

Abstract
Renewable CH4 produced from electrocatalytic CO2 reduction is viewed as a sustainable and versatile energy carrier, compatible with existing infrastructure. However, conventional alkaline and neutral CO2-to-CH4 systems suffer CO2 loss to carbonates, and recovering the lost CO2 requires input energy exceeding the heating value of the produced CH4. Here we pursue CH4-selective electrocatalysis in acidic conditions via a coordination method, stabilizing free Cu ions by bonding Cu with multidentate donor sites. We find that hexadentate donor sites in ethylenediaminetetraacetic acid enable the chelation of Cu ions, regulating Cu cluster size and forming Cu-N/O single sites that achieve high CH4 selectivity in acidic conditions. We report a CH4 Faradaic efficiency of 71% (at 100 mA cm−2) with <3% loss in total input CO2 that results in an overall energy intensity (254 GJ/tonne CH4), half that of existing electroproduction routes.
Subject terms: Electrocatalysis, Energy, Energy science and technology
Conventional alkaline and neutral CO2-to-CH4 systems suffer carbon loss, and recovering the lost carbon requires input energy exceeding the heating value of CH4. Here, the authors report a chelating strategy to obtain Cu-N/O single sites decorated Cu clusters, which enables energy- and carbon-efficient CH4 electroproduction in an acidic system.
Introduction
Renewable fuels are a critical component of global net-zero emission scenarios and offer high-density long-term energy storage. CO2 electrochemical reduction (CO2R) provides a decarbonized path to a variety of chemicals and fuels when powered by renewable electricity1. Of the various CO2R products (mainly carbon monoxide, methane, ethylene, ethanol and n-propanol)2, methane (CH4) has the highest energy density of 55.5 GJ/tonne3, and is a key input for hard-to-decarbonize industries. Renewable synthetic CO2-derived CH4 avoids emissions associated with the extraction of fossil-CH4 (natural gas, NG) and does not add to the natural carbon cycle. As a result, CH4 produced from captured CO2 and renewable electricity could provide a pathway to decarbonize existing NG supplies ( > 30% of the world’s fossil energy consumption4,5), compatible with vast NG storage and distribution infrastructure6–12.
Present day CO2R catalysts have achieved Faradaic efficiencies (FEs) of 70-80% towards CH4 at practical current densities (>100 mA cm−2) in alkaline and neutral mediums13–20. However, these systems suffer from CO2 loss to (bi)carbonates, and regenerating the CO2 is costly13,14. In alkaline systems, CO2 reacts rapidly with excess hydroxides in the electrolytes, consuming 20-fold that reacted productively via CO2R21,22. Recovering the CO2 requires an energy input of 289 GJth/tonne CH4—over 5x the heating value of CH4 (higher heating value, HHV, 55.5 GJ/tonne, Fig. 1a, c; Supplementary Notes 1–3; Table S1). In neutral CO2R electrolyzers, CO2 loss to (bi)carbonates is 4-fold that converted to CH4. The CO2 converted to (bi)carbonates (Eq. 1–3), migrates across the anion-selective membrane, combines with the protons from the oxygen evolution reaction (OER) on the anode, reverts to CO2, and mixes with produced O223,24. We performed neutral-medium CO2R and found the anode tail gas to consist of 67 v/v % CO2 and 33 v/v % O2 (Supplementary Fig. 1). Separating CO2 from this stream costs 55-73 GJth/tonne CH4, exceeding the CH4 heating value (Fig. 1b, c; Supplementary Notes 4; Table S1). The loss of CO2 fundamentally limits the single-pass conversion (SPC, here defined as the fraction of input CO2 that is reduced to the target product) to <20% in alkaline and neutral electrolyzers21,25,26. Achieving high SPC to CH4 will requires carbon efficient systems with minimal CO2 loss27–29.
| 1 |
| 2 |
| 3 |
Fig. 1. Schematic illustration and techno-energy analysis of different systems.

Schematic figure of (a) alkaline flow-cell system and (b) neutral MEA system. AEM, anion exchange membrane. MEA, membrane electrode assembly. c Energy penalty of CO2 regeneration from carbonate and anodic CO2/O2 separation. d Scheme of the acidic microchanneled MEA system used in this work. e Comparison of electrolyzer-specific energy distribution in different systems. Details of the energy analysis are provided in Supplementary Notes 1–4.
Here, we demonstrate a CH4-producing membrane electrode assembly (MEA) system that operates in acidic conditions (Fig. 1d). Internal recapture and recycling of CO2, via an internally channeled bipolar membrane, eliminates the need for downstream CO2 regeneration or separation. To enhance CH4 selectivity and minimize hydrogen evolution reaction (HER, Supplementary Fig. 2) in this acidic system, we pursued an in-situ multidentate coordinating strategy, using molecules with multi-teeth as decorations, to constrain Cu(II) from the copper phthalocyanine (CuPc) precursor and regulate Cu cluster size14,30. We screened a range of candidates with various multidentate sites as the decorations and found ethylenediaminetetraacetic acid (EDTA) chelated Cu ions stronger through hexadentate coordination compared with the lower multidentate coordinated molecules. With EDTA decoration, we obtained low-coordinated Cu clusters decorated by Cu-N/O single sites - that facilitate CO2R to produce CH4. Density functional theory (DFT) computations indicate that these N/O coordinated Cu decoration sites enhance CH4 selectivity by stabilizing the adsorption of *CHO and *O key intermediates. With this strategy we achieve a CH4 FE of 71% at a current density of 100 mA cm-2 and a CH4 energy efficiency (EE) of 21%. By eliminating CO2 loss, we achieve a single-pass CO2 conversion of 78%, 5× higher than neutral electrolyzers, and an energy intensity of 254 GJ/tonne CH4 (Fig. 1e). The produced CH4 has 50% the energy intensity of that produced in the best prior electrolyzers.
Results and discussion
Carbon-efficient CO2-to-CH4 system optimization
We integrated a cation exchange membrane (CEM) and an anion exchange membrane (AEM) combination in a zero-gap manner as applied previously to achieve high single pass conversion in the generation of multicarbon products31 (Supplementary Fig. 3). H2SO4 was employed as the anolyte, providing protons to regenerate CO2 within the MEA cell. We further incorporated various ionomers in the catalyst layer to tune the cathodic local microenvironment (local alkalinity, ion migration and CO2 mass transport)32–34. The operating conditions and binder materials were optimized for each case and PiperIon ionomer performed best, with a moderate CH4 FE of 25% and an H2 FE of 45% at a current density of 100 mA cm-2 (Supplementary Fig. 4).
Catalyst performance
To enhance the selectivity of CH4, we deployed the low-coordination Cu strategy that is selective for CO2 electrochemical methanation14,30. Low-coordinated Cu sites can be produced from the in-situ reduction of Cu(II)Pc precursors during CO2R and using conductive carbon nanoparticles (CNP) as modulators, confining the Cu cluster size14. Without the constraining effect of CNP, free Cu ions readily agglomerates into large Cu clusters, forming high-coordination number sites14,35 that shift the reaction from CO2 hydrogenation to C-C coupling14,17,36. CNP can sterically distribute the metallic Cu clusters and restrict the Cu agglomeration size, which is essential to preserve CO2 hydrogenation activity14. Varying the CuPc/CNP ratio from 5:1 to 1.5:1, CH4 FE increased from 21% to 46%. Further increasing CNP content elevated H2 FE (Supplementary Fig. 5). Within the initial hour of the electrolysis, we observed a rapid decay of CH4 FE (from 46% to 25%) accompanied by an increase in C2H4 FE (from 8% to 19%), which we attributed to the continuous leaching of Cu ions and agglomeration in this acid system (Supplementary Fig. 6)37,38.
To further increase CH4 selectivity, we designed a multidentate chelating strategy that captures and constrain free Cu ions14,39–41. We screened several typical molecules that enable bonding Cu ions through multidentate donor sites. EDTA presents a stronger chelating effect on Cu through hexadentate coordination compared with the lower multidentate coordinated complexes (Supplementary Fig. 7). We fabricated the molecule decorated CuPc/CNP composite catalysts by spray-coating the mixture onto the gas diffusion layers (GDLs). After the initial hour of electrolysis, the ethylenediamine (ED, bidentate coordinated with Cu) and ethylenediamine-N, N′-diacetic acid (EDDA, tetradentate coordinated with Cu) decorated samples showed lower ethylene FE (17% and 11% respectively) than the sample without decorations (19%). The CH4 FE was 32% for the ED decorated sample and 41% for EDDA decorated sample, slightly improved over the no-decoration case. For the EDTA decorated sample, CH4 FE remained >65% after the initial hour of electrolysis with minimal increase in C2H4 FE (5%, Supplementary Fig. 8). We attribute this improvement in CH4 production to the hexadentate coordinated sites of EDTA that more intensely chelate Cu ions than the other two complexes (Supplementary Fig. 9).
The EDTA loading was screened with a fixed CuPc/CNP ratio of 1.5:1 (Supplementary Fig. 10). At 100 mA cm-2, the EDTA/CuPc/CNP attained a CH4 FE of 71%, a 20% improvement over the CuPc/CNP case (Fig. 2a). The CH4 FE of > 60% was maintained over a wide current window from 50 to 200 mA cm-2 with full cell potentials <4.2 V. The CH4 FE remained constant during the initial hour (Supplementary Fig. 11), indicating the regulation of Cu ions by the multidentate chelation effect. The control sample EDTA/CNP showed only a trace CH4 FE of ~4% and an H2 FE of ~90% (Fig. 2b), indicating that in the absence of Cu sites, EDTA is not an active catalyst for CO2-to-CH4 conversion. Without CNP, EDTA-decorated CuPc (0.1 mg/cm2) showed a CH4 FE of 48.9% (Fig. 2c), 20% higher than the pristine CuPc electrode with the same loading (Supplementary Fig. 12). These results evidence the critical role of multidentate chelating effect of EDTA in enhancing CH4 FE. However, in the absence of CNP conductors and Cu modulators, the full cell voltage was high ( > 6 V at 200 mA cm-2, Fig. 2c). The CH4 selectivity also suffered without CNP regulators and production shifted to C2H4 FE (CH4:C2H4 shifted from 20:1 to 4:1). The FE of liquid products were quantified, and the total measured FE approached 100% at the same current density range (50 to 200 mA cm-2) in all three cases, within experimental error. (Supplementary Fig. 13). Control experiments were carried out under Ar conditions to rule out EDTA and CNP as the potential carbon sources in the production of carbon-based products. The exclusive H2 production under such conditions indicates that EDTA and CNP were not reactive carbon sources (Supplementary Fig. 14).
Fig. 2. Electrocatalytic performance of electrocatalysts in carbon-efficiency system.

Products distribution of (a) EDTA/CuPc/CNP, (b) EDTA/CNP and (c) EDTA/CuPc at current range from 50 to 200 mA cm−2. FE, Faradaic Efficiency. Values are means, and error bars indicate SD (n = 3 replicates). d Stability test of CO2 methanation during 5 h of electrolysis under the current density of 100 mA cm−2. e Single pass conversion of CO2 at different flow rates. The SPC results were obtained at a constant current density of 100 mA cm−2.SPC, CO2 single pass conversion efficiency.
We performed a durability test of EDTA/CuPc/CNP in the acidic MEA with 5-mM H2SO4 anolyte. The CH4 FE remained over 50% with a steady full cell potential of 3.6 V for 5 h (Fig. 2d). We compared the CO2R performances (Supplementary Fig. 15) and CO2 single pass conversion (SPC) in our acidic system with the conventional neutral (0.5 M KHCO3 anolyte) and alkaline systems (0.5 M KOH anolyte). With a total CO2R FE > 50%, the total CO2 SPC achieved 78%, 4-fold the theoretical maximum of 20% for neutral and alkaline systems (Fig. 2e, Supplementary Fig. 16). The SPC towards CH4 attained a record-high value of 51%, 3.6-fold higher that of neutral medium electrolyzers with the same catalyst (14%, Supplementary Fig. 17). In neutral/alkaline systems, the (bi)carbonates cross through the AEM, leading to the CO2 loss. The CEM in the acidic MEA provided a locally acidic domain for CO2 regeneration within the cell and thereby minimized CO2 loss ( < 3 v/v % CO2 detected in the anode tail gas, Supplementary Fig. 1) and achieved high CO2 single pass conversion. The CEM and the integrated microchannels do not add significant ohmic resistance to the overall system31, as indicated by the comparable voltage with the neutral system (Supplementary Fig. 15).
Surface characterizations of catalysts
To investigate the multidentate chelating effect and probe the mechanism behind the enhanced CH4 selectivity, we investigated the electronic state and coordination number of Cu at a current density of 100 mA cm-2 with in-situ X-ray absorption spectroscopy (XAS). The CuPc/CNP samples were analyzed, with and without EDTA, and metallic Cu, CuO, Na2[Cu(EDTA)] samples were taken as references. We found from the X-ray absorption near-edge structure (XANES) spectra that both with and without EDTA decoration the original Cu(II) peaks present at 8991 eV. These Cu(II) original peaks shifted to 8980 eV during CO2R, indicating the electronic state of Cu reduced from Cu(II) to the lower state of Cu(0) (Fig. 3a), as expected for Cu clusters were formed by CuPc reduction during CO2R14. The EDTA decorated CuPc/CNP sample showed a slightly higher energy position between 8980 ev and 8991 eV (compared to the bare CuPc/CNP sample decoration), indicating the preservation of oxidized states of Cu species during CO2R (Fig. 3a)42,43.
Fig. 3. Surface characterizations of catalysts.

a The in-situ XANES spectra of CuPc/CNP and EDTA/CuPc/CNP samples before and during CO2R. The metallic Cu was shown as reference. XANES, X-ray absorption near-edge structure. The in-situ EXAFS spectra of (b) CuPc/CNP and (c) EDTA/CuPc/CNP before and during CO2R. The metallic Cu, CuO, and Na2[Cu(EDTA)] samples are shown as references. EXAFS, extended X-ray absorption fine structure. The in-situ experiments were all performed at 100 mA cm−2, the current condition for the best CO2R performance. XPS Cu 2p spectra of (d) CuPc/CNP, and (e) EDTA/CuPc/CNP before CO2R. XPS Cu 2p spectra of (f) CuPc/CNP, and (g)EDTA/CuPc/CNP after CO2R. XPS, X-ray photoelectron spectra.
We then obtained the in-situ extended X-ray absorption fine structure (EXAFS) spectra to investigate the Cu coordination environments. The sample without decoration showed an increase in Cu-Cu peak in the initial hour, and a sharp drop in Cu-N/O peak (coordination number dropped from 3.8 to 0.6) during the CO2R process, indicating the Cu agglomeration (Fig. 3b and supplementary Fig. 18, 19, Table S2). This Cu agglomeration leads to decline of CH4 selectivity during CO2R (Supplementary Fig. 6)30. With EDTA decoration, the Cu-Cu peak increased and Cu-N/O peak declined in the initial 30 min, then remained stable for the rest of the process (Supplementary Fig. 20), demonstrating the regulation of Cu ions via the chelating effect (Fig. 3c). We obtained small Cu clusters decorated with additional Cu-N/O sites. The fitted Cu-Cu bond coordination number of the EDTA decorated sample is smaller (5.4) than that of pristine CuPc/CNP (6.7), demonstrating the multidentate chelation constraining effect on Cu cluster size (Supplementary Table S2) The fitted Cu-N/O coordination number of the EDTA decorated sample was larger (2.5, Supplementary Fig. 21, Table S2) than the sample without EDTA decoration (0.6). We attributed the enhanced and maintained CH4 FE (Supplementary Fig. 11) to the EDTA chelating effect on Cu ions -that confined Cu cluster size and generated additional Cu-N/O active sites13,35.
To further investigate the catalyst surface structures, we performed X-ray photoelectron spectra (XPS) and scanning electron microscope (SEM) before and after CO2 electrolysis. All samples were processed in a glove box after CO2R to protect them from being oxidized in the air. The Cu 2p XPS spectra of both samples before CO2R, with and without EDTA decoration, showed the peaks at 955.0 eV (2p/1/2) and 935.1 eV (2p3/2), which are associated with the Cu(II) state (Fig. 3d, e). For the post-electrolysis samples, the deconvoluted Cu 2p spectra show Cu(0)/(I) peaks located at 944.4 (2p/1/2) and 932.9 eV (2p3/2), further confirming that Cu(0) sites were formed in the CO2R process (Fig. 3f, g). However, the EDTA decorated sample presented a smaller Cu(0)/(I) peak ratio than the sample without decoration. We quantified the Cu(0)/(I):Cu(II) peak ratio by integrating the peak area for both Cu(0)/(I) and Cu(II). The EDTA decorated sample showed lower Cu(0)/(I):Cu(II) peak ratios compared to the sample without decoration (Supplementary Table S3), indicating that multidentate chelating decoration is essential to regulation of Cu ions—a finding consistent with the in-situ XAS results. The deconvoluted N 1 s spectra also showed that the Cu-N bond was decomposed in the post-electrolysis states of both samples (Supplementary Fig. 22, 23). To further prove the multidentate chelating effect on the stabilization of the Cu-N bond, we normalized the Cu-N bond with reference to inert pyridinic N to calculate the Cu-N loss ratio (Supplementary Table S4). The sample with EDTA decoration demonstrated a lower Cu-N loss (35%) compared to the one without decoration (57%), consistent with the in-situ XAS results, further confirming the formation and preservation of Cu-N sites facilitated by multidentate coordination of EDTA. We then assessed catalyst structure change before and after CO2R using SEM (Supplementary Fig. 24, 25). The sample without any decoration presented a needle-like structure attributed to the unconstrained Cu deposition when reducing from CuPc during CO2R. In contrast, the EDTA decorated sample was composed of uniformly distributed particles, attributed here to effect of the multidentate coordination in preventing non-regulated Cu deposition during CO2R.
DFT calculations
To further probe the effect of the decorated Cu-N/O sites on boosting the CH4 selectivity, we performed DFT calculations on a series of [C10H14+/–nCuN2O8]n+/– (n = 0, 1, or 2) complex structures. We presented the free energy diagram of the lowest-energy pathway for CO2-to-CH4 on Cu active site in the complex structures of [C10H14CuN2O8] and [C10H15CuN2O8]+ (Fig. 4a) and the corresponding atomic configurations of each elementary step (Fig. 4b). The CH4 production initiates from thermodynamically inhibited adsorption and protonation of CO2 on [C10H14CuN2O8] (Fig. 4a), similar to the results on Cu(111) facet44, with free energy changes of 0.28 eV and 0.63 eV, respectively. The potential-determining step (PDS) is the protonation of *OCH3 to *O + CH4, exhibiting a free energy change of 0.80 eV. We noted that the protonation/deprotonation between [C10H14CuN2O8] and [C10H15CuN2O8]+ (Fig. 4a, inset) is a thermal-neutral step (with a free energy of 0.08 eV for Step 5 and −0.02 eV for Step 9 to Step 0). Such a configuration of [C10H14CuN2O8] and [C10H15CuN2O8]+ can stabilize the adsorption of *CHO and *O, which decreases the free energy needed for the *CO protonation to *CHO and *OCH3 protonation to *O. The PDS on Cu(111) facet is the protonation of *CO species (i.e., *CO-to-*CHO) with a free energy change of 0.85 eV. Compared to Cu(111) facet, we found that [C10H14CuN2O8] enables CO2 adsorption and exhibits a comparable free energy change for the PDS, indicating that [C10H14CuN2O8] offers extra active sites for CO2-to-CH4 process. The formation of [C10H14CuN2O8] sites also prevents the agglomeration of Cu clusters, lowers the *CO coverage on the Cu(111) facet, and inhibits C-C coupling – collectively enhancing CH4 selectivity.
Fig. 4. DFT calculations on CO2 protonation to CH4.
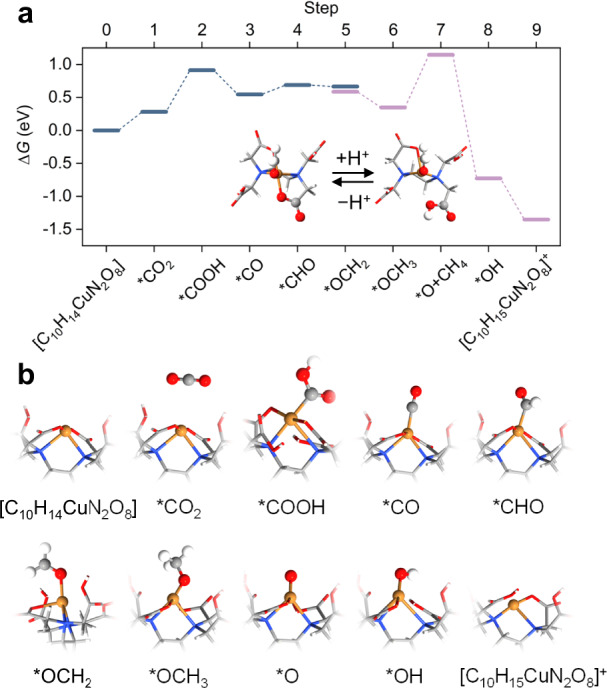
a Free energy diagram for CH4 production on Cu active site in the complex structures of [C10H14CuN2O8] and [C10H15CuN2O8]+. The inserted figures represent the protonation/deprotonation between [C10H14CuN2O8] and [C10H15CuN2O8]+. b Corresponding atomic configurations for each elementary step, including [C10H14CuN2O8], *CO2, *COOH, *CO, *CHO, *OCH2, *OCH3, *O, *OH, and [C10H15CuN2O8]+. Orange, red, gray, white, blue sphere represent Cu, O, C, H, N atoms, respectively.
In summary, this work presented a CH4-selective single-site decorated Cu strategy compatible with a carbon-efficient system. Employing acidic conditions in a structured MEA electrolyzer eliminated CO2 loss and the associated energy cost of CO2 regeneration. We developed an multidentate chelating strategy to obtain Cu-N/O single sites decorated low-coordinated Cu that enables 71% FE of CH4 in this carbon-efficient system. We obtained a full cell potential of 3.6 V at 100 mA cm-2 and a record-high SPC towards the CH4 of 51% and an energy efficiency for CH4 production of 21%. By avoiding the additional energy consumption of CO2 regeneration and improving the energy efficiency of CO2-to-CH4, renewable CO2-derived CH4 is produced at an overall energy cost of ~254 GJ/tonne—50% less than the conventional alkaline and neutral approaches. This study demonstrates a strategy to simultaneously achieve carbon- and energy-efficient CO2 methanation.
Methods
Preparation of electrodes
The electrodes were prepared by air-bushing the CuPc/CNP or EDTA/CuPc/CNP inks onto hydrophobic carbon papers. The spray density was kept at 0.1 mL cm-2. The CuPc/CNP catalyst inks were prepared by dispersing 64 mg CuPc ( > 99.5%, Sigma-Aldrich) and 42 mg CNP (Vulcan XC 72, Fuel Cell Store) mixture in 30 mL methanol (>99.5%, Fisher chemical) with 150 µL 5 wt% PiperIon (Fuel Cell Store) anion exchange ionomer as the binder. We obtained different CuPc/CNP ratios by varying the CNP quantity in the mixture, and the CuPc/CNP ratio was ranged from 1:4 to 5:1. The molecule (ED, EDDA and EDTA) decorated CuPc/CNP inks were prepared by adding 16 mg decoration molecule (ED > 99.5% Sigma-Aldrich; EDDA > 98% Sigma-Aldrich; EDTA > 99.5%, Sigma-Aldrich) into 2 mL fully dispersed CuPc/CNP solutions, followed by sonicating for 24 h. The EDTA and CuPc ratio were optimized from 16:1 to 2:1 by tuning the EDTA weight in the CuPc/CNP dispersed solution.
Acidic MEA configuration
The cathodes for the acidic MEA were based on either CuPc/CNP or EDTA decorated CuPc/CNP electrodes with the catalyst loading of 0.15 mg cm-2. The anodes were based on Ti felt (0.3 mm thickness) loaded with 1 mg cm-2 IrO2. 0.005 M H2SO4 was used as anolyte circulated with a flow rate of 5 mL min-1. A microchanneled cation exchange membrane (Nafion 117, Fuel Cell Store) facing the anodic side was used for transporting proton and locally regenerated CO2. The channeled CEM was prepared by hot embossing under a temperature of 220 °F and a pressure of 1.25 MPa for 5min31. An anion exchange membrane (Sustainion X37-50 Grade RT, Dioxide Materials, USA) facing the cathodic side was used to facilitate CO2R activation. DI water was circulated in the middle channel layer at a constant flow rate of 0.5 mL min-2 using a syringe pump.
Electroreduction of CO2
The CO2R was carried out at constant current densities ranging from 50 to 200 mA cm-2. The gas products were analyzed in 1 mL volume through a gas chromatograph (GC, Perkin Elmer Clarus 590) equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID). The Faradaic efficiency was calculated via the following equation:
| 4 |
where z represents the number of electrons required to produce the product, F represents the Faraday constant, P represents the atmosphere pressure, R represents the ideal gas constant, T represents the temperature, v represents the gas flow rate at the gas, and I represents the total current.
The full cell voltage was obtained during CO2R and the energy efficiency was calculated using the following equation:
| 5 |
where is the thermodynamic potential, is the full cell potential voltage during the experiments, and FE is the Faradaic efficiency of each product.
The single-pass CO2 conversion efficiency (SPC) of CO2 was calculated using the following equation:45
| 6 |
where j represents the partial current density of a specific product, z represents the number of electrons required for the specific product, F represents the Faraday constant, Vm represents the molar volume.
Characterizations of catalysts
Cu catalyst electronic state and the local coordination environment were investigated by in-situ XAS measurements, which were performed at beamline 9BM of the Advanced Photon Source (APS, Argonne National Laboratory, Lemont, Illinois, United States) and the silicon drift detector at the 17 C beamline of National Synchrotron Radiation Research Center (NSRRC, Hsinchu, Taiwan)18. The ex-situ XPS spectra were obtained through a Thermo Scientific K-Alpha spectrophotometer with the monochromated Al Kα X-ray radiation source. The ex-situ samples were treated and stored strictly under the N2 condition to reduce the possible oxidation of Cu. SEM characterizations were conducted with a high-resolution scanning electron microscope (HR-SEM, Hitachi S-5200).
DFT calculations
First-principles calculations based on DFT46,47 were performed using the projector-augmented wave method48,49 as implemented in the Vienna ab initio simulation package (VASP). Electron exchange and correlation terms were treated50 by generalized gradient approximation which is parametrized by Perdew-Burke-Ernzerhof with long-range dispersion correction derived from the DFT-D2 method of Grimme51. [C10H14+/–nCuN2O8]n+/– (n = 0, 1, or 2) was modeled in a supercell with a vacuum thickness >20 Å in each direction. Cut-off energy was set to 450 eV and the Brillouin zone was sampled by gamma-centered 1 × 1 × 1 k-points generated by the Monkhorst-Pack scheme52. Structural optimization was considered to reach the convergence when the residual force on each ion was <0.01 eV Å−1 and the energy difference between the two iterations was <10−5 eV per atom. A Fermi-level smearing width of 0.05 eV was used for the calculations of adsorbates, whereas 0.01 eV for non-adsorbed species, to improve the convergence.
Supplementary information
Acknowledgements
The authors acknowledge support from the Natural Sciences and Engineering Research Council (NSERC) of Canada, Natural Resources Canada—Clean Growth Program, and the Natural Gas Innovation Fund (NGIF). The infrastructure provided through the Canada Foundation for Innovation and the Ontario Research Fund supported the work. R.K.M. thanks NSERC, Hatch, and the Government of Ontario for their support through graduate scholarships. P.O. thanks the Climate Positive Energy for its support through Rising Stars in Clean Energy Postdoctoral Fellowship. Synchrotron experiments were carried out at SXRMB beamline at the Canadian Light Source (CLS). S.F.H. thanks for the supports from the National Science and Technology Council, Taiwan (Contract No. NSTC 111−2628-M-A49-007) and the support from the Yushan Young Scholar Program, Ministry of Education, Taiwan. W.N. acknowledges the financial support of the Postdoc. Mobility Fellowship from the Swiss National Science Foundation (SNSF) (No. P500PN_202906). We acknowledge the help from Dr. Qunfeng Xiao, Dr. Mohsen Shakouri, and Dr. Alisa Paterson for their kind technical assistance.
Author contributions
D.S. and E.H.S. supervised the project. M.F. and Y.X. designed and carried out all the experiments. M.F. designed the catalysts and performed catalysts characterizations. R.K.M., A.O., and J.W. carried out the TEA analysis. P.O. carried out the DFT simulation. M.F., R.K.M, and K.X. analyzed the experimental data and prepared the manuscript. R.K.M. performed the SPC experiments. Y.C. synthesized the catalysts and Y.Z. carried out SEM characterizations, Z.-Y.L., T.-J.L., W.N., J.A., and E.S. carried out XAS characterization. S.-F.H., J.L., and X.W. analysed the XAS data. J.E.H., C.P.O., S.L., and Y.C.X. performed product analysis. All authors discussed the results and assisted during manuscript preparation.
Peer review
Peer review information
Nature Communications thanks the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
Data that support the findings of this study can be found in the article and the Supplementary information. Source data are available from the corresponding author upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Mengyang Fan, Rui Kai Miao, Pengfei Ou, and Yi Xu.
Contributor Information
Edward H. Sargent, Email: ted.sargent@utoronto.ca
David Sinton, Email: sinton@mie.utoronto.ca.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-023-38935-2.
References
- 1.Artz J, et al. Sustainable conversion of carbon dioxide: an integrated review of catalysis and life cycle assessment. Chem. Rev. 2018;118:434–504. doi: 10.1021/acs.chemrev.7b00435. [DOI] [PubMed] [Google Scholar]
- 2.Shin H, Hansen KU, Jiao F. Techno-economic assessment of low-temperature carbon dioxide electrolysis. Nat. Sustain. 2021;4:911–919. doi: 10.1038/s41893-021-00739-x. [DOI] [Google Scholar]
- 3.Engineering ToolBox. Fuel Gases—Heating Values. https://www.engineeringtoolbox.com/heating-values-fuel-gases-d_823.html (2005).
- 4.IEA (2021). Key World Energy Statistics, IEA, Paris. https://www.iea.org/reports/key-world-energy-statistics-2021 (2021).
- 5.Litvinenko V. The role of hydrocarbons in the global energy agenda: The focus on liquefied natural gas. Resources. 2020;9:59. doi: 10.3390/resources9050059. [DOI] [Google Scholar]
- 6.van der Zwaan, B., Detz, R., Meulendijks, N. & Buskens, P. Renewable natural gas as climate-neutral energy carrier? Fuel311, 122547 (2022).
- 7.Likhanov VA, Lopatin OP. Use of natural gas, methanol, and ethanol fuel emulsions as environmentally friendly energy carriers for mobile heat power plants. Therm. Eng. 2017;64:935–944. doi: 10.1134/S0040601517120060. [DOI] [Google Scholar]
- 8.Danieli P, et al. The potential of the natural gas grid to accommodate hydrogen as an energy vector in transition towards a fully renewable energy system. Appl. Energy. 2022;313:118843. doi: 10.1016/j.apenergy.2022.118843. [DOI] [Google Scholar]
- 9.Yao JG, Bui M, Dowell NM. Grid-scale energy storage with net-zero emissions: comparing the options. Sustain. Energy Fuels. 2019;3:3147–3162. doi: 10.1039/C9SE00689C. [DOI] [Google Scholar]
- 10.Gorre, J., Ortloff, F. & van Leeuwen, C. Production costs for synthetic methane in 2030 and 2050 of an optimized Power-to-Gas plant with intermediate hydrogen storage. Appl. Energy253, 113594 (2019).
- 11.Ghaib K, Ben-Fares F-Z. Power-to-methane: A state-of-the-art review. Renew. Sustain. Energy Rev. 2018;81:433–446. doi: 10.1016/j.rser.2017.08.004. [DOI] [Google Scholar]
- 12.Fernández-González, J., Rumayor, M., Domínguez-Ramos, A. & Irabien, Á. CO2 electroreduction: Sustainability analysis of the renewable synthetic natural gas. Int. J. Greenh. Gas Control. 114, 103549 (2022).
- 13.Zhang L, et al. Enhanced cuprophilic interactions in crystalline catalysts facilitate the highly selective electroreduction of CO2 to CH4. J. Am. Chem. Soc. 2021;143:3808–3816. doi: 10.1021/jacs.0c11450. [DOI] [PubMed] [Google Scholar]
- 14.Xu Y, et al. Low coordination number copper catalysts for electrochemical CO2 methanation in a membrane electrode assembly. Nat. Commun. 2021;12:2932. doi: 10.1038/s41467-021-23065-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang, Y. R. et al. Implanting numerous hydrogen-bonding networks in a Cu-porphyrin-based nanosheet to boost CH4 selectivity in neutral-media CO2 electroreduction. Angew. Chem. Int. Ed. Engl. 60, 21952–21958 (2021). [DOI] [PubMed]
- 16.Liu Y, et al. The synthesis of hexaazatrinaphthylene-based 2D conjugated copper metal-organic framework for highly selective and stable electroreduction of CO2 to methane. Angew. Chem. Int Ed. Engl. 2021;60:16409–16415. doi: 10.1002/anie.202105966. [DOI] [PubMed] [Google Scholar]
- 17.Li Y, et al. Promoting CO2 methanation via ligand-stabilized metal oxide clusters as hydrogen-donating motifs. Nat. Commun. 2020;11:6190. doi: 10.1038/s41467-020-20004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hung SF, et al. A metal-supported single-atom catalytic site enables carbon dioxide hydrogenation. Nat. Commun. 2022;13:819. doi: 10.1038/s41467-022-28456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen S, et al. Engineering water molecules activation center on multisite electrocatalysts for enhanced CO2 methanation. J. Am. Chem. Soc. 2022;144:12807–12815. doi: 10.1021/jacs.2c03875. [DOI] [PubMed] [Google Scholar]
- 20.Han Z, et al. Steering surface reconstruction of copper with electrolyte additives for CO2 electroreduction. Nat. Commun. 2022;13:3158. doi: 10.1038/s41467-022-30819-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma M, et al. Insights into the carbon balance for CO2 electroreduction on Cu using gas diffusion electrode reactor designs. Energy Environ. Sci. 2020;13:977–985. doi: 10.1039/D0EE00047G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sisler J, et al. Ethylene electrosynthesis: A comparative techno-economic analysis of alkaline vs membrane electrode assembly vs CO2–CO–C2H4 tandems. ACS Energy Lett. 2021;6:997–1002. doi: 10.1021/acsenergylett.0c02633. [DOI] [Google Scholar]
- 23.Lin M, Han L, Singh MR, Xiang C. An experimental- and simulation-based evaluation of the CO2 utilization efficiency of aqueous-based electrochemical CO2 reduction reactors with ion-selective membranes. ACS Appl. Energy Mater. 2019;2:5843–5850. doi: 10.1021/acsaem.9b00986. [DOI] [Google Scholar]
- 24.Ma M, Kim S, Chorkendorff I, Seger B. Role of ion-selective membranes in the carbon balance for CO2 electroreduction via gas diffusion electrode reactor designs. Chem. Sci. 2020;11:8854–8861. doi: 10.1039/D0SC03047C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dinh C-T, Li YC, Sargent EH. Boosting the single-pass conversion for renewable chemical electrosynthesis. Joule. 2019;3:13–15. doi: 10.1016/j.joule.2018.10.021. [DOI] [Google Scholar]
- 26.Jeng E, Jiao F. Investigation of CO2 single-pass conversion in a flow electrolyzer. React. Chem. Eng. 2020;5:1768–1775. doi: 10.1039/D0RE00261E. [DOI] [Google Scholar]
- 27.O’Brien CP, et al. Single pass CO2 conversion exceeding 85% in the electrosynthesis of multicarbon products via local CO2 regeneration. ACS Energy Lett. 2021;6:2952–2959. doi: 10.1021/acsenergylett.1c01122. [DOI] [Google Scholar]
- 28.Kim JYT, et al. Recovering carbon losses in CO2 electrolysis using a solid electrolyte reactor. Nat. Catal. 2022;5:288–299. doi: 10.1038/s41929-022-00763-w. [DOI] [Google Scholar]
- 29.Ozden A, et al. Carbon-efficient carbon dioxide electrolysers. Nat. Sustain. 2022;5:563–573. doi: 10.1038/s41893-022-00879-8. [DOI] [Google Scholar]
- 30.Weng Z, et al. Active sites of copper-complex catalytic materials for electrochemical carbon dioxide reduction. Nat. Commun. 2018;9:415. doi: 10.1038/s41467-018-02819-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu Y, et al. A microchanneled solid electrolyte for carbon-efficient CO2 electrolysis. Joule. 2022;6:1–11. doi: 10.1016/j.joule.2022.04.023. [DOI] [Google Scholar]
- 32.Mayerhöfer B, et al. On the effect of anion exchange ionomer binders in bipolar electrode membrane interface water electrolysis. J. Mater. Chem. A. 2021;9:14285–14295. doi: 10.1039/D1TA00747E. [DOI] [Google Scholar]
- 33.Cao X, Novitski D, Holdcroft S. Visualization of hydroxide ion formation upon electrolytic water splitting in an anion exchange membrane. ACS Mater. Lett. 2019;1:362–366. doi: 10.1021/acsmaterialslett.9b00195. [DOI] [Google Scholar]
- 34.Kim C, et al. Tailored catalyst microenvironments for CO2 electroreduction to multicarbon products on copper using bilayer ionomer coatings. Nat. Energy. 2021;6:1026–1034. doi: 10.1038/s41560-021-00920-8. [DOI] [Google Scholar]
- 35.Cai Y, et al. Insights on forming N,O-coordinated Cu single-atom catalysts for electrochemical reduction CO2 to methane. Nat. Commun. 2021;12:586. doi: 10.1038/s41467-020-20769-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou X, et al. Stabilizing Cu2+ ions by solid solutions to promote CO2 electroreduction to methane. J. Am. Chem. Soc. 2022;144:2079–2084. doi: 10.1021/jacs.1c12212. [DOI] [PubMed] [Google Scholar]
- 37.Boutin E, Salame A, Robert M. Confined molecular catalysts provide an alternative interpretation to the electrochemically reversible demetallation of copper complexes. Nat. Commun. 2022;13:4190. doi: 10.1038/s41467-022-31661-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar P, et al. Cobalt phthalocyanine immobilized on graphene oxide: an efficient visible-active catalyst for the photoreduction of carbon dioxide. Chemistry. 2014;20:6154–6161. doi: 10.1002/chem.201304189. [DOI] [PubMed] [Google Scholar]
- 39.Chen S, et al. MOF encapsulating N-heterocyclic carbene-ligated copper single-atom site catalyst towards efficient methane electrosynthesis. Angew. Chem. Int. Ed. Engl. 2022;61:e202114450. doi: 10.1002/anie.202114450. [DOI] [PubMed] [Google Scholar]
- 40.Yang, J. et al. Beyond single-atom catalysts: exploration of Cu dimer and trimer for CO2 reduction to methane. Appl. Catal. A-Gen. 642, 118708 (2022).
- 41.Jovanov ZP, Ferreira de Araujo J, Li S, Strasser P. Catalyst preoxidation and EDTA electrolyte additive remedy activity and selectivity declines during electrochemical CO2 reduction. J. Phys. Chem. C. 2019;123:2165–2174. doi: 10.1021/acs.jpcc.8b08794. [DOI] [Google Scholar]
- 42.Frank P, Benfatto M, Hedman B, Hodgson KO. The X-ray absorption spectroscopic model of the copper(II) Imidazole complex ion in liquid aqueous solution: a strongly solvated square pyramid. Inorg. Chem. 2012;51:2086–2096. doi: 10.1021/ic2017819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaur A, Shrivastava BD, Joshi SKJ. Copper K-edge XANES of Cu(I) and Cu(II) oxide mixtures. Phys. Conf. Ser. 2009;190:012084. doi: 10.1088/1742-6596/190/1/012084. [DOI] [Google Scholar]
- 44.Zhou B, et al. Highly efficient binary copper-iron catalyst for photoelectrochemical carbon dioxide reduction toward methane. Proc. Natl Acad. Sci. USA. 2020;117:1330–1338. doi: 10.1073/pnas.1911159117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang JE, et al. CO2 electrolysis to multicarbon products in strong acid. Science. 2021;372:1074–1078. doi: 10.1126/science.abg6582. [DOI] [PubMed] [Google Scholar]
- 46.Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965;140:A1133–A1138. doi: 10.1103/PhysRev.140.A1133. [DOI] [Google Scholar]
- 47.Kresse G. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B. 1996;54:11169–11186. doi: 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- 48.Blochl PE. Projector augmented-wave method. Phys. Rev. B. 1994;50:17953–17979. doi: 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- 49.Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B59, 1758–1775 (1999).
- 50.Wellendorff J, et al. Density functionals for surface science: exchange-correlation model development with Bayesian error estimation. Phy. Rev. B. 2012;85:235149. doi: 10.1103/PhysRevB.85.235149. [DOI] [Google Scholar]
- 51.Grimme S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006;27:1787–1799. doi: 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- 52.Monkhorst HJ, Pack JD. Special points for Brillouin-zone integrations. Phys. Rev. B. 1976;13:5188–5192. doi: 10.1103/PhysRevB.13.5188. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data that support the findings of this study can be found in the article and the Supplementary information. Source data are available from the corresponding author upon request.