


Abstract
The c-Myc oncoprotein and its dimerization partner Max bind the DNA core consensus sequence CACGTG (E-box) and activate gene transcription. However, the low levels of induction have hindered the identification of novel Myc target genes by differential screening techniques. Here, we describe a computer-based pre-selection of candidate Myc/Max target genes, based on two restrictive criteria: an extended E-box consensus sequence for Myc/Max binding and the occurrence of this sequence within a potential genomic CpG island. Candidate genes selected by these criteria were evaluated experimentally for their response to Myc. Two Myc target genes are characterized here in detail. These encode nucleolin, an abundant nucleolar protein, and BN51, a co-factor of RNA polymerase III. Myc activates transcription of both genes via E-boxes located in their first introns, as seen for several well-characterized Myc targets. For both genes, mutation of the E-boxes abolishes transcriptional activation by Myc as well as repression by Mad1. In addition, the BN51 promoter is selectively activated by Myc and not by USF, another E-box-binding factor. Both nucleolin and BN51 are implicated in the maturation of ribosomal RNAs, albeit in different ways. We propose that Myc, via regulation of these and probably many other transcriptional targets, may be an important regulator of ribosome biogenesis.
INTRODUCTION
The myc family of mammalian proto-oncogenes includes three evolutionarily conserved genes c-, N- and L-myc, which encode related proteins. myc genes are differentially expressed during embryonic development (1) and proliferating post-natal tissues, with few exceptions, express c-myc (2). Oncogenic activation of these genes, in particular c-myc, is observed in a wide range of human and animal neoplasias and generally results in their constitutive expression (2). In normal cells, expression of c-myc is strictly dependent on mitogenic signals and is suppressed by growth inhibitory and differentiation inducing signals (2,3). c-myc expression is also regulated by the products of other oncogenes or tumor suppressor genes, such as tyrosine kinase receptors (4) or the APC/β-catenin/TCF pathway (5). This may explain why c-myc is sometimes overexpressed in tumors without being mutated.
The c-myc protein product (Myc) conveys strong mitogenic and apoptotic stimuli. Constitutive expression of Myc reduces growth factor requirements, prevents growth arrest by a variety of growth inhibitory signals, and can block cellular differentiation (reviewed in 2,6). Conversely, activation of a conditional Myc–estrogen receptor chimera (Myc-ER) (7) in quiescent cells induces entry into the cell cycle in the absence of mitogens (8,9). However, concomitant with their mitogenic action, Myc or Myc-ER can induce apoptosis if survival factors are absent from the extracellular environment (10,11; reviewed in 2,3). Myc-induced apoptosis has been linked to activation of the p19ARF1–p53 pathway and is separable from mitogenesis (12).
Myc is a transcription factor of the basic helix–loop–helix leucine zipper (bHLH-Zip) family. Myc dimerizes with another bHLH-Zip protein, Max, to bind the specific DNA sequence CACGTG (the E-box) and activate transcription (13,14; reviewed in 3,15). Transcription-competent Myc/Max dimers are the active form of Myc in inducing cell cycle progression, apoptosis and malignant transformation (3,15). In addition to its function as a transactivator, Myc can also repress transcription of several genes that are clearly relevant to its biological function (16–18). In fact, some reports have suggested that transcription repression, rather than activation, correlates with the biological activity of Myc (19,20; reviewed in 3,21,22). There is, however, no clear demonstration that gene repression is a direct action of Myc in vivo, rather than the indirect consequence of activating as yet unknown Myc target genes (see also Discussion). The molecular mechanisms proposed to explain transcription repression by Myc (21,22) therefore remain speculative.
Max also forms heterodimers with the other bHLH-Zip proteins Mad1, Mxi-1 (or Mad2), Mad3, Mad4 and Mnt (or Rox) (23–27). These alternative dimers also bind the E-box and actively repress transcription and can therefore antagonize both the transcription and transforming activities of Myc. Furthermore, the myc and mad family genes are generally regulated in opposite modes in growth control and development (25,28; reviewed in 3). In summary, Myc, Max and Mad proteins form a network that regulates gene expression, proliferation, apoptosis and differentiation.
To further understand the function of Myc, Max and Mad, it is necessary to identify the genes that are directly regulated by these proteins, in particular, Myc-induced and/or Mad-repressed genes. Known Myc target genes include α-prothymosin (αPT), ornithine decarboxylase (ODC), Cdc25A, cad, MrDb, ECA39, EIF-4E, LDH-A, rcl, E2F-2, RCC1, the telomerase catalytic subunit hTERT and others (reviewed in 3,29,30). Although ODC, Cdc25A, LDH-A, rcl, E2F-2, αPT and hTERT share some of the potential of Myc to induce cell cycle progression, apoptosis, immortalization and/or transformation (31–35), additional target genes must be identified to explain the whole range of biological effects controlled by Myc (6). A common feature of these genes is the low magnitude of their response to Myc. Generally, induction by Myc results in only 2- to 5-fold increases, making identification of novel targets difficult. Indeed, only a few of the known targets have been identified by differential screening techniques, the majority of Myc-regulated genes being postulated by the presence of E-box sequences (30). In the present study, we describe a computerized screening strategy so as to facilitate initial identification of potential Myc-regulated genes, and characterize two novel Myc targets, nucleolin and BN51.
MATERIALS AND METHODS
Computer-based analysis of sequences in the GenBank databases
The program (cpg) was written in C and used the GCG package library functions (Wisconsin Package v.8, Genetics Computer Group, Madison, WI) for the input of parameters, nucleotide query sequences and GCG formatted database access (EMBL). With each perfect match for VCACGTGB in any nucleotide sequence entry of the database, the program computed three values from within windows of determined length around the detected match. First, the average GC content expressed as the global percentage of G and C in the window, secondly, the CpG/GpC ratio and, finally, observed/expected CpG, defined as (no. CpG/length)/[(no. C × no. G)/length]. If the 5′- or 3′-end of the sequence did not permit application of the predetermined window length, the computation was made on the available sequence. The searches presented here used the following criteria: a window size of 100 nt in both directions and a GC content ≥65% (in both directions from the hit).
Induction of Myc-ER cells
We used Myc-ER clone 3, a Rat1 cell line constitutively expressing a Myc–ER fusion protein (7,10). Cells were grown to ~40% confluence in phenol red-free DMEM medium (Gibco BRL no. 11880-028) supplemented with 10% charcoal-stripped fetal calf serum to prevent activation of Myc–ER. Subconfluent cultures were serum-starved for 24 h prior to activation of Myc–ER by addition of 200 nM trans-4-hydroxytamoxifen (OHT) (Laboratoires Besins-Isovesco, Paris, France). Cells were harvested at various time points by scraping and stored frozen until required for nuclear run-on or RNA preparation.
Preparation of filters for nuclear run-on and reverse northern analysis
Plasmid DNAs containing the genes of interest were isolated from Escherichia coli using the Promega Wizard miniprep DNA extraction kit. Aliquots of 12 µg of each DNA were ethanol precipitated, resuspended in 200 µl H2O and denatured by addition of 20 µl 2 N NaOH and heating at 95°C for 10 min. Denatured DNA was then transferred to a second tube containing 600 µl ice-cold 10× SSC (1.5 M NaCl, 0.15 M Na- citrate, pH 7.0). The solution was neutralized by the addition of 44 µl 2 M HEPES (free acid) and divided equally between two wells on a 48-well slot blotter, so as to produce two identical replica blots. Each blot contained actin, tubulin, GAPDH and 36B4 as internal standards, and ODC as a positive control for a Myc-regulated gene. The filters were washed twice in 6× SSC and allowed to air dry. After drying the filters were baked under vacuum for 2 h at 80°C.
Nuclear run-on
Cells from two 15 cm dishes were resuspended in 8 ml ice-cold buffer A (10 mM Tris–HCl, pH 7.5, 5 mM MgCl2, 10 mM NaCl, 0.5 mM DTT), incubated on ice for 20 min, centrifuged, resuspended in 2 ml buffer A and lysed by rapid pipetting of the suspension. Nuclei were recovered by centrifugation at 2000 r.p.m. for 5 min, washed once in 2 ml buffer B (20 mM Tris pH 7.5, 10 mM MgCl2, 140 mM KCl, 20% glycerol, 0.5 mM DTT), resuspended in buffer B supplemented with 0.5 mM ATP, 0.5 mM CTP, 0.5 mM GTP, 5 µM UTP and 200 µCi [α-32P]UTP, and incubated at 30°C for 15 min. The reaction was stopped with 1 ml ice-cold buffer C (10 mM Tris–HCl, pH 7.5, 50 mM MgCl2, 500 mM NaCl, 2 mM CaCl2). RNase-free DNase (200 U) was added to the mixture, followed by incubation at room temperature for 5 min. The reaction mix was then passed twice through a QIAshredder™ unit (Qiagen) and RNA was extracted using the RNeasy™ midi-prep system (Qiagen), according to the manufacturer’s instructions. Prehybridization of filters was performed in TESS2D (20 mM TES, pH 7.4, 2× Denhardt’s solution, 400 mM NaCl, 2.6 mM EDTA and 0.07% SDS) supplemented with 0.4 mg/ml yeast RNA at 65°C for 4 h. The eluant (200 µl) from the RNeasy™ preparation of nuclear run-on RNA was added to 1 ml of TESS2D and hybridization allowed to take place for 24 h at 65°C. The blots were then washed three times for 30 min in 1× SSC, 0.1% SDS at 65°C. Run-on signals were quantified with a phosphorimager.
Reverse northern and northern blotting
Total cellular RNA was isolated using the RNeasy™ mini prep kit (Qiagen). An aliquot of 1 µg of RNA was labeled by random priming using a mixture of [α-32P]dATP and [α-32P]dCTP. The labeled probes were isolated by spin filtration using Sephadex G-50. Hybridization of slot-blotted DNAs was performed as for nuclear run-on, apart from the use of denatured salmon sperm DNA as non-specific competitor. Northern blotting was performed by standard methods using random primed DNA probes. All signals were quantified with a phosphorimager.
Plasmid constructions
To generate the reporter pNucL14 (Fig. 2A), the promoter, exon 1, intron 1 and the first 8 bp of exon 2 were cloned in pGL3basic (Promega). Digestion with NcoI and subsequent treatment with S1 nuclease followed by religation destroyed the natural translation start codon located in exon 1. The product of this reaction was confirmed by sequencing. To generate the mutants pNucL14mut1, pNucL14mut2 and pNucL14mut1/2 we first subcloned the EcoRV–HindIII fragment of pNucL14 into pBluescript to generate pBSnuc. This construct was used for PCR mutagenesis of the nucleolin gene by mutating the respective CACGTG sequences to TACGTG. Subcloning the various mutants back into pNucL14 using EcoRV and HindIII then produced each reporter construct. To generate pGL3-BN51 (Fig. 3A), a genomic DNA fragment spanning the promoter, exon 1, intron 1 and part of exon 2 of BN51 was recovered from plasmid pBN51eco (36) with XbaI and AflIII. This fragment was subcloned in pGL3-basic (Promega) cleaved with NheI and NcoI. The AflIII + NcoI ligation allowed fusion of BN51 and luciferase on the natural BN51 start codon. Mutagenesis of the E-box (pGL3-BN51-mut1, Fig. 3A) was performed by PCR using native Pfu polymerase (Stratagene) in a modified buffer (30 mM Tricine pH 8.4, 2 mM MgCl2, 5 mM 2-mercaptoethanol, 0.01% Thesit). We amplified a 500 bp fragment of intron 1 encompassing the NarI and AflIII sites and engineered new EcoRI and BamHI sites on each side of this fragment. The PCR product was cloned into pBluescript and sequenced (pBS-BN51). The mutant BN51 fragment was recovered (NarI–AflIII) and subcloned in pGL3basic (XbaI–AflIII) in a three-way ligation together with the XbaI–NarI fragment from pBN51eco. cDNAs expressing human Myc, Mad1 or USF were subcloned in the expression vector BJ3 (13).
Figure 2.

Myc activates the nucleolin promoter via E-boxes located in intron 1. (A) Structure of the nucleolin reporter gene constructs. The mouse nucleolin promoter, exon 1, intron 1 and the first 8 nt of exon 2 were fused to the 5′-end of the luciferase gene in pGL3-basic to generate the reporter construct pNucL14. The ATG in exon 1 was deleted (see Materials and Methods). In pNucL14-mut1 and pNucL14-mut2, two groups of E-boxes located in intron 1 were mutated as shown (black dots). All E-boxes were mutated in pNucL14-mut1/2. The reporters were transiently transfected into (B) Rat1 and (C) 293T cells, together with plasmids expressing Myc, Mad or USF, as indicated. Luciferase activities are expressed relative to pNucL14 alone. The data are averaged from three independent experiments.
Figure 3.
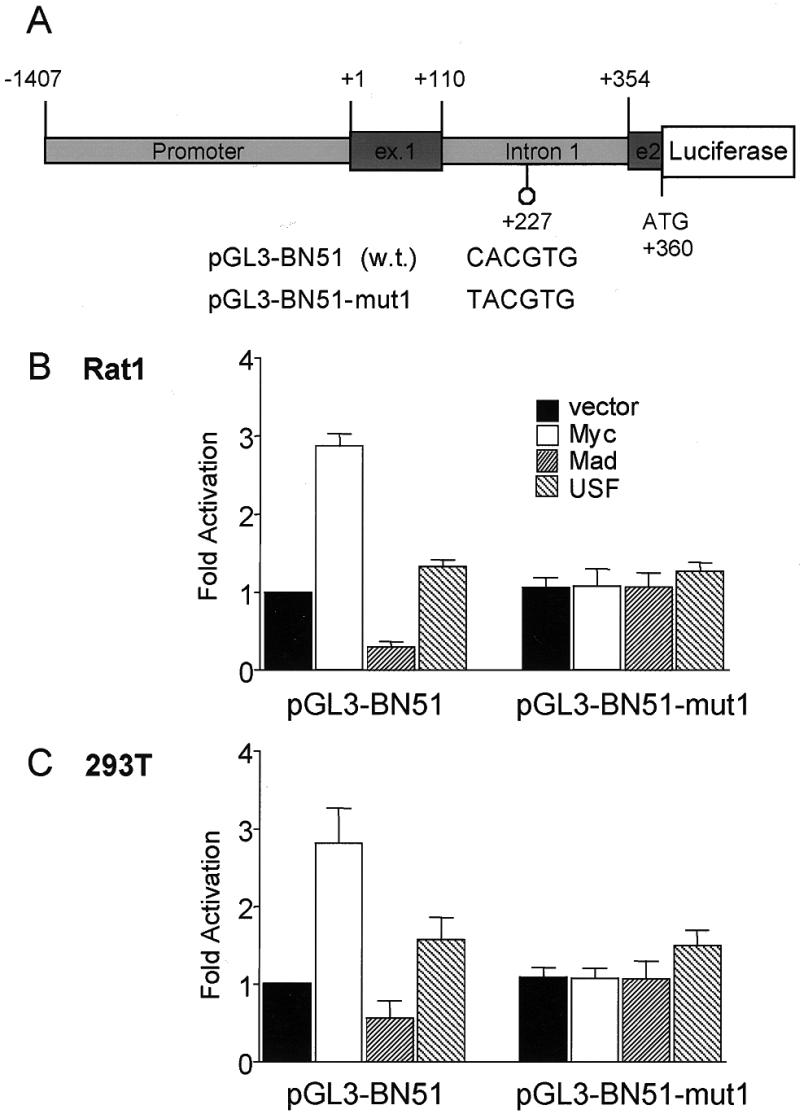
Myc activates the BN51 promoter via a single E-box located in intron 1. (A) Structure of the BN51–reporter gene constructs. The human BN51 promoter, exon 1, intron 1 and the first 6 nt of exon 2 were fused to the 5′-end of the luciferase gene in pGL3-basic (pGL3-BN51). The natural BN51 ATG in exon 2 was fused in-frame to luciferase. The E-box located in intron 1 was mutated to TACGTG (pGL3-BN51-mut). During construction of the reporters, we resequenced the exon 1–intron 1 region of the BN51 gene, revealing several errors in the published sequence (GenBank accession no. L15301) (73). These included the omission of 81 nt at the 5′-end of intron 1 and several nucleotide substitutions. In the published sequence there were two E-boxes, one in intron 1 and one spanning the exon 1–intron 1 boundary. Resequencing demonstrated that the latter E-box does not exist (GenBank accession no. for the corrected sequence AF142779). The reporters were transiently transfected in (B) Rat1 and (C) 293T cells, together with plasmids expressing Myc, Mad or USF, as indicated. Luciferase activities are expressed relative to pGL3-BN51 alone. The data are averaged from three independent experiments.
Transient transfections and reporter gene assays
293T cells were transiently transfected with a standard calcium phosphate DNA precipitation procedure. Plasmid quantities used in the transfections were 10 ng of nucleolin or BN51 reporters driving expression of firefly luciferase (FL), 10 ng of a CMV-driven Renilla-luciferase (RL) construct (Promega) and 0.1 µg of BJ3 derivatives (empty BJ3 vector or BJ3 expressing Myc, Mad1 or USF). Rat1 fibroblasts were transfected with Superfect™ (Qiagen AG) with 0.1 µg of reporter, 0.1 µg of CMV-RL and 1 µg of BJ3 derivatives. All transfections were performed in 12-well tissue culture plates. Transfected cells were incubated in DMEM (Gibco BRL catalog no. 31966-021) with 10% fetal calf serum and harvested 40 h post-transfection. Cells were lysed and luciferase activities were measured with the Dual Luciferase assay (Promega) using a Lumac Biocounter M2500. RL activity was used to normalize FL activity within each sample.
RESULTS
Principle of the screen
Owing to their low level of induction, identification of novel Myc targets by differential screening strategies has been very limited in its success. We therefore developed an alternative approach, aimed at identifying potential targets for which sequence information was already available in the databases. Such a strategy is dependent upon defining the nucleotide sequence preferences of Myc/Max dimers. We have previously identified the extended consensus VCACGTGB (where V = A, C or G and B = T, C or G), as the optimal E-box site for Myc/Max, both in vitro and in vivo (37). This E-box sequence contains a central CpG dinucleotide. CpG pairs throughout mammalian genomes are generally methylated at position N7 of cytosine, except in small regions of the genome (~0.3–1.5 kb) called CpG islands. In vitro Myc/Max dimers fail to recognize the E-box if the central CpG dinucleotide is methylated (38). By inference, most functional E-boxes should be located within CpG islands. In fact, a feature that characterizes several Myc target genes, including ODC, cad and LDH-A, is the location of the E-boxes within CpG islands. Furthermore, CpG islands are characterized by a distinct sequence composition (39; see below) and are generally located at the 5′-end of genes, frequently encompassing the promoter, exon 1 and intron 1. We thus designed a screen based on three main steps: (i) computer-based identification of VCACGTGB motifs located within potential CpG islands among mammalian genomic or cDNA sequences; (ii) large-scale experimental analysis of the computer-selected population to identify candidate Myc-regulated genes; (iii) detailed characterization of individual genes.
Computer-based identification of putative Myc target sequences
We developed a program that analyses the base composition of DNA sequences surrounding E-boxes, permitting the selection of potential CpG islands. The criteria adopted were a G+C content >65% (compare with <50% in bulk genomic DNA) and a ratio of CpG to GpC >0.6 (compare with <0.5 in bulk genomic DNA) (39–41). Our first search (June 1995, used as a basis for this work) resulted in the selection of 977 E-boxes within potential CpG islands from a total of 42 971 E-box-containing sequences identified in the databases (Table 1). This represents 2.27% of the E-box-containing sequences and 0.33% of the total number of sequences analyzed. Included in the selected population were the E-boxes of known Myc target genes such as ODC and cad. Not all E-box-containing CpG islands will correspond to bona fide Myc-regulated genes. In order to identify those that are indeed regulated by Myc in cultured cells, the selected genes were requested from original sources: 316 were obtained and analyzed experimentally as described below.
Table 1. Number of sequences selected and screened by the program cpg.
| Species | Sequences analyzed | Sequences with E-boxes | Sequences with E-boxes in CpG islands |
|---|---|---|---|
| Primate | 36 442 | 12 775 (35%) | 473 (1.29%) |
| Rodent | 24 375 | 11 324 (46%) | 142 (0.58%) |
| Mammalian | 6483 | 2200 (34%) | 130 (2.01%) |
| EST | 224 784 | 12 327 (5.5%) | 124 (0.06%) |
| Vertebrate | 7544 | 4345 (58%) | 108 (1.43%) |
| Total | 299 628 | 42 971(14%) | 977 (0.33%) |
The number of sequences fulfilling the requirement of an E-box located within a potential CpG island was determined on the June 1995 EMBL release. The fraction of each population selected is shown in parentheses.
Experimental screens of candidate Myc target genes
Upon Myc–ER activation, Myc target genes are induced, as are their transcription rates and mRNA levels (8). These two parameters were evaluated for our collection of computer-identified clones by nuclear run-on and reverse northern blotting, respectively (see Materials and Methods). The screen consisted of 316 genes, as well as internal controls for genes previously shown to be induced (ODC, αPT and cdc25a) and not induced (36B4, tubulin, actin and GAPDH) by Myc. Plasmids were replica slot-blotted on duplicate nylon filters and probed in parallel with radiolabeled nuclear run-on RNA or first strand cDNA probes, derived from parallel cultures of Rat1 Myc–ER cells, before and 4 h after activation with OHT. Each signal was normalized to that of the 36B4 gene under the same conditions. Table 2 gives the fold induction for various genes upon Myc–ER activation, as described below. 36B4 encodes acidic ribosomal phosphoprotein P0 and is not modulated by Myc, estradiol, tamoxifen, the Rb–E2F pathway or upon cell cycle entry (42–44; Table 2, row 1). Actin and tubulin were also not induced (Table 2, rows 2 and 3). GAPDH was consistently induced by Myc–ER by a factor of 2.5 and was therefore no longer used as a reference. Of the known Myc target genes that yielded a detectable signal in run-on and/or reverse northern, ODC was induced ~2-fold, αPT to a lesser extent and cdc25A not at all (Table 2, rows 4–6).
Table 2. Run-on and reverse northern analysis identifies a number of potential targets of Myc.
| Gene | Nuclear run-on | Reverse northern | |
|---|---|---|---|
| 1 | 36B4 | 1.00 | l.07 ± 0.19 (n = 8) |
| 2 | Tubulin | n.d. | l.14 ± 0.21 (n = 5) |
| 3 | Actin | n.d. | 1.14 ± 0.28 (n = 8) |
| 4 | ODC | 2.77, 1.l1 | 2.30 ± 1.11 (n = 6) |
| 5 | α-Prothymosin | n.s. | 1.37 ± 0.83 (n = 8) |
| 6 | cdc25a | n.s. | 1.06 ± 0.31 (n = 8) |
| 7 | Nucleolin | 2.56, 2.01 | 2.98 ± 1.86 (n = 7) |
| 8 | BN51 | 2.16 ± 0.78 (n = 4) | 2.53 ± 0.71 (n = 7) |
| 9 | Cyclin D2 | 8.33, 1.91 | 2.09 ± 0.46 (n = 4) |
| 10 | Nucleolar protein p120 | 7.97, 2.41 | 3.27 ± 1.54 (n = 6) |
| 11 | wee I kinase | 1.91, 1.88 | 3.20 ± 1.29 (n = 4) |
| 12 | a05 | 2.42, 0.95 | 1.94 ± 0.87 (n = 7) |
| 13 | HSC70 | 1.34,1.03 | 2.95 ± 0.41 (n = 4) |
| 14 | c02 | n.s. | 1.94 ± 0.54 (n = 4) |
| 15 | f11 | n.s. | 3.66 ± 0.89 (n = 4) |
| 16 | pl(mcm3) | n.s. | 1.96 ± 0.37 (n = 4) |
| 17 | c-met | n.s. | 2.34 ± 0.47 (n = 4) |
| 18 | Topoisomerase I | n.s. | 2.02 ± 0.98 (n = 6) |
| 19 | Nucleolar antigen Ki67 | n.s. | 2.00 ± 0.44 (n = 4) |
Rows 1–3 show the internal controls, rows 4–6 positive controls and rows 7–19 genes identified as potential targets of Myc. Serum-starved Rat1 Myc–ER cells were treated with OHT for 4 h or left untreated prior to analysis of gene expression by run-on or reverse northern blotting. All values were calculated relative to the expression of 36B4. n.d., not determined; n.s., no signal.
Six of the candidate genes showed increases in run-on signals (Table 2, rows 7–13), although the results obtained with this technique were prone to variability. Figure 1A shows an example of run-on analysis for nucleolin and BN51. The genes that had shown no response to Myc–ER in the nuclear run-on also showed no changes by reverse northern. However, those genes that had shown an apparent increase in transcription levels also showed increases in mRNA levels (Table 2, rows 7–13, and Fig. 1B). Furthermore, a number of the genes that gave no signal in nuclear run-on were detectable by reverse northern. Of these, seven were induced upon Myc–ER activation (Table 2, rows 13–19). In summary, 316 clones were tested, 123 gave a detectable signal with run-on and/or reverse northern, 110 were invariant upon Myc–ER activation and 13 were induced in several experiments (Table 2, rows 7–19). We also attempted to see whether the sequences surrounding these potential novel targets allowed, in conjunction with those of already established targets, the definition of a more precise consensus sequence surrounding the E-box. However, no additional similarity could be found beyond the VCACGTGB of our original search.
Figure 1.
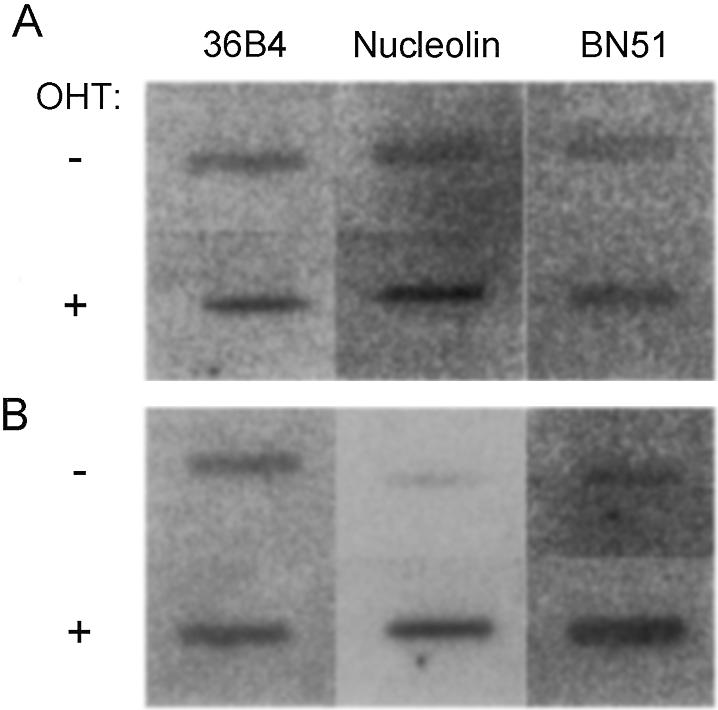
Run-on and reverse northern analysis of nucleolin and BN51 gene expression in Rat1 Myc–ER cells. Serum-starved Rat1 Myc–ER cells were treated with OHT for 4 h (+) or left untreated (–) prior to analysis of gene expression by (A) run-on or (B) reverse northern analysis (see Materials and Methods). The genes analyzed are indicated on top.
At this stage, the 13 genes must still be considered as potential Myc target genes and should be studied in more detail to confirm their response to Myc. In particular: (i) direct and more quantitative analysis of mRNA levels should confirm induction of these genes by Myc–ER with, if possible, induction observed in the presence of the protein synthesis inhibitor cycloheximide, demonstrating that the gene is a direct transcriptional target of Myc; (ii) E-boxes in the regulatory regions of these genes should confer Myc-dependent transcriptional activation in transient transfection assays. Of the 13 genes identified, two, nucleolin and BN51, were studied in further detail and met these criteria.
RNA was prepared from Rat1 Myc–ER cells before and after induction with OHT, and in the presence or absence of cycloheximide, and analyzed by northern blotting. Six hours following Myc–ER activation, nucleolin and BN51 mRNAs were induced ~2-fold (data not shown), consistent with the reverse northern data (Table 2). Although modest, these levels of induction by Myc–ER are similar to those observed for other Myc-induced genes in this system. The effect of cycloheximide on Myc–ER-induced transcription could not be tested, since expression of these genes was induced by cycloheximide alone, a phenomenon that has already been seen for other targets of Myc (45).
Nucleolin and BN51 are induced by Myc in an E-box-dependent manner
Nucleolin genes and/or cDNAs have been cloned from human (46,47), mouse (48), rat (49) and hamster (50), and related proteins have been identified in budding and fission yeasts (51,52). All the mammalian genes contain a first intron that overlaps with a highly conserved CpG island (53). The mouse gene contains five E-boxes spanning 150 bp of intron 1, within the CpG island (Fig. 2A). The first two and the fourth E-boxes are conserved in the human and hamster genes. We generated a reporter construct containing the promoter, exon 1, intron 1 and the first 8 bp of exon 2 of mouse nucleolin driving expression of luciferase (pNucL14, Fig. 2A). The nucleolin ATG in exon 1 was mutated. When transfected into either 293T or Rat1 cells, pNucL14 showed high basal transcriptional activity, comparable to a CMV–luciferase reporter (data not shown). Co-transfection of a Myc expression construct (BJ3–myc3) activated the reporter ~4-fold in Rat1 and 293T cells (Fig. 2B and C). A Myc mutant deficient in DNA binding (MycR392/394/396A; 13) had no effect (data not shown). In contrast to Myc, a construct expressing Mad1 repressed pNucL14 activity (Fig. 2B and C). The reporter was also activated by USF, which also recognizes and transactivates from E-box elements, albeit to a lesser extent.
A deletion mutant of the reporter was made in which the region encompassing the five E-boxes was deleted (pNucL8). This reporter had a decreased basal transcriptional activity (40% of that of the native reporter) and was unresponsive to co-transfection of Myc or Mad expression plasmids (data not shown). To further address which of the E-boxes were responsible for mediating the effects of Myc and Mad we constructed three series of point mutations. pNucL14-mut1 had the first three CACGTG motifs mutated to TACGTG, pNucL14-mut2 had the last two E-boxes mutated and pNucL14-mut1/2 had all five E-boxes mutated. Mutation of CACGTG to TACGTG eliminates all binding and responsiveness to Myc/Max complexes (13,39,54). pNucL14-mut1 had reduced basal activity compared with the wild-type construct and was neither activated by Myc nor repressed by Mad1 (Fig. 2B and C). pNucL14-mut2 had a basal level close to that of the native reporter and was still induced by Myc and repressed by Mad1 (Fig. 2B and C). pNucL14-mut1/2 behaved very similarly to pNucL14mut1. The pNucL14 reporter was also induced by USF. In Rat1 cells, this response was largely dependent upon the first three E-boxes, although a residual response of pNucL14-mut1 and mut1/2 to USF was observed (Fig. 2B). In 293T cells, the response to USF was largely independent of all E-boxes (Fig. 2C). Altogether, our data show that E-boxes 1, 2 and/or 3 of intron 1 are responsible for activation of the nucleolin promoter by Myc, its repression by Mad and are limiting for the basal activity of the promoter. In contrast, E-boxes 4 and 5 play a minimal role, if any. Given that E-box 3 is not conserved in all species (see above), we surmise that E-boxes 1 and 2 are critical for regulation by Myc and Mad1.
The human BN51 gene contains a single E-box within intron 1 (Fig. 3A legend). We constructed a reporter gene (pGL3-BN51) containing the BN51 promoter, exon 1, intron 1 and the first 6 bp of exon 2, allowing the use of the natural BN51 start codon to initiate translation of luciferase. This reporter showed a significant level of basal activity (~20% of CMV–luciferase), was induced by Myc ~3-fold, and repressed by Mad1 in both Rat1 and 293T cells (Fig. 3B and C). Mutation of the E-box (pGL3-BN51mut) abolished the response to both Myc and Mad (Fig. 3B and C). Neither pGL3-BN51wt nor pGL3-BN51mut was significantly induced by USF. Thus, BN51 appears to be a Myc-specific target, as has been previously shown for cad and αPT.
DISCUSSION
We have shown here that the application of a computer-based analysis of DNA databases using restrictive sequence-dependent criteria permits the selection of potential Myc target genes. Furthermore, several criteria establish that the genes for BN51 and nucleolin are bona fide Myc targets: (i) BN51 and nucleolin gene transcription (as assessed by run-on) and mRNA levels (as assessed by reverse northern) are induced upon activation of Myc–ER in Rat1 cells; (ii) Myc activates transcription from the BN51 and nucleolin promoters via one and three (probably two) E-boxes, respectively; (iii) in either case, the E-boxes are located in the first intron and within a CpG island, as defined by the initial conditions of our screen.
Consistent with our identification of BN51 as a Myc target, BN51 transcription is induced after serum stimulation of quiescent fibroblasts, but this induction is abolished by cycloheximide (36). Thus, BN51 is a ‘delayed early’ serum response gene that must be induced by the product of an ‘immediate early’ gene, such as c-myc (reviewed in 2). Nucleolin expression is enhanced in tumors and in proliferating cells, is low in serum-deprived cells, increases upon cell cycle re-entry (55–58) and is induced by interleukin-2 in T cells (59). In human neuroblastoma cells, nucleolin and N-myc expression decrease concomitantly during differentiation (60).
Nucleolin is an abundant nucleolar and multifunctional protein involved in rRNA processing and ribosome assembly (61–65 and references therein). The human BN51 gene was cloned by virtue of its ability to suppress G1 arrest in a temperature-sensitive hamster cell line (tsBN51) (66). BN51 encodes a subunit of RNA polymerase (pol) III homologous to the RPC53 subunit of Saccharomyces cerevisiae RNA pol C (the pol III counterpart in yeast; 67–69). RNA pol III activity is temperature sensitive in extracts of tsBN51 cells, suggesting that loss of BN51/pol III function is directly responsible for G1 arrest (68). Consistent with this notion, tsRPC53 mutants of S.cerevisiae also arrest in G1 at the restrictive temperature (67). It will be interesting to address whether Myc is rate limiting for RNA pol III transcription in vivo. RNA pol III is involved in transcription of the 5S rRNA and tRNAs. While RNA pol I transcribes the 45S precursor of the 28S, 18S and 5.8S rRNAs, the maturation of these rRNAs is defective in tsBN51 cells at the restrictive temperature (69).
Recent data suggest that Myc function is critical not only for cell cycle progression, but also for cell growth, i.e. the accumulation of cell mass in both Drosophila (70) and mammalian cells (discussed in 6). Deletion of both c-myc alleles by homologous recombination in a diploid clone of Rat1 fibroblasts impaired, but surprisingly did not abrogate, cell proliferation (71). These Rat1 myc–/– cells grew and proliferated very slowly, with prolonged G1 and G2 phases and decreased rates of total RNA and protein accumulation (as well as protein degradation), whilst maintaining a normal cell size. This is the phenotype expected if the rates of cell proliferation and cell growth are concomitantly reduced. We speculate that two of the main defects in Rat1 myc–/– cells are decreases in RNA pol III activity and in the production of ribosomes, leading to the observed generalized defects in cellular protein synthesis.
It was recently reported that most known Myc-induced genes, with the exception of cad, were not down-regulated in Rat1 myc–/– cells (72). It should be recalled, however, that those cells were derived from immortalized Rat1 cells and were selected in culture to grow without myc. Thus, Myc-dependent genes that are essential for cell proliferation might have been re-expressed in Rat1 myc–/– cells, owing to genetic or epigenetic alterations that might have occurred either before of after disruption of c-myc. It will be important to address whether BN51, nucleolin or other genes involved in ribosome biogenesis are down-regulated like cad in Rat1 myc–/– cells, accounting for their slow growth phenotype.
A parallel report (20) also showed that c-MycS, an N-terminally truncated form of Myc supposedly defective in transactivation (73), was capable of restoring a normal proliferation rate in Rat1 myc–/– cells and retained some of the transforming and apoptotic functions of c-Myc. It was not checked, however, whether c-MycS up-regulated expression of cad in Rat1 myc–/– cells. The same question may apply to BN51, nucleolin or other Myc-induced genes.
In addition to nucleolin and BN51, other products of Myc-induced genes might be involved either in ribosome biogenesis or in additional steps of protein biosynthesis. The translation factors eIF4E and eIF2α are induced by Myc (74,75). The human gene MrDb and its Drosophila homolog Pitchoune are both Myc targets and encode a Dead-box helicase (76). Proteins of this class function in rRNA maturation and ribosome assembly in S.cerevisiae (77–79). Other genes that contain E-boxes and were induced by Myc–ER in our preliminary run-on or reverse northern screens (Table 2) might function in ribosome biogenesis. These include topoisomerase I (80), which also interacts with nucleolin (81), nucleolar protein p120 (82), ribosomal protein L13 (EST clone c02) and Hsc70. The latter is closely related to Hsp70, which might also be induced by Myc (55,83) and contribute to ribosome assembly (84). Furthermore, some newly identified CpG island-associated E-boxes are within additional genes involved in ribosome biogenesis (our unpublished observations). Finally, it is noteworthy that small nucleolar RNAs (snoRNAs) are processed from the introns of primary RNA pol II transcripts. Among the protein-encoding genes that host snoRNA precursors in their introns, we find Hsc70 (snoRNA U14), RCC1 (U17), nucleolin (U20), elongation factor 2 (U37), ribosomal protein L5 (U21) and several other ribosomal protein genes (e.g. RP S8, U38b and U40) (85–89). Some of these are known Myc targets and/or contain E-boxes in intron 1. By coordinately regulating these numerous genes, Myc may act as a central regulator of ribosome production in cells.
NOTE ADDED IN PROOF
One of the other 11 genes that tested positive, cyclin D2, has also recently been published as a Myc target gene. [C.Bouchard, K.Thieke, A.Maier, R.Saffrich, J.Hanley-Hyde, W.Ansorge, S.Reed, P.Sicinski, J.Bartek and M.Eilers (1999) Direct induction of cycling D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J., 18, 5321–5333.]
Acknowledgments
ACKNOWLEDGEMENTS
We are indebted to the many people who supplied the DNAs used in this project. We would like to thank in particular Dr Michael Ittmann and Dr Henri-Marc Bourbon for the various reagents they supplied relating to BN51 and nucleolin, respectively. Many thanks go to Jaromir Vlach for reconstructing the promoter, exon 1 and intron 1 fragments of the nucleolin gene. We are grateful to all members of the Amati laboratory and colleagues at ISREC for the many fruitful discussions that took place in the course of this work. Our thanks also go to Dr Phil Shaw and Prof. Susanna Cotecchia for helpful comments on the manuscript. We would also like to thank Prof. Susanna Cotecchia for her consideration during the preparation of this manuscript. P.J.G. was supported by fellowships from EMBO and the European Community. B.A. was the recipient of a START fellowship and of a research grant from the Swiss National Science Foundation.
DDBJ/EMBL/GenBank accession no. AF142779
REFERENCES
- 1.Downs K.M., Martin,G.R. and Bishop,J.M. (1989) Genes Dev., 3, 860–869. [DOI] [PubMed] [Google Scholar]
- 2.Marcu K.B., Bossone,S.A. and Patel,A.J. (1992) Annu. Rev. Biochem., 61, 809–860. [DOI] [PubMed] [Google Scholar]
- 3.Henriksson M. and Luscher,B. (1996) Adv. Cancer Res., 68, 109–182. [DOI] [PubMed] [Google Scholar]
- 4.Roussel M.F., Cleveland,J.L., Shurtleff,S.A. and Sherr,C.J. (1991) Nature, 353, 361–363. [DOI] [PubMed] [Google Scholar]
- 5.He T.C., Sparks,A.B., Rago,C., Hermeking,H., Zawel,L., da Costa,L.T., Morin,P.J., Vogelstein,B. and Kinzler,K.W. (1998) Science, 281, 1509–1512. [DOI] [PubMed] [Google Scholar]
- 6.Amati B., Alevizopoulos,K. and Vlach,J. (1998) Front. Biosci., 3, D250–D268. [DOI] [PubMed] [Google Scholar]
- 7.Eilers M., Picard,D., Yamamoto,K.R. and Bishop,J.M. (1989) Nature, 340, 66–68. [DOI] [PubMed] [Google Scholar]
- 8.Eilers M., Schirm,S. and Bishop,J.M. (1991) EMBO J., 10, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Muller D., Bouchard,C., Rudolph,B., Steiner,P., Stuckmann,I., Saffrich,R., Ansorge,W., Huttner,W. and Eilers,M. (1997) Oncogene, 15, 2561–2576. [DOI] [PubMed] [Google Scholar]
- 10.Evan G.I., Wyllie,A.H., Gilbert,C.S., Littlewood,T.D., Land,H., Brooks,M., Waters,C.M., Penn,L.Z. and Hancock,D.C. (1992) Cell, 69, 119–128. [DOI] [PubMed] [Google Scholar]
- 11.Harrington E.A., Bennett,M.R., Fanidi,A. and Evan,G.I. (1994) EMBO J., 13, 3286–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherr C.J. (1998) Genes Dev., 12, 2984–2991. [DOI] [PubMed] [Google Scholar]
- 13.Amati B., Dalton,S., Brooks,M.W., Littlewood,T.D., Evan,G.I. and Land,H. (1992) Nature, 359, 423–426. [DOI] [PubMed] [Google Scholar]
- 14.Kretzner L., Blackwood,E.M. and Eisenman,R.N. (1992) Nature, 359, 426–429. [DOI] [PubMed] [Google Scholar]
- 15.Amati B. and Land,H. (1994) Curr. Opin. Genet. Dev., 4, 102–108. [DOI] [PubMed] [Google Scholar]
- 16.Lee T.C., Li,L., Philipson,L. and Ziff,E.B. (1997) Proc. Natl Acad. Sci. USA, 94, 12886–12891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marhin W.W., Chen,S., Facchini,L.M., Fornace,A.J.,Jr and Penn,L.Z. (1997) Oncogene, 14, 2825–2834. [DOI] [PubMed] [Google Scholar]
- 18.Amundson S.A., Zhan,Q., Penn,L.Z. and Fornace,A.J.,Jr (1998) Oncogene, 17, 2149–2154. [DOI] [PubMed] [Google Scholar]
- 19.Li L.H., Nerlov,C., Prendergast,G., MacGregor,D. and Ziff,E.B. (1994) EMBO J., 13, 4070–4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao Q., Claassen,G., Shi,J., Adachi,S., Sedivy,J. and Hann,S.R. (1998) Genes Dev., 12, 3803–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bouchard C., Staller,P. and Eilers,M. (1998) Trends Cell Biol., 8, 202–206. [DOI] [PubMed] [Google Scholar]
- 22.Facchini L.M. and Penn,L.Z. (1998) FASEB J., 12, 633–651. [PubMed] [Google Scholar]
- 23.Ayer D.E., Kretzner,L. and Eisenman,R.N. (1993) Cell, 72, 211–222. [DOI] [PubMed] [Google Scholar]
- 24.Zervos A.S., Gyuris,J. and Brent,R. (1993) Cell, 72, 223–232. [DOI] [PubMed] [Google Scholar]
- 25.Hurlin P.J., Queva,C., Koskinen,P.J., Steingrimsson,E., Ayer,D.E., Copeland,N.G., Jenkins,N.A. and Eisenman,R.N. (1995) EMBO J., 14, 5646–5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hurlin P., Queva,C. and Eisenman,R. (1997) Genes Dev., 11, 44–58. [DOI] [PubMed] [Google Scholar]
- 27.Meroni G., Reymond,A., Alcalay,M., Borsani,G., Tanigami,A., Tonlorenzi,R., Nigro,C.L., Messali,S., Zollo,M., Ledbetter,D.H., Brent,R., Ballabio,A. and Carrozzo,R. (1997) EMBO J., 16, 2892–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chin L., Schreiber,A.N., Pellicer,I., Chen,K., Lee,H.W., Dudast,M., Cordon,C.C. and DePinho,R.A. (1995) Proc. Natl Acad. Sci. USA, 92, 8488–8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grandori C. and Eisenman,R.N. (1997) Trends Biochem. Sci., 22, 177–181. [DOI] [PubMed] [Google Scholar]
- 30.Dang C.V. (1999) Mol. Cell. Biol., 19, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Packham G., Porter,C.W. and Cleveland,J.L. (1996) Oncogene, 13, 461–469. [PubMed] [Google Scholar]
- 32.Galaktionov K., Lee,A.K., Eckstein,J., Draetta,G., Meckler,J., Loda,M. and Beach,D. (1995) Science, 269, 1575–1577. [DOI] [PubMed] [Google Scholar]
- 33.Bodnar A.G., Ouellette,M., Frolkis,M., Holt,S.E., Chiu,C.P., Morin,G.B., Harley,C.B., Shay,J.W., Lichtsteiner,S. and Wright,W.E. (1998) Science, 279, 349–352. [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez P., Vinuela,J.E., Alvarez-Fernandez,L., Buceta,M., Vidal,A., Dominguez,F. and Gomez-Marquez,J. (1998) Biochem. J., 331, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shim H., Chun,Y.S., Lewis,B.C. and Dang,C.V. (1998) Proc. Natl Acad. Sci. USA, 95, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ittmann M.M. (1994) Cell Growth Differ., 5, 783–788. [PubMed] [Google Scholar]
- 37.Solomon D.L.C., Amati,B. and Land,H. (1993) Nucleic Acids Res., 21, 5372–5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prendergast G.C., Lawe,D. and Ziff,E.B. (1991) Cell, 65, 395–407. [DOI] [PubMed] [Google Scholar]
- 39.Cross S.H. and Bird,A.P. (1995) Curr. Opin. Genet. Dev., 5, 309–314. [DOI] [PubMed] [Google Scholar]
- 40.Cross S.H., Charlton,J.A., Nan,X. and Bird,A.P. (1994) Nature Genet., 6, 236–244. [DOI] [PubMed] [Google Scholar]
- 41.McQueen H.A., Clark,V.H., Bird,A.P., Yerle,M. and Archibald,A.L. (1997) Genome Res., 7, 924–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laborda J. (1991) Nucleic Acids Res., 19, 3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brunner N., Yee,D., Kern,F.G., Spang-Thomsen,M., Lippman,M.E. and Cullen,K.J. (1993) Eur. J. Cancer, 29A, 562–569. [DOI] [PubMed] [Google Scholar]
- 44.Hurford R.J., Cobrinik,D., Lee,M. and Dyson,N. (1997) Genes Dev., 11, 1447–1463. [DOI] [PubMed] [Google Scholar]
- 45.Packham G. and Cleveland,J.L. (1997) Oncogene, 15, 1219–1232. [DOI] [PubMed] [Google Scholar]
- 46.Srivastava M., Fleming,P.J., Pollard,H.B. and Burns,A.L. (1989) FEBS Lett., 250, 99–105. [DOI] [PubMed] [Google Scholar]
- 47.Srivastava M., McBride,O.W., Fleming,P.J., Pollard,H.B. and Burns,A.L. (1990) J. Biol. Chem., 265, 14922–14931. [PubMed] [Google Scholar]
- 48.Bourbon H.M., Lapeyre,B. and Amalric,F. (1988) J. Mol. Biol., 200, 627–638. [DOI] [PubMed] [Google Scholar]
- 49.Bourbon H.M. and Amalric,F. (1990) Gene, 88, 187–196. [DOI] [PubMed] [Google Scholar]
- 50.Lapeyre B., Bourbon,H. and Amalric,F. (1987) Proc. Natl Acad. Sci. USA, 84, 1472–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kondo K. and Inouye,M. (1992) J. Biol. Chem., 267, 16252–16258. [PubMed] [Google Scholar]
- 52.Gulli M.P., Girard,J.P., Zabetakis,D., Lapeyre,B., Melese,T. and Caizergues Ferrer,M. (1995) Nucleic Acids Res., 23, 1912–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bourbon H.M., Prudhomme,M. and Amalric,F. (1988) Gene, 68, 73–84. [DOI] [PubMed] [Google Scholar]
- 54.Littlewood T.D., Amati,B., Land,H. and Evan,G.I. (1992) Oncogene, 7, 1783–1792. [PubMed] [Google Scholar]
- 55.Ohmori H., Murakami,T., Furutani,A., Higashi,K., Hirano,H., Gotoh,S., Kuroiwa,A., Masui,A., Nakamura,T. and Amalric,F. (1990) Exp. Cell Res., 189, 227–232. [DOI] [PubMed] [Google Scholar]
- 56.Derenzini M., Sirri,V., Trere,D. and Ochs,R.L. (1995) Lab. Invest., 73, 497–502. [PubMed] [Google Scholar]
- 57.Mehes G. and Pajor,L. (1995) Cell Prolif., 28, 329–336. [DOI] [PubMed] [Google Scholar]
- 58.Sirri V., Roussel,P., Gendron,M.C. and Hernandez-Verdun,D. (1997) Cytometry, 28, 147–156. [PubMed] [Google Scholar]
- 59.Herblot S., Chastagner,P., Samady,L., Moreau,J.L., Demaison,C., Froussard,P., Liu,X., Bonnet,J. and Theze,J. (1999) J. Immunol., 162, 3280–3288. [PubMed] [Google Scholar]
- 60.Murakami T., Ohmori,H., Gotoh,S., Tsuda,T., Ohya,R., Akiya,S. and Higashi,K. (1991) J. Biochem. Tokyo, 110, 146–150. [DOI] [PubMed] [Google Scholar]
- 61.Bouvet P., Diaz,J.J., Kindbeiter,K., Madjar,J.J. and Amalric,F. (1998) J. Biol. Chem., 273, 19025–19029. [DOI] [PubMed] [Google Scholar]
- 62.Ginisty H., Amalric,F. and Bouvet,P. (1998) EMBO J., 17, 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tuteja R. and Tuteja,N. (1998) Crit. Rev. Biochem. Mol. Biol., 33, 407–436. [DOI] [PubMed] [Google Scholar]
- 64.Ginisty H., Sicard,H., Roger,B. and Bouvet,P. (1999) J. Cell Sci., 112, 761–772. [DOI] [PubMed] [Google Scholar]
- 65.Pinol-Roma S. (1999) Mol. Biol. Cell, 10, 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ittmann M., Greco,A. and Basilico,C. (1987) Mol. Cell. Biol., 7, 3386–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mann C., Micouin,J.Y., Chiannilkulchai,N., Treich,I., Buhler,J.M. and Sentenac,A. (1992) Mol. Cell. Biol., 12, 4314–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ittmann M., Ali,J., Greco,A. and Basilico,C. (1993) Cell Growth Differ., 4, 503–511. [PubMed] [Google Scholar]
- 69.Jackson A.J., Ittmann,M. and Pugh,B.F. (1995) Mol. Cell. Biol., 15, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Johnston L.A., Prober,D.A., Edgar,B.A., Eisenman,R.N. and Gallant,P. (1999) Cell, 98, 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mateyak M.K., Obaya,A.J., Adachi,S. and Sedivy,J.M. (1997) Cell Growth Differ., 8, 1039–1048. [PubMed] [Google Scholar]
- 72.Bush A., Mateyak,M., Dugan,K., Obaya,A., Adachi,S., Sedivy,J. and Cole,M. (1998) Genes Dev., 12, 3797–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Spotts G.D., Patel,S.V., Xiao,Q. and Hann,S.R. (1997) Mol. Cell. Biol., 17, 1459–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rosenwald I.B., Rhoads,D.B., Callanan,L.D., Isselbacher,K.J. and Schmidt,E.V. (1993) Proc. Natl Acad. Sci. USA, 90, 6175–6178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jones R.M., Branda,J., Johnston,K.A., Polymenis,M., Gadd,M., Rustgi,A., Callanan,L. and Schmidt,E.V. (1996) Mol. Cell. Biol., 16, 4754–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zaffran S., Chartier,A., Gallant,P., Astier,M., Arquier,N., Doherty,D., Gratecos,D. and Semeriva,M. (1998) Development, 125, 3571–3584. [DOI] [PubMed] [Google Scholar]
- 77.Daugeron M.C. and Linder,P. (1998) RNA, 4, 566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.de la Cruz J., Kressler,D., Tollervey,D. and Linder,P. (1998) EMBO J., 17, 1128–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kressler D., de la Cruz,J., Rojo,M. and Linder,P. (1998) Mol. Cell. Biol., 18, 1855–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Muller M.T., Pfund,W.P., Mehta,V.B. and Trask,D.K. (1985) EMBO J., 4, 1237–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bharti A.K., Olson,M.O., Kufe,D.W. and Rubin,E.H. (1996) J. Biol. Chem., 271, 1993–1997. [DOI] [PubMed] [Google Scholar]
- 82.Gustafson W.C., Taylor,C.W., Valdez,B.C., Henning,D., Phippard,A., Ren,Y., Busch,H. and Durban,E. (1998) Biochem. J., 331, 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lin H., Head,M., Blank,M., Han,L., Jin,M. and Goodman,R. (1998) J. Cell. Biochem., 69, 181–188. [DOI] [PubMed] [Google Scholar]
- 84.Pelham H.R.B. (1984) EMBO J., 3, 3095–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kiss T. and Filipowicz,W. (1993) EMBO J., 12, 2913–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Barbhaiya H., Leverette,R.D., Liu,J. and Maxwell,E.S. (1994) Eur. J. Biochem., 226, 765–771. [DOI] [PubMed] [Google Scholar]
- 87.Nicoloso M., Caizergues-Ferrer,M., Michot,B., Azum,M.C. and Bachellerie,J.P. (1994) Mol. Cell. Biol., 14, 5766–5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nicoloso M., Qu,L.H., Michot,B. and Bachellerie,J.P. (1996) J. Mol. Biol., 260, 178–195. [DOI] [PubMed] [Google Scholar]
- 89.Qu L.H., Nicoloso,M., Michot,B., Azum,M.C., Caizergues-Ferrer,M., Renalier,M.H. and Bachellerie,J.P. (1994) Nucleic Acids Res., 22, 4073–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]