

Abstract
Type 2 diabetes mellitus (T2DM) represents one of the fastest growing epidemic metabolic disorders worldwide and is a strong contributor for a broad range of comorbidities, including vascular, visual, neurological, kidney, and liver diseases. Moreover, recent data suggest a mutual interplay between T2DM and Corona Virus Disease 2019 (COVID‐19). T2DM is characterized by insulin resistance (IR) and pancreatic β cell dysfunction. Pioneering discoveries throughout the past few decades have established notable links between signaling pathways and T2DM pathogenesis and therapy. Importantly, a number of signaling pathways substantially control the advancement of core pathological changes in T2DM, including IR and β cell dysfunction, as well as additional pathogenic disturbances. Accordingly, an improved understanding of these signaling pathways sheds light on tractable targets and strategies for developing and repurposing critical therapies to treat T2DM and its complications. In this review, we provide a brief overview of the history of T2DM and signaling pathways, and offer a systematic update on the role and mechanism of key signaling pathways underlying the onset, development, and progression of T2DM. In this content, we also summarize current therapeutic drugs/agents associated with signaling pathways for the treatment of T2DM and its complications, and discuss some implications and directions to the future of this field.
Keywords: β cell dysfunction, antidiabetic drug, diabetes complications, insulin resistance, pathology, therapeutic target
Cao et al. provide a comprehensive overview of signaling pathways in the pathogenesis and therapy of type 2 diabetes mellitus (T2DM) and its complications.

1. INTRODUCTION
Type 2 diabetes mellitus (T2DM), a chronic noncommunicable disease that is diagnosed by aberrant high blood glucose levels, has attracted increasing attention due to its high prevalence and enormous health burden. Currently, there are more than 537 million patients with diabetes, most of which are T2DM, and this number is projected to reach at 783 million by 2045, 1 accompanied by a younger trend in the onset age around the world. 2 , 3 Beyond a straightforward rise in blood glucose, T2DM may cause a series of complications that are linked to the vascular and neural damages triggered by hyperglycemia, such as diabetic nephropathy (DN), diabetic retinopathy (DR), diabetic neuropathy, and cardiovascular disease (CVD). 3 Of note, T2DM is emerging as an increased risk of severe COVID‐19, the recent novel coronavirus pandemic worldwide. For a long duration, T2DM and its complications have been witnessed to impose substantial effects in the quality of life and socioeconomic burden, 4 which inspires the incredible progress in mechanism exploration and drug intervention for T2DM. However, although current antidiabetic drugs, insulin therapy, and lifestyle interventions, for example, metformin administration, carbohydrate restriction, and/or endurance exercise, have warranted decent control of T2DM progression, implementing and maintaining these changes for prolonged periods are still challenging, especially given the pervasiveness of drug side‐effects and the accessibility of calorically dense foods and sedentary lifestyle. To further develop new pharmacological strategies that could potently target pathological mechanisms and avoid side‐effects, it is still urgent and important to unceasingly disclose the mechanistic underpinnings of T2DM.
Numerous signaling pathways play essential roles in the development of T2DM and are implicated in its therapy. From a pathological view, insulin resistance (IR) and subsequent insulin deficiency due to pancreatic β cell damage are two main pathological features of T2DM, and their variable combination further contributes to the complexity of T2DM and the diversity in the patients' conditions. 1 In addition, other pathological processes, including chronic inflammation, incretin dysregulation, hyperglucagonemia, lipolysis, central appetite dysregulation, abnormal gastric emptying, gut dysbiosis, and islet amyloid polypeptide (IAPP) deposition, are also regarded as key regulators in the pathophysiology of T2DM. 1 The endeavors throughout the past decades have uncovered that a series of signaling pathways play important roles in controlling these pathological changes. For example, phosphoinositide 3‐kinase (PI3K)/protein kinase B (PKB, also known as AKT) signaling cascade regulates insulin response, 5 and AMP‐activated protein kinase (AMPK) pathway prevents β cell dysfunction. 6
From a therapeutic view, most of current glucose‐lowering drugs have been found to exert their pharmaceutical effects dependently of signaling pathways, more or less. For instance, glucagon‐like peptide‐1 (GLP‐1) receptor agonists bind to their receptors on β cells and promote insulin exocytosis by increasing intracellular Ca2+ levels via the protein kinase C (PKC)/cyclic adenosine monophosphate (cAMP) signaling pathway. 7 , 8 Recently, four categories of potential hypoglycemic drugs with new mechanisms of action have been proposed, which may stimulate insulin secretion, utilize the incretin axis, maintain hepatic glucose homeostasis, and improve insulin sensitivity, repsectively. 9 Interestingly, these potential drugs target a number of key receptors, vital enzymes, or ion channels, such as glucokinase (GK) activators, G‐protein‐coupled receptor 40 (GPCR40), GLP‐1 receptor (GLP‐1R), and so on, which are involved in numerous signaling pathways associated with T2DM. 9 Therefore, a better understanding of the signaling pathways is of importance for in‐depth dissecting the pathological mechanisms for T2DM and facilitating the development of targeted drugs and interventions against T2DM and its complications.
In this review, focusing on the two most important pathological features of T2DM, that is, IR and impaired β cells, align with simplified mention of other pathological changes, we elaborated the roles of signaling pathways in pathological changes of T2DM and its complications on the one hand, and expatiated the intervention mechanisms of important glucose‐lowing drugs on these signaling pathways and emphasized the important targets on the other hand.
2. HISTORY OF T2DM AND SIGNALING PATHWAYS
Historically, the progresses of T2DM in pathogenesis and therapy are closely related to the discovery and elucidation of signaling pathways (Figure 1). As one of the oldest diseases in human history, diabetes was first mentioned at 1552 BC, 10 but it was not until the discovery of insulin in 1921 that understanding of the disease reached a milestone stage. 11 The notion that serum Ca2+ concentration changes with insulin levels was established as early as 1930 and Ca2+ signaling was linked into insulin secretion in1976. 12 Time lapsing to 1990, the intracellular transmitter and effector of insulin were discovered and the PI3K/AKT signaling was identified as the chief pathway to mediate the insulin action. 13 Since then, mechanism of insulin action has been gradually revealed, leading to the discoveries of several classical signaling pathways related to T2DM, such as the phosphorylation of mitogen‐activated protein kinase (MAPK), AMPK, WNT, and transforming growth factor β (TGFβ) pathways. 14 Furthermore, in recent years, several novel signaling pathways have also been linked to insulin production and action, such as fibroblast growth factors (FGFs), hypoxia‐inducible factors (HIFs), yes‐associated protein (YAP), and the unfold protein response (UPR) signaling pathways.
FIGURE 1.

Timeline of key scientific and pharmaceutical discoveries in T2DM.
Continuous revelation of the pathogenesis and signaling pathways of T2DM also provides a basis for the development of drugs that target T2DM, despite the confusion of the study path. Metformin, the first‐line glucose‐lowering drug, was first synthesized in 1922, but it was not until 2022 that metformin was confirmed to exert a hypoglycemic effect by activating the presenilin enhancer 2 (PEN2)‐mediated AMPK protein 15 , 16 signaling pathway. Akin to metformin, sulfonylureas (SUs) were found to have hypoglycemic effects accidentally in 1942, but it was until 1979 when researchers truly found the underlying mechanism, by which acting on β cells to stimulate insulin secretion through closing K+ channels and activating Ca2+ signal. 17 In the latest years, novel hypoglycemic drugs, such as GLP‐1 and sodium‐glucose cotransporter 2 (SGLT2) inhibitor, are effective in reducing fasting blood glucose 18 and preventing the reabsorption of urine glucose, 19 regardless of their unspecified mechanisms. Collectively, since these drugs have different safety, tolerability, and availability, hindering their best clinical use, it is particularly important to explore the pathogenesis and precise signaling pathways of T2DM, which would drive the identification of the targets of existing and novel drugs.
3. PATHOLOGICAL FEATURES OF T2DM
T2DM is conventionally featured with two pathological traits: IR and subsequent β cell dysfunction, which are the consequences of the feedback loops between disordered insulin secretion and insulin action (Figure 2).
FIGURE 2.

Pathological features of T2DM. T2DM is characterized by insulin resistance and β cell dysregulation, along with various pathological disturbances, which are coordinately regulated by multiple tissues and organs, including the pancreas, liver, adipose, muscle, gut, and brain. As the major characteristic of T2DM, insulin resistance occurs when the tissues become less sensitive to insulin, leading to increased glucose production and lipogenesis in the liver, enhanced lipolysis and reduced triglyceride synthesis in the white adipose tissue (WAT), and decreased glucose uptake and elevated fatty acid oxidation in the skeletal muscle, and so on. Failure of pancreatic β cells that are responsible for the production and secretion of insulin results in decreased cell number and impair insulin secretion. The vicious cycle of insulin resistance, hyperglycemia, inflammation, and so on, aggravate the onset, development, and progression of T2DM. The online resource inside this figure was quoted or modified from Scienceslide2016 plug‐in.
IR, defined as the impairment of insulin sensitivity, is a predictor of T2DM and describes the failure of target organs/tissues to answer insulin stimulation. 20 Previous studies have reported that numerous factors, including obesity, overnutrition, physical inactivity, gastrointestinal microbial disturbance, and family history of T2DM could drive IR. 21 , 22 Insulin is the only one hormone that actively lowers blood glucose by acting on its target organs/tissues, including the hypothalamus, liver, adipose tissue, and skeletal muscle. The hypothalamus is regarded as the major regulator of appetite, and the signaling pathways triggered by insulin, as well as leptin, play key roles in maintaining energy expenditure, glucose homeostasis and insulin sensitivity in peripheral tissues. 23 Insulin stimulation can inspire divergent physiological responses in the peripheral tissues, including increased glycogenesis and de novo lipogenesis (DNL) but decreased gluconeogenesis in the liver, 24 enhanced glucose uptake in the skeletal muscle and adipose tissue, suppressed lipolysis in the adipose tissue, and so on. Dysregulation of these responses, at least a part of them, is therefore to be the major consequence of IR. Additional pathological changes may be also intimately associated with IR. For example, compositional and functional alterations in gut microbiota have been observed in patients with IR and metabolic dysfunction, 25 which may modulate cellular metabolism and energy homeostasis through homeostatic and pathogenic microbiota‐host interactions. In addition, a decrease in circulating adiponectin, an insulin‐sensitizing, anti‐inflammatory, antiatherosclerotic, and hepato‐protective factor predominantly produced by adipocytes, 26 has been shown to extensively promote hepatic and muscular IR. 27 , 28 , 29 , 30
Pancreatic β cells function as the hub for insulin secretion (Figure 3), therefore declining β cell function due to dysregulated genetic and external factors is key to T2DM progression. 31 β Cell dysfunction is clearly presented in the patients with hyperglycemia, but whether this trait occurs early or late in the T2DM remains controversial. The current conclusion is more inclined to that β cell function may be weakened early in T2DM progression and gradually decline as glucose tolerance deteriorates. 32 Under normal conditions, β cells are inactive in secreting insulin during fasting period, and postprandial transient hyperglycemia can stimulate β cells to enhance glucose‐stimulated insulin secretion (GSIS), thereby increasing the blood insulin to meet the demands for lowering blood glucose. Generally, β cells are capable of producing sufficient insulin to compensate for IR and maintaining euglycemia. However, chronic exposure to excess circulating nutrients, along with the consequent changed epigenetic factors, could induce a toxic state in the pancreatic islet, resulting in β cell dysfunction and compensatory failure followed by insulin deficiency, eventually causing hyperglycemia and T2DM. 33
FIGURE 3.

Multiple signaling pathways regulate β cell function. Elevated glucose enters the β cell via GLUT1/2 transporters to produce ATP through glycolysis in the cytoplasm and oxidative metabolism in the mitochondria, resulting in an increase in the ratio of ATP to ADP. Elevated cytoplasmic ATP leads to the close of ATP‐sensitive potassium channel, membrane depolarization, and subsequent Ca2+ influx, triggering the release of insulin from insulin granules. Meanwhile, multiple signaling pathways participate in mediating β cell function and insulin secretion, including AMPK, MAPK, WNT, PI3K/AKT, TGFβ, YAP/TAZ, and so on.
Substantial evidence supports that metabolic stress could compel the progression of multiple cellular events that drive or aggravate IR and β cell dysfunction. Despite of the biological differences among divergent peripheral metabolic tissues/organs, these cellular events have notable resemblance. Roughly, they include low inflammation state, 34 , 35 endoplasmic reticulum (ER) stress, 36 , 37 mitochondrial dysfunction, 38 lipotoxicity damage, cell death, and so on, whose interaction could cause inflammatory attack, 35 protein turbulence, 36 , 39 , 40 reactive oxygen species (ROS) accumulation, 41 ATP deficiency, 42 , 43 , 44 and ceramide overload, 45 accelerating the pathogenesis of IR and β cell dysfunction. In particular, mitochondria serve as the prime core of glucose metabolism and ATP production in cells. Mitochondrial dysfunction, characterized as a defect in mitochondrial dynamics and thus cellular bioenergetics, is highly implicated in T2DM progression, 42 as hyperglycemia can compel the mitochondria to enhance ROS production. 42 , 46 Physiologically, mitochondria can rely on its powerful plasticity of dynamic structures to restore ROS and ATP imbalances, while, as metabolic pathology proceeds, such self‐regulating mechanisms might be compromised, therefore advancing IR, β cell dysfunction and T2DM. 42 , 47 , 48 Altogether, the pathological crosstalk between metabolic stresses and cellular events is a cause for the progression of IR, β cell dysfunction, and consequent T2DM.
A bevy of complicated and interdependent mechanisms have been conceptualized to dictate these cellular events, T2DM pathological features and their vicious cycles, which are primarily mediated and executed by signaling pathways. Therefore, although new identifications involving signaling pathways remain to be completely validated, it is urgent to summarize which signaling pathways and how they contribute to these pathological cellular events and T2DM.
4. SIGNALING PATHWAYS IN T2DM
Numerous prominent signaling pathways are involved in IR and/or β cell dysfunction, including PI3K/AKT, AMPK, MAPK, WNT, UPR, Hippo, HIFs, TGFβ, FGFs, bile acids (BAs), Ca2+‐related signaling pathways, and others. These signaling pathways act through their coiled interactions to enable the regulation of various biological processes regarding insulin action and production, as well as other pathophysiological modules controlling overall glucose metabolism. Adherent to this, extensive investigations have revealed that a remarkable level of dysregulations in these pathways proceed in the related cells and tissues from patients and animals with T2DM, IR, and obesity. Despite a fraction of mechanistic underpinnings are still entangled, such shifts in signaling pathways, on the one hand, relate in a large part to the metabolic stress proceeding T2DM, and on the other hand, reciprocally disrupt glucose homeostasis and thus participate in T2DM progression. Herein, we will discuss the physio‐pathological switches, roles, and mechanisms of diverse signaling pathways in T2DM development, primarily in terms of insulin action and production, along with other related biological processes.
4.1. PI3K/AKT pathway
Over the past decades, the PI3K/AKT pathway has been identified as the prime effector pathway in response to insulin action on the liver, adipose tissue and skeletal muscle (Figure 4). Basically, insulin binds to the extracellular domain of insulin receptor tyrosine kinase (IRTK) at cell surface, 49 , 50 and rapidly activates insulin receptor substrate (IRS), which then recruits and phosphorylates PI3K, and thus activate AKT. 51 , 52 AKTs are divided into three homologous isoforms, namely AKT1, AKT2, and AKT3. 53 The predominantly expressed isoform in peripheral tissues is AKT2, while in pancreas, it is the AKT1, implying the tissue specificity of AKT isoforms. 53
FIGURE 4.

Role of the PI3K/AKT signaling pathway in different metabolic tissues. In response to insulin (or IGF1 in the skeletal muscle), the INSR (or IGFR in the skeletal muscle) is activated, causing tyrosine phosphorylation of IRS1/2. Phosphotyrosine sites on IRSs allow binding of the lipid kinase PI3K, which synthesizes PIP3 at the plasma membrane, leading to AKT phosphorylation by PDK1 and mTORC2 to fully activate the PI3K/AKT pathway. In the liver, skeletal muscle and adipose tissue, activated AKT (especially AKT2) can phosphorylate a number of substrates at Ser/Thr residues, including: (1) GSK3β, which stimulates glycogen synthesis via regulating G6P and maintaining mRNA translation by modulating Atrogin1 and MURF1; (2) TSC2, which permits activation of mTORC1 and its downstream targets 4E‐BP1, S6K1 and SREBP1c to increase protein synthesis and lipogenesis; (3) FoxOs, which decrease gluconeogenesis by suppressing gluconeogenic gene expression. In both skeletal muscle and adipose tissue, AKT inactivates AS160 to promote glucose uptake via the translocation of GSVs to the plasma membrane. In the adipose tissue, AKT also suppresses lipolysis that is mediated by PDE3B, and occurs largely through attenuation of cAMP‐stimulated events and phosphorylation of HSL and ATGL. In the pancreas, activated AKT (especially AKT1) promotes PDX1 activation and nuclear translocation by relieving FoxO1 induced FoxA2 inhibition, thereby maintaining β cell mass and insulin secretion.
Although the process of insulin‐induced PI3K/AKT activation is similar, the distal steps of its activation vary across different peripheral tissues, leading to distinct biological outcomes. In the liver, the activation of PI3K/AKT pathway is responsible for decreasing hepatic glucose production (HGP), promoting glycogen synthesis, and increasing lipid biosynthesis. Activated AKT2 inhibits gluconeogenesis by phosphorylating forkhead box O families (FoxOs) and subsequently preventing the transcription of key gluconeogenic enzymes, including phosphoenolpyruvate carboxykinase (PEPCK) and glucose 6‐phosphatase (G6Pase). Meanwhile, AKT2 activation can induce the phosphorylation of glycogen synthase kinase 3 (GSK3) to promote glycogen synthesis. 54 Additionally, AKT2 activation can promote hepatic DNL by transcriptionally enhancing the levels of several lipogenic proteins, such as acetyl‐CoA carboxylase 1 (ACC1), fatty acid synthase, and glycerol‐3‐phosphate acyltransferase 1. 50 , 55 Furthermore, the upstream mechanisms dictating such transcriptional regulation include the increases in mammalian target of rapamycin complex 1 (mTORC1)‐dependent sterol regulatory element binding protein (SREBP) transcription, 56 ribosomal S6 protein kinase 1 (S6K1)‐dependent SREBP maturation, 57 and SREBP stabilization. 58 While in the adipose tissue and skeletal muscle, enhanced glucose uptake is a shared outcome of insulin‐induced AKT2 activation, which is mainly related to an increase in the density of glucose transporter 4 (GLUT4) in the plasma membrane. 59 , 60 Moreover, AKT2 activation in the adipose tissue can promote DNL via the mechanisms similar to those in the liver and suppress the lipolysis through multiple downstream mechanisms. These mechanisms involve the suppression of cAMP‐dependent protein kinase A (PKA) activity 61 and the regulation of lipolytic regulatory proteins, such as mTORC1, 62 S6K1, 63 protein phosphatase 1 (PPI), 64 protein phosphatase 2A (PP2A), 65 and interferon regulatory factor 4 (IRF4). 66 In summary, the well‐proceeding of these downstream branches of AKT confers the peripheral insulin action with the power to reduce the blood glucose, or in other words, maintains insulin sensitivity, thereby contributing to glucose homeostasis.
Consistent with its protective effect on insulin sensitivity, the PI3K/AKT pathway is usually impaired in the insulin‐resistant tissues, thus disrupting the key metabolic actions of insulin. 38 , 67 Actually, AKT2 knockout mice usually exhibit T2DM phenotype with IR and glucose intolerance. 46 , 47 The decreased ability to activate PI3K/AKT pathway could derive from several pathological facets related to IR and T2DM, including the mutation of this pathway itself, dysregulation of some regulator proteins or other signaling pathways, lipid accumulation or alteration, reduced circulating adiponectin, and enhanced inflammation state. In T2DM, these factors together with the impaired AKT pathway, control IR progression and thus systemic glucose state in a complicated way.
Alterations of both the upstream and downstream components in the PI3K/AKT pathway itself have been found to aggravate peripheral IR. For example, insulin receptor (INSR) with a single mutation at leucine 973 significantly attenuated insulin/PI3K/AKT activation in the liver and white adipose tissue (WAT), impaired systemic insulin sensitivity, and thus decreased insulin‐induced glucose uptake. 68 In parallel, mice with knockout of insulin and/or insulin like growth factor 1 (IGF1) receptors developed IR, glucose intolerance, and islet hyperplasia with hyperinsulinemia, accompanied with increased lipolysis and adipocyte apoptosis. 69 Meanwhile, fat‐specific disruption of the downstream effectors of AKT pathway, FoxOs, can result in a reversal of IR in the liver, an exacerbation of hyperinsulinemia, but a maintenance of normal glucose tolerance. 70
Dysregulation of certain regulators might also compromise insulin sensitivity and glucose homeostasis through altering AKT pathway. For instance, phosphatase and tensin homolog (PTEN), a negative regulator that induces IR by converting PIP3 back to PIP2 via dephosphorylation, is often overexpressed in T2DM patients, 71 contributing to the termination of PI3K/AKT2 signaling network and the progression of IR. 72 , 73 Liver‐specific deletion of sirtuin 1 (SIRT1) has also been pointed out to cause ROS accumulation and thus impair AKT signaling, eventually inducing IR, hepatic glucose overproduction and chronic hyperglycemia. 74 Consistently, the activation of ApoM/S1P complex, which also activates the AKT pathway, can prevent IR progression through upregulating SIRT1. 75 More recently, it was shown that TGFβ1 stimulated clone 22 D4 (TSC22D4) is a novel interaction partner for AKT, and its dysregulation is able to disrupt insulin sensitivity and glucose disposal in mice. 76
It is well known that lipid metabolism and chronic inflammation could modulate AKT‐associated insulin sensitivity and IR. Diacylglycerol accumulation in the liver impairs the AKT activity through PKC, 77 , 78 , 79 , 80 an inhibitor of the PI3K/AKT2 pathway. 81 Similarly, under T2DM conditions, exosomes with altered lipid composition can be produced by the intestine and taken up by the macrophages and hepatocytes to repress the hepatic insulin/PI3K/AKT signaling pathway. 82 Reduced circulating adiponectin, 29 , 30 such as globular adiponectin (gAcrp30), could also cause hepatic and muscular IR by increasing ectopic lipid storage in these organs. 83 Furthermore, enhanced inflammation state, caused by certain dysregulated inflammatory modulators, such as protease‐activated receptor 2 (PAR2), 84 and neurite outgrowth inhibitor (Nogo), 85 have also been linked to impaired AKT activation and thus peripheral IR. Besides, numerous microRNAs (miRNAs), 86 for example, miR‐26a, can interfere with many proximal PI3K/AKT pathway components to regulate insulin sensitivity and glucose metabolism. 87 In addition, a wide scope of signaling pathways have been characterized to modulate the consequence of insulin‐triggered AKT activation through directly regulating AKT or indirectly affecting AKT upstream and downstream branches, thereby influencing the progression of IR and T2DM (Figure 5). These signaling pathways, as well as their interplay with the PI3K/AKT cascade, will be introduced in the below sections.
FIGURE 5.

Crosstalk between PI3K/AKT and various pathways. The PI3K/AKT signaling inactivator PKC is promoted by the BMP family via the TGFβ pathway and Ca2+ overloading, while PTEN is inhibited by MAPK and YAP pathways. IRS2 is activated by the YAP pathway, but repressed by multiple signals, including MAPK, UPR, and WNT pathways, and proteins, such as IL‐6 and IKKβ. Both AKT and PDK1, are activated by BMPs, while AKT is in turn dephosphorylated by the UPR pathway and Ca2+ overloading. The AKT downstream GSK3β can be activated by the UPR pathway, but blocked by the AMPK signaling and FGF family; FoxOs are also stimulated by the UPR pathway in line with β‐catenin and SIRTs, while inhibited by the MAPK signaling and FGF family; mTORC1 can be motivated by MAPK, AMPK, and YAP pathways, but repressed by FGFs pathways. The glucose transporter GLUTs expression and membrane translocation can be improved by the AMPK signaling and FGF family, while inhibited by the MAPK and YAP pathways, as well as Ca2+ overloading.
In the pancreatic islet, PI3K/AKT1 activation can increase β cell mass and stimulate insulin production, indicating another link between the dysregulation of PI3K/AKT pathway and T2DM progression. Increasing evidence indicates that β cell‐specific overexpression or constitutive activation of AKT1, as well as knockout of PTEN, 88 FoxO1, 89 tuberous sclerosis complexes (TSCs) 90 or mTORC1, 91 are able to increase β cell mass, proliferation, neogenesis, and cell size, thereby improving glucose tolerance. On the contrary, specific knockout of 3‐phosphoinositol dependent protein kinase 1 (PDK1), 92 IRS2, 93 , 94 INSR, 95 IGF1, 96 or S6K1, 97 can repress AKT1 98 signaling transduction, leading to the decreases in insulin content and secretion, β‐cell mass and proliferation, and glucose tolerance, and eventually facilitating the development of hyperglycemia and T2DM. Mechanistically, the PI3K/AKT1 pathway relies on the transcriptional factor pancreatic and duodenal homeobox 1 (PDX1) to control β cell differentiation, function, survival and proliferation. 53 PDX1 controls the expression of multiple key genes for β cell fate, such as insulin (Ins1 and Ins2), neurogenin 3 (Ngn3), SRY‐box transcription factor 9 (Sox9), v‐mafmusculoaponeurotic fibrosarcomaoncogene homolog A (MafA), Glut2, GK (Gck), iapp, cyclin D1/2 (Ccnd1/2), and transient receptor potential canonical 3/6 (Trpc3/6). 99 All in all, as the core signaling pathway downstream of peripheral insulin action or a potent regulator of β cell function, the PI3K/AKT pathway holds a great power in the regulation of T2DM progression, and a notable potential to be a drug target for effectively treating T2DM.
4.2. AMPK pathway
The AMPK pathway is famous for its critical role in sensing cellular energy status (Figure 6). 100 AMPK can be canonically activated by the increasing AMP and/or ADP along with declining ATP through the upstream kinase liver kinase B1 (LKB1)‐dependent Thr172 phosphorylation, or noncanonically activated by other stimulations through several recently described pathways that are independent of AMP/ADP, including those related to the lysosome, mitochondrion, Ca2+/calmodulin‐dependent protein kinase kinase 2 (CaMKK2) and TGFβ‐activated kinase 1 (TAK1). 101 , 102 , 103 Owing to these mechanisms, AMPK can sense the availability of glucose, glycogen, FAs, Ca2+, leptin, and adiponectin, and the damage to lysosomes and nuclear DNA, as well as the stimulation of multiple drugs. 15 , 102 Basically, activated AMPK pathway is able to endorse ATP‐producing catabolic pathways via phosphorylating and activating certain proteins, while curb energy consumption via phosphorylating and inactivating proteins involved in anabolic (biosynthetic) pathways, thereby balancing cellular metabolism and functions. 102 , 104 In this regard, the downstream events of the AMPK pathway encompass: carbohydrate or glucose metabolism, FA and cholesterol metabolism, protein synthesis, the counteracting effects on mTORCs, mitochondrial biogenesis, mitophagy, and autophagy. 100 , 105 Such potent effects of the AMPK pathway in cellular metabolism and functions are prone to endow it with an incredible significance in the peripheral insulin action, glucose uptake, nutrient intake, lipid metabolism, inflammation, insulin secretion, and thus systematic homeostasis of glucose and lipids, warranting the vigorous attention of its therapeutic potential in the area of T2DM. 104
FIGURE 6.

The AMPK pathway regulates glycolipid metabolism in T2DM. Under conditions of energy requirement, such as starvation, exercise and OS, the decrease of ATP/AMP ratio activates AMPK directly. Meanwhile, low glucose decreases FBP level, which is sensed by aldolase. Reduced FBP only binds to a small number of aldolases, the remaining which occupy TRPV. Aldolase strongly interacts with TRPV channels and blocks Ca2+ release, suppressing v‐ATPase activity and stimulating AMPK activity. Metformin interacts with PEN2 and inhibits v‐ATPase activity, thereby activating AMPK. Besides, obesity‐induced hyperinsulinemia activates protein kinases to suppress AMPK activity. Upon activation, AMPK acts on multiple downstream targets to increase ATP generation and decrease ATP consumption by inhibiting anabolic processes and increasing catabolic processes, subsequently regulating a series of downstream targets to affect glucose and lipid metabolism.
Degrading of AMPK activity is widely observed in the skeletal muscle from mice and humans with obesity and T2DM, probably due to that high glucose could inhibit the AMPK activity by breaking the active LKB1 complex, 106 activating the protein phosphatase PP2A, 107 upregulating the phosphatase PH domain and leucine rich repeat protein phosphatase 2 (PHLPP2), 108 and inducing ubiquitination degradation of AMPK subunits. 109 , 110 AMPK activation has been found to exert constructive effects on the glucose uptake of skeletal muscle, and regarding this, the deficiency of muscular AMPK activity might compromise the whole‐body glucose homeostasis during T2DM. In detail, the activation of AMPK increases glucose uptake through the mechanisms by which involve the inhibited sequestering of GLUT4 to Golgi apparatus, the enhancement of GLUT4 translocation to plasma membrane, and the upregulation of GLUT4 expression. 103 In the cells that hinge on GLUT1, AMPK also boosts their glucose uptake through promoting the translocation of GLUT1 to the plasma membrane and increasing GLUT1 expression. 111 Since glucose uptake is a key step in the downstream of insulin action, such physiological supportive effect of AMPK in glucose uptake might bypass the PI3K/AKT pathway to link AMPK activation with IR inhibition. 112 In fact, muscle contraction, hypoxia and adiponectin stimulation have been found to initiate GLUT4 translocation via the activation of AMPK in the skeletal muscle, which might counteract muscular IR. 20 Supporting this, multiple drugs or natural products that activate AMPK can improve IR and glucose homeostasis in the mice and patients with obesity or T2DM, including metformin, 15 O304, 113 MK‐8722, 114 Canagliflozin, 115 and PF‐739. 116 Interestingly, although AMPK does not directly regulate the insulin/AKT pathway, the ATK pathway has instead been identified to inhibit AMPK activation, 117 which may complicate the relationship between the AMPK pathway and insulin action. Collectively, identification of accurate alterations, detailed mechanistic cues, and pharmaceutical activators regarding the skeletal muscle‐specific AMPK pathway might confer a basis for the advancement of T2DM mechanistic investigation and drugs intervention.
In recent perspectives, hepatic AMPK activation appears to indirectly inhibit hepatic gluconeogenesis, refreshing the previous understanding of its direct effect on HGP. 103 Nevertheless, genetically or pharmacologically induced activation of AMPK in hepatocytes can still indirectly suppress gluconeogenesis, alleviate the liver IR, lower HGP, and improve glucose parameters in the human and animals with obesity and T2DM. 103 , 118 These effects are primarily mediated through the inhibition of DNL, the depression of inflammatory makers, and the promotion of mitochondrial function in the liver and other organs. 119 Hence, it is of importance for future study to deepen our knowledge and understanding of the exact role of AMPK in hepatic glucose metabolism.
The AMPK pathway can also participate in T2DM progression by controlling a wide scope of metabolic processes that are not directly linked to peripheral IR. First, hypothalamic AMPK activation under ghrelin or low glucose stimulation can improve appetite via inhibiting ACC1, 120 and activating autophagy 121 and p21‐activated kinases, 122 to enhance energy supply. Conversely, AMPK inactivation in response to leptin and insulin suppresses appetite, preventing obesity and T2DM. Second, AMPK activation is able to widely promote FA oxidation in the skeletal muscle and liver through inhibiting the phosphorylation of ACC1/2, thus indirectly amending hyperinsulinemia, glucose intolerance and IR. 119 Meanwhile, the suppression of cholesterol synthesis also responds to AMPK activation, in which involves the inhibition of 3‐hydroxy‐3‐methylglutaryl (HMG) coenzyme A (CoA) reductase (HMGCR). 123 Third, AMPK activation can stimulate glycolysis through the phosphorylation and activation of phosphofructokinase (PFK), and repress the glucose storage by inhibiting multiple isoforms of glycogen synthase (GS). 105 , 124 Forth, the activation of AMPK pathway has been linked to the depression of inflammation in macrophages, adipose tissue, liver and skeletal muscle, thereby ameliorating systemic IR and improving glucose homeostasis. 125 Finally, the important role of AMPK activation in maintaining mitochondrial homeostasis may also prime its indirect role in ensuring metabolic efficiency of cells and tissues. 105 In line with this, adipocyte‐specific deficiency of AMPK in mice worsens HFD‐induced systemic IR through disrupting mitochondrial integrity in adipocytes, in terms of the mitochondrial function, structure, and markers of mitophagy. 126
In the pancreatic β cells, the AMPK/mTOR pathway may affect T2DM progression by modulating β cell mass and insulin secretion. 127 It has been reported that the switch from mTORC1 to AMPK is the basis for the growth of β cells during embryonic and early postnatal life, including promoting β cell mitochondrial biogenesis and functional maturation of oxidative metabolism. 128 In detail, the AMPK pathway represses mTORC1 activation to confer β cells with functional maturation, and therefore decreased AMPK activation in diabetic islets may be prime for enhanced mTORC1 signaling. 129 Despite physiological mTORC1 activation has positive effects on β cell survival, proliferation, and homeostasis, 130 sustained mTORC1 activation in the cases of overnutrition or hyperinsulinemia, conversely leads to β cell failure and impaired GSIS, which can be inversed by a short‐term inhibition on mTORC1 activation. 131 In addition, it has been showed that abnormally increased glycolytic metabolites caused by chronic hyperglycemia can engender an inhibition of AMPK but an activation of mTORC1 to reprogram metabolic gene expression, weaken mitochondrial glucose metabolism and ultimately impair GSIS. 132 In summary, since switches between mTORC1 and AMPK underlie β‐cell metabolic plasticity, the AMPK/mTOR pathway might play prestigious roles than previous thought in the β cell function and T2DM progression.
Although the AMPK pathway has been regarded as a potent target for T2DM treatment, its effect on GSIS is still entangled. For instance, LKB1, the key upstream activator of AMPK, has debatable roles in insulin secretion. On the one hand, LKB1 deficiency in β cells could promote insulin secretion by elevating ACC1 activity and plasma membrane excitability. 133 , 134 On the other hand, the absence of LKB1/AMPK activity also promotes the mitochondrial impairment that compromises GSIS. Notably, a recent study reported that AMPK activation influences GSIS in β cells in the manners dependent of action duration and glucose concentration. 135 Specifically, drug activation of AMPK primes GSIS in a short duration, while a long‐term AMPK activation represses insulin secretion. 135 Meanwhile, only a high level of glucose action can potentiate insulin secretion. 135 Additionally, the promoting effect of AMPK on GSIS has been recently reported. It has shown that β cell‐specific deletion of AMPK increases the levels of miR‐125b‐5p, which could subsequently impair GSIS in both MIN6 cells and human islets. 136 As well, silencing of the metal‐dependent protein phosphatase 1E (PPM1E), the most markedly downregulated protein phosphatase in T2DM patients’ islets, can promote GSIS through increasing the phosphorylation of CaMKII, AMPK, and ACC. 137 Altogether, these conflicting results warrant further investigations to provide more reliable information on the role of AMPK in T2DM and its clinical application.
4.3. MAPK pathway
The MAPK signaling pathways contain three major subclasses, namely the extracellular signal‐regulated kinases 1/2 (ERK1/2), c‐Jun N‐terminal kinases (JNKs), and p38 family (Figure 7A). 138 , 139 , 140 Multiple stimuli, such as hormones, growth factors, and TGFβ‐related agents, have been identified to activate MAPK pathways via a dedicated three‐tiered protein kinase cascade that is comprised of a MAPK kinase kinase (MAPKKK), a MAPK kinase (MAPKK), and the MAPK. 141 Activated MAPK pathways indorse selective phosphorylation of transcriptional factors, for example, nuclear factor of activated T cells (NFAT), activator protein 1 (AP‐1), and C/EBP‐homologous protein (CHOP)/DNA damage‐inducing protein 34 (GADD34), as well as protein kinases, for example, ribosomal s6 kinases (RSKS) and eukaryotic initiation factor 4E (eIF4E), thus controlling gene transcription and signaling transduction. 141 As such, they are capable of connecting extracellular stimuli to cellular events such as proliferation, inflammation, differentiation and apoptosis. Accumulating evidence suggests that MAPK pathways are altered in several metabolic tissues during T2DM progression and play significant roles in peripheral IR and β cell fate, despite of the discrepancies among the three subclasses. 142
FIGURE 7.

Involvement of MAPK, YAP, Wnt, and TGFβ pathways in T2DM. (A) The classic MAPKKK–MAPKK–MAPK or Raf–Ras–MAPK signaling pathway can be activated by various metabolic signals. Upon being activated, MAPKs, comprising of three subtypes (ERK1/2, JNK1/2/3, and p38 family), induce diverse metabolic responses, including chronic inflammation state, insulin resistance, and altered gluconeogenesis in metabolic tissues, as well as improved insulin secretion and survival of pancreatic β cells. (B) The Hippo pathway is mainly composed of MST1/2 and LATS1/2. Active MST1/2 phosphorylates LATS1/2, which in turn phosphorylates YAP/TAZ and thus inhibits YAP/TAZ activity. Metabolites and hormones could act through GPCRs to regulate the Hippo pathway and YAP/TAZ. Deficient glucose metabolism decreases YAP/TAZ activity and prevents the formation of YAP–TEAD complex. Subsequently, YAP/TAZ could regulate various glycolipid metabolism processes in different metabolic organs. (C) WNT pathway encompasses three different branches, including WNT/β‐catenin, WNT/PCP, and WNT/Ca2+ pathways. Under metabolic stress, WNT/β‐catenin pathway could aggravate hepatic insulin resistance and enhance hepatic glucogenesis. WNT/PCP pathway increases hepatic insulin resistance via activating JNK pathway, whereas activated WNT10b/β‐catenin and WNT/PCP pathways improve the insulin sensitivity of skeletal muscle. Moreover, WNT/Ca2+ pathway could regulate insulin secretion in β cell. (D) The TGFβ pathway could influence T2DM development by affecting the function of pancreatic islet as well as the insulin signaling of nonislet tissues. TGFβ inhibits β cell proliferation, facilitates the generation of β cell, and enhances GSIS. Moreover, different WNT ligands have specific effects in adipose insulin resistance and hepatic gluconeogenesis.
It appears that ERK1/2, JNKs, and p38s pathways are all basically activated in the liver of mice and humans with metabolic stress, and contribute to impaired hepatic insulin sensitivity and glucose metabolism, exacerbating the progression of T2DM. Hepatic ERK1/2 activities have been found to be increased in both genetic and diet‐induced obesity mouse models, 143 triggering overall IR and impaired glucose homeostasis, while obese mice with decreased hepatic ERK1/2 showed better systemic insulin and glucose tolerance. Mechanistically, activation of ERK1/2 could participate in a range of biological activities that function individually or interdependently in aggravating IR progression. These biological activities roughly include the serine phosphorylation of IRS proteins, 143 the connection between the CBA stimulation and impaired hepatic glucose metabolism, 144 , 145 the negative effect of hepatocyte‐derived fibrinogen‐related protein 1 (HFREP1) in hepatic insulin sensitivity, 146 the positive feedback between cytokine secretion of macrophages and IR of hepatocytes, 147 and the gluconeogenic response of FGF21 stimulation on the liver. 148 Additionally, the protective effect of serum‐ and glucocorticoid‐regulated kinase 1 (SGK1) in hepatic insulin sensitivity relies on the inhibition of ERK1/2 activity. 149
Similar to ERK1/2, hepatic JNKs are also activated by obesity. Hepatic activation of JNK is believed to reduce the expression of peroxisome proliferator‐activated receptor α (PPARα) target genes, for example, FGF21, to block FA oxidation and thus aggravate IR. 150 Correspondingly, liver‐specific deficiency of functional JNKs in mice ameliorates diet‐induced IR and hyperglycemia. 151 Akin to ERK1/2 and JNKs, hepatic activation of p38 family is also linked to glucose intolerance and hyperinsulinemia. Supporting this, either expression of dominant‐negative p38α or inhibition of p38α in the liver could decrease fasting insulin levels and recuperate glucose tolerance in obese mice. 152 In detail, p38α activation induces the serine phosphorylation of IRS1 to compromise hepatic insulin sensitivity 152 and upregulates gluconeogenesis by blunting the activation of AMPK pathway 153 and instigating the expression of genes including CREB, C/EBPα, PPARα, and PGC1α. 142 However, one important study once noted that hepatic p38 activity is reduced in the livers of obese mice, which is contrast to the previous findings, and hepatic activation of p38 can attenuate ER stress and reset glycemia in diabetic mice by enhancing nuclear translocation of X‐box binding protein 1s (XBP1s). 154 Future studies are hence required for identifying the real role of p38 signaling pathways in hepatic glucose metabolism.
Multiple studies delineated that JNK1 is usually activated in the adipose tissue of high‐fat diet (HFD)‐fed mice to aggravate IR. Inversely, adipocyte‐specific deletion of JNK1 155 , 156 or JNK interacting protein 1 (JIP1), 157 a key protein activating JNK, could restore insulin action in the adipose tissue with metabolic stress. The contribution of JNKs pathway in establishing adipocyte IR springs from its promotive role in the inflammation of adipose tissue. Activated JNKs can trigger the secretion of HMGB1, a proinflammatory adipocytokine, and thus promote WAT inflammation and IR in obese patients. 158 Meanwhile, macrophage‐specific JNK deficiency was shown to reduce the polarization of proinflammatory macrophages in the adipose tissue. 155 Additionally, several studies unconcealed that ERK1/2 is also abnormally activated in the diabetic adipose tissue to facilitate adipocyte IR by inducing adipogenesis 159 and local inflammation state, 160 , 161 despite of the obscure molecular mechanisms.
In the skeletal muscle, MAPK pathways are also deviant in obese or diabetic mice, 142 , 162 suggesting their potential roles in modulating muscular IR and glucose metabolism. 163 In fact, ERK1/2 pathway has been observed to negatively control the action of GS in myotubes in a manner independent of GSK3. 164 The abnormal activation of JNK1 in the skeletal muscle was also detected in the HFD‐fed mice, and muscular blocking of JNK significantly reduced obesity‐induced hyperglycemia by halting inflammation and IR, as well as enhancing glucose uptake of skeletal muscle. 165 Basically, mice with increased activity of overall p38 in the skeletal muscle could prevent the development of diet‐induced obesity and IR by enhancing miR‐21 expression and repressing PTEN expression. 166 It is of interest to note that distinct p38 isoforms might have differential metabolic functions in the skeletal muscle. For example, p38α/β were apprised to advocate the inflammatory state by initiating inflammatory cytokine expression and infiltration of proinflammatory macrophages, 167 while p38γ was demonstrated to elevate basal glucose uptake but decrease contraction‐stimulated glucose uptake, partially by changing the expression of GLUT4 in skeletal muscle. 163 Meanwhile, it appears that the p38β is responsible for the major catabolic action of p38 family by affecting the C/EBPβ activity. 168
In the pancreas, the activity of ERK1/2 is also obviously upregulated by hyperglycemia, 169 thereby promoting GSIS and survival of pancreatic islets during T2DM progression. Indeed, ERK1/2 activation in the β cells is sensitive to the glucose and GLP‐1 action, 170 and further upregulates the transcription of genes related to insulin production and secretion 171 , 172 by adjusting the formation of transcriptional complex composed of NFAT and its partners. 173 In parallel, ERK1/2 may contribute to the exocytosis of insulin granules via inducing phosphorylation of synapsin I (a key protein in exocytosis), 174 and the first phase of GSIS showed a reduction of 40% in mice that ERK1 and ERK2 are inhibited simultaneously. 175 Moreover, many in vitro and in vivo studies 176 also indicate that ERK1 is indispensable for the glucose‐induced activation of genes responsible for β cell survival, such as mitogen‐ and stress‐activated kinase 1 (MSK1) and CREB. 175 However, other MAPK pathways might differentially affect the biology of β cells. One significant instance is that suppression of p38 and JNK pathways is essential for metformin to upregulate the expression of pancreatic aquaporin 7 (AQP7) and subsequently induce glycerol influx and insulin secretion of β cells in T2DM. 177 Therefore, there is a calling for more investigations about the accurate roles of different MAPK pathways in the pancreas biology.
What's more, sustained activation of MAPK/ERK signal transduction in hypothalamus has also been uncovered to be key to the antidiabetic action of intracerebroventricular injected FGF1 and subsequent remission of hyperglycemia T2DM rodents. 178 Taken together, one interesting aspect that emerged from the existing findings of MAPK pathways in T2DM is that targeting MAPK signaling potentially broaden the scope of antidiabetic interventions.
4.4. WNT pathway
The WNT pathways are classified into two major groups, β‐catenin‐dependent (canonical) or β‐catenin‐independent (noncanonical) (Figure 7C). 179 The nuclear translocation of β‐catenin is key to activating canonical WNT pathway, where it binds to the transcription factor T‐cell factor/lymphoid enhancer‐binding factor (TCF/LEF) and consequently controls the transcription of target genes. 179 The noncanonical pathways are subdivided into the WNT/Ca2+ pathway and WNT/planar cell polarity (PCP) pathway. 180 Activated WNT/Ca2+ pathway enhances Ca2+ influx and initiates various signaling pathways that phosphorylate RORα and induce nuclear translocation of NFAT and nemo like kinase. 180 The WNT/PCP pathway works in both dishevelled (DVL)‐independent and ‐dependent ways: the binding of WNT to the receptor like tyrosine kinase (RYK) is capable of activating protein tyrosine kinase (SRC) independently of DVL, while its binding to the ROR1/2‐Fzd complex can activate DVL, a central mediator of WNT/PCP pathway, and then trigger the activation of Ras‐related C3 botulinum toxin substrate 1 (RAC1), Ras homolog gene family (RhoA), and cell division cycle 42 (CDC42), thereby governing the cytoskeleton remodeling and the activation of AP‐1 and NFAT. 181 , 182 Additionally, some novel noncanonical WNT pathways, such as WNT/mTOR, WNT/YAP/TAZ, WNT/LRP5/mTOR/AKT and WNT/Hippo, have been delineated. 182 , 183 It is warranted that aberrant WNT pathways play important roles in multiple pathological processes, including IR and β cell dysfunction, 184 , 185 , 186 and are causative to the progression of T2DM, 185 , 187 yielding a latent therapeutic strategy for treating this disease. 184 , 188
Dysregulated WNT pathways has been implicated in hepatic IR. It has shown that overexpression of β‐catenin is correlated with a rise in fasting glucose concentrations, while specific knock‐out of β‐catenin in the liver is sufficient to improve hepatic insulin sensitivity and decrease blood glucose concentrations in obese mice. 189 Mechanistically, decreased phosphorylation of hepatic IRS1/2 and GSK3β may mediate the inhibitory effect of WNT/β‐catenin pathway in insulin sensitivity. Meanwhile, the abundant FoxO1 nuclear accumulation connects WNT/β‐catenin activation with hepatic glucogenesis. Consistent with these findings, mice with a knockdown of LDL receptor‐related protein 6 (LRP6), a WNT coreceptor, were resistant to HFD‐induced hyperglycemia and hepatic IR, probably due to the enhanced transcription of leptin receptor. 189 Furthermore, aberrant expression of the key effector of WNT/β‐catenin pathway, transcription factor 7 like 2 (TCF7L2), has been witnessed to induce the transcription of gluconeogenic enzymes (e.g., FBP1, PCK1, and G6Pase) and insulin signaling proteins (e.g., IRS1/2 and AKT2). 190 , 191 Intriguingly, a number of gene TCF7L2 variants are correlated with the susceptibility of T2DM, which primes them as effective predictors of T2DM risk. 192 Hepatic activation of WNT/PCP pathway was also observed to trigger the serine phosphorylation of IRS1 via activating the JNK signaling. 193 In addition, intercellular and interorgan communications might contribute to the hepatic consequences of WNT. For example, secreted frizzled‐related protein 4 (sFRP4), an adipokine with elevated expression in obese WAT, can function as a WNT antagonist and promote hepatic DNL and IR, 194 adding an additional layer of complexity to the role and mechanism of WNT pathway in hepatic function and metabolism.
The WNT pathways also modulate adipose IR 185 and adipogenesis, 195 notwithstanding the diverse regulatory roles of different WNT pathways. It has been disclosed that the activation of WNT5a/PCP pathway could promote adipose IR via inducing adipose inflammation. 196 Concomitant with this, sFRP5, a protein counteracting WNT5a/PCP activation, is able to suppress inflammation by blocking the JNK pathway, and thus improve glucose and insulin intoleration in obese mice. 197 Additionally, WNT pathways in the brown adipose tissue (BAT) also regulate IR progression. For instance, knockdown of LRP6, a receptor of WNTs, was shown to improve BAT insulin sensitivity through increasing the expression of PGC1α and uncoupling protein 1 (UCP1). 198 In terms of adipogenesis, it appears that different WNT members have distinct functional outcomes. Basically, several WNT members, including WNT3a, WNT6, WNT8, WNT10a, and WNT10b, have been observed to suppress adipogenesis in the β‐catenin‐ or PCP‐dependent ways, 184 , 199 while others, such as WNT4, WNT5a, WNT5b and WNT11, are capable of stimulating adipogenesis. 195 , 200 , 201 Given the complexity of WNT members, it is of interest to dissect their real roles in adipogenesis under diverse physiological and pathological conditions.
The WNT10b/β‐catenin pathway is depressed in the skeletal muscle tissues of overweight and prediabetes. Therefore, not surprisingly, activating WNT10b/β‐catenin pathway could improve insulin sensitivity in the skeletal muscle, which is dependent on a reduction in the lipid deposition of myoblasts, which is regulated by SREBP1c. 202 Similarly, activation of WNT/PCP pathway is also related to an improved insulin sensitivity of skeletal muscle. 193 However, another WNT antagonist, secreted frizzled‐related protein 3 (sFRP3), was significantly reduced in the skeletal muscle of prediabetes and T2DM, leading to impaired insulin sensitivity in T2DM, 203 recapitulating the distinct roles of different WNT members in adipogenesis. In summary, the functions of WNT pathways in skeletal muscle IR may vary with different pathways, as well as with different contexts.
The WNT pathways are also important for β cell fate and insulin secretion. 204 Several WNT pathways, such as WNT3a/β‐catenin 204 , 205 , 206 and WNT4/PCP, 207 could improve β cell proliferation and insulin secretion. In‐depth dissections revealed that the activation of TCF7L2 and FoxO1 is key to this proproliferation effect of WNT3a/β‐catenin pathway. 187 , 208 , 209 TCF7L2 is crucial to maintaining the normal functions of β cells, and its silencing can inhibit GSIS through regulating the expression of genes that control the fusion of secretory granule, such as syntaxin 1A, and syntaxin‐binding protein 1. 210 As to the WNT4/PCP pathway, its promotive effect on β cell proliferation relies on JNK activation and the increment of NK6 homeobox 1 and PDX1 protein. 207 In addition, the WNT/Ca2+ pathway may contribute to insulin secretion, as inactivation of Ca2+ and NAFT could diminish the biosynthesis of dense core granule. 211 Nonetheless, some WNTs ligands might exert counteracting effects on β cell proliferation and function, raising the possibility that the uncontrolled imbalance of WNTs might be responsible for the deficiency of functional β cells during T2DM progression. 185 , 212
4.5. UPR pathway
The UPR pathway can be activated by the perturbation of ER homeostasis, which is characterized by the accumulation of unfolded/misfolded proteins, to alleviate the stress of the ER or cause cell death. The UPR cascade encompasses three upstream branches to sense the ER transmembrane stress: inositol‐requiring enzyme 1 (IRE1), protein kinase R‐like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Figure 8A). 213 , 214 Activated IRE1α could produce a transcriptionally active XBP1s, 215 , 216 which further overcomes the ER turbulence by inducing the transcription of genes associated with protein folding, translocating, trafficking, and ER‐associated degradation, 217 as well as by interacting with several signaling pathways, such as p38/MAPK and PI3K pathways. 213 If ER stress is not mitigated, IRE1α would become hyperactivated and oligomerized to degrade hundreds of ER‐localized mRNAs for relieving the folding burden on ER, 213 , 214 or induce cell death 213 , 214 by degrading certain miRNAs targeting proapoptotic genes and activating apoptosis signal‐regulating kinases. 218 Activated PERK could phosphorylate eukaryotic translation initiation factor 2 (eIF2α) to attenuate global protein translation. 213 , 214 By selectively upregulating the expression of activating transcription factor 4 (ATF4), the PERK pathway enhances the transcription of growth arrest and CHOP/GADD34 to negatively regulate itself and induce cell death respectively. 214 , 219 During ER stress, ATF6 can be transported to the Golgi apparatus and then be cleaved to release the transcriptionally active ATF6(p50) cytosolic fragment. 220 The active fragment is translocated to nucleus and then regulates the transcription of multiple genes involved in increasing ER protein‐folding capacity, including XBP1s, to relieve ER stress. 221 To date, the importance of ER stress in T2DM progression 37 has inspired a consensus that activation of UPR pathway is an emblematic phenomenon in T2DM‐associated dysmetabolic outcomes, which in turn controls peripheral IR and β cell dysfunction via various mechanisms.
FIGURE 8.

The UPR pathway in T2DM. (A) Beneficial and harmful effects of UPR activation in regulating β cell function and survival. Adaptive UPR plays a role in β cell survival, proliferation, and identity by promoting the transcription of related genes through ATF4/6 and XBP1s, and by suppressing global translation. Black lines indicate beneficial effects of UPR. Chronical or terminal UPR leads to β cell dysfunction and failure. Chronical PERK activation results in translational suppression of required genes, including chaperones, insulin, and ER enzymes, as well as induction of cell death through CHOP. Besides, ATF4 can interact with its target TRB3 to inhibit the transcriptional activity of CREB, leading to the reduction of exocytotic gene expression. Hyperactivation of IRE1α leads to RIDD process, which cleaves numerous mRNAs including genes related to protein folding, β cell maturation and cell survival, and the activation of JNK and p38. All these finally lead to excess β cell death. Red lines indicate harmful effects of UPR. (B) Model recapitulating the interplay between UPR and insulin response in peripheral tissue. During T2DM, elevated circulating factors, such as glucose, cytokines, and FFAs, can trigger ER stress, which activates the three UPR branches. Activated PERK/eIF2α/ATF4 arm affects insulin sensitivity. On the one hand, the UPR may modulate the transcription of genes associated with inflammation, ER stress and insulin response, such as TRB3, an endogenous inhibitor of AKT. On the other hand, activated PERK can directly regulate the expression and activity of FoxO1, thereby enhancing insulin resistance. IRE1/XBP1 impairs insulin signaling through enhancing the activation of JNK and transcription of P300. Both JNK and P300 blunt the activation of IRS1/2. Inhibition of IRE1α activity by BI‐1 increases hepatic insulin sensitivity and glucose homeostasis. ATF6 branch protects organs from insulin resistance by inhibiting CREB activity, increasing PPARα transcriptional capacity and the expression of chaperones and PERK arm inhibitor P58(IPK).
Most UPR components, induced by increased ER stress, have been observed to be upregulated in peripheral tissues during obesity and T2DM (Figure 8B). 39 , 222 , 223 Subsequently, the UPR pathway is able to modulate IR in the liver, 224 adipose tissue, and skeletal muscle, 224 as well as adipogenesis, 225 despite certain inconsistent roles among different branches. Activated PERK pathway probably increases hepatic IR, as evidenced by that liver‐specific depression of the PERK/eIF2α/ATF4 pathway by GADD34 overexpression or ATF4 depletion could improve IR and glucose intolerance in diet‐induced obese (DIO) mice. 226 , 227 The underlying mechanisms might be associated with transcriptional regulation of numerous targets, such as tribbles homolog 3 (TRB3) (an endogenous inhibitor of AKT), 228 , 229 PPARγ, 230 and glutamic pyruvate transaminase 2 (GPT2) (a promotor of gluconeogenesis). 231 In addition, PERK may counteract the effect of AKT by potentiating FoxO1 activity. 232 Notably, PERK also acts as a receptor for the gut‐microbe‐derived metabolite, trimethylamine N‐oxide, 233 which is increased in IR and associated with several complications of metabolic syndrome in human. 234 In contrast, it has also shown that activation of the eIF2α/ATF4 pathway by the heme‐regulated eIF2α kinase (HRI) can promote the expression of FGF21, a metabolism‐beneficial liver hormone, thereby reducing glucose intolerance in DIO mice, 235 and ensuring the effects of metformin on appetite and weight loss, 236 , 237 , 238 indicative of the ambiguous roles of PERK pathway in T2DM.
The IRE1α branch is also abnormally activated in the liver with IR/hyperinsulinemia, along with increased XBP1s splicing and nuclear localization. 239 Intriguingly, activation of IRE1α by insulin plays protective effects on hepatic insulin action and glucose homeostasis, 240 which may be related to activating growth differentiation factor 15 (GDF15) transcription, 241 driving hepatic autophagy, 242 relieving ER stress, and/or decreasing FoxO1 expression. 243 However, IRE1α/XBP1s activation was also found to blunt insulin signaling and exacerbate IR in the liver 244 by reducing expression of the proinflammation factor, Bax inhibitor 1 (BI‐1), 245 and inducing the acetylation of IRS1/2. 246
Distinct to the PERK and IRE1α branches, the ATF6 is decreased in the liver of diabetic mice, 247 and potentially functions protectively in hepatic glucose metabolism. Indeed, hepatocyte‐specific overexpression of ATF6α reduced hepatic glucose output and steatosis, 248 while its whole‐body deletion exacerbated glucose intolerance. 249 Mechanistically, ATF6 can suppress the transcription of gluconeogenic genes by disrupting the interaction between CREB and CREB‐regulated transcription coactivator 2 (CRTC2), 250 promote hepatic FA oxidation by interacting with PPARα, 248 and suppress the PERK/eIF2α/ATF4 branch by promoting P58(IPK) expression. 228
In obese individuals, chronic ER stress and UPR pathways in the adipose tissue can be sustainedly activated by increased circulating levels of FFAs 251 and insulin, 252 which in turn impairs insulin signaling of adipose tissue. For example, the IRE1α/JNK1 axis inactivates IRS1, 253 and the PERK/ATF4/TRB3 axis induces AKT suppression, 222 abolishing insulin sensitivity and glucose transport in the adipose tissue. Adipose UPR pathways also contribute to insulin desensitization in other organs, for one reason that ER stress is linked to reduced adiponectin secretion, 254 , 255 , 256 and for another reason that the PERK arm can elevate circulating levels of tumor necrosis factor α (TNFα), interleukin‐6 (IL‐6), and IL‐1β. 251 , 257 All of these impose a vicious ER stress feedback and further exacerbate the adipose tissue per se and systemic IR. 258
In the skeletal muscle, aberrant activation of UPR pathways is also found in patients with T2DM and pregnant women with obesity or gestational diabetes. 223 , 259 These abnormalities impair muscular insulin action via the mechanisms that resemble those in the adipose tissue. 259 That is, both PERK‐induced TRB3 activation 259 and IRE1α/JNK‐triggered IRS1 phosphorylation can contribute to muscular IR. 40 Furthermore, transcriptionally activated ATF6/XBP1s pathway warrants increased expression of skeletal muscle kidney‐enriched inositol polyphosphate phosphatase (SKIP) to induce muscular IR. 260 Of note, activation of IRE1α/JNK axis also bridges the increased release of IL‐6 and TNFα from skeletal muscle and the disrupted systemic insulin sensitivity in T2DM. 261 , 262 In addition, activated PERK is implicated in the secretion of myokines, including musclin and ceramides, 263 , 264 which are key to insulin desensitization in peripheral organs. 265 In contrast, inhibition of ER stress significantly enhances the expression of UCP1 in the inguinal WAT and improves metabolic phenotypes in DIO mice, 266 which is linked into a decrease in the JNK‐mediated degradation of PPARγ. Moreover, the ATF6 branch might regulate multiple processes in the skeletal muscle, including exercise training adaption, 267 glucosamine‐induced disruption of glucose uptake 268 and apoptosis, 269 all of which are closely related to T2DM.
On the road to T2DM, proinsulin is prone to misfolding, and excess insulin production can aggravate ER stress in β cells. 37 In parallel, dysregulations of UPR pathways, for example, transcriptional dysregulation related to pancreatic aging, 270 local inflammation 271 and glucotoxicity, 37 are also observed in the islets of T2DM patients and rodents, 272 which possibly exert effects on β cell dysfunction by regulating insulin production and cell fate. Supporting this notion, the PERK pathway is considered to act as a metabolic sensor to modulate insulin production and secretion in a delicate way. In detail, although ablating PERK is shown to impair insulin trafficking and β cell survival, leading to insulin insufficiency and hyperglycemia, 273 partial attenuation of PERK activity instead enhances GSIS through regulating ER chaperones and Ca2+ transit. 274 , 275 Moreover, both β cell‐specific ablating 276 and enhancing phosphorylation 277 of eIF2α cause reduced insulin secretion, increased β cell apoptosis and thus severe diabetes. Besides, PERK/ATF4/TRB3 axis acts through inducting CREB inhibition to depress the transcription of key exocytosis genes, and consequently reduce insulin secretion. 278 Meanwhile, the IRE1α pathway also appears to ensure β cell function. The IRE1α/XBP1s axis promotes ER protein folding capacity by regulating the transcription of genes involved in insulin folding, process and degradation, 279 , 280 and thus improves insulin production and secretion. 281 , 282 Supporting this, XBP1s deficiency in β cells markedly blunts GSIS, 280 increases β cell apoptosis by deactivating β cell identity genes, 283 and enhances inflammation and oxidative stress. 281 Conversely, prolonged XBP1s production in rat β cells can inhibit the expression of β cell markers, and eventually lead to β cell apoptosis. 284 , 285 The ATF6 pathway has an essential role in supporting β cell function and survival by inducing the transcription of target genes, including ER chaperones, 275 disulfide redox enzymes, and several quality control and degradation factors, as well as XBP1s. 37 In line with this, whole‐body deletion of ATF6 impaired glucose intolerance and blunted insulin secretion, 249 whereas ATF6 induction improved β cell insulin secretion and viability under ER stress conditions. 286 Furthermore, inhibiting ATF6 could result in embryonic lethality and β cell receding by arresting cell cycle entry, 287 while upregulating ATF6α activity is able to markedly expand β cell mass in db/db mice, 288 suggesting its importance in the fate control of β cells. 289 , 290 Of note, ATF6 also participates in β cell proliferation induced by the salt‐inducible kinases inhibitor, known as HG‐9‐91‐01, and knockdown ATF6 can efficiently reverse such proproliferating effect. 291
4.6. Hippo pathway
The Hippo signaling pathway, mainly comprising macrophage stimulating 1/2 (MST1/2), large tumor suppressor 1/2 (LATS1/2), and their cofactors such as salvador homolog (SAV) and MOB kinase activator 1A/B (Mob1A/B), predominantly controls the activity of YAP/PDZ‐binding motif (TAZ), two closely related mammalian transcriptional coactivators that shuttle between the cytoplasm and nucleus (Figure 7B). 292 , 293 , 294 Generally, active MST1/2 phosphorylates LATS1/2, which in turn phosphorylates YAP/TAZ and thus inhibits the activity of YAP/TAZ. 292 In contrast, dephosphorylation allows YAP/TAZ to translocate into the nucleus and combine with its partner transcriptional enhanced association domain (TEAD) to reprogram gene expression. 292 However, the upstream regulation of YAP/TAZ is beyond the Hippo pathway, especially when cells are stimulated by those uncanonical signals such as mechanical stimuli, G protein‐coupled receptor (GPCR) ligands, metabolites, and cell stresses. 292 , 293 , 294 Currently, deciphered regulatory loops that involve many kinds of metabolic responses gradually related YAP/TAZ and Hippo pathway to the peripheral glucose metabolism, insulin signaling, the fate of β cells, and thus pathological processes of T2DM. 293 , 294 , 295
Cells may employ the YAP/TAZ as a coordinator to balance glucose energy supply and consumption. 295 Glucose deprivation, reduced glucose uptake and inhibited glycolysis could repress the activity of YAP/TAZ, the formation of the YAP/TEAD complex, and thus suppress the transcription of target genes, 296 , 297 , 298 such as GLUT3, hexokinase 2 (HK2) and phosphofructokinase B3 (PFKB3), as well as to diminish glucose consumption. Mechanistically, deficient glucose metabolism could act through the activation of AMPK and LAST1/2 to inhibit YAP/TAZ activity, and could also depress the phosphofructokinase 1 (PFK1) to prevent the formation of the YAP/TEAD complex. 296 In contrast, high glucose could upregulate HBP‐dependent O‐GlcNAcylation of YAP, which enhances YAP activity by restraining LATS‐dependent phosphorylation and proteasomal degradation. 299 Given that blood glucose level after long‐standing fasting and postprandial during T2DM is associated with dysregulated cellular glucose metabolism, it is possible that the Hippo pathway may modulate T2DM progression via regulating cellular glucose metabolism.
YAP is decreased in the skeletal muscle of obese patients and mice with IR. 300 Consistent with this, the YAP/TAZ pathway might promote peripheral insulin signaling and repress glucogenesis, whereas the Hippo pathway works inversely. For example, YAP/TAZ might upregulate the insulin/AKT pathway by potentiating IRS2 transcription in the liver of mice with codeleted PTEN and SAV1. 301 Moreover, the mutually concordant positive effects between YAP/TAZ and mTOR pathways also underpin the connection between YAP/TAZ and the insulin/AKT signaling. 302 On the contrary, the Hippo pathway exerts converse effects on insulin signaling and glucose metabolism. One evidence is that upregulating the upstream Hippo kinase MST3 could exacerbate IR, hyperglycemia and hyperinsulinemia by depressing IRS1 and upregulating the transcription of gluconeogenic regulators and enzymes. 303 YAP can also act through PGC1α to suppress the transcription of hepatic gluconeogenic genes, lower plasma glucose level and improve glucose tolerance. 304 Additionally, glucagon activates LATS and restrains YAP, 305 which in return eradicates upregulation of hepatic gluconeogenic genes and represses gluconeogenesis. 304
The Hippo pathway may also serve as an essential regulator in the capacity of adipose tissue to endure metabolic stress. 306 To deal with the increasing stress during obesity, WAT might rely on YAP/TAZ to resist apoptosis and ameliorate T2DM. 38 , 306 Additionally, it has shown that the impairments of FA oxidation and consequent enhanced adiposity of skeletal muscle in obese patients or prediabetic mice are caused partially by reduced YAP. 300 Intriguingly, the Hippo pathway may also participate in inflammatory responses 300 , 307 and mitochondrial maintenance, bringing about an attractive notion that dysregulation of YAP/TAZ and Hippo pathway might influence the advancement of T2DM in the manners beyond our imagination.
The Hippo pathway and YAP/TAZ also modulate pancreatic cell differentiation, proliferation and apoptosis. 293 , 308 Overall, YAP/TAZ could render expansion of early embryonic pancreas epithelium, but hamper endocrinogenesis of pancreatic progenitor cells. 308 In harmony with this, YAP/TAZ in adult pancreatic β cells remains low expression level, 308 indicating that silencing of YAP/TAZ is critical for the maturation of β cells. In contrast, the Hippo pathway 309 tends to induce apoptosis of β cells, as MST1 is activated to empower β cell apoptosis, while its deletion pronouncedly restores β cell function and mass, attenuating diabetic conditions in mice with T2DM.
4.7. HIFs pathway
HIFs are a family of DNA binding transcription factors activated by hypoxia in mammalian, among which HIF1α and HIF2α are best‐studied and have been reported to play critical roles in several diseases, such as T2DM, atherosclerosis and cancer. 310 Under hypoxic conditions, HIF1α maintains stability via limited oxygen and could be translocated to the nucleus to bind to HIF1β and other response elements, 311 thus augmenting the activation of its target genes transcriptionally, such as vascular endothelial growth factor (VEGF), angiopoietin, and platelet‐derived growth factor.
Previous evidence demonstrated that the impaired HIFs signaling pathway acts as one of the key pathogenic factors of T2DM, and is involved in IR. 310 , 312 In hepatocytes, HIF1α modulates glucose transport and fructose production by regulating its downstream targets, such as GLUT1 and PDK1. 310 HIF2α could regulate gluconeogenesis and HGP through the IRS2/PI3K/AKT pathway. 313 Activated HIF2α by refeeding is also able to attenuate postprandial glucagon signaling through upregulating cAMP level and inhibiting CREB activity, ultimately repressing the expression of PEPCK and G6Pase. 314 Meanwhile, intestine HIF2α could inhibit the expression of neuraminidase 3 (Neu3), thus substantially ameliorating hepatic steatosis, glucose intolerance and IR. 315 Adipocyte‐specific knockout HIF1α and HIF1β in HFD‐fed mice can enhance glucose tolerance and insulin sensitivity by inducing tyrosine phosphorylation of signal transducer and activator of transcription 3 (STAT3) and suppressor of cytokine signaling 3 (SOCS3)‐mediated increase of adiponectin. 316 Consistently, decreased insulin sensitivity and glucose intolerance could be observed in mice with WAT‐specific HIF1α overexpression. 317 Notably, as essential mediators of adaptation to hypoxia, HIFs pathways also play a key role in controlling mitochondrial functions and ROS production, which potentially complicate the interactions between mitochondrial dysfunction and glucose metabolism and T2DM. 318
Disruption of HIFs homeostasis affects insulin secretion. 319 HIF1β is significantly down‐regulated in islets of T2DM. 320 Specific knockout of HIF1α in β cells impaired insulin secretion and decreased glucose‐stimulated ATP production. 319 However, overload of HIFs could also lead to β cell dysfunction. For example, deletion of von Hippel–Lindau factor, a regulator of HIF hydrolysis, markedly impaired insulin secretion and glucose homeostasis in mice, accompanied with increased HIFs levels. 321 These results suggest that proper levels of HIFs might be critical to maintaining β cell homeostasis and function, which awaits further investigation.
4.8. TGFβ pathway
The TGFβ superfamily contains a number of subfamily proteins, such as bone morphogenetic proteins (BMPs), activins and TGFβs (Figure 7D). Activation of TGFβ receptors could further activate small mothers against decapentaplegic homolog (SMADs), a class of second messengers, and other signaling pathways, such as MAPK, RHO GTPase, and PI3K/AKT pathways. 322 The TGFβ pathway connects contextual determinants with specific cellular responses by controlling SMAD‐dependent transcription programming and integrating with other pathways. These fundamental mechanisms underlie the function of TGFβ pathway in controlling peripheral insulin signaling and pancreatic β cell biology during T2DM pathogenesis.
An increasing body of evidence supports that the TGFβ pathway can affect glucose homeostasis. It is increasingly clear that several members of the TGFβ superfamily, including activin A and B, GDF11, BMP2‐4, and TGFβ1‐3, have emerged as novel regulators in the insulin signaling of adipose tissue, skeletal muscle, and liver. 323 Among which, the bioactivity of the activin/GDF11 is reported to be modulated by the antagonists follistatin (FST) and follistatin like 3 (FSTL3), which could increase fat mass and adipose IR, and therefore disrupt glucose homeostasis. 324 Specifically, overexpression of fstl3 in obese mice is capable of improving muscle insulin sensitivity and reducing fat accumulation, but enhancing hepatic glucagon sensitivity. 325 By contrast, genetic removal of fst enhanced WAT insulin sensitivity and suppressed HGP, thereby ameliorating glucose tolerance in obese mice. 326 BMPs, particularly BMP4, 6, and 7, are also involved in the control of glucose homeostasis. Specifically, BMP7 promotes, while BMP4 decreases insulin sensitivity in the adipose and muscle of T2DM mice, respectively. 327 Mechanistically, BMP7 can activate PDK1 and AKT to boost GLUT4 translocation to the plasma membrane and in turn increase glucose uptake, while BMP4 counts on the activation of PKCθ to induce IR. BMP6 may restore the levels of blood glucose and lipids in T2DM mice via reducing hepatic gluconeogenesis and glucose output. 328 However, the dysregulation of TGFβ pathway in the diabetic peripheral tissues remains incompletely understood and awaits further study.
The discoveries about the roles of TGFβ in pancreatic development and functions establish another strong link between the TGFβ pathway and T2DM, 329 regardless of the diversity of TGFβ pathway in controlling the proliferation, apoptosis and differentiation of β cells. To be specific, activin A could inhibit β cell proliferation, but facilitate the generation of β cells from adult stem cells, human embryonic stem cells, ductal cells, human amniotic epithelial cells), 330 and α cells. 331 Activin A and activin B potentiate the dedifferentiation of β cells by depressing and upregulating the transcription of crucial genes associated with β cell maturity and immaturity, respectively. 332 Activation of SMADs is key to the regulation of TGFβ signaling on β cell fate. In detail, SMAD7 is believed to enhance proliferation, 333 while SMAD2 and SMAD3 block proliferation of β cells by regulating the nuclear localization of p27, a cell‐cycle modulator inhibiting cyclin‐dependent kinase (CDK). Otherwise, activation of TGFβ/SMAD3 signaling also has a role in β cell apoptosis. 334 Furthermore, SMAD2, 3, and 7, particularly SAMD7, connect the dedifferentiation and proliferation of β cells together. 335 Recently, the roles of TGFβ pathway in controlling β cell functions are also emerging. For instance, activin A/B, TGFβ1, FST, GDF11, and BMPs, have been demonstrated to enhance GSIS, 336 , 337 , 338 while the addition of FSTL3 is able to reverse these effects in both functional and nonfunctional islets. 338 Mechanistically, these effects may be predominantly attributed to the activation of SMAD2, since its disruption could compromise insulin secretion under high glucose and diabetic conditions. 337 Furthermore, the effects of TGFβ pathway on GSIS may be associated with the modulation of KATP and calcium channels activity, 337 as well as the altered expression of essential genes governing insulin secretion, such as the GLUT2 and calcium voltage‐gated channel subunit alpha1 D genes. 338
4.9. FGFs pathway
The human FGF superfamily consists of 22 structurally related signaling molecules. 339 FGFs exert their pleiotropic effects by dimerizing, activating and phosphorylating FGF receptors, leading to the activation of the RAS/MAPK, PI3K/AKT, Ca2+, PKC, and STAT signaling cascades in a cellular context‐dependent manner (Figure 9). 340 Most FGFs family members canonically present in the extracellular matrix (ECM) and act in autocrine and/or paracrine manners to activate FGFRs. 341 While FGF19 subfamily members, including FGF19 (the mouse ortholog FGF15), FGF21, and FGF23, are liberated from the ECM into the bloodstream and thus work in an endocrine manner. 342 Tremendous efforts throughout the past decades have ascertained the pleiotropic effects of FGFs pathways on T2DM progression, potentiating the development of engineered FGF analogs and mimetics targeting T2DM. 343 , 344 , 345
FIGURE 9.
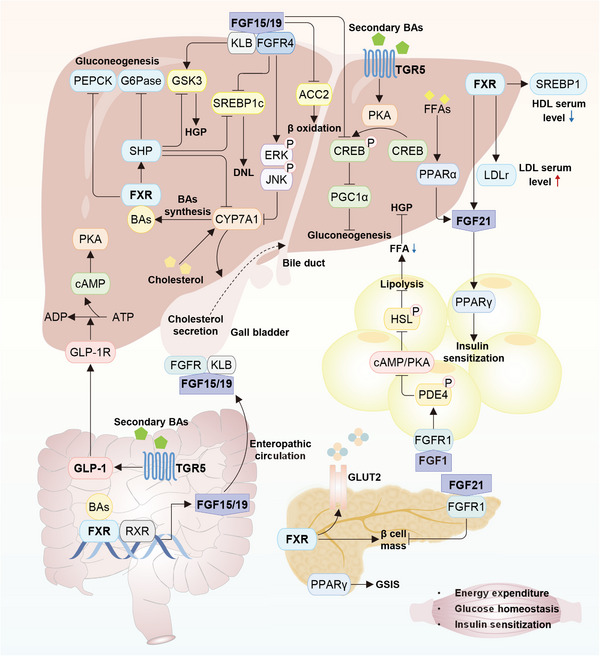
Crosstalk among bile acids, FGFs, and GLP‐1 pathways in T2DM. Several important signaling pathways are interacted in multiple metabolic tissues. In hepatocytes, FXR induces SHP, subsequently inhibits CYP7A1 transcription and BA synthesis. Under BAs stimuli, FXR inhibits the transcription of PEPCK and G6Pase to suppress gluconeogenesis, represses GSK3 to suppress hepatic glycogen production (HGP), and suppresses SREBP1C to regulate de novo lipid synthesis (DNL). In the ileum, BAs are reabsorbed into enterocytes and then enter circulation induced by intestinal FXR. Together, activated FXR could reduce serum glucose level. In addition, FGF15/19 is secreted to the blood and then circulates to the liver, where FGF15/19 binds to its receptors, subsequently inhibiting BA synthesis and meanwhile regulating downstream molecules in metabolic tissues, including ACC1, GSK3, CREB, and SREBP1c. However, FGF21 stimulates glucose uptake in the adipose tissue, but inhibits β‑cell proliferation in pancreatic islets. Interestingly, activated TGR5 promotes the secretion of FGF19 and GLP‐1 to blood and mediates downstream functions. PPARs have been characterized as core lipid sensors that modulate whole‐body energy metabolism. Collectively, the cooperation among BAs receptors, FGFs, GLP‐1, and PPARs maintains the glycolipid metabolic responses in T2DM. The online resource inside this figure was quoted or modified from Scienceslide2016 plug‐in.
Several subclasses of FGFs pathways have been disclosed to impose beneficial impacts on peripheral insulin action and glucose homeostasis. Among them, FGF21 has obtained the most interests owing to its powerful effects on favoring insulin sensitivity and glucose metabolism within the liver and adipose tissue. 343 , 346 As a stress‐inducible hormone, FGF21 is strongly induced by starvation, amino acid restriction, ketogenic diet and HFD treatment in the liver. In the liver, by binding to FGF receptor 1c (FGFR1c) and the coreceptor β‐Klotho, 347 FGF21 can elicit the FGF signaling to increase insulin sensitivity by suppressing mTORC1, 348 reduce lipid accumulation by promoting PGC1α‐induced FA oxidation 349 and repressing SREBPB1c‐mediated lipogenesis, 350 and block TG‐enriched very‐low‐density lipoprotein (VLDL) uptake by decreasing VLDL receptor expression. 351 In the adipose tissue, FGF21 may stimulate glucose uptake in an insulin‐independent manner but instead through inducing GLUT1 expression, inhibit lipolysis by sequential activation of ERK1/2 and serum response factor/Ets‐like protein‐1 (SRF/Elk‐1), 352 , 353 , 354 and promote adiponectin production and secretion from adipocytes through a PPARγ‐dependent mechanism. 355 , 356 Paradoxically, the endogenous expression and circulating level of FGF21 are increased in obese humans and mice, which is likely linked to the chronic mild mitochondrial dysfunction. 357
The FGF15/19 signaling is another important pathway for peripheral insulin action and glucose metabolism. After feeding, FGF15/19 is released postprandially from the small intestine to serum and arrived at other organs. 358 In the liver, FGF15/19 binds to its coreceptors, FGFR4 and β‐Klotho, to trigger ERK pathway and consequently phosphorylate and inactivate GSK3 in an insulin‐independent manner, ultimately increasing the hepatic GS activity. 359 Meanwhile, FGF15/19 can suppress gluconeogenesis through a mechanism involving the inactivation of CREB and subsequent downregulation of PGC1α. 358 Additionally, FGF15/19 is strongly induced by the nuclear receptor farnesoid X receptor (FXR) in the small intestine to repress hepatic BAs synthesis. 360 Beyond these, FGF15/19 also regulates postprandial glucose and energy homeostasis 361 through modulating the synthesis of proteins, glycogen and glucose.
In addition to aforementioned FGFs, FGF1 is emerged as a potent regulator in peripheral glucose homeostasis. 344 Both acute and chronic treatment with recombinant FGF1 can normalize blood glucose concentration, at least partially attributed to suppressing HGP and promoting insulin‐dependent glucose uptake in skeletal muscle. 362 At the same time, FGF1 may activate the phosphodiesterase 4D (PED4D) and then repress cAMP/PKA axis to inhibit lipolysis in adipose tissue and suppress HGP. 363 Of note, FGF1 expression in adipose tissue has been reported to be controlled by PPARγ, 364 , 365 suggesting that the PPARγ/FGF1 axis might be another crucial mechanism for systemic insulin sensitivity. Similar to FGF1, FGF4 may also function as a potent anti‐hyperglycemic factor, and paracrine FGF4 exhibits higher hypoglycemic capacity even than endocrine FGF21. 366 Correspondingly, FGF4 treatment is able to effectively improve IR in genetic‐induced obese mice through many potential mechanisms, for example, activating AMPK in the skeletal muscle to promote GLUT4 expression and its translocation to cell membrane. 366
The FGF pathway also confers metabolic benefits in β cells function and survival, as well as central neural regulation. 344 For instance, FGF21 knockout mice display islet hyperplasia and distort morphology, along with increased β cell proliferation and a larger α‐cell population, while islets and cultured β cells treated with FGF21 are partially protected from apoptosis possibly due to the activation of PI3K/AKT signaling. 367 In addition, FGF15/19 could reduce food intake and improve glucose tolerance by inhibiting the hypothalamic–pituitary–adrenal axis. 368 , 369 , 370 Moreover, glucose can induce the release of FGF1 into the cerebrospinal fluid, which further initiates feeding suppression and glycemic control by interrelating with tanycytes, astrocytes and glucose‐sensing neurons of the hypothalamus. 344
4.10. Bile acids
BAs, the major components of bile, are synthesized from cholesterol in liver via the classical pathway regulated by cholesterol‐7α‐hydroxylase (CYP7A1). 371 BAs play important roles in facilitating the absorption of dietary lipids, maintaining cholesterol homeostasis and activating hormones (Figure 9). Over the past decades, BAs have been regarded as critical regulators in glucose, lipid and energy metabolism, as well as potential targets for preventing cardiovascular complications and improving glycemic control in T2DM patients. 372
BAs act primarily through activating intracellular ligand‐activated receptors, among which the best‐studied are FXR and G protein‐coupled BA receptor (TGR5). FXR (also known as NR1H4) is a member of nuclear receptor family and expressed in many tissues. 373 FXR located in the intestine and liver, could be activated directly by BAs and forms a heterodimer with retinoid‐X‐receptor (RXR). FXR exerts effects on the expression of downstream genes and the inhibition of BAs synthesis through two ways: (1) inducing the small heterodimer partner (SHP) to inhibit CYP7A1 in the liver; (2) increasing circulating FGF19/15 in the gut to repress CYP7A1. TGR5, belonging to the rhodopsin‐like superfamily of G protein‐coupled receptors, is a cell membrane receptor for secondary BAs and moderately expressed in nearly all tissues. BAs could regulate glucolipid metabolism via binding to FXR and TGR5. 374 For example, activated FXR is able to repress hepatic DNL 375 and gluconeogenesis 376 by inhibiting the expression of SREBP1c, PEPCK, and G6Pase. Moreover, systematic knockout of TGR5 could aggravate IR and glucose intolerance through miR‐26a and the subsequent cAMP/PKA pathway. 377 , 378 Furthermore, both FXR and TGR5 are capable to regulate GSIS in pancreatic β cells through inducing the relocalization of GLUT2 on the membrane. 379
TGR5 is coexpressed with FXR on L cells in the intestine, and the activation of FXR could facilitate the release of GLP‐1, 380 which then enhances insulin secretion and improves glucose homeostasis. Intriguingly, hyocholic acid has been shown to promote GLP‐1 secretion through synchronously activating TGR5 and inhibiting FXR, which exerts profound effects on glucose metabolism in the pig and diabetic mouse models. 381 As well, a recent study showed that soluble dietary fiber oligofructose could activate TGR5 to enhance GLP‐1 activity by stimulating 6α‐hydroxylated BAs, thus improving host glucose metabolism and maintaining glucose homeostasis. 382 All of above suggest that BAs signaling pathways mediated by intestinal GLP‐1 have strong potential for T2DM therapeutic applications.
The gut microbiota has been showed to regulate the level and composition of BA metabolites through secreting enzymes to catalyze the dehydroxylation of BAs, thus influencing the function of BAs in regulating glycolipid metabolism. In turn, BAs also interact with and affect the gut microbiota, 383 thus creating a dynamic equilibrium between them. Interestingly, metformin has been reported to decrease the abundance of species of B. fragilis in the intestine to upregulate the bile acid glycoursodeoxycholic acid in the gut, thus improving metabolic dysfunction and hyperglycemia in T2DM patients. 384 Adjustment of BAs homeostasis has been applied to optimize glycaemia parameters and improve pathological conditions in various diabetic animal models and human patients, 385 , 386 which implies potential therapeutic roles for BAs signaling in the treatment of T2DM.
4.11. Ca2+ signals
Ca2+ signals are finely tuned by a huge group of proteins, including channels, pumps, transporters, and binding proteins, 387 , 388 which underpin the accurate influx and reflux of Ca2+. 387 Ca2+ homeostasis is key to the well‐proceeding of many cellular processes, 387 , 388 including insulin secretion, β cell mass, insulin sensitivity, and glucose sensing and disposing. Disruption of Ca2+ homeostasis, which has been witnessed in the peripheral tissues and β cells of the diabetic mice and human patients, is widely considered to be a critical regulator of IR, β cell dysfunction, and T2DM.
Cellular Ca2+ pool encompasses several subcellular pools, primarily including the cytosolic, ER, mitochondria, Golgi apparatus, and even lysosomes ones, which intimately and dynamically communicate with each other as well as extracellular space. 387 , 389 , 390 A range of extra‐ and intracellular signal stimuli could act through various signaling pathways to activate divergent Ca2+ channels on different membranes, resulting in Ca2+ influxes and thus producing specific Ca2+ signatures in distinct subcellular compartments. 387 , 388 Among them, several Ca2+ channels are extensively studied, such as channels voltage‐dependent Ca2+ channels (VDCCs) and ligand‐gated channels and transient receptor potential (TRP) channels on plasma membrane, 387 , 388 ORAI1 channel and sarco/ER Ca2+‐ATPase (SERCA) pumps on ER membrane, 391 and voltage‐dependent anion channels (VDACs) 387 , 389 , 392 and mitochondrial calcium uniporter complex on the inner and outer mitochondrial membranes. 392 , 393 , 394 , 395 Elevated Ca2+ concentration could initiate multiple signaling cascades by interacting with and activating the Ca2+ sensor protein calmodulin (CaM), which subsequently induces numerous downstream targets in various ways. The major downstream targets of Ca2+‐bound CaM are Ca2+/calmodulin‐dependent protein kinases (CaMKs), which in turn elicit the phosphorylation of CERB or the activation of calcineurin (CaN)/NFAT1 pathway, 396 , 397 thereby regulating glucose sensing, insulin exocytosis, and insulin transcription. 398 In addition, many other metabolism‐related pathways and transcription factors are tightly regulated by the Ca2+ signaling, such as MAPK, nuclear factor‐κB (NF‐κB), myocyte enhancer factor 2, and FoxO1. 399 , 400 , 401 Meanwhile, to avoid overflow of Ca2+ and cytotoxicity, other signaling pathways, such as the PLC/PIP2/IP3 pathway, could trigger various pumps and exchangerfFs, including the Na+/Ca2+ exchanger, 392 , 402 the plasma membrane Ca2+ ATPase, 389 , 391 IP3 receptor (IP3R), and ryanodine receptor (RyR), 387 , 389 to remove Ca2+ outward, thus restoring Ca2+ concentrations. Additionally, mitochondria‐associated ER membranes (MAMs) hinge on an IP3R1/GRP75/VDAC1 complex‐dependent mechanism to take charge of Ca2+ transport from the ER to mitochondria. 403 Besides, the Golgi apparatus and lysosomes also participate in Ca2+ homeostasis by acting as Ca2+ stores, which have been elaborately reviewed elsewhere. 404 Cumulatively, attributed to this subtle regulatory network, cellular Ca2+ concentrations remain lively balanced, which is central to the fine progressing of many cellular behaviors and states.
Actually, previous studies suggested that hyperglycemia relies on a Ca2+‐dependent mechanism to activate glucose transport in skeletal muscle. 405 Additionally, it is well established that insulin stimulation induces a rapid and transient increase in cytoplasmic and mitochondrial Ca2+ through RyR and IP3R activation, which is required for GLUT4 translocation and glucose uptake. 406 , 407 Ca2+ signals also play important roles in controlling insulin secretion 408 , 409 and β cell proliferation. 410 , 411 , 412 For example, during GSIS, fusion of insulin granules with the plasma membrane requires an increase in intracellular Ca2+. 413 Consistent with the essential role of Ca2+ in insulin action and secretion, it was defined that Ca2+ dysregulation is linked to the progression of T2DM.
Dysregulation of ER Ca2+ concentration is notably implicated in the progression of IR in peripheral tissues. For instance, SERCA2b has been widely observed to be dramatically reduced in the liver, 414 WAT, 415 and skeletal muscle 416 from obese and diabetic mice, causing the reduction of ER luminal Ca2+ concentration and therefore aggravating IR. 417 , 418 Consistently, liver‐, 417 adipocyte‐, 415 or muscle‐specific 419 restoring SERCA in obese and diabetic mice could improve systemic insulin sensitivity and glucose homeostasis, ameliorate hepatosteatosis, reduce adipose mass, but increase BAT energy expenditure and UCP1/3 expression, in which reduced ER stress and decreased PKCδ activation may contribute to these observed effects. Reduced intracellular and ER Ca2+ storage capacity, resulting from the accumulation of cholesterol, 420 , 421 , 422 has also shown to be linked to PKC activation, 423 , 424 as well as the development of hepatic IR. 425 , 426 Mechanistically, activated PKC may mediate the phosphorylation of ORAI1 and inhibit its activity, thereby decreasing the refilling of the ER Ca2+ stores and further aggravating hepatic IR. 427 Of note, it has been reported that ER‐mitochondria interactions and interorganelle Ca2+ exchange are reduced in high‐fat high‐sucrose diet‐induced obese mice, while a healthy diet could effectively restore the communication between ER and mitochondria that improves hepatic insulin sensitivity and glucose homeostasis, 428 which might depend on the activation of PKCε. 429
Obesity and diabetic conditions bring about mitochondria Ca2+ turbulence and thus resulting in mitochondria dysfunction, 430 , 431 which collectively aggravate T2DM progression by abolishing insulin signaling. 430 , 431 , 432 For example, obesity is reported to drive the increased localization of IP3R1 at the MAMs, leading to an overload of mitochondrial Ca2+ influx, grievous oxidative stress and IR in the liver. 433 , 434 Furthermore, disruption of MAMs and mitochondrial Ca2+ have been viewed as early events preceding mitochondrial dysfunction and IR in the liver and skeletal muscle. 430 , 432 For example, in mice fed a HFD, the mitochondrial dysfunction arising from the downregulation of glucose‐regulated protein 75 (GRP75), an important regulator for mitochondrial Ca2+ homeostasis, can aggravate systemic IR, while induction of GRP75 in mice could inverse such effects. 435 Furthermore, reduced mitochondrial Ca2+, resulting from the mutation and acute deletion of seipin, a protein modulating mitochondrial Ca2+ by interacting with SERCA, 436 , 437 has been pointed out to impair tricarboxylic acid cycle cycles and subsequent reduction in citrate, 438 leading to the defects in lipid storage and lipogenesis. Similar to mitochondrial Ca2+ deficiency, mitochondrial Ca2+ overload caused by excessive MAM content has also been shown to induce IR and fat deposition in hepatocytes. 439 , 440 Meanwhile, in the skeletal muscle, MAM formation is also found to be augmented by obesity in a PDK4‐dependent way, thereby leading to increased mitochondrial Ca2+ accumulation, mitochondrial dysfunction, ER stress, and consequent IR. 434
Furthermore, other regulators and signaling pathways responsible for whole‐cellular Ca2+ homeostasis also play critical roles in regulating peripheral IR progression. For instance, obesity can upregulate CaMKII activity via enhancing ER stress, 441 , 442 which further compromises the insulin signaling in the liver. 228 While, abolishing CaMK1D significantly improves peripheral insulin sensitivity and glucose control in DIO or HFD‐fed mice by reprograming glyceraldehyde 3‐phosphate dehydrogenase‐/peroxisomal biogenesis factor 3 (PEX3)‐mediated metabolic processes. 443 Besides, the capsaicin‐activated TRPV1 is repressed in the adipose tissue from HFD‐fed mice, 444 , 445 and involved in CaMKII/AMPK/SIRT1/PPARγ‐mediated WAT browning. 444 Moreover, activation of TRPV1 may also contribute to enhanced metabolic function of BAT, 445 possibly through mediating SIRT1/PPARγ axis, 445 as well as controlling clock gene oscillations, indicating a protective role of TRPV1 in metabolic diseases. 446 However, these studies might be challenged by other results that genetic deletion or pharmacological inhibition of TRPV1 protected mice from obesity, IR, hypertension, inflammation, or leptin resistance. 447 , 448 Additionally, another TRP channel, TRPM2, is able to mediate angiotensin II‐induced IR in adipocytes. 449 Moreover, silencing of herpud1, an ER membrane protein maintaining intracellular Ca2+ homeostasis under stress conditions, has an ability to decrease insulin‐dependent glucose uptake, GLUT4 translocation, and AKT activation in myotubes via increasing the IP3R‐dependent cytosolic Ca2+ response and CaN activity. 450 Accordingly, correcting the SR lipid composition and thus Ca2+ handling in the skeletal muscle can lead to an improvement in IR in DIO mice. 451
During the pathogenesis of T2DM, Ca2+ homeostasis in the pancreatic β cells is also discerned to be broken, which in turn aggravates the dysfunction of β cells in terms of their insulin secretion and fate decision. For example, due to reduced transcription, 408 , 409 changed location 452 and depressed activity of VDCCs, 453 , 454 Ca2+ influx across VDCCs is decreased in the diabetic β cells, leading to reduced Ca2+ influx near insulin granules docked sites, inhibited fusion of insulin granule with the plasma membrane, and thus impaired GSIS. 452 Specifically, it is reported that IR can induce a reduction in PIP2, and impair VDCC activity and subsequent insulin secretion. 453 Furthermore, depletion of other critical Ca2+ signaling mediators, including CaMKII, CREB, CaM, CaN, and CRTC2, in mouse β cells, can impair insulin secretion and systemic glucose homeostasis. 455 , 456 , 457 On the contrary, specific deletion of Ca2+/CaM‐dependent serine protein kinase in mouse β cells results in reduced blood glucose, hyperinsulinemia, and IR in HFD‐fed mice. Short‐term stimulation of Ca2+ signaling pathways might yield positive effects for β cells, whereas chronic Ca2+ stimulation instead presents deleterious effects on β cell mass. Transient activation of CaMKIV/CREB, 458 , 459 , 460 CaN/NFAT, 461 and CaN/TFEB 462 pathways promote glucose‐mediated β cell proliferation and survival by reprogramming gene transcription, consistent with the positive roles of WNT/Ca2+ pathways in β cell survival. However, mice overexpressing constitutively active CaN or CaMKIIα in β cells instead, develop glucose intolerance and diabetes with decreased β cell mass, 463 which is due to increased apoptosis and decreased proliferation of β cells. Furthermore, aberrantly elevated cytosolic Ca2+ is also required for β cell de‐ or trans‐differentiation processes, resulting in the exacerbation of T2DM. 464 One notable example is that overexpression of CaM caused the trans‐differentiation of β cells into glucagon‐expressing cells and thus T2DM progression, 464 while blocking β cell depolarization or Ca2+ influx markedly reduced trans‐differentiation of β cells into gastrin‐expressing cells in both diabetic mice and isolated islets. 465 Additionally, β cell deletion of ATP binding cassette subfamily C member 8 (ABCC8), a subunit of the ATP‐sensitive potassium (KATP) channel, results in increased cytosolic Ca2+ and loss of β cell maturation, accompanied by enhanced expression of dedifferentiation marker aldehyde dehydrogenase 1 family member A3 (ALDH1a3). 466
4.12. Other pathways
4.12.1. PPARs pathway
PPARs, including PPARα, PPARβ/δ, and PPARγ, have been characterized as core lipid sensors that modulate whole‐body energy metabolism (Figure 9). 467 PPARα is the predominant PPAR isoform in the liver and plays an important role in regulating fatty acid transport, ketogenesis, and β‐oxidation. 468 In addition to lipid regulation, while, PPARβ/δ could increase the production of GLP‐1 to stimulate insulin secretion and reduce IR. 469 PPARγ mainly functions as a core regulator of adipogenesis, improving insulin sensitivity and glucose metabolism. In terms of mechanisms, deletion of IRF3 can increase the expression of PPARγ, leading to severe IR and glucose intolerance. 470 As opposed to IRF3, TAZ could protect mice from HFD‐induced IR and glucose intolerance through upregulating the expression of PPARγ. 471 Furthermore, PPARγ may induce the enhanced level of FGF1 in the adipose tissue, and considering the aforementioned protective function of FGF1 in insulin sensitivity and glucose metablism, 363 PPARγ/FGF1 axis may be indispensable in maintaining insulin sensitization an metabolic homeostasis. 364
4.12.2. SIRTs pathway
As highly conserved NAD+‐dependent protein deacetylases, 472 SIRTs are composed of seven subtypes and have been shown to regulate insulin secretion and affect hepatic insulin signaling and glucose homeostasis. SIRT3 expression is significantly decreased in the islets of T2DM patients and diabetic mice, which contributes to increased ROS level, mitochondrial dysfunction, and impaired insulin secretion. 473 Furthermore, SIRT1 could repress the transcription of UCP2, which promotes the conversion from glucose to ATP and Ca2+ influx, thus initiating insulin secretion and augmenting the sensitivity of pancreas to glucose. 474 What's more, SIRT1 and SIRT6 may deacetylate PGC1α and FoxO1 to facilitate the transcription of gluconeogenic genes, 475 , 476 ultimately regulating GSIS in the islets 477 and ameliorating glucose homeostasis in the liver. Of note, SIRT1 could also react with PPARγ to reduce the activity of PPARγ in WAT, thus inhibiting insulin signaling and adipogenesis. 478
4.12.3. IL‐6 pathway
By binding to membrane receptors, IL‐6 leads to the activation of multiple downstream signaling pathways inside target cells, such as PI3K/AKT, MAPK, and janus kinase (JAK)/STAT. 479 IL‐6 is reported to inhibit IRS, AKT2 and ERK phosphorylation directly or indirectly in the liver, leading to impaired insulin signaling pathway and IR. 480 , 481 , 482 , 483 Besides, IL‐6 is able to suppress glucose transport by affecting adiponectin and GLUT4, 484 repress lipogenesis by inhibiting PPARγ, 485 and stimulates lipolysis by activating AMPK in the adipose tissue, 486 which may link the IL‐6 pathway to the progression of IR and T2DM. However, there are also some conflicting results. For instance, in the case of HFD feeding, deletion of IL‐6 and IL‐6R promoted the development of hepatic IR. 487 , 488 In addition, the role of IL‐6 in pancreatic islets appears to be contradictory. Several studies have shown that IL‐6 contributes to the induction of low grade inflammation in the islets, which ultimately results in impaired insulin secretion. 489 , 490 , 491 Paradoxically, acute exposure of IL‐6 does not seem to exert its potential hazardous effects on the islets, 492 or affect the normal functioning of β cells. 480 , 493 , 494 , 495
4.12.4. JAK/STAT pathway
Depending on the cytokine or growth factor signals, different combinations of JAKs and STATs are activated with a high degree of specificity. Multiple JAK/STAT pathways have been implicated in glucose metabolism, including the IL‐6/STAT3, GH/JAK2/STAT5/IGF1, and IL‐4/STAT6 axes. Specifically, liver‐specific knockout of STAT3 in mice blocked the IL‐6 signaling pathway, leading to increased IR and gluconeogenesis, 496 , 497 via the induction of toll‐like receptor 4 expression. 498 Knockout of the genes encoding growth hormone receptor, JAK2 or STAT5 in the liver and muscle also enhanced lipid accumulation and IR, 499 , 500 possibly due to increases in FFAs flux and DNL. 500 However, the role of JAK2/STAT5 in the adipose tissue still remains controversial. 501 , 502 What we do know is that the IL‐4/STAT6 pathway has been shown to increase glucose oxidation by inhibiting PPARα activity in hepatocytes, while knockout of STAT6 promotes hepatic steatosis and IR. 503
The JAK/STAT pathway also plays a role in lipid metabolism. Mice with adipose‐specific jak2 knockout had impaired lipolysis in response to growth hormones and leptin. 501 , 504 Likewise, loss of either STAT3 or STAT5 505 , 506 significantly impaired lipolysis, probably resulting from repression of essential lipolytic genes. Muscle‐specific deletion of stat5 leads to increased accumulation of lipids in skeletal muscle, dyslipidemia, hepatic steatosis, and hyperglycemia, accompanied by altered expression of genes involved in lipogenesis, lipid uptake, lipolysis, insulin signaling, and glucose uptake. 507
4.12.5. NLRP3 inflammasome
The NOD‐like receptor (NLR) family pyrin domain‐containing 3 (NLRP3) inflammasome is a cytosolic multiprotein complex composed of the innate immune receptor NLRP3, the adapter apoptosis‐associated speck‐like protein containing card (ASC), and the inflammatory protease caspase‐1. Several endogenous ligands related to metabolic stress, such as lipopolysaccharide (LPS), 508 , 509 palmitic acids, oleic acid, 510 homocysteine and ROS, 511 , 512 have been reported to activate NLRP3 inflammasome in metabolic organs. Activated NLRP3 inflammasome induces the maturation and release of IL‐1β and IL‐18, responding to microbial infection, endogenous danger signals, and environmental stimuli. 513 IL‐1β has been demonstrated to induce IR by reducing IRS1 phosphorylation and expression 514 and inducing IL‐6 release. 515 The deficiency of NLRP3, caspase‐1, or IL‐1β in the liver and adipose tissue can result in reduced inflammasome activation, improved lipid metabolism, enhanced insulin sensitivity, and reduced blood glucose in DIO mouse models. 516 , 517 , 518 Additionally, IL‐1β can influence the activation of MAPK 519 and NF‐κB, 520 which in turn dysregulates the expression of genes involved in β cell death. Likewise, NLRP3 knockout is able to protect β cells from damage, rescue HFD‐induced β cell loss, and increase insulin secretion. 521 , 522
4.12.6. NF‐κB pathway
NF‐κB is an important transcription factor controlling different biological processes, such as immune activation, cell survival and stress response. 523 In canonical NF‐kB signaling, cytokines and PAMPs stimulate cell surface receptors to initiate activation of the inhibitory κB proteins (IκB) kinase (IKK) complex composed of IKK1 (IKKα) and IKK2 (IKKβ) and the regulatory subunit NEMO (IKKγ), ultimately driving transcription of target genes. IKKβ has been shown to phosphorylate and inactivate TSC1 in the mTOR pathway, 524 as well as IRS, 525 and S6K1 526 in the PI3K/AKT pathway, to promote IR. In both genetic‐ and DIO mice, deficiency of IKKB can prevent the advancement of IR and glucose intolerance. 527 , 528 Besides, NF‐kB may drive the expression of a plethora of inflammatory chemokines and cytokines in multiple metabolic tissues 529 and regulate numerous genes involved in insulin signaling, such as SOCS3 and protein tyrosine phosphatase 1B (PTP1B). 530 , 531 In the pancreas, inhibition of IKKβ is sufficient to provide protection against cytokine‐induced β cell apoptosis in both human and mouse diabetic islets, 532 thus regulating β cell mass and insulin secretion. 533
5. THERAPEUTIC TARGETS IN T2DM COMPLICATIONS
T2DM causes a number of long‐term complications in the neural, macrovascular and microvascular systems, which are instrumental for death and disability of patients with T2DM (Figure 10). 10 , 534 These complications encompass the damages to the coronary and cerebrovascular arteries, the kidneys, the eyes, and the nerves. 10 , 534 In the following section, we will revisit the potential signaling pathways, as well as their associated therapeutic targets, underlying T2DM complications.
FIGURE 10.

Signaling pathways in T2DM complications. Hyperglycemia causes macrovascular and microvascular lesions, contributing to diabetic complications such as diabetic retinopathy, diabetic nephropathy, diabetic neuropathy, and diabetic cardiovascular disease. Targeting the major pathological features of different complications, some signaling pathways have been discovered. These signaling pathways play important roles in promotion or inhibition of disease development, contributing to the treatment of diabetic complications.
5.1. Diabetic nephropathy
Diabetic nephropathy is perhaps among the most devastating T2DM complications, which bedevils up to 40% of T2DM patients and leads to kidney failure, CVD, and premature death. 535 The pathological changes of DN mainly include renal fibrosis and podocyte injury, 536 which are linked to various signaling pathways, particularly the TGFβ and mTOR pathways.
Dysregulation of TGFβ pathway is widely considered playing a key role in renal fibrosis, which is characterized by excessive deposition of ECM. Correspondingly, targeting TGFβ was also identified to be of therapeutic potential for DN. 537 , 538 Considerable evidence has revealed increased expression and activation of TGFβ pathway components, such as TGFβ1 and SMAD2/3, in the fibrotic kidney. 537 , 538 Consistently, inhibition of TGFβ1 or its downstream pathways substantially restrained renal fibrosis in DN, whereas overexpression of TGFβ1 prompted renal fibrosis, suggesting a profibrotic role of TGFβ pathway in DN. 537 , 538 TGFβ pathways can induce the activation of myofibroblasts, apoptosis of endothelial cells and podocytes, overproduction of ECM, and impairment of ECM degradation via multiple downstream mechanisms. 538 , 539 , 540 , 541 These downstream mechanisms include the imbalance between upregulation of profibrotic SMAD3 and downregulation of antifibrotic SMAD7, interplays with other signaling pathways (e.g., MAPK, WNT/β‐catenin, ERK, PI3K/AKT, mTOR pathways), and the involvement of miRNAs, lncRNAs and epigenetic modifications. 538 Accordingly, numerous approaches blocking TGFβ signaling, including TGFβ neutralizing antibodies, antisense TGFβ oligodeoxynucleotides, soluble human TGFR2, and specific inhibitors to TGFR1 kinases can effectively halt the progression of renal fibrosis and DN. 537 , 538 , 542 However, due to the side‐effects of TGFβ inhibition in autoimmune diseases, direct targeting of TGFβ1 is barely possible to be a viable strategy against renal fibrosis and DN. 538 Alternatively, indirect approaches to antagonizing TGFβ signaling might hence be promising to yield effective therapeutic agents for DN. In addition to TGFβ signaling, other signaling pathways may be of therapeutic potential in renal fibrosis and DN. For example, it has shown that high glucose can trigger the JAK/STAT signaling cascade to stimulate excessive proliferation of glomerular mesangial cells and aggravate DN, 543 and the small‐molecule inhibitor of JAK/STAT, baricitinib, has been demonstrated to decrease albuminuria in patients with T2DM and DN. 544
Podocyte injury is another important pathological feature of DN, in which the mTOR pathway is hyper‐activated due to high concentrations of glucose. 545 , 546 Enhanced mTORC1 signaling may trigger podocyte injury and DN by stimulating podocyte hypertrophy, mesangial expansion, glomerular basement membrane thickening, foot process effacement, podocyte loss, and albuminuria. 546 , 547 In line with this, restoration of mTORC1 activity is a critical therapeutic approach for the prevention of DN. For example, rapamycin, a specific and potent inhibitor of mTOR, markedly ameliorated mesangial expansion and proteinuria. 548 Moreover, the mechanism by which metformin and SGLT2 inhibitors exert kidney‐protective effects may be also related to the mTOR pathway, albeit it is currently unclear. 546
5.2. Diabetic retinopathy
Nearly 60% of T2DM patients are expected to have DR in the first decade after diagnosis. 549 , 550 DR is characterized by early pericyte loss, vascular leakage, retinal ischemia, and an overcompensatory retinal neovascularization, which is highly related to the regulation of signaling pathways. 551
Due to the local ischemia/hypoxia, the expression of VEGF is enhanced in ocular fluid of patients with DR. 552 , 553 , 554 Upregulated VEGF can increase the phosphorylation of occludin/zonula occludens‐1 (ZO‐1) to augment endothelial paracellular permeability, and cause activation of MAP to promote the proliferation of endothelial cells, thereby exacerbating the development of DR. 555 , 556 In parallel, intravitreal injection neutralizing antibody of VEGF, such as Ranibizumab, Pegaptanib, and Aflibercept, has been clinically used to treat proliferative diabetic retinopathy. 550 , 557 , 558
As endogenous ligands for the vascular endothelial receptor tyrosine kinase 2 (Tie2), angiopoietin1/2 (Ang1/2) are involved in the regulation of vascular permeability and angiogenesis in DR. 559 Current evidence showed that Ang1 promotes the formation of Tie2/PTP/VE‐cadherin complexes at cellular membrane to decrease vascular permeability, 560 but Ang2 enhances vascular leakage in DR through activating VEGF/β1‐integrin and regulating VE‐cadherin‐containing cell‐cell junctions. 561 , 562 , 563 Meanwhile, Ang2 could reduce pericyte capillary coverage and increase intraretinal neovascularization via the inhibition of Ang1/Tie2 interactions, suggesting that modulation of Ang1/2 is an attractive therapeutic target for the prevention and treatment of DR. 564
Emerging evidence suggests an essential role of circular RNAs (circRNAs) and miRNAs in DR. 565 , 566 For example, diabetes‐related stress upregulates the expression of cPWWP2A, a novel circRNA, to sequester and inhibit miR‐579 activity, thus alleviating T2DM‐induced retinal vascular dysfunction. 565 cZNF532 is another circRNA that could regulate pericyte biology by sequestering miR‐29a‐3p and activating chondroitin sulfate proteoglycan 4 (CSPG4), lysyl oxidase like 2 (LOXL2), and CDK2, which indicates that overexpression of cZNF532 or inhibition of miR‐29a‐3p could ameliorate DR. 567
5.3. Diabetic neuropathy
Among diabetes neuropathy, distal symmetric polyneuropathy is very common and defined as a loss of sensory function beginning distally in the lower extremities. 568 Approximately 30–50% of patients with diabetic neuropathy develop neuropathic pain called painful diabetic neuropathy (PDN). 569 PDN is characterized by neuropathic pain, small‐fiber degeneration, and dorsal root ganglion (DRG) nociceptor hyperexcitability. 570
DRGs are important neurons that comprise thermoreceptors, mechanoreceptors, and itch sensors. Nav1.8, an α‐subunits of voltage‐gated sodium channels, is important for the development of abnormal pain sensation. 571 Methylglyoxal, a reactive metabolite increased in diabetes, could posttranslationally modify Nav1.8 and enhance the activity of the nonselective cation channel TRPA1, respectively resulting in Nav1.8 gain of function pain and neuron hyperexcitability. 572 , 573 In addition, excitatory CXCR4/CXCL12 signaling in Nav1.8‐positive DRG neurons plays a critical role in the pathogenesis of mechanical allodynia and small‐fiber degeneration in a mouse model of PDN. 570 In T2DM, Ca2+ channels have also been implicated in PDN. For example, CaV3.2 activity is enhanced through the glycosylation of extracellular arginine residues, resulting in hyperexcitability pain in DRG neurons. 574
5.4. Cardiovascular complications
Macrovascular complications of T2DM include coronary heart disease, cardiomyopathy, arrhythmias and sudden death, cerebrovascular disease, and peripheral artery disease, among which CVD is the primary cause for death in diabetic patients. The development of diabetic cardiovascular complications also relates to the dysregulation of signaling pathways. A prominent example is that hyperglycemia can induce the formation of nonenzymatically glycated proteins or lipids, called advanced glycation end‐products (AGEs), which is closely related to the pathogenesis of CVD. AGEs have been shown to activate NF‐κB and increase vascular cell adhesion molecule1 (VCAM1) expression, initiating the first step of atherogenesis. 575 Besides, glycated low‐density lipoprotein (LDL) is recognized and taken up by AGE receptors or macrophage scavenger receptors, resulting in lipid‐laden foam cells in the arterial intima and the promotion of atherosclerosis. 576 In vascular smooth muscle cells (VSMC) and aortas from db/db mice, miR‐504 might participate in metabolic memory of CVD, 577 as evidenced by that miR‐504 in VSMC can inhibit contractile genes and enhance the activation of ERK1/2 and its target genes (Grb10 and Egr2), inhibiting proinflammatory response. Additionally, quaking (QKI) is one of the RNA‐binding proteins related to diabetic cardiomyopathy and atherosclerosis. QKI‐7 is highly expressed in human coronary arterial ECs in T2DM patients and is able to disrupt cell barrier, compromise angiogenesis, and enhance monocyte adhesion by binding and promoting mRNA degradation of downstream targets. 578
5.5. T2DM and COVID‐19
Recent evidence showed a mutual interplay between COVID‐19 and T2DM. 579 On the one hand, COVID‐19 patients with diabetes are more likely to develop severe condition of higher death incidence compared with those without diabetes. 580 , 581 On the other hand, new‐onset hyperglycemia, ketoacidosis, diabetes, and severe metabolic complications of preexisting diabetes have been observed in patients with COVID‐19. 582 , 583 , 584 There is an intricate interaction network existing between diabetes and COVID‐19. Common pathogenetic processes between COVID‐19 and diabetes mellitus discovered by differential gene expressions pattern analysis are related to the mRNA metabolism, subcellular organelle organization, nucleotide synthesis, immune responses, and autophagy process. Other studies revealed that five biomarker genes (CP, SOCS3, AGT, PSMB8, and CFB) and 4 miRNAs (hsa‐miR‐298, hsa‐miR‐3925‐5p, hsa‐miR‐4691‐3p, and hsa‐miR‐5196‐5p) closely bridge T2DM and COVID‐19. 585 , 586 , 587
Severe COVID‐19 is associated with worse glycemic control, 588 and the reason for this may partly lie in increased expression of ACE2, a key receptor for severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2), in lungs of T2DM patients. 589 Growing evidence suggests that elevated glucose level and glycolysis may directly increase and sustain SARS‐CoV‐2 replication. 590 Meanwhile, diabetes can affect immune response to SARS‐CoV‐2. Hyperglycemia and IR are capable of increasing synthesis of AGEs and proinflammatory cytokines to mediate tissue inflammation. 591 Additionally, hyperglycemia may also impede type I interferon production and signaling 591 to augment inflammatory responses to SARS‐CoV‐2 infection, leading to extreme systemic immune responses. 592 , 593
The precise mechanisms underlying the development of new‐onset T2DM in people with COVID‐19 are still not known, but may involve a number of complex etiologies. On the one hand, SARS‐CoV‐2 could directly infect β cells and lead to β cell dysfunction and metabolic dysregulation. 594 SARS‐CoV‐2 could also bind to NRP1, thus attenuating insulin secretion and inducing β cell apoptosis through the JNK/MAPK pathway. 595 Meanwhile, SARS‐CoV‐2 might downregulate ACE2 activity and subsequently activate macrophages and trigger NF‐κB signaling in the pancreas, leading to excessive synthesis and secretion of inflammatory cytokines, eventually damaging islets and β cells. 596 , 597 On the other hand, IR, resulting from viral infection seems to be another cause of hyperglycemia upon COVID‐19. Infection of SARS‐CoV‐2 may induce an inflammatory state, 598 , 599 in which IL‐6 might play a primary albeit not exclusive role in inducing IR and β cell dysfunction. 579 , 600
6. ANTIDIABETIC DRUGS
Poor control of hyperglycemia results in markedly increased microvascular, macrovascular and metabolic complications, and therefore, optimal glycemic management is the first priority in T2DM patients. 601 Although diet control and exercise could partly alleviate hyperglycemia, almost all T2DM patients still need antidiabetic drugs and/or insulin to maintain standard blood glucose. With the in‐depth study of T2DM signaling pathways, effective glucose‐lowering drugs often work by acting on important targets in signaling pathways. In the following section, we will summarize glucose‐lowering drugs in clinical application and clinical trials, as well as promising agents and targets, describing their action mechanisms and related signaling pathways (Figure 11 and Table 1).
FIGURE 11.
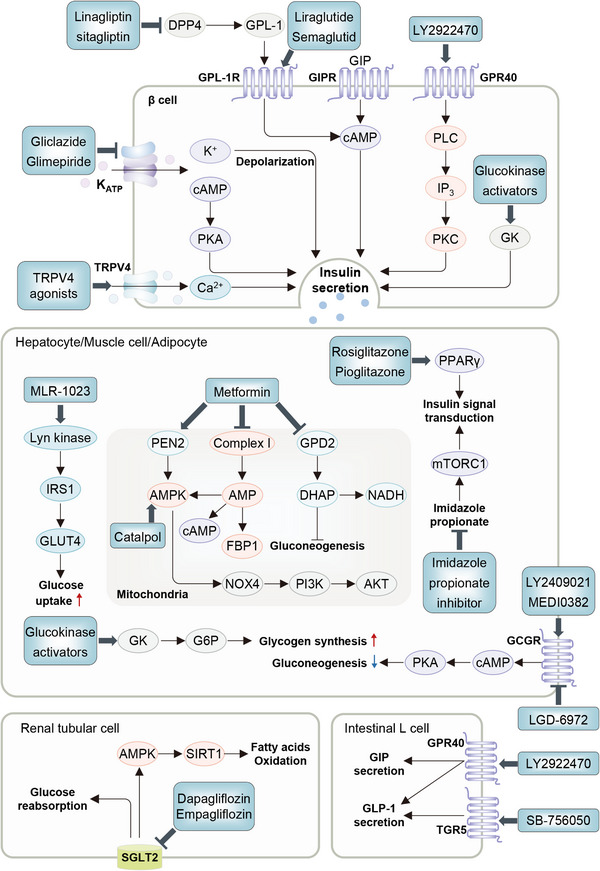
Clinical and potential drugs in T2DM therapy. In the plasma, DPP4 inhibitors, such as Linagliptin, increase the plasma content of GLP‐1 by inhibiting the degradation of GLP‐1 by DPP4. In pancreatic β cells, the agonists of GLP‐1R (GLP‐1, Liraglutide), GIPR, and GPR40 (LY2922470) promote insulin secretion through different signaling pathways. The inhibitor of KATP (Gliclazide) and the agonist of Ca2+ channel also promote the secretion of insulin. In metabolic organs, MLR‐1023 activates the Lyn kinase, leading to tyrosine phosphorylation of IRS1. Then a cascade of signaling events lead to increased glucose uptake and utilization. Metformin could bind to PEN2 to forms a complex with ATP6AP1, a subunit of the v‐ATPase8, leading to the inhibition of v‐ATPase and the activation of AMPK. Inhibition of the mTORC1/2 pathway may enhance insulin signaling at the IRS level and improve glucose tolerance. The activation of glucagon receptor results in increased glycogenolysis and gluconeogenesis via cAMP/PKA pathway. In the kidney, Ertugliflozin and Dapagliflozin promote urinary glucose excretion by inhibiting SGLT2. In the intestine, the agonist of TGR5 (SB‐756050) and GPR40 (LY2922470) promotes the secretion of GIP and GLP‐1.
TABLE 1.
Agents for T2DM in clinical or clinical trials
| Agent | Target or pathway | Phase | NCT number |
|---|---|---|---|
| Metformin hydrochloride | AMPK/PI3K/AKT/mTOR; autophagy; mitophagy | US FDA approved | NA |
| Ertugliflozin | SGLT2 | US FDA approved | NA |
| Dapagliflozin | US FDA approved | NA | |
| Chlorpropamide | Na+ K+‐ATPase | US FDA approved | NA |
| Acarbose | Glucosidase inhibitor | US FDA approved | NA |
| Voglibose | US FDA approved | NA | |
| Miglitol | US FDA approved | NA | |
| Saxagliptin | DPP4 | US FDA approved | NA |
| Linagliptin | US FDA approved | NA | |
| Alogliptin | IV | NCT03042325 | |
| Trelagliptin | IV | NCT02771093 | |
| DBPR108 | III | NCT04218734 | |
| Retagliptin | III | NCT05054842 | |
| Gliclazide | K+ channel | US FDA approved | NA |
| Glipizide | US FDA approved | NA | |
| Glyburide | US FDA approved | NA | |
| Glimepiride | US FDA approved | NA | |
| Repaglinide | US FDA approved | NA | |
| Mitiglinide | IV | NCT02143765 | |
| MSDC‐0160 | II | NCT00760578 | |
| Liraglutide | GLP‐1R | US FDA approved | NA |
| Semaglutide | US FDA approved | NA | |
| Taspoglutide | III | NCT00909597 | |
| Pioglitazone | PPAR | US FDA approved | NA |
| Rosiglitazone | US FDA approved | NA | |
| FK614 | II | NCT00036192 | |
| LY3298176 | GLP‐1R/GIPR | III | NCT03882970 |
| AZD1656 | Glucokinase | II | NCT01152385 |
| Dorzagliatin | II | NCT04531631 | |
| MK‐0941 | II | NCT00824616 | |
| PF‐04937319 | II | NCT01475461 | |
| LGD‐6972 | GCGR | II | NCT02851849 |
| LY2409021 | II | NCT01241448 | |
| PF‐06291874 | II | NCT02554877 | |
| Cotadutide | GLP‐1R/GCGR | II | NCT04208620 |
| MEDI0382 | II | NCT03745937 | |
| SAR425899 | II | NCT02973321 | |
| GSK1292263 | GPR119 | II | NCT01119846 |
| GSK256073 | GPR109A | II | NCT01376323 |
| MLR‐1023 | Insulin receptor | II | NCT02317796 |
| AZD7687 | Acyltransferase | I | NCT01217905 |
| LY2922470 | GPR40 | I | NCT01746017 |
| MK‐8666 | I | NCT01971554 | |
| LY2405319 | FGF21 receptor | I | NCT01869959 |
| SB756050 | Bile acid receptor | I | NCT00607906 |
| Coenzyme Q | Electron transport chain | II | NCT00703482 |
| MitoQ | NA | NCT04558190 |
This table summarizes agents used to treat diabetes by targeting different signaling pathways, in clinical and preclinical.
Data source: U.S. National Library of Medicine ClinicalTrials.gov.
6.1. Clinical drugs
6.1.1. Biguanides
As a first‐line drug for T2DM, metformin has been shown to have pleiotropic effects on glucose metabolism. 602 The core glucose‐lowering effect of metformin is the inhibition of hepatic gluconeogenesis by repressing mitochondrial respiratory chain complex 1 and increasing AMP level. 603 , 604 In addition, metformin can bound to PEN2 and form a complex with ATP6AP1 to inhibit v‐ATPase and activate AMPK. 15 Metformin can also directly act on the intestine to activate muscarinic M3 receptor, WNT and AMPK to stimulate GLP‐1 secretion, thereby altering glucose absorption. 602 Additionally, metformin could prevent the reabsorption of active BAs by modulation of the transcription of FXR via an AMPK‐mediated mechanism in enterocytes, ultimately increasing GLP‐1 secretion. 605 Moreover, metformin has beneficial effects on alleviating chronic low‐grade inflammation in T2DM. 606 It not only inhibits monocyte‐to‐macrophage differentiation via inhibiting STAT3, 607 but also suppresses LPS‐induced inflammatory response in macrophages via the AMPK/ATF3 pathway. 608
6.1.2. PPARγ agonists
Thiazolidinediones (TZDs), a kind of classic glucose‐lowering drugs, are the ligands for PPARγ. PPAR agonists primarily stimulate WAT remodeling and modulate lipid flux to improve insulin signaling and glucose homeostasis. TZDs binding to PPARγ could promote FFAs storage and reduce lipid ectopic accumulation in subcutaneous fat tissue. 609 Rosiglitazone and pioglitazone are the representatives of TZDs. Due to the potential for serious adverse cardiovascular effects, the United States Food and Drug Administration adds a "black box warning" to the rosiglitazone label, 610 while pioglitazone can significantly lower risk of death, myocardial infarction, or stroke among patients with diabetes. 611 In order to make good use of PPARγ, some studies focused on the potential effects of posttranslational modifications (PTMs) of PPARγ on treating T2DM in terms of phosphorylation, acetylation, ubiquitination, SUMOylation, O‐GlcNAcylation, and S‐nitrosylation. PTMs have shed light on selective activation of PPARγ, which shows great potential to circumvent TZDs' side effects while maintaining insulin sensitization. 612
6.1.3. K+ channel inhibitors
Changes in membrane potential are critical for insulin secretion, and SUs are a class of drugs that can depolarize cell membrane and promote insulin secretion. 613 By binding to the sulfonylurea receptor that is tightly linked to K+ channels, SUs can lead to the closure of KATP channels and the depolarization of β cell membrane, thereby stimulating insulin secretion. 614 , 615 In addition, SUs may enhance the GSIS by increasing intracellular cAMP level, 613 which further activates Epac2, a protein that has an ability to exchange guanine nucleotide with Rap. 616 Gliclazide is a representative of SUs, while Repaglinide is a representative drug in non‐SUs insulin secretagogues. With a different structure with SUs, 617 Repaglinide binds to its receptors and closes KATP channels to depolarize β cells, inducing Ca2+ influx and promoting Ca2+‐dependent insulin granules exocytosis. 618
6.1.4. GLP‐1R agonists and DPP4 inhibitors
GLP‐1 is an incretin hormone and exerts its action by binding to GPCRs to stimulate insulin secretion through rapid increases of cAMP and intracellular Ca2+. 619 In β cells, the binding of GLP‐1 and its receptor leads to activation of adenylate cyclase (AC) and a subsequent increase in cAMP. cAMP activates PKA, thereby inducing the closure of K+ channel and the opening of Ca2+. The subsequent Ca2+ influx promotes exocytosis of insulin granules and acute secretion of insulin into the circulation. 620 GLP‐1 also contributes to the control of blood glucose by inhibiting glucagon secretion, gastric emptying, and food ingestion. 619 , 621 GLP‐1R agonists also have a role in the treatment of neurodegenerative diseases, 622 nonalcoholic fatty liver disease 623 and obesity. 624 Given that GLP‐1 is mainly inactivated by dipeptidyl peptidase4 (DPP4) within 3 minutes in the circulation, 625 DPP4 inhibitors are applied to provide prolonged effects in vivo. 626 Notably, recent studies suggested DPP4 as a functional receptor for MERS‐CoV and SARS‐COV‐2, and its inhibitors have been found to inhibit infection with coronavirus. 627 , 628
6.1.5. SGLT2 inhibitors
SGLT2, a high‐capacity transporter in the proximal tubule, is the major pathway for renal glucose reabsorption. SGLT2‐inhibition prevents the reabsorption of filtered glucose and sodium, resulting in glycosuria and natriuresis. 629 SGLT2 inhibitors (SGLT2is) therefore are developed as a new class of antidiabetic drugs in T2DM, reducing plasma glucose levels in an insulin‐independent manner. 630 Canagliflozin, a representative SGLT2i, was reported to promote mitochondrial oxidative phosphorylation and FA oxidation via the AMPK/SIRT1/PGC1α pathway. 631 SGLT2is could also reduce cardiovascular events and protect kidney, 632 , 633 but increase the risk for diabetic ketoacidosis by promoting the production and reabsorption of ketone. 634
6.1.6. AGIs
Alpha‐glucosidase is an intestinal brush border enzyme responsible for the hydrolysis of disaccharides into monosaccharides, which is necessary for carbohydrate absorption. 635 Inhibition of alpha‐glucosidase results in reduced carbohydrates absorption and increased GLP‐1, thus decreasing the postprandial blood glucose. 636 , 637 Alpha‐glucosidase inhibitors that are currently used in clinic include acarbose, voglibose, and miglitol, as well as a variety of natural products such as hypericin, oleanolic acid and ursolic acid. 638
6.2. Preclinical drugs
6.2.1. GK activators
GK elicits GSIS in β cells and promotes hepatic glycogen production and storage. 639 Under raised glucose concentrations, GK dissociates from the GK regulatory protein, a competitive inhibitor of glucose, and phosphorylates glucose to G6P, thus reducing serum glucose and promoting HGP. 24 Thus targeting GK may have therapeutic effects on T2DM, and several GK activators have been tested in animal experiments, in which a few have reached the clinical trials phase. 639
6.2.2. GCGR antagonists and agonists
The glucagon receptor (GCGR) is a GPCR. Its activation results in increased glycogenolysis and gluconeogenesis via the cAMP/PKA pathway, 640 while its antagonization improves glucose control in T2DM. Nevertheless, adverse events stopped the development of GCGR antagonists (GRA). Although with new approaches, none of GRA has progressed to phase III clinical trials so far. 641 Additionally, glucagon signaling has been associated with increased energy expenditure. GCGR agonists can increase energy expenditure and reduce hepatic fat by promoting HGP. Although this may cause a spike in blood glucose, which can be effectively offset by GLP‐1R agonists. 642 Thus, GCGR has become a focus as a pharmaceutical target in the context of bi‐ or tri‐modal peptide agonists for the treatment of metabolic diseases. 643
6.2.3. TGR5 agonists
TGR5 agonists have been proposed as a potential treatment for T2DM. 644 , 645 Activated TGR5 not only stimulates GLP‐1 secretion, but also induces activity of type 2 iodothyronine deiodinase, resulting in increased thermogenesis and energy expenditure. 646 , 647 SB‐756050, a selective TGR5 agonist, could produce a significant enhancement of glucose‐induced GLP‐1 secretion in combination with DPP4, improve glucose disposal rate, and enhance insulin secretion. 648 Unfortunately, SB‐756050 did not show consistent efficacy in clinical trials. 648 Interestingly, it has recently been reported that a FXR/TGR5 dual agonist prevents progression of nephropathy in diabetes and obesity, 649 suggesting an attractive strategy for the therapy of T2DM and its complications.
6.2.4. GPR40 agonists
The G protein‐coupled receptor 40 (GPR40), also known as free fatty acid receptor 1 (FFAR1), is highly expressed in the pancreas and enteroendocrine cells in the gastrointestinal tract. When glucose level elevates, GPR40 facilitates insulin secretion through the PLC/IP3/PKC pathway. 650 , 651 GPR40 could also promote incretin secretion, such as GLP‐1 and gastric inhibitory polypeptide. 652 LY2922470 is a GPR40 agonist that has shown an effective and persistent dose‐dependent reduction in glucose levels and a significant increase in insulin and GLP‐1 secretion in preclinical trials. 653 GPR40 agonists might also regulate inflammatory responses and thus play a role in improving the development of diabetes complications. 654 , 655
6.2.5. Lyn kinase activators
Activation of Lyn kinase can directly induce tyrosine phosphorylation of IRS1, resulting in increased GLUT4 translocation, and enhanced glucose uptake and utilization. 656 MLR‐1023 is a highly potent and selective Lyn kinase activator, as well as a novel non‐PPARγ insulin sensitizer, which could reduce plasma glucose levels without the risk of hypoglycemia or weight gain. 656 , 657
6.2.6. Mitochondria‐targeted antioxidants
Several mitochondria‐targeted antioxidants and peptides have been found to reduce oxidative stress and mitochondrial damage, so as to exert therapeutic effect on some chronic diseases such as diabetes and Alzheimer's disease. These antioxidants include SkQ1, MitoQ, and Coenzyme Q. 658 Both SkQ1 and MitoQ are ubiquinone derivatives. Ubiquinone can serve as electron carriers and antioxidants to profoundly prevent lipid peroxidation. 659 Interestingly, Coenzyme Q is a lipid‐soluble molecule, one of the key components of electron transport chain. After being oxidized, Coenzyme Q needs to undergo a self‐reducing process before it can continue to function as an antioxidant. 660 Szeto‐Schiller (SS) peptide is a novel mitochondrial‐targeted peptide. 661 The water‐soluble tetrapeptide SS‐31 concentrates in the inner mitochondrial membrane and may exert radical‐sweeping ability without reliance on mitochondrial membrane potential and energy. 662 Given that administration of mitochondria‐targeted antioxidants may restore mitochondrial function and alleviate symptoms, further examination and analysis of treatment efficacy and safety are suggested to prepare for clinical trials.
6.3. Potential therapeutic targets and drugs
In addition to the antidiabetic drugs mentioned above, some potential targets and compounds are emerging to improve T2DM pathology. Although no effective target drugs have been developed, targeting these potential targets or modifying compounds could provide new ideas and possible therapeutic strategies for treating T2DM and its complications. For example, FGFs, especially FGF1 and FGF21, have emerged as a promising solution to the diabetes dilemma. Central injection of FGF1 can improve central glucose sensing and peripheral glucose uptake through restoring glucose‐sensing neurons, inducing neurogenesis, suppressing reactive astrocytes and restoring synaptic functionality. 344 While FGF21 plays important roles in regulating energy balance and glucose and lipid homeostasis through a heterodimeric receptor complex comprising FGFR1 and β‐klotho. 343 Imidazole propionate is a microbially produced histidine‐derived metabolite that impairs glucose tolerance by inhibiting insulin signaling at the level of IRS through activation of the p38γ/p62/mTORC1 pathway. 663 Inhibition of imidazole propionate may be effective in controlling blood glucose. Similarly, β‐aminoisobutyric acid (BAIBA) is a natural catabolite of thymine. It was previously reported that BAIBA attenuated inflammation and IR in HFD‐fed mice, and these effects were negated by siRNA‐mediated suppression of AMPK. 664 Other potential drugs/targets, such as catalpol (a natural product isolated from the root of rehmannia glutinosa), thioredoxin‐interacting protein (TXNIP, a cellular redox regulator upregulated in diabetes), 665 and transient receptor potential vanilloid 4 (TRPV4, a Ca2+‐permeable nonselective cation channel) 666 have been reported to be involved in the development of T2DM, including but not limited to affecting insulin production and secretion, 667 β cell function, 143 IR, 668 and glucose homeostasis. 665 , 669
7. CONCLUSION AND PERSPECTIVES
Throughout the past decades, the surged prevalence of T2DM and its vicious complications has coined an urgent craving for better understanding the mechanisms underpining the pathogenesis of T2DM and how to manage T2DM efficiently, particularly through drug administration. As a multifactoral disease, T2DM is driven by genetic, epigenetic, and nongenetic mechanisms, among which a substantial fraction are interdependent signaling pathways. A highly possible paradigm for various signaling pathways acting in T2DM, is where they serve as both the causes and consequences of T2DM progression and, function in an interacted manner rather than separately. In particular, environmental factors, together with genetic risks correlated to T2DM, are prone to elicit vibrations of the signaling pathways in the peripheral tissues and pancreatic islets. Interfering with these signaling pathways seems to engender outcomes that are conducive or obstructive to the pathological pertubances of T2DM, especially IR and β cell dysfunction. In terms of IR, modifications in these signaling pathways are likely to rely on their multifaceted interplays with the insulin/AKT to fluctuate the peripheral susceptibility to insulin action, despite the discrepancies in the specific roles of individual pathways. In terms of β cell dysfunction, signaling pathways also converge to regulating insulin secretion and fate of β cells, so intrinsic or acquired traits of these signaling pathways appear to partially dictate the adaptive capacity of β cells in response to metabolic demands. Importantly, many medications that normalize blood glucose levels and prevent or inverse diabetic complications have been identified to establish their pharmaceutical benefits via the mechanisms involving signaling pathways. One prominent example is the extensively used clinical antidiabetic drug, metformin, which acts through the AMPK pathway to powerfully improve metabolic disoders. 15 Nevertheless, a fraction of drugs or compounds targeting signaling pathways have unexpected side‐effects or inadequate effectiveness, owing to additional pathophysiological roles or minor metabolic effects of signaling pathways. For example, TGFβ‐specific therapies have been witnessed to exert inevitable effects on immune system, thus slowering the progress in employing them for treating DN. For these reasons, discerning the functions and mechanisms of signaling pathways in T2DM would help ease the road to well‐managing T2DM with drugs.
However, there are several obstacles in studying signaling pathways and interventions of T2DM. Firstly, an unfortunate trend is excessively highlighting the pathological significance of a sole molecule in signaling pathways, which is intensified by the dramatic progress in genetic manipulation technologies. This misfortune of trend in large part relates to concerns that manipulating genes could inevitably cause adaptive or compensatory effects concealing the real effects of certain molecules or pathways in T2DM progression. Luckily, considerable progresses in omics methods, such as, spatially resolved genomics, transcriptomics, and metabonomics, make it available to rectify this trend. In particular, devising ways to assess genomic, proteomic, and metabolic status in prediabetic and diabetic tissues while preserving spatial information and single cell resolution, is of great propensity to delineate the inclusive but accurate molecular mechanisms of T2DM. Furthermore, biomolecular condensates or droplets have been increasingly identified as an interaction basis for the molecules in signaling pathways, 670 which is less emphasized in the past studies of T2DM. Hence, it might be urgent to highlight the roles of liquid condensates or liquid–liquid phase separation in T2DM, which is possibly linked to abnormal protein aggregation. In addition, another important question to purse for the accurate roles of signaling pathways in T2DM, is to create in vitro models that sincerely mimic the real progression of T2DM. Nevertheless, feasible approaches to address this problem might be involved in the organoid technologies, which may largely recapitulate essential features of in vivo organ development and biological function. As such, they offer tractable and faithful in vitro tools for disentangling molecular mechanisms of T2DM and developing regenerative pancreatic islets, as well as confer a promising strategy that compensates for pharmacological therapies in advanced T2DM. Importantly, we anticipate that systematic application of these novel methodologies and notions in basic research and clinical translation of signaling pathways in T2DM would have a promising impact in contributing to the discovery of antidiabetic drugs with enhanced effectiveness and safety. This is possibly because precise interventions usually derive from precise investigations.
In summary, remarkable insights over the past few decades have gained vital understandings of signaling pathways related to T2DM and therapeutic interventions. Efficient management of T2DM and its complications is still challenging, but these understandings available now, together with more discoveries in the future, hold the potential to achieve more potent and specific drug interventions for T2DM and its complications. Hence, we hope that the knowledges summarized here can provide different ideas for researchers working in the field of T2DM, ultimately helping identify new therapeutic targets that could break the vicious development of this disease.
AUTHOR CONTRIBUTIONS
Y. T. and X. F. conceived the idea; R. C., H. T., Y. Z., G. L., H. X., and G. R. performed the literature search and drafted the manuscript; R. C., H. T., and Y. Z. revised and edited the manuscript; X. F. and Y. T. supervised and revised the manuscript. All the authors have read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
All the authors declared no conflict of interest.
ETHICS STATEMENT
Not applicable.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (92157205, 81970561 and 82172986), the Ministry of Science and Technology of China (2018ZX09201018‐005), and the 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYJC18049).
Cao R, Tian H, Zhang Y, et al. Signaling pathways and intervention for therapy of type 2 diabetes mellitus. MedComm. 2023;4:e283. 10.1002/mco2.283
Contributor Information
Yan Tian, Email: tyfxh@163.com.
Xianghui Fu, Email: xfu@scu.edu.cn.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Ahmad E, Lim S, Lamptey R, Webb DR, Davies MJ. Type 2 diabetes. Lancet. 2022;400(10365):1803‐1820. [DOI] [PubMed] [Google Scholar]
- 2. Bullock A, Sheff K. Incidence trends of type 1 and type 2 diabetes among youths, 2002–2012. N Engl J Med. 2017;377(3):301. [DOI] [PubMed] [Google Scholar]
- 3. Lawrence JM, Divers J, Isom S, et al. Trends in prevalence of type 1 and type 2 diabetes in children and adolescents in the US, 2001–2017. Jama. 2021;326(8):717‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scott ES, Januszewski AS, O'Connell R, et al. Long‐term glycemic variability and vascular complications in type 2 diabetes: post hoc analysis of the FIELD study. J Clin Endocrinol Metab. 2020;105(10). [DOI] [PubMed] [Google Scholar]
- 5. Nagao H, Cai W, Brandao BB, et al. Leucine‐973 is a crucial residue differentiating insulin and IGF‐1 receptor signaling. J Clin Invest. 2023;133(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gleason CE, Lu D, Witters LA, Newgard CB, Birnbaum MJ. The role of AMPK and mTOR in nutrient sensing in pancreatic beta‐cells. J Biol Chem. 2007;282(14):10341‐10351. [DOI] [PubMed] [Google Scholar]
- 7. Lin H, Smith N, Spigelman AF, et al. Beta‐cell knockout of SENP1 reduces responses to incretins and worsens oral glucose tolerance in high‐fat diet‐fed mice. Diabetes. 2021;70(11):2626‐2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF. Glucagon‐like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci USA. 1987;84(10):3434‐3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perreault L, Skyler JS, Rosenstock J. Novel therapies with precision mechanisms for type 2 diabetes mellitus. Nat Rev Endocrinol. 2021;17(6):364‐377. [DOI] [PubMed] [Google Scholar]
- 10. Demir S, Nawroth PP, Herzig S, Ekim Üstünel B. Emerging targets in type 2 diabetes and diabetic complications. Adv Sci (Weinh). 2021;8(18):e2100275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lewis GF, Brubaker PL. The discovery of insulin revisited: lessons for the modern era. J Clin Invest. 2021;131(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Somers G, Devis G, van Obberghen E, Malaisse WJ. Calcium‐antagonists and islet function. VI. Effects of barium. Pflugers Arch. 1976;365(1):21‐28. [DOI] [PubMed] [Google Scholar]
- 13. Ruderman NB, Kapeller R, White MF, Cantley LC. Activation of phosphatidylinositol 3‐kinase by insulin. Proc Natl Acad Sci USA. 1990;87(4):1411‐1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rossomando AJ, Payne DM, Weber MJ, Sturgill TW. Evidence that pp42, a major tyrosine kinase target protein, is a mitogen‐activated serine/threonine protein kinase. Proc Natl Acad Sci USA. 1989;86(18):6940‐6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ma T, Tian X, Zhang B, et al. Low‐dose metformin targets the lysosomal AMPK pathway through PEN2. Nature. 2022;603(7899):159‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou G, Myers R, Li Y, et al. Role of AMP‐activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167‐1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tomlinson B, Patil NG, Fok M, Chan P, Lam CWK. The role of sulfonylureas in the treatment of type 2 diabetes. Expert Opin Pharmacother. 2022;23(3):387‐403. [DOI] [PubMed] [Google Scholar]
- 18. Brown E, Heerspink HJL, Cuthbertson DJ, Wilding JPH. SGLT2 inhibitors and GLP‐1 receptor agonists: established and emerging indications. Lancet. 2021;398(10296):262‐276. [DOI] [PubMed] [Google Scholar]
- 19. Xu B, Li S, Kang B, Zhou J. The current role of sodium‐glucose cotransporter 2 inhibitors in type 2 diabetes mellitus management. Cardiovasc Diabetol. 2022;21(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. James DE, Stöckli J, Birnbaum MJ. The aetiology and molecular landscape of insulin resistance. Nat Rev Mol Cell Biol. 2021;22(11):751‐771. [DOI] [PubMed] [Google Scholar]
- 21. Johnson AM, Olefsky JM. The origins and drivers of insulin resistance. Cell. 2013;152(4):673‐684. [DOI] [PubMed] [Google Scholar]
- 22. Khalid M, Alkaabi J, Khan MAB, Adem A. Insulin signal transduction perturbations in insulin resistance. Int J Mol Sci. 2021;22(16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Herrera Moro Chao D, Kirchner MK, Pham C, et al. Hypothalamic astrocytes control systemic glucose metabolism and energy balance. Cell Metab. 2022;34(10):1532‐1547 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. 2017;13(10):572‐587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mayneris‐Perxachs J, Moreno‐Navarrete JM, Fernandez‐Real JM. The role of iron in host‐microbiota crosstalk and its effects on systemic glucose metabolism. Nat Rev Endocrinol. 2022;18(11):683‐698. [DOI] [PubMed] [Google Scholar]
- 26. Yamauchi T, Kamon J, Waki H, et al. The fat‐derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7(8):941‐946. [DOI] [PubMed] [Google Scholar]
- 27. Ikeda T, Kajita K, Zhiliang W, et al. Effects of phorbol ester‐sensitive PKC (c/nPKC) activation on the production of adiponectin in 3T3‐L1 adipocytes. IUBMB Life. 2009;61(6):644‐650. [DOI] [PubMed] [Google Scholar]
- 28. Mao X, Kikani CK, Riojas RA, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8(5):516‐523. [DOI] [PubMed] [Google Scholar]
- 29. Cheng KK, Lam KS, Wang B, Xu A. Signaling mechanisms underlying the insulin‐sensitizing effects of adiponectin. Best Pract Res Clin Endocrinol Metab. 2014;28(1):3‐13. [DOI] [PubMed] [Google Scholar]
- 30. Kojta I, Chacińska M, Błachnio‐Zabielska A. Obesity, bioactive lipids, and adipose tissue inflammation in insulin resistance. Nutrients. 2020;12(5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ling C, Bacos K, Rönn T. Epigenetics of type 2 diabetes mellitus and weight change ‐ a tool for precision medicine? Nat Rev Endocrinol. 2022;18(7):433‐448. [DOI] [PubMed] [Google Scholar]
- 32. Kahn SE. The relative contributions of insulin resistance and beta‐cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia. 2003;46(1):3‐19. [DOI] [PubMed] [Google Scholar]
- 33. Eizirik DL, Pasquali L, Cnop M. Pancreatic β‐cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat Rev Endocrinol. 2020;16(7):349‐362. [DOI] [PubMed] [Google Scholar]
- 34. Wensveen F, Jelenčić V, Valentić S, et al. NK cells link obesity‐induced adipose stress to inflammation and insulin resistance. Nat Immunol. 2015;16(4):376‐385. [DOI] [PubMed] [Google Scholar]
- 35. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98‐107. [DOI] [PubMed] [Google Scholar]
- 36. Arunagiri A, Haataja L, Pottekat A, et al. Proinsulin misfolding is an early event in the progression to type 2 diabetes. eLife. 2019;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yong J, Johnson JD, Arvan P, Han J, Kaufman RJ. Therapeutic opportunities for pancreatic β‐cell ER stress in diabetes mellitus. Nat Rev Endocrinol. 2021;17(8):455‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roden M, Shulman GI. The integrative biology of type 2 diabetes. Nature. 2019;576(7785):51‐60. [DOI] [PubMed] [Google Scholar]
- 39. Xu H, Tian Y, Tang D, et al. An endoplasmic reticulum stress‐microRNA‐26a feedback circuit in NAFLD. Hepatology. 2021;73(4):1327‐1345. [DOI] [PubMed] [Google Scholar]
- 40. Salvadó L, Palomer X, Barroso E, Vázquez‐Carrera M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol Metab. 2015;26(8):438‐448. [DOI] [PubMed] [Google Scholar]
- 41. Angelova PR, Abramov AY. Functional role of mitochondrial reactive oxygen species in physiology. Free Radic Biol Med. 2016;100:81‐85. [DOI] [PubMed] [Google Scholar]
- 42. Rovira‐Llopis S, Bañuls C, Diaz‐Morales N, Hernandez‐Mijares A, Rocha M, Victor VM. Mitochondrial dynamics in type 2 diabetes: Pathophysiological implications. Redox Biol. 2017;11:637‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Supale S, Li N, Brun T, Maechler P. Mitochondrial dysfunction in pancreatic β cells. Trends Endocrinol Metab. 2012;23(9):477‐487. [DOI] [PubMed] [Google Scholar]
- 44. Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta‐cell dysfunction in diabetes mellitus. Antioxid Redox Signal. 2017;26(10):501‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yazıcı D, Sezer H. Insulin resistance, obesity and lipotoxicity. Adv Exp Med Biol. 2017;960:277‐304. [DOI] [PubMed] [Google Scholar]
- 46. Gerber P, Rutter G. The role of oxidative stress and hypoxia in pancreatic beta‐cell dysfunction in diabetes mellitus. Antioxid Redox Signal. 2017;26(10):501‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pinti MV, Fink GK, Hathaway QA, Durr AJ, Kunovac A, Hollander JM. Mitochondrial dysfunction in type 2 diabetes mellitus: an organ‐based analysis. Am J Physiol Endocrinol Metab. 2019;316(2):E268‐E285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Boardman NT, Trani G, Scalabrin M, Romanello V, Wust RCI. Intra‐cellular to inter‐organ mitochondrial communication in striated muscle in health and disease. Endocr Rev. 2023; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Najjar SM, Perdomo G. Hepatic insulin clearance: mechanism and physiology. Physiology (Bethesda). 2019;34(3):198‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Duvel K, Yecies JL, Menon S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39(2):171‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto‐oncogene product by phosphatidylinositol‐3,4‐bisphosphate. Science. 1997;275(5300):665‐668. [DOI] [PubMed] [Google Scholar]
- 52. Pei J, Wang B, Wang D. Current studies on molecular mechanisms of insulin resistance. J Diabetes Res. 2022;2022:1863429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169(3):381‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu H, Stepicheva NA, Ghosh S, et al. Reducing Akt2 in retinal pigment epithelial cells causes a compensatory increase in Akt1 and attenuates diabetic retinopathy. Nat Commun. 2022;13(1):6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab. 2012;23(2):65‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170(4):605‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Owen JL, Zhang Y, Bae SH, et al. Insulin stimulation of SREBP‐1c processing in transgenic rat hepatocytes requires p70 S6‐kinase. Proc Natl Acad Sci USA. 2012;109(40):16184‐16189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bengoechea‐Alonso MT, Ericsson J. A phosphorylation cascade controls the degradation of active SREBP1. J Biol Chem. 2009;284(9):5885‐5895. [DOI] [PubMed] [Google Scholar]
- 59. Ng Y, Ramm G, Lopez JA, James DE. Rapid activation of Akt2 is sufficient to stimulate GLUT4 translocation in 3T3‐L1 adipocytes. Cell Metab. 2008;7(4):348‐356. [DOI] [PubMed] [Google Scholar]
- 60. Leto D, Saltiel AR. Regulation of glucose transport by insulin: traffic control of GLUT4. Nat Rev Mol Cell Biol. 2012;13(6):383‐396. [DOI] [PubMed] [Google Scholar]
- 61. Koren S, DiPilato LM, Emmett MJ, et al. The role of mouse Akt2 in insulin‐dependent suppression of adipocyte lipolysis in vivo. Diabetologia. 2015;58(5):1063‐1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chakrabarti P, English T, Shi J, Smas CM, Kandror KV. Mammalian target of rapamycin complex 1 suppresses lipolysis, stimulates lipogenesis, and promotes fat storage. Diabetes. 2010;59(4):775‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee G, Zheng Y, Cho S, et al. Post‐transcriptional regulation of de novo lipogenesis by mTORC1‐S6K1‐SRPK2 signaling. Cell. 2017;171(7):1545‐1558 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sanders FWB, Acharjee A, Walker C, et al. Hepatic steatosis risk is partly driven by increased de novo lipogenesis following carbohydrate consumption. Genome Biol. 2018;19(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Filali‐Mouncef Y, Hunter C, Roccio F, et al. The menage a trois of autophagy, lipid droplets and liver disease. Autophagy. 2022;18(1):50‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Eguchi J, Wang X, Yu S, et al. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011;13(3):249‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huang X, Liu G, Guo J, Su Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci. 2018;14(11):1483‐1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nagao H, Cai W, Brasil Brandão B, et al. Leucine‐973 is a crucial residue differentiating insulin and IGF‐1 receptor signaling. J Clin Invest. 2022; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sakaguchi M, Fujisaka S, Cai W, et al. Adipocyte dynamics and reversible metabolic syndrome in mice with an inducible adipocyte‐specific deletion of the insulin receptor. Cell Metab. 2017;25(2):448‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Homan EP, Brandão BB, Softic S, et al. Differential roles of FOXO transcription factors on insulin action in brown and white adipose tissue. J Clin Invest. 2021;131(19) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yin L, Cai WJ, Chang XY, et al. Analysis of PTEN expression and promoter methylation in Uyghur patients with mild type 2 diabetes mellitus. Medicine (Baltimore). 2018;97(49):e13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mukherjee R, Vanaja KG, Boyer JA, et al. Regulation of PTEN translation by PI3K signaling maintains pathway homeostasis. Mol Cell. 2021;81(4):708‐723 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Brunner JS, Vogel A, Lercher A, et al. The PI3K pathway preserves metabolic health through MARCO‐dependent lipid uptake by adipose tissue macrophages. Nat Metab. 2020;2(12):1427‐1442. [DOI] [PubMed] [Google Scholar]
- 74. Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, Deng CX. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest. 2011;121(11):4477‐4490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kurano M, Tsukamoto K, Shimizu T, et al. Protection against insulin resistance by apolipoprotein M/sphingosine‐1‐phosphate. Diabetes. 2020;69(5):867‐881. [DOI] [PubMed] [Google Scholar]
- 76. Demir S, Wolff G, Wieder A, et al. TSC22D4 interacts with Akt1 to regulate glucose metabolism. Sci Adv. 2022;8(42):eabo5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Petersen MC, Madiraju AK, Gassaway BM, et al. Insulin receptor Thr1160 phosphorylation mediates lipid‐induced hepatic insulin resistance. J Clin Invest. 2016;126(11):4361‐4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sano H, Kane S, Sano E, et al. Insulin‐stimulated phosphorylation of a Rab GTPase‐activating protein regulates GLUT4 translocation. J Biol Chem. 2003;278(17):14599‐14602. [DOI] [PubMed] [Google Scholar]
- 79. Ter Horst KW, Gilijamse PW, Versteeg RI, et al. Hepatic diacylglycerol‐associated protein kinase Cε translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep. 2017;19(10):1997‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Perry RJ, Peng L, Cline GW, et al. Mechanisms by which a very‐low‐calorie diet reverses hyperglycemia in a rat model of type 2 diabetes. Cell Metab. 2018;27(1):210‐217.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hagiwara A, Cornu M, Cybulski N, et al. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012;15(5):725‐738. [DOI] [PubMed] [Google Scholar]
- 82. Kumar A, Sundaram K, Mu J, et al. High‐fat diet‐induced upregulation of exosomal phosphatidylcholine contributes to insulin resistance. Nat Commun. 2021;12(1):213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li X, Zhang D, Vatner DF, et al. Mechanisms by which adiponectin reverses high fat diet‐induced insulin resistance in mice. Proc Natl Acad Sci USA. 2020;117(51):32584‐32593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shearer AM, Wang Y, Fletcher EK, et al. PAR2 promotes impaired glucose uptake and insulin resistance in NAFLD through GLUT2 and Akt interference. Hepatology. 2022;76(6):1778‐1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wang X, Yang Y, Zhao D, et al. Inhibition of high‐fat diet‐induced obesity via reduction of ER‐resident protein Nogo occurs through multiple mechanisms. J Biol Chem. 2022;298(2):101561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tian Y, Xu J, Du X, Fu X. The interplay between noncoding RNAs and insulin in diabetes. Cancer Lett. 2018;419:53‐63. [DOI] [PubMed] [Google Scholar]
- 87. Fu X, Dong B, Tian Y, et al. MicroRNA‐26a regulates insulin sensitivity and metabolism of glucose and lipids. J Clin Invest. 2015;125(6):2497‐2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Stiles BL, Kuralwalla‐Martinez C, Guo W, et al. Selective deletion of Pten in pancreatic beta cells leads to increased islet mass and resistance to STZ‐induced diabetes. Mol Cell Biol. 2006;26(7):2772‐2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kitamura T, Nakae J, Kitamura Y, et al. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. J Clin Invest. 2002;110(12):1839‐1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Howell JJ, Manning BD. mTOR couples cellular nutrient sensing to organismal metabolic homeostasis. Trends Endocrinol Metab. 2011;22(3):94‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yin Q, Ni Q, Wang Y, et al. Raptor determines beta‐cell identity and plasticity independent of hyperglycemia in mice. Nat Commun. 2020;11(1):2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hashimoto N, Kido Y, Uchida T, et al. Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nat Genet. 2006;38(5):589‐593. [DOI] [PubMed] [Google Scholar]
- 93. Bouzakri K, Zachrisson A, Al‐Khalili L, et al. siRNA‐based gene silencing reveals specialized roles of IRS‐1/Akt2 and IRS‐2/Akt1 in glucose and lipid metabolism in human skeletal muscle. Cell Metab. 2006;4(1):89‐96. [DOI] [PubMed] [Google Scholar]
- 94. Withers DJ, Gutierrez JS, Towery H, et al. Disruption of IRS‐2 causes type 2 diabetes in mice. Nature. 1998;391(6670):900‐904. [DOI] [PubMed] [Google Scholar]
- 95. Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue‐specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96(3):329‐339. [DOI] [PubMed] [Google Scholar]
- 96. Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF. Irs‐2 coordinates Igf‐1 receptor‐mediated beta‐cell development and peripheral insulin signalling. Nat Genet. 1999;23(1):32‐40. [DOI] [PubMed] [Google Scholar]
- 97. Pende M, Kozma SC, Jaquet M, et al. Hypoinsulinaemia, glucose intolerance and diminished beta‐cell size in S6K1‐deficient mice. Nature. 2000;408(6815):994‐997. [DOI] [PubMed] [Google Scholar]
- 98. Bernal‐Mizrachi E, Fatrai S, Johnson JD, et al. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J Clin Invest. 2004;114(7):928‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Baumel‐Alterzon S, Scott DK. Regulation of Pdx1 by oxidative stress and Nrf2 in pancreatic beta‐cells. Front Endocrinol (Lausanne). 2022;13:1011187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab. 2018;27(2):299‐313. [DOI] [PubMed] [Google Scholar]
- 101. Zhang CS, Hawley SA, Zong Y, et al. Fructose‐1,6‐bisphosphate and aldolase mediate glucose sensing by AMPK. Nature. 2017;548(7665):112‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Steinberg GR, Hardie DG. New insights into activation and function of the AMPK. Nat Rev Mol Cell Biol. 2022; [DOI] [PubMed] [Google Scholar]
- 103. Steinberg GR, Carling D. AMP‐activated protein kinase: the current landscape for drug development. Nat Rev Drug Discov. 2019;18(7):527‐551. [DOI] [PubMed] [Google Scholar]
- 104. González A, Hall MN, Lin SC, Hardie DG. AMPK and TOR: the yin and yang of cellular nutrient sensing and growth control. Cell Metab. 2020;31(3):472‐492. [DOI] [PubMed] [Google Scholar]
- 105. Garcia D, Shaw RJ. AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. 2017;66(6):789‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lin R, Elf S, Shan C, et al. 6‐Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1‐AMPK signalling. Nat Cell Biol. 2015;17(11):1484‐1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gao X, Zhao L, Liu S, et al. γ‐6‐phosphogluconolactone, a byproduct of the oxidative pentose phosphate pathway, contributes to AMPK activation through inhibition of PP2A. Mol Cell. 2019;76(6):857‐871. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yan Y, Krecke KN, Bapat AS, et al. Phosphatase PHLPP2 regulates the cellular response to metabolic stress through AMPK. Cell Death Dis. 2021;12(10):904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Liu Y, Jurczak MJ, Lear TB, et al. A Fbxo48 inhibitor prevents pAMPKα degradation and ameliorates insulin resistance. Nat Chem Biol. 2021;17(3):298‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Jiang P, Ren L, Zhi L, et al. Negative regulation of AMPK signaling by high glucose via E3 ubiquitin ligase MG53. Mol Cell. 2021;81(3):629‐637. e5. [DOI] [PubMed] [Google Scholar]
- 111. Wu N, Zheng B, Shaywitz A, et al. AMPK‐dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell. 2013;49(6):1167‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123(7):2764‐2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Steneberg P, Lindahl E, Dahl U, et al. PAN‐AMPK activator O304 improves glucose homeostasis and microvascular perfusion in mice and type 2 diabetes patients. JCI Insight. 2018;3(12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Myers RW, Guan HP, Ehrhart J, et al. Systemic pan‐AMPK activator MK‐8722 improves glucose homeostasis but induces cardiac hypertrophy. Science. 2017;357(6350):507‐511. [DOI] [PubMed] [Google Scholar]
- 115. Hawley SA, Ford RJ, Smith BK, et al. The Na+/glucose cotransporter inhibitor canagliflozin activates AMPK by inhibiting mitochondrial function and increasing cellular AMP levels. Diabetes. 2016;65(9):2784‐2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Cokorinos EC, Delmore J, Reyes AR, et al. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non‐human primates and mice. Cell Metab. 2017;25(5):1147‐1159.e10. [DOI] [PubMed] [Google Scholar]
- 117. Koh A, Mannerås‐Holm L, Yunn NO, et al. Microbial imidazole propionate affects responses to metformin through p38γ‐dependent inhibitory AMPK phosphorylation. Cell Metab. 2020;32(4):643‐653. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Garcia D, Hellberg K, Chaix A, et al. Genetic liver‐specific AMPK activation protects against diet‐induced obesity and NAFLD. Cell Rep. 2019;26(1):192‐208. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Fullerton MD, Galic S, Marcinko K, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin‐sensitizing effects of metformin. Nat Med. 2013;19(12):1649‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Galic S, Loh K, Murray‐Segal L, Steinberg GR, Andrews ZB, Kemp BE. AMPK signaling to acetyl‐CoA carboxylase is required for fasting‐ and cold‐induced appetite but not thermogenesis. Elife. 2018;7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Oh TS, Cho H, Cho JH, Yu SW, Kim EK. Hypothalamic AMPK‐induced autophagy increases food intake by regulating NPY and POMC expression. Autophagy. 2016;12(11):2009‐2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kong D, Dagon Y, Campbell JN, et al. A postsynaptic AMPK→p21‐activated kinase pathway drives fasting‐induced synaptic plasticity in AgRP neurons. Neuron. 2016;91(1):25‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Loh K, Tam S, Murray‐Segal L, et al. Inhibition of adenosine monophosphate‐activated protein kinase‐3‐hydroxy‐3‐methylglutaryl coenzyme a reductase signaling leads to hypercholesterolemia and promotes hepatic steatosis and insulin resistance. Hepatol Commun. 2019;3(1):84‐98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Bultot L, Guigas B, Von Wilamowitz‐Moellendorff A, et al. AMP‐activated protein kinase phosphorylates and inactivates liver glycogen synthase. Biochem J. 2012;443(1):193‐203. [DOI] [PubMed] [Google Scholar]
- 125. Gauthier MS, O'Brien EL, Bigornia S, et al. Decreased AMP‐activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole‐body insulin resistance in morbidly obese humans. Biochem Biophys Res Commun. 2011;404(1):382‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Mottillo EP, Desjardins EM, Crane JD, et al. Lack of adipocyte AMPK exacerbates insulin resistance and hepatic steatosis through brown and beige adipose tissue function. Cell Metab. 2016;24(1):118‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Rourke JL, Hu Q, Screaton RA. AMPK and friends: central regulators of β cell biology. Trends Endocrinol Metab. 2018;29(2):111‐122. [DOI] [PubMed] [Google Scholar]
- 128. Jaafar R, Tran S, Shah AN, et al. mTORC1 to AMPK switching underlies beta‐cell metabolic plasticity during maturation and diabetes. J Clin Invest. 2019;129(10):4124‐4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Howell JJ, Hellberg K, Turner M, et al. Metformin inhibits hepatic mTORC1 signaling via dose‐dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017;25(2):463‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ardestani A, Lupse B, Kido Y, Leibowitz G, Maedler K. mTORC1 signaling: a double‐edged sword in diabetic beta cells. Cell Metab. 2018;27(2):314‐331. [DOI] [PubMed] [Google Scholar]
- 131. Yuan T, Rafizadeh S, Gorrepati KD, et al. Reciprocal regulation of mTOR complexes in pancreatic islets from humans with type 2 diabetes. Diabetologia. 2017;60(4):668‐678. [DOI] [PubMed] [Google Scholar]
- 132. Haythorne E, Lloyd M, Walsby‐Tickle J, et al. Altered glycolysis triggers impaired mitochondrial metabolism and mTORC1 activation in diabetic beta‐cells. Nat Commun. 2022;13(1):6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Fu A, Robitaille K, Faubert B, et al. LKB1 couples glucose metabolism to insulin secretion in mice. Diabetologia. 2015;58(7):1513‐1522. [DOI] [PubMed] [Google Scholar]
- 134. Swisa A, Granot Z, Tamarina N, et al. Loss of liver kinase B1 (LKB1) in beta cells enhances glucose‐stimulated insulin secretion despite profound mitochondrial defects. J Biol Chem. 2015;290(34):20934‐20946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Nguyen‐Tu MS, Harris J, Martinez‐Sanchez A, et al. Opposing effects on regulated insulin secretion of acute vs chronic stimulation of AMP‐activated protein kinase. Diabetologia. 2022;65(6):997‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Cheung R, Pizza G, Chabosseau P, et al. Glucose‐dependent miR‐125b is a negative regulator of beta‐cell function. Diabetes. 2022;71(7):1525‐1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Gheibi S, Cataldo LR, Hamilton A, et al. Reduced expression level of protein phosphatase PPM1E serves to maintain insulin secretion in type 2 diabetes. Diabetes. 2023;72(4):455‐466. [DOI] [PubMed] [Google Scholar]
- 138. Wen X, Zhang B, Wu B, et al. Signaling pathways in obesity: mechanisms and therapeutic interventions. Signal Transduct Target Ther. 2022;7(1):298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kassouf T, Sumara G. Impact of conventional and atypical MAPKs on the development of metabolic diseases. Biomolecules. 2020;10(9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. De Felice FG, Gonçalves RA, Ferreira ST. Impaired insulin signalling and allostatic load in Alzheimer disease. Nat Rev Neurosci. 2022;23(4):215‐230. [DOI] [PubMed] [Google Scholar]
- 141. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10‐year update. Physiol Rev. 2012;92(2):689‐737. [DOI] [PubMed] [Google Scholar]
- 142. Lawan A, Bennett AM. Mitogen‐activated protein kinase regulation in hepatic metabolism. Trends Endocrinol Metab. 2017;28(12):868‐878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Zheng Y, Zhang W, Pendleton E, et al. Improved insulin sensitivity by calorie restriction is associated with reduction of ERK and p70S6K activities in the liver of obese Zucker rats. J Endocrinol. 2009;203(3):337‐347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Kwong E, Li Y, Hylemon PB, Zhou H. Bile acids and sphingosine‐1‐phosphate receptor 2 in hepatic lipid metabolism. Acta Pharm Sin B. 2015;5(2):151‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Studer E, Zhou X, Zhao R, et al. Conjugated bile acids activate the sphingosine‐1‐phosphate receptor 2 in primary rodent hepatocytes. Hepatology. 2012;55(1):267‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wu H‐T, Ou H‐Y, Hung H‐C, et al. A novel hepatokine, HFREP1, plays a crucial role in the development of insulin resistance and type 2 diabetes. Diabetologia. 2016;59(8):1732‐1742. [DOI] [PubMed] [Google Scholar]
- 147. Manowsky J, Camargo RG, Kipp AP, Henkel J, Püschel GP. Insulin‐induced cytokine production in macrophages causes insulin resistance in hepatocytes. Am J Physiol Endocrinol Metab. 2016;310(11):E938‐E946. [DOI] [PubMed] [Google Scholar]
- 148. Fisher FM, Estall JL, Adams AC, et al. Integrated regulation of hepatic metabolism by fibroblast growth factor 21 (FGF21) in vivo. Endocrinology. 2011;152(8):2996‐3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Liu H, Yu J, Xia T, et al. Hepatic serum‐ and glucocorticoid‐regulated protein kinase 1 (SGK1) regulates insulin sensitivity in mice via extracellular‐signal‐regulated kinase 1/2 (ERK1/2). Biochem J. 2014;464(2):281‐289. [DOI] [PubMed] [Google Scholar]
- 150. Vernia S, Cavanagh‐Kyros J, Garcia‐Haro L, et al. The PPARα‐FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 2014;20(3):512‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Pal M, Febbraio MA, Lancaster GI. The roles of c‐Jun NH2‐terminal kinases (JNKs) in obesity and insulin resistance. J Physiol. 2016;594(2):267‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Hemi R, Yochananov Y, Barhod E, et al. p38 mitogen‐activated protein kinase‐dependent transactivation of ErbB receptor family: a novel common mechanism for stress‐induced IRS‐1 serine phosphorylation and insulin resistance. Diabetes. 2011;60(4):1134‐1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Jing Y, Liu W, Cao H, et al. Hepatic p38α regulates gluconeogenesis by suppressing AMPK. J Hepatol. 2015;62(6):1319‐1327. [DOI] [PubMed] [Google Scholar]
- 154. Lee J, Sun C, Zhou Y, et al. p38 MAPK–mediated regulation of Xbp1s is crucial for glucose homeostasis. Nature Medicine. 2011;17(10):1251‐1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Han MS, Jung DY, Morel C, et al. JNK expression by macrophages promotes obesity‐induced insulin resistance and inflammation. Science. 2013;339(6116):218‐222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Sabio G, Das M, Mora A, et al. A stress signaling pathway in adipose tissue regulates hepatic insulin resistance. Science. 2008;322(5907):1539‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Kant S, Standen CL, Morel C, et al. A protein scaffold coordinates SRC‐mediated JNK activation in response to metabolic stress. Cell Rep. 2017;20(12):2775‐2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Shimizu T, Yamakuchi M, Biswas KK, et al. HMGB1 is secreted by 3T3‐L1 adipocytes through JNK signaling and the secretion is partially inhibited by adiponectin. Obesity. 2016;24(9):1913‐1921. [DOI] [PubMed] [Google Scholar]
- 159. Bost F, Aouadi M, Caron L, et al. The extracellular signal‐regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes. 2005;54(2):402‐411. [DOI] [PubMed] [Google Scholar]
- 160. Jager J, Corcelle V, Grémeaux T, et al. Deficiency in the extracellular signal‐regulated kinase 1 (ERK1) protects leptin‐deficient mice from insulin resistance without affecting obesity. Diabetologia. 2011;54(1):180‐189. [DOI] [PubMed] [Google Scholar]
- 161. Wang N, Zhao TT, Li SM, et al. Fibroblast growth factor 21 exerts its anti‐inflammatory effects on multiple cell types of adipose tissue in obesity. Obesity (Silver Spring). 2019;27(3):399‐408. [DOI] [PubMed] [Google Scholar]
- 162. Hirosumi J, Tuncman G, Chang L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333‐336. [DOI] [PubMed] [Google Scholar]
- 163. Bengal E, Aviram S, Hayek T. p38 MAPK in glucose metabolism of skeletal muscle: beneficial or harmful? Int J Mol Sci. 2020;21(18) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Montori‐Grau M, Tarrats N, Osorio‐Conles O, et al. Glucose dependence of glycogen synthase activity regulation by GSK3 and MEK/ERK inhibitors and angiotensin‐(1‐7) action on these pathways in cultured human myotubes. Cell Signal. 2013;25(5):1318‐1327. [DOI] [PubMed] [Google Scholar]
- 165. Sabio G, Kennedy NJ, Cavanagh‐Kyros J, et al. Role of muscle c‐Jun NH2‐terminal kinase 1 in obesity‐induced insulin resistance. Mol Cell Biol. 2010;30(1):106‐115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Lawan A, Min K, Zhang L, et al. Skeletal muscle‐specific deletion of MKP‐1 reveals a p38 MAPK/JNK/Akt signaling node that regulates obesity‐induced insulin resistance. Diabetes. 2018;67(4):624‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Wang X, Zhao D, Cui Y, Lu S, Gao D, Liu J. Proinflammatory macrophages impair skeletal muscle differentiation in obesity through secretion of tumor necrosis factor‐α via sustained activation of p38 mitogen‐activated protein kinase. J Cell Physiol. 2019;234(3):2566‐2580. [DOI] [PubMed] [Google Scholar]
- 168. Zhang G, Li Y‐P. p38β MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPβ. Skelet Muscle. 2012;2(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Henkel J, Neuschafer‐Rube F, Pathe‐Neuschafer‐Rube A, Puschel GP. Aggravation by prostaglandin E2 of interleukin‐6‐dependent insulin resistance in hepatocytes. Hepatology. 2009;50(3):781‐790. [DOI] [PubMed] [Google Scholar]
- 170. Arnette D, Gibson TB, Lawrence MC, et al. Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic beta cells. J Biol Chem. 2003;278(35):32517‐32525. [DOI] [PubMed] [Google Scholar]
- 171. Benes C, Poitout V, Marie JC, Martin‐Perez J, Roisin MP, Fagard R. Mode of regulation of the extracellular signal‐regulated kinases in the pancreatic beta‐cell line MIN6 and their implication in the regulation of insulin gene transcription. Biochem J. 1999;340(Pt 1):219‐225. [PMC free article] [PubMed] [Google Scholar]
- 172. Mayer SI, Thiel G. Calcium influx into MIN6 insulinoma cells induces expression of Egr‐1 involving extracellular signal‐regulated protein kinase and the transcription factors Elk‐1 and CREB. Eur J Cell Biol. 2009;88(1):19‐33. [DOI] [PubMed] [Google Scholar]
- 173. Lawrence MC, McGlynn K, Park BH, Cobb MH. ERK1/2‐dependent activation of transcription factors required for acute and chronic effects of glucose on the insulin gene promoter. J Biol Chem. 2005;280(29):26751‐26759. [DOI] [PubMed] [Google Scholar]
- 174. Longuet C, Broca C, Costes S, Hani EH, Bataille D, Dalle S. Extracellularly regulated kinases 1/2 (p44/42 mitogen‐activated protein kinases) phosphorylate synapsin I and regulate insulin secretion in the MIN6 beta‐cell line and islets of Langerhans. Endocrinology. 2005;146(2):643‐654. [DOI] [PubMed] [Google Scholar]
- 175. Leduc M, Richard J, Costes S, et al. ERK1 is dispensable for mouse pancreatic beta cell function but is necessary for glucose‐induced full activation of MSK1 and CREB. Diabetologia. 2017;60(10):1999‐2010. [DOI] [PubMed] [Google Scholar]
- 176. Yeo RW, Yang K, Li G, Lim SK. High glucose predisposes gene expression and ERK phosphorylation to apoptosis and impaired glucose‐stimulated insulin secretion via the cytoskeleton. PLoS One. 2012;7(9):e44988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. He X, Gao F, Hou J, et al. Metformin inhibits MAPK signaling and rescues pancreatic aquaporin 7 expression to induce insulin secretion in type 2 diabetes mellitus. J Biol Chem. 2021;297(2):101002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Brown JM, Bentsen MA, Rausch DM, et al. Role of hypothalamic MAPK/ERK signaling and central action of FGF1 in diabetes remission. iScience. 2021;24(9):102944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Nusse R, Clevers H. Wnt/β‐Catenin signaling, disease, and emerging therapeutic modalities. Cell. 2017;169(6):985‐999. [DOI] [PubMed] [Google Scholar]
- 180. Abou Ziki MD, Mani A. The interplay of canonical and noncanonical Wnt signaling in metabolic syndrome. Nutr Res. 2019;70:18‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Park HW, Kim YC, Yu B, et al. Alternative Wnt signaling activates YAP/TAZ. Cell. 2015;162(4):780‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, Long F. WNT‐LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. 2013;17(5):745‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Heallen T, Zhang M, Wang J, et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332(6028):458‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Aamir K, Khan HU, Sethi G, Hossain MA, Arya A. Wnt signaling mediates TLR pathway and promote unrestrained adipogenesis and metaflammation: Therapeutic targets for obesity and type 2 diabetes. Pharmacol Res. 2020;152:104602. [DOI] [PubMed] [Google Scholar]
- 185. Nie X, Wei X, Ma H, Fan L, Chen WD. The complex role of Wnt ligands in type 2 diabetes mellitus and related complications. J Cell Mol Med. 2021;25(14):6479‐6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Chen J, Ning C, Mu J, Li D, Ma Y, Meng X. Role of Wnt signaling pathways in type 2 diabetes mellitus. Mol Cell Biochem. 2021;476(5):2219‐2232. [DOI] [PubMed] [Google Scholar]
- 187. Welters HJ, Kulkarni RN. Wnt signaling: relevance to beta‐cell biology and diabetes. Trends Endocrinol Metab. 2008;19(10):349‐355. [DOI] [PubMed] [Google Scholar]
- 188. Nair GG, Tzanakakis ES, Hebrok M. Emerging routes to the generation of functional β‐cells for diabetes mellitus cell therapy. Nat Rev Endocrinol. 2020;16(9):506‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Liu H, Fergusson MM, Wu JJ, et al. Wnt signaling regulates hepatic metabolism. Sci Signal. 2011;4(158):ra6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Norton L, Fourcaudot M, Abdul‐Ghani MA, et al. Chromatin occupancy of transcription factor 7‐like 2 (TCF7L2) and its role in hepatic glucose metabolism. Diabetologia. 2011;54(12):3132‐3142. [DOI] [PubMed] [Google Scholar]
- 191. Ip W, Shao W, Song Z, Chen Z, Wheeler MB, Jin T. Liver‐specific expression of dominant‐negative transcription factor 7‐like 2 causes progressive impairment in glucose homeostasis. Diabetes. 2015;64(6):1923‐1932. [DOI] [PubMed] [Google Scholar]
- 192. Del Bosque‐Plata L, Martínez‐Martínez E, Espinoza‐Camacho M, Gragnoli C. The role of TCF7L2 in type 2 diabetes. Diabetes. 2021;70(6):1220‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Almario RU, Karakas SE. Roles of circulating WNT‐signaling proteins and WNT‐inhibitors in human adiposity, insulin resistance, insulin secretion, and inflammation. Horm Metab Res. 2015;47(2):152‐157. [DOI] [PubMed] [Google Scholar]
- 194. Hörbelt T, Knebel B, Fahlbusch P, et al. The adipokine sFRP4 induces insulin resistance and lipogenesis in the liver. Biochim Biophys Acta Mol Basis Dis. 2019;1865(10):2671‐2684. [DOI] [PubMed] [Google Scholar]
- 195. Reggio A, Rosina M, Palma A, et al. Adipogenesis of skeletal muscle fibro/adipogenic progenitors is affected by the WNT5a/GSK3/beta‐catenin axis. Cell Death Differ. 2020;27(10):2921‐2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Catalán V, Gómez‐Ambrosi J, Rodríguez A, et al. Activation of noncanonical Wnt signaling through WNT5A in visceral adipose tissue of obese subjects is related to inflammation. J Clin Endocrinol Metab. 2014;99(8):E1407‐E1417. [DOI] [PubMed] [Google Scholar]
- 197. Ouchi N, Higuchi A, Ohashi K, et al. Sfrp5 is an anti‐inflammatory adipokine that modulates metabolic dysfunction in obesity. Science. 2010;329(5990):454‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Liu W, Singh R, Choi CS, et al. Low density lipoprotein (LDL) receptor‐related protein 6 (LRP6) regulates body fat and glucose homeostasis by modulating nutrient sensing pathways and mitochondrial energy expenditure. J Biol Chem. 2012;287(10):7213‐7223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Cawthorn WP, Bree AJ, Yao Y, et al. Wnt6, Wnt10a and Wnt10b inhibit adipogenesis and stimulate osteoblastogenesis through a β‐catenin‐dependent mechanism. Bone. 2012;50(2):477‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Keats EC, Dominguez JM, 2nd , Grant MB, Khan ZA. Switch from canonical to noncanonical Wnt signaling mediates high glucose‐induced adipogenesis. Stem Cells. 2014;32(6):1649‐1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. van Tienen FH, Laeremans H, van der Kallen CJ, Smeets HJ. Wnt5b stimulates adipogenesis by activating PPARgamma, and inhibiting the beta‐catenin dependent Wnt signaling pathway together with Wnt5a. Biochem Biophys Res Commun. 2009;387(1):207‐211. [DOI] [PubMed] [Google Scholar]
- 202. Abiola M, Favier M, Christodoulou‐Vafeiadou E, Pichard AL, Martelly I, Guillet‐Deniau I. Activation of Wnt/beta‐catenin signaling increases insulin sensitivity through a reciprocal regulation of Wnt10b and SREBP‐1c in skeletal muscle cells. PLoS One. 2009;4(12):e8509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Pachori AS, Madan M, Nunez Lopez YO, Yi F, Meyer C, Seyhan AA. Reduced skeletal muscle secreted frizzled‐related protein 3 is associated with inflammation and insulin resistance. Obesity (Silver Spring). 2017;25(4):697‐703. [DOI] [PubMed] [Google Scholar]
- 204. Wilson C. Diabetes: human β‐cell proliferation by promoting Wnt signalling. Nat Rev Endocrinol. 2013;9(9):502. [DOI] [PubMed] [Google Scholar]
- 205. Chen X, Wei R, Jin T, Du H. Notoginsenoside R1 alleviates TNF‐alpha‐induced pancreatic beta‐cell min6 apoptosis and dysfunction through up‐regulation of miR‐29a. Artif Cells Nanomed Biotechnol. 2019;47(1):2379‐2388. [DOI] [PubMed] [Google Scholar]
- 206. Welters HJ, Oknianska A, Erdmann KS, Ryffel GU, Morgan NG. The protein tyrosine phosphatase‐BL, modulates pancreatic beta‐cell proliferation by interaction with the Wnt signalling pathway. J Endocrinol. 2008;197(3):543‐552. [DOI] [PubMed] [Google Scholar]
- 207. Bader E, Migliorini A, Gegg M, et al. Identification of proliferative and mature β‐cells in the islets of Langerhans. Nature. 2016;535(7612):430‐434. [DOI] [PubMed] [Google Scholar]
- 208. Mohan S, Kesavan C. T‐cell factor 7L2 is a novel regulator of osteoblast functions that acts in part by modulation of hypoxia signaling. Am J Physiol Endocrinol Metab. 2022;322(6):E528‐E539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Cui J, Duan J, Chu J, et al. Chikusetsu saponin IVa protects pancreatic beta cell against intermittent high glucose‐induced injury by activating Wnt/beta‐catenin/TCF7L2 pathway. Aging (Albany NY). 2020;12(2):1591‐1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. da Silva Xavier G, Loder MK, McDonald A, et al. TCF7L2 regulates late events in insulin secretion from pancreatic islet beta‐cells. Diabetes. 2009;58(4):894‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Goodyer WR, Gu X, Liu Y, Bottino R, Crabtree GR, Kim SK. Neonatal β cell development in mice and humans is regulated by calcineurin/NFAT. Dev Cell. 2012;23(1):21‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Kozinski K, Jazurek M, Dobrzyn P, et al. Adipose‐ and muscle‐derived Wnts trigger pancreatic β‐cell adaptation to systemic insulin resistance. Sci Rep. 2016;6:31553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Lemmer IL, Willemsen N, Hilal N, Bartelt A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol Metab. 2021;47:101169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Khanna M, Agrawal N, Chandra R, Dhawan G. Targeting unfolded protein response: a new horizon for disease control. Expert Rev Mol Med. 2021;23:e1. [DOI] [PubMed] [Google Scholar]
- 215. LaMarche NM, Kane H, Kohlgruber AC, Dong H, Lynch L, Brenner MB. Distinct iNKT cell populations use IFNgamma or ER stress‐induced IL‐10 to control adipose tissue homeostasis. Cell Metab. 2020;32(2):243‐258 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Cho YM, Kim DH, Lee KH, Jeong SW, Kwon OJ. The IRE1alpha‐XBP1s pathway promotes insulin‐stimulated glucose uptake in adipocytes by increasing PPARgamma activity. Exp Mol Med. 2018;50(8):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Liu X, Beaudoin JD, Davison CA, et al. A functional non‐coding RNA is produced from xbp‐1 mRNA. Neuron. 2020;107(5):854‐863. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Lee JH, Lee J. Endoplasmic reticulum (ER) stress and its role in pancreatic β‐Cell dysfunction and senescence in type 2 diabetes. Int J Mol Sci. 2022;23(9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Iurlaro R, Muñoz‐Pinedo C. Cell death induced by endoplasmic reticulum stress. Febs j. 2016;283(14):2640‐2652. [DOI] [PubMed] [Google Scholar]
- 220. Westrate LM, Hoyer MJ, Nash MJ, Voeltz GK. Vesicular and uncoated Rab1‐dependent cargo carriers facilitate ER to Golgi transport. J Cell Sci. 2020;133(14) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. 2020;21(8):421‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Villalobos‐Labra R, Subiabre M, Toledo F, Pardo F, Sobrevia L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol Aspects Med. 2019;66:49‐61. [DOI] [PubMed] [Google Scholar]
- 223. Liong S, Lappas M. Endoplasmic reticulum stress regulates inflammation and insulin resistance in skeletal muscle from pregnant women. Mol Cell Endocrinol. 2016;425:11‐25. [DOI] [PubMed] [Google Scholar]
- 224. Herrema H, Guan D, Choi JW, et al. FKBP11 rewires UPR signaling to promote glucose homeostasis in type 2 diabetes and obesity. Cell Metab. 2022;34(7):1004‐1022 e8. [DOI] [PubMed] [Google Scholar]
- 225. Basseri S, Lhoták S, Sharma AM, Austin RC. The chemical chaperone 4‐phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J Lipid Res. 2009;50(12):2486‐2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Oyadomari S, Harding HP, Zhang Y, Oyadomari M, Ron D. Dephosphorylation of translation initiation factor 2alpha enhances glucose tolerance and attenuates hepatosteatosis in mice. Cell Metab. 2008;7(6):520‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Seo J, Fortuno ES, 3rd , Suh JM, et al. Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes. 2009;58(11):2565‐2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Ozcan L, Cristina de Souza J, Harari AA, Backs J, Olson EN, Tabas I. Activation of calcium/calmodulin‐dependent protein kinase II in obesity mediates suppression of hepatic insulin signaling. Cell Metab. 2013;18(6):803‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress‐inducible gene, is induced via ATF4‐CHOP pathway and is involved in cell death. Embo J. 2005;24(6):1243‐1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Xiao G, Zhang T, Yu S, et al. ATF4 protein deficiency protects against high fructose‐induced hypertriglyceridemia in mice. J Biol Chem. 2013;288(35):25350‐25361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Martino MR, Gutiérrez‐Aguilar M, Yiew NKH, et al. Silencing alanine transaminase 2 in diabetic liver attenuates hyperglycemia by reducing gluconeogenesis from amino acids. Cell Rep. 2022;39(4):110733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Zhang W, Hietakangas V, Wee S, Lim SC, Gunaratne J, Cohen SM. ER stress potentiates insulin resistance through PERK‐mediated FOXO phosphorylation. Genes Dev. 2013;27(4):441‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Chen S, Henderson A, Petriello MC, et al. Trimethylamine N‐oxide binds and activates PERK to promote metabolic dysfunction. Cell Metab. 2019;30(6):1141‐1151. e5. [DOI] [PubMed] [Google Scholar]
- 234. Tang WH, Wang Z, Li XS, et al. Increased trimethylamine N‐oxide portends high mortality risk independent of glycemic control in patients with type 2 diabetes mellitus. Clin Chem. 2017;63(1):297‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Zarei M, Barroso E, Leiva R, et al. Heme‐regulated eIF2α kinase modulates hepatic FGF21 and is activated by PPARβ/δ deficiency. Diabetes. 2016;65(10):3185‐3199. [DOI] [PubMed] [Google Scholar]
- 236. Hsu JY, Crawley S, Chen M, et al. Non‐homeostatic body weight regulation through a brainstem‐restricted receptor for GDF15. Nature. 2017;550(7675):255‐259. [DOI] [PubMed] [Google Scholar]
- 237. Emmerson PJ, Wang F, Du Y, et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat Med. 2017;23(10):1215‐1219. [DOI] [PubMed] [Google Scholar]
- 238. Day EA, Ford RJ, Smith BK, et al. Metformin‐induced increases in GDF15 are important for suppressing appetite and promoting weight loss. Nat Metab. 2019;1(12):1202‐1208. [DOI] [PubMed] [Google Scholar]
- 239. Ning J, Hong T, Ward A, et al. Constitutive role for IRE1α‐XBP1 signaling pathway in the insulin‐mediated hepatic lipogenic program. Endocrinology. 2011;152(6):2247‐2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Shrestha N, Torres M, Zhang J, et al. Integration of ER protein quality control mechanisms defines beta cell function and ER architecture. J Clin Invest. 2023;133(1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Zhang M, Sun W, Qian J, Tang Y. Fasting exacerbates hepatic growth differentiation factor 15 to promote fatty acid β‐oxidation and ketogenesis via activating XBP1 signaling in liver. Redox Biol. 2018;16:87‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Zhang Z, Qian Q, Li M, et al. The unfolded protein response regulates hepatic autophagy by sXBP1‐mediated activation of TFEB. Autophagy. 2021;17(8):1841‐1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Sun H, Wei G, Liu H, et al. Inhibition of XBP1s ubiquitination enhances its protein stability and improves glucose homeostasis. Metabolism. 2020;105:154046. [DOI] [PubMed] [Google Scholar]
- 244. Lebeaupin C, Vallée D, Rousseau D, et al. Bax inhibitor‐1 protects from nonalcoholic steatohepatitis by limiting inositol‐requiring enzyme 1 alpha signaling in mice. Hepatology. 2018;68(2):515‐532. [DOI] [PubMed] [Google Scholar]
- 245. Lisbona F, Rojas‐Rivera D, Thielen P, et al. BAX inhibitor‐1 is a negative regulator of the ER stress sensor IRE1alpha. Mol Cell. 2009;33(6):679‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Cao J, Peng J, An H, et al. Endotoxemia‐mediated activation of acetyltransferase P300 impairs insulin signaling in obesity. Nat Commun. 2017;8(1):131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Wang Y, Vera L, Fischer WH, Montminy M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009;460(7254):534‐537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Chen X, Zhang F, Gong Q, et al. Hepatic ATF6 increases fatty acid oxidation to attenuate hepatic steatosis in mice through peroxisome proliferator‐activated receptor α. Diabetes. 2016;65(7):1904‐1915. [DOI] [PubMed] [Google Scholar]
- 249. Usui M, Yamaguchi S, Tanji Y, et al. Atf6α‐null mice are glucose intolerant due to pancreatic β‐cell failure on a high‐fat diet but partially resistant to diet‐induced insulin resistance. Metabolism. 2012;61(8):1118‐1128. [DOI] [PubMed] [Google Scholar]
- 250. Chen Y, Wang J, Wang Y, et al. A propolis‐derived small molecule ameliorates metabolic syndrome in obese mice by targeting the CREB/CRTC2 transcriptional complex. Nat Commun. 2022;13(1):246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K. Obesity‐induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep. 2012;2:799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Boden G, Cheung P, Salehi S, et al. Insulin regulates the unfolded protein response in human adipose tissue. Diabetes. 2014;63(3):912‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Li H, Zhou B, Xu L, et al. The reciprocal interaction between autophagic dysfunction and ER stress in adipose insulin resistance. Cell Cycle. 2014;13(4):565‐579. [DOI] [PubMed] [Google Scholar]
- 254. Mondal AK, Das SK, Varma V, et al. Effect of endoplasmic reticulum stress on inflammation and adiponectin regulation in human adipocytes. Metab Syndr Relat Disord. 2012;10(4):297‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Liu Z, Gan L, Wu T, et al. Adiponectin reduces ER stress‐induced apoptosis through PPARα transcriptional regulation of ATF2 in mouse adipose. Cell Death Dis. 2016;7(11):e2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Torre‐Villalvazo I, Bunt AE, Alemán G, et al. Adiponectin synthesis and secretion by subcutaneous adipose tissue is impaired during obesity by endoplasmic reticulum stress. J Cell Biochem. 2018;119(7):5970‐5984. [DOI] [PubMed] [Google Scholar]
- 257. Jiao P, Ma J, Feng B, et al. FFA‐induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKβ pathways. Obesity (Silver Spring). 2011;19(3):483‐491. [DOI] [PubMed] [Google Scholar]
- 258. Perkins ND. Integrating cell‐signalling pathways with NF‐kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8(1):49‐62. [DOI] [PubMed] [Google Scholar]
- 259. Koh HJ, Toyoda T, Didesch MM, et al. Tribbles 3 mediates endoplasmic reticulum stress‐induced insulin resistance in skeletal muscle. Nat Commun. 2013;4:1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Ijuin T, Hosooka T, Takenawa T. Phosphatidylinositol 3,4,5‐trisphosphate phosphatase SKIP links endoplasmic reticulum stress in skeletal muscle to insulin resistance. Mol Cell Biol. 2016;36(1):108‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest. 2017;127(1):43‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Reyna SM, Ghosh S, Tantiwong P, et al. Elevated toll‐like receptor 4 expression and signaling in muscle from insulin‐resistant subjects. Diabetes. 2008;57(10):2595‐2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Guo Q, Hu H, Liu X, et al. C/EBPβ mediates palmitate‐induced musclin expression via the regulation of PERK/ATF4 pathways in myotubes. Am J Physiol Endocrinol Metab. 2019;316(6):E1081‐E1092. [DOI] [PubMed] [Google Scholar]
- 264. McNally BD, Ashley DF, Hänschke L, et al. Long‐chain ceramides are cell non‐autonomous signals linking lipotoxicity to endoplasmic reticulum stress in skeletal muscle. Nat Commun. 2022;13(1):1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Flamment M, Hajduch E, Ferré P, Foufelle F. New insights into ER stress‐induced insulin resistance. Trends Endocrinol Metab. 2012;23(8):381‐390. [DOI] [PubMed] [Google Scholar]
- 266. Lee JM, Park S, Lee D, et al. Reduction in endoplasmic reticulum stress activates beige adipocytes differentiation and alleviates high fat diet‐induced metabolic phenotypes. Biochim Biophys Acta Mol Basis Dis. 2021;1867(5):166099. [DOI] [PubMed] [Google Scholar]
- 267. Wu J, Ruas JL, Estall JL, et al. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC‐1α/ATF6α complex. Cell Metab. 2011;13(2):160‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Raciti GA, Iadicicco C, Ulianich L, et al. Glucosamine‐induced endoplasmic reticulum stress affects GLUT4 expression via activating transcription factor 6 in rat and human skeletal muscle cells. Diabetologia. 2010;53(5):955‐965. [DOI] [PubMed] [Google Scholar]
- 269. Nakanishi K, Sudo T, Morishima N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol. 2005;169(4):555‐560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Li J, Zheng Y, Yan P, et al. A single‐cell transcriptomic atlas of primate pancreatic islet aging. Natl Sci Rev. 2021;8(2):nwaa127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. O'Neill CM, Lu C, Corbin KL, et al. Circulating levels of IL‐1B+IL‐6 cause ER stress and dysfunction in islets from prediabetic male mice. Endocrinology. 2013;154(9):3077‐3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Cnop M, Toivonen S, Igoillo‐Esteve M, Salpea P. Endoplasmic reticulum stress and eIF2α phosphorylation: The Achilles heel of pancreatic β cells. Mol Metab. 2017;6(9):1024‐1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Gupta S, McGrath B, Cavener DR. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes. 2010;59(8):1937‐1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Kim MJ, Kim MN, Min SH, et al. Specific PERK inhibitors enhanced glucose‐stimulated insulin secretion in a mouse model of type 2 diabetes. Metabolism. 2019;97:87‐91. [DOI] [PubMed] [Google Scholar]
- 275. Sowers CR, Wang R, Bourne RA, et al. The protein kinase PERK/EIF2AK3 regulates proinsulin processing not via protein synthesis but by controlling endoplasmic reticulum chaperones. J Biol Chem. 2018;293(14):5134‐5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Back SH, Scheuner D, Han J, et al. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009;10(1):13‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Cnop M, Ladriere L, Hekerman P, et al. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid‐induced endoplasmic reticulum stress and causes pancreatic beta‐cell dysfunction and apoptosis. J Biol Chem. 2007;282(6):3989‐3997. [DOI] [PubMed] [Google Scholar]
- 278. Liew CW, Bochenski J, Kawamori D, et al. The pseudokinase tribbles homolog 3 interacts with ATF4 to negatively regulate insulin exocytosis in human and mouse beta cells. J Clin Invest. 2010;120(8):2876‐2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Shoulders MD, Ryno LM, Genereux JC, et al. Stress‐independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013;3(4):1279‐1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Lee AH, Heidtman K, Hotamisligil GS, Glimcher LH. Dual and opposing roles of the unfolded protein response regulated by IRE1alpha and XBP1 in proinsulin processing and insulin secretion. Proc Natl Acad Sci USA. 2011;108(21):8885‐8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Hassler JR, Scheuner DL, Wang S, et al. The IRE1α/XBP1s pathway is essential for the glucose response and protection of β cells. PLoS Biol. 2015;13(10):e1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Tsuchiya Y, Saito M, Kadokura H, et al. IRE1‐XBP1 pathway regulates oxidative proinsulin folding in pancreatic β cells. J Cell Biol. 2018;217(4):1287‐1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Lee K, Chan JY, Liang C, et al. XBP1 maintains beta cell identity, represses beta‐to‐alpha cell transdifferentiation and protects against diabetic beta cell failure during metabolic stress in mice. Diabetologia. 2022;65(6):984‐996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Allagnat F, Christulia F, Ortis F, et al. Sustained production of spliced X‐box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia. 2010;53(6):1120‐1130. [DOI] [PubMed] [Google Scholar]
- 285. Cunha DA, Gurzov EN, Naamane N, et al. JunB protects beta‐cells from lipotoxicity via the XBP1‐AKT pathway. Cell Death Differ. 2014;21(8):1313‐1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Marrocco V, Tran T, Zhu S, et al. A small molecule UPR modulator for diabetes identified by high throughput screening. Acta Pharm Sin B. 2021;11(12):3983‐3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Sharma RB, O'Donnell AC, Stamateris RE, et al. Insulin demand regulates β cell number via the unfolded protein response. J Clin Invest. 2015;125(10):3831‐3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Matsuda T, Kido Y, Asahara S, et al. Ablation of C/EBPbeta alleviates ER stress and pancreatic beta cell failure through the GRP78 chaperone in mice. J Clin Invest. 2010;120(1):115‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Yamamoto K, Sato T, Matsui T, et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13(3):365‐376. [DOI] [PubMed] [Google Scholar]
- 290. Sharma RB, O'Donnell AC, Stamateris RE, et al. Insulin demand regulates beta cell number via the unfolded protein response. J Clin Invest. 2015;125(10):3831‐3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Charbord J, Ren L, Sharma RB, et al. In vivo screen identifies a SIK inhibitor that induces β cell proliferation through a transient UPR. Nat Metab. 2021;3(5):682‐700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Totaro A, Panciera T, Piccolo S. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol. 2018;20(8):888‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Ardestani A, Lupse B, Maedler K. Hippo signaling: key emerging pathway in cellular and whole‐body metabolism. Trends Endocrinol Metab. 2018;29(7):492‐509. [DOI] [PubMed] [Google Scholar]
- 294. Koo JH, Guan KL. Interplay between YAP/TAZ and metabolism. Cell Metab. 2018;28(2):196‐206. [DOI] [PubMed] [Google Scholar]
- 295. Ibar C, Irvine KD. Integration of Hippo‐YAP signaling with metabolism. Dev Cell. 2020;54(2):256‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Enzo E, Santinon G, Pocaterra A, et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. Embo J. 2015;34(10):1349‐1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Mo JS, Meng Z, Kim YC, et al. Cellular energy stress induces AMPK‐mediated regulation of YAP and the Hippo pathway. Nat Cell Biol. 2015;17(4):500‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Wang W, Xiao ZD, Li X, et al. AMPK modulates hippo pathway activity to regulate energy homeostasis. Nat Cell Biol. 2015;17(4):490‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Peng C, Zhu Y, Zhang W, et al. Regulation of the hippo‐YAP pathway by glucose sensor o‐GlcNAcylation. Mol Cell. 2017;68(3):591‐604. e5. [DOI] [PubMed] [Google Scholar]
- 300. Watt KI, Henstridge DC, Ziemann M, et al. Yap regulates skeletal muscle fatty acid oxidation and adiposity in metabolic disease. Nat Commun. 2021;12(1):2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Jeong SH, Kim HB, Kim MC, et al. Hippo‐mediated suppression of IRS2/AKT signaling prevents hepatic steatosis and liver cancer. J Clin Invest. 2018;128(3):1010‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9(3):316‐323. [DOI] [PubMed] [Google Scholar]
- 303. Iglesias C, Floridia E, Sartages M, et al. The MST3/STK24 kinase mediates impaired fasting blood glucose after a high‐fat diet. Diabetologia. 2017;60(12):2453‐2462. [DOI] [PubMed] [Google Scholar]
- 304. Hu Y, Shin DJ, Pan H, et al. YAP suppresses gluconeogenic gene expression through PGC1α. Hepatology. 2017;66(6):2029‐2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Yu FX, Zhang Y, Park HW, et al. Protein kinase A activates the hippo pathway to modulate cell proliferation and differentiation. Genes Dev. 2013;27(11):1223‐1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306. Wang L, Wang S, Shi Y, et al. YAP and TAZ protect against white adipocyte cell death during obesity. Nat Commun. 2020;11(1):5455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Zhou Y, Huang T, Zhang J, et al. Emerging roles of Hippo signaling in inflammation and YAP‐driven tumor immunity. Cancer Lett. 2018;426:73‐79. [DOI] [PubMed] [Google Scholar]
- 308. Mamidi A, Prawiro C, Seymour PA, et al. Mechanosignalling via integrins directs fate decisions of pancreatic progenitors. Nature. 2018;564(7734):114‐118. [DOI] [PubMed] [Google Scholar]
- 309. Ardestani A, Maedler K. The Hippo signaling pathway in pancreatic β‐cells: functions and regulations. Endocr Rev. 2018;39(1):21‐35. [DOI] [PubMed] [Google Scholar]
- 310. Gonzalez FJ, Xie C, Jiang C. The role of hypoxia‐inducible factors in metabolic diseases. Nat Rev Endocrinol. 2018;15(1):21‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Jiang X, Zhang D, Zhang H, Huang Y, Teng M. Role of Ran‐regulated nuclear‐cytoplasmic trafficking of pVHL in the regulation of microtubular stability‐mediated HIF‐1alpha in hypoxic cardiomyocytes. Sci Rep. 2015;5:9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Lee YS, Kim JW, Osborne O, et al. Increased adipocyte O2 consumption triggers HIF‐1alpha, causing inflammation and insulin resistance in obesity. Cell. 2014;157(6):1339‐1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Taniguchi CM, Finger EC, Krieg AJ, et al. Cross‐talk between hypoxia and insulin signaling through Phd3 regulates hepatic glucose and lipid metabolism and ameliorates diabetes. Nat Med. 2013;19(10):1325‐1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Ramakrishnan SK, Zhang H, Takahashi S, et al. HIF2α is an essential molecular brake for postprandial hepatic glucagon response independent of insulin signaling. Cell Metab. 2016;23(3):505‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Xie C, Yagai T, Luo Y, et al. Activation of intestinal hypoxia‐inducible factor 2alpha during obesity contributes to hepatic steatosis. Nat Med. 2017;23(11):1298‐1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Jiang C, Qu A, Matsubara T, et al. Disruption of hypoxia‐inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high‐fat diet‐fed mice. Diabetes. 2011;60(10):2484‐2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Takikawa A, Mahmood A, Nawaz A, et al. HIF‐1α in myeloid cells promotes adipose tissue remodeling toward insulin resistance. Diabetes. 2016;65(12):3649‐3659. [DOI] [PubMed] [Google Scholar]
- 318. Li HS, Zhou YN, Li L, et al. HIF‐1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019;25:101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Cheng K, Ho K, Stokes R, et al. Hypoxia‐inducible factor‐1alpha regulates beta cell function in mouse and human islets. J Clin Invest. 2010;120(6):2171‐2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Gunton JE, Kulkarni RN, Yim S, et al. Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic‐islet dysfunction in human type 2 diabetes. Cell. 2005;122(3):337‐349. [DOI] [PubMed] [Google Scholar]
- 321. Choi D, Cai EP, Schroer SA, Wang L, Woo M. Vhl is required for normal pancreatic beta cell function and the maintenance of beta cell mass with age in mice. Lab Invest. 2011;91(4):527‐538. [DOI] [PubMed] [Google Scholar]
- 322. Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11(10):790‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Brown ML, Schneyer A. A decade later: revisiting the TGFβ family's role in diabetes. Trends Endocrinol Metab. 2021;32(1):36‐47. [DOI] [PubMed] [Google Scholar]
- 324. Brown ML, Bonomi L, Ungerleider N, et al. Follistatin and follistatin like‐3 differentially regulate adiposity and glucose homeostasis. Obesity (Silver Spring). 2011;19(10):1940‐1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Brandt C, Hansen RH, Hansen JB, et al. Over‐expression of follistatin‐like 3 attenuates fat accumulation and improves insulin sensitivity in mice. Metabolism. 2015;64(2):283‐295. [DOI] [PubMed] [Google Scholar]
- 326. Tao R, Wang C, Stöhr O, et al. Inactivating hepatic follistatin alleviates hyperglycemia. Nat Med. 2018;24(7):1058‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Chattopadhyay T, Singh RR, Gupta S, Surolia A. Bone morphogenetic protein‐7 (BMP‐7) augments insulin sensitivity in mice with type II diabetes mellitus by potentiating PI3K/AKT pathway. Biofactors. 2017;43(2):195‐209. [DOI] [PubMed] [Google Scholar]
- 328. Pauk M, Bordukalo‐Niksic T, Brkljacic J, et al. A novel role of bone morphogenetic protein 6 (BMP6) in glucose homeostasis. Acta Diabetologica. 2019;56(3):365‐371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Wang HL, Wang L, Zhao CY, Lan HY. Role of TGF‐beta signaling in beta cell proliferation and function in diabetes. Biomolecules. 2022;12(3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Kim HS, Hong SH, Oh SH, Kim JH, Lee MS, Lee MK. Activin A, exendin‐4, and glucose stimulate differentiation of human pancreatic ductal cells. J Endocrinol. 2013;217(3):241‐252. [DOI] [PubMed] [Google Scholar]
- 331. Brown ML, Andrzejewski D, Burnside A, Schneyer AL. Activin enhances α‐ to β‐Cell transdifferentiation as a source for β‐cells in male FSTL3 knockout mice. Endocrinology. 2016;157(3):1043‐1054. [DOI] [PubMed] [Google Scholar]
- 332. Szabat M, Johnson JD, Piret JM. Reciprocal modulation of adult beta cell maturity by activin A and follistatin. Diabetologia. 2010;53(8):1680‐1689. [DOI] [PubMed] [Google Scholar]
- 333. Sehrawat A, Shiota C, Mohamed N, et al. SMAD7 enhances adult β‐cell proliferation without significantly affecting β‐cell function in mice. J Biol Chem. 2020;295(15):4858‐4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Lee JH, Mellado‐Gil JM, Bahn YJ, Pathy SM, Zhang YE, Rane SG. Protection from β‐cell apoptosis by inhibition of TGF‐β/Smad3 signaling. Cell Death Dis. 2020;11(3):184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. El‐Gohary Y, Tulachan S, Wiersch J, et al. A smad signaling network regulates islet cell proliferation. Diabetes. 2014;63(1):224‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Wu H, Mezghenna K, Marmol P, et al. Differential regulation of mouse pancreatic islet insulin secretion and Smad proteins by activin ligands. Diabetologia. 2014;57(1):148‐156. [DOI] [PubMed] [Google Scholar]
- 337. Nomura M, Zhu HL, Wang L, Morinaga H, Takayanagi R, Teramoto N. SMAD2 disruption in mouse pancreatic beta cells leads to islet hyperplasia and impaired insulin secretion due to the attenuation of ATP‐sensitive K+ channel activity. Diabetologia. 2014;57(1):157‐166. [DOI] [PubMed] [Google Scholar]
- 338. Brown ML, Ungerleider N, Bonomi L, Andrzejewski D, Burnside A, Schneyer A. Effects of activin A on survival, function and gene expression of pancreatic islets from non‐diabetic and diabetic human donors. Islets. 2014;6(5‐6):e1017226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Jin L, Yang R, Geng L, Xu A. Fibroblast growth factor‐based pharmacotherapies for the treatment of obesity‐related metabolic complications. Annu Rev Pharmacol Toxicol. 2022; [DOI] [PubMed] [Google Scholar]
- 340. Giacomini A, Grillo E, Rezzola S, et al. The FGF/FGFR system in the physiopathology of the prostate gland. Physiol Rev. 2021;101(2):569‐610. [DOI] [PubMed] [Google Scholar]
- 341. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8(3):235‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Degirolamo C, Sabba C, Moschetta A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat Rev Drug Discov. 2016;15(1):51‐69. [DOI] [PubMed] [Google Scholar]
- 343. Geng L, Lam KSL, Xu A. The therapeutic potential of FGF21 in metabolic diseases: from bench to clinic. Nat Rev Endocrinol. 2020;16(11):654‐667. [DOI] [PubMed] [Google Scholar]
- 344. Gasser E, Moutos CP, Downes M, Evans RM. FGF1 ‐ a new weapon to control type 2 diabetes mellitus. Nat Rev Endocrinol. 2017;13(10):599‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Kliewer SA, Mangelsdorf DJ. A dozen years of discovery: insights into the physiology and pharmacology of FGF21. Cell Metab. 2019;29(2):246‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346. Markan KR, Naber MC, Ameka MK, et al. Circulating FGF21 is liver derived and enhances glucose uptake during refeeding and overfeeding. Diabetes. 2014;63(12):4057‐4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Kurosu H, Choi M, Ogawa Y, et al. Tissue‐specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem. 2007;282(37):26687‐26695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Gong Q, Hu Z, Zhang F, et al. Fibroblast growth factor 21 improves hepatic insulin sensitivity by inhibiting mammalian target of rapamycin complex 1 in mice. Hepatology. 2016;64(2):425‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Potthoff MJ, Inagaki T, Satapati S, et al. FGF21 induces PGC‐1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci USA. 2009;106(26):10853‐10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Zhang Y, Lei T, Huang JF, et al. The link between fibroblast growth factor 21 and sterol regulatory element binding protein 1c during lipogenesis in hepatocytes. Mol Cell Endocrinol. 2011;342(1‐2):41‐47. [DOI] [PubMed] [Google Scholar]
- 351. Zarei M, Barroso E, Palomer X, et al. Hepatic regulation of VLDL receptor by PPARbeta/delta and FGF21 modulates non‐alcoholic fatty liver disease. Mol Metab. 2018;8:117‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Ogawa Y, Kurosu H, Yamamoto M, et al. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc Natl Acad Sci USA. 2007;104(18):7432‐7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Ge X, Chen C, Hui X, Wang Y, Lam KS, Xu A. Fibroblast growth factor 21 induces glucose transporter‐1 expression through activation of the serum response factor/Ets‐like protein‐1 in adipocytes. J Biol Chem. 2011;286(40):34533‐34541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. BonDurant LD, Ameka M, Naber MC, et al. FGF21 regulates metabolism through adipose‐dependent and ‐independent mechanisms. Cell Metab. 2017;25(4):935‐944. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Lin Z, Tian H, Lam KS, et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013;17(5):779‐789. [DOI] [PubMed] [Google Scholar]
- 356. Dutchak PA, Katafuchi T, Bookout AL, et al. Fibroblast growth factor‐21 regulates PPARgamma activity and the antidiabetic actions of thiazolidinediones. Cell. 2012;148(3):556‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357. Keipert S, Ost M. Stress‐induced FGF21 and GDF15 in obesity and obesity resistance. Trends Endocrinol Metab. 2021;32(11):904‐915. [DOI] [PubMed] [Google Scholar]
- 358. Potthoff MJ, Boney‐Montoya J, Choi M, et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB‐PGC‐1alpha pathway. Cell Metab. 2011;13(6):729‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. Kir S, Beddow SA, Samuel VT, et al. FGF19 as a postprandial, insulin‐independent activator of hepatic protein and glycogen synthesis. Science. 2011;331(6024):1621‐1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360. Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2(4):217‐225. [DOI] [PubMed] [Google Scholar]
- 361. Gadaleta RM, Moschetta A. Metabolic messengers: fibroblast growth factor 15/19. Nat Metab. 2019;1(6):588‐594. [DOI] [PubMed] [Google Scholar]
- 362. Suh JM, Jonker JW, Ahmadian M, et al. Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature. 2014;513(7518):436‐439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Sancar G, Liu S, Gasser E, et al. FGF1 and insulin control lipolysis by convergent pathways. Cell Metab. 2022;34(1):171‐183 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Jonker JW, Suh JM, Atkins AR, et al. A PPARgamma‐FGF1 axis is required for adaptive adipose remodelling and metabolic homeostasis. Nature. 2012;485(7398):391‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365. Choi Y, Jang S, Choi MS, Ryoo ZY, Park T. Increased expression of FGF1‐mediated signaling molecules in adipose tissue of obese mice. J Physiol Biochem. 2016;72(2):157‐167. [DOI] [PubMed] [Google Scholar]
- 366. Ying L, Wang L, Guo K, et al. Paracrine FGFs target skeletal muscle to exert potent anti‐hyperglycemic effects. Nat Commun. 2021;12(1):7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367. Wente W, Efanov AM, Brenner M, et al. Fibroblast growth factor‐21 improves pancreatic beta‐cell function and survival by activation of extracellular signal‐regulated kinase 1/2 and Akt signaling pathways. Diabetes. 2006;55(9):2470‐2478. [DOI] [PubMed] [Google Scholar]
- 368. Morton GJ, Matsen ME, Bracy DP, et al. FGF19 action in the brain induces insulin‐independent glucose lowering. J Clin Invest. 2013;123(11):4799‐4808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Bookout AL, de Groot MH, Owen BM, et al. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat Med. 2013;19(9):1147‐1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Perry RJ, Lee S, Ma L, Zhang D, Schlessinger J, Shulman GI. FGF1 and FGF19 reverse diabetes by suppression of the hypothalamic‐pituitary‐adrenal axis. Nat Commun. 2015;6:6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Fuchs CD, Trauner M. Role of bile acids and their receptors in gastrointestinal and hepatic pathophysiology. Nat Rev Gastroenterol Hepatol. 2022;19(7):432‐450. [DOI] [PubMed] [Google Scholar]
- 372. Perino A, Demagny H, Velazquez‐Villegas L, Schoonjans K. Molecular physiology of bile acid signaling in health, disease, and aging. Physiol Rev. 2021;101(2):683‐731. [DOI] [PubMed] [Google Scholar]
- 373. Porez G, Prawitt J, Gross B, Staels B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J Lipid Res. 2012;53(9):1723‐1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Thomas C, Gioiello A, Noriega L, et al. TGR5‐mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10(3):167‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Rao A, Kosters A, Mells JE, et al. Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high‐fat diet‐fed mice. Sci Transl Med. 2016;8(357):357ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376. Gao F, Zhang X, Zhou L, et al. Type 2 diabetes mitigation in the diabetic Goto‐Kakizaki rat by elevated bile acids following a common‐bile‐duct surgery. Metabolism. 2016;65(2):78‐88. [DOI] [PubMed] [Google Scholar]
- 377. Chen X, Xu H, Ding L, et al. Identification of miR‐26a as a target gene of bile acid receptor GPBAR‐1/TGR5. PLoS One. 2015;10(6):e0131294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378. Li H, Perino A, Huang Q, et al. Integrative systems analysis identifies genetic and dietary modulators of bile acid homeostasis. Cell Metab. 2022;34(10):1594‐1610 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Seyer P, Vallois D, Poitry‐Yamate C, et al. Hepatic glucose sensing is required to preserve beta cell glucose competence. J Clin Invest. 2013;123(4):1662‐1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Pathak P, Liu H, Boehme S, et al. Farnesoid X receptor induces Takeda G‐protein receptor 5 cross‐talk to regulate bile acid synthesis and hepatic metabolism. J Biol Chem. 2017;292(26):11055‐11069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Zheng X, Chen T, Jiang R, et al. Hyocholic acid species improve glucose homeostasis through a distinct TGR5 and FXR signaling mechanism. Cell Metab. 2021;33(4):791‐803 e7. [DOI] [PubMed] [Google Scholar]
- 382. Makki K, Brolin H, Petersen N, et al. 6alpha‐hydroxylated bile acids mediate TGR5 signalling to improve glucose metabolism upon dietary fiber supplementation in mice. Gut. 2022; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Chiang JYL, Ferrell JM. Bile acids as metabolic regulators and nutrient sensors. Annu Rev Nutr. 2019;39:175‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384. Sun L, Xie C, Wang G, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med. 2018;24(12):1919‐1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Trabelsi MS, Lestavel S, Staels B, Collet X. Intestinal bile acid receptors are key regulators of glucose homeostasis. Proc Nutr Soc. 2017;76(3):192‐202. [DOI] [PubMed] [Google Scholar]
- 386. Fang S, Suh JM, Reilly SM, et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med. 2015;21(2):159‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Giorgi C, Danese A, Missiroli S, Patergnani S, Pinton P. Calcium dynamics as a machine for decoding signals. Trends Cell Biol. 2018;28(4):258‐273. [DOI] [PubMed] [Google Scholar]
- 388. Luan S, Wang C. Calcium signaling mechanisms across kingdoms. Annu Rev Cell Dev Biol. 2021;37:311‐340. [DOI] [PubMed] [Google Scholar]
- 389. Madreiter‐Sokolowski CT, Thomas C, Ristow M. Interrelation between ROS and Ca(2+) in aging and age‐related diseases. Redox Biol. 2020;36:101678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Pierro C, Sneyers F, Bultynck G, Roderick HL. ER Ca(2+) release and store‐operated Ca(2+) entry ‐ partners in crime or independent actors in oncogenic transformation? Cell Calcium. 2019;82:102061. [DOI] [PubMed] [Google Scholar]
- 391. Vandecaetsbeek I, Vangheluwe P, Raeymaekers L, Wuytack F, Vanoevelen J. The Ca2+ pumps of the endoplasmic reticulum and Golgi apparatus. Cold Spring Harb Perspect Biol. 2011;3(5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Garbincius JF, Elrod JW. Mitochondrial calcium exchange in physiology and disease. Physiol Rev. 2022;102(2):893‐992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Sancak Y, Markhard AL, Kitami T, et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science. 2013;342(6164):1379‐1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Raffaello A, De Stefani D, Sabbadin D, et al. The mitochondrial calcium uniporter is a multimer that can include a dominant‐negative pore‐forming subunit. Embo J. 2013;32(17):2362‐2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Patron M, Granatiero V, Espino J, Rizzuto R, De Stefani D. MICU3 is a tissue‐specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019;26(1):179‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. Screaton RA, Conkright MD, Katoh Y, et al. The CREB coactivator TORC2 functions as a calcium‐ and cAMP‐sensitive coincidence detector. Cell. 2004;119(1):61‐74. [DOI] [PubMed] [Google Scholar]
- 397. Macian F. NFAT proteins: key regulators of T‐cell development and function. Nat Rev Immunol. 2005;5(6):472‐484. [DOI] [PubMed] [Google Scholar]
- 398. Sabatini PV, Speckmann T, Lynn FC. Friend and foe: beta‐cell Ca(2+) signaling and the development of diabetes. Mol Metab. 2019;21:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Bernal‐Mizrachi E, Wen W, Shornick M, Permutt MA. Activation of nuclear factor‐kappaB by depolarization and Ca(2+) influx in MIN6 insulinoma cells. Diabetes. 2002;51(Suppl 3):S484‐S488. [DOI] [PubMed] [Google Scholar]
- 400. Duan L, Cobb MH. Calcineurin increases glucose activation of ERK1/2 by reversing negative feedback. Proc Natl Acad Sci USA. 2010;107(51):22314‐22319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401. Wu H, Rothermel B, Kanatous S, et al. Activation of MEF2 by muscle activity is mediated through a calcineurin‐dependent pathway. Embo J. 2001;20(22):6414‐6423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Samanta K, Mirams GR, Parekh AB. Sequential forward and reverse transport of the Na(+) Ca(2+) exchanger generates Ca(2+) oscillations within mitochondria. Nat Commun. 2018;9(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Liu Y, Ma X, Fujioka H, Liu J, Chen S, Zhu X. DJ‐1 regulates the integrity and function of ER‐mitochondria association through interaction with IP3R3‐Grp75‐VDAC1. Proc Natl Acad Sci USA. 2019;116(50):25322‐25328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404. Yang J, Zhao Z, Gu M, Feng X, Xu H. Release and uptake mechanisms of vesicular Ca(2+) stores. Protein Cell. 2019;10(1):8‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Nolte LA, Rincón J, Wahlström EO, Craig BW, Zierath JR, Wallberg‐Henriksson H. Hyperglycemia activates glucose transport in rat skeletal muscle via a Ca(2+)‐dependent mechanism. Diabetes. 1995;44(11):1345‐1348. [DOI] [PubMed] [Google Scholar]
- 406. Contreras‐Ferrat A, Lavandero S, Jaimovich E, Klip A. Calcium signaling in insulin action on striated muscle. Cell Calcium. 2014;56(5):390‐396. [DOI] [PubMed] [Google Scholar]
- 407. Contreras‐Ferrat A, Llanos P, Vasquez C, et al. Insulin elicits a ROS‐activated and an IP(3)‐dependent Ca(2)(+) release, which both impinge on GLUT4 translocation. J Cell Sci. 2014;127(Pt 9):1911‐1923. [DOI] [PubMed] [Google Scholar]
- 408. Bysani M, Agren R, Davegårdh C, et al. ATAC‐seq reveals alterations in open chromatin in pancreatic islets from subjects with type 2 diabetes. Sci Rep. 2019;9(1):7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Araki K, Araki A, Honda D, et al. TDP‐43 regulates early‐phase insulin secretion via CaV1.2‐mediated exocytosis in islets. J Clin Invest. 2019;129(9):3578‐3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Hija A, Salpeter S, Klochendler A, et al. G0‐G1 transition and the restriction point in pancreatic β‐cells in vivo. Diabetes. 2014;63(2):578‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Porat S, Weinberg‐Corem N, Tornovsky‐Babaey S, et al. Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab. 2011;13(4):440‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Tuduri E, Soriano S, Almagro L, et al. The pancreatic beta‐cell in ageing: implications in age‐related diabetes. Ageing Res Rev. 2022;80:101674. [DOI] [PubMed] [Google Scholar]
- 413. Vakilian M, Tahamtani Y, Ghaedi K. A review on insulin trafficking and exocytosis. Gene. 2019;706:52‐61. [DOI] [PubMed] [Google Scholar]
- 414. Park SW, Zhou Y, Lee J, Lee J, Ozcan U. Sarco(endo)plasmic reticulum Ca2+‐ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc Natl Acad Sci USA. 2010;107(45):19320‐19325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Bauzá‐Thorbrügge M, Banke E, Chanclón B, et al. Adipocyte‐specific ablation of the Ca(2+) pump SERCA2 impairs whole‐body metabolic function and reveals the diverse metabolic flexibility of white and brown adipose tissue. Mol Metab. 2022;63:101535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Jung TW, Kim HC, Kim HU, et al. Asprosin attenuates insulin signaling pathway through PKCδ‐activated ER stress and inflammation in skeletal muscle. J Cell Physiol. 2019;234(11):20888‐20899. [DOI] [PubMed] [Google Scholar]
- 417. Fu S, Yang L, Li P, et al. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature. 2011;473(7348):528‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418. Ikeda K, Kang Q, Yoneshiro T, et al. UCP1‐independent signaling involving SERCA2b‐mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nat Med. 2017;23(12):1454‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419. Ouyang Z, Li W, Meng Q, et al. A natural compound jaceosidin ameliorates endoplasmic reticulum stress and insulin resistance via upregulation of SERCA2b. Biomed Pharmacother. 2017;89:1286‐1296. [DOI] [PubMed] [Google Scholar]
- 420. Wang W, Zhang X, Gao Q, et al. A voltage‐dependent K(+) channel in the lysosome is required for refilling lysosomal Ca(2+) stores. J Cell Biol. 2017;216(6):1715‐1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Lebeau PF, Byun JH, Platko K, et al. Caffeine blocks SREBP2‐induced hepatic PCSK9 expression to enhance LDLR‐mediated cholesterol clearance. Nat Commun. 2022;13(1):770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Li Y, Ge M, Ciani L, et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic‐endoplasmic reticulum calcium ATPase‐2b activity in parallel with increased order of membrane lipids: implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol‐loaded macrophages. J Biol Chem. 2004;279(35):37030‐37039. [DOI] [PubMed] [Google Scholar]
- 423. Ali ES, Petrovsky N. Calcium signaling as a therapeutic target for liver steatosis. Trends Endocrinol Metab. 2019;30(4):270‐281. [DOI] [PubMed] [Google Scholar]
- 424. Ribeiro CM, McKay RR, Hosoki E, Bird GS, Putney JW, Jr . Effects of elevated cytoplasmic calcium and protein kinase C on endoplasmic reticulum structure and function in HEK293 cells. Cell Calcium. 2000;27(3):175‐185. [DOI] [PubMed] [Google Scholar]
- 425. Samuel VT, Shulman GI. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018;27(1):22‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Schmitz‐Peiffer C. Deconstructing the role of PKC epsilon in glucose homeostasis. Trends Endocrinol Metab. 2020;31(5):344‐356. [DOI] [PubMed] [Google Scholar]
- 427. Kawasaki T, Ueyama T, Lange I, Feske S, Saito N. Protein kinase C‐induced phosphorylation of Orai1 regulates the intracellular Ca2+ level via the store‐operated Ca2+ channel. J Biol Chem. 2010;285(33):25720‐25730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Beaulant A, Dia M, Pillot B, et al. Endoplasmic reticulum‐mitochondria miscommunication is an early and causal trigger of hepatic insulin resistance and steatosis. J Hepatol. 2022;77(3):710‐722. [DOI] [PubMed] [Google Scholar]
- 429. Rieusset J, Fauconnier J, Paillard M, et al. Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria‐associated ER membrane integrity to hepatic insulin resistance. Diabetologia. 2016;59(3):614‐623. [DOI] [PubMed] [Google Scholar]
- 430. Hernández‐Alvarez MI, Sebastián D, Vives S, et al. Deficient endoplasmic reticulum‐mitochondrial phosphatidylserine transfer causes liver disease. Cell. 2019;177(4):881‐895. e17. [DOI] [PubMed] [Google Scholar]
- 431. Tubbs E, Chanon S, Robert M, et al. Disruption of mitochondria‐associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes. 2018;67(4):636‐650. [DOI] [PubMed] [Google Scholar]
- 432. Theurey P, Tubbs E, Vial G, et al. Mitochondria‐associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J Mol Cell Biol. 2016;8(2):129‐143. [DOI] [PubMed] [Google Scholar]
- 433. Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, Hotamisligil GS. Chronic enrichment of hepatic endoplasmic reticulum‐mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. 2014;20(12):1427‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434. Thoudam T, Ha CM, Leem J, et al. PDK4 Augments ER‐Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling During Obesity. Diabetes. 2019;68(3):571‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Zhao Q, Luo T, Gao F, et al. GRP75 regulates mitochondrial‐supercomplex turnover to modulate insulin sensitivity. Diabetes. 2022;71(2):233‐248. [DOI] [PubMed] [Google Scholar]
- 436. Bi J, Wang W, Liu Z, et al. Seipin promotes adipose tissue fat storage through the ER Ca2⁺‐ATPase SERCA. Cell Metab. 2014;19(5):861‐871. [DOI] [PubMed] [Google Scholar]
- 437. Combot Y, Salo VT, Chadeuf G, et al. Seipin localizes at endoplasmic‐reticulum‐mitochondria contact sites to control mitochondrial calcium import and metabolism in adipocytes. Cell Rep. 2022;38(2):110213. [DOI] [PubMed] [Google Scholar]
- 438. Ding L, Yang X, Tian H, et al. Seipin regulates lipid homeostasis by ensuring calcium‐dependent mitochondrial metabolism. Embo J. 2018;37(17) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439. Dong Z, Wang J, Qiu T, et al. Perfluorooctane sulfonate induces mitochondrial calcium overload and early hepatic insulin resistance via autophagy/detyrosinated alpha‐tubulin‐regulated IP3R2‐VDAC1‐MICU1 interaction. Sci Total Environ. 2022;825:153933. [DOI] [PubMed] [Google Scholar]
- 440. Zhang Z, Luo Z, Yu L, et al. Ruthenium 360 and mitoxantrone inhibit mitochondrial calcium uniporter channel to prevent liver steatosis induced by high‐fat diet. Br J Pharmacol. 2022;179(11):2678‐2696. [DOI] [PubMed] [Google Scholar]
- 441. Qiu X, Li J, Lv S, et al. HDAC5 integrates ER stress and fasting signals to regulate hepatic fatty acid oxidation. J Lipid Res. 2018;59(2):330‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442. Ersoy BA, Maner‐Smith KM, Li Y, Alpertunga I, Cohen DE. Thioesterase‐mediated control of cellular calcium homeostasis enables hepatic ER stress. J Clin Invest. 2018;128(1):141‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443. Stork BA, Dean A, Ortiz AR, et al. Calcium/calmodulin‐dependent protein kinase kinase 2 regulates hepatic fuel metabolism. Mol Metab. 2022;62:101513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Baskaran P, Krishnan V, Ren J, Thyagarajan B. Capsaicin induces browning of white adipose tissue and counters obesity by activating TRPV1 channel‐dependent mechanisms. Br J Pharmacol. 2016;173(15):2369‐2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Baskaran P, Krishnan V, Fettel K, et al. TRPV1 activation counters diet‐induced obesity through sirtuin‐1 activation and PRDM‐16 deacetylation in brown adipose tissue. Int J Obes (Lond). 2017;41(5):739‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Moraes MN, Mezzalira N, de Assis LV, Menaker M, Guler A, Castrucci AM. TRPV1 participates in the activation of clock molecular machinery in the brown adipose tissue in response to light‐dark cycle. Biochim Biophys Acta Mol Cell Res. 2017;1864(2):324‐335. [DOI] [PubMed] [Google Scholar]
- 447. Marshall NJ, Liang L, Bodkin J, et al. A role for TRPV1 in influencing the onset of cardiovascular disease in obesity. Hypertension. 2013;61(1):246‐252. [DOI] [PubMed] [Google Scholar]
- 448. Lee E, Jung DY, Kim JH, et al. Transient receptor potential vanilloid type‐1 channel regulates diet‐induced obesity, insulin resistance, and leptin resistance. Faseb j. 2015;29(8):3182‐3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Gao M, Du Y, Xie JW, et al. Redox signal‐mediated TRPM2 promotes Ang II‐induced adipocyte insulin resistance via Ca(2+)‐dependent CaMKII/JNK cascade. Metabolism. 2018;85:313‐324. [DOI] [PubMed] [Google Scholar]
- 450. Navarro‐Marquez M, Torrealba N, Troncoso R, et al. Herpud1 impacts insulin‐dependent glucose uptake in skeletal muscle cells by controlling the Ca(2+)‐calcineurin‐Akt axis. Biochim Biophys Acta Mol Basis Dis. 2018;1864(5 Pt A):1653‐1662. [DOI] [PubMed] [Google Scholar]
- 451. Funai K, Lodhi IJ, Spears LD, et al. Skeletal muscle phospholipid metabolism regulates insulin sensitivity and contractile function. Diabetes. 2016;65(2):358‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452. Gandasi NR, Yin P, Riz M, et al. Ca2+ channel clustering with insulin‐containing granules is disturbed in type 2 diabetes. J Clin Invest. 2017;127(6):2353‐2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453. Kolic J, Manning Fox JE, Chepurny OG, et al. PI3 kinases p110α and PI3K‐C2β negatively regulate cAMP via PDE3/8 to control insulin secretion in mouse and human islets. Mol Metab. 2016;5(7):459‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. Xie B, Nguyen PM, Guček A, Thonig A, Barg S, Idevall‐Hagren O. Plasma membrane phosphatidylinositol 4,5‐bisphosphate regulates Ca(2+)‐influx and insulin secretion from pancreatic β cells. Cell Chem Biol. 2016;23(7):816‐826. [DOI] [PubMed] [Google Scholar]
- 455. Shin S, Le Lay J, Everett LJ, Gupta R, Rafiq K, Kaestner KH. CREB mediates the insulinotropic and anti‐apoptotic effects of GLP‐1 signaling in adult mouse β‐cells. Mol Metab. 2014;3(8):803‐812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456. Blanchet E, Van de Velde S, Matsumura S, et al. Feedback inhibition of CREB signaling promotes beta cell dysfunction in insulin resistance. Cell Rep. 2015;10(7):1149‐1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Santos GJ, Ferreira SM, Ortis F, et al. Metabolic memory of ß‐cells controls insulin secretion and is mediated by CaMKII. Mol Metab. 2014;3(4):484‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458. Persaud SJ, Liu B, Sampaio HB, Jones PM, Muller DS. Calcium/calmodulin‐dependent kinase IV controls glucose‐induced Irs2 expression in mouse beta cells via activation of cAMP response element‐binding protein. Diabetologia. 2011;54(5):1109‐1120. [DOI] [PubMed] [Google Scholar]
- 459. Tessem JS, Moss LG, Chao LC, et al. Nkx6.1 regulates islet β‐cell proliferation via Nr4a1 and Nr4a3 nuclear receptors. Proc Natl Acad Sci USA. 2014;111(14):5242‐5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460. Liu B, Barbosa‐Sampaio H, Jones PM, Persaud SJ, Muller DS. The CaMK4/CREB/IRS‐2 cascade stimulates proliferation and inhibits apoptosis of β‐cells. PLoS One. 2012;7(9):e45711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461. Heit JJ, Apelqvist AA, Gu X, et al. Calcineurin/NFAT signalling regulates pancreatic beta‐cell growth and function. Nature. 2006;443(7109):345‐349. [DOI] [PubMed] [Google Scholar]
- 462. Zummo FP, Krishnanda SI, Georgiou M, et al. Exendin‐4 stimulates autophagy in pancreatic β‐cells via the RAPGEF/EPAC‐Ca(2+)‐PPP3/calcineurin‐TFEB axis. Autophagy. 2022;18(4):799‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463. Bernal‐Mizrachi E, Cras‐Méneur C, Ye BR, Johnson JD, Permutt MA. Transgenic overexpression of active calcineurin in beta‐cells results in decreased beta‐cell mass and hyperglycemia. PLoS One. 2010;5(8):e11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464. Epstein PN, Overbeek PA, Means AR. Calmodulin‐induced early‐onset diabetes in transgenic mice. Cell. 1989;58(6):1067‐1073. [DOI] [PubMed] [Google Scholar]
- 465. Dahan T, Ziv O, Horwitz E, et al. Pancreatic β‐cells express the fetal islet hormone gastrin in rodent and human diabetes. Diabetes. 2017;66(2):426‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466. Stancill JS, Cartailler JP, Clayton HW, et al. Chronic beta‐cell depolarization impairs beta‐cell Identity by disrupting a network of Ca(2+)‐regulated genes. Diabetes. 2017;66(8):2175‐2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. Montaigne D, Butruille L, Staels B. PPAR control of metabolism and cardiovascular functions. Nat Rev Cardiol. 2021;18(12):809‐823. [DOI] [PubMed] [Google Scholar]
- 468. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non‐alcoholic fatty liver disease. J Hepatol. 2015;62(3):720‐733. [DOI] [PubMed] [Google Scholar]
- 469. Daoudi M, Hennuyer N, Borland MG, et al. PPARbeta/delta activation induces enteroendocrine L cell GLP‐1 production. Gastroenterology. 2011;140(5):1564‐1574. [DOI] [PubMed] [Google Scholar]
- 470. Tang P, Virtue S, Goie JYG, et al. Regulation of adipogenic differentiation and adipose tissue inflammation by interferon regulatory factor 3. Cell Death Differ. 2021;28(11):3022‐3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471. El Ouarrat D, Isaac R, Lee YS, et al. TAZ Is a negative regulator of PPARγ activity in adipocytes and TAZ deletion improves insulin sensitivity and glucose tolerance. Cell Metab. 2020;31(1):162‐173. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472. Yang T, Fu M, Pestell R, Sauve AA. SIRT1 and endocrine signaling. Trends Endocrinol Metab. 2006;17(5):186‐191. [DOI] [PubMed] [Google Scholar]
- 473. Caton PW, Richardson SJ, Kieswich J, et al. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia. 2013;56(5):1068‐1077. [DOI] [PubMed] [Google Scholar]
- 474. Bordone L, Motta MC, Picard F, et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS Biol. 2006;4(2):e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475. Nie Y, Erion DM, Yuan Z, et al. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol. 2009;11(4):492‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476. Shi MY, Bang IH, Han CY, Lee DH, Park BH, Bae EJ. Statin suppresses sirtuin 6 through miR‐495, increasing FoxO1‐dependent hepatic gluconeogenesis. Theranostics. 2020;10(25):11416‐11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477. Wang X, Gao H, Wu W, et al. The zinc transporter Slc39a5 controls glucose sensing and insulin secretion in pancreatic beta‐cells via Sirt1‐ and Pgc‐1alpha‐mediated regulation of Glut2. Protein Cell. 2019;10(6):436‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478. Qiang L, Wang L, Kon N, et al. Brown remodeling of white adipose tissue by SirT1‐dependent deacetylation of Ppargamma. Cell. 2012;150(3):620‐632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479. Hunter CA, Jones SA. IL‐6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16(5):448‐457. [DOI] [PubMed] [Google Scholar]
- 480. Kim HJ, Higashimori T, Park SY, et al. Differential effects of interleukin‐6 and ‐10 on skeletal muscle and liver insulin action in vivo. Diabetes. 2004;53(4):1060‐1067. [DOI] [PubMed] [Google Scholar]
- 481. Klover PJ, Zimmers TA, Koniaris LG, Mooney RA. Chronic exposure to interleukin‐6 causes hepatic insulin resistance in mice. Diabetes. 2003;52(11):2784‐2789. [DOI] [PubMed] [Google Scholar]
- 482. Senn JJ, Klover PJ, Nowak IA, Mooney RA. Interleukin‐6 induces cellular insulin resistance in hepatocytes. Diabetes. 2002;51(12):3391‐3399. [DOI] [PubMed] [Google Scholar]
- 483. Kim JH, Kim JE, Liu HY, Cao W, Chen J. Regulation of interleukin‐6‐induced hepatic insulin resistance by mammalian target of rapamycin through the STAT3‐SOCS3 pathway. J Biol Chem. 2008;283(2):708‐715. [DOI] [PubMed] [Google Scholar]
- 484. Lagathu C, Bastard JP, Auclair M, Maachi M, Capeau J, Caron M. Chronic interleukin‐6 (IL‐6) treatment increased IL‐6 secretion and induced insulin resistance in adipocyte: prevention by rosiglitazone. Biochem Biophys Res Commun. 2003;311(2):372‐379. [DOI] [PubMed] [Google Scholar]
- 485. Li H, Dong M, Liu W, et al. Peripheral IL‐6/STAT3 signaling promotes beiging of white fat. Biochim Biophys Acta Mol Cell Res. 2021;1868(10):119080. [DOI] [PubMed] [Google Scholar]
- 486. Ruderman NB, Keller C, Richard AM, et al. Interleukin‐6 regulation of AMP‐activated protein kinase. Potential role in the systemic response to exercise and prevention of the metabolic syndrome. Diabetes. 2006;55(Suppl 2):S48‐S54. [DOI] [PubMed] [Google Scholar]
- 487. Wunderlich FT, Strohle P, Konner AC, et al. Interleukin‐6 signaling in liver‐parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. 2010;12(3):237‐249. [DOI] [PubMed] [Google Scholar]
- 488. Matthews VB, Allen TL, Risis S, et al. Interleukin‐6‐deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia. 2010;53(11):2431‐2441. [DOI] [PubMed] [Google Scholar]
- 489. Hu FB, Meigs JB, Li TY, Rifai N, Manson JE. Inflammatory markers and risk of developing type 2 diabetes in women. Diabetes. 2004;53(3):693‐700. [DOI] [PubMed] [Google Scholar]
- 490. Spranger J, Kroke A, Mohlig M, et al. Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population‐based european prospective investigation into cancer and nutrition (EPIC)‐potsdam study. Diabetes. 2003;52(3):812‐817. [DOI] [PubMed] [Google Scholar]
- 491. Ellingsgaard H, Ehses JA, Hammar EB, et al. Interleukin‐6 regulates pancreatic alpha‐cell mass expansion. Proc Natl Acad Sci USA. 2008;105(35):13163‐13168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492. Akiyama T, Takasawa S, Nata K, et al. Activation of Reg gene, a gene for insulin‐producing beta ‐cell regeneration: poly(ADP‐ribose) polymerase binds Reg promoter and regulates the transcription by autopoly(ADP‐ribosyl)ation. Proc Natl Acad Sci USA. 2001;98(1):48‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493. Paula FM, Leite NC, Vanzela EC, et al. Exercise increases pancreatic beta‐cell viability in a model of type 1 diabetes through IL‐6 signaling. FASEB J. 2015;29(5):1805‐1816. [DOI] [PubMed] [Google Scholar]
- 494. Allen TL, Whitham M, Febbraio MA. IL‐6 muscles in on the gut and pancreas to enhance insulin secretion. Cell Metab. 2012;15(1):8‐9. [DOI] [PubMed] [Google Scholar]
- 495. Timper K, Dalmas E, Dror E, et al. Glucose‐dependent insulinotropic peptide stimulates glucagon‐like peptide 1 production by pancreatic islets via interleukin 6, produced by alpha cells. Gastroenterology. 2016;151(1):165‐179. [DOI] [PubMed] [Google Scholar]
- 496. Ramadoss P, Unger‐Smith NE, Lam FS, Hollenberg AN. STAT3 targets the regulatory regions of gluconeogenic genes in vivo. Mol Endocrinol. 2009;23(6):827‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497. Luan D, Dadpey B, Zaid J, et al. Adipocyte‐secreted IL‐6 sensitizes macrophages to IL‐4 signaling. Diabetes. 2023;72(3):367‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498. Kim TH, Choi SE, Ha ES, et al. IL‐6 induction of TLR‐4 gene expression via STAT3 has an effect on insulin resistance in human skeletal muscle. Acta Diabetol. 2013;50(2):189‐200. [DOI] [PubMed] [Google Scholar]
- 499. Sos BC, Harris C, Nordstrom SM, et al. Abrogation of growth hormone secretion rescues fatty liver in mice with hepatocyte‐specific deletion of JAK2. J Clin Invest. 2011;121(4):1412‐1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500. Hosui A, Tatsumi T, Hikita H, et al. Signal transducer and activator of transcription 5 plays a crucial role in hepatic lipid metabolism through regulation of CD36 expression. Hepatol Res. 2017;47(8):813‐825. [DOI] [PubMed] [Google Scholar]
- 501. Corbit KC, Camporez JPG, Tran JL, et al. Adipocyte JAK2 mediates growth hormone‐induced hepatic insulin resistance. JCI Insight. 2017;2(3):e91001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502. Nordstrom SM, Tran JL, Sos BC, Wagner KU, Weiss EJ. Disruption of JAK2 in adipocytes impairs lipolysis and improves fatty liver in mice with elevated GH. Mol Endocrinol. 2013;27(8):1333‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503. Ricardo‐Gonzalez RR, Red Eagle A, Odegaard JI, et al. IL‐4/STAT6 immune axis regulates peripheral nutrient metabolism and insulin sensitivity. Proc Natl Acad Sci USA. 2010;107(52):22617‐22622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504. Shi SY, Luk CT, Brunt JJ, et al. Adipocyte‐specific deficiency of Janus kinase (JAK) 2 in mice impairs lipolysis and increases body weight, and leads to insulin resistance with ageing. Diabetologia. 2014;57(5):1016‐1026. [DOI] [PubMed] [Google Scholar]
- 505. Kaltenecker D, Mueller KM, Benedikt P, et al. Adipocyte STAT5 deficiency promotes adiposity and impairs lipid mobilisation in mice. Diabetologia. 2017;60(2):296‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506. Mathur M, Yeh YT, Arya RK, et al. Adipose lipolysis is important for ethanol to induce fatty liver in the National Institute on Alcohol Abuse and Alcoholism murine model of chronic and binge ethanol feeding. Hepatology. 2022; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507. Baik M, Lee MS, Kang HJ, et al. Muscle‐specific deletion of signal transducer and activator of transcription 5 augments lipid accumulation in skeletal muscle and liver of mice in response to high‐fat diet. Eur J Nutr. 2017;56(2):569‐579. [DOI] [PubMed] [Google Scholar]
- 508. Kovtonyuk LV, Caiado F, Garcia‐Martin S, et al. IL‐1 mediates microbiome‐induced inflammaging of hematopoietic stem cells in mice. Blood. 2022;139(1):44‐58. [DOI] [PubMed] [Google Scholar]
- 509. Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007;56(7):1761‐1772. [DOI] [PubMed] [Google Scholar]
- 510. Finucane OM, Lyons CL, Murphy AM, et al. Monounsaturated fatty acid‐enriched high‐fat diets impede adipose NLRP3 inflammasome‐mediated IL‐1beta secretion and insulin resistance despite obesity. Diabetes. 2015;64(6):2116‐2128. [DOI] [PubMed] [Google Scholar]
- 511. Zhang SY, Dong YQ, Wang P, et al. Adipocyte‐derived lysophosphatidylcholine activates adipocyte and adipose tissue macrophage nod‐like receptor protein 3 inflammasomes mediating homocysteine‐induced insulin resistance. EBioMedicine. 2018;31:202‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512. Wen H, Gris D, Lei Y, et al. Fatty acid‐induced NLRP3‐ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12(5):408‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513. Sharma BR, Kanneganti TD. NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol. 2021;22(5):550‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514. Jager J, Gremeaux T, Cormont M, Le Marchand‐Brustel Y, Tanti JF. Interleukin‐1beta‐induced insulin resistance in adipocytes through down‐regulation of insulin receptor substrate‐1 expression. Endocrinology. 2007;148(1):241‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515. Chevrel G, Granet C, Miossec P. Contribution of tumour necrosis factor alpha and interleukin (IL) 1beta to IL6 production, NF‐kappaB nuclear translocation, and class I MHC expression in muscle cells: in vitro regulation with specific cytokine inhibitors. Ann Rheum Dis. 2005;64(9):1257‐1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516. Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity‐induced inflammation and insulin resistance. Nat Med. 2011;17(2):179‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517. Dror E, Dalmas E, Meier DT, et al. Postprandial macrophage‐derived IL‐1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nat Immunol. 2017;18(3):283‐292. [DOI] [PubMed] [Google Scholar]
- 518. Kotas ME, Jurczak MJ, Annicelli C, et al. Role of caspase‐1 in regulation of triglyceride metabolism. Proc Natl Acad Sci USA. 2013;110(12):4810‐4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519. Ou Y, Zheng Z, Niu B, Su J, Su H. Different MAPK signal transduction pathways play different roles in the impairment of glucosestimulated insulin secretion in response to IL1beta. Mol Med Rep. 2020;22(4):2973‐2980. [DOI] [PubMed] [Google Scholar]
- 520. Maedler K, Sergeev P, Ris F, et al. Glucose‐induced beta cell production of IL‐1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2017;127(4):1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 521. Chen C, Ma X, Yang C, et al. Hypoxia potentiates LPS‐induced inflammatory response and increases cell death by promoting NLRP3 inflammasome activation in pancreatic beta cells. Biochem Biophys Res Commun. 2018;495(4):2512‐2518. [DOI] [PubMed] [Google Scholar]
- 522. Sokolova M, Sahraoui A, Hoyem M, et al. NLRP3 inflammasome mediates oxidative stress‐induced pancreatic islet dysfunction. Am J Physiol Endocrinol Metab. 2018;315(5):E912‐E923. [DOI] [PubMed] [Google Scholar]
- 523. Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF‐κB: a blossoming of relevance to human pathobiology. Cell. 2017;168(1‐2):37‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524. Lee DF, Kuo HP, Chen CT, et al. IKKbeta suppression of TSC1 function links the mTOR pathway with insulin resistance. Int J Mol Med. 2008;22(5):633‐638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525. Hasegawa Y, Saito T, Ogihara T, et al. Blockade of the nuclear factor‐kappaB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation. 2012;125(9):1122‐1133. [DOI] [PubMed] [Google Scholar]
- 526. Zhang J, Gao Z, Yin J, Quon MJ, Ye J. S6K directly phosphorylates IRS‐1 on Ser‐270 to promote insulin resistance in response to TNF‐(alpha) signaling through IKK2. J Biol Chem. 2008;283(51):35375‐35382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527. Arkan MC, Hevener AL, Greten FR, et al. IKK‐beta links inflammation to obesity‐induced insulin resistance. Nat Med. 2005;11(2):191‐198. [DOI] [PubMed] [Google Scholar]
- 528. Liu J, Ibi D, Taniguchi K, et al. Inflammation improves glucose homeostasis through IKKbeta‐XBP1s interaction. Cell. 2016;167(4):1052‐1066 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529. Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science. 2013;339(6116):172‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 530. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF‐kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135(1):61‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531. Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein‐tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem. 2008;283(21):14230‐14241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Ho E, Quan N, Tsai YH, Lai W, Bray TM. Dietary zinc supplementation inhibits NFkappaB activation and protects against chemically induced diabetes in CD1 mice. Exp Biol Med (Maywood). 2001;226(2):103‐111. [DOI] [PubMed] [Google Scholar]
- 533. Heimberg H, Heremans Y, Jobin C, et al. Inhibition of cytokine‐induced NF‐kappaB activation by adenovirus‐mediated expression of a NF‐kappaB super‐repressor prevents beta‐cell apoptosis. Diabetes. 2001;50(10):2219‐2224. [DOI] [PubMed] [Google Scholar]
- 534. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14(2):88‐98. [DOI] [PubMed] [Google Scholar]
- 535. Zoungas S, Arima H, Gerstein HC, et al. Effects of intensive glucose control on microvascular outcomes in patients with type 2 diabetes: a meta‐analysis of individual participant data from randomised controlled trials. Lancet Diabetes Endocrinol. 2017;5(6):431‐437. [DOI] [PubMed] [Google Scholar]
- 536. Anders HJ, Huber TB, Isermann B, Schiffer M. CKD in diabetes: diabetic kidney disease versus nondiabetic kidney disease. Nat Rev Nephrol. 2018;14(6):361‐377. [DOI] [PubMed] [Google Scholar]
- 537. Meng XM, Tang PM, Li J, Lan HY. TGF‐β/Smad signaling in renal fibrosis. Front Physiol. 2015;6:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538. Meng XM, Nikolic‐Paterson DJ, Lan HY. TGF‐β: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325‐338. [DOI] [PubMed] [Google Scholar]
- 539. Vindevoghel L, Lechleider RJ, Kon A, et al. SMAD3/4‐dependent transcriptional activation of the human type VII collagen gene (COL7A1) promoter by transforming growth factor beta. Proc Natl Acad Sci USA. 1998;95(25):14769‐14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540. Chen SJ, Yuan W, Mori Y, Levenson A, Trojanowska M, Varga J. Stimulation of type I collagen transcription in human skin fibroblasts by TGF‐beta: involvement of Smad 3. J Invest Dermatol. 1999;112(1):49‐57. [DOI] [PubMed] [Google Scholar]
- 541. Yuan W, Varga J. Transforming growth factor‐beta repression of matrix metalloproteinase‐1 in dermal fibroblasts involves Smad3. J Biol Chem. 2001;276(42):38502‐38510. [DOI] [PubMed] [Google Scholar]
- 542. Chang AS, Hathaway CK, Smithies O, Kakoki M. Transforming growth factor‐β1 and diabetic nephropathy. Am J Physiol Renal Physiol. 2016;310(8):F689‐F696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543. Marrero MB, Banes‐Berceli AK, Stern DM, Eaton DC. Role of the JAK/STAT signaling pathway in diabetic nephropathy. Am J Physiol Renal Physiol. 2006;290(4):F762‐F768. [DOI] [PubMed] [Google Scholar]
- 544. Tuttle KR, Brosius FC, 3rd , Adler SG, et al. JAK1/JAK2 inhibition by baricitinib in diabetic kidney disease: results from a Phase 2 randomized controlled clinical trial. Nephrol Dial Transplant. 2018;33(11):1950‐1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545. Gödel M, Hartleben B, Herbach N, et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest. 2011;121(6):2197‐2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546. Huynh C, Ryu J, Lee J, Inoki A, Inoki K. Nutrient‐sensing mTORC1 and AMPK pathways in chronic kidney diseases. Nat Rev Nephrol. 2022; [DOI] [PubMed] [Google Scholar]
- 547. Leibowitz G, Cerasi E, Ketzinel‐Gilad M. The role of mTOR in the adaptation and failure of beta‐cells in type 2 diabetes. Diabetes Obes Metab. 2008;10(Suppl 4):157‐169. [DOI] [PubMed] [Google Scholar]
- 548. Mori H, Inoki K, Masutani K, et al. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem Biophys Res Commun. 2009;384(4):471‐475. [DOI] [PubMed] [Google Scholar]
- 549. Zhang Y, Sun X, Icli B, Feinberg MW. Emerging roles for microRNAs in diabetic microvascular disease: novel targets for therapy. Endocr Rev. 2017;38(2):145‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 550. Wang W, Lo ACY. Diabetic retinopathy: pathophysiology and treatments. Int J Mol Sci. 2018;19(6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551. Wu JH, Li YN, Chen AQ, et al. Inhibition of Sema4D/PlexinB1 signaling alleviates vascular dysfunction in diabetic retinopathy. EMBO Mol Med. 2020;12(2):e10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552. Romeo G, Liu WH, Asnaghi V, Kern TS, Lorenzi M. Activation of nuclear factor‐kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51(7):2241‐2248. [DOI] [PubMed] [Google Scholar]
- 553. Huang H, He J, Johnson D, et al. Deletion of placental growth factor prevents diabetic retinopathy and is associated with Akt activation and HIF1α‐VEGF pathway inhibition. Diabetes. 2015;64(1):200‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554. Aiello LP, Avery RL, Arrigg PG, et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med. 1994;331(22):1480‐1487. [DOI] [PubMed] [Google Scholar]
- 555. Antonetti DA, Barber AJ, Hollinger LA, Wolpert EB, Gardner TW. Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J Biol Chem. 1999;274(33):23463‐23467. [DOI] [PubMed] [Google Scholar]
- 556. Rousseau S, Houle F, Landry J, Huot J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene. 1997;15(18):2169‐2177. [DOI] [PubMed] [Google Scholar]
- 557. Wells JA, Glassman AR, Ayala AR, et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N Engl J Med. 2015;372(13):1193‐1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558. Sivaprasad S, Prevost AT, Vasconcelos JC, et al. Clinical efficacy of intravitreal aflibercept versus panretinal photocoagulation for best corrected visual acuity in patients with proliferative diabetic retinopathy at 52 weeks (CLARITY): a multicentre, single‐blinded, randomised, controlled, phase 2b, non‐inferiority trial. Lancet. 2017;389(10085):2193‐2203. [DOI] [PubMed] [Google Scholar]
- 559. Koh GY. Orchestral actions of angiopoietin‐1 in vascular regeneration. Trends Mol Med. 2013;19(1):31‐39. [DOI] [PubMed] [Google Scholar]
- 560. Saharinen P, Eklund L, Miettinen J, et al. Angiopoietins assemble distinct Tie2 signalling complexes in endothelial cell‐cell and cell‐matrix contacts. Nat Cell Biol. 2008;10(5):527‐537. [DOI] [PubMed] [Google Scholar]
- 561. Rangasamy S, Srinivasan R, Maestas J, McGuire PG, Das A. A potential role for angiopoietin 2 in the regulation of the blood‐retinal barrier in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2011;52(6):3784‐3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562. Feng Y, vom Hagen F, Pfister F, et al. Impaired pericyte recruitment and abnormal retinal angiogenesis as a result of angiopoietin‐2 overexpression. Thromb Haemost. 2007;97(1):99‐108. [PubMed] [Google Scholar]
- 563. Hakanpaa L, Sipila T, Leppanen VM, et al. Endothelial destabilization by angiopoietin‐2 via integrin β1 activation. Nat Commun. 2015;6:5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564. Moss A. The angiopoietin:Tie 2 interaction: a potential target for future therapies in human vascular disease. Cytokine Growth Factor Rev. 2013;24(6):579‐592. [DOI] [PubMed] [Google Scholar]
- 565. Liu C, Ge HM, Liu BH, et al. Targeting pericyte‐endothelial cell crosstalk by circular RNA‐cPWWP2A inhibition aggravates diabetes‐induced microvascular dysfunction. Proc Natl Acad Sci USA. 2019;116(15):7455‐7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566. Salzman J. Circular RNA expression: its potential regulation and function. Trends Genet. 2016;32(5):309‐316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567. Jiang Q, Liu C, Li CP, et al. Circular RNA‐ZNF532 regulates diabetes‐induced retinal pericyte degeneration and vascular dysfunction. J Clin Invest. 2020;130(7):3833‐3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568. Feldman EL, Callaghan BC, Pop‐Busui R, et al.Diabetic neuropathy. Nat Rev Dis Primers. 2019;5(1):41. [DOI] [PubMed] [Google Scholar]
- 569. Elafros MA, Andersen H, Bennett DL, et al. Towards prevention of diabetic peripheral neuropathy: clinical presentation, pathogenesis, and new treatments. Lancet Neurol. 2022;21(10):922‐936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570. Jayaraj ND, Bhattacharyya BJ, Belmadani AA, et al. Reducing CXCR4‐mediated nociceptor hyperexcitability reverses painful diabetic neuropathy. J Clin Invest. 2018;128(6):2205‐2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571. Blair NT, Bean BP. Roles of tetrodotoxin (TTX)‐sensitive Na+ current, TTX‐resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci. 2002;22(23):10277‐10290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572. Andersson DA, Gentry C, Light E, et al. Methylglyoxal evokes pain by stimulating TRPA1. PLoS One. 2013;8(10):e77986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573. Bierhaus A, Fleming T, Stoyanov S, et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat Med. 2012;18(6):926‐933. [DOI] [PubMed] [Google Scholar]
- 574. Orestes P, Osuru HP, McIntire WE, et al. Reversal of neuropathic pain in diabetes by targeting glycosylation of Ca(V)3.2 T‐type calcium channels. Diabetes. 2013;62(11):3828‐3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575. Kunt T, Forst T, Wilhelm A, et al. Alpha‐lipoic acid reduces expression of vascular cell adhesion molecule‐1 and endothelial adhesion of human monocytes after stimulation with advanced glycation end products. Clin Sci (Lond). 1999;96(1):75‐82. [PubMed] [Google Scholar]
- 576. Singh R, Barden A, Mori T, Beilin L. Advanced glycation end‐products: a review. Diabetologia. 2001;44(2):129‐146. [DOI] [PubMed] [Google Scholar]
- 577. Reddy MA, Das S, Zhuo C, et al. Regulation of vascular smooth muscle cell dysfunction under diabetic conditions by miR‐504. Arterioscler Thromb Vasc Biol. 2016;36(5):864‐873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578. Yang C, Eleftheriadou M, Kelaini S, et al. Targeting QKI‐7 in vivo restores endothelial cell function in diabetes. Nat Commun. 2020;11(1):3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579. Montefusco L, Ben Nasr M, D'Addio F, et al. Acute and long‐term disruption of glycometabolic control after SARS‐CoV‐2 infection. Nat Metab. 2021;3(6):774‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 580. Yang X, Yu Y, Xu J, et al. Clinical course and outcomes of critically ill patients with SARS‐CoV‐2 pneumonia in Wuhan, China: a single‐centered, retrospective, observational study. Lancet Respir Med. 2020;8(5):475‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581. Remuzzi A, Remuzzi G. COVID‐19 and Italy: what next? Lancet. 2020;395(10231):1225‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582. Heaney AI, Griffin GD, Simon EL. Newly diagnosed diabetes and diabetic ketoacidosis precipitated by COVID‐19 infection. Am J Emerg Med. 2020;38(11):2491. e3‐2491.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583. Hollstein T, Schulte DM, Schulz J, et al. Autoantibody‐negative insulin‐dependent diabetes mellitus after SARS‐CoV‐2 infection: a case report. Nat Metab. 2020;2(10):1021‐1024. [DOI] [PubMed] [Google Scholar]
- 584. Li J, Wang X, Chen J, Zuo X, Zhang H, Deng A. COVID‐19 infection may cause ketosis and ketoacidosis. Diabetes Obes Metab. 2020;22(10):1935‐1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585. Bhavya, Pathak E, Mishra R. Deciphering the link between diabetes mellitus and SARS‐CoV‐2 infection through differential targeting of microRNAs in the human pancreas. J Endocrinol Invest. 2022;45(3):537‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586. AmeliMojarad M, AmeliMojarad M, Pourmadian A. Simvastatin Therapy Increased miR‐150‐5p Expression in the Patients with Type 2 Diabetes and COVID‐19. Cell Physiol Biochem. 2022;56(6):685‐691. [DOI] [PubMed] [Google Scholar]
- 587. Behera J, Ison J, Voor MJ, Tyagi SC, Tyagi N. Diabetic Covid‐19 severity: Impaired glucose tolerance and pathologic bone loss. Biochem Biophys Res Commun. 2022;620:180‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588. Unnikrishnan R, Misra A. Diabetes and COVID19: a bidirectional relationship. Eur J Clin Nutr. 2021;75(9):1332‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589. Bornstein SR, Dalan R, Hopkins D, Mingrone G, Boehm BO. Endocrine and metabolic link to coronavirus infection. Nat Rev Endocrinol. 2020;16(6):297‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590. Codo AC, Davanzo GG, Monteiro LB, et al. Elevated glucose levels favor SARS‐CoV‐2 infection and monocyte response through a HIF‐1α/glycolysis‐dependent axis. Cell Metab. 2020;32(3):437‐446. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 591. Hu R, Xia CQ, Butfiloski E, Clare‐Salzler M. Effect of high glucose on cytokine production by human peripheral blood immune cells and type I interferon signaling in monocytes: Implications for the role of hyperglycemia in the diabetes inflammatory process and host defense against infection. Clin Immunol. 2018;195:139‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592. Hussain A, Bhowmik B, do Vale Moreira NC. COVID‐19 and diabetes: knowledge in progress. Diabetes Res Clin Pract. 2020;162:108142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593. Rajpal A, Rahimi L, Ismail‐Beigi F. Factors leading to high morbidity and mortality of COVID‐19 in patients with type 2 diabetes. J Diabetes. 2020;12(12):895‐908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594. Steenblock C, Richter S, Berger I, et al. Viral infiltration of pancreatic islets in patients with COVID‐19. Nat Commun. 2021;12(1):3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 595. Wu CT, Lidsky PV, Xiao Y, et al. SARS‐CoV‐2 infects human pancreatic β cells and elicits β cell impairment. Cell Metab. 2021;33(8):1565‐1576. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596. Ni W, Yang X, Yang D, et al. Role of angiotensin‐converting enzyme 2 (ACE2) in COVID‐19. Crit Care. 2020;24(1):422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597. Accili D. Can COVID‐19 cause diabetes? Nat Metab. 2021;3(2):123‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598. Solerte SB, Di Sabatino A, Galli M, Fiorina P. Dipeptidyl peptidase‐4 (DPP4) inhibition in COVID‐19. Acta Diabetol. 2020;57(7):779‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599. Naguib MN, Raymond JK, Vidmar AP. New onset diabetes with diabetic ketoacidosis in a child with multisystem inflammatory syndrome due to COVID‐19. J Pediatr Endocrinol Metab. 2021;34(1):147‐150. [DOI] [PubMed] [Google Scholar]
- 600. Reiterer M, Rajan M, Gómez‐Banoy N, et al. Hyperglycemia in acute COVID‐19 is characterized by insulin resistance and adipose tissue infectivity by SARS‐CoV‐2. Cell Metab. 2021;33(11):2174‐2188. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601. Nauck MA, Wefers J, Meier JJ. Treatment of type 2 diabetes: challenges, hopes, and anticipated successes. Lancet Diabetes Endocrinol. 2021;9(8):525‐544. [DOI] [PubMed] [Google Scholar]
- 602. Foretz M, Guigas B, Viollet B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat Rev Endocrinol. 2019;15(10):569‐589. [DOI] [PubMed] [Google Scholar]
- 603. Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494(7436):256‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 604. LaMoia TE, Shulman GI. Cellular and molecular mechanisms of metformin action. Endocr Rev. 2021;42(1):77‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605. Lien F, Berthier A, Bouchaert E, et al. Metformin interferes with bile acid homeostasis through AMPK‐FXR crosstalk. J Clin Invest. 2014;124(3):1037‐1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606. Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542(7640):177‐185. [DOI] [PubMed] [Google Scholar]
- 607. Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte‐to‐macrophage differentiation via AMPK‐mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes. 2015;64(6):2028‐2041. [DOI] [PubMed] [Google Scholar]
- 608. Kim J, Kwak HJ, Cha JY, et al. Metformin suppresses lipopolysaccharide (LPS)‐induced inflammatory response in murine macrophages via activating transcription factor‐3 (ATF‐3) induction. J Biol Chem. 2014;289(33):23246‐23255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609. Mastrototaro L, Roden M. Insulin resistance and insulin sensitizing agents. Metabolism. 2021;125:154892. [DOI] [PubMed] [Google Scholar]
- 610. Nissen SE. The rise and fall of rosiglitazone. Eur Heart J. 2010;31(7):773‐776. [DOI] [PubMed] [Google Scholar]
- 611. Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta‐analysis of randomized trials. Jama. 2007;298(10):1180‐1188. [DOI] [PubMed] [Google Scholar]
- 612. Ahmadian M, Suh JM, Hah N, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19(5):557‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613. Lv W, Wang X, Xu Q, Lu W. Mechanisms and characteristics of sulfonylureas and glinides. Curr Top Med Chem. 2020;20(1):37‐56. [DOI] [PubMed] [Google Scholar]
- 614. Burke MA, Mutharasan RK, Ardehali H. The sulfonylurea receptor, an atypical ATP‐binding cassette protein, and its regulation of the KATP channel. Circ Res. 2008;102(2):164‐176. [DOI] [PubMed] [Google Scholar]
- 615. Ashcroft SJ, Niki I, Kenna S, et al. The beta‐cell sulfonylurea receptor. Adv Exp Med Biol. 1993;334:47‐61. [DOI] [PubMed] [Google Scholar]
- 616. Zhang CL, Katoh M, Shibasaki T, et al. The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science. 2009;325(5940):607‐610. [DOI] [PubMed] [Google Scholar]
- 617. Wang LC, Fang FS, Gong YP, Yang G, Li CL. Characteristics of repaglinide and its mechanism of action on insulin secretion in patients with newly diagnosed type‐2 diabetes mellitus. Medicine (Baltimore). 2018;97(38):e12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 618. Quast U, Stephan D, Bieger S, Russ U. The impact of ATP‐sensitive K+ channel subtype selectivity of insulin secretagogues for the coronary vasculature and the myocardium. Diabetes. 2004;53(Suppl 3):S156‐S164. [DOI] [PubMed] [Google Scholar]
- 619. Drucker DJ. Mechanisms of action and therapeutic application of glucagon‐like peptide‐1. Cell Metab. 2018;27(4):740‐756. [DOI] [PubMed] [Google Scholar]
- 620. Müller TD, Finan B, Bloom SR, et al. Glucagon‐like peptide 1 (GLP‐1). Mol Metab. 2019;30:72‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 621. Shao S, Zhang X, Xu Q, Pan R, Chen Y. Emerging roles of glucagon like peptide‐1 in the management of autoimmune diseases and diabetes‐associated comorbidities. Pharmacol Ther. 2022;239:108270. [DOI] [PubMed] [Google Scholar]
- 622. Kopp KO, Glotfelty EJ, Li Y, Greig NH. Glucagon‐like peptide‐1 (GLP‐1) receptor agonists and neuroinflammation: Implications for neurodegenerative disease treatment. Pharmacol Res. 2022;186:106550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 623. Pan Q, Lin S, Li Y, et al. A novel GLP‐1 and FGF21 dual agonist has therapeutic potential for diabetes and non‐alcoholic steatohepatitis. EBioMedicine. 2021;63:103202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624. Drucker DJ. GLP‐1 physiology informs the pharmacotherapy of obesity. Mol Metab. 2022;57:101351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625. Drucker DJ, Nauck MA. The incretin system: glucagon‐like peptide‐1 receptor agonists and dipeptidyl peptidase‐4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696‐1705. [DOI] [PubMed] [Google Scholar]
- 626. Deacon CF. Dipeptidyl peptidase 4 inhibitors in the treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2020;16(11):642‐653. [DOI] [PubMed] [Google Scholar]
- 627. Raj VS, Mou H, Smits SL, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus‐EMC. Nature. 2013;495(7440):251‐254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 628. Vankadari N, Wilce JA. Emerging WuHan (COVID‐19) coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect. 2020;9(1):601‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 629. Fathi A, Vickneson K, Singh JS. SGLT2‐inhibitors; more than just glycosuria and diuresis. Heart Fail Rev. 2021;26(3):623‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 630. Ferrannini E, Solini A. SGLT2 inhibition in diabetes mellitus: rationale and clinical prospects. Nat Rev Endocrinol. 2012;8(8):495‐502. [DOI] [PubMed] [Google Scholar]
- 631. Yang X, Liu Q, Li Y, et al. The diabetes medication canagliflozin promotes mitochondrial remodelling of adipocyte via the AMPK‐Sirt1‐Pgc‐1α signalling pathway. Adipocyte. 2020;9(1):484‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632. Verma S, McMurray JJV. SGLT2 inhibitors and mechanisms of cardiovascular benefit: a state‐of‐the‐art review. Diabetologia. 2018;61(10):2108‐2117. [DOI] [PubMed] [Google Scholar]
- 633. Nespoux J, Vallon V. SGLT2 inhibition and kidney protection. Clin Sci (Lond). 2018;132(12):1329‐1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 634. Musso G, Saba F, Cassader M, Gambino R. Diabetic ketoacidosis with SGLT2 inhibitors. Bmj. 2020;371:m4147. [DOI] [PubMed] [Google Scholar]
- 635. Kalra S. Alpha glucosidase inhibitors. J Pak Med Assoc. 2014;64(4):474‐476. [PubMed] [Google Scholar]
- 636. Proença C, Ribeiro D, Freitas M, Fernandes E. Flavonoids as potential agents in the management of type 2 diabetes through the modulation of α‐amylase and α‐glucosidase activity: a review. Crit Rev Food Sci Nutr. 2022;62(12):3137‐3207. [DOI] [PubMed] [Google Scholar]
- 637. Dalsgaard NB, Gasbjerg LS, Hansen LS, et al. The role of GLP‐1 in the postprandial effects of acarbose in type 2 diabetes. Eur J Endocrinol. 2021;184(3):383‐394. [DOI] [PubMed] [Google Scholar]
- 638. Ding H, Hu X, Xu X, Zhang G, Gong D. Inhibitory mechanism of two allosteric inhibitors, oleanolic acid and ursolic acid on α‐glucosidase. Int J Biol Macromol. 2018;107(Pt B):1844‐1855. [DOI] [PubMed] [Google Scholar]
- 639. Toulis KA, Nirantharakumar K, Pourzitaki C, Barnett AH, Tahrani AA. Glucokinase activators for type 2 diabetes: challenges and future developments. Drugs. 2020;80(5):467‐475. [DOI] [PubMed] [Google Scholar]
- 640. Janah L, Kjeldsen S, Galsgaard KD, et al. Glucagon receptor signaling and glucagon resistance. Int J Mol Sci. 2019;20(13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641. Scheen AJ, Paquot N, Lefebvre PJ. Investigational glucagon receptor antagonists in Phase I and II clinical trials for diabetes. Expert Opin Investig Drugs. 2017;26(12):1373‐1389. [DOI] [PubMed] [Google Scholar]
- 642. Boland ML, Laker RC, Mather K, et al. Resolution of NASH and hepatic fibrosis by the GLP‐1R/GcgR dual‐agonist Cotadutide via modulating mitochondrial function and lipogenesis. Nat Metab. 2020;2(5):413‐431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643. Eriksson O, Velikyan I, Haack T, et al. Imaging of the glucagon receptor in subjects with type 2 diabetes. J Nucl Med. 2021;62(6):833‐838. [DOI] [PubMed] [Google Scholar]
- 644. Ding L, Yang Q, Zhang E, et al. Notoginsenoside Ft1 acts as a TGR5 agonist but FXR antagonist to alleviate high fat diet‐induced obesity and insulin resistance in mice. Acta Pharm Sin B. 2021;11(6):1541‐1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645. Huang S, Ma S, Ning M, et al. TGR5 agonist ameliorates insulin resistance in the skeletal muscles and improves glucose homeostasis in diabetic mice. Metabolism. 2019;99:45‐56. [DOI] [PubMed] [Google Scholar]
- 646. Watanabe M, Houten SM, Mataki C, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439(7075):484‐489. [DOI] [PubMed] [Google Scholar]
- 647. Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon‐like peptide‐1 secretion through TGR5 in a murine enteroendocrine cell line STC‐1. Biochem Biophys Res Commun. 2005;329(1):386‐390. [DOI] [PubMed] [Google Scholar]
- 648. Hodge RJ, Lin J, Vasist Johnson LS, Gould EP, Bowers GD, Nunez DJ. Safety, pharmacokinetics, and pharmacodynamic effects of a selective TGR5 agonist, SB‐756050, in type 2 diabetes. Clin Pharmacol Drug Dev. 2013;2(3):213‐222. [DOI] [PubMed] [Google Scholar]
- 649. Wang XX, Wang D, Luo Y, et al. FXR/TGR5 dual agonist prevents progression of nephropathy in diabetes and obesity. J Am Soc Nephrol. 2018;29(1):118‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650. Schnell S, Schaefer M, Schöfl C. Free fatty acids increase cytosolic free calcium and stimulate insulin secretion from beta‐cells through activation of GPR40. Mol Cell Endocrinol. 2007;263(1‐2):173‐180. [DOI] [PubMed] [Google Scholar]
- 651. Prentki M, Tornheim K, Corkey BE. Signal transduction mechanisms in nutrient‐induced insulin secretion. Diabetologia. 1997;40(Suppl 2):S32‐S41. [DOI] [PubMed] [Google Scholar]
- 652. Bianchini G, Nigro C, Sirico A, et al. A new synthetic dual agonist of GPR120/GPR40 induces GLP‐1 secretion and improves glucose homeostasis in mice. Biomed Pharmacother. 2021;139:111613. [DOI] [PubMed] [Google Scholar]
- 653. Hamdouchi C, Kahl SD, Patel Lewis A, et al. The Discovery, Preclinical, and Early Clinical Development of Potent and Selective GPR40 Agonists for the Treatment of Type 2 Diabetes Mellitus (LY2881835, LY2922083, and LY2922470). J Med Chem. 2016;59(24):10891‐10916. [DOI] [PubMed] [Google Scholar]
- 654. Kim JW, Roh E, Choi KM, Yoo HJ, Hwang HJ, Baik SH. GPR40 agonism modulates inflammatory reactions in vascular endothelial cells. Diabetes Metab J. 2022;46(3):506‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 655. Park J, Lee MY, Seo YS, Kang B, Lim SC, Kang KW. GPR40 agonist inhibits NLRP3 inflammasome activation via modulation of nuclear factor‐κB and sarco/endoplasmic reticulum Ca(2+)‐ATPase. Life Sci. 2021;287:120127. [DOI] [PubMed] [Google Scholar]
- 656. Lee MK, Kim SG, Watkins E, et al. A novel non‐PPARgamma insulin sensitizer: MLR‐1023 clinicalproof‐of‐concept in type 2 diabetes mellitus. J Diabetes Complications. 2020;34(5):107555. [DOI] [PubMed] [Google Scholar]
- 657. Ochman AR, Lipinski CA, Handler JA, Reaume AG, Saporito MS. The Lyn kinase activator MLR‐1023 is a novel insulin receptor potentiator that elicits a rapid‐onset and durable improvement in glucose homeostasis in animal models of type 2 diabetes. J Pharmacol Exp Ther. 2012;342(1):23‐32. [DOI] [PubMed] [Google Scholar]
- 658. Ding XW, Robinson M, Li R, Aldhowayan H, Geetha T, Babu JR. Mitochondrial dysfunction and beneficial effects of mitochondria‐targeted small peptide SS‐31 in Diabetes Mellitus and Alzheimer's disease. Pharmacol Res. 2021;171:105783. [DOI] [PubMed] [Google Scholar]
- 659. Zinovkin RA, Zamyatnin AA. Mitochondria‐targeted drugs. Curr Mol Pharmacol. 2019;12(3):202‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 660. Hidalgo‐Gutiérrez A, González‐García P, Díaz‐Casado ME, et al. Metabolic targets of coenzyme Q10 in mitochondria. Antioxidants (Basel). 2021;10(4) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 661. Battogtokh G, Choi YS, Kang DS, et al. Mitochondria‐targeting drug conjugates for cytotoxic, anti‐oxidizing and sensing purposes: current strategies and future perspectives. Acta Pharm Sin B. 2018;8(6):862‐880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 662. Wu X, Pang Y, Zhang Z, et al. Mitochondria‐targeted antioxidant peptide SS‐31 mediates neuroprotection in a rat experimental glaucoma model. Acta Biochim Biophys Sin (Shanghai). 2019;51(4):411‐421. [DOI] [PubMed] [Google Scholar]
- 663. Koh A, Molinaro A, Ståhlman M, et al. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell. 2018;175(4):947‐961. e17. [DOI] [PubMed] [Google Scholar]
- 664. Jung TW, Hwang HJ, Hong HC, Yoo HJ, Baik SH, Choi KM. BAIBA attenuates insulin resistance and inflammation induced by palmitate or a high fat diet via an AMPK‐PPARδ‐dependent pathway in mice. Diabetologia. 2015;58(9):2096‐2105. [DOI] [PubMed] [Google Scholar]
- 665. Thielen LA, Chen J, Jing G, et al. Identification of an anti‐diabetic, orally available small molecule that regulates TXNIP expression and glucagon action. Cell Metab. 2020;32(3):353‐365. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 666. Hu W, Ding Y, Li Q, Shi R, He Y. Transient receptor potential vanilloid 4 channels as therapeutic targets in diabetes and diabetes‐related complications. J Diabetes Investig. 2020;11(4):757‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 667. Xu G, Chen J, Jing G, Shalev A. Thioredoxin‐interacting protein regulates insulin transcription through microRNA‐204. Nat Med. 2013;19(9):1141‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 668. Sun K, Halberg N, Khan M, Magalang UJ, Scherer PE. Selective inhibition of hypoxia‐inducible factor 1α ameliorates adipose tissue dysfunction. Mol Cell Biol. 2013;33(5):904‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669. Yan J, Wang C, Jin Y, et al. Catalpol ameliorates hepatic insulin resistance in type 2 diabetes through acting on AMPK/NOX4/PI3K/AKT pathway. Pharmacol Res. 2018;130:466‐480. [DOI] [PubMed] [Google Scholar]
- 670. Wang B, Zhang L, Dai T, et al. Liquid‐liquid phase separation in human health and diseases. Signal Transduct Target Ther. 2021;6(1):290. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.