


Abstract
Liquid–liquid phase separation (LLPS) plays a critical role in regulating gene transcription via the formation of transcriptional condensates. However, LLPS has not been reported to be engineered as a tool to activate endogenous gene expression in mammalian cells or in vivo. Here, we developed a droplet-forming CRISPR (clustered regularly interspaced short palindromic repeats) gene activation system (DropCRISPRa) to activate transcription with high efficiency via combining the CRISPR-SunTag system with FETIDR–AD fusion proteins, which contain an N-terminal intrinsically disordered region (IDR) of a FET protein (FUS or TAF15) and a transcription activation domain (AD, VP64/P65/VPR). In this system, the FETIDR–AD fusion protein formed phase separation condensates at the target sites, which could recruit endogenous BRD4 and RNA polymerase II with an S2 phosphorylated C-terminal domain (CTD) to enhance transcription elongation. IDR-FUS9Y>S and IDR-FUSG156E, two mutants with deficient and aberrant phase separation respectively, confirmed that appropriate phase separation was required for efficient gene activation. Further, the DropCRISPRa system was compatible with a broad set of CRISPR-associated (Cas) proteins and ADs, including dLbCas12a, dAsCas12a, dSpCas9 and the miniature dUnCas12f1, and VP64, P65 and VPR. Finally, the DropCRISPRa system could activate target genes in mice. Therefore, this study provides a robust tool to activate gene expression for foundational research and potential therapeutics.
Graphical Abstract
Graphical Abstract.
Phase-separated DropCRISPRa system for efficient gene activation.
INTRODUCTION
The phase separation of substances is a common natural physical phenomenon, and usually refers to the spontaneous formation of different phases in a homogeneously mixed liquid, thus termed liquid–liquid phase separation (LLPS) (1). In cells, LLPS is driven by multiple folded domains (2) or multivalent interactions among intrinsically disordered regions (IDRs) of proteins (3,4). It has been reported that LLPS is the basis of membraneless compartments for intracellular organization and plays an important role in a spectrum of biological processes (5). In recent years, a number of studies have proposed that dynamic compartmentalization of transcription factors (TFs) and coactivators can undergo LLPS to regulate transcription and transcription-coupled processes, including transcription initiation, elongation, co-transcriptional RNA splicing and mRNA modification (6–10).
The FET family is composed of highly conserved RNA-binding proteins, FUS, EWS and TAF15, which are implicated in multiple cellular processes, such as transcription regulation, microRNA processing and maintenance of genomic integrity (11). The FET proteins contain an N-terminal IDR and a C-terminal RNA-binding domain (12). The IDR of FET can form biomolecular condensates in vitro (13) and in vivo (14), and these condensates can recruit RNA polymerase II (RNAPII) to promote gene expression (15,16). Some natural and artificial protein fusions, such as EWS–FLI1 (17), FUS–Gal4 (18), FUS–TetR (19) and FUS–LacI (20) fusions, which comprise FETIDR linked to a DNA-binding domain (DBD) of a TF, have been reported to recognize the cognate DNA motif of the TF near transcription start sites and subsequently activate gene expression. However, only EWS–FLI1 target genes or exogenous reporter genes containing the binding sites of Gal4, TetR or LacI could be activated by these protein fusions, and the activation of endogenous genes across the genome has not been reported by FETIDR–TF fusions.
The clustered regularly interspaced short palindromic repeats activation (CRISPRa) system is a platform for gene expression activation without editing the primary sequence of the genome (21). Most CRISPRa systems use a catalytically inactive or ‘dead’ CRISP-associated protein (dCas), when complexed with a guide RNA (gRNA), to target a specific gene and recruit a transcription activation domain (AD) module to activate gene expression. Currently, a spectrum of CRISPRa systems have been reported, such as dCas–AD fusions (22–24), CRISPR/gRNA-directed synergistic activation mediator (dCas/gRNA-SAM) (25,26) and a signal amplification CRISPR-SunTag system (27). Although these CRISPRa systems have already conferred high transcriptional activation, it is a therapeutic requirement to develop more potent systems to easily activate genes in settings with extremely low delivery efficiency in vivo. The previous strategies to improve CRISPRa systems are mainly based on CRISPR–Cas protein engineering, gRNA modification and AD module combination (28). However, whether an LLPS-based CRISPRa system could potently activate gene expression has not been reported.
In this study, we combined the CRISPR-SunTag system and droplet-forming FETIDR–AD fusions to develop a DropCRISPRa system for highly efficient gene activation in mammalian cells and mice. In this system, the dCas protein was fused to a tandem array of GCN4 peptides that could recruit the scFvGCN4-FETIDR–AD fusions and form LLPS droplets. Subsequently, these phase-separated droplets were able to enrich RNAPII and BRD4, increased the transcription elongation rate, and thus improved endogenous gene activation. The DropCRISPRa system could be constructed with a variety of Cas proteins and AD modules and could activate target genes in mammalian cells and in mice in vivo. Thus, it is a promising tool for future synthetic biology and therapeutic applications.
MATERIALS AND METHODS
Plasmid construction
Constructs for the scFv-GCN4-IDR–VP64 fusion proteins
Syn-IDR (DDX4/FUS/TAF15/FUS9YS/FUSG156E)–VP64 were synthesized (Genewiz, China) and used for subsequent cloning.
Pblu-EF1α-scFv-GCN4-sfGFP-IDR (DDX4/FUS/TAF15/FUS9YS/FUSG156E)–VP64-GB1-NLS-PA: Syn-IDR (DDX4/FUS/TAF15/FUS9YS/FUSG156E)–VP64 and Pblu-EF1α-scFv-GCN4-sfGFP–VP64-GB1-NLS-PA were digested with AgeI and SpeI and ligated to each other.
Constructs for the scFv-GCN4-IDR-P65/VPR fusion proteins
Pblu-EF1α-scFv-GCN4-sfGFP-P65-GB1-NLS-PA: a DNA fragment containing P65 was amplified from the VPR template and inserted into the AgeI and SpeI sites in the Pblu-EF1α-scFv-GCN4-sfGFP–VP64-GB1-NLS-PA plasmid.
Pblu-EF1α-scFv-GCN4-sfGFP-IDR (DDX4/FUS/TAF15)-P65-GB1-NLS-PA: a DNA fragment containing DDX4/FUS/TAF15IDR was amplified and inserted into the AgeI and NheI sites in the Pblu-EF1α-scFv-GCN4-sfGFP-P65-GB1-NLS-PA plasmid.
Pblu-EF1α-scFv-GCN4-sfGFP-IDR (DDX4/FUS/TAF15)-VPR-GB1-NLS-PA: DNA fragment 1 containing DX4/FUS/TAF15IDR, DNA fragment 2 containing VPR and the Pblu-EF1α-scFv-GCN4-sfGFP-VPR-GB1-NLS-PA plasmid backbone digested with AgeI and SpeI were joined together by DNA assembly.
Constructs for the Cas–GCN4 fusion proteins and the luciferase reporter (SunTag system)
pEF1α-dLbCas12a-10×GCN4, pEF1α-dLbCas12a-3×GCN4 and pEF1α-dLbCas12a-24×GCN4: DNA fragment 1 containing dLbCas12a, DNA fragment 2 containing 10×GCN4, 3×GCN4 or 24×GCN4, and the Pblu-EF1α-NLS-PA plasmid backbone digested with EcoRI and AgeI were joined together by DNA assembly.
pEF1α-dLbCas12a-10×GCN4-GS2P2A-TagBlue: a DNA fragment containing GSGP2A-TagBlue and the pEF1α-dLbCas12a-10×GCN4 plasmid digested with AgeI and BsrGI were joined together by DNA assembly.
pEF1α-dUnCas12f1-10×GCN4: a DNA fragment containing dUnCas12f1 was amplified from a synthesized plasmid containing dUnCas12f1 (Genewiz, China) and inserted into the EcoRI and XhoI sites in the pEF1α-dLbCas12a-10×GCN4 plasmid.
pBluSKP-TRE-Luciferase–GSGP2A–mCherry: DNA fragment 1 containing luciferase-GSGP2A-mCherry, DNA fragment 2 containing the TRE promoter, and the pBluSKP plasmid digested with SalI and SacI were joined together by DNA assembly.
pCAG-dSpCas9-24×GCN4 and pCAG-dAsCas12a-10×GCN4 have been described previously (29,30).
Constructs for the Cas–gp41 fusion proteins (MoonTag system)
The pEF1a-dLbCpf1E925A-24×gp41-NLS and Pblu-EF1-Nb-gp41-mCherry-GB1-NLS-PA were synthesized (Genewiz, China) and used for subsequent clones.
The pEF1a-dLbCpf1E925A-10×gp41-NLS: a DNA fragment containing 10×gp41 and the pEF1a-dLbCpf1E925A-24×gp41-NLS plasmid backbone digested with XhoI and AgeI were joined together by DNA assembly.
Pblu-EF1-Nb-gp41-mCherry-IDR (DDX4/FUS)-VP64-GB1-NLS-PA: VP64 or IDR (DDX4/FUS)-VP64 and Pblu-EF1-Nb-gp41-mCherry-GB1-NLS-PA digested with BamHI and SpeI were joined together by DNA assembly.
Single guide RNA (sgRNA) constructs
sgRNA target sites were designed through https://www.benchling.com/. An sgRNA expression plasmid was constructed by ligating the corresponding annealed oligos to the basic plasmid under the human U6 promoter (synthesized from Genewiz of China). The sequences of all sgRNAs are listed in Supplementary Table S1.
Cell culture and transfection
All cells were obtained from the ATCC. HEK293T cells and B16 were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. MCF7 cells were cultured in RPMI-1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin. All cells were cultured at 37 °C under 5% CO2.
All cells were transfected with polyethyleneimine (PEI, Sigma). The total amount of DNA was 0.5 μg per well for a 24-well plate, and plasmids encoding the DropCRISPRa system and sgRNA were transfected at a 3:2:5 ratio. Approximately 1 × 105 cells were plated in 24-well plates 1 day before transfection. Ten hours after transfection, the culture medium was replaced with fresh DMEM and then incubated for 48 h.
Immunofluorescence staining
Cells were seeded on glass coverslips, fixed with 4% paraformaldehyde for 20 min then permeabilized with phosphate-buffered saline (PBS) containing 0.25% Triton X-100 for 30 min at room temperature. After permeabilization, cells were blocked in PBST containing 5% bovine serum albumin (BSA) at room temperature for 1–2 h and incubated with Alexa Fluor® 647-conjugated antibodies overnight at 4°C. After washing three times with PBST, cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). The coverslips were mounted with DAPI and sealed. Images were captured on a Nikon A1 confocal microscope.
FRAP and live-cell imaging
HEK293T cells grown on a glass bottom microwell dish were transfected with 2 μg of plasmids encoding the DropCRISPRa system. At 36 h after transfection, cells were imaged. During image acquisition, cells were maintained in DMEM supplemented with 25 mM HEPES. Images were acquired at intervals of 3 s and were analyzed with NIS-Elements software to identify fusion and fission events.
Fluorescence recovery after photobleaching (FRAP) experiments were performed on a Nikon A1 confocal microscope with a ×60 oil objective. For each experiment, one droplet with a diameter of 2.5–3.5 μm was selected. A square region of interest (ROI) was bleached once using a 488 nm line at 100% laser power. Fluorescence intensity changes with time were recorded for two pre-bleaching frames and 50 post-bleaching frames. At each time point, mean intensities of the bleached region (Fs), neighboring unbleached region (control, Fc) and a background region (Fb) were analyzed using NIS-Elements software. The photobleaching rate (r) by comparing the fluorescence of the control region before (Fc0) and after (Fc) photobleaching was calculated as (r = Fc/Fc0). Normalized fluorescence intensity (F) was calculated as (F = [Fs – Fb]/r). The mobile fraction (Fm) was calculated as (Fm = F∞/F0).
We constructed a CRISPRTag–TRE-H2B reporter cell line, in which a DNA fragment containing a CRISPRTag-V1 fragment and a TRE-driven H2B expression cassette were inserted into the ACTB site in the HEK293T cells using our previously developed highly effective knock-in approach (31). We used the same method as above to construct the CRISPRTag–TRE-Luciferase-24×MS2 reporter cell line. The CRISPRTag–TRE-H2B HEK293T reporter cells grown on a glass bottom microwell dish were transfected with 2 μg of plasmids encoding the dSpCas9-SunTag system targeting the CRISPRTag for target site labeling and dLbCas12a–mCherry-MoonTag systems targeting the TRE for gene activation. At 48 h after transfection, cells were stained with Hoechst 33342 (beyotime, C1022) and incubated at 37°C for 15 min. After staining, the culture medium was replaced with fresh DMEM supplemented with 25 mM HEPES and then imaged. The fluorescence intensity was analyzed with ImageJ software. The CRISPRTag–TRE-luciferase-24×MS2 HEK293T reporter cells grown on a glass bottom microwell dish were transfected with 2 μg of plasmids encoding the dSpCas9-SunTag system for target site labeling, the dLbCas12a–mCherry-MoonTag system for gene activation and MCP–blue fluorescent protein (BFP) to visualize MS2-tagged mRNA. At 48 h after transfection, the culture medium was replaced with fresh DMEM supplemented with 25 mM HEPES and then imaged. The fluorescence intensity was analyzed with ImageJ software. For bioluminescence detection, 50 μg/ml d-luciferin was added to cells which were then captured using the IVIS Lumina II In Vivo Imaging System within 5 min of addition of d-luciferin. Radiance values were calculated for the ROI using Living Image 3.1 software.
Quantitative reverse transcrition polymerase chain reaction (RT–qPCR)
RNA was isolated using Trizol Reagent and reverse-transcribed into cDNA with the Prime Script RT Reagent Kit (TAKARA, RR047A). RT–qPCR was performed by the LightCycler 96 System (Roche, Switzerland) using SYBR Premix Ex Taq II (Accurate Biology, AG11701). The ΔΔCt method was applied to show relative gene expression after normalizing to glyceraldehyde phosphate dehydrogenase (GAPDH) expression. All qPCR primers are listed in Supplementary Table S2.
RNA-seq and bioinformatics analysis
Total RNA was isolated with TRIzol reagent and mRNA was enriched and fragmented for library construction. The constructed sequencing libraries were sequenced on the Illumina HiSeq Platform with 150 bp paired-end reads, and the paired-end clean reads were aligned to the GRCh38.104 reference genome using Hisat2 (v2.0.1). Htseq (V 0.6.1) was used to count the read numbers mapped to each gene. Fragments per kilobase per million (FPKM) of each gene were calculated based on the length of the gene and the read count mapped to this gene. Differential expression analysis between the two groups was performed using the DESeq2 R package (v1.26.0). The resulting P-value [q value|false discovery rate (FDR)] of <0.05 and fold change of >2. All figures were plotted using the R package.
ChIP-PCR
Cells were grown on 10 cm plates to 60% confluency and transfected with the DropCRISPRa system targeting CD69. At 48 h after transfection, the chromatin immunoprecipitation (ChIP)-PCR assay was performed using a ChIP Kit (Abcam, ab270816) as per the manufacturer's instructions. A total of 8 × 106 cells were used for ChIP enrichment, and chromatin fragments were sheared to 100–600 bp using the Q800R2 DNA Shearing Sonicator. Immunoprecipitation was performed using an antibody against active RNAPII (RNAPII S2P) (Abcam, ab238146) and with isotype-matched IgG control (ChIP Kit, ab270816). The immunoprecipitated DNA was analyzed by qPCR using SYBR Premix Ex Taq II on a LightCycler 96 System (Roche, Switzerland) using the following program: initial denaturation at 95°C for 30 s followed by 45 cycles of 95°C for 10 s and 60°C for 34 s. Relative enrichment for each target was calculated by normalization to the input control.
Transcription elongation rate assay
The HEK293T cells were transfected with the DropCRISPRa system targeting CD69. After incubation for 48 h, the cells were treated with 100 μM DRB (Sigma, D1916-10MG) for 3 h and washed twice with PBS to remove the DRB. After incubation in fresh medium for various periods, the cells were harvested and total RNA was isolated for RT–qPCR.
Gene activation in vivo
Female BALB/c mice aged 6 weeks were hydrodynamically injected with plasmids encoding the DropCRISPRa system and the luciferase reporter. A total of 10 μg of Pblu-EF1α-scFv-GCN4-sfGFP-GB1-NLS-PA/Pblu-EF1α-scFv-GCN4-sfGFP-VP64-GB1-NLS-PA/Pblu-EF1α-scFv-GCN4-sfGFP-FUS-VP64-GB1-NLS-PA plasmids, 15 μg pEF1α-dLbCas12a-10×GCN4, 15 μg of pBluSKP-TRE-luciferase-GSGP2A–mCherry and 10 μg of sgRNA targeting the reporter were mixed with 1.5–1.8 ml of 0.9% sterile saline (10% of body weight) and then delivered to each mouse by hydrodynamic tail vein injection lasting 7 s. For bioluminescence detection, mice were injected intraperitoneally with 100 μl of 20 mg/ml d-luciferin and then anesthetized with isoflurane. Bioluminescence images were captured using the IVIS Lumina II In Vivo Imaging System within 10 min of d-luciferin injection. Radiance values were calculated for the ROI using Living Image 3.1 software.
Statistics
Gene expression analyses were conducted using one-way analysis of variance (ANOVA) test. The results were considered statistically significant if the P-value was <0.05.
RESULTS
Development of the DropCRISPRa system
To develop a phase-separated gene activation system (DropCRISPRa), we capitalized on the dLbCas12a-SunTag system (10×GCN4) for targeting and IDR–AD (scFvGCN4-sfGFP-IDR–VP64-NLS) for gene activation (Figure 1A). In the design, the Cas–gRNA complex is responsible for target site recognition and binding, multivalent interactions between 10×GCN4 and scFv-GCN4 directly recruit multiple copies of ADs (VP64) to the target site, and IDR–IDR interactions drive LLPS transcription condensate formation to further recruit more ADs and endogenous transcriptional regulators (Figure 1A). In this system, the IDR functions as an essential component and its property determines transcription output. Therefore, to develop this system, the first step is to find a suitable IDR.
Figure 1.

Development of the DropCRISPRa system. (A) Schematic of the DropCRISPRa system. The dLbCas12a-SunTag system for targeting and IDR–AD (scFvGCN4-sfGFP-IDR–AD) undergoing LLPS for gene activation. (B) FRAP analysis of scFvGCN4-sfGFP-IDRs (DDX4, FUS and TAF15)–VP64 droplets in the HEK293T cells transiently transfected with the DropCRISPRa system targeting the HBG1 gene. Data are plotted as means ± standard deviation (SD) (n = 7). Scale bar, 5 μm. (C) Time-lapse imaging of droplet fusion and fission events. Scale bar, 5 μm. (D) RT–qPCR revealed relative mRNA expression of IL1RN, HBG, TTN and CD69 in the HEK293T cells transfected with the DropCRISPRa systems targeting the promoter region of each gene. Blank, HEK293T cells without transfection; Ctrl, HEK293T cells transfected with the plasmids encoding Cas–GCN4 protein fusion, scFvGCN4–sfGFP and gRNA. Mean values are presented with SD, n = 3 biological triplicates. *P < 0.05, **P < 0.01, one-way ANOVA test versus VP64. (E and F) The specificity of gene activation with the DropCRISPRa system was revealed by RNA-seq in the HEK293T cells transfected with the FUSIDR–VP64 system targeting IL1RN. The average of three biological replicates is shown.
FUS and TAF15 contain an N-terminal IDR domain and have been shown to form biomolecular condensates in cells (16) (Supplementary Figure S1A). The RNA helicase DDX4 is unable to regulate transcription but also contains an N-terminal IDR domain. To test whether these IDR domains could undergo phase separation in cells, we ectopically expressed the scFvGCN4-sfGFP-IDR-NLS (termed GFP–IDR) in HEK293T cells and found that all of them could form droplets (Supplementary Figure S1B). To assess the liquidity inside the droplets, we performed FRAP experiments and found that the GFP–IDR fluorescence signal exhibited fast recovery after photobleaching (Supplementary Figure S1B). The time-lapse imaging study revealed that GFP–IDR droplets underwent fusion and fission (Supplementary Figure S1C; Supplementary movies S1–S6). Addition of 2.5% 1,6-hexanediol, a compound that disrupts weak hydrophobic interactions and LLPS, completely dissolved the GFP–IDR droplets (Supplementary Figure S1D). Therefore, we found three IDR candidates for the DropCRISPRa system.
To test whether these IDRs could function in the system, we constructed IDR–VP64 plasmids and transfected them with the dLbCas12a-GCN4 and sgRNA plasmids targeting the promoter of endogenous HBG1 in HEK293T cells. The FRAP and time-lapse imaging experiments showed that DropCRISPRa could form liquid-like condensates (Figure 1B, C; Supplementary movies S7–S12). To determine whether the DropCRISPRa system could activate gene expression, we transfected HEK293T and MCF7 cells with the DropCRISPRa system targeting the endogenous promoters of HBG1, IL1RN, TTN and CD69. The results revealed that the FUSIDR–VP64-based DropCRISPRa system consistently showed high gene activation efficiency in all the tested cell lines and genes (Figure 1D; Supplementary Figure S2). Next, we performed transcriptome profiling by RNA-seq in HEK293T cells co-transfected with the DropCRISPRa (FUSIDR–VP64) system targeting IL1RN for assessing the gene activation specificity. As shown in Figure 1E and F, IL1RN was the most activated gene with statistical significance and only a few genes showed expression change, indicating high genome-wide targeted transcriptional specificity and robust gene activation compared with the control group transfected with dCas12a-SunTag, IL1RN-targeted gRNA and scFvGCN4–sfGFP without IDR and AD. Taken together, we developed a DropCRISPRa system with phase separation property, which could achieve highly efficient and specific endogenous gene activation in cells.
We also tested whether the DropCRISPRa system was compatible with other common ADs, such as the natural P65 AD and the synthetic VPR AD. Then, we generated the DropCRISPRa-P65 and the DropCRISPRa-VPR systems (Supplementary Figure S3A, D). After transfection into HEK293T cells, the two systems could form droplets (Supplementary Figure S3B, E) and the DropCRISPRa-P65 (FUSIDR and TAF15IDR based) showed significantly higher activity than the SunTag-P65 system without an IDR (Supplementary Figure S3C) and the DropCRISPRa-VPR showed mild enhanced activity compared with the SunTag-VPR system without an IDR (Supplementary Figure S3F). In addition, we found that DropCRISPRa-P65 had better activity than DropCRISPRa-VPR by activating HBG1, IL1RN and TTN in HEK293T cells (Supplementary Figure S3C, F). Therefore, we developed multiple platforms for gene activation.
Additionally, Cas12a exhibits self-processing capability for crRNA (gRNA), which means that multiple crRNAs within a single transcript can be cut out and processed into a mature form by Cas12a itself, and thus it allows for multiplexed genome editing with a single crRNA array transcript (32). So, we tested the DropCRISPRa system for multiplexed gene activation. We generated a 3-crRNA array to target the promoter regions of HBG1, IL1RN and TTN (Supplementary Figure S4A), transfected HEK293T cells with the DropCRISPRa (VP64, P65 and VPR) systems and the 3-crRNA array, and found that all the three genes were significantly activated (Supplementary Figure S4B–D). These results indicated that DropCRISPRa could be used for multiplexed gene activation.
Phase separation is required for efficient gene activation
Next, we investigated whether the high gene activation capability was dependent on the phase separation property. To this end, we generated mdFUSIDR–VP64 and maFUSIDR–VP64 mutants, which were based on FUS9Y>S and FUSG156E; the former was reported to be unable to undergo LLPS (16) and the latter was reported to form more insoluble aggregates (14) (Figure 2A). As expected, mdFUSIDR–VP64 lost its phase separation property when expressed in HEK293T cells (Figure 2B). The FRAP and time-lapse imaging experiments demonstrated that the liquid-like phase separation property of maFUSIDR–VP64 formed condensates (Figure 2C; Supplementary movies S13 and S14). 1,6-Hexanediol is widely used to dissect condensates in liquid- or solid-like states (33), and ∼75% of cells transfected with maFUSIDR–VP64 formed droplets and ∼42% of cells maintained at least one droplet upon 2.5% 1,6-hexanediol treatment (Figure 2D). In contrast, ∼70% of cells transfected with FUSIDR–VP64 formed droplets and almost all of the cells lost droplets upon 2.5% 1,6-hexanediol treatment (Figure 2D). These observations indicated that maFUSIDR–VP64 could form both liquid- and solid-like condensates and FUSIDR–VP64 formed liquid-like condensates (Figure 2C, D). To evaluate the activity of FUSIDR–VP64 mutants in gene activation, we transfected FUSIDR–VP64 mutant-based DropCRISPRa systems in HEK293T cells and found that mdFUSIDR–VP64 and maFUSIDR–VP64 reduced gene activation efficiency compared with FUSIDR–VP64 (Figure 2E). In detail, the mdFUSIDR–VP64 mutant induced obviously decreased gene activation (Figure 2E). On the other hand, the maFUSIDR–VP64 mutant displayed less decreased gene activation activity (Figure 2E), which was consistent with the finding that maFUSIDR–VP64 could form both liquid- and solid-like droplets and the liquid-like droplets might partially maintain transcription promotion activity (34).
Figure 2.

Phase separation is required for efficient gene activation. (A) Schematic of the mdFUSIDR–VP64 (FUS9Y>S) and maFUSIDR–VP64 (FUSG156E) DropCRISPRa mutants. (B) GFP fluorescence in HEK293T cells ectopically expressing DropCRISPRa mutants and gRNA targeting HBG1. Scale bar, 5 μm. (C) The FRAP and time-lapse imaging experiments revealed the liquid-like feature of part of the maFUS–VP64 condensates. Scale bar, 5 μm. (D) GFP fluorescence of condensates in HEK293T cells treated or not (Ctrl) with 2.5% 1,6-hexanediol for 5 min. Quantification of the cells with droplets is shown on the right, n ≥ 100 cells. Scale bar, 5 μm. (E) Relative mRNA expression of HBG1, IL1RN, TTN and CD69 in HEK293T cells transfected with the indicated gene activation systems. Mean values are presented with the SD, n = 3 biological triplicates. *P < 0.05, **P < 0.01, one-way ANOVA test. Scale bar, 5 μm.
It has been reported that TFs require a narrow optimum of IDR concentration and interactions for inducing endogenous gene activation, such as EWS–FLI1 (35). Therefore, we performed a dose-dependent assay of IDR–VP64 to activate transcription in HEK293T cells. As shown in Supplementary Figure S5A, FUSIDR–VP64 increased the expression of HBG1, IL1RN, TTN and CD69 when compared with VP64 without an IDR across all the tested doses (from 25 to 100 ng per well of a 24-well plate). In contrast, similarly to EWS–FLI1 (35), DDX4IDR–VP64 increased gene expression at the low dose (25 ng) but decreased gene expression at the high dose (100 ng). In addition, we optimized the GCN4–scFv interaction strength to change the amount of directly recruited IDR–VP64 by varying the copy number of GCN4 fused to Cas protein. As shown in Supplementary Figure S5B, more interactions (in the 24×GCN4 system) led to less gene activation, which was similar to EWS–FLI1 (35), and the 3×GCN4 system exhibited the best IDR-promoted gene activation effect. It is more interesting that FUSIDR–VP64 induced comparable gene expression levels between the 3×GCN4 and 10×GCN4 systems, but because VP64 without an IDR induced much weaker gene activation in the 3×GCN4 system, FUSIDR–VP64 displayed a much clearer and obvious enhancement effect, indicating that IDR–IDR interaction-mediated phase separation played a major role in such a system (Supplementary Figure S5B). These results also showed that FUSIDR–VP64 could further boost gene expression in the 10×GCN4 system, although VP64 without an IDR already activated gene transcription robustly in such a system.
Together, all the above observations indicated that LLPS itself and its characteristics, including its liquid-like property, IDR concentration and IDR–IDR interactions, could affect target gene activation in the DropCRISPRa system. Among all the tested systems, FUSIDR outperformed TAF15IDR and DDX4IDR.
The DropCRISPRa system forms a droplet at the target site to drive RNA synthesis
IDR–IDR interactions could drive condensation and there were several droplet puncta in a single cell after transfection with the DropCRISPRa system for target gene activation, but not all droplets were involved in target gene activation. Therefore, it was essential to check whether a droplet could form at the target DNA site. To this end, we constructed a reporter cell line, namely CRISPRTag-TRE-H2B, in which a DNA fragment containing a CRISPR targetable DNA sequence with six repeats (CRISPRTag-V1) (36) and a Tet-inducible promoter (TRE)-driven H2B expression cassette was inserted into the ACTB site in HEK293T cells. The ‘CRISPRTag-TRE-H2B’ DNA site could be labeled and visualized live by a dSpCas9-sfGFP-SunTag system (green) via targeting the CRISPRTag sequence, and the expression of exogenous H2B could be activated by a MoonTag-based DropCRISPRa system (dLbCas12a-mCherry-MoonTag, red) via targeting the TRE sequence (37), which contained dLbCas12a-10×gp41 and Nb-mCherry-FUS-VP64 (Supplementary Figure S6A, B). Next, we co-transfected the dSpCas9–sfGFP-SunTag and dLbCas12a–mCherry-MoonTag systems into the reporter cells and found that one green and red merged focus was found within ∼50% of the cells with both red and green puncta, indicating that the DropCRISPRa system formed a droplet at the target site (Supplementary Figure S6C). When the sgRNA targeting the TRE region or dLbCas12a-10×gp41 was absent, no merged foci could be observed, suggesting that dCas protein and sgRNA were essential for the droplet formation at the target site (Supplementary Figure S6C). Consistently, when one of the DropCRISPRa system components, sgRNA, Cas protein or IDR–AD was absent, the target gene could not be activated (Supplementary Figure S6D). Further, the FRAP and time-lapse imaging experiments with mCherry quenching at the merged foci demonstrated the liquid-like feature of the FUSIDR–VP64 condensates (Supplementary Figure S6E–G). All the above results suggested that the DropCRISPRa system could form an LLPS droplet at the target site.
We next asked whether the LLPS droplet at the target site could drive RNA synthesis. Similar to the above-described approach, we constructed a reporter cell line, namely CRISPRTag-TRE-luciferase-24×MS2. The genomic site of the exogenous CRISPRTag-TRE-luciferase-24×MS2 reporter DNA fragment could be visualized live by a dSpCas9–sfGFP-SunTag system (green), the expression of luciferase could be activated by a DropCRISPRa-MoonTag system (red) and the nascent MS2-tagged luciferase mRNA could be labeled and visualized by MCP–BFP (blue) (Figure 3A). Luciferase assay results confirmed that the DropCRISPRa–VP64-MoonTag systems could activate the expression of the exogenous luciferase gene (Figure 3B). Next, we co-transfected the DropCRISPRa-MoonTag (mCherry–FUSIDR–VP64) system and the MCP–BFP system into the reporter cell line and found that the MCP–BFP–mRNA puncta co-localized with the FUSIDR–VP64 droplet (Figure 3C). Quantitative assay revealed that ∼88.4% of the co-transfected cells showed a single blue nascent mRNA punctum and ∼73.8% of these blue mRNA puncta overlapped with a red FUSIDR–VP64 droplet (Figure 3D), indicating that most luciferase mRNA synthesis was driven by an LLPS droplet (∼73.8% merged puncta) and only a small portion was driven in a droplet-independent way (such as directly activated by VP64). Further, we also observed co-localization of the target gene, the FUSIDR–VP64 droplet and the nascent luciferase mRNA in the nucleus (Figure 3E). Together, all the above observations indicated that the DropCRISPRa system forms a droplet at the target site to drive RNA synthesis.
Figure 3.

The DropCRISPRa system forms a droplet at the target site to drive RNA synthesis. (A) Schematic of the experimental design. The HEK293T reporter cell line contained a CRISPRTag-TRE-luciferase-24×MS2 DNA fragment at the ACTB locus. The dSpCas9–sfGFP-SunTag system (green) labeled the target site via targeting the CRISPRTag sequence, the dLbCas12a–mCherry-MoonTag system (red) activated luciferase expression via targeting the TRE promotor and MCP–BFP was used to visualize the MS2-tagged luciferase mRNA. (B) Luciferase activity in the reporter cell line after transfection with the indicated gene activation systems. Mean values are presented with the SD, n = 3 biological triplicates. ***P < 0.001, **P < 0.01, one-way ANOVA test versus VP64. (C) Representative image of reporter cells transfected with the indicated systems, including dLbCas12a-VPR-2A–mCherry or DropCRISPRa-MoonTag gene activation systems and MCP–BFP for labeling of nascent local mRNA. (D) Quantifications of local mRNA labeling efficiency and the merge efficiency of local mRNA and the FUSIDR–VP64 droplet, n ≥ 80 cells. (E) Co-localization of the target gene, the FUSIDR–VP64 droplet and the nascent luciferase mRNA in the nucleus. The line scan result along the white lines in the merged droplet is shown. Scale bar, 5 μm.
The DropCRISPRa systems based on variant Cas proteins activate gene transcription
SpCas9 and AsCas12a are two widely used Cas nucleases, and thus DropCRISPRa systems based on these two proteins would have more expanded applications. As expected, we found that the DropCRISPRa-SpCas9 and DropCRISPRa-AsCas12a systems formed phase-separated condensates and significantly activated transcription of HBG1 and IL1RN compared with the SunTag–VP64 system in transiently transfected HEK293T cells (Figure 4A–D). More importantly, UnCas12f1 (529 amino acids), a type V-F Cas12f nuclease much smaller than Cas9 or Cas12a (usually 1000–1500 amino acids), is good for constructing an easily delivered small gene activation system, which is particularly adapted for therapeutic applications (38). As shown in Figure 4E and F, the DropCRISPRa-UnCas12f1 system formed LLPS droplets and significantly activated the expression of HBG1 and IL1RN in HEK293T cells. Furthermore, we also tested these DropCRISPRa systems based on P65 and VPR, and the results were consistent with the DropCRISPRa-LbCas12a system (Supplementary Figure S7).
Figure 4.

The DropCRISPRa systems based on variant Cas proteins activate gene transcription. (A, C and E) GFP fluorescence images of condensates in HEK293T cells transfected with the indicated DropCRISPRa (-SpCas9, -AsCas12a and -Un1Cas12f1) systems targeting HBG1. Scale bar, 5 μm. (B, D and F) RT–qPCR revealed relative mRNA expression of HBG1 and IL1RN in HEK293T cells transfected with the indicated systems. Mean values are presented with the SD, n = 3 biological triplicates. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA test versus VP64.
Comparison of the DropCRISPRa systems with the dCas12a–VPR and dCas9–VPR systems
It has been reported that dCas protein and VPR directly fused proteins could potently activate gene expression (23,39,40). Therefore, we compared the dLbCas12a–VPR and dSpCas9–VPR direct fusion systems with the DropCRISPRa systems based on VP64, P65 and VPR. As shown in Supplementary Figure S8, all the systems were able to activate HBG1, IL1RN, TTN and CD69 in both HEK293T and MCF7 cells, although to a different extent. The DropCRISPRa system (based on VP64, P65 or VPR) outperformed other systems at six out of the eight tested sites (all the four genes in HEK293T cells and HBG1 and CD69 in MCF7 cells) and the dLbCas12a–VPR system was the best one for the remaining two sites (IL1RN and TTN in MCF7 cells) and equal best with the FUS–VP64 system for HBG1 activation in MCF7 cells (Supplementary Figure S8C). It was noted that the dSpCas9–VPR system did not outperform other systems in any tested sites even though we tried four sgRNAs at each gene and used the best one. We might have failed to find an optimal gRNA for each of the four genes as gRNA played an essential role in gene activation (29,41–43). We believed that, at some sites in certain cell types or tissues, the dSpCas9–VPR system was able to outperform other systems. Our results, together with previous reports (29,41,44,45), suggested that the activation system itself, the gRNA targeting site, the context of the target gene, the cell type and other factors could affect the performance of the CRISPR-based gene activation system. Therefore, for a given gene, we need to screen out an optimal system and an optimal gRNA to achieve the best activation, and the DropCRISPRa system served as a good tool with potent and stable activity.
The DropCRISPRa system compartmentalizes RNAPII and BRD4 to the droplets and enhances the transcription rate
Next, we investigated how the DropCRISPRa system induced gene activation. It has been reported that FUSIDR condensates can compartmentalize the C-terminal domain of RNAPII (RNAPII-CTD) for transcription activation (18). To test this possibility, we transfected HEK293T cells with VP64-, DDX4IDR–VP64- or FUSIDR–VP64-based gene activation systems targeting HBG1, and performed immunofluorescence staining of post-translationally modified CTDs representing three functional transcription states, namely all CTDs, transcription initiating CTDs with phosphorylated Ser5 (CTD-S5) and elongating CTDs with phosphorylated Ser2 (CTD-S2) (46). The DDX4IDR–VP64 and FUSIDR–VP64 systems but not the VP64 system could form LLPS droplets (Figure 5A), and CTD, CTD-S2 and CTD-S5 were recruited into ∼65.7, 93.8 and 10.4% of the droplets formed by FUSIDR–VP64, and ∼6.5, 5.8 and 6.1% of the droplets formed by DDX4IDR–VP64, respectively (Figure 5A). BRD4 has been reported to regulate the phosphorylation of S2 within the CTD to promote the transition from transcription pausing to elongation (47,48). Therefore, we stained the transfected HEK293T cells and found that BRD4 was recruited into ∼95.3% of the FUSIDR–VP64 and 5.1% of DDX4IDR–VP64 formed droplets (Figure 5B). These results suggested that the major role of the droplets formed via phase separation could be in transcription elongation instead of initiation regulation.
Figure 5.

The DropCRISPRa system compartmentalizes RNAPII and BRD4 to the droplets and enhances the transcription rate. (A and B) GFP fluorescence and immunofluorescence staining of transcriptional regulators, i.e. RNAPII-CTD, Ser2 phosphorylated CTD (CTD-S2), Ser5 phosphorylated CTD (CTD-S5) and BRD4, in the HEK293T cells transfected with the indicated systems targeting HBG1. Quantification of co-localization efficiency is shown on the right, n ≥ 50 cells. Scale bar, 5 μm. (C) Analyses of the transcription elongation rate of the target gene activated by the indicated systems. The genome structure of the CD69 gene and the primer pairs used to amplify the regions spanning the 4.5 kb intron 1 are shown on the top. The HEK293T cells were transfected with the indicated systems targeting CD69 and, 48 h later, treated with DRB for 3 h to reversibly block transcription elongation. Total RNA was harvested at the indicated intervals after the removal of DRB and used for RT–qPCR with the indicated primer pairs. The expression values were plotted relative to the DRB-untreated group. (D) ChIP-PCR revealed the RNAPII-CTD-S2 occupancy along the CD69 gene. Schematic diagram showing the primer sets covering the promoter-distal region (p1), promoter-proximal region (p2) and gene body region (p3–p7) (top). Relative enrichment for each site was calculated by normalization to input control (bottom). **P < 0.01, ***P < 0.001, one-way ANOVA test versus VP64.
To examine whether the DropCRISPRa system accelerated transcription elongation, we activated the CD69 gene via the DropCRISPRa (FUSIDR–VP64) system. Because the CD69 gene contains a long intron 1 of 4.5 kb, it is relatively easy to calculate the elongation rate by examining the nascent pre-mRNA with two pairs of primers spanning exon 1–intron 1 and intron 1–exon 2, respectively (blue and red arrows in Figure 5C). Thus, we transfected HEK293T cells with the CD69 targeting system and treated the cells for 3 h with DRB, a drug that can lock RNAPII at the initiation stage and reversibly block transcription elongation (49). We harvested samples at different intervals after the removal of DRB and performed RT–qPCR using the indicated primer pairs. The exon 1 region of the CD69 gene was transcriptionally recovered immediately after DRB removal (Figure 5C). In contrast, the transcription recovery of the exon 2 region showed a delay after drug release (Figure 5C). The VP64, DDX4IDR–VP64 and FUSIDR–VP64 systems were delayed for ∼6, 6 and 4 min, suggesting an elongation rate of ∼0.7, ∼0.7 and ∼1.1 kb/min, respectively (Figure 5C). Furthermore, we performed ChIP-qPCR using an antibody against CTD-S2 and primers covering a promoter-distal region (p1), a promoter-proximal region (p2) and the gene body region (p3–p7). The results showed that CTD-S2 in the cells transfected with the FUSIDR–VP64 system was significantly more enriched at the promoter-proximal and gene body regions than that in the cells transfected with the VP64 and DDX4IDR–VP64 systems, indicating that more RNAPII-CTD-S2 molecules were driving transcription elongation of the CD69 gene in the DropCRISPRa system (Figure 5D).
Taken together, these results suggested that both BRD4 and RNAPII were recruited into the FUSIDR–VP64 formed droplets, and BRD4 might accelerate the phosphorylation of CTD-S2 and promote productive transcription elongation.
In vivo gene activation using the DropCRISPRa system
To test the DropCRISPRa system in vivo, we delivered the plasmids encoding the system and a TRE-driven luciferase reporter to the liver in adult mice via hydrodynamic tail vein injection (Figure 6A). At 12, 24, 48 and 72 h post-injection, the bioluminescence was measured and a time-dependent decrease in the signal was observed (Figure 6B). At 12 h and 24 h, we observed a robust luciferase reporter expression in mice to which the FUSIDR–VP64 system was delivered (Figure 6B, C). In contrast, the VP64 system showed only weak bioluminescence (Figure 6B, C). Last, we tested whether the DropCRISPRa system could also efficiently activate endogenous target genes in living mice. The mouse liver expressing the DropCRISPRa system targeting Fgf21 showed higher gene activation than the VP64 system (Figure 6D) at 24 h after plasmid injection. These results demonstrated that the DropCRISPRa system can provide an approach for robust expression of target genes in living mice.
Figure 6.

In vivo gene activation using the DropCRISPRa system. (A) Schematic of the TRE-driven luciferase reporter and the DropCRISPRa system used for in vivo gene activation, together with the experimental design. (B and C) Time-course bioluminescence imaging. The mice were anesthetized and bioluminescence was measured at 12, 24, 48 and 72 h post-plasmid injection. Radiance values were calculated for the ROI using Living Image 3.1 software. Data are plotted as means ± SD (n= 6), Student's t-test *P < 0.05, **P < 0.01. (D) Endogenous Fgf21 activation in vivo using the DropCRISPRa system 24 h after plasmid injection. n = 6 mice. Student's t-test *P < 0.05
DISCUSSION
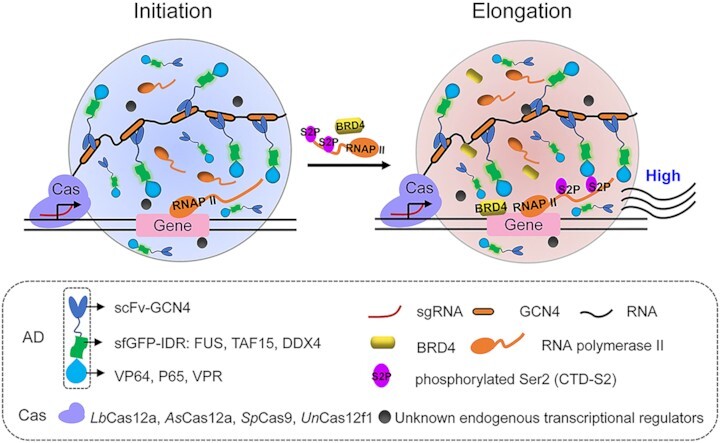
In this study, we engineered a DropCRISPRa system capable of highly activating endogenous genes in an LLPS-dependent manner (Figure 7). The DropCRISPRa system consists of a dCas protein fused to a tandem array of GCN4, a phase-separated scFv-GCN4-sfGFP-IDR–AD (IDR, FUS and TAF15; AD, VP64, VPR and P65) module and a gene targeting gRNA. In this system, the dCas–gRNA complex can recognize and bind to the target site, the multivalent GCN4–scFv interactions recruit multiple copies of the ADs to activate gene expression, possibly via regulating transcription initiation, and the IDR–IDR interactions drive LLPS transcription condensate formation to further recruit more ADs and endogenous RNAPII and BRD4 to enhance the transcription elongation rate and finally boost gene activation (Figure 7).
Figure 7.

Model of the DropCRISPRa system for efficient gene activation. The DropCRISPRa system consists of a dCas protein fused to a tandem array of GCN4, an scFv-GCN4-sfGFP-IDR–AD module and a targeting gRNA. The Cas–gRNA complex recognizes and binds to the target site, then multivalent interactions between GCN4 repeats and scFv–GCN4 directly recruit multiple copies of ADs to the target site, and the IDR–IDR interactions drive transcription condensate formation via LLPS to further recruit more ADs and endogenous RNAPII and BRD4 to enhance the gene transcription rate, and finally results in robust transcriptional activation of the target gene.
The engineered DropCRISPRa system exhibits compatibility with various Cas family nucleases. In this study, LbCas12a, AsCas12a, SpCas9 and UnCas12f1 have been successfully used to construct DropCRISPRa systems (Figure 4; Supplementary Figure S7). With unique features, such as PAM preference, size and activity, these nucleases can expand the scope and versatility of the DropCRISPRa system. First, the T-rich PAM of Cas12a and Cas12f1 and the G-rich PAM of Cas9 can cover most of the gene regions and facilitate the targeting of different genes. Second, Cas12a can self-process crRNA, which allows a crRNA array to target multiple genes, and thus facilitates multiplex gene activation. Third, SpCas9 is the best characterized and most widely used Cas protein, and currently available engineered Cas9 variants, such as high-fidelity eCas9 (50) and HFCas9 (51), broad PAM compatibility xCas9 (52) (NG, GAA and GAT) and PAMless SpRY (53), can provide a broad application of the DropCRISPRa system. Last, the small size of UnCas12f1 can facilitate therapeutic applications when delivered by cargo size-limited lentiviral and adeno-associated viral (AAV) vectors.
Similarly, the DropCRISPRa system is also compatible with various ADs, such as VP64, VPR and P65. Among these systems, DropCRISPRa-VP64 and DropCRISPRa-P65 demonstrated high gene activation in all four tested genes compared with the VP64 and P65 SunTag counterpart systems (Supplementary Figures S3, S4 and S7). DropCRISPRa-VPR showed a mild advantage although without a statistically significant difference (Supplementary Figures S3, S4 and S7). In addition, it is noteworthy that different ADs exhibit different gene activation power for different target genes in different cells, and no particular DropCRISPRa system performs consistently best for any target genes in any given cell. For example, DropCRISPRa-VP64 performed better than DropCRISPRa-p65 to activate CD69, but worse to activate IL1RN in HEK293T cells (Figure 1D; Supplementary Figure S3C). Furthermore, when compared with already existing direct fusion systems including dLbCas12a-VPR and dSpCas9-VPR, the DropCRISPRa system also showed high gene activation (Supplementary Figure S8). Therefore, for a particular purpose to activate a given gene in a given tissue, variant DropCRISPRa systems with different Cas proteins and different ADs should be screened to determine which is the best one.
In the DropCRISPRa system, the LLPS of FUSIDR–AD could recruit RNAPII and BRD4 to induce a high gene activation, while DDX4IDR–TF generally did not (Figures 1D, 2 and 4). These results remind us that the content and property of the LLPS droplets formed by different IDRs may dramatically affect the transcriptional outcome. In theory, any LLPS condensate involved in transcription could be a target for engineering with the DropCRISPRa system. For example, the transcription co-activators BRD4 and MED1 (9), the transcription elongation factor P-TEFb (54) as well as the enhancer–promoter structuring factor YY1 (55) have been reported to undergo LLPS to regulate transcription, and the IDRs of these proteins are worth testing in the future. Moreover, the strategy based on the combination of LLPS and the CRISPR system can be expanded beyond gene activation. Since LLPS has been reported to be involved in diverse biological processes, including DNA damage repair (56) and heterochromatin formation (57), it will be extremely interesting to engineer LLPS–CRISPR systems to manipulate gene editing, base editing, gene expression inhibition and other functions.
Recent studies, in which droplets were formed by FUS–Gal4 (18), FUS–TetR (19), FUS–LacI (20) and EWS–FLI1 (17), have demonstrated that the AD phase separation can amplify gene expression in the reporter system, but not for activation of endogenous genes in cells. In the DropCRISPRa system, we combined the CRISPR-SunTag system and the FETIDR–AD droplets to achieve endogenous target gene transcription activation. We further applied the system to target transcriptional activation in living mice.
A number of reports have provided numerous pieces of evidence to support that transcription factors and cofactors form transcription condensates with LLPS properties to activate gene expression (6,7,9,10,16). Consistently, in this study, we found that the DropCRISPRa system could form a transcriptional condensate at the target gene to increase RNAPII S2 phosphorylation and thus promote transcription elongation (Figures 3 and 5). These observations are consistent with a recent report showing that mutant ENL employed the same transcriptional condensate-dependent mechanism to drive oncogenic gene activation (58). Furthermore, we demonstrated that the high activation of endogenous genes depended on a phase separation property, as evidenced by the observations that the mdFUSIDR–VP64 (unable to form droplets) and maFUSIDR–VP64 mutants (which form insoluble aggregates) reduced gene activation efficiency compared with FUSIDR–VP64 (Figure 2). However, a recent article reported that transcription activation was enhanced by multivalent interactions independent of phase separation (20). There are two major differences between that article and our study. First, the target genes and cell lines are different. In that article, the target is a reporter gene with a synthetic promoter containing 256 repeats of lacO and 96 repeats of tetO sites followed by a CMV core promoter in human U2OS cells. Also promoter–AD interaction is mediated by lacO–LacI interactions (256×) or tetO–TetR interactions (96×), or dCas9/sgRNA targeting the lacO/tetO repeats (256×/96×). In our study, the targets are four endogenous genes (HBG1, IL1RN, TTN and CD69) in human HEK293T and MCF7 cells, the TRE-luciferase reporter (6×gRNA target site) and the Fgf21 gene in mice. The promoter–AD interaction is mediated by the DropCRISPRa system targeting a single site for endogenous genes or targeting six sites for the TRE promoter (6×) in the luciferase reporter. Second, the transcription activation AD modules are different. In the previously reported study, VP16, P65, VPR and Rta were used as AD modules. In our study, the AD modules contain an IDR of FUS or TAF15 and VP64, VPR or the activation domain of P65. From the two major differences, in the previous study, multivalent interactions between the AD modules and the promoter (256× or 96×) are the major force to recruit enough ADs for gene activation. In contrast, in our study, the interactions between the AD modules and the promoters (1× or 6×) are relatively weak and the IDR–-IDR interactions are the major force to recruit enough ADs and other factors (e.g. BRD4) to drive gene expression. Therefore, it is not surprising that phase separation plays a different role in the two investigations. We believe that multivalent interactions between TFs and the promoter (and enhancer) could be the major force to drive some endogenous gene expression in certain cells or tissues; on the other hand, phase separation driven by IDR–IDR interactions among TFs and cofactors could play a critical role for the expression of other genes in different cells or tissues.
In conclusion, we developed a highly efficient DropCRISPRa system with phase separation properties for gene activation in both mammalian cells and living mice. This system holds great potential for broad use in synthetic biology and therapeutic applications.
DATA AVAILABILITY
The accession number for RNA-seq is PRJNA844422 in NCBI BioProject at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA844422.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Professor Baohui Chen from Zhejiang University for providing CRISPRTag plasmids.
Author contributions: Z.R., S.M. and Y.L. conceived the idea, designed the experiments, analyzed the data and wrote the manuscript. S.M performed most experiments. K.L. and M.L. constructed plasmids and gene activation. X.W., J.L., X.Z. and H.H. constructed plasmids. L.L. and T.H. performed some FRAP and time-lapse imaging experiments. X.G. read the manuscript and gave helpful advice to improve the manuscript.
Contributor Information
Shufeng Ma, Department of Nephrology, Shenzhen Hospital, Southern Medical University, Shenzhen 518110, China; Cancer Research Institute, School of Basic Medical Sciences, State Key Laboratory of Organ Failure Research, National Clinical Research Center of Kidney Disease, Key Laboratory of Organ Failure Research (Ministry of Education), Southern Medical University, Guangzhou 510515, China.
Kaitong Liao, Cancer Research Institute, School of Basic Medical Sciences, State Key Laboratory of Organ Failure Research, National Clinical Research Center of Kidney Disease, Key Laboratory of Organ Failure Research (Ministry of Education), Southern Medical University, Guangzhou 510515, China.
Mengrao Li, Cancer Research Institute, School of Basic Medical Sciences, State Key Laboratory of Organ Failure Research, National Clinical Research Center of Kidney Disease, Key Laboratory of Organ Failure Research (Ministry of Education), Southern Medical University, Guangzhou 510515, China.
Xinlong Wang, Cancer Research Institute, School of Basic Medical Sciences, State Key Laboratory of Organ Failure Research, National Clinical Research Center of Kidney Disease, Key Laboratory of Organ Failure Research (Ministry of Education), Southern Medical University, Guangzhou 510515, China.
Jie Lv, Cancer Research Institute, School of Basic Medical Sciences, State Key Laboratory of Organ Failure Research, National Clinical Research Center of Kidney Disease, Key Laboratory of Organ Failure Research (Ministry of Education), Southern Medical University, Guangzhou 510515, China.
Xin Zhang, Cancer Research Institute, School of Basic Medical Sciences, State Key Laboratory of Organ Failure Research, National Clinical Research Center of Kidney Disease, Key Laboratory of Organ Failure Research (Ministry of Education), Southern Medical University, Guangzhou 510515, China; Affiliated Dongguan Hospital, Southern Medical University, (Dongguan People's Hospital), Dongguan 523058, China.
Hongxin Huang, Dermatology Hospital, Southern Medical University, Guangzhou 510091, China.
Lian Li, Cancer Research Institute, School of Basic Medical Sciences, State Key Laboratory of Organ Failure Research, National Clinical Research Center of Kidney Disease, Key Laboratory of Organ Failure Research (Ministry of Education), Southern Medical University, Guangzhou 510515, China.
Tao Huang, Cancer Research Institute, School of Basic Medical Sciences, State Key Laboratory of Organ Failure Research, National Clinical Research Center of Kidney Disease, Key Laboratory of Organ Failure Research (Ministry of Education), Southern Medical University, Guangzhou 510515, China; Dermatology Hospital, Southern Medical University, Guangzhou 510091, China.
Xiaohua Guo, Department of Nephrology, Shenzhen Hospital, Southern Medical University, Shenzhen 518110, China.
Ying Lin, Cancer Research Institute, School of Basic Medical Sciences, State Key Laboratory of Organ Failure Research, National Clinical Research Center of Kidney Disease, Key Laboratory of Organ Failure Research (Ministry of Education), Southern Medical University, Guangzhou 510515, China; Experimental Education/Administration Center, School of Basic Medical Science, Southern Medical University, Guangzhou 510515, China.
Zhili Rong, Cancer Research Institute, School of Basic Medical Sciences, State Key Laboratory of Organ Failure Research, National Clinical Research Center of Kidney Disease, Key Laboratory of Organ Failure Research (Ministry of Education), Southern Medical University, Guangzhou 510515, China; Dermatology Hospital, Southern Medical University, Guangzhou 510091, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The National Natural Science Foundation of China [82072329 to Y.L., 82070002 to Z.R., 82200072 to S.M. and 82102443 to T.H.]; The National Key R&D Program of China [2022YFA0806300 to Z.R.]; the Natural Science Foundation of Guangdong Province [2023A1515012269 and 2022A1515011091 to Y.L.]; the GuangDong Basic and Applied Basic Research Foundation [2021A1515110878 to S.M., 2022A1515111046 to Z.X. and 2021B1515140031 to Z.R.]; and the Fellowship of China Postdoctoral Science Foundation [2022T150300 and 2021M691473 to S.M. and 2021M701634 to X.Z.].
Conflict of interest statement. Z.R., Y.L., S.M., K.L. and M.L. have filed a patent application for the DropCRISPRa system.
REFERENCES
- 1. Hyman A.A., Weber C.A., Julicher F.. Liquid–liquid phase separation in biology. Annu. Rev. Cell Dev. Biol. 2014; 30:39–58. [DOI] [PubMed] [Google Scholar]
- 2. Li P., Banjade S., Cheng H.C., Kim S., Chen B., Guo L., Llaguno M., Hollingsworth J.V., King D.S., Banani S.F.et al.. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012; 483:336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nott T.J., Petsalaki E., Farber P., Jervis D., Fussner E., Plochowietz A., Craggs T.D., Bazett-Jones D.P., Pawson T., Forman-Kay J.D.et al.. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell. 2015; 57:936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Smith J., Calidas D., Schmidt H., Lu T., Rasoloson D., Seydoux G.. Spatial patterning of P granules by RNA-induced phase separation of the intrinsically-disordered protein MEG-3. Elife. 2016; 5:e21337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alberti S., Gladfelter A., Mittag T.. Considerations and challenges in studying liquid–liquid phase separation and biomolecular condensates. Cell. 2019; 176:419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boija A., Klein I.A., Sabari B.R., Dall’Agnese A., Coffey E.L., Zamudio A.V., Li C.H., Shrinivas K., Manteiga J.C., Hannett N.M.et al.. Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell. 2018; 175:1842–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Henninger J.E., Oksuz O., Shrinivas K., Sagi I., LeRoy G., Zheng M.M., Andrews J.O., Zamudio A.V., Lazaris C., Hannett N.M.et al.. RNA-mediated feedback control of transcriptional condensates. Cell. 2021; 184:207–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Murthy A.C., Tang W.S., Jovic N., Janke A.M., Seo D.H., Perdikari T.M., Mittal J., Fawzi N.L.. Molecular interactions contributing to FUS SYGQ LC-RGG phase separation and co-partitioning with RNA polymerase II heptads. Nat. Struct. Mol. Biol. 2021; 28:923–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sabari B.R., Dall’Agnese A., Boija A., Klein I.A., Coffey E.L., Shrinivas K., Abraham B.J., Hannett N.M., Zamudio A.V., Manteiga J.C.et al.. Coactivator condensation at super-enhancers links phase separation and gene control. Science. 2018; 361:eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shao W., Bi X., Pan Y., Gao B., Wu J., Yin Y., Liu Z., Peng M., Zhang W., Jiang X.et al.. Phase separation of RNA-binding protein promotes polymerase binding and transcription. Nat. Chem. Biol. 2022; 18:70–80. [DOI] [PubMed] [Google Scholar]
- 11. Andersson M.K., Stahlberg A., Arvidsson Y., Olofsson A., Semb H., Stenman G., Nilsson O., Aman P.. The multifunctional FUS, EWS and TAF15 proto-oncoproteins show cell type-specific expression patterns and involvement in cell spreading and stress response. BMC Cell Biol. 2008; 9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thomsen C., Grundevik P., Elias P., Stahlberg A., Aman P.. A conserved N-terminal motif is required for complex formation between FUS, EWSR1, TAF15 and their oncogenic fusion proteins. FASEB J. 2013; 27:4965–4974. [DOI] [PubMed] [Google Scholar]
- 13. Qamar S., Wang G., Randle S.J., Ruggeri F.S., Varela J.A., Lin J.Q., Phillips E.C., Miyashita A., Williams D., Strohl F.et al.. FUS phase separation is modulated by a molecular chaperone and methylation of arginine cation–pi interactions. Cell. 2018; 173:720–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Patel A., Lee H.O., Jawerth L., Maharana S., Jahnel M., Hein M.Y., Stoynov S., Mahamid J., Saha S., Franzmann T.M.et al.. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell. 2015; 162:1066–1077. [DOI] [PubMed] [Google Scholar]
- 15. Burke K.A., Janke A.M., Rhine C.L., Fawzi N.L.. Residue-by-residue view of in vitro FUS granules that bind the C-terminal domain of RNA polymerase II. Mol. Cell. 2015; 60:231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wei M.T., Chang Y.C., Shimobayashi S.F., Shin Y., Strom A.R., Brangwynne C.P.. Nucleated transcriptional condensates amplify gene expression. Nat. Cell Biol. 2020; 22:1187–1196. [DOI] [PubMed] [Google Scholar]
- 17. Johnson K.M., Mahler N.R., Saund R.S., Theisen E.R., Taslim C., Callender N.W., Crow J.C., Miller K.R., Lessnick S.L.. Role for the EWS domain of EWS/FLI in binding GGAA-microsatellites required for Ewing sarcoma anchorage independent growth. Proc. Natl Acad. Sci. USA. 2017; 114:9870–9875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zuo L., Zhang G., Massett M., Cheng J., Guo Z., Wang L., Gao Y., Li R., Huang X., Li P.et al.. Loci-specific phase separation of FET fusion oncoproteins promotes gene transcription. Nat. Commun. 2021; 12:1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schneider N., Wieland F.G., Kong D., Fischer A.A.M., Horner M., Timmer J., Ye H., Weber W.. Liquid–liquid phase separation of light-inducible transcription factors increases transcription activation in mammalian cells and mice. Sci. Adv. 2021; 7:eabd3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trojanowski J., Frank L., Rademacher A., Mucke N., Grigaitis P., Rippe K.. Transcription activation is enhanced by multivalent interactions independent of phase separation. Mol. Cell. 2022; 82:1878–1893. [DOI] [PubMed] [Google Scholar]
- 21. Kampmann M. CRISPRi and CRISPRa screens in mammalian cells for precision biology and medicine. ACS Chem. Biol. 2018; 13:406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maeder M.L., Linder S.J., Cascio V.M., Fu Y., Ho Q.H., Joung J.K.. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods. 2013; 10:977–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chavez A., Scheiman J., Vora S., Pruitt B.W., Tuttle M., E P.R.I., Lin S., Kiani S., Guzman C.D., Wiegand D.Jet al.. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods. 2015; 12:326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zetsche B., Volz S.E., Zhang F.. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015; 33:139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Konermann S., Brigham M.D., Trevino A.E., Joung J., Abudayyeh O.O., Barcena C., Hsu P.D., Habib N., Gootenberg J.S., Nishimasu H.et al.. Genome-scale transcriptional activation by an engineered CRISPR–Cas9 complex. Nature. 2015; 517:583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dahlman J.E., Abudayyeh O.O., Joung J., Gootenberg J.S., Zhang F., Konermann S.. Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat. Biotechnol. 2015; 33:1159–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tanenbaum M.E., Gilbert L.A., Qi L.S., Weissman J.S., Vale R.D.. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014; 159:635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Casas-Mollano J.A., Zinselmeier M.H., Erickson S.E., Smanski M.J.. CRISPR–Cas activators for engineering gene expression in higher eukaryotes. CRISPR J. 2020; 3:350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang X., Wang W., Shan L., Han L., Ma S., Zhang Y., Hao B., Lin Y., Rong Z.. Gene activation in human cells using CRISPR/Cpf1-p300 and CRISPR/Cpf1-SunTag systems. Protein Cell. 2018; 9:380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ye H., Rong Z., Lin Y.. Live cell imaging of genomic loci using dCas9-SunTag system and a bright fluorescent protein. Protein Cell. 2017; 8:853–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma S., Wang X., Hu Y., Lv J., Liu C., Liao K., Guo X., Wang D., Lin Y., Rong Z.. Enhancing site-specific DNA integration by a Cas9 nuclease fused with a DNA donor-binding domain. Nucleic Acids Res. 2020; 48:10590–10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Campa C.C., Weisbach N.R., Santinha A.J., Incarnato D., Platt R.J.. Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat. Methods. 2019; 16:887–893. [DOI] [PubMed] [Google Scholar]
- 33. Ulianov S.V., Velichko A.K., Magnitov M.D., Luzhin A.V., Golov A.K., Ovsyannikova N., Kireev I.I., Gavrikov A.S., Mishin A.S., Garaev A.Ket al.. Suppression of liquid-liquid phase separation by 1,6-hexanediol partially compromises the 3D genome organization in living cells. Nucleic. Acids. Res. 2021; 49:10524–10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yang L., Gal J., Chen J., Zhu H.. Self-assembled FUS binds active chromatin and regulates gene transcription. Proc. Natl Acad. Sci. USA. 2014; 111:17809–17814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chong S., Graham T.G.W., Dugast-Darzacq C., Dailey G.M., Darzacq X., Tjian R.. Tuning levels of low-complexity domain interactions to modulate endogenous oncogenic transcription. Mol. Cell. 2022; 82:2084–2097. [DOI] [PubMed] [Google Scholar]
- 36. Chen B., Zou W., Xu H., Liang Y., Huang B.. Efficient labeling and imaging of protein-coding genes in living cells using CRISPR-Tag. Nat. Commun. 2018; 9:5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boersma S., Khuperkar D., Verhagen B.M.P., Sonneveld S., Grimm J.B., Lavis L.D., Tanenbaum M.E.. Multi-color single-molecule imaging uncovers extensive heterogeneity in mRNA decoding. Cell. 2019; 178:458–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Anguela X.M., High K.A.. Entering the modern era of gene therapy. Annu. Rev. Med. 2019; 70:273–288. [DOI] [PubMed] [Google Scholar]
- 39. Kleinstiver B.P., Sousa A.A., Walton R.T., Tak Y.E., Hsu J.Y., Clement K., Welch M.M., Horng J.E., Malagon-Lopez J., Scarfo I.et al.. Engineered CRISPR-Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol. 2019; 37:276–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Escobar M., Li J., Patel A., Liu S., Xu Q., Hilton I.B.. Quantification of genome editing and transcriptional control capabilities reveals hierarchies among diverse CRISPR/Cas systems in human cells. ACS Synth. Biol. 2022; 11:3239–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tak Y.E., Horng J.E., Perry N.T., Schultz H.T., Iyer S., Yao Q., Zou L.S., Aryee M.J., Pinello L., Joung J.K.. Augmenting and directing long-range CRISPR-mediated activation in human cells. Nat. Methods. 2021; 18:1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang X., Lv S., Luo Z., Hu Y., Peng X., Lv J., Zhao S., Feng J., Huang G., Wan Q.L.et al.. MiniCAFE, a CRISPR/Cas9-based compact and potent transcriptional activator, elicits gene expression in vivo. Nucleic Acids Res. 2021; 49:4171–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ma S., Lv J., Sun J., Tang P., Li H., Zhou H., Zhang Z., Lin Y., Rong Z.. iKA-CRISPR hESCs for inducible and multiplex orthogonal gene knockout and activation. FEBS Lett. 2018; 592:2238–2247. [DOI] [PubMed] [Google Scholar]
- 44. Chavez A., Tuttle M., Pruitt B.W., Ewen-Campen B., Chari R., Ter-Ovanesyan D., Haque S.J., Cecchi R.J., Kowal E.J.K., Buchthal J.et al.. Comparison of Cas9 activators in multiple species. Nat. Methods. 2016; 13:563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lebar T., Lainscek D., Merljak E., Aupic J., Jerala R.. A tunable orthogonal coiled-coil interaction toolbox for engineering mammalian cells. Nat. Chem. Biol. 2020; 16:513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Eick D., Geyer M.. The RNA polymerase II carboxy-terminal domain (CTD) code. Chem. Rev. 2013; 113:8456–8490. [DOI] [PubMed] [Google Scholar]
- 47. Jang M.K., Mochizuki K., Zhou M., Jeong H.S., Brady J.N., Ozato K.. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell. 2005; 19:523–534. [DOI] [PubMed] [Google Scholar]
- 48. Devaiah B.N., Lewis B.A., Cherman N., Hewitt M.C., Albrecht B.K., Robey P.G., Ozato K., Sims R.J. 3rd, Singer D.S.. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc. Natl Acad. Sci. USA. 2012; 109:6927–6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Singh J., Padgett R.A.. Rates of in situ transcription and splicing in large human genes. Nat. Struct. Mol. Biol. 2009; 16:1128–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Slaymaker I.M., Gao L., Zetsche B., Scott D.A., Yan W.X., Zhang F.. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016; 351:84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K.. High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016; 529:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z.et al.. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018; 556:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Walton R.T., Christie K.A., Whittaker M.N., Kleinstiver B.P.. Unconstrained genome targeting with near-PAMless engineered CRISPR–Cas9 variants. Science. 2020; 368:290–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fu H., Liu R., Jia Z., Li R., Zhu F., Zhu W., Shao Y., Jin Y., Xue Y., Huang J.et al.. Poly(ADP-ribosylation) of P-TEFb by PARP1 disrupts phase separation to inhibit global transcription after DNA damage. Nat. Cell Biol. 2022; 24:513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang W., Qiao S., Li G., Cheng J., Yang C., Zhong C., Stovall D.B., Shi J., Teng C., Li D.et al.. A histidine cluster determines YY1-compartmentalized coactivators and chromatin elements in phase-separated enhancer clusters. Nucleic Acids Res. 2022; 50:4917–4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kilic S., Lezaja A., Gatti M., Bianco E., Michelena J., Imhof R., Altmeyer M.. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J. 2019; 38:e101379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Strom A.R., Emelyanov A.V., Mir M., Fyodorov D.V., Darzacq X., Karpen G.H.. Phase separation drives heterochromatin domain formation. Nature. 2017; 547:241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Song L., Yao X., Li H., Peng B., Boka A.P., Liu Y., Chen G., Liu Z., Mathias K.M., Xia L.et al.. Hotspot mutations in the structured ENL YEATS domain link aberrant transcriptional condensates and cancer. Mol. Cell. 2022; 82:4080–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for RNA-seq is PRJNA844422 in NCBI BioProject at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA844422.