


Abstract
The transcriptional activity of the p53 tumor suppressor protein is crucial for the regulation of cell growth, apoptosis and tumor progression. The first identified p53 relative, p73, was reported to be monoallelically expressed in normal tissues. In some tumors, loss of heterozygosity was associated with overexpression of the silent allele. Human p73α was transfected into the wild-type p53-expressing human ovarian carcinoma cell line A2780. Unlike human osteosarcoma Saos-2 cells, A2780 cells could tolerate hyperexpression of p73α and clones overexpressing p73α could be isolated. No p53–p73 protein–protein interaction was found in these clones in co-immunoprecipitation experiments. Endogenous p53 transcriptional activity was markedly decreased both when p73 was integrated into the genome and in transient transfections using a reporter plasmid containing the p53 binding site linked to luciferase. Transient transfection of p73 with a mutation in the DNA-binding domain did not show these effects. The competition for p53 DNA binding by p73α was also evident in gel shift experiments. The results suggest that p73 can modulate p53 function by inhibiting its DNA binding and that overexpression of p73 in tumors might be a novel mechanism of inactivation of p53.
INTRODUCTION
Mutations in the p53 tumor suppressor protein play an important role in the origin of cancer (1–3). More than 90% of the mutations found in p53 are located within the DNA-binding domain, between amino acids 102 and 292 (4–6). Functional analysis of mutant proteins has shown that the common biological effect of mutations in human tumors is to inactivate p53 as a transcriptional factor (7,8). This leads to an inability to transactivate downstream genes such as p21/WAF1 and BAX, which are key executors of p53 activity (9–11).
A p53-related gene, p73, coding for a very similar amino acid sequence in the DNA-binding domain, has recently been cloned and characterized (12). p73 can transcriptionally activate p53 target genes such as p21 or BAX and induces apoptosis in p53-null Saos-2 cells (12,13).
The structural similarities between p53 and p73, at least in the DNA-binding domain, suggest that they might have similar roles in the cell (14,15). There are, however, significant differences: p73 was reported not to be inducible by exposing cells to DNA-damaging agents such as UV irradiation (12,13), although recently evidence of p73 induction has been reported (16–18); mutational analysis of p73 in human cancer failed to reveal significant tumor-associated mutations (19–23); p73 knock-out mice show specific neuronal disorders and a defective immunological response but do not develop spontaneous tumors, which are invariably found in p53-null mice (24,25).
Increased expression of p73 has been reported in some tumors (19,26–28). The observation that wild-type p73 may be activated in tumors suggests it might be important for tumor progression rather than suppression and that its role, if any, in cancer remains to be elucidated.
We examined the consequences of hyperexpression of p73 in wild-type p53-expressing human ovarian carcinoma cells. This study reports the activity of endogenous p53 and the possible interference between these two genes in clones obtained after transfection of p73 in the wild-type p53-expressing human ovarian cancer cell line A2780.
MATERIALS AND METHODS
Cell culture and treatment
The human ovarian carcinoma cell line A2780 and its subline A2780/E6 (29), the human osteosarcoma cell line Saos-2, the human ovarian cancer cell line SK23 (30) (obtained from the p53-null SKOV-3 cell line after transfection with temperature-sensitive Val135 mutant p53) and the human erythroleukemia cell line K562 were grown in RPMI 1640 supplemented with 10% fetal calf serum. p53–/– mouse embryonic fibroblasts (MEF cells) (kindly supplied by Dr T. Jacks, MIT, Cambridge, MA) were grown in DMEM supplemented with 10% heat-inactivated fetal calf serum. cis-Dichlorodiaminoplatinum (DDP) and taxol were obtained from Bristol Myers Squibb (USA), doxorubicin (DX) from Pharmacia & Upjohn (Milan, Italy).
Transfections
p73α-HA in pCDNA-3 was a gift from Dr D. Caput (Sanofi, Labege, France). pCDNA3-HA was constructed by inserting the HA epitope coding sequence in the multiple cloning site of pCDNA-3 (Invitrogen). p73αmut-HA (Arg292→His) was obtained by site-directed mutagenesis (GeneEditor; Promega, Italy). pG13Luc containing 13 copies of the RGC p53 site upstream of the luciferase gene was a gift from Prof B. Vogelstein (John Hopkins Oncology Center, Baltimore, MD). The human cyclin D1 promoter/luciferase construct (cycD1-Luc, D1-973pXP2) was obtained from Dr Muller (IMT, Marburg, Germany). The PGL2-control vector was from Promega, Italy.
The calcium phosphate precipitation method was used for stable and transient transfections (except for K562 cells, which were electroporated) and p73-expressing clones were selected in the presence of 500 µg/ml G418. Resistant clones were analyzed by western blotting and the results confirmed by northern blot analysis.
Luciferase activity was measured using a luciferase reporter kit (Dual System; Promega, Italy) following the manufacturer’s instructions. In transient transfection experiments 0.1–20 µg of pG13Luc, p73α-HA, p73αmut-HA, pCDNA3-HA, cycD1-Luc, pGL2-control vector and p53 plasmids were used and 2 µg of β-gal- or 0.5 µg of pRL-CMV-expressing plasmids were included as internal standards.
Immunoblotting and immunoprecipitation
Cell extracts were prepared by lysing cells in 50 mM Tris–HCl pH 7.4, 250 mM NaCl, 0.1% Nonidet P-40, 5 mM EDTA, 50 mM NaF, in the presence of aprotinin, leupeptin and PMSF as proteases inhibitors, for 30 min on ice.
Insoluble material was pelleted at 13 000 r.p.m. for 10 min at 4°C and the protein concentration was determined using a Bio-Rad assay kit (Bio-Rad, Richmond, CA). Thirty micrograms of total cellular proteins were separated by SDS–PAGE and electrotransferred to nitrocellulose. Immunoblotting was carried out with p53 monoclonal antibodies (DO-1; Santa Cruz Biotechnology) for p53, anti-p21 polyclonal antibodies (Santa Cruz Biotechnology), anti-MDM2 monoclonal antibodies (Calbiochem) and an anti-HA monoclonal antibody (12Ca5; Boehringer Mannheim, Monza Italy). Antibody binding was revealed by peroxidase secondary antibodies and visualized using ECL (Amersham, Italy).
Two hundred micrograms of protein were immunoprecipated using DO-1, anti-MDM2 and anti-HA monoclonal antibodies and protein A/G–Sepharose (Santa Cruz, Heidelberg, Germany). Immunoprecipitated proteins were separated using SDS–PAGE and p73HA and MDM2 proteins were revealed by western blotting using the corresponding antibody.
Northern blot analysis
Total RNA was isolated from cells growing in culture by the guanidine thiocyanate method according to standard procedures, fractionated by electrophoresis on a formaldehyde–agarose gel and transferred to nylon membranes (31).
Filters were hybridized with cDNAs 32P-labeled using a Rediprime kit (Amersham, Italy). Hybridizations were done in 50% formamide, 10% dextran sulfate, 1% SDS, 1 M NaCl at 42°C for 16 h, followed by two 10 min washes at room temperature with 2× SSC (150 mM NaCl, 15 mM sodium citrate) and one 30 min wash at 65°C in 2× SSC, 1% SDS.
Nucleic extracts and EMSA
Cells (106) were lysed on ice in buffer containing 10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, 0.8% Nonidet P-40. Nuclei were pelleted, extracted for 1 h in extraction buffer (20 mM HEPES pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF) and cleared by centrifugation at 12 000 g for 15 min. Ten micrograms of nuclear extracts were incubated on ice for 1 h in 15 µl of buffer containing 20 mM HEPES pH 7.5, 100 mM NaCl, 1 mM MgCl2, 0.1% Nonidet P-40, 5% sucrose, 1 µg poly(dI·dC), 2 µl of pAb421 hybridoma supernatant and 1 ng of 32P-end-labeled CON p53 binding site oligonucleotide (5′-GGACATGCCCGGGCATGTCG). As a non-specific competitor, a MYC oligonucleotide (5′-CCCCCACCACGTGGTGCCTGA) was used. DNA–protein complexes were separated by electrophoresis through a 5% native polyacrylamide gel, dried and visualized. In some experiments anti-p73 antibody (clone ER-13; NeoMarkers, Union City, CA) was added to the reaction mixture in an attempt to identify p73 bound to DNA.
RESULTS
The wild-type p53-expressing A2780 cell line can tolerate hyperexpression of p73α
Using a standard colony assay it has been shown that wild-type p73α exogenously expressed in SK-N-AS neuroblastoma and Saos-2 osteosarcoma cells suppressed growth (12,13). Both cell lines were also sensitive to wild-type p53 expression. Taking into account that the final effect of growth suppression induced by wild-type p53 often depends on the particular cellular context, and that the same could be true for wild-type p73, we transfected wild-type p73α into the human ovarian cancer cell line A2780 which expresses wild-type p53. In parallel we transfected p73α into Saos-2 cells. After 2 weeks there were no colonies in Saos-2 cells (results similar to data previously reported). However, there was colony production in the A2780 cell line. Clones were analyzed by anti-HA western blot and northern blot analysis (Fig. 1A and B). Two clones were selected which expressed p73 protein and RNA of the expected size. As a control for further experiments we used a clone (A2780/HA) obtained after transfection with plasmid pCDNA3-HA.
Figure 1.
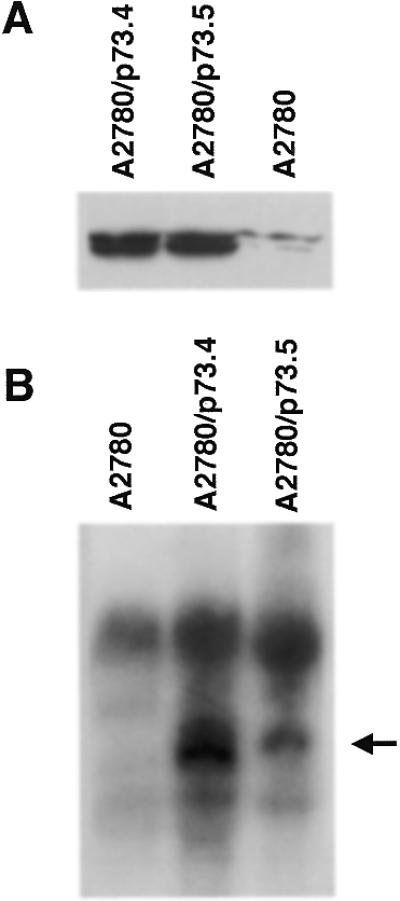
Expression of p73 in clones from A2780 cells. (A) Anti-HA western blot of whole cell extracts. (B) Northern blot analysis of p73 transcripts in selected clones.
p73 is not activated by doxorubicin, DDP and taxol
We analyzed three anticancer drugs, doxorubicin, taxol and DDP, for their ability to increase p53 and p73 in A2780/p73 clones. We did not find any further accumulation of p73 after drug treatment (Fig. 2A) under conditions in which there was marked accumulation of p53. Furthermore, this accumulation of p53 was restricted to the nucleus (Fig. 2B) in A2780/p73 clones, thus excluding cytoplasmic sequestration of p53 in the two A2780/p73 clones.
Figure 2.
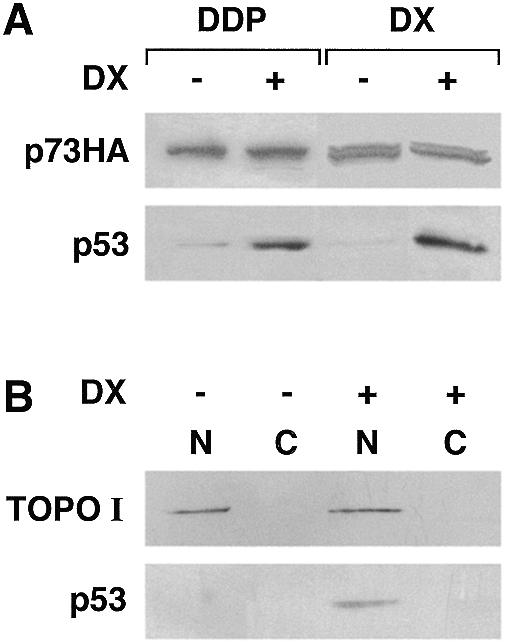
(A) p53 but not p73 is responsive to DNA damage. Blot of whole cell extracts obtained before and after treatment with DDP (15 µM for 2 h followed by 24 h incubation in drug-free medium) or DX (0.2 µM for 24 h) in clone A2780/p73.4. (B) Nuclear accumulation of p53 in A2780/p73.4 cells after DX treatment (conditions as in A). Blots were probed with antibodies to topoisomerase I (TOPO I) to verify the separation of nuclear and cytoplasmic fractions.
Reduced activation of p53 downstream genes after DDP treatment in A2780/p73 clones
We used the A2780/p73 clones to check whether activation of p53 was different in the presence or absence of p73. In A2780/p73 clones there was an increase in the basal levels of p21, bax and 14-3-3σ compared with the A2780 parental cells (Fig. 3A and B). However after drug-induced activation of p53, the consequent induction of p21, bax and 14-3-3σ was lower in the two A2780/p73 clones than in the A2780 parental cells or in the HA-transfected clone.
Figure 3.
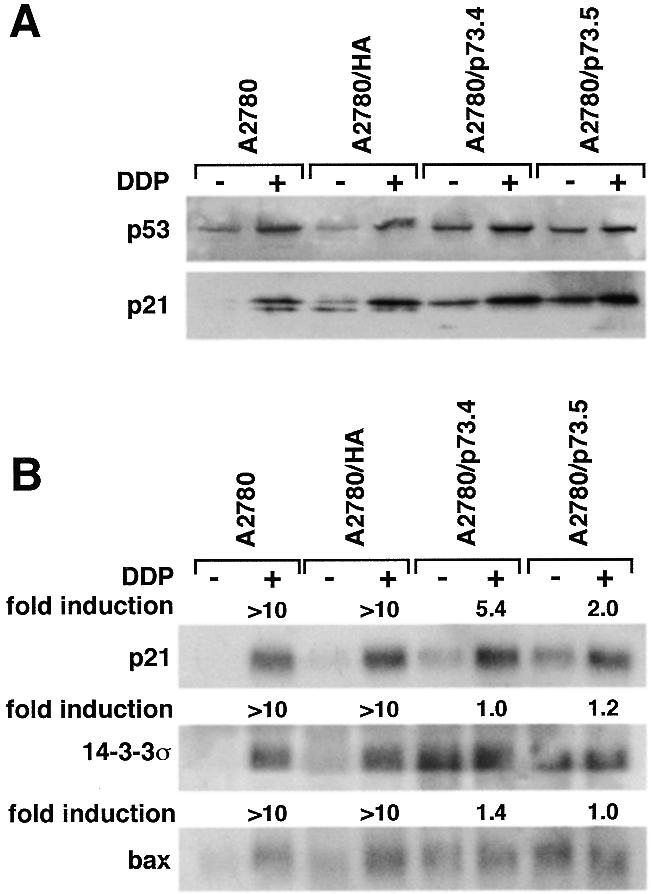
(A) Western blot analysis of p53 and p21 levels and (B) northern blot analysis of p21, 14-3-3σ and bax expression in cells untreated (–) or treated (+) for 2 h with 15 µM DDP followed by 24 h incubation in drug-free medium.
p53 and p73 do not co-immunoprecipitate
We examined whether the altered function of p53 in A2780/p73 was connected with an interaction with p73, by immunoprecipitation/western blot analysis of proteins from A2780 and A2780/p73 cells. Proteins immunoprecipitated by mAb DO-1, pAb421 (for p53) or HA (for p73) were fractionated by SDS–PAGE gel and western blot analysis was carried out with the anti-HA tag antibody. We found no HA tag response in immunoprecipitates with DO-1 and pAb421 from A2780 and A2780/p73 cells (data not shown).
Decreased transcriptional activity of p53 in A2780/p73 clones
In the next set of experiments, A2780 and A2780/p73 cells were transfected with a luciferase reporter plasmid containing 13 copies of the p53 binding sites (a β-gal plasmid was included in each transfection for normalization). The introduction of equal amounts of reporter plasmid showed that the transactivation capacity of p53 in A2780/p73 clones was much lower than in the A2780/HA clone (Fig. 4A).
Figure 4.
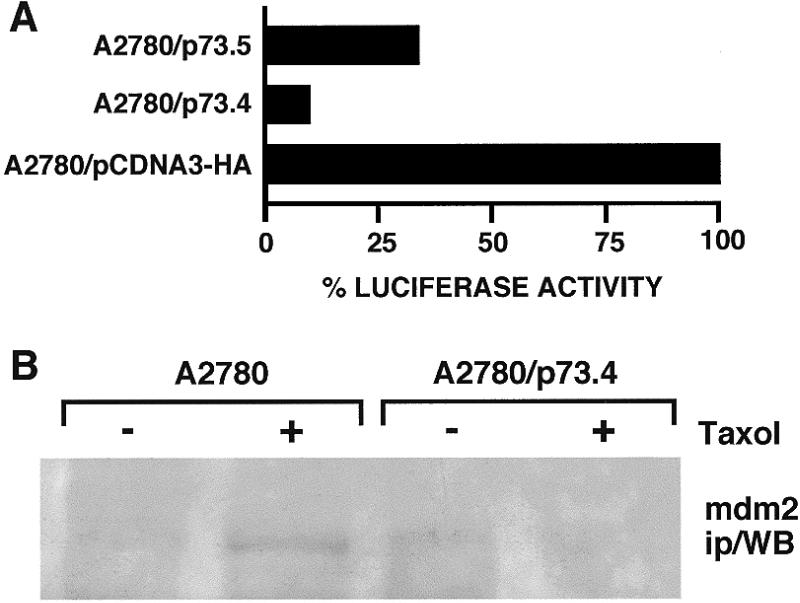
(A) Activation of p53 response plasmid pG13Luc transfected into clones A2780/HA and A2780/p73 (10 µg of pG13 luc were used). (B) MDM2 immunoprecipitates from clones A2780 (lanes 1 and 2) and A2780/p73.4 (lanes 3 and 4) without treatment (lanes 1 and 3) and after treatment with 100 nM taxol for 24 h (lanes 2 and 4). Aliquots of 200 µg of protein were immunoprecipitated with anti-MDM2 mAb and analyzed by SDS–PAGE. Western blot analysis was carried out with anti-MDM2 antibody.
To check whether the high expression of p73, which has a strong similarity in the DNA-binding domain to p53, induces accumulation of MDM2 protein, which in turn inactivates p53 (32–34), we used immunoprecipitation/western blot analysis to measure the amount of MDM2 in A2780 and A2780/p73 (Fig. 4B). We found only traces of MDM2 protein after induction of p53 with taxol in A2780 but not in A2780/p73 clones. Similar results were obtained by northern blot analysis (data not shown).
Transcriptional activity of p53 and p73 in transient transfection experiments
We initially examined whether p73 activates luciferase expression under the control of a p53-responsive element in Saos-2, K562, SKOV3, A2780/E6 and p53–/– MEF cells, all cell lines lacking p53 expression. The data obtained, in agreement with that previously reported (13,35), indicate that p73 can bind the same sequences recognized by p53 and therefore can work as a transcriptional factor (data not shown). We next performed transient transfections in A2780 and SK23 cells with reporter plasmids and a p73 expression plasmid. Increasing the amount of p73 transfected led to a decrease in p53-dependent luciferase activity in A2780 cells (Fig. 5A). In contrast, transfection of p73αmut-HA did not alter p53-dependent luciferase activity. In the same system, co-transfection of increasing amounts of p53 expression plasmid induced accumulation of luciferase (Fig. 5B). In SK23 cells, which contain a temperature-sensitive mutant murine p53, shifting the temperature to 32°C increased luciferase activity. Co-transfection with p73 again strongly reduced this activity (Fig. 5C) while the transfection of similar amounts of p53 expression plasmid slightly increased luciferase activity.
Figure 5.

Activation of p53-responsive plasmid pG13luc co-transfected into A2780 with increasing amounts of p73wt or mut (A) or p53 (B) expression plasmid. (C) Activation of p53 responsive plasmid pG13Luc co-transfected into SK23 cells with 10 µg of p73 or p53 expression plasmid at either 37 or 32°C.
Co-transfection of p53- and p73-expressing plasmids in the p53-null cell line SKOV3 resulted in similar findings (Fig. 6). Both p73 and p53 alone induced luciferase activity when pG13-Luc was used, although again p73 was much less active. In the presence of p73, p53 transcriptional activity was reduced, and only by increasing the ratio of p53 to p73 to 20:1 could we see an increase in luciferase activity over p73 alone. Co-transfection of p53 and a mutant p73 did not significantly change the transcriptional activity of p53.
Figure 6.

Co-transfection of a p53-expressing plasmid and wild-type or mut p73-expressing plasmids in p53-null SKOV3 cells. Increasing amounts of p53 were co-transfected with 1 µg of p73-expressing plasmid.
When under the same experimental conditions the p73 expression plasmid was co-transfected with the p53-unrelated reporter plasmid pGL2 or cycD1-Luc, no decrease in luciferase activity was found (data not shown).
Loss of transactivation capability of p53 in the presence of p73 is correlated with changes in specific DNA-binding activity of p53
Using the gel shift assay, we analyzed the ability of p53 to specifically bind DNA in clone A2780/p73 in comparison with A2780 (Fig. 7A, lanes 4–6). In our hands, neither p73 nor p53 bound to DNA could be detected in the absence of specific antibodies. We could detect binding of p53 to DNA, after addition of anti-p53 antibody (pAb421). Using an anti-p73 monoclonal antibody (clone ER-13), reported to induce a supershift when used in combination with recombinant p73 (36), we could not detect any binding. It should be noted, however, that no data are as yet available in the literature showing a retarded band attributable to p73 in extracts prepared from cells growing in culture.
Figure 7.
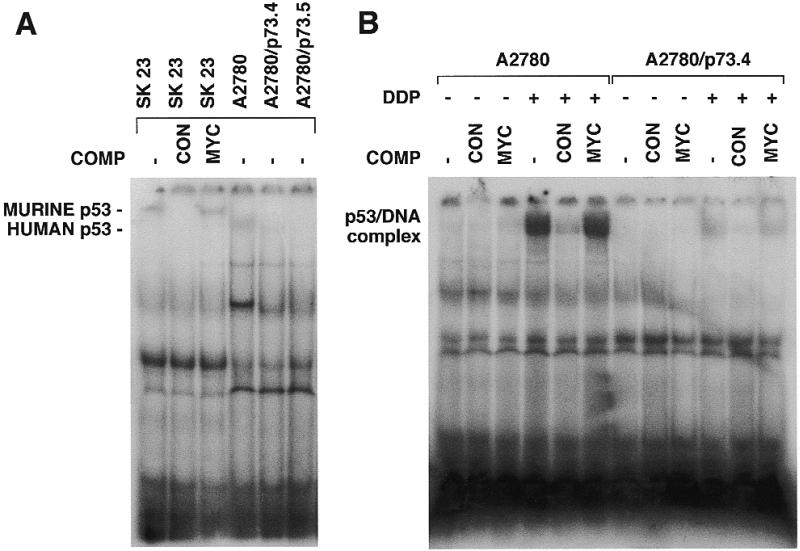
(A) DNA binding activity of wild-type p53 lysates from SK23, A2780, A2780/p73.4 or A2780/p73.5 cells. A 50-fold molar excess of specific (CON) or non-specific (MYC) unlabeled oligonucleotide was added to the reaction. Protein complexes were separated in 5% TBE–polyacrylamide gels. (B) DNA-binding activity of wild-type p53 after treatment with DDP. Lysates from A2780 and A2780/p73 cells untreated (–) or treated (+) with 15 µM DDP for 24 h. Competitions with a 50-fold molar excess of specific (CON) or non-specific (MYC) unlabeled oligonucleotide were included in the reaction.
p53 binding to its consensus site was strongly reduced in extracts from p73-expressing clones. After treatment of A2780 and A2780/p73 cells with DDP, these differences were even more pronounced (Fig. 7B).
Possible sequestration of p53 from its DNA binding site through competitive binding with p73
Nuclear extracts from A2780/p73 had less p53-binding activity than extracts from wild-type A2780. One possibility is that nuclear extracts from A2780/p73 may contain some factor (p73) which competes for the formation of p53–DNA complexes. We mixed nuclear extracts from A2780 treated with DDP with nuclear extracts from A2780/p73 (both clones gave the same result) before and after treatment. Extracts from A2780/p73 may possess a factor that inhibits p53–DNA complex formation (Fig. 8A, compare lane 2 with lanes 3–6).
Figure 8.
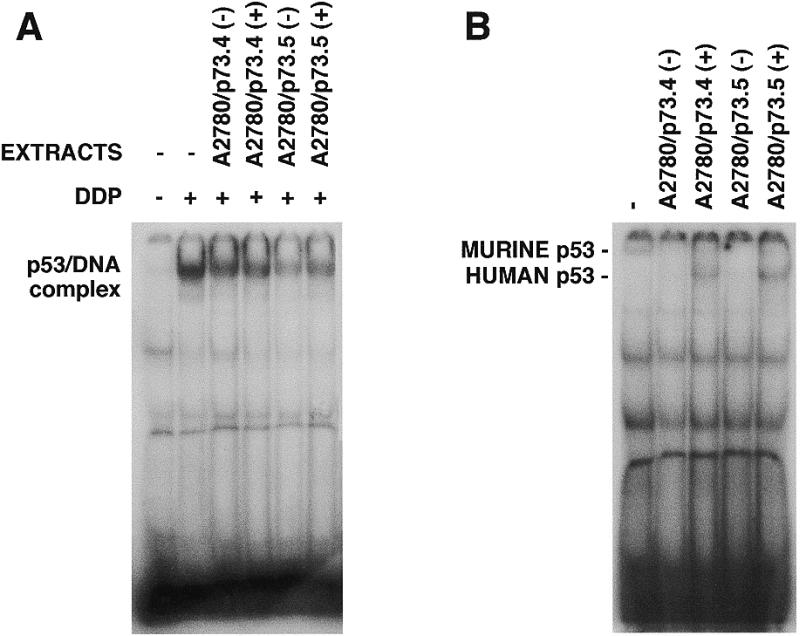
DNA-binding activity of p53 in competitive experiments with proteins obtained from A2780/p73 clones. (A) DNA-binding activity of p53 from A2780 without treatment (–) or after treatment with 15 µM DDP for 24 h (+). Nucleic extracts from DDP-treated A2780 cells were mixed with extracts from untreated (–) or DDP-treated (+) A2780/p73.4 or A2780/p73.5 cells. (B) DNA-binding activity of p53 from SK23 cells at 32°C (–); the same mixed with extracts from A2780/p73.4 or A2780/p73.5 cells untreated (–) or treated with DDP (+).
Similar experiments were done with extracts from SK23 cells (Fig. 8B). Results with mouse wild-type p53-binding activity in the presence of extracts from A2780/p73 cells were similar and even clearer because of differences in the molecular weights of DNA–protein complexes formed by mouse and human wild-type p53.
DISCUSSION
Loss or inactivation of p53 is thought to contribute to the development of a large proportion of human cancers. The typical mechanism of p53 inactivation is multiple mutations, most of them generally concentrated in the DNA-binding domain. However, many other factors can contribute to p53 inactivation, such as cytoplasmic sequestration, overexpression of viral or cellular proteins which can bind and block p53 function, and others (37–40).
Many p53 functions are mediated by its transcriptional activity, which can be induced in response to a variety of DNA-damaging agents. Induction of transcriptional activity is thought to be due to the accumulation of p53, mostly by stabilization, and to post-translational modifications, mainly through phosphorylation at the N- or C-terminus (39,41–44).
Recently new genes have been cloned which code for proteins having very high homology with p53 in the DNA-binding site. p73 is the first of these relatives which binds and activates the same sequences recognized by p53 (12).
We investigated how hyperexpression of p73α can influence p53 activity in the human ovarian carcinoma cell line A2780 expressing wild-type p53.
We could isolate clones of A2780 overexpressing p73α which grow in culture like the parental cell line. The presence of p73α did not significantly change the basal levels of p53. The two p73α-expressing clones had higher basal levels of bax, p21 and 14-3-3σ mRNAs than parental A2780 and A2780/HA cells but these levels did not further increase after DDP treatment under conditions in which a >10-fold induction of these mRNAs is observed in A2780 and A2780/HA cells. Only p21 showed, in A2780/p73 clones, a moderate increase over basal levels. This is likely to be due to the known ability of DDP or other stimuli to increase in a p53-independent way the levels of p21 (45,46). The fact that the two p73α-expressing clones have higher basal levels of p53 downstream genes suggests that in some way they adapt to grow in the presence of relatively high levels of genes normally expressed at very low levels. It should be noted, however, that after DNA damage, i.e. when activation of these genes should result in cell cycle arrest or apoptosis, there is no further increase in their level and this could result in an altered cellular response to stress. Since we could not detect direct binding of p73 to p53 recognition sequences in cell extracts, we cannot exclude that other mechanisms, different from direct inhibition of p53 by p73, could be responsible for the lack of induction of p53 target genes after damage in p73-overexpressing cells. In both parental cells and in p73α-expressing clones, p53 was present in the nucleus, thus excluding cytoplasmic sequestration as a possible mechanism of inactivation. Similarly, the levels of MDM2, which might be activated by p73 and could be responsible for the inactivation of p53 (33,34,37), were very low in A2780 cells and did not change significantly in p73α-expressing clones, thus excluding the possibility that MDM2 plays a major role in this cellular system.
We therefore concentrated on p53 transcriptional ability in the presence of p73 hyperexpression. We present several pieces of evidence for a decrease in p53 transcriptional activity. Introduction of reporter plasmid pG13Luc (containing p53-specific DNA binding sites) in A2780/HA and A2780/p73 clones led to a very pronounced reduction in luciferase activity in A2780/p73 in comparison with A2780/HA cells. This basal luciferase activity reflects calcium phosphate transfection-induced stress, which has been reported to activate p53 (47). The same results were obtained by co-transfecting pG13Luc and p73-HA in A2780 cells, strongly suggesting that the results found in A2780/p73 clones are not due to clonal selection. Increasing the amount of p73 transfected reduced the p53-dependent luciferase activity. This is not due to a general transcriptional repression by p73, since transfection experiments with mutp73 (not able to bind DNA) did not lead to a reduction in activity, even at the highest concentration of plasmid tested. These results, which were confirmed in transfection experiments in p53-null cells, indicate that an intact DNA-binding domain is necessary for p73 to reduce p53 activity, suggesting that ‘occupation’ of the p53-binding site by p73 is the possible mechanism accounting for these effects. Moreover, A2780/p73 clones transfected with the PGL2 or cyc-D1 reporter plasmid did not show the reduced luciferase activity observed with PG13Luc, indicating that in these clones no general transcription/translation defects are present and the observed p73 repression is ‘p53 specific’.
The results are quite unexpected, p73, which has a strong homology with p53 in the DNA-binding domain and may bind the same DNA-specific elements (12,36), reduces binding of p53 to this sequence and activation of pG13Luc.
As previously reported (24), we could also not detect any direct p53–p73 binding in these cells, at least in co-precipitation experiments, in agreement with recent reports (48), again suggesting that the p73-induced decrease in p53 transcriptional activity might be due to competition for the same DNA binding site.
If this were true, as indicated by gel shift competition experiments in which nuclear extracts from p73-overexpressing clones were mixed with extracts of wild-type p53-expressing cells (of human or murine origin), p73 should have a similar affinity for the DNA binding site but should lack the activation ability of p53 or, at least, should have less transactivation ability. This could be due to a difference in the post-translational modifications between p73 and p53.
Although at this stage of knowledge it is entirely speculative, the recent finding that in some tumors the silent allele of p73 may be activated suggests that this protein might act as an oncogene. The activation of p73 might therefore inactivate p53 by competing for its DNA binding site, thus preventing the activation of p53 downstream genes.
Acknowledgments
ACKNOWLEDGEMENTS
We are particularly grateful to Dr Daniel Caput for the generous gift of p73 cDNA. The generous contribution of the Italian Association for Cancer Research is gratefully acknowledged. Faina Vikhanskaya is a visiting scientist from the Institute of Cytology (Russian Accademy of Sciences), St Petersburg, Russia.
REFERENCES
- 1.Hollstein M., Sidransky,D., Vogelstein,B. and Harris,C.C. (1991) Science, 253, 49–53. [DOI] [PubMed] [Google Scholar]
- 2.Harris C.C. (1996) Br. J. Cancer, 73, 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harris C.C. and Hollstein,M. (1993) N. Engl. J. Med., 329, 1318–1327. [DOI] [PubMed] [Google Scholar]
- 4.Levine A.J., Momand,J. and Finlay,C.A. (1991) Nature, 351, 453–456. [DOI] [PubMed] [Google Scholar]
- 5.Hainaut P., Soussi,T., Shomer,B., Hollstein,M., Greenblatt,M., Hovig,E., Harris,C.C. and Montesano,R. (1997) Nucleic Acids Res., 25, 151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beroud C., Verdier,F. and Soussi,T. (1996) Nucleic Acids Res., 24, 147–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kern S.E., Pietenpol,J.A., Thiagalingam,S., Seymour,A., Kinzler,K.W. and Vogelstein,B. (1992) Science, 256, 827–830. [DOI] [PubMed] [Google Scholar]
- 8.Chen J.Y., Funk,W.D., Wright,W.E., Shay,J.W. and Minna,J.D. (1993) Oncogene, 8, 2159–2166. [PubMed] [Google Scholar]
- 9.Ludwig R.L., Bates,S. and Vousden,K.H. (1996) Mol. Cell. Biol., 16, 4952–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine A.J. (1997) Cell, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- 11.el Deiry W.S., Tokino,T., Velculescu,V.E., Levy,D.B., Parsons,R., Trent,J.M., Lin,D., Mercer,W.E., Kinzler,K.W. and Vogelstein,B. (1993) Cell, 75, 817–825. [DOI] [PubMed] [Google Scholar]
- 12.Kaghad M., Bonnet,H., Yang,A., Creancier,L., Biscan,J.C., Valent,A., Minty,A., Chalon,P., Lelias,J.M., Dumont,X. et al. (1997) Cell, 90, 809–819. [DOI] [PubMed] [Google Scholar]
- 13.Jost C.A., Marin,M.C. and Kaelin,W.G.,Jr (1997) Nature, 389, 191–194. [DOI] [PubMed] [Google Scholar]
- 14.Dickman S. (1997) Science, 277, 1605–1606. [DOI] [PubMed] [Google Scholar]
- 15.Oren M. (1997) Cell, 90, 829–832. [DOI] [PubMed] [Google Scholar]
- 16.Yuan Z.M., Shioya,H., Ishiko,T., Sun,X., Gu,J., Huang,Y.Y., Lu,H., Kharbanda,S., Weichselbaum,R. and Kufe,D. (1999) Nature, 399, 814–817. [DOI] [PubMed] [Google Scholar]
- 17.Agami R., Blandino,G., Oren,M. and Shaul,Y. (1999) Nature, 399, 809–813. [DOI] [PubMed] [Google Scholar]
- 18.Gong J.G., Costanzo,A., Yang,H.Q., Melino,G., Kaelin,W.G.,Jr, Levrero,M. and Wang,J.Y. (1999) Nature, 399, 806–809. [DOI] [PubMed] [Google Scholar]
- 19.Takahashi H., Ichimiya,S., Nimura,Y., Watanabe,M., Furusato,M., Wakui,S., Yatani,R., Aizawa,S. and Nakagawara,A. (1998) Cancer Res., 58, 2076–2077. [PubMed] [Google Scholar]
- 20.Nomoto S., Haruki,N., Kondo,M., Konishi,H. and Takahashi,T. (1998) Cancer Res., 58, 1380–1383. [PubMed] [Google Scholar]
- 21.Han S., Semba,S., Abe,T., Makino,N., Furukawa,T., Fukushige,S., Takahashi,H., Sakurada,A., Sato,M., Shiiba,K. et al. (1999) Eur. J. Surg. Oncol., 25, 194–198. [DOI] [PubMed] [Google Scholar]
- 22.Chi S.G., Chang,S.G., Lee,S.J., Lee,C.H., Kim,J.I. and Park,J.H. (1999) Cancer Res., 59, 2791–2793. [PubMed] [Google Scholar]
- 23.Tokuchi Y., Hashimoto,T., Kobayashi,Y., Hayashi,M., Nishida,K., Hayashi,S., Imai,K., Nakachi,K., Ishikawa,Y., Nakagawa,K. et al. (1999) Br. J. Cancer, 80, 1623–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaghad M., Yang,A., Bonnet,H., Walker,N., Sharpe,A., McKeon,F. and Caput,D. (1998) In Proceedings of the 9th p53 Workshop, 1998. Elounda Beach, Crete, Greece, p. 65.
- 25.Donehower L.A., Harvey,M., Slagle,B.L., McArthur,M.J., Montgomery,C.A.J., Butel,J.S. and Bradley,A. (1992) Nature, 356, 215–221. [DOI] [PubMed] [Google Scholar]
- 26.Mai M., Yokomizo,A., Qian,C., Yang,P., Tindall,D.J., Smith,D.I. and Liu,W. (1998) Cancer Res., 58, 2347–2349. [PubMed] [Google Scholar]
- 27.Yokomizo A., Mai,M., Tindall,D.J., Cheng,L., Bostwick,D.G., Naito,S., Smith,D.I. and Liu,W. (1999) Oncogene., 18, 1629–1633. [DOI] [PubMed] [Google Scholar]
- 28.Loiseau H., Arsaut,J. and Demotes-Mainard,J. (1999) Neurosci. Lett., 263, 173–176. [DOI] [PubMed] [Google Scholar]
- 29.Vikhanskaya F., Vignati,S., Beccaglia,P., Ottoboni,C., Russo,P., D’Incalci,M. and Broggini,M. (1998) Exp. Cell Res., 241, 96–101. [DOI] [PubMed] [Google Scholar]
- 30.Vikhanskaya F., Erba,E., D’Incalci,M. and Broggini,M. (1994) Nucleic Acids Res., 22, 1012–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 32.Lane D.P. and Hall,P.A. (1997) Trends Biochem. Sci., 22, 372–374. [DOI] [PubMed] [Google Scholar]
- 33.Haupt Y., Maya,R., Kazaz,A. and Oren,M. (1997) Nature, 387, 296–299. [DOI] [PubMed] [Google Scholar]
- 34.Kubbutat M.H., Jones,S.N. and Vousden,K.H. (1997) Nature, 387, 299–303. [DOI] [PubMed] [Google Scholar]
- 35.Di Como C.J., Gaiddon,C. and Prives,C. (1999) Mol. Cell. Biol., 19, 1438–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marin M.C., Jost,C.A., Irwin,M.S., DeCaprio,J.A., Caput,D. and Kaelin,W.G.,Jr (1998) Mol. Cell. Biol., 18, 6316–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Momand J., Zambetti,G.P., Olson,D.C., George,D. and Levine,A.J. (1992) Cell, 69, 1237–1245. [DOI] [PubMed] [Google Scholar]
- 38.Werness B.A., Levine,A.J. and Howley,P.M. (1990) Science, 248, 76–79. [DOI] [PubMed] [Google Scholar]
- 39.Meek D.W. (1998) Cell Signal., 10, 159–166. [DOI] [PubMed] [Google Scholar]
- 40.Vogelstein B. and Kinzler,K.W. (1992) Cell, 70, 523–526. [DOI] [PubMed] [Google Scholar]
- 41.Shieh S.Y., Ikeda,M., Taya,Y. and Prives,C. (1997) Cell, 91, 325–334. [DOI] [PubMed] [Google Scholar]
- 42.Gottlieb T.M. and Oren,M. (1996) Biochim. Biophys. Acta, 1287, 77–102. [DOI] [PubMed] [Google Scholar]
- 43.Siliciano J.D., Canman,C.E., Taya,Y., Sakaguchi,K., Appella,E. and Kastan,M.B. (1997) Genes Dev. 11, 3471–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kapoor M. and Lozano,G. (1998) Proc. Natl Acad. Sci. USA, 95, 2834–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vikhanskaya F., D’Incalci,M. and Broggini,M. (1995) Int. J. Cancer, 61, 397–401. [DOI] [PubMed] [Google Scholar]
- 46.Michieli P., Chedid,M., Lin,D., Pierce,J.H., Mercer,W.E. and Givol,D. (1994) Cancer Res., 54, 3391–3395. [PubMed] [Google Scholar]
- 47.Renzing J. and Lane,D.P. (1995) Oncogene, 10, 1865–1868. [PubMed] [Google Scholar]
- 48.Davison T.S., Vagner,C., Kaghad,M., Ayed,A., Caput,D. and Arrowsmith,C.H. (1999) J. Biol. Chem., 274, 18709–18714. [DOI] [PubMed] [Google Scholar]