


Abstract
Initiation of DNA replication occurs with high frequency within oriβ, a short region 3′ to the Chinese hamster dhfr gene. Homodimers of RIP60 (replication initiation-region protein 60 kDA) purified from nuclear extract bind two ATT-rich sites in oriβ and foster the formation of a twisted 720 bp DNA loop in vitro. Using a one hybrid screen in yeast, we have cloned the cDNA for human RIP60. RIP60 contains 15 C2H2 zinc finger (ZF) DNA binding motifs organized in three clusters, termed hand Z1 (ZFs 1–5), hand Z2 (ZFs 6–8) and hand Z3 (ZFs 9–15). A proline-rich region is located between hands Z2 and Z3. Gel mobility shift and DNase I footprinting experiments show hands Z1 and Z2 independently bind the oriβ RIP60 sites specifically, but with different affinities. Hand Z3 binds DNA, but displays no specificity for RIP60 sites. Ligation enhancement, DNase I footprinting, and atomic force microscopy assays show that hand Z2 and a portion of the associated proline-rich region is sufficient for protein multimerization on DNA and DNA looping in vitro. Polyomavirus origin-dependent plasmid replication assays show RIP60 has weak replication enhancer activity, suggesting that RIP60 does not harbor a transcriptional transactivation domain. Because vertebrate origins of replication have no known consensus sequence, we suggest that sequence-specific DNA binding proteins such as RIP60 may act as accessory factors in origin identification prior to the assembly of pre-initiation complexes.
INTRODUCTION
In the yeast Saccharomyces cerevisiae, a pathway for the assembly of replication initiation complexes at chromosomal origins of replication (or ARS elements) has been described (reviewed in 1–3). The origin recognition complex (ORC) binds in a sequence-specific and ATP-dependent manner to the conserved ARS-consensus sequence (ACS) throughout the cell cycle, providing a protein–DNA complex that recruits replication proteins such as Cdc6p, Mcm proteins and Cdc45p to a pre-replication complex (pre-RC) in a cell cycle-specific manner. During S phase entry, the pre-RC is activated by at least two cell cycle-regulated kinases (4,5). While these and other proteins are known to be involved in the assembly and activation of replication complexes in S.cerevisiae, the mechanism by which replication forks are established by ORC and its associated factors is not known.
Homologs of ORC, Cdc6 and Mcm proteins have been identified in other yeasts, plants and vertebrate organisms, suggesting the pathway for assembly of replication complexes is highly conserved in eukaryotes (1). However, significant differences between origin structure in S.cerevisiae and other organisms suggest that additional steps in regulation of the initiation pathway have emerged during evolution. For example, binding of Schizosaccharomyces pombe ORC to origin DNA is mediated by AT-hooks fused to ORC4p (6). In Xenopus egg extract replication of DNA molecules requires ORC (7,8), Cdc6 (9) and Mcm proteins (10), but not specific ACS-like DNA sequences (11–13). Moreover, in contrast to S.cerevisiae, Xenopus ORC does not appear to be bound to mitotic chromatin in cycling egg extracts (7). Comparison of the properties of CDC6 homologs in S.cerevisiae, S.pombe and human cells also indicates significant differences occur in the down-regulation of initiation pathways after S phase entry (14–16).
In mammalian cells, lack of a definitive genetic assay has precluded identification of the cis-acting elements required for replicator function (reviewed in 17). Comparison of initiation sites mapped by biochemical and physical methods has failed to identify an ACS-like sequence, nor has vertebrate ORC been reported to bind DNA in a sequence-specific manner. Nonetheless, numerous studies show replication initiates at preferred sites in mammalian chromosomes (reviewed in 18), and recent work demonstrates that these preferred sites are used in multiple cell cycles (19). Examination of origin selection during the cell cycle indicates that specific initiation sites are identified after mitosis during early G1 by factors that are subject to regulation by cell cycle-dependent kinases (20,21). Elements of nuclear structure, including attachment to the nuclear matrix, have been proposed to provide the critical determinants that lead to selection of specific initiation sites (22). Based on these and other studies, we have proposed that transcription factors and other DNA binding proteins expressed upon exit from mitosis act upstream of ORC to identify those sites in mammalian chromatin where initiation complexes are assembled (23).
To address this possibility we have searched for proteins that interact with oriβ, a short region located 17 kb downstream from the 3′ end of the Chinese hamster dhfr gene (24). Biochemical and physical mapping studies, including those using 2D gel analysis of replication intermediates, have shown initiation of DNA replication begins with high frequency at oriβ (reviewed in 18). We have sequenced oriβ (25) and characterized it with regard to structural features such as bent DNA (26), an unusual GC-rich Z-triplex motif (27,28) and DNA unwinding elements (25). Protein binding studies in vitro show oriβ contains binding sites for Oct1, AP-1 and RIP60 (replication initiation-region protein 60 kDa) (29,30). In addition to inducing DNA bending (26), dimers of RIP60 purified from HeLa cell nuclear extract bind to two inverted ATT-rich sites within oriβ and dimer–dimer interactions mediate the formation of a 720 bp twisted DNA loop in vitro (31).
Because DNA looping has been implicated in the regulation of initiation of DNA replication in both plasmids and viruses (32–34), we have cloned the cDNA for RIP60 and characterized its DNA binding and DNA looping activities. RIP60 contains three clusters of C2H2 zinc finger (ZF) motifs, two of which bind RIP60 sites specifically in vitro. Here we show that the central cluster (hand Z2) and a portion of an associated proline-rich protein multimerization domain are sufficient for both high affinity DNA binding and DNA looping by RIP60. Using activation of the polyomavirus origin of replication as a functional assay, we also show that RIP60 is not a replication enhancer akin to upstream transcription factors.
MATERIALS AND METHODS
Oligonucleotide primers
The following oligonucleotides were synthesized and purified by a combination of denaturing gel electrophoresis and gel elution:
p512 (5′-GGCAGATCTGGCCTGTCTGTGAAT-3′);
p520 (5′-CCTCTAGATCTGTTCTATATCAGATTG-3′);
p521 (5′-GGTCCGGATCCCTAGTTTTGATGAGGG-3′);
OCH7 (5′-GATCTTTTATTATTATTATTAGTTCG-3′);
OCH8 (5′-GATCCGAACTAATAATAATAATAAAA-3′);
OCH13 (5′–GATCCGGGAAGGCGGGCGCTGGGGGCGCTGC-GGCGCTGCGCTCCACCT-3′);
OCH14 (5′-GATCTAGGTGGAGCGCAGCGCCGCAGCGCCCC-CAGCGCCCGCCTTCCCG-3′);
OCH35 (5′-GCATAATAAAAAAAATTAGT-3′);
OCH36 (5′-ACTAATTTTTTTTATTTATGC-3′);
OCH37 (5′-CTGTTTTTTTTAGTATTAAGC-3′);
OCH38 (5′-GCTTAATACTAAAAAAAACAG-3′);
OCH39 (5′-GATCTTAACAGTAATAATAAATATCT-3′);
OCH40 (5′-GATCAGATATTTATTATTACTGTTAA-3′);
RIP1 (5′-GACCCGGGATCCATGCTGGAACGTCGTTGCAGG-3′);
RIP2 (5′-GACCCGGGATCCGGGAGATGCCGTCGACCGC-3′);
RIP3 (5′-GACCCGGGATCCGTACCTCTGAAACCGGCCCAG-3′);
RIP4 (5′-GACCCGGGATCCTCAGACATCGTGCTTCTTCTG-3′);
RIP5 (5′-GACCCGGGATCCTGGGCCGGTTCCAGAGGTACC-3′);
RIP6 (5′-GACCCGGGATCCGCGGTCGACGGCATCTCCACC-3′);
RIP7 (5′-GACCCGGGATCCGAATTCTCGCTTGTGAATCTTGCT-GTGAGACAGC-3′);
RIP8 (5′-GACCCGGGATCCTCCGAGGGGTCGGCCCAGGCG-3′);
RIP9 (5′-GACCCGGGATCCGAGGGAGGGGGGGGCTTCGA-TCGG-3′);
RIP10 (5′-GACCCGGGATCCGAATTCTACAGCTGCGACGAC-TGCGGCAGGAGC-3′).
Construction of plasmids and strains for the one hybrid screen in yeast
Plasmid pBM2389 contains a histidine reporter gene regulated by an enhancerless PGAL1-UAS promoter (35). pBM2389 contains a TRP1 selectable marker and CEN/ARS sequences for plasmid maintenance. Plasmid pJL638 contains a lacZ reporter gene that also is regulated by an enhancerless PGal1-uasA promoter (36). pJL638 contains a URA3 selectable marker but lacks a yeast origin to facilitate chromosomal integration. The pACT expression library contains random human B-cell cDNAs fused to sequences encoding the GAL4 activation domain under the control of a constitutive ADH promoter (37). The vector pACT contains a selectable LEU2 marker and the 2µ origin of replication. Two oligonucleotides representing the downstream RIP60-binding site (OCH7 and OCH8) were annealed, multimerized by ligation, and cloned into the BamHI site located upstream of the HIS3 reporter gene in pBM2389. Clone pCH14 contains eight repeats of OCH7/OCH8 at the BamHI site of pBM2389. Five tandem repeats of OCH7/OCH8 (5x-DSR) were removed from pCH14 as a BamHI/BglII fragment and inserted into the BamHI site of pBM2389 to generate pCH25. The TRP1 marker gene was removed from pCH25 as a NcoI fragment and replaced with the ADE2 marker from pADE2 (provided by J. Kurjan) to generate pCH47. The 5x-DSR fragment was cloned into the BglII site located upstream of the lacZ reporter gene in pJL638 to generate pCH33. Fragment E, which contains the downstream RIP60 binding site and surrounding bent DNA sequences (nucleotides 3382–3536 in ref. 25), was amplified by PCR and cloned into the BamHI and BglII sites of pUC19 to generate pUC/E (provided by K. Murakami). Fragment E was removed from pUC/E as a BamHI/BglII fragment and inserted into the BglII site of pJL638 to generate pCH36. pJL638, pCH33 and pCH36 were each linearized at the StuI site located in the URA3 marker and integrated at the URA3 gene in yeast strain GGY1 (MATα Δgal80 Δura3 leu2 his3 ade2 tyr) by homologous recombination to generate yeast strains YCH3, YCH4 and YCH5, respectively. Integration of the lacZ reporter plasmids were confirmed by Southern blot analysis. YCH4 was transformed with pCH47 to generate the yeast one hybrid reporter strain, YCH4/pCH47.
One hybrid screen for RIP60
YEPD (rich media) and SD (synthetic dropout media) were prepared as described (38). High efficiency yeast transformations were performed by the method of Scheistel and Geitz (39). YCH4/pCH47 was transformed with B-cell pACT library DNA and transformants were selected on SD plates lacking histidine, adenine and leucine. Histidine prototrophs from the transformation (1.9 × 107 total) were assayed for β-galactosidase production (40) on Protran nitrocellulose filters (Schleicher & Schuell). Transformants which tested positive for lacZ expression were isolated on SD plates lacking leucine and re-tested for β-galactosidase production. pACT plasmids were isolated from lacZ-positive transformants and purified through bacterial transformation and plasmid isolation. The purified pACT plasmids were retested in YCH4 for β-galactosidase production and assayed for target sequence specificity in YCH3 and YCH5. Isolated plasmids that tested positive for lacZ expression in YCH4 and/or YCH5, but not YCH3, were sequenced and analyzed in further detail. Clone 146A-1, isolated in the one hybrid screen with YCH4/pCH47, was sequenced on both strands with custom oligonucleotide primers.
Isolation of full-length RIP60 cDNA
To isolate full-length clones that encompass the 146A-1 cDNA, XL1-Blue bacterial cells (Stratagene) were infected with a HeLa cDNA library constructed in Lamda Zap (Stratagene) and viral plaques were transferred to Hybond N+ nylon membranes (Amersham). The filters were probed with a random-primed (Life Technologies) radiolabeled probe from the 143A-1 cDNA, which overlaps clone 146A-1. Positive plaques were purified in a secondary screen and pBluescript plasmids excised from isolated Lamda Zap viral particles were sequenced with M13/pUC forward and reverse primers. Of several positive full-length clones, pBS-27 was sequenced on both strands (GenBank accession no. AF201303).
Construction of expression plasmids for GST-, HA- and GFP-tagged fusion proteins
pGEX-2T and pGEX-5X-1 (Pharmacia Biotech) were used for expressing GST-tagged fusion proteins, pCMV-HA (41) for expressing HA-tagged fusion proteins, and pK7-GFP (provided by I. Macara) for expressing GFP-tagged fusion proteins. The following RIP60 cDNA fragments were amplified by PCR with the indicated primer sets using pBS-27 as a template: Z123 (RIP1/RIP4, nucleotides 1–1704), Z12 (RIP1/RIP5, nucleotides 1–1058), Z1 (RIP1/RIP6, nucleotides 1–702), Z23 (RIP2/RIP4, nucleotides 682–1704), Z3 (RIP3/RIP4, nucleotides 1039–1704), Z2 (RIP2/RIP5, nucleotides 682–1058), Z12P (RIP1/RIP9, nucleotides 1–1122), Z12ΔP (RIP1/RIP7, nucleotides 1–948), Z2P (RIP2/RIP9, nucleotides 682–1122), Z2ΔP (RIP2/RIP7, nucleotides 682–948), PZ3 (RIP8/RIP4, nucleotides 949–1704) and ΔPZ3 (RIP10/RIP4, nucleotides 1123–1704). The following PCR products were digested with BamHI and inserted in frame into the BamHI site of pGEX-2T to generate the indicated plasmids for the purification of GST fusion proteins: Z123 (pCH59), Z23 (pCH61), Z3 (pCH126), Z12 (pCH65), Z2 (pCH67) and Z1 (pCH69). The following PCR products were digested with BamHI and inserted in frame into the BamHI site of pCMV-HA to generate the indicated plasmids for the expression of HA-tagged fusion proteins: Z123 (pCH71), Z23 (pCH73), Z3 (pCH75), Z12 (pCH76), Z2 (pCH78), Z1 (pCH79), Z12ΔP (pCH89), Z12P (pCH91), Z2ΔP (pCH93), Z2P (pCH94), ΔPZ3 (pCH96) and PZ3 (pCH108). The following PCR products were digested with BamHI and inserted in frame into the BamHI site of pK7-GFP to generate the indicated plasmids for the expression of GFP fusion proteins: Z12 (pCH83), Z1 (pCH84), Z2 (pCH85), Z123 (pCH86) and Z23 (pCH88).
Purification of GST-tagged RIP60 fusion proteins
BL21 bacterial cultures (400 ml) were grown in LB media containing ampicillin (100 µg/ml) in the presence of 50 µM ZnCl2 to 1.0 OD600 and 1 mM IPTG was then added for 5 h at 37°C. Cells were pelleted by centrifugation, resuspended in 10 ml lysis buffer (1× PBS, 10 mM β-mercaptoethanol, 1 mM PMSF, 25 µg/ml aprotinin, 25 µg/ml leupeptin, 1 mg/ml lysozyme), and incubated on ice for 20 min. Cells were lysed by sonication on ice, Triton X-100 was added to 1%, and the cell lysate was cleared by centrifugation before addition of glutathione sepharose beads (Pharmacia Biotech) for 2 h on ice with rocking. The beads were washed several times with ice-cold PBS with 1% Triton X-100 and then PBS before elution with 10 serial additions of glutathione elution buffer [20 mM reduced glutathione, 100 mM Tris–Cl (pH 8.0), 120 mM NaCl, 0.1% Triton X-100]. Protein fractions were flash frozen in liquid nitrogen and stored at –80°C. Protein concentrations were estimated by comparison to known protein standards on Coomassie Blue stained protein gels.
Electrophoretic gel mobility shift assays
The following oligonucleotides were annealed (10 pmol/µl final concentration) in the presence of 1× STE buffer [100 mM NaCl, 10 mM Tris–Cl (pH 8.0), 1 mM EDTA] to generate the indicated double-stranded probes: OCH7/OCH8 (DSR), OCH13/OCH14 (IBF), OCH35/OCH36 (SV-AT), OCH37/OCH38 (Py-AT) and OCH39/OCH40 (USR). DSR (50 pmol) was labeled by Klenow fill-in reaction of 5′ overhangs in the presence of [α-32P]dATP; other probes were end-labeled with polynucleotide kinase and [γ-32P] binding reactions (30 µl) contained 0.05 pmol labeled DSR probe, 40 mM KCl, 10 mM HEPES (pH 8.0), 4% Ficoll, 33 ng/µl poly-dIdC, 1 mM DTT, 16.7 µM ZnCl2 and 0.5–10 µg of protein. Binding reactions were incubated for 20 min at 37°C, 15 min on ice, and then resolved on 6% native polyacrylamide gels in 0.25× TBE buffer. Gels were dried, exposed to Kodak XAR film at –80°C, and signals were detected by autoradiography. Competition binding reactions were assembled as described above with the addition of 0.5, 5 or 50 pmol of unlabeled double-stranded competitor DNA to the reactions prior to the addition of protein. For time course competition assays, 60 µl binding reactions were assembled and incubated as above. Following the incubation step on ice, 50 pmol of unlabeled DSR competitor DNA was added to the binding reactions and samples where removed at the indicated time points, loaded on a 6% native PAGE gel, and resolved by electrophoresis at 25 V until all samples were loaded. After the final sample was loaded, the voltage was increased to 100 V and samples were resolved by electrophoresis for an additional 1.5 h. Signals on the dried gels were quantified on a phosphoimager (BioRad model GS 525). A value for percentage probe bound was calculated as a ratio of protein-bound probe to total probe (bound and unbound probe) per sample and values were plotted against time.
In vitro DNase I footprinting
Probe DHFRE-top was generated by digesting pUC/E with BamHI, end-labeling by Klenow fill-in in the presence of [α-32P]dATP, and digesting with BglII. Probe DHFR-E-bottom was generated by digesting pUC/E with BglII, end-labeling by Klenow fill-in in the presence of [α-32P]dATP, and digesting with BamHI. Footprinting probes were purified by gel electrophoresis and Geneclean (Bio 101), and resuspended in distilled water at a specific activity of 50 000 d.p.m./µl. Binding reactions (50 µl) that contained 50 000 d.p.m. of labeled probe were assembled as for gel mobility shift assays. After incubation at 37°C for 20 min and on ice for 15 min, 1 vol of a 5 mM CaCl2, 10 mM MgCl2 solution was added for 1 min at room temperature, followed by the addition of 0.02 U of DNase I for 1 min. Footprinting reactions were stopped with the addition of 90 µl prewarmed stop buffer (200 mM NaCl, 30 mM EDTA, 1% SDS), extracted with phenol, ethanol precipitated, and resuspended in sequencing dye buffer. Samples were resolved by electrophoresis at 65 W for 1.5–3.0 h on denaturing 8% polyacryamide gels. The gel was dried, exposed to Kodak XAR film at –80°C, and signals were detected by autoradiography.
Transfection of mammalian cells
Mouse NIH 3T3 cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% calf serum. CHOC 400 and HeLa cells were maintained in DMEM media supplemented with 5% fetal bovine serum. CHO K1 cells were maintained in F12 media supplemented with 5% fetal bovine serum. Cells were at 50% density and incubated for 24 h prior to transfection by calcium phosphate coprecipitation as described previously (42).
Polyoma virus origin-dependent replication assays
The following reporter plasmids were used in replication assays: pPyOICAT is a reporter plasmid that contains the polyomavirus core origin of replication but lacks the enhancer region (43); pPy(AM)6OICAT contains six AP-1 binding sites in the enhancer region of pPyOICAT (44); pBOS-LT is a polyomavirus large T-antigen expression (44). Plasmids pRSV-FOS and pRSV-JUN are mammalian expression plasmids for the c-Fos and c-Jun proteins. Fragment E was removed from pUC/E as a BamHI/BglII fragment and inserted into the BglII site of pPyOICAT to generate pPy(DHFR-E)OICAT. 5x-DSR was removed from pCH14 as a BamHI/BglII fragment and inserted into the BglII site of pPyOICAT to generate pCH30. Unmethylated pUC19 was isolated and purifed from pUC19 transformed dam– Escherichia coli cells. For replication assays, NIH 3T3 cells were transfected by calcium phosphate coprecipitation with DNA samples that contained reporter plasmid DNA (500 ng), pEF-BOS-LT (4 ug), effector plasmid DNA (4 µg), unmethylated pUC19 (200 ng) and sheared salmon sperm DNA (12.7 µg total DNA). Forty-eight hours after transfection, cells were scraped into 15 ml polypropylene tubes, pelleted by centrifugation, and washed twice with PBS. Cells were resuspended in PBS and a sample of the cell suspension was removed for protein analysis. The cell samples for protein analysis were pelleted by centrifugation, resuspended in SDS gel-loading buffer, boiled for 10 min, and assayed for protein expression by western blot analysis using anti-HA monoclonal antibody 12CA5.
The remainder of the cell samples for DNA analysis were pelleted by centrifugation, and lysed by pipetting in the presence of 500 µl HIRT neutral lysis buffer [0.5% SDS, 100 mM NaCl, 5 mM EDTA (pH 8.0), 10 mM Tris–Cl (pH 8.0) (45)]. The samples stored overnight at 4°C, the precipitate then was pelleted by centrifugation for 30 min at 14 000 r.p.m. at 4°C, and the soluble DNA in the supernatant was extracted with phenol and chloroform, precipitated with ethanol, and resuspended in 60 µl distilled water. DNA samples (20 µl) were digested for 2 h with EcoRI and DpnI and resolved by electrophoresis on a 1% agarose in 1× TBE. DNA was transferred to a nitrocellulose membrane by Southern blotting. The Southern blots were probed with a random-primed radiolabeled pPyOICAT probe specific for the reporter plasmid and pUC19, washed and exposed to Kodak XAR film. Probe-specific signals were quantified by phosphoimager analysis and a replication efficiency value was determined from two independent transfections as the ratio of signal from replicated reporter DNA relative to pUC19 DNA.
Preparation of mammalian cell nuclear extracts
Nuclear extracts were prepared as described (46). Briefly, cells on 100 mM dishes were washed twice with ice-cold PBS and buffer A [10 mM HEPES (pH 8.0), 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA (pH 8.0), 1 mM DTT, 0.2 mM sodium vanadate, 0.4 mM PMSF, 0.3 µg/ml leupeptin, 0.2 mM NaF] was added. The cells were scraped with buffer into microfuge tubes and incubated on ice for 15 min. Buffer B (10% Nonidet P-40) was added, the cell lysate was vortexed for 15 s, and then centrifuged for 30 s at 14 000 r.p.m. in a microfuge at 4°C. The supernatant (cytoplasmic fraction) was discarded and the intact nuclei pellet was resuspended in an ice-cold mixture of 800 µl buffer A and 50 µl buffer B. The nuclear suspension was vortexed for 15 s, centrifuged for 30 s at 14 000 r.p.m. in a microfuge at 4°C, and the supernatant was removed. The nuclear pellet was resuspended in 100 µl ice-cold buffer C [50 mM HEPES (pH 7.8), 50 mM KCl, 300 mM NaCl, 0.1 M EDTA (pH 8.0), 10% glycerol, 1 mM DTT, 0.2 mM sodium vanandate, 0.667 mM PMSF, 0.2 mM NaF], mixed gently at 4°C for 20 min, and centrifuged for 5 min at 14 000 r.p.m. in a microfuge at 4°C. The supernatant (nuclear extract) was transferred to a new microfuge tube, flash frozen in liquid nitrogen, and stored at –80°C.
Ligation enhancement assays
Ligation enhancement assays were performed as described (47), with some modifications. Primers p512 and p521 were used to amplify a 1118 bp fragment of dhfr oriβ from pMC-D (nucleotides 2434–3536, ref. 25). The oriβ fragment was cut with BamHI and BglII and cloned into the BglII site of pPyOICAT to generate pCH127. pCH127 was linearized at the AccI site and purified to generate the pCH127(AccI) substrate. Where indicated, pCH127(AccI) was dephosphorylated by treatment with calf intestine alkaline phosphatase (CIP). Binding reactions (50 µl) were assembled that contained 100 ng pCH127(AccI), 2 mM DTT, 2 mM ATP, 20 µM ZnCl2, 20 mM MgCl2, 40 mM KCl, 10 mM HEPES (pH 8.0), 4% Ficoll and 100 ng GST-fusion protein (where indicated, 5.0 pmol of competitor DNA was added prior to the addition of protein). Binding reactions were incubated at room temperature for 15 min and diluted with the addition of 50 µl distilled water. One Weiss unit of T4 DNA ligase was added and reactions were incubated for 5 min at 15°C. An equal volume of PK buffer [10 mM Tris–Cl (pH 7.8), 5 mM EDTA, 0.5% SDS] containing 20 µg Proteinase K was then added to the samples and protein was digested for 1 h at 37°C. Samples were extracted with phenol, ethanol precipitated, resuspended in 30 µl distilled water, resolved on 1% agarose gels in TAE buffer, transferred to nitrocellulose, and probed with radiolabeled pPyOICAT. Blots were washed and exposed to Kodak XAR film for 1–10 min at room temperature.
Atomic force microscopy (AFM)
AFM studies were performed using a Nanoscope III AFM (Digital Instruments) equipped with a Plexiglass tapping-mode fluid cell. The microscope was operated in fluid tapping mode using cantilever frequencies between 12 and 24 kHz. Triangular silicon nitride cantilevers (100 µm) with oxide sharpened oriented twin-tips having a normal spring constant of 0.1 N/m were used. Linear and supercoiled DNA was diluted to 100 ng/µl in TE. Binding reactions (5 µl) were assembled that contained 1 µl template DNA, 1 µl GST-RIP60 fusion protein (100 ng/µl), and 2 µl holding buffer [20 mM Tris–Cl (pH 8), 50 mM KCl, 5 mM MgCl2, 1 mM β-mercaptoethanol] then incubated for 20 min at room temperature. Binding reactions were diluted (1:16) in deposition buffer (20 mM Tris–HCl, pH 7.5, 5 mM KCl, 5 mM MgCl2, 1 mM β-mercaptoethanol, 2 mM ZnCl2) and 7 µl was deposited on freshly cleaved mica chips. Samples were then imaged by AFM while still wet.
RESULTS
RIP60 is a polydactyl zinc finger protein
RIP60 was originally purified from HeLa cell nuclear extract as a DNA-binding activity that specifically recognized an ATT-rich sequence located within the Chinese hamster cell dhfr origin of replication, oriβ (30). Sequence analysis of tryptic RIP60 fragments identified two amino acid sequences, VAEALEEAAAK and NLVSHRRIHTGERPYA, the second of which is similar to a Kruppel C2H2-type zinc finger (ZF) DNA-binding motif (L.Dailey and L.Zeltser, unpublished results). Repeated attempts to clone the cDNA for RIP60 by PCR based on the peptide sequences failed. Because scanning transmission electron microscopy indicated RIP60 binds DNA as a homodimer (31), we reasoned a one hybrid screen in yeast might provide the specificity necessary to clone the RIP60 cDNA. The screen in yeast was devised to identify proteins that bind the RIP60 DSR target sequence and activate expression of a linked reporter gene (lacZ or histidine) by recruiting a fused GAL4 transcriptional activation domain (GAD) to the promoter (Fig. 1A).
Figure 1.
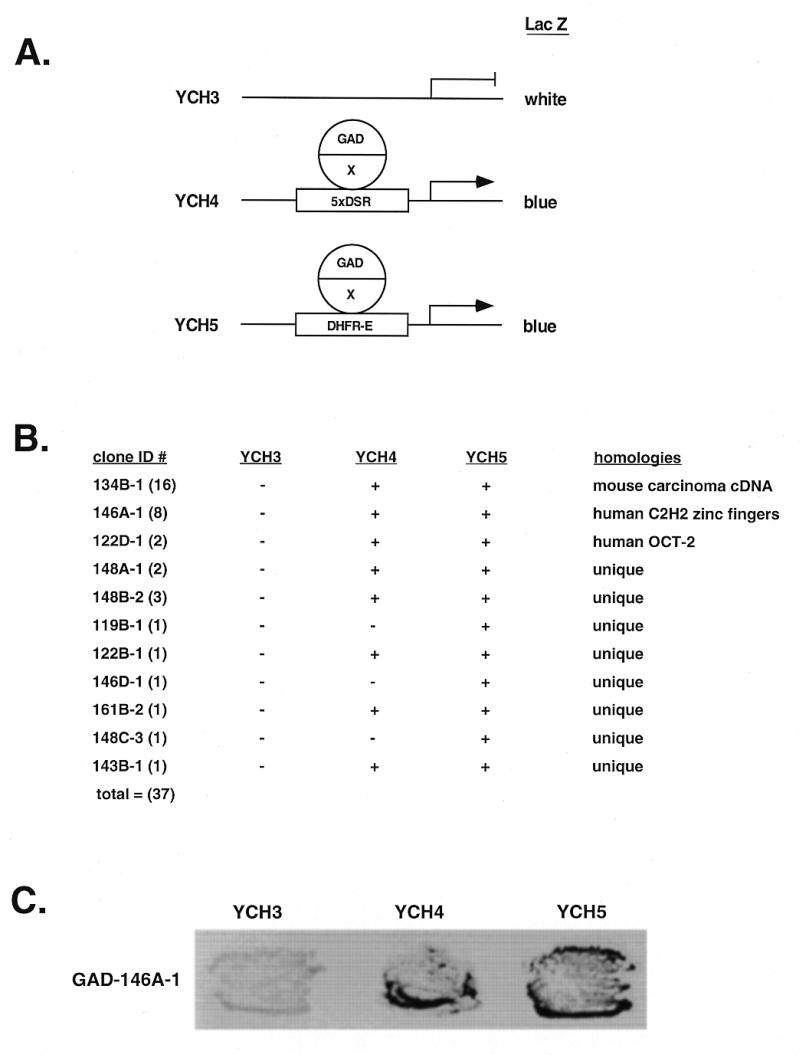
One hybrid screen in yeast for RIP60. (A) A genetic screen in S.cerevisiae was used to identify cDNA-encoded fusion proteins that bind the RIP60 target sequence (DSR) and activate expression of a linked reporter gene (LacZ or histidine) by recruiting a fused GAL4 activation domain (GAD) to the promoter. Shown are lacZ reporter strains in which lacZ expression is controlled by five copies of the DSR target sequence (YCH4) or a single copy of the DSR embedded in its native flanking sequences (YCH5). YCH3 was used as a control. (B) Summary of the one hybrid screen. From 14 million transformants, 37 clones were found that activated expression in YCH4 and/or YCH5 but not in YCH3. (C) Specificity test in yeast for RIP60 binding. In the one hybrid screen multiple in-frame fusions were identified that overlapped with clone 146A-1. Each of these fusions contained the Z2 and PRR region of RIP60 and activated lacZ expression in both YCH4 and YCH5, but not in YCH3.
From 14 × 106 primary transformants, 37 fusion proteins that specifically activate lacZ expression in yeast strains YCH4 and YCH5, but not YCH3, were identified (Fig. 1B and C). Sequence analysis indicated that 16 overlapping clones (clone class 134B) share significant similarity to a cDNA for an HMG protein expressed in a mouse carcinoma cell line, two encode the human Oct2 transcription factor, and eight overlapping clones (class 146A) encode a protein with multiple C2H2 ZF motifs. Specificity tests indicated that each of the eight clones from the latter group encode fusion proteins that require RIP60 target sequences to induce lacZ reporter expression (Fig. 1C and data not shown).
Using clone 146A-1 as probe, a HeLa cDNA phage library was used to isolate the full-length cDNA for the ZF protein. Two clones with poly(A) tails of different lengths but otherwise identical cDNA sequence were isolated. The 2.9 kb cDNA of pBS-27 has 129 bp of untranslated 5′ sequence, an open reading frame that encodes a 567 amino acid protein of 63 kDa, and 1124 bp of 3′ non-translated sequence including the poly(A) tail. Sequence analysis shows the open reading frame includes the two RIP60 peptides (underlined), 15 Kruppel-like C2H2 ZF motifs (in bold), and a proline-rich region between ZFs 8 and 9 (Fig. 2A). The 15 ZFs are organized in three clusters, which we refer to as hand Z1 (ZF 1–5), Z2 (ZF 6–8) and Z3 (ZF 9–15) (Fig. 2B). Other than the ZF motifs, no regions of homology to other proteins were identified. Based on the DNA binding specificity (see below), predicted molecular weight, and the presence of the two novel peptide sequences, we concluded that the 2.9 kb cDNA of clone pBS-27 encodes RIP60.
Figure 2.

RIP60 is a polydactyl ZF protein. Two otherwise identical cDNAs with different poly(A) tails were isolated from a HeLa cell lambda phage library using a clone related to 146A as probe. (A) The open reading frame of both clones predicts a 567 amino acid protein that includes 15 ZFs (bold) and both RIP60 peptide sequences obtained by microsequencing (underlined). (B) RIP60 contains 15 C2H2 Kruppel-like ZFs organized in three hands termed Z1 (ZFs 1–5), Z2 (ZFs 6–8) and Z3 (ZFs 9–15). A proline-rich region (PRR) predicted to form three polyproline helices separates hands Z2 and Z3.
Sequence-specific binding by hands Z1 and Z2
The RIP60 cDNA and various truncated versions of the RIP60 open reading frame were inserted into several expression vectors for production of GST fusion proteins in bacteria, and HA and GFP-tagged proteins in mammalian cells (Fig. 3A). We first examined the expression and localization of the HA- and GFP-tagged fusion proteins in mammalian cells. Western blot analysis demonstrated that each recombinant HA-tagged fusion protein was expressed in mouse NIH 3T3 cells in accord with its predicted size (data not shown). Despite the lack of an obvious nuclear localization signal, fluorescence microscopy of transfected CHOC 400 and NIH 3T3 cells also showed that each RIP60 GFP fusion protein was localized primarily in the cell nucleus (data not shown).
Figure 3.
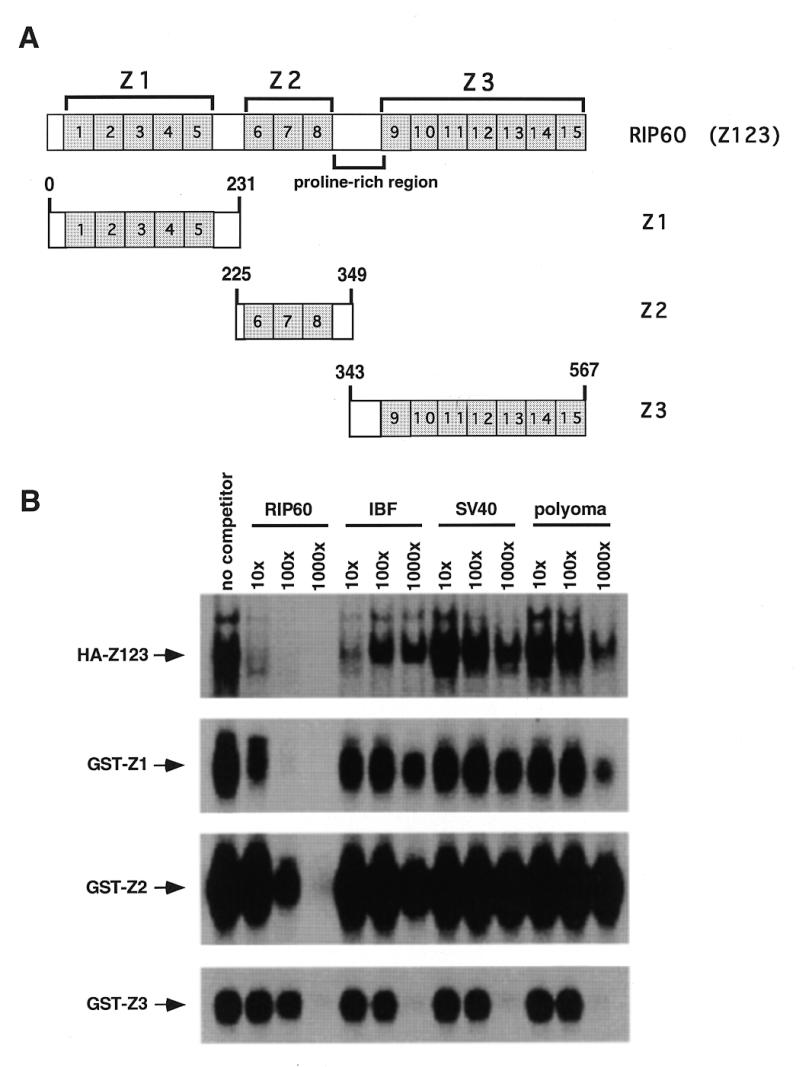
Specific DNA binding by RIP60 is located in both hands Z1 and Z2. (A) Schematic representation of RIP60 (Z123) showing the organization of hands Z1, Z2, Z3 and the proline-rich region. (B) Analysis of RIP60 DNA binding activity by gel mobility shift assays. GST fusion proteins containing hands Z1, Z2 or Z3 were expressed in E.coli, purified and analyzed for DNA binding activity. Because expression of full length RIP60 in E.coli was difficult, analysis of RIP60 binding activity was conducted with nuclear extracts from cells transfected with pCMV-HA-RIP60 (see Results). Binding specificity was assessed by competition with the DSR RIP60 binding site, an intron binding factor site (IBF), or the AT-rich regions from either the SV40 or polyomavirus origins of replication.
RIP60 purified from HeLa nuclear extract binds specifically to two inverted ATT-rich sequences within oriβ, as determined by DNase I footprinting, competitive gel shift analysis and scanning transmission electron microscopy (30,31). To map the DNA binding domains of RIP60, GST fusion proteins containing hands Z1, Z2 and Z3 (and combinations thereof) were used in gel mobility shift and DNase I footprinting assays. Expression of full-length RIP60 (Z123) was difficult in bacteria: <10% of the full-length RIP60 GST-fusion protein was observed after induction as compared to constructs for Z1, Z2 or Z3 (data not shown). Therefore binding studies were also performed with nuclear extracts from mouse NIH 3T3 cells transfected with a mammalian expression vector containing the human RIP60 coding sequence (pCMV-HA-Z123).
Competitive gel shift analyses showed that full-length RIP60 (HA-Z123) binds to the DSR probe in the presence of a 1000-fold molar excess of IBF, SV-AT and Py-AT competitors, and that binding is competed by a 10-fold excess of DSR DNA (Fig. 3B). Because the AT-rich sequences from the SV40 and polyomavirus origins of replication (SV-AT and Py-AT) failed to compete for binding, we conclude that RIP60 binds the ATT-rich DSR specifically, and is not a general AT-rich DNA binding protein. Addition of anti-HA monoclonal antibody 12CA5 to nuclear extract from cells transfected with pCMV-HA-Z123 supershifted the putative HA-Z123/DSR complex, verifying that the specific DNA-binding activity resulted from expression of HA-tagged RIP60 (data not shown).
Binding of GST-Z1 to the DSR probe was eliminated by a 100-fold excess of DSR competitor DNA, but was not significantly affected by large molar excesses of the IBF, SV-AT or Py-AT competitors (Fig. 3B). A slight competitive effect was observed in the presence of 1000-fold excess of Py-AT competitor DNA. Binding of the GST-Z2 to the DSR probe was stable in the presence of 1000-fold excess of IBF, SV-AT or Py-AT competitors, but was competed with 100-fold excess of unlabeled DSR DNA. Similar binding specificities were observed for GST-Z12 and GST-Z23 (data not shown). In contrast, binding of GST-Z3 to the DSR probe was eliminated by a 1000-fold excess of all unlabeled competitors tested. From these results we conclude that GST-Z123, GST-Z1, GST-Z12, GST-Z2 and GST-Z23 all bind specifically to the DSR from oriβ. In a similar manner, the USR was also able to compete for binding of these proteins to the DSR (data not shown). These results are consistent with our previous findings using purified RIP60 and indicate that RIP60 binds specifically to the USR and DSR ATT-rich target sites, but not all AT-rich sequences.
GST-Z2 forms multimers on DNA
To examine the interaction of RIP60 and hands Z1 and Z2 with the DSR in more detail, in vitro DNase I footprinting assays were performed with end-labeled DHFR-E, a 250 bp bent DNA fragment which contains the DSR and a neighboring consensus AP-1 binding site. DNase I footprinting assays with RIP60 purified from HeLa cell nuclear extract showed that the protein protects the ATT-rich DSR sequence on the top strand of DHFR-E from nucleotide position 3461 to position 3481, and from position 3476 to position 3461 on the bottom strand (30). Binding of RIP60 to the DSR also induces a characteristic nuclease hypersensitive site at position 3483 that borders the protected sequences on the top strand.
GST-RIP60 protected the DSR sequences from DNase I digestion on both the top strand (position 3461–3481) and bottom strand (position 3476–3461) of the DHFR-E probe (Fig. 4A and B), while little protection from nuclease cleavage was observed outside of the DSR target (Fig. 4A and B, lane 20). While the region of nuclease protection provided by GST-RIP60 mapped to the same nucleotide residues observed in footprinting experiments using native RIP60, the recombinant fusion protein did not induce hypersensitive sites within flanking sequences on either the top or bottom strands of the DHFR-E probe.
Figure 4.
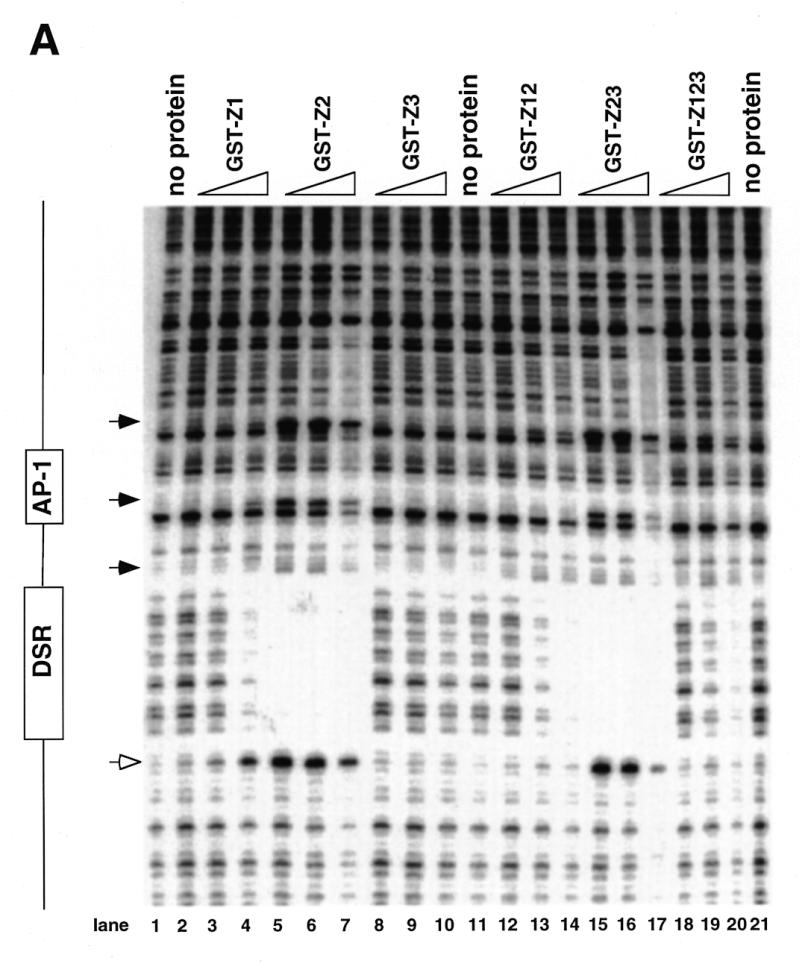
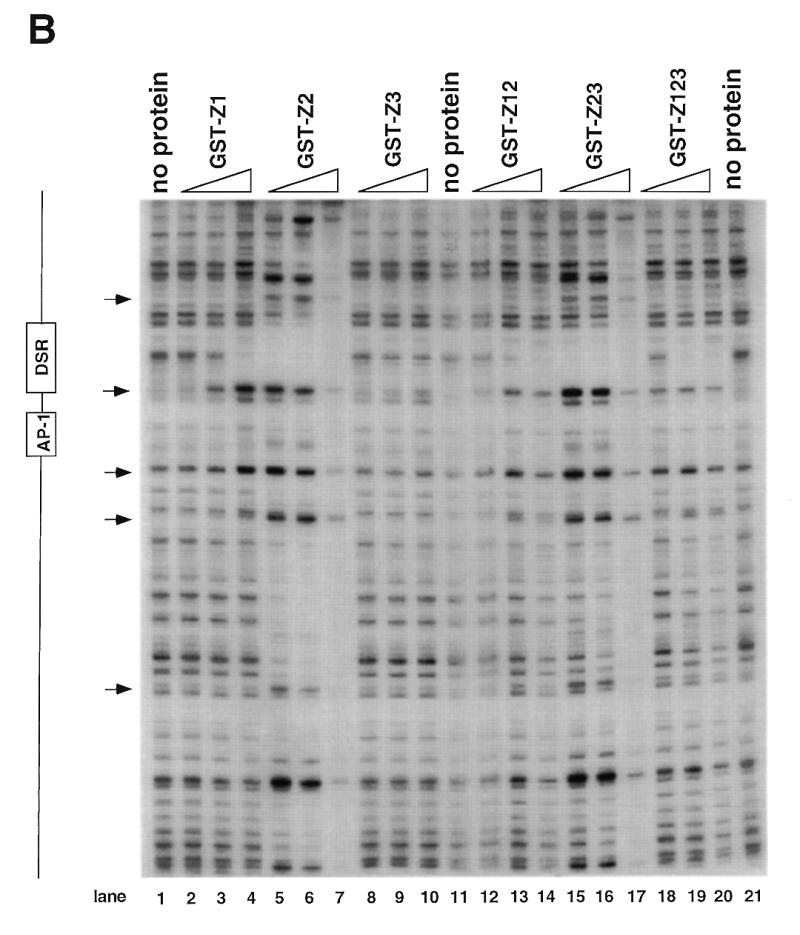
DNase I footprinting of RIP60 on the DHFR-E bent DNA fragment. The indicated RIP60 GST fusion proteins were incubated with end labeled DHFR-E probes and digested with DNase I as described in the Materials and Methods. The digestion products were resolved on sequencing gels and the footprinting patterns were visualized by autoradiography. (A) Nuclease protection patterns for the top strand of DHFR-E. Hypersensitive sites are indicated by arrows. The open arrow indicates a prominent hypersensitive site observed with RIP60 purified from cell extract and by in vivo genomic footprinting of the bent DNA motif in CHO cells (not shown). (B) Nuclease protection patterns for the bottom strand of DHFR-E. Hypersensitive sites are indicated by arrows.
GST fusion proteins possessing either the Z1 or Z2 domains (GST-Z1, GST-Z2, GST-Z12, GST-Z23) protected the same DSR sequences on both strands of the DHFR-E probe from nuclease digestion as did GST-RIP60 and native RIP60 (Fig. 4A and B, and ref. 30). Fusion proteins containing hand Z2 induced multiple hypersensitive sites on the top strand of DHFR-E (arrows), including the prominent site at position 3483 (Fig. 4A, open arrow). At higher protein concentrations both GST-Z2 (lanes 5–7) and GST-Z23 (lanes 15–17) protected sequences extending through the AP-1 site toward the end of DHFR-E. In contrast, GST-Z3 did not protect any sequence on either strand of the DHFR-E probe from DNase I cleavage (Fig. 4A and B, lanes 8–10). These results are consistent with the gel shift experiments which indicate that Z3 domain of RIP60 does not recognize the DSR. GST-Z12 bound the DSR and produced the same nuclease protection patterns as GST-Z1 or GST-Z2, but it did not induce nuclease hypersensitive sites on either strand of the DHFR-E probe that were observed with GST-Z2 or GST-Z23. Rather the pattern of nuclease protection and hypersensitivity induced by GST-Z12 was virtually identical to that of GST-RIP60 (or Z123). Failure to observe extended footprints by GST-RIP60 may have resulted from difficulty in producing the full-length protein in bacteria.
The footprinting results demonstrated that GST-RIP60, and truncated forms of RIP60 that contained either domains Z1 or Z2, bound specifically to the DSR target, protecting both strands of the bent DNA region from DNase I cleavage in a manner similar to the full length protein. The observation that GST-Z2 and GST-Z23, at higher protein concentrations, protected sequences adjacent to the DSR also suggests that sequences within hand Z2 are sufficient for multimerization of RIP60 on DNA.
Specific binding of GST-Z2 is more stable than that of GST-Z1
Both GST-Z1 and GST-Z2 bound the DSR and USR specifically in footprinting and gel shift experiments, suggesting that RIP60 contains two independent domains capable of recognizing ATT-rich binding sites. To ascertain if Z1 and Z2 show any differences in binding activity, we examined the stability of Z1 and Z2 complexes with the DSR in competition gel shift assays. Identical amounts of GST-Z1 and GST-Z2 were incubated with 32P-labeled probe for 20 min, and then a 1000-fold molar excess of unlabeled competitor DSR was added. Samples were removed from the binding reactions at various times thereafter and loaded directly onto non-denaturing gels running at low voltage. After all the samples were loaded, the voltage was increased to 100 V for 1 h. After drying the gels (Fig. 5A and B), the signals in the shifted complexes were quantified by phosphoimaging. In Figure 5C the CPM within the protein/DNA complexes are expressed as a percentage of the total signal per lane (i.e. bound probe over bound plus free probe). Under these binding conditions, binding of GST-Z2 to the DSR appeared significantly more stable over time than that of GST-Z1.
Figure 5.
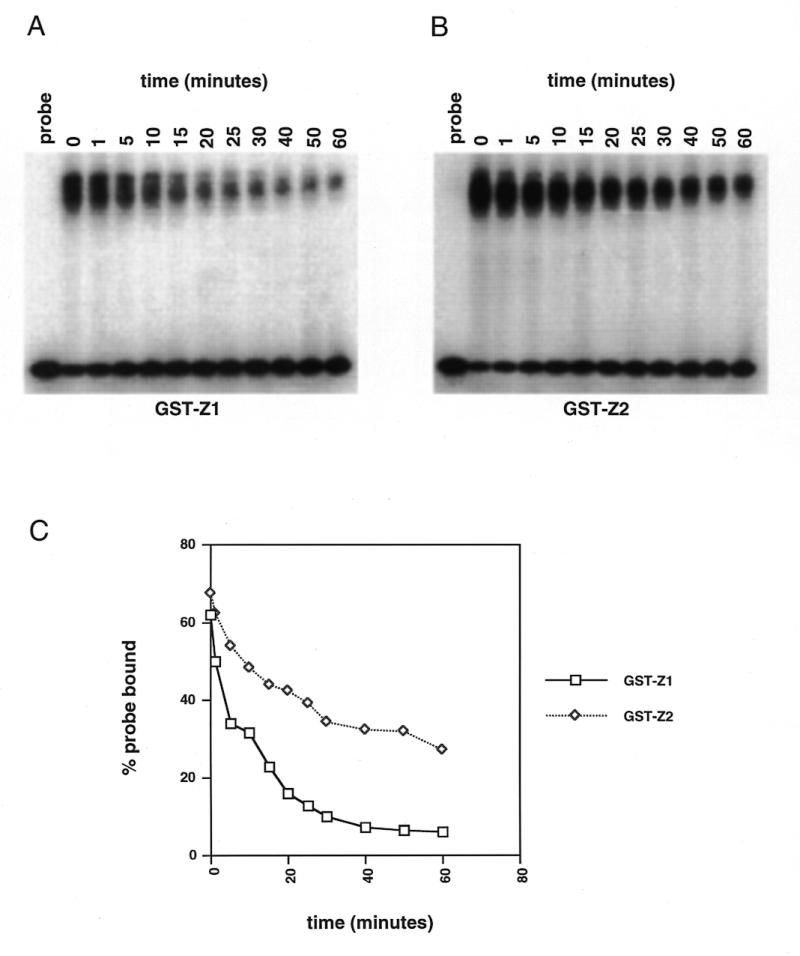
GST-Z2 binds the DSR more avidly than does GST-Z1. GST-Z1 and GST-Z2 were bound to labeled DSR probe at room temperature under standard gel shift conditions. After 20 min a 1000-fold molar excess of unlabeled probe was added, the incubation was continued and samples were withdrawn at the indicated time points and loaded directly onto gels as described in the Materials and Methods. (A) Autoradiograph of GST-Z1 binding after addition of unlabeled probe (time 0). (B) Autoradiograph of GST-Z2 binding after addition of unlabeled probe. (C) Quantification of percent probe bound as a function of time after the addition of unlabeled probe.
Hand Z2 is sufficient for DNA looping by RIP60
DNA looping by origin binding proteins has been shown to be functionally important in both plasmid and viral DNA replication. To begin mapping of the domains involved in DNA looping by RIP60, we used a ligation enhancement assay. The assay detects changes in the efficiency of end-to-end ligation of a linear plasmid when proteins bound near the ends of the DNA template interact to bring the plasmid ends in close proximity to one another (47). For ligation enhancement assays, the URS and DRS RIP60 binding sites were separated by ~6 kb by linearizing pCH127 with AccI. In the linear pCH127 substrate, the USR and DSR sites are located 300 and 450 bp from the DNA ends, respectively (Fig. 6A).
Figure 6.
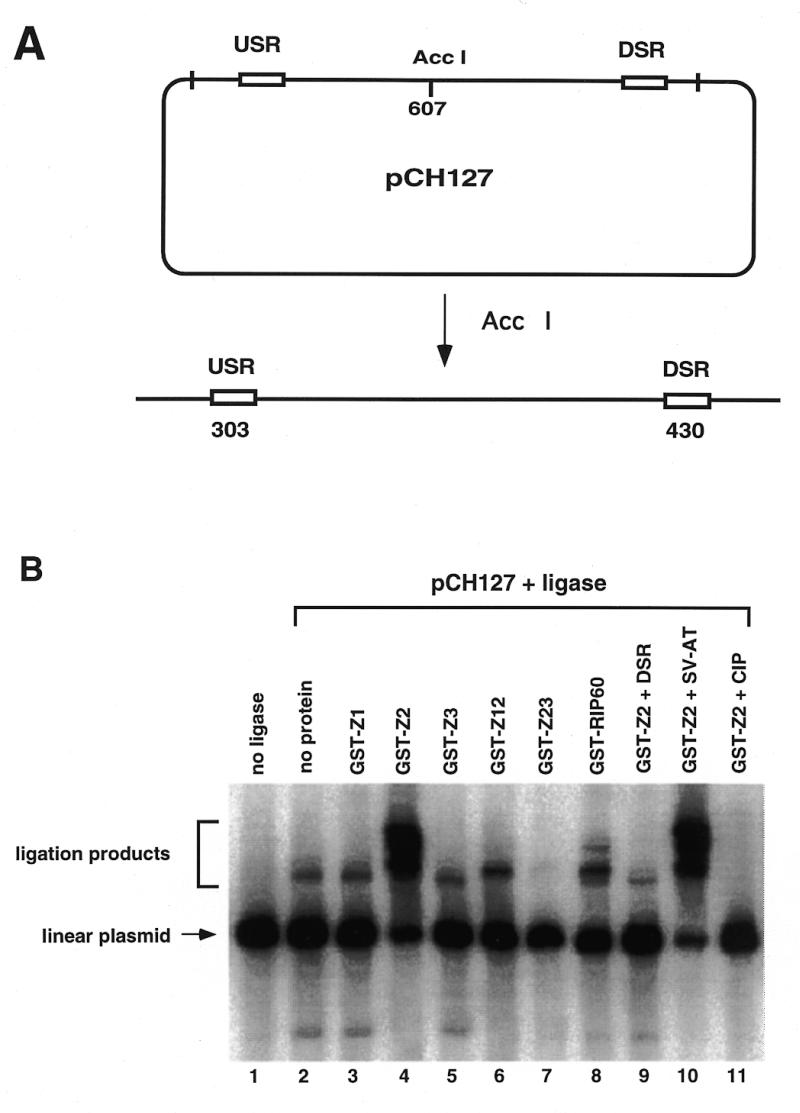
Binding of RIP60 GST-Z2 to the USR and DSR enhances ligation of linear pCH127. A ligation enhancement assay was used to test for the ability of RIP60 fusion proteins to mediate DNA looping between the USR and DSR. (A) pCH127 contains a 1.1 kb fragment of the DHFR ori-β region that includes both the USR and DSR RIP60 binding sites. (B) pCH127 was linearized with AccI and incubated with the indicated protein factors for 15 min prior to the addition of ligase and ATP for 5 min. The products of the reactions were resolved by gel electrophoresis, blotted to nylon, and visualized by hybridization with 32P-labeled pCH127. Ligation products representing open circles or linear dimers are indicated by the square bracket. Addition of DSR competitor DNA eliminated increased ligation by GST-Z2 (lane 9) whereas the AT-rich region from the SV40 origin of replication had no effect (lane 10). Dephosphorylated linear pCH127 did not support ligation with (lane 11) or without (not shown) GST-Z2.
In the absence of protein or ligase, only the linear pCH127 plasmid template was observed (Fig. 6B, lane 1), while the addition of ligase alone for 5 min (lane 2) generated two ligation products. The addition of GST-Z1 or GST-Z3 had little effect on the plasmid ligation products when compared to ligase alone (compare lanes 3 and 5 to lane 2 in Fig. 6B). The addition of GST-Z2 (lane 4), GST-Z12 (lane 6) or GST-RIP60 (lane 8) altered the distribution of ligation products, resulting in the formation of more slowly migrating bands. The addition of GST-Z23 (lane 7) had no effect in this experiment. The presence of DSR competitor inhibited the ability of GST-Z2 to enhance plasmid ligation (lane 9), while the presence of an excess amount of the SV-AT competitor DNA had no effect (lane 10). These results indicate that binding of GST-Z2 to the linear pCH127 plasmid at the USR and DSR was required to enhance ligation of the template under these conditions. The ligation products induced by GST-Z2 also required 5′-phosphates on the substrate, as prior dephosphorylation of the linear pCH127 template with calf intestinal phosphatase prevented GST-Z2 ability to enhance plasmid ligation (lane 11).
To assess the role of the PRR in DNA looping, we compared the binding activity of GST-Z2 to GST-Z2ΔP in gel mobility shift, ligation enhancement and AFM DNA looping assays (Fig. 7). At equivalent protein concentrations GST-Z2 bound the DSR probe more avidly than GST-Z2ΔP (compare lanes 2–4 to lanes 5–7, Fig. 7A). While the weak binding of GST-Z2ΔP was specific (data not shown), it did not form multimers on the gel shift probe as did GST-Z2 (arrows, Fig. 7A). In ligation enhancement assays, addition of ligase alone for 20 min resulted in a broader spectrum of ligation products (Fig. 7B, lane 3) as compared to the addition of ligase for 5 min (see Fig. 6B, lane 2). As before, addition of GST-Z2 markedly increased the efficiency of end-to-end ligation of linear pCH127 (lanes 6–8), yielding products similar to those observed with ligase alone after 20 min (lane 3). GST-Z2ΔP, at any protein concentration tested, had no effect (lanes 9–11). These results suggest that the PRR is involved in protein–DNA and/or protein–protein interactions required for DNA looping by RIP60.
Figure 7.
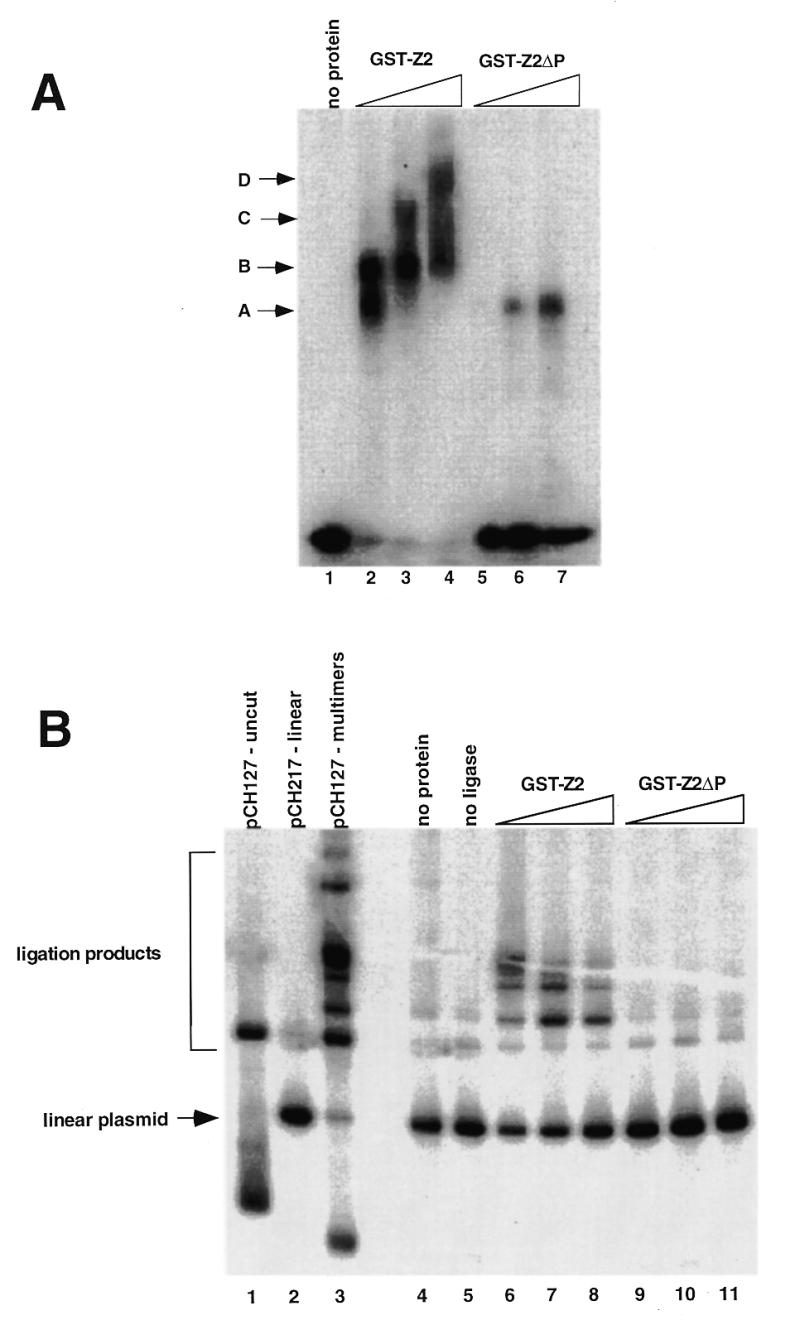
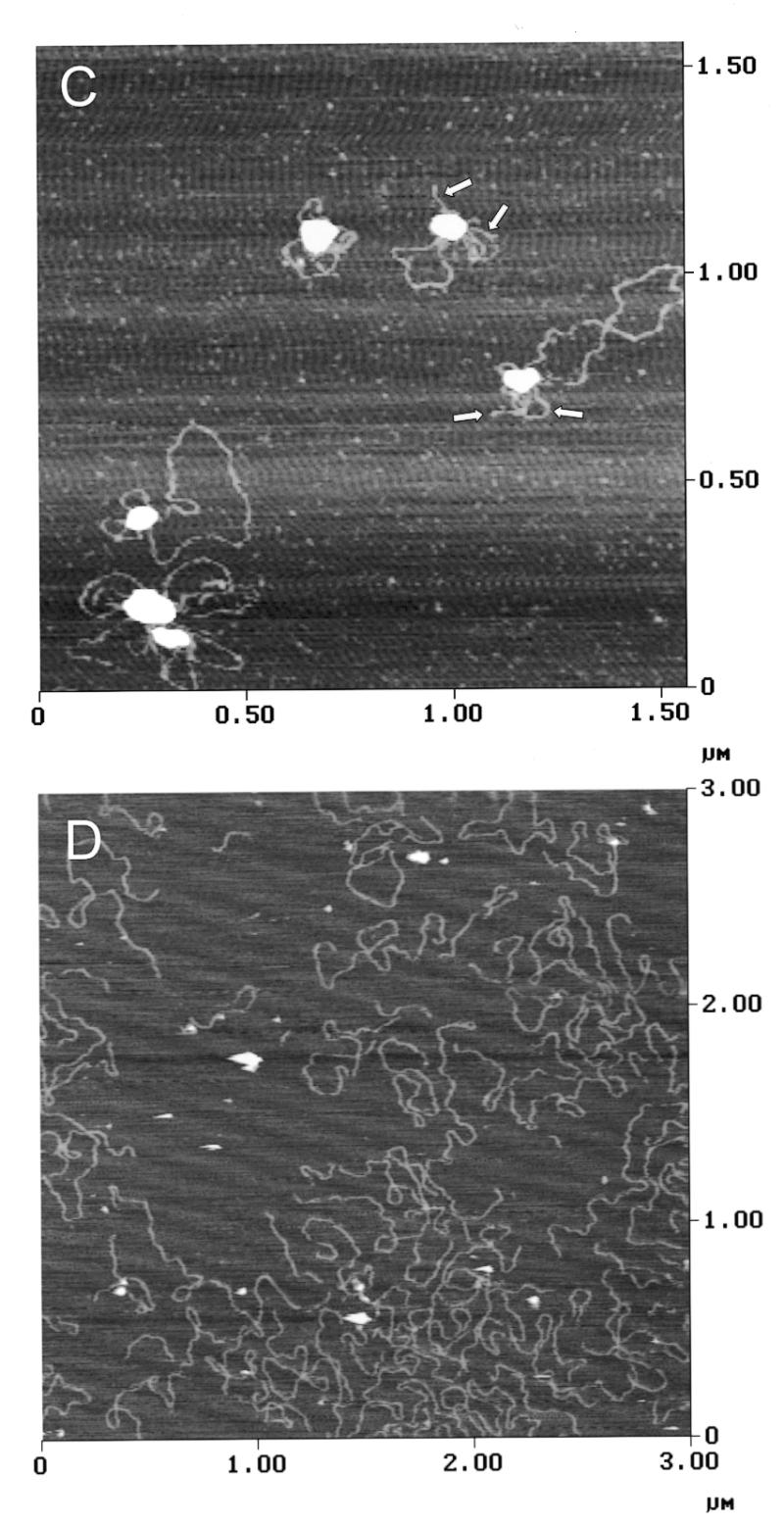
The proline-rich region stimulates DNA binding and DNA looping by hand Z2. (A) Gel mobility shift assays were used to assess the effect of the proline-rich region of DNA binding by GST-Z2. Equivalent amounts of GST-Z2 and GST-Z2ΔP were incubated with 32P-labeled DSR probe, the products were resolved by non-denaturing electrophoresis, and binding was visualized by autoradiography. GST-Z2 forms at least four gel shift complexes with the DSR (A–D) whereas GST-Z2ΔP forms a single complex. Competition assays showed binding by GST-Z2ΔP was specific for the DSR (data not shown). (B) GST-Z2 and GST-Z2ΔP were tested for their ability to mediate DNA looping using a ligation enhancement assay. In this instance ligation of the linear pCH127 control was allowed to proceed for 20 min in order to visualize a more complex array of ligation products. Lanes 6–8 and 9–11 contained 5, 50 or 500 ng of the indicated protein, respectively. (C) DNA looping between the USR and DSR by GST-Z2. The arrows indicate free ends of pCH127 that protrude from the loop complex. (D) The PRR is required for DNA looping. When incubated with GST-Z2ΔP looping between the USR and DSR in linear pCH127 was not observed by AFM.
To confirm these results, we examined DNA binding of GST-Z2 and GST-Z2ΔP to linear pCH127 by AFM. In the presence of GST-Z2, looping between the upstream and downstream binding sites was readily evident (Fig. 7C). In several instances the ends of the linear pCH127 DNA were observed to protrude from the looped DNA complex (arrows, Fig. 7C). When GST-Z2ΔP was used in AFM experiments, stable binding to the USR or DSR on the linear DNA substrate was rarely observed, and loops between the two sites were not detected (Fig. 7D).
Effects of RIP60 on activation of the polyomavirus origin of replication
In an attempt to study the effects of RIP60 expression on DNA replication, we used a polyomavirus (Py)-based transient plasmid replication assay. The Py core origin requires an enhancer for activity (reviewed in 48). When the enhancer element is replaced with binding sites for a variety transcription factors, the cognate transcription factors are able to stimulate Py origin-dependent plasmid replication, providing the transcription factors contain a transcriptional activation domain (48). For example, the heterodimeric transcription factor complex of Fos and Jun (AP-1) has been shown to enhance replication from the polyomavirus origin of replication through the agency of AP-1 binding sites located in the enhancer (44,49). The polyomavirus origin-dependent plasmid replication assay utilized four plasmid components: (i) a reporter plasmid containing the Py core origin of replication that harbors protein recognition sequences in place of the native viral enhancer region; (ii) a Py large T-antigen expression plasmid required for replication of the reporter plasmid; (iii) effector plasmids expressing HA-tagged RIP60 fusion proteins or control transcription factors; and (iv) unmethylated pUC19 plasmid DNA (pUC19–) which serves as an internal control for transfection efficiency and plasmid recovery. As shown in Figure 8A, the reporter plasmids used here were derived from pPyOICAT (44) and contained either six AP-1 binding sites (pPy-AM6-OICAT), the 250 bp DHFR-E fragment (pPy-DHFR-E-OICAT), or the 5x-DSR fragment (pCH30).
Figure 8.
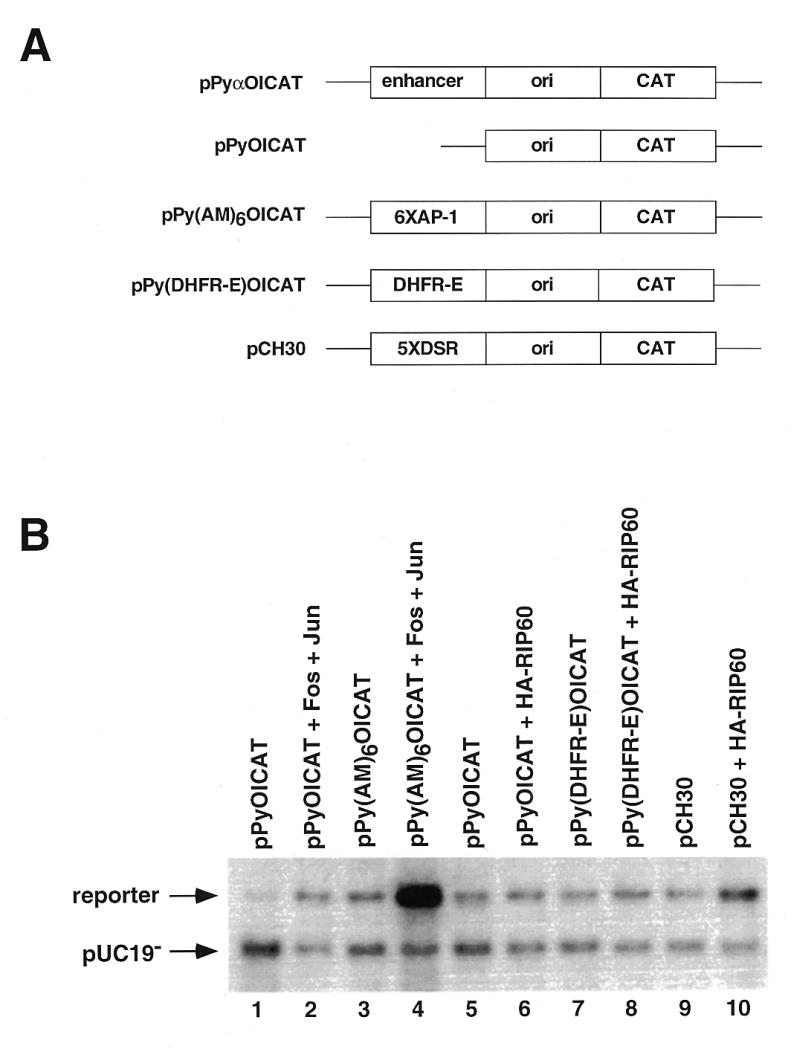
Polyomavirus origin-dependent replication assay for replication enhancer activity. (A) A series of plasmids containing the wild type polyomavirus (Py) core origin of replication were used in transient replication assays to test RIP60 for replication enhancer activity. pPyOICAT contains the Py core origin and the native α-enhancer region. The negative control plasmid pPyOICAT lacks the α-enhancer, whereas the positive control plasmid pPy(AM)6OICAT contains six AP-1 sites in place of the α-enhancer. Test plasmids pPy(DHFR-E)OICAT and pCH30 contained the DHFR-E fragment or five copies of the DSR in place of the α-enhancer, respectively. (B) Representative results of transient Py-origin dependent plasmid replication assays. The indicated reporter plasmids were cotransfected into NIH 3T3 cells with unmethylated pUC19 plasmid DNA, an expression vector for Py large T antigen, and the indicated expression vectors for c-fos, c-jun, or RIP60. After imaging by autoradiography, the signals in each band were quantified in a phosphoimager and the ratio of signal between the reporter and the unmethylated pUC19 control plasmid was calculated. Coexpression of Fos and Jun stimulated replication of pPy(AM)6OICAT ~40-fold in this experiment.
As a positive control, NIH 3T3 cells were transfected with the pPy(AM)6OICAT reporter plasmid in the absence or presence of Fos and Jun effector plasmids. Consistent with the results of others (44,49), we observed that coexpression of Fos and Jun stimulated replication of the pPy(AM)6OICAT reporter by ~40-fold over control (Fig. 8B, lane 4). In contrast, expression of RIP60 increased replication of a reporter plasmid with five copies of the DSR in the enhancer region (pCH30) only ~3.5-fold (Fig. 8B, lane 10). RIP60 had no effect on replication of the enhancerless plasmid OICAT (lane 6) or on the DHFR-E plasmid with a single binding site in context of the bent DNA sequence (lane 8). These results and sequence analysis indicate that RIP60 does not contain a potent transcriptional transactivation domain, which was independently confirmed in transcription enhancer assays (P.Held and N.Heintz, unpublished data).
DISCUSSION
The steps involved in origin recognition in animal cells are unknown. We have proposed that mammalian replication origins are multipartite regulatory elements that resemble transcriptional promoters (23). In this model, ORC and its associated factors are considered the functional counterparts of basal transcription factors in that these proteins are used for all replication initiation events, regardless of cell type or time of activation during the S phase. As cells exit mitosis, we suggest that transcription factors and other DNA binding proteins establish a coordinated program of transcription and replication for the ensuing cell cycle. Similar to their effects on gene promoters, the binding of sequence-specific factors may modify local chromatin environment, distort DNA structure, or promote the binding of ORC or other replication factors. Introducing an origin identification step upstream of ORC that depends on site-specific DNA binding proteins would provide for flexibility in origin selection in different cell types, facilitate coordination between gene transcription and replication, and account for the heterogeneity in origin sequences that have been described to date.
To identify proteins that might participate in establishment of the replication program for the dhfr gene, footprinting and gel shift experiments were used to identify protein binding sites in the dhfr ori-β region. These studies showed oriβ binds AP-1 and Oct1 in vitro (29) and led to the identification of RIP60 (30). Recently, high resolution mapping of short replication intermediates have placed the USR RIP60 site in the precise center of a dhfr origin of bidirectional DNA replication (50). At this time, however, there is no formal evidence that RIP60 or any other sequence-specific DNA binding protein is involved in oriβ activity.
Here we describe the cloning and initial characterization of RIP60. Due to the high number of ZFs in RIP60, PCR-based cloning from RIP60 peptide sequences was unsuccessful. Because RIP60 binds the DSR as a homodimer (31), we reasoned a one hybrid screen in yeast might provide the specificity required to distinguish RIP60 from the hundreds of other ZF proteins expressed in mammalian cells. Indeed, the one hybrid screen proved remarkably efficient, resulting in the isolation of multiple overlapping cDNAs for RIP60, each of which encoded fusion proteins that included hand Z2 (ZFs 6–8) and the PRR. More important, no other ZF protein was recovered in the screen, suggesting that the screen was both sensitive and specific.
RIP60 purified from HeLa cell nuclear extract produces a footprint of ~20 bp over both strands of the DSR (29,30). Footprinting and gel shift experiments indicate that the N-terminal five ZFs of hand Z1 and the central three ZFs of hand Z2 are sufficient for specific binding to both the DSR and USR sites in oriβ (Figs 3 and 4). While GST-Z1 and GST-Z2 independently recapitulate many of the features of the RIP60 footprint on the DSR, binding of GST-Z1 to either the USR or the DSR is unstable relative to GST-Z2, GST-Z1 does not form multimers on DNA or mediate DNA looping (not shown), and GST-Z1 has no activity in ligation enhancement assays. In contrast, the GST-Z2 fusion that contains 37 amino acids from the PRR is capable of binding both the USR and DSR in vitro, multimerizing on DNA, and forming a DNA loop in excess of 6 kb (Figs 6 and 7). GST-Z3 binds DNA, but does not bind the USR or DSR in vitro.
Based on the studies with the GST fusion proteins, we conclude that GST-Z2 supports both the sequence-specific recognition of the USR and DSR and the protein–protein interactions required for looping in vitro. Because deletion of the PRR reduces the stability of DNA binding by hand Z2 as well as eliminates looping, at this time we cannot ascribe the DNA looping activity directly to the PRR. However, the PRR does contain three consensus sequences for polyproline helices, which have been implicated in protein–protein interactions (51). We are presently examining how binding to DNA facilitates the protein–protein interactions required for looping. Because the PRR appears to be required for both stable binding and multimerization on DNA, our present data suggests binding and looping includes the following steps. First, GST-Z2 binds the USR and DSR sites in a site-specific manner and nucleates the assembly of protein multimers on neighboring DNA sequences. It is evident from the footprinting experiments that ZFs 6–8 in hand Z2 are able to stably bind DNA sequences that border the DSR once Z2 has bound the DSR (Fig. 4A and B); these multimers may occupy several hundred base pairs of DNA. A specific binding site appears to be required for nucleating multimerization, as addition of specific DSR competitor to the ligation enhancement assays inhibits looping. Second, because GST-Z2 does not aggregate in solution (data not shown), DNA binding may alter the conformation of ZFs 6–8 and/or the PRR such that the inter-molecular protein–protein interactions between distantly bound multimers required for DNA looping are favored. Finally, competition assays suggest formation of the DNA loop stabilizes both protein–DNA and protein–protein interactions in the loop, as has been observed for EBNA1 (33,52,53).
Despite strong DNA binding and looping in vitro, full length RIP60 and various derivatives thereof do not have significant replication enhancer activity in the polyoma origin-dependent plasmid replication assay. Other work has shown that neither RIP60 binding sites nor RIP60 itself stimulate the expression of reporter genes in transient transfection assays (P.Held and N.H.Heintz, unpublished data). Together with sequence analysis, these results indicate that RIP60 does not contain a transcription transactivation domain. Thus, like many other polydactyl ZF proteins of unknown function, RIP60 does not appear to be a transcription factor.
Despite lack of a transactivation domain, RIP60 has several properties similar to the origin binding protein EBNA1, which is required for the replication and maintenance of Epstein Barr Virus (EBV). EBNA1, which has no inherent enzymatic activity involved in replication, binds as a homodimer to two sets of related sequences in EBV oriP in vitro and fosters the formation of a 900 bp DNA loop (33,34). Presumably EBNA1 acts in replication by interacting with cellular replication factors such as RPA (54). Like EBNA1, RIP60 appears to have no inherent enzymatic activity, binds as a homodimer to two sets of sequences within an origin region, and induces the formation of a 720 bp DNA loop.
Few of the hundreds of polydactyl ZF proteins that have been sequenced have been assigned a function, and given that RIP60 was not found in the public databases, clearly more remain to be discovered. While originally described as a DNA binding motif, ZFs have been implicated in binding RNA, binding RNA/DNA hybrids, and protein–protein interactions (55). A homodimer of RIP60 bound to DNA would include 30 ZFs, only a portion of which are likely to be in contact with DNA. The remaining ZFs could be involved in binding RNA, other distal DNA sequences, or even other proteins. Dissection of the cellular function of RIP60 will require further work, such as genomic footprinting and chromatin immunoprecipitation experiments to ascertain the binding activity of RIP60 at oriβ during the cell cycle.
Acknowledgments
ACKNOWLEDGEMENTS
We thank J. Li and J. Milbrandt for assistance with the one hybrid screen, J. Kurjan for yeast plasmids, A. Quinn and D. Taatjes for assistance with AFM and L. Frappier for advice concerning ligation enhancement assays. We would also like to thank Y. Ito and colleagues for the pPyOICAT vectors and assistance with the Py replication assay, B. Brinton for NIH 3T3 cell transfection parameters, N. Heintz for assistance with protein purification, and S. Illenye for technical assistance. This work was supported by the Lake Champlain Cancer Research Organization and the Vermont Cancer Center.
DDBJ/EMBL/GenBank accession nos AF201303
REFERENCES
- 1.Stillman B. (1996) Science, 274, 1659–1664. [DOI] [PubMed] [Google Scholar]
- 2.Newlon C. (1997) Cell, 91, 717–720. [DOI] [PubMed] [Google Scholar]
- 3.Bell S.P. and Dutta,A. (1997) Annu. Rev. Cell Dev. Biol., 13, 293–332. [DOI] [PubMed] [Google Scholar]
- 4.Diffley J.F. (1998) Curr. Biol., 8, 771–773.9651681 [Google Scholar]
- 5.Bousett K. and Diffley,J.F. (1998) Genes Dev., 12, 480–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chuang R.Y. and Kelly,T.J. (1999) Proc. Natl Acad. Sci. USA, 96, 2656–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carpenter P.B., Mueller,P.R. and Dunphy,W.G. (1996) Nature, 379, 357–360. [DOI] [PubMed] [Google Scholar]
- 8.Yu G., Wu,J.R. and Gilbert,D.M. (1998) Genes Cells, 3, 709–720. [DOI] [PubMed] [Google Scholar]
- 9.Coleman T.R., Carpenter,P.B. and Dunphy,W.G. (1996) Cell, 87, 53–63. [DOI] [PubMed] [Google Scholar]
- 10.Chong J.P.J., Mahbubani,H.M., Khoo,C.-Y. and Blow,J.J. (1995) Nature, 375, 418–421. [DOI] [PubMed] [Google Scholar]
- 11.Harland R.M. and Laskey,R.A. (1980) Cell, 21, 761–777. [DOI] [PubMed] [Google Scholar]
- 12.Blow J.J. and Laskey,R.A. (1986) Cell, 47, 577–587. [DOI] [PubMed] [Google Scholar]
- 13.Walter J. and Newport,J.W. (1997) Science, 275, 993–995. [DOI] [PubMed] [Google Scholar]
- 14.Liang C. and Stillman,B. (1997) Genes Dev., 11, 3375–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jallepalli P.V. and Kelly,T.J. (1996) Genes Dev., 10, 541–552. [DOI] [PubMed] [Google Scholar]
- 16.Saha P., Chen,J., Thome,K.C., Lawlis,S.J., Hou,Z.H., Hendricks,M., Parvin,J.D. and Dutta,A. (1998) Mol. Cell. Biol., 18, 2758–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DePamphilis M. (1996) DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 45–86.
- 18.Heintz N.H. (1996) DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 983–1004.
- 19.Dimitrova D.S. and Gilbert,D.M. (1998) J. Cell Sci., 111, 2989–2998. [DOI] [PubMed] [Google Scholar]
- 20.Wu J.R. and Gilbert,D.M. (1996) Science, 271, 2170–1272. [Google Scholar]
- 21.Wu J.R. and Gilbert,D.M. (1997) Mol. Cell. Biol., 17, 4312–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilbert D.M., Miyazawa,H. and DePamphilis,M.L. (1995) Mol. Cell. Biol., 15, 2942–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heintz N.H., Dailey,L., Held,P. and Heintz,N. (1992) Trends Genet., 8, 376–381. [DOI] [PubMed] [Google Scholar]
- 24.Burhans W.C., Vassilev,L., Caddle,M.S., Heintz,N.H. and DePamphilis,M.L. (1990) Cell, 62, 955–965. [DOI] [PubMed] [Google Scholar]
- 25.Caddle M.S., Lussier,R.H. and Heintz,N.H. (1990) J. Mol. Biol., 211, 19–33. [DOI] [PubMed] [Google Scholar]
- 26.Caddle M.S., Dailey,L. and Heintz,N.H. (1990) Mol. Cell. Biol., 10, 6236–6243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bianchi A., Wells,R.D., Heintz,N.H. and Caddle,M.S. (1990) J. Biol. Chem., 265, 21789–21796. [PubMed] [Google Scholar]
- 28.Brinton B., Caddle,M.S. and Heintz,N.H. (1991) J. Biol. Chem., 266, 5153–5161. [PubMed] [Google Scholar]
- 29.Held P., Soultanakis,E., Dailey,L., Kouzarides,T., Heintz,N. and Heintz,N.H. (1992) DNA Replication and the Cell Cycle. Springer-Verlag, Berlin.
- 30.Dailey L., Caddle,M.S., Heintz,N. and Heintz,N.H. (1990) Mol. Cell. Biol., 10, 6225–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mastrangelo I.A., Held,P.G., Dailey,L., Wall,J.S., Hough,P.V.C., Heintz,N. and Heintz,N.H. (1993) J. Mol. Biol., 232, 766–778. [DOI] [PubMed] [Google Scholar]
- 32.Miron A., Mukherjee,S. and Bastia,D. (1992) EMBO J., 11, 1205–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frappier L. and O’Donnell,M. (1991) Proc. Natl Acad. Sci. USA, 88, 10875–10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su W., Middleton,T., Sugden,B. and Echols,H. (1991) Proc. Natl Acad. Sci. USA, 88, 10870–10874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J., Wilson,T.E., Milbrandt,J. and Johnston,M. (1993) Methods Enzymol., 6, 1–13. [Google Scholar]
- 36.Li J.J. and Herskowitz,I. (1993) Science, 262, 1870–1874. [DOI] [PubMed] [Google Scholar]
- 37.Durfee T., Becherer,K., Chen,P.L., Yeh,S.H., Yang,Y., Kilburn,A.E., Lee,W.H. and Elledge,S.J. (1993) Genes Dev., 7, 555–559. [DOI] [PubMed] [Google Scholar]
- 38.Gutherie C. and Fink,G.R. (1991) Guide to Yeast Genetics and Molecular Biology. Academic Press, San Diego, CA.
- 39.Schiestl R.H. and Gietz,R.D. (1989) Curr. Genet., 16, 339–346. [DOI] [PubMed] [Google Scholar]
- 40.Breeden L. and Nasmyth,K. (1985) Cold Spring Harbor Symp. Quant. Biol., 50, 643–650. [DOI] [PubMed] [Google Scholar]
- 41.Baker S.J., Markowitz,S., Fearon,E.R., Wilson,J.K. and Vogelstein,B. (1990) Science, 249, 912–915. [DOI] [PubMed] [Google Scholar]
- 42.Magae J., Wu,C.-L., Illenye,S., Harlow,E. and Heintz,N.H. (1996) J. Cell Sci., 109, 1717–1726. [DOI] [PubMed] [Google Scholar]
- 43.Murakami Y., Satake,M., Yamaguchi-Iwai,Y., Sakai,M., Muramatsu,M. and Ito,Y. (1991) Proc. Natl Acad. Sci. USA, 88, 3947–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ito K., Asano,M., Hughes,P., Kohzaki,H., Masutani,C., Hanaoka,F., Kerppola,T., Curran,T., Murakami,Y. and Ito,Y. (1996) EMBO J., 15, 5636–5646. [PMC free article] [PubMed] [Google Scholar]
- 45.Hirt B. (1967) J. Mol. Biol., 26, 365–369. [DOI] [PubMed] [Google Scholar]
- 46.Staal F.J., Roederer,M., Herzenberg,L.A. and Herzenberg,L. (1990) Proc. Natl Acad. Sci. USA, 87, 9943–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goldsmith K., Bendell,L. and Frappier,L. (1993) J. Virol., 67, 3418–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hassell J.A. and Brinton,B.T. (1996) DNA Replication in Eukaryotic Cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 639–677.
- 49.Guo Z.S. and DePamphilis,M.L. (1992) Mol. Cell. Biol., 12, 2514–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kobayasi T., Rein,T. and DePamphilis,M.L. (1998) Mol. Cell. Biol., 18, 3266–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Williamson M.P. (1994) Biochem. J., 297, 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frappier L., Goldsmith,K. and Bendell,L. (1994) J. Biol. Chem., 269, 1057–1062. [PubMed] [Google Scholar]
- 53.Laine A. and Frappier,L. (1995) J. Biol. Chem., 270, 30914–30918. [DOI] [PubMed] [Google Scholar]
- 54.Zhang D., Frappier,L., Gibbs,E., Hurwitz,J. and O’Donnell,M. (1998) Nucleic Acids Res., 26, 631–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mackay J.P. and Crossley,M. (1998) Trends Biochem. Sci., 23, 1–4. [DOI] [PubMed] [Google Scholar]