sequences were manually removed from the final genome assembly and the flagged genes filtered from the final gene set. The resulting 12B1 assembly and gene annotations following these steps were designated as final (v1).
Repeat prediction.
De novo repeat identification was separately performed on the scaffolded assemblies of strains 12B1 and UTEX 2797 using RepeatModeler v2.0.196. The resulting modeled libraries were used to inform repeat masking the assemblies with RepeatMasker v4.0.797. Repeats were also masked using the UTEX 2797 de novo repeat library for the short-read only assemblies. Total repetitive sequence made up 29.4% and 35.5% of the 12B1 and UTEX 2797 genome assemblies, respectively, which are within the range of values (22.9% - 64%) reported for other haptophytes48,98,99.
Gene Prediction.
To maximize capture of the P. parvum transcriptome for gene calling, we performed RNA-Seq of UTEX 2797 cultures grown in six different conditions: low salinity (2 PSU), medium salinity (11 PSU), high salinity (32 PSU), low vitamin (1/10th the standard concentration of L1 media), low phosphorus (1/25th the standard concentration of L1 media), and low light (30 μmol photos m−2 s−1). Additional cultures in standard media conditions (see Table S1) were sampled at four diurnal timepoints: T0 (onset of light cycle), T6 (6 hrs after onset of light cycle), T12 (onset of dark cycle), and T18 (6 hrs after onset of dark cycle). Starting 100 mL cultures were inoculated at 10,000 cells/mL and grown at 20 °C using a 12:12 light dark cycle. Beginning five days post inoculation, cultures were maintained using semi-continuous replacement every three days by discarding 10% of the culture and replacing with fresh media. Cell densities were measured every three days to track culture growth. Upon reaching densities of ~1×106 cells/mL, cultures were harvested by centrifugation at 4500 × g for 5 minutes and snap freezing in liquid nitrogen. Total RNA was extracted from pelleted cells using the following protocol: https://dx.doi.org/10.17504/protocols.io.bv3hn8j6100. Stranded RNA-Seq libraries were constructed and sequenced by Novogene Corporation Inc. (Sacramento, CA) using a NEBNext Ultra TM RNA Library Prep Kit (NEB, USA) following manufacturer’s recommendations. Libraires were sequenced on the Illumina NovaSeq 600 platform to produce 150 bp paired-end reads. RNA-Seq reads were aligned to the UTEX 2797 scaffolded assembly using STAR v2.7.8a101 with the maximum intron length set to 10 kbp.
Gene model and protein prediction was first conducted on the UTEX 2797 scaffolded assembly with BRAKER2 v2.1.5102,103. BRAKER2 was supplied the UTEX 2797 scaffolded assembly with repeats soft-masked, a custom protein database comprised of Swiss-Prot and all haptophyte predicted proteins from MMETSP80, and the UTEX 2797 Illumina RNA-Seq data aligned to the soft-masked genome. All subsequent predictions for other strains via BRAKER2 utilized the resulting Augustus species-specific training configuration file104 and the same custom protein databases.
Characterization of Assembly Completeness.
Telomeric repeats were identified from scanning the final assemblies of 12B1 and UTEX 2797 with TRFFinder v4.09105 using the following parameters: 2 7 7 80 10 50 500 -f -d -m -h. Any tandem repeat within the first or last 2 kbp of a scaffold’s length and whose repeat block was any permutation of the conserved Haptophyta telomere repeat TTAGGG106 was selected. Chromosomal end-to-end assembly was accomplished for 29.4% (n = 10) of 12B1 and 7.6% (n = 5) of UTEX 2797 scaffolds, evidenced by the presence of telomeric sequence on both scaffold ends (Table S2). A single telomere repeat could be identified in an additional 44.1% (n = 15) of 12B1 scaffolds and 34.8% (n = 23) of UTEX 2797 scaffolds (Table S2). Overall, 35 and 33 predicted telomeres were identified on 12B1 and UTEX 2797 scaffolds, respectively. Although a rough estimate, this suggests a haploid chromosome count for these strains of approximately n = 17. Conservation of core genes was performed using BUSCO v4.0.6 using the eukaryota_odb10 dataset, created on 2019–11-20 and consisting of 70 genomes and 255 conserved gene families107,108.
Synteny.
Pairwise synteny between the Hi-C scaffolded genomes of UTEX 2797 and 12B1 was identified and visualized with the JCVI pipeline109. Syntenic blocks within and between genome assemblies were identified with SynMap2 on the online Comparative Genomics Platform (CoGe) using the Relative Gene Order algorithm and Quota Align Merge with default settings to merge syntenic blocks110. Synteny visualizations between UTEX 2797 haplotypes on syntenic scaffolds were performed with XMatchView111. Shared synteny in the region surrounding HGT11 was visualized with pyGenomeViz112.
QUANTIFICATION AND STATISTICAL ANALYSIS
Phylogenomic analysis.
Orthologous sequences were identified using OrthoFinder v2.4.1113 with the single longest predicted coding sequence (CDS) per gene as input and using the blast_nucl sequence search option. Gene trees were constructed for every orthogroup containing sequences from at least 10 strains. Orthogroup CDS nucleotide sequences were aligned with MAFFT v7.471114 in the GUIDANCE v2.02 alignment software suite using the codon aware method115. The length of the ungapped alignment was evaluated using TrimAL v1.4.rev15116, and orthogroups with ungapped alignments less than 150 bp were excluded from further analysis. Maximum likelihood (ML) gene phylogenies were constructed with IQ-TREE v1.6.12117 using the trimmed multiple sequence alignment as input. ModelFinder118 was used to determine the best-fit nucleic acid substitution model, and 1000 replicates of both SH-aLRT and ultrafast bootstrapping analyses were performed. Gene trees were rooted based on the species tree using Notung v2.9.1.5 with its duplication, transfer, loss and ILS aware parsimony-based root optimization algorithm using default costs for all events119,120. In total, gene trees were built for 15,074 orthogroups that passed the strain count and alignment length filtering thresholds. The number of synonymous substitutions per synonymous site (Ks) was assessed for each pair of sequences within each orthogroup using the ungapped alignment as input. Ks was calculated according to LPB93 method using the yn00 module in PAML v4.9121–123.
The P. parvum species tree was constructed based on the combined signal from 2699 single-copy orthogroups (SCOGs) using both concatenation- and coalescence-based approaches. For the concatenation approach, the ML phylogeny was constructed in IQ-TREE v2.2.0124 based on a concatenated nucleotide data matrix consisting of 96 partitions and 2,982,918 sites with no missing data. The best partition model was selected using a relaxed hierarchical clustering algorithm as implemented by ModelFinder in IQ-TREE v2.2.0. The coalescence-based phylogeny estimation was conducted using ASTRAL v5.7.1125. Gene and site concordance factors were calculated for both species trees with IQ-TREE v2.2.0 using the 2699 single-copy genes used to construct the species trees.
Because the most closely related haptophyte species with sequenced genomes were too divergent to serve as outgroups for a nucleotide-based phylogeny, support for different root locations for the species phylogeny were evaluated using IQ-TREE v2.2.0 using the same best partition scheme as above but with a linked non-reversible DNA substitution model 12.12 across all partitions. The --root-test option was enabled to perform a tree topology test and compare the log-likelihoods of the trees based on every possible root location126. Both concatenation- and coalescent-based species trees supported the monophyletic origin of A-type and B-type prymnesins (Figure 2A, Figure S2). The root with the highest likelihood score caused the C-type prymnesin producers to be paraphyletic, which is a phylogenetic pattern also observed in a prior 18S rDNA phylogeny20. However, ‘rootstrap’ support for this root location was low (68%), and two alternative root locations, including one in which C-type producers were monophyletic, could not be rejected (AU tree topology test P-values > 0.1; Figure 2A, Figure S2). Support for the species tree topology was assessed using gene and site concordance factors (gCF/sCF)127. Support was generally high across the tree but low within A-type and B-type clades (Figure 2A, Figure S2). These low support values also correspond to areas of disagreement between the concatenation and coalescent based phylogenies (Figure S2).
Multi-labeled (MUL) species trees were built using GRAMPA v1.3 with UTEX 2797 as the h1 polyploid clade39. Two MUL tree analyses were run using different sets of Notung rooted gene trees as input: SCOGs only (2699 trees) and all orthogroups (15,040 trees). The MUL tree with the best parsimony score was the same using either gene tree set (Table S4). Sister taxa of each UTEX 2797 gene were identified using the Bio.Phylo Biopython toolkit128. The proportion of genes along each UTEX 2797 scaffold that grouped with subclade A1 or A2 were calculated with BEDTools intersect129 using 500 kbp windows and requiring a 50% minimum overlap for each gene. Subclade proportions and scaffold synteny were visualized using Circos v0.69–9130.
Breadth of coverage.
Breadth of coverage was calculated from alignments of the filtered Illumina reads to the 12B1 and UTEX 2797 scaffolded assemblies. To control for library differences between strains, the filtered Illumina reads were randomly subsampled to equal read counts (n = 44 million pairs) using reformat.sh, a tool of the BBTools software suite v38.8782, with sample seed set to 13. Alignments were generated using BWA-MEM v0.7.1576. Depth of coverage was calculated on a per-base level for both 12B1 and UTEX 2797 assemblies using samtools. Breadth of coverage was calculated as the proportion of base-pairs in the final assembly that had coverage greater than N coverage. BOC using coverage n = 0, 5, 10, 20, and 50 were all evaluated.
Heterozygosity and read coverage analyses.
Heterozygosity and coverage of maximal unique k-mers were estimated using the subsampled, filtered Illumina gDNA reads (see Characterization of Assembly Completeness Methods section) using KMC v3.1.1131 with a k-mer length (-k) of 21, minimal k-mer occurrence (-ci) of 1, and maximal k-mer occurrence (-cs) of 10,000.
Read coverage of single copy orthogroups (SCOGs) was calculated by first aligning the subsampled, filtered Illumina gDNA reads to each strain’s assembly using BWA-MEM v0.7.1576. Mean read depths were calculated for each SCOG with BEDTools intersect129 with the -mean option enabled. The median read depth of the 2699 SCOGs was then used as an estimate of the average coverage of the genome. The haploid genome size (G) was calculated using the Lander-Waterman equation40: where LN is the total combined length (bp) of the input Illumina reads and C is coverage as estimated by SCOG read depth41.
Functional annotation and enrichment tests.
Gene functional annotations were assigned via InterProScan v5.50–84.0132 using default settings and KofamScan133 with the threshold-scale set to 0.9. In total, 86.2% and 86.4% of the 12B1 and UTEX 2797 genes could be assigned predicted functional annotations. KinFin v1.0134 was used to transfer gene functional annotations to the orthogroup level if the annotation was assigned to 50% or more of strains in an orthogroup. All gene and orthogroup functional annotations are available for download from the project’s FigShare data repository (see Data Availability). Tests for enrichment of higher-level functional categories were performed using the core go.obo ontology (Gene Ontology Consortium) and the KEGG PATHWAY metabolic hierarchy downloaded via the KEGG API. Hypergeometric tests were performed in python using the SciPy library hypergeom135, and p-values were adjusted for multiple comparisons using the StatsModels library multitest136 with the Benjamini & Hochberg (BH) method137.
Horizontal gene transfer.
We assessed the genomes for possible HGT events using a modified Alien Index (AI) score138, which was calculated as previously described43. Briefly, each predicted protein sequence was queried against the same custom protein database used for BlobTools (see above) with DIAMOND (v2.0.8.146)78. A custom python script sorted the DIAMOND results based on the normalized bitscore (nbs), where nbs was calculated as the bitscore of the single best scoring HSP to the subject sequence divided by the best bitscore possible for the query sequence (i.e., the bitscore of the query aligned to itself). The AI score is given by the formula: , where nbsO is the normalized bit score of the best hit to a species outside of the Haptista lineage (NCBI:txid2608109), nbsH is the normalized bit score of the best hit to a species within the Haptista lineage skipping all hits to the Prymnesium genus (NCBI:txid35143). AI scores range from −1 to 1, being greater than 0 if the predicted protein sequence had a better hit to a non-Haptista sequence, suggestive of either horizontal gene transfer (HGT) or contamination43. Because donor and recipient lineages cannot be differentiated based on AI score alone, and given the relatively common occurrence of HGT from haptophytes to dinoflagellates (particularly dinoflagellates containing haptophyte-derived chloroplasts)139, we also skipped all hits to the Dinophyceae lineage (NCBI:txid 2864) when calculating AI scores. We filtered our HGT candidates (AI > 0) to those that were most likely to be phylogenetically informative by requiring AI > 0.1 and total hits ≥ 50. In addition, if the top hit was to a eukaryote, we further required than ≥ 5 haptophytes from outside the Prymnesium genus be present in the top 200 hits. This extra requirement allowed us to evaluate whether haptophytes formed a monophyletic clade or whether Prymnesium grouped separately, which would provide stronger support for HGT140. Lastly, due to the risk of bacterial contamination in the Illumina-only assemblies, all candidate HGTs had to be present in one or both scaffolded assemblies of 12B1 and UTEX 2797. The AI screen flagged 95 orthogroups as candidate HGTs. See Data Availability for access to the database and scripts.
Phylogenetic trees of protein sequences were constructed for all filtered AI-flagged HGT candidates. Full-length proteins corresponding to the top 200 hits (E-value < 1 × 10−10) to each query sequence were extracted from the local database using esl-sfetch141. Protein queries with less than 50 significant hits were skipped. Protein sequences were aligned with MAFFT v7.471 using the E-INS-i strategy and the BLOSUM30 amino acid scoring matrix114 and trimmed with trimAL v1.4.rev15 using its gappyout strategy116. Proteins with trimmed alignments < 150 amino acids in length were excluded. The topologies of the remaining genes were inferred using maximum likelihood as implemented in IQ-TREE v1.6.12117 using an empirically determined substitution model and 1000 rapid bootstrap replications. The phylogenies were midpoint rooted and branches with local support < 95 were collapsed using the ape and phangorn R packages142,143. Phylogenies were visualized using ITOL version 4144 and inspected manually to identify phylogenetically supported HGT candidate proteins. Most AI-flagged HGT candidates (n = 61) were phylogenetically inconclusive or lacked support for HGT. Of the 34 HGT candidates that passed manual inspection, we focused on the eleven HGTs with clear donor lineages with strong node support (Fig. S4, Table S6).
Gene expression.
Quantification of gene expression was performed using Kallisto v0.46.2145. The Kallisto index was built using all BRAKER predicted transcripts with the default k-mer size of 31. Transcripts per million (TPM) gene abundance values from Kallisto were scaled using the average transcript length, averaged over samples and to library size, using the lengthScaledTPM option in tximport v1.18.0146. The full matrix of gene expression can be downloaded from the project’s FigShare data repository.
Supplementary Material
Table S1. Summary of Prymnesium parvum strains, genome assemblies, gene models, and genome sizes. Related to Figure 1 and Figure 4.
Summary of strains and their respective prymnesin types, sources, culturing conditions are provided in rows 2-8. Cultures were kept at 20 °C using a 12:12 light dark cycle and irradiance of 40-200 μmol photos m-2 s-1. N50 is defined as the sequence length of the shortest contig at 50% of the total genome length. L50 is defined as count of smallest number of contigs whose length sum makes up half of genome size. CMUK = coverage of maximal unique k-mers; larger CMUKs indicative of smaller haploid genomes and vice versa
Table S2. Coordinates of each predicted telomeric repeat for 12B1 and UTEX 2797 assemblies. Related to Figure 1.
Relative position of repeat on either the beginning (1-) or end (2-) of scaffold terminus is also denoted, as well as the distance from this terminus.
Table S3. Summary of BUSCO analysis performed on haptophyte assemblies. Related to Figure 1.
Ten BUSCO genes were missing in every haptophyte assembly investigated: 1030907at2759, 1094121at2759, 1304061at2759, 1364586at2759, 1442062at2759, 261419at2759, 270107at2759, 331411at2759, 453044at2759, 975158at2759.
Table S4. GRAMPA gene tree-Species tree reconciliation. Related to Figure 3.
ST = Single-labeled starting tree, MUL = Multi-labeled tree.
Table S5. Tests for enrichment of GO and KEGG gene annotation terms. Related to Figure 5.
Table S6. Eleven HGTs with strong phylogenetic support. Related to Figure 6.
Numbers in parentheses following gene IDs in UTEX 2797 indicate maximum length-scaled TPMs. Full matrix of gene expression can be downloaded from the project’s FigShare data repository.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical Commercial Assays | ||
| Proximo Hi-C 2.0 Kit | Lieberman-Aiden et al88 | N/A |
| Deposited Data | ||
| Raw data | This paper | SRA: PRJNA807128; EBI MetaboLights: MTBLS5893; |
| All genome assemblies, predicted CDS and protein sequences, multiple sequence alignments, tree files, and other related data files | This paper | https://doi.org/10.6084/m9.figshare.21376500 |
| Experimental Models: Organisms/Strains | ||
| Prymnesium parvum: 12B1 | Dr. William Driscoll | UTEX Culture Collection of Algae: UTEX LB ZZ1299 |
| Prymnesium parvum: 12A1 | Dr. William Driscoll | UTEX Culture Collection of Algae: UTEX LB ZZ1300 |
| Prymnesium parvum: CCMP2941 | NCMA at Bigelow Laboratory | NCMA: CCMP2941 |
| Prymnesium parvum: CCMP3037 | NCMA at Bigelow Laboratory | NCMA: CCMP3037 |
| Prymnesium parvum: K-0081 | NORCCA: The Norwegian Culture Collection of Algae | NORCCA: K-0081 |
| Prymnesium parvum: K-0374 | NORCCA: The Norwegian Culture Collection of Algae | NORCCA: K-0374 |
| Prymnesium parvum: K-0252 | NORCCA: The Norwegian Culture Collection of Algae | NORCCA: K-0252; UIO55; Roscoff: RCC3427 |
| Prymnesium parvum: RCC3703 | Roscoff Culture Collection | Roscoff: RCC3703; CCAP 946/6; NCMA: CCMP708 |
| Prymnesium parvum: RCC3426 | Roscoff Culture Collection | Roscoff: RCC3426; UIO54 |
| Prymnesium parvum: RCC1433 | Roscoff Culture Collection | Roscoff: RCC1433; AC36 |
| Prymnesium parvum: RCC191 | Roscoff Culture Collection | Roscoff: RCC191; PLY527; PCC 527 |
| Prymnesium parvum: RCC1436 | Roscoff Culture Collection | Roscoff: RCC1436; AC45 |
| Prymnesium parvum: UTEX 2797 | UTEX Culture Collection of Algae | UTEX: UTEX LB 2797 |
| Prymnesium parvum: UTEX 995 | UTEX Culture Collection of Algae | UTEX: UTEX LB 995; PLY94 |
| Prymnesium parvum: KAC-39 | Kalmar Algae Collection | Kalmar: KAC-39 |
| Software and Algorithms | ||
| Scripts for data analysis and visualization | This paper | https://github.com/WisecaverLab/Pparvum_genome_diversity |
| FlowCal Python library | Castillo-Hair et al62 | https://github.com/taborlab/FlowCal |
| Proteowizard v3.0.20303 | Chambers et al.66 | https://proteowizard.sourceforge.io |
| MZmine2 v2.53 | Pluskal et al.67 | https://github.com/mzmine/mzmine2 |
| enviPat Web 2.4 tool | Loos et al.68 | https://www.envipat.eawag.ch |
| pymzML | Kösters et al.69 | https://github.com/pymzml/pymzML |
| matplotlib | Hunter70 | https://matplotlib.org |
| svgutils | Telenczuk71 | https://github.com/btel/svg_utils |
| FastQC v0.10.0 | Babraham Bioinformatics73 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Abyss v2.2.4 | Simpson et al74 | https://github.com/bcgsc/abyss |
| BlobTools v1.1.1 | Laetsch and Blaxter75 | https://github.com/DRL/blobtools |
| BWA-MEM v0.7.15 | Li and Durbin76 | https://github.com/lh3/bwa |
| blastn v2.11.0 | Camacho et al77 | https://blast.ncbi.nlm.nih.gov/doc/blast-help/downloadblastdata.html |
| DIAMOND v2.0.8.146 | Buchfink et al78 | https://github.com/bbuchfink/diamond |
| BBTools v38.87 | Bushnell82 | https://jgi.doe.gov/data-and-tools/software-tools/bbtools/ |
| REVIGO | Supek et al59 | http://revigo.irb.hr |
| Guppy v2.3.5 | Oxford Nanopore Technologies84 | https://nanoporetech.com |
| MaSuRCA v3.3.1 | Zimin et al85 | https://github.com/alekseyzimin/masurca |
| Canu v2.1.1 | Koren et al86 | https://github.com/marbl/canu |
| Pilon v1.23 | Walker et al87 | https://github.com/broadinstitute/pilon |
| SAMBLASTER | Faust and Hall89 | https://github.com/GregoryFaust/samblaster |
| SAMtools | Li et al90 | https://github.com/samtools/samtools |
| Kraken v2 | Wood and Salzberg93 | https://ccb.jhu.edu/software/kraken/ |
| Juicebox | Rao et al91 and Durand et al92 | https://github.com/aidenlab/Juicebox |
| LACHESIS | Burton et al95 | N/A |
| RepeatModeler v2.0.1 | Flynn et al96 | http://repeatmasker.org |
| RepeatMasker v4.0.7 | Smit et al97 | http://repeatmasker.org |
| STAR v2.7.8a | Sobin et al101 | https://github.com/alexdobin/STAR |
| BRAKER2 v2.1.5 | Hott et al102 and Bruna et al103 | https://github.com/Gaius-Augustus/BRAKER |
| Augustus | Stanke et al104 | https://github.com/Gaius-Augustus/Augustus |
| TRFFinder v4.09 | Benson105 | https://github.com/Benson-Genomics-Lab/TRF |
| BUSCO v4.0.6 | Simão et al107 and Manni et al108 | https://busco.ezlab.org |
| JCVI pipeline | Tang et al109 | https://pypi.org/project/jcvi/ |
| Comparative Genomics Platform (CoGe) | Haug-Baltzell et al110 | https://genomevolution.org/CoGe/ |
| XMatchView | Warren111 | https://github.com/bcgsc/xmatchview |
| pyGenomeViz | Shimoyama112 | https://moshi4.github.io/pyGenomeViz/ |
| OrthoFinder v2.4.1 | Emms and Kelly113 | https://github.com/davidemms/OrthoFinder |
| MAFFT v7.471 | Katoh and Standley114 | https://mafft.cbrc.jp/alignment/software/ |
| GUIDANCE v2.02 | Sela et al115 | http://guidance.tau.ac.il/source |
| TrimAL v1.4.rev15 | Capella-Gutierrez et al116 | http://trimal.cgenomics.org |
| IQ-TREE v1.6.12 and v2.2.0 | Nguyen et al117; Minh et al124; Naser-Khdour et al126; Minh et al127 | http://www.iqtree.org |
| ModelFinder | Kalyaanamoorthy et al118 | http://www.iqtree.org |
| Notung v2.9.1.5 | Chen et al119 and Stolzer et al120 | https://www.cs.cmu.edu/~durand/Notung/ |
| PAML v4.9 | Li121; Pamilo and Bianchi122; Yang123 | http://abacus.gene.ucl.ac.uk/software/paml.html |
| ASTRAL v5.7.1 | Zhang et al125 | https://github.com/smirarab/ASTRAL |
| GRAMPA v1.3 | Thomas et al39 | https://gwct.github.io/grampa/ |
| Bio.Phylo Biopython toolkit | Talevich et al128 | https://biopython.org |
| BEDTools | Quinlan and Hall129 | https://github.com/arq5x/bedtools2 |
| Circos v0.69–9 | Krzywinski et al130 | http://circos.ca |
| KMC v3.1.1 | Kokot et al131 | https://github.com/refresh-bio/KMC |
| InterProScan v5.50–84.0 | Jones et al132 | https://github.com/ebi-pf-team/interproscan |
| KofamScan | Aramaki et al133 | https://github.com/takaram/kofam_scan |
| KinFin v1.0 | Laetsch and Blaxter134 | https://kinfin.readme.io/docs |
| SciPy | Virtanen et al135 | https://scipy.org |
| StatsModels | Seabold and Perktold136 | https://www.statsmodels.org/stable/index.html |
| esl-sfetch | Eddy141 | https://github.com/EddyRivasLab/easel |
| ape R package | Paradis et al142 | https://cran.r-project.org/web/packages/ape/ |
| phangorn R package | Schliep143 | https://cran.r-project.org/web/packages/phangorn/ |
| ITOL version 4 | Letunic and Bork144 | https://itol.embl.de |
| Kallisto v0.46.2 | Bray et al145 | https://github.com/pachterlab/kallisto |
| tximport v1.18.0 | Soneson et al146 | https://bioconductor.org/packages/release/bioc/html/tximport.html |
| Other | ||
| Protocol for total DNA extraction from plant tissue using CTAB method V.2 | Auber72 | https://www.protocols.io/view/total-dna-extraction-from-plant-tissue-using-ctab-b5qhq5t6 |
| custom protein database used in the BlobTools analysis | Dr. Jennifer Wisecaver, Purdue University | https://www.datadepot.rcac.purdue.edu/jwisecav/custom-refseq/2021-08-02/ |
| Protocol for algal nuclei isolation for Nanopore sequencing of HMW DNA V.3 | Auber and Wisecaver83 | https://www.protocols.io/view/algal-nuclei-isolation-for-nanopore-sequencing-of-8epv568jdg1b/v3 |
| Wisecaver Lab algal RNA extraction protocol using Ambion TRI Reagent | Auber et al100 | https://www.protocols.io/view/wisecaver-lab-algal-rna-extraction-protocol-using-3byl4k6r8vo5/v1 |
Highlights.
The protist morphospecies Prymnesium parvum contains at least three cryptic species
Haploid genome size differs dramatically between these cryptic species
Strains can be hybrids that retain two phylogenetically distinct haplotypes
Variable gene families include candidates for the biosynthesis of toxic metabolites
ACKNOWLEDGEMENTS
We thank members of the Wisecaver lab, Dr. Jody Banks, and Dr. Brian Dilkes for helpful discussions as well as Dr. Clint Chapple and Dr. Scott McAdam for plant tissue for flow cytometry. This work was conducted in part using the resources of the Rosen Center for Advanced Computing at Purdue University and well as through use of the Flow Cytometry and Cell Separation Facility at the Bindley Bioscience Center at Purdue, a core facility of the NIH-funded Indiana Clinical and Translational Sciences Institute. This work was supported by the National Institute for Environmental Health Sciences under grants F32-ES032276 to TRF and R21-ES032056 to BSM and the National Science Foundation under grant DEB-1831493 to JHW and WWD.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Roelke DL, Barkoh A, Brooks BW, Grover JP, Hambright KD, LaClaire JW, Moeller PDR, and Patino R (2016). A chronicle of a killer alga in the west: ecology, assessment, and management of Prymnesium parvum blooms. Hydrobiologia 764, 29–50. 10.1007/s10750-015-2273-6. [DOI] [Google Scholar]
- 2.Gobler CJ, Doherty OM, Hattenrath-Lehmann TK, Griffith AW, Kang Y, and Litaker RW (2017). Ocean warming since 1982 has expanded the niche of toxic algal blooms in the North Atlantic and North Pacific oceans. Proc. Natl. Acad. Sci. U. S. A. 114, 4975–4980. 10.1073/pnas.1619575114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hallegraeff GM, Anderson DM, Belin C, Bottein M-YD, Bresnan E, Chinain M, Enevoldsen H, Iwataki M, Karlson B, McKenzie CH, et al. (2021). Perceived global increase in algal blooms is attributable to intensified monitoring and emerging bloom impacts. Commun. Earth Environ. 2, 1–10. 10.1038/s43247-021-00178-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wells ML, Karlson B, Wulff A, Kudela R, Trick C, Asnaghi V, Berdalet E, Cochlan W, Davidson K, De Rijcke M, et al. (2020). Future HAB science: Directions and challenges in a changing climate. Harmful Algae 91, 101632. 10.1016/j.hal.2019.101632. [DOI] [PubMed] [Google Scholar]
- 5.Roelke DL, and Manning SR (2018). Harmful algal species fact sheet: Prymnesium parvum (Carter) “golden algae.” In Harmful Algal Blooms (John Wiley & Sons, Ltd; ), pp. 629–632. 10.1002/9781118994672.ch16q. [DOI] [Google Scholar]
- 6.Southard GM, Fries LT, and Barkoh A (2010). Prymnesium parvum: The Texas experience. JAWRA J. Am. Water Resour. Assoc. 46, 14–23. 10.1111/j.1752-1688.2009.00387.x. [DOI] [Google Scholar]
- 7.Tábora-Sarmiento S, Patiño R, Portillo-Quintero C, and Coldren C (2022). Air, land, and water variables associated with the first appearance and current spatial distribution of toxic Prymnesium parvum blooms in reservoirs of the Southern Great Plains, USA. Sci. Total Environ. 836, 155567. 10.1016/j.scitotenv.2022.155567. [DOI] [PubMed] [Google Scholar]
- 8.Baker JW, Grover JP, Brooks BW, Ureña-Boeck F, Roelke DL, Errera R, and Kiesling RL (2007). Growth and toxicity of Prymnesium parvum (Haptophyta) as a function of salinity, light, and temperature. J. Phycol. 43, 219–227. 10.1111/j.1529-8817.2007.00323.x. [DOI] [Google Scholar]
- 9.Brutemark A, and Granéli E (2011). Role of mixotrophy and light for growth and survival of the toxic haptophyte Prymnesium parvum. Harmful Algae 10, 388–394. 10.1016/j.hal.2011.01.005. [DOI] [Google Scholar]
- 10.Granéli E, Edvardsen B, Roelke DL, and Hagström JA (2012). The ecophysiology and bloom dynamics of Prymnesium spp. Harmful Algae 14, 260–270. 10.1016/j.hal.2011.10.024. [DOI] [Google Scholar]
- 11.Nygaard K, and Tobiesen A (1993). Bacterivory in algae: A survival strategy during nutrient limitation. Limnol. Oceanogr. 38, 273–279. 10.4319/lo.1993.38.2.0273. [DOI] [Google Scholar]
- 12.Tillmann U (1998). Phagotrophy by a plastidic haptophyte, Prymnesium patelliferum. Aquat. Microb. Ecol. 14, 155–160. [Google Scholar]
- 13.Tillmann U (2003). Kill and eat your predator: a winning strategy of the planktonic flagellate Prymnesium parvum. Aquat. Microb. Ecol. 32, 73–84. 10.3354/ame032073. [DOI] [Google Scholar]
- 14.Remmel EJ, and Hambright KD (2012). Toxin-assisted micropredation: experimental evidence shows that contact micropredation rather than exotoxicity is the role of Prymnesium toxins. Ecol. Lett. 15, 126–132. 10.1111/j.1461-0248.2011.01718.x. [DOI] [PubMed] [Google Scholar]
- 15.Carvalho WF, and Granéli E (2010). Contribution of phagotrophy versus autotrophy to Prymnesium parvum growth under nitrogen and phosphorus sufficiency and deficiency. Harmful Algae 9, 105–115. 10.1016/j.hal.2009.08.007. [DOI] [Google Scholar]
- 16.Manning SR, and La Claire JW (2010). Prymnesins: toxic metabolites of the golden alga, Prymnesium parvum Carter (Haptophyta). Mar. Drugs 8, 678–704. 10.3390/md8030678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasmussen SA, Andersen AJC, Andersen NG, Nielsen KF, Hansen PJ, and Larsen TO (2016). Chemical diversity, origin, and analysis of phycotoxins. J. Nat. Prod. 79, 662–673. 10.1021/acs.jnatprod.5b01066. [DOI] [PubMed] [Google Scholar]
- 18.Rasmussen SA, Meier S, Andersen NG, Blossom HE, Duus JØ, Nielsen KF, Hansen PJ, and Larsen TO (2016). Chemodiversity of ladder-frame prymnesin polyethers in Prymnesium parvum. J. Nat. Prod. 79, 2250–2256. 10.1021/acs.jnatprod.6b00345. [DOI] [PubMed] [Google Scholar]
- 19.Binzer SB, Svenssen DK, Daugbjerg N, Alves-de-Souza C, Pinto E, Hansen PJ, Larsen TO, and Varga E (2019). A-, B- and C-type prymnesins are clade specific compounds and chemotaxonomic markers in Prymnesium parvum. Harmful Algae 81, 10–17. 10.1016/j.hal.2018.11.010. [DOI] [PubMed] [Google Scholar]
- 20.Anestis K, Kohli GS, Wohlrab S, Varga E, Larsen TO, Hansen PJ, and John U (2021). Polyketide synthase genes and molecular trade-offs in the ichthyotoxic species Prymnesium parvum. Sci. Total Environ. 795, 148878. 10.1016/j.scitotenv.2021.148878. [DOI] [PubMed] [Google Scholar]
- 21.Driscoll WW, Espinosa NJ, Eldakar OT, and Hackett JD (2013). Allelopathy as an emergent, exploitable public good in the bloom-forming microalga Prymnesium parvum. Evolution 67, 1582–1590. 10.1111/evo.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blossom HE, Rasmussen SA, Andersen NG, Larsen TO, Nielsen KF, and Hansen PJ (2014). Prymnesium parvum revisited: relationship between allelopathy, ichthyotoxicity, and chemical profiles in 5 strains. Aquat. Toxicol. Amst. Neth. 157, 159–166. [DOI] [PubMed] [Google Scholar]
- 23.Medić N, Varga E, Waal D.B.V. de, Larsen TO, and Hansen PJ (2022). The coupling between irradiance, growth, photosynthesis and prymnesin cell quota and production in two strains of the bloom-forming haptophyte, Prymnesium parvum. Harmful Algae 112, 102173. 10.1016/j.hal.2022.102173. [DOI] [PubMed] [Google Scholar]
- 24.Driscoll WW, Wisecaver JH, Hackett JD, Espinosa NJ, Padway J, Engers JE, and Bower JA (2022). Behavioral differences underlie toxicity and predation variation in blooms of Prymnesium parvum. Ecol. Lett. in press. [DOI] [PubMed] [Google Scholar]
- 25.Larsen A, Eikrem W, and Paasche E (1993). Growth and toxicity in Prymnesium patelliferum (Prymnesiophyceae) isolated from Norwegian waters. Can. J. Bot. 71, 1357–1362. 10.1139/b93-161. [DOI] [Google Scholar]
- 26.Larsen A, and Bryant S (1998). Growth rate and toxicity of Prymnesium parvum and Prymnesium patelliferum (haptophyta) in response to changes in salinity, light and temperature. Sarsia 83, 409–418. 10.1080/00364827.1998.10413700. [DOI] [Google Scholar]
- 27.Lysgaard ML, Eckford-Soper L, and Daugbjerg N (2018). Growth rates of three geographically separated strains of the ichthyotoxic Prymnesium parvum (Prymnesiophyceae) in response to six different pH levels. Estuar. Coast. Shelf Sci. 204, 98–102. 10.1016/j.ecss.2018.02.030. [DOI] [Google Scholar]
- 28.Talarski A, Manning SR, and La Claire JW (2016). Transcriptome analysis of the euryhaline alga, Prymnesium parvum (Prymnesiophyceae): Effects of salinity on differential gene expression. Phycologia 55, 33–44. 10.2216/15-74.1. [DOI] [Google Scholar]
- 29.Rashel RH, and Patiño R (2017). Influence of genetic background, salinity, and inoculum size on growth of the ichthyotoxic golden alga (Prymnesium parvum). Harmful Algae 66, 97–104. 10.1016/j.hal.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 30.Taylor RB, Hill BN, Bobbitt JM, Hering AS, Brooks BW, and Chambliss CK (2020). Suspect and non-target screening of acutely toxic Prymnesium parvum. Sci. Total Environ. 715, 136835. 10.1016/j.scitotenv.2020.136835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richardson ET, and Patiño R (2021). Growth of the harmful alga, Prymnesium parvum (Prymnesiophyceae), after gradual and abrupt increases in salinity. J. Phycol. 57, 1335–1344. 10.1111/jpy.13172. [DOI] [PubMed] [Google Scholar]
- 32.Taylor RB, Hill BN, Langan LM, Chambliss CK, and Brooks BW (2021). Sunlight concurrently reduces Prymnesium parvum elicited acute toxicity to fish and prymnesins. Chemosphere 263, 127927. 10.1016/j.chemosphere.2020.127927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larsen A, and Edvardsen B (1998). Relative ploidy levels in Prymnesium parvum and P. patelliferum (Haptophyta) analyzed by flow cytometry. Phycologia 37, 412–424. 10.2216/i0031-8884-37-6-412.1. [DOI] [Google Scholar]
- 34.Tanaka T, Maeda Y, Veluchamy A, Tanaka M, Abida H, Maréchal E, Bowler C, Muto M, Sunaga Y, Tanaka M, et al. (2015). Oil Accumulation by the Oleaginous Diatom Fistulifera solaris as Revealed by the Genome and Transcriptome. Plant Cell 27, 162–176. 10.1105/tpc.114.135194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steenwyk JL, Lind AL, Ries LNA, dos Reis TF, Silva LP, Almeida F, Bastos RW, Fraga da Silva TF de C, Bonato VLD, Pessoni AM, et al. (2020). Pathogenic Allodiploid Hybrids of Aspergillus Fungi. Curr. Biol. 30, 2495–2507.e7. 10.1016/j.cub.2020.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ortiz-Merino RA, Braun-Galleani S, Byrne KP, Porro D, Branduardi P, and Wolfe KH (2017). Evolutionary restoration of fertility in an interspecies hybrid yeast, by whole-genome duplication after a failed mating-type switch. PLOS Biol. 15, e2002128. 10.1371/journal.pbio.2002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.VanBuren R, Man Wai C, Wang X, Pardo J, Yocca AE, Wang H, Chaluvadi SR, Han G, Bryant D, Edger PP, et al. (2020). Exceptional subgenome stability and functional divergence in the allotetraploid Ethiopian cereal teff. Nat. Commun. 11, 884. 10.1038/s41467-020-14724-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Novikova PY, Hohmann N, Nizhynska V, Tsuchimatsu T, Ali J, Muir G, Guggisberg A, Paape T, Schmid K, Fedorenko OM, et al. (2016). Sequencing of the genus Arabidopsis identifies a complex history of nonbifurcating speciation and abundant trans-specific polymorphism. Nat. Genet. 48, 1077–1082. 10.1038/ng.3617. [DOI] [PubMed] [Google Scholar]
- 39.Thomas GWC, Ather SH, and Hahn MW (2017). Gene-tree reconciliation with MUL-trees to resolve polyploidy events. Syst. Biol. 66, 1007–1018. 10.1093/sysbio/syx044. [DOI] [PubMed] [Google Scholar]
- 40.Lander ES, and Waterman MS (1988). Genomic mapping by fingerprinting random clones: A mathematical analysis. Genomics 2, 231–239. 10.1016/0888-7543(88)90007-9. [DOI] [PubMed] [Google Scholar]
- 41.Pflug JM, Holmes VR, Burrus C, Johnston JS, and Maddison DR (2020). Measuring genome sizes using read-depth, k-mers, and flow cytometry: methodological comparisons in beetles (Coleoptera). G3 Genes Genomes Genet. 10, 3047–3060. 10.1534/g3.120.401028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sibbald SJ, Eme L, Archibald JM, and Roger AJ (2020). Lateral gene transfer mechanisms and pan-genomes in eukaryotes. Trends Parasitol. 36, 927–941. 10.1016/j.pt.2020.07.014. [DOI] [PubMed] [Google Scholar]
- 43.Wisecaver JH, Alexander WG, King SB, Todd Hittinger C, and Rokas A (2016). Dynamic evolution of nitric oxide detoxifying flavohemoglobins, a family of single-protein metabolic modules in bacteria and eukaryotes. Mol. Biol. Evol, msw073. [DOI] [PubMed] [Google Scholar]
- 44.Agarwal V, El Gamal AA, Yamanaka K, Poth D, Kersten RD, Schorn M, Allen EE, and Moore BS (2014). Biosynthesis of polybrominated aromatic organic compounds by marine bacteria. Nat. Chem. Biol. 10, 640–647. 10.1038/nchembio.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schroeder DC, Oke J, Malin G, and Wilson WH (2002). Coccolithovirus (Phycodnaviridae): characterisation of a new large dsDNA algal virus that infects Emiliana huxleyi. Arch. Virol. 147, 1685–1698. 10.1007/s00705-002-0841-3. [DOI] [PubMed] [Google Scholar]
- 46.Wagstaff BA, Vladu IC, Barclay JE, Schroeder DC, Malin G, and Field RA (2017). Isolation and Characterization of a Double Stranded DNA Megavirus Infecting the Toxin-Producing Haptophyte Prymnesium parvum. Viruses 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dunigan DD, Fitzgerald LA, and Van Etten JL (2006). Phycodnaviruses: a peek at genetic diversity. Virus Res. 117, 119–132. 10.1016/j.virusres.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 48.Read BA, Kegel J, Klute MJ, Kuo A, Lefebvre SC, Maumus F, Mayer C, Miller J, Monier A, Salamov A, et al. (2013). Pan genome of the phytoplankton Emiliania underpins its global distribution. Nature 499, 209–213. 10.1038/nature12221. [DOI] [PubMed] [Google Scholar]
- 49.Green JC, Hibberd DJ, and Pienaar RN (1982). The taxonomy of Prymnesium (Prymnesiophyceae) including a description of a new cosmopolitan species, P. patellifera sp. nov., and further observations on P. parvum N. carter. Br. Phycol. J. 17, 363–382. 10.1080/00071618200650381. [DOI] [Google Scholar]
- 50.Larsen A, and Medlin LK (1997). Inter- and intraspecific genetic variation in twelve Prymnesium (Haptophyceae) clones. J. Phycol. 33, 1007–1015. 10.1111/j.0022-3646.1997.01007.x. [DOI] [Google Scholar]
- 51.Hovde BT, Deodato CR, Andersen RA, Starkenburg SR, Barlow SB, and Cattolico RA (2019). Chrysochromulina: Genomic assessment and taxonomic diagnosis of the type species for an oleaginous algal clade. Algal Res. 37, 307–319. 10.1016/j.algal.2018.11.023. [DOI] [Google Scholar]
- 52.Sonneborn TM (1975). The Paramecium aurelia complex of fourteen sibling species. Trans. Am. Microsc. Soc. 94, 155–178. 10.2307/3224977. [DOI] [Google Scholar]
- 53.Simon EM, Nanney DL, and Doerder FP (2008). The “Tetrahymena pyriformis” complex of cryptic species. Biodivers. Conserv. 17, 365–380. 10.1007/s10531-007-9255-6. [DOI] [Google Scholar]
- 54.John U, Litaker RW, Montresor M, Murray S, Brosnahan ML, and Anderson DM (2014). Formal revision of the Alexandrium tamarense species complex (Dinophyceae) taxonomy: The introduction of five species with emphasis on molecular-based (rDNA) classification. Protist 165, 779–804. 10.1016/j.protis.2014.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barreto FS, Tomas CR, and McCartney MA (2011). AFLP fingerprinting shows that a single Prymnesium parvum harmful algal bloom consists of multiple clones. J. Hered. 102, 747–752. 10.1093/jhered/esr081. [DOI] [PubMed] [Google Scholar]
- 56.Verster KI, Wisecaver JH, Karageorgi M, Duncan RP, Gloss AD, Armstrong EE, Price DK, Menon AR, Ali ZM, and Whiteman NK (2019). Horizontal Transfer of Bacterial Cytolethal Distending Toxin B Genes to Insects. Mol. Biol. Evol. 36. 10.1093/molbev/msz146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Richards TA, Soanes DM, Jones MDM, Vasieva O, Leonard G, Paszkiewicz K, Foster PG, Hall N, and Talbot NJ (2011). Horizontal gene transfer facilitated the evolution of plant parasitic mechanisms in the oomycetes. Proc. Natl. Acad. Sci. 108, 15258–15263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Slot JC, and Hibbett DS (2007). Horizontal transfer of a nitrate assimilation gene cluster and ecological transitions in fungi: a phylogenetic study. PLoS ONE 2, e1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Supek F, Bošnjak M, Škunca N, and Šmuc T (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLOS ONE 6, e21800. 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dpooležel J, Binarová P, and Lcretti S (1989). Analysis of Nuclear DNA content in plant cells by Flow cytometry. Biol. Plant. 31, 113–120. 10.1007/BF02907241. [DOI] [Google Scholar]
- 61.Čertnerová D (2021). Nuclei isolation protocols for flow cytometry allowing nuclear DNA content estimation in problematic microalgal groups. J. Appl. Phycol. 33, 2057–2067. 10.1007/s10811-021-02433-z. [DOI] [Google Scholar]
- 62.Castillo-Hair SM, Sexton JT, Landry BP, Olson EJ, Igoshin OA, and Tabor JJ (2016). FlowCal: A user-friendly, open source software tool for automatically converting flow cytometry Data from arbitrary to calibrated units. ACS Synth. Biol. 5, 774–780. 10.1021/acssynbio.5b00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galbraith DW (2014). Endoreduplicative standards for calibration of flow cytometric C-Value measurements. Cytometry A 85, 368–374. 10.1002/cyto.a.22440. [DOI] [PubMed] [Google Scholar]
- 64.Cavaller-Smith T (1985). The evolution of genome size (John Wiley and Sons Inc.,New York, NY: ). [Google Scholar]
- 65.Svenssen DK, Binzer SB, Medić N, Hansen PJ, Larsen TO, and Varga E (2019). Development of an indirect quantitation method to assess ichthyotoxic B-type prymnesins from Prymnesium parvum. Toxins 11, 251. 10.3390/toxins11050251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, et al. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920. 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pluskal T, Castillo S, Villar-Briones A, and Oresic M (2010). MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395. 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Loos M, Gerber C, Corona F, Hollender J, and Singer H (2015). Accelerated isotope fine structure calculation using pruned transition trees. Anal. Chem. 87, 5738–5744. 10.1021/acs.analchem.5b00941. [DOI] [PubMed] [Google Scholar]
- 69.Kösters M, Leufken J, Schulze S, Sugimoto K, Klein J, Zahedi RP, Hippler M, Leidel SA, and Fufezan C (2018). pymzML v2.0: introducing a highly compressed and seekable gzip format. Bioinformatics 34, 2513–2514. 10.1093/bioinformatics/bty046. [DOI] [PubMed] [Google Scholar]
- 70.Hunter JD (2007). Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 9, 90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- 71.Telenczuk B (2023). svg_utils: A python-based SVG editor.
- 72.Auber R (2019). Total DNA extraction from plant tissue using CTAB method. protocols.io. 10.17504/protocols.io.bamnic5e. [DOI] [Google Scholar]
- 73.Babraham Bioinformatics (2011). FastQC A quality control tool for high throughput sequence data.
- 74.Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJM, and Birol İ (2009). ABySS: A parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Laetsch DR, and Blaxter ML (2017). BlobTools: Interrogation of genome assemblies. F1000Research 6, 1287. 10.12688/f1000research.12232.1. [DOI] [Google Scholar]
- 76.Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760. 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421. 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Buchfink B, Xie C, and Huson DH (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 79.O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, et al. (2016). Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 44, D733–D745. 10.1093/nar/gkv1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Keeling PJ, Burki F, Wilcox HM, Allam B, Allen EE, Amaral-Zettler LA, Armbrust EV, Archibald JM, Bharti AK, Bell CJ, et al. (2014). The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): Illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLOS Biol. 12, e1001889. 10.1371/journal.pbio.1001889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Matasci N, Hung L-H, Yan Z, Carpenter EJ, Wickett NJ, Mirarab S, Nguyen N, Warnow T, Ayyampalayam S, Barker M, et al. (2014). Data access for the 1,000 Plants (1KP) project. GigaScience 3, 17. 10.1186/2047-217X-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bushnell B (2017). BBTools Software Package.
- 83.Auber R, and Wisecaver J (2019). Algal nuclei isolation for Nanopore sequencing of HMW DNA. protocols.io. 10.17504/protocols.io.7b7hirn. [DOI] [Google Scholar]
- 84.Oxford Nanopore Technologies (2019). Guppy, local accelerated basecalling for Nanopore data.
- 85.Zimin AV, Marçais G, Puiu D, Roberts M, Salzberg SL, and Yorke JA (2013). The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677. 10.1093/bioinformatics/btt476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, and Phillippy AM (2017). Canu: scalable and accurate long-read assembly via adaptive k -mer weighting and repeat separation. Genome Res. 27, 722–736. 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. (2014). Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963. 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Faust GG, and Hall IM (2014). SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505. 10.1093/bioinformatics/btu314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinforma. Oxf. Engl. 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016). Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101. 10.1016/j.cels.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wood DE, and Salzberg SL (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, R46. 10.1186/gb-2014-15-3-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bickhart DM, Rosen BD, Koren S, Sayre BL, Hastie AR, Chan S, Lee J, Lam ET, Liachko I, Sullivan ST, et al. (2017). Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 49, 643–650. 10.1038/ng.3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Burton JN, Adey A, Patwardhan RP, Qiu R, Kitzman JO, and Shendure J (2013). Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125. 10.1038/nbt.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, and Smit AF (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457. 10.1073/pnas.1921046117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Smit AF, Hubley R, and Green P (2017). RepeatMasker Open-4.0.
- 98.Hulatt CJ, Wijffels RH, and Posewitz MC (2021). The genome of the Haptophyte Diacronema lutheri (Pavlova lutheri, Pavlovales): A model for lipid biosynthesis in eukaryotic algae. Genome Biol. Evol. 13, evab178. 10.1093/gbe/evab178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen D, Yuan X, Zheng X, Fang J, Lin G, Li R, Chen J, He W, Huang Z, Fan W, et al. (2022). Multi-omics analyses provide insight into the biosynthesis pathways of fucoxanthin in Isochrysis galbana. Genomics Proteomics Bioinformatics. 10.1016/j.gpb.2022.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Auber R, Estep G, and Wisecaver J (2022). Wisecaver Lab algal RNA extraction protocol using Ambion TRI Reagent. protocols.io. https://www.protocols.io/view/wisecaver-lab-algal-rna-extraction-protocol-using-bv3hn8j6.
- 101.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hoff K, Lomsadze A, Borodovsky M, and Stanke M (2019). Whole-genome annotation with BRAKER. Methods Mol. Biol. Clifton NJ 1962, 65–95. 10.1007/978-1-4939-9173-0_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brůna T, Hoff KJ, Lomsadze A, Stanke M, and Borodovsky M (2021). BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinforma. 3, lqaa108. 10.1093/nargab/lqaa108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stanke M, Keller O, Gunduz I, Hayes A, Waack S, and Morgenstern B (2006). AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Benson G (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fulnecková J, Sevcíková T, Fajkus J, Lukesová A, Lukes M, Vlcek C, Lang BF, Kim E, Eliás M, and Sykorová E (2013). A broad phylogenetic survey unveils the diversity and evolution of telomeres in eukaryotes. Genome Biol. Evol. 5, 468–483. 10.1093/gbe/evt019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, and Zdobnov EM (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 108.Manni M, Berkeley MR, Seppey M, Simão FA, and Zdobnov EM (2021). BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654. 10.1093/molbev/msab199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tang H, Bowers JE, Wang X, Ming R, Alam M, and Paterson AH (2008). Synteny and collinearity in plant genomes. Science 320, 486–488. 10.1126/science.1153917. [DOI] [PubMed] [Google Scholar]
- 110.Haug-Baltzell A, Stephens SA, Davey S, Scheidegger CE, and Lyons E (2017). SynMap2 and SynMap3D: web-based whole-genome synteny browsers. Bioinforma. Oxf. Engl. 33, 2197–2198. 10.1093/bioinformatics/btx144. [DOI] [PubMed] [Google Scholar]
- 111.Warren RL (2018). Visualizing genome synteny with xmatchview. J. Open Source Softw. 3, 497. 10.21105/joss.00497. [DOI] [Google Scholar]
- 112.Shimoyama Y (2022). pyGenomeViz: A genome visualization python package for comparative genomics.
- 113.Emms DM, and Kelly S (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16, 157. 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Katoh K, and Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sela I, Ashkenazy H, Katoh K, and Pupko T (2015). GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 43, W7–W14. 10.1093/nar/gkv318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Capella-Gutierrez S, Silla-Martinez JM, and Gabaldon T (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Nguyen LT, Schmidt HA, Von Haeseler A, and Minh BQ (2015). IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, and Jermiin LS (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen K, Durand D, and Farach-Colton M (2000). NOTUNG: a program for dating gene duplications and optimizing gene family trees. J. Comput. Biol. J. Comput. Mol. Cell Biol. 7, 429–447. 10.1089/106652700750050871. [DOI] [PubMed] [Google Scholar]
- 120.Stolzer M, Lai H, Xu M, Sathaye D, Vernot B, and Durand D (2012). Inferring duplications, losses, transfers and incomplete lineage sorting with nonbinary species trees. Bioinformatics 28, i409–i415. 10.1093/bioinformatics/bts386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li W-H (1993). Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J. Mol. Evol. 36, 96–99. 10.1007/BF02407308. [DOI] [PubMed] [Google Scholar]
- 122.Pamilo P, and Bianchi NO (1993). Evolution of the Zfx and Zfy genes: rates and interdependence between the genes. Mol. Biol. Evol. 10, 271–281. 10.1093/oxfordjournals.molbev.a040003. [DOI] [PubMed] [Google Scholar]
- 123.Yang Z (2007). PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 124.Minh BQ, Schmidt HA, Chernomor O, Schrempf Dominik, Woodhams MD, von Haeseler A, and Lanfear R (2020). IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang C, Rabiee M, Sayyari E, and Mirarab S (2018). ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinformatics 19, 153. 10.1186/s12859-018-2129-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Naser-Khdour S, Minh BQ, and Lanfear R (2021). Assessing confidence in root placement on phylogenies: An empirical study using nonreversible models for mammals. Syst. Biol, syab067. 10.1093/sysbio/syab067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Minh BQ, Hahn MW, Lanfear R. 2020. New Methods to Calculate Concordance Factors for Phylogenomic Datasets. Mol. Biol. Evol. 37:2727–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Talevich E, Invergo BM, Cock PJ, and Chapman BA (2012). Bio.Phylo: A unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython. BMC Bioinformatics 13, 209. 10.1186/1471-2105-13-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Krzywinski M, Schein J, Birol İ, Connors J, Gascoyne R, Horsman D, Jones SJ, and Marra MA (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kokot M, Długosz M, and Deorowicz S (2017). KMC 3: counting and manipulating k-mer statistics. Bioinformatics 33, 2759–2761. 10.1093/bioinformatics/btx304. [DOI] [PubMed] [Google Scholar]
- 132.Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, and Ogata H (2020). KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 36, 2251–2252. 10.1093/bioinformatics/btz859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Laetsch DR, and Blaxter ML (2017). KinFin: Software for Taxon-Aware Analysis of Clustered Protein Sequences. G3 Genes Genomes Genet. 7, 3349–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, et al. (2020). SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17, 261–272. 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Seabold S, and Perktold J (2010). Statsmodels: Econometric and Statistical Modeling with Python. Proc. 9th Python Sci. Conf., 92–96. 10.25080/Majora-92bf1922-011. [DOI] [Google Scholar]
- 137.Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 57, 289–300. 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 138.Gladyshev EA, Meselson M, and Arkhipova IR (2008). Massive horizontal gene transfer in bdelloid rotifers. Science 320, 1210–1213. [DOI] [PubMed] [Google Scholar]
- 139.Hehenberger E, Gast RJ, and Keeling PJ (2019). A kleptoplastidic dinoflagellate and the tipping point between transient and fully integrated plastid endosymbiosis. Proc. Natl. Acad. Sci. 116, 17934–17942. 10.1073/pnas.1910121116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Soanes D, and Richards TA (2014). Horizontal gene transfer in eukaryotic plant pathogens. Annu. Rev. Phytopathol. 52, 583–614. [DOI] [PubMed] [Google Scholar]
- 141.Eddy SR (2009). A new generation of homology search tools based on probabilistic inference. Genome Inf. 23, 205–211. [PubMed] [Google Scholar]
- 142.Paradis E, Claude J, and Strimmer K (2004). APE: analyses of phylogenetics and evolution in R language. Bioinforma. Oxf. Engl. 20, 289–290. [DOI] [PubMed] [Google Scholar]
- 143.Schliep KP (2011). phangorn: phylogenetic analysis in R. Bioinforma. Oxf. Engl. 27, 592–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Letunic I, and Bork P (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47, W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 146.Soneson C, Love MI, and Robinson MD (2016). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research. 10.12688/f1000research.7563.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of Prymnesium parvum strains, genome assemblies, gene models, and genome sizes. Related to Figure 1 and Figure 4.
Summary of strains and their respective prymnesin types, sources, culturing conditions are provided in rows 2-8. Cultures were kept at 20 °C using a 12:12 light dark cycle and irradiance of 40-200 μmol photos m-2 s-1. N50 is defined as the sequence length of the shortest contig at 50% of the total genome length. L50 is defined as count of smallest number of contigs whose length sum makes up half of genome size. CMUK = coverage of maximal unique k-mers; larger CMUKs indicative of smaller haploid genomes and vice versa
Table S2. Coordinates of each predicted telomeric repeat for 12B1 and UTEX 2797 assemblies. Related to Figure 1.
Relative position of repeat on either the beginning (1-) or end (2-) of scaffold terminus is also denoted, as well as the distance from this terminus.
Table S3. Summary of BUSCO analysis performed on haptophyte assemblies. Related to Figure 1.
Ten BUSCO genes were missing in every haptophyte assembly investigated: 1030907at2759, 1094121at2759, 1304061at2759, 1364586at2759, 1442062at2759, 261419at2759, 270107at2759, 331411at2759, 453044at2759, 975158at2759.
Table S4. GRAMPA gene tree-Species tree reconciliation. Related to Figure 3.
ST = Single-labeled starting tree, MUL = Multi-labeled tree.
Table S5. Tests for enrichment of GO and KEGG gene annotation terms. Related to Figure 5.
Table S6. Eleven HGTs with strong phylogenetic support. Related to Figure 6.
Numbers in parentheses following gene IDs in UTEX 2797 indicate maximum length-scaled TPMs. Full matrix of gene expression can be downloaded from the project’s FigShare data repository.
Data Availability Statement
All genome assemblies, predicted CDS and protein sequences, multiple sequence alignments, tree files, and other related data files are available through FigShare (https://doi.org/10.6084/m9.figshare.21376500). Scripts for chromatogram production are available on Figshare https://doi.org/10.6084/m9.figshare.22267066.v1. All other scripts are available through GitHub (https://github.com/WisecaverLab/Pparvum_genome_diversity). Raw sequencing reads have been deposited in the Sequence Read Archive database under accession number PRJNA807128. LC-MS data from chemotyping is available in the EBI MetaboLights database under accession MTBLS5893.