


Abstract
Most eubacteria, and all eukaryotes examined thus far, encode homologs of the DNA mismatch repair protein MutS. Although eubacteria encode only one or two MutS-like proteins, eukaryotes encode at least six distinct MutS homolog (MSH) proteins, corresponding to conserved (orthologous) gene families. This suggests evolution of individual gene family lines of descent by several duplication/specialization events. Using quantitative phylogenetic analyses (RASA, or relative apparent synapomorphy analysis), we demonstrate that comparison of complete MutS protein sequences, rather than highly conserved C-terminal domains only, maximizes information about evolutionary relationships. We identify a novel, highly conserved middle domain, as well as clearly delineate an N-terminal domain, previously implicated in mismatch recognition, that shows family-specific patterns of aromatic and charged amino acids. Our final analysis, in contrast to previous analyses of MutS-like sequences, yields a stable phylogenetic tree consistent with the known biochemical functions of MutS/MSH proteins, that now assigns all known eukaryotic MSH proteins to a monophyletic group, whose branches correspond to the respective specialized gene families. The rooted phylogenetic tree suggests their derivation from a mitochondrial MSH1-like protein, itself the descendent of the MutS of a symbiont in a primitive eukaryotic precursor.
INTRODUCTION
In most eukaryotes and eubacteria, evolutionarily conserved long patch mismatch repair systems increase DNA replication fidelity 100- to 1000-fold, by removing base substitution and frameshift mismatches that escape polymerase proofreading (1,2). In Escherichia coli, MutS protein homodimers bind preferentially to base mispairs (e.g. G/T) and to insertion/deletion loop-outs (e.g. AAAA/TTT) in DNA. MutS and MutL then activate the MutH protein to make the excision-initiating nick in the unmethylated strand at the nearest hemimethylated d(GATC) sequence, most likely via a translocation/search process that requires ATP hydrolysis (3). Delay in methylation of newly replicated d(GATC) sequences by the E.coli DNA-adenine methylase results in direction of MutHLS-dependent mismatch-provoked nicking to the nascent DNA strand, providing essential strand specificity.
Both eubacteria and eukaryotes express strongly conserved MutS and MutL homologs, but the E.coli MutH/d(GATC) methylation mechanism is not found in eukaryotes, or even in all eubacteria; the mechanism of strand discrimination here remains poorly understood. Strikingly, the single eubacterial MutS is replaced by at least six MutS homologs in eukaryotes and MutL by at least five MutL homologs (1,4). These homolog families show strong conservation of amino acids in certain common domains. The members of eukaryotic MSH families also show strong family-specific conservation of sequence and function.
Genetic and biochemical analyses have demonstrated that evolution of distinct eukaryotic MSH families has been accompanied by acquisition of new functions. Mitochondrially targeted MSH1 is necessary for mitochondrial stability in yeast (5), MSH2, MSH3 and MSH6 correct replication errors in nuclear DNA (1,2), and MSH4 and MSH5 play positive roles in meiotic recombination (6,7). Unlike prokaryotic MutS homodimers, eukaryotic mismatch recognition proteins function as heterodimers, with distinct but overlapping mismatch specificities: MSH2·MSH6 heterodimers recognize base/base mismatches and small insertion/deletion loop-outs, whereas MSH2·MSH3 heterodimers recognize loop-outs of various sizes, but not base mispairs (8–11).
MSH proteins thus pose interesting questions for phylogenetic/evolutionary analysis. Was there a single or multiple evolutionary precursor(s) of MSH subfamilies? What was the source of the precursor(s)? What was the order of occurrence of the diversification/specialization steps: acquisition of nuclear replication fidelity and meiotic recombination functions, shifting to heterodimeric structures, development of substrate recognition specificities? We have used a phylogenetic approach, in conjunction with genetic and biochemical information, to address these questions. This requires analysis of a wide range of sequences from highly diverse organisms and extraction of the maximum amount of phylogenetic information. Some earlier studies of MSH phylogenies were incomplete with respect to the range of groups of MutS-like proteins analyzed (12,13), descriptions of methods (14,15) or definition of the phylogenetic root. A more complete study focused only on a very highly conserved MutS/MSH region (16). We have used alignments and analyses that extend over complete protein sequences, in order to capture as much phylogenetic information as possible. We also used a tree-independent analysis of the phylogenetic signal termed RASA (relative apparent synapomorphy analysis) to identify phylogenetically problematical sequences; some of these were then excluded from the analysis and, as a result, we have maximized tree stability. The results of our analysis appear more consistent with known biochemical functions of MutS and MSH proteins than results of previous studies. Rooted phylogenetic analysis of complete MutS-like sequences indicates that all eukaryotic MSH proteins are monophyletic and originated from a eubacterial endosymbiont.
MATERIALS AND METHODS
Alignments and phylogenetic methods
Several different MutS-like protein sequences were used to search the latest version of the SWISSPROT protein database, using Blast 2.0 (17). Using a method for the creation of multiple alignments described previously (18), ClustalW and Blast 2.0 alignments of all available MutS-like protein sequences were combined into a final representative alignment (available upon request) employing the Genetic Data Environment (GDE) (19), with the PAM 250 protein similarity matrix, and both full-length and individual domains used to identify homologous regions. We excluded some putative proteins identified in eukaryotic genomes (i.e. Caenorhabditis elegans MSH2, MSH4 and MSH6) but not confirmed by cDNA sequences. Differential shadings of alignments were carried out with BOXSHADE version 3.03. Bootstrap and phylogenetic reconstruction methods were performed with PHYLIP version 3.51 (20).
RASA
Phylogenetic signal and taxon-variance ratios were determined using RASA v.2.2 (21,22). RASA software and documentation for the Macintosh were downloaded from the internet at http://loco.biology.unr.edu/archives/rasa/rasa.html and used to identify long branches, as follows. First, we calculated for every pair (i,j) of protein sequences two parameters: Eij, the phenetic similarity, a measure of the similarity of the i,j pair of sequences, corresponding here to the total number of identical amino acids (character states) at variable sites in a given alignment of the two proteins; RASij, the pairwise cladistic uniqueness, which reflects the uniqueness of the i,j pair with respect to the other sequences, corresponding to the total number of sequences k ≠ i,j not showing the shared i,j amino acid, summed over all positions of i,j identity. For the set of all i,j pairs, RASij is on average expected to be a linear function of Eij (21), since the more positions of identity scored, the higher the total RASij score. Second, in order to define a statistical measure of the phylogenetic signal, the pair of RASij,Eij matrices were studied in the RASA regression and two taxon-variance terms for each protein sequence (taxon) i were calculated. The phenetic-variance term VarE(i) is the statistical variance for the set of Eij values for fixed i, all j ≠ i, relative to the mean of those values. The phylogenetic (cladistic) variance term VarRAS(i) reflects the summed squared deviations of RASij values from the linear regression line with respect to the Eij values, for all (n – 1) j ≠ i. Comparison of the ratios VarRAS(i)/VarE(i) for all i sequences provides a means to diagnose long phylogenetic branches, because these two variance measures are proportionate when the amount of branch length heterogeneity on the true tree is low. These measures lose proportionality for long-branch taxa (22), because the phenetic variance VarE(i) contributed by the long-branch taxon is low, but its cladistic variance VarRAS(i) is inflated.
We also used an independent parameter, the test statistic tRASA, to measure the phylogenetic signal itself (21). The tRASA statistic is generated using Student’s t-test to assess the significance of the deviation of the observed slope of RASij versus Eij from a null slope, corresponding to a null hypothesis that considers the possibility that the character states are distributed randomly among the taxa. The null slope is calculated by assuming equiprobable distributions of E and RAS among all taxa. While significantly positive tRASA values are usually associated with hierarchical patterns in a character state matrix of the type that is expected when truly phylogenetic patterns predominate in the matrix, significantly negative tRASA values usually indicate some source of disruption of the hierarchy, such as long-branch taxa (22).
RESULTS AND CONCLUSIONS
Classification of MutS-like proteins according to sequence organization and co-occurrence with MutL proteins
Inspection of initial alignments of all available MutS-like protein sequences identified two clearly distinct groups of proteins. In a representative list (Table 1) of MutS-like sequences used in this study, the two groups show clear differences in organization of primary structure and genomic context (Table 1, columns 5 and 6). For example, Bacillus subtilis contains two mutS-like genes (here called mutS and msp, see below) and only one mutL. Each corresponding MutS-like protein sequence contains a C-terminal conserved domain, however, the protein designated MutS contains two other conserved domains not present in the protein designated MSP (Table 1 and Fig. 1A). Furthermore, in the case of Helicobacter pylori, msp is present, but both mutS and mutL are absent. A previous analysis also identified two groups (lineages) of MutS-like proteins (16), but the proposed composition of these differs in important ways from the two groups identified here. We argue below that because of their gross differences in functional domain structure the two groups delineated by our analysis most likely have different biological functions, and therefore suggest the designation MutS/MSH for eubacterial MutS proteins previously designated MutS1 plus all eukaryotic MSH proteins, and MSP (MutS paralog) for the novel eubacterial open reading frames previously designated MutS2 (16,23). Thus, these designations discriminate between genes that function similarly to MutS and those that simply contain similar domains. We consider below and in the next section whether MSP proteins should be included in our analyses that address the questions of MSH origin and diversification.
Table 1. Sequences used in this study.
| Protein | Organisma | Class | Length | Conserved domains (coordinates)b |
Genomic repertoire | ||
|---|---|---|---|---|---|---|---|
| (amino acids) | N | M | C | ||||
| Prokaryotes | |||||||
| MutS | Streptococcus pneumoniaec | MutS/MSH | 844 | 9–47 | 250–342 | 539–782 | mutS, mutL |
| MutS | Bacillus subtilisc | MuLS/MSH | 852 | 1–39 | 244–336 | 534–776 | mutS, mutL, msp |
| MutS | Escherichia colid | MutS/MSH | 853 | 13–51 | 267–355 | 552–794 | mutS, mutL |
| MutS | Haemophilus influenzaed | MutS/MSH | 854 | 13–51 | 267–357 | 554–796 | mutS, mutL |
| MutS | Azotobacier vinelandiid | MutS/MSH | 855 | 10–48 | 264–354 | 551–792 | mutS, mutL |
| MutS | Thermus aquaticusd | MutS/MSH | 811 | 16–54 | 250–339 | 527–760 | mutS, mutL |
| MutS | Synechocystis sp.d | MutS/MSH | 912 | 62–100 | 334–426 | 623–870 | mutS, mutL, msp |
| MutS | Rickettsia prowazekiid | MutS/MSH | 891 | 23–61 | 288–379 | 581–826 | mutS, mutL |
| MSP | Bacillus subtilisd | MSP | 785 | – | – | 276–513 | mutS, mutL, msp |
| MSP | Synechocystis sp.d | MSP | 822 | – | – | 279–544 | mutS, mutL, msp |
| MSP | Helicobacter pylorid | MSP | 762 | – | – | 277–510 | msp |
| Eukaryotes | |||||||
| MSH1 (mtMutS) | Sarcophytom glaucume | MutS/MSH | 982 | 5–47 | 334–423 | 631–876 | msh1 and other? |
| MSH1 | Saccharomyces cerevisiae | MutS/MSH | 959 | 61–99 | 342–434 | 685–938 | msh1/2/3/4/5/6, mlh1/2/3, pms1/2 |
| MSH2 | Saccharomyces cerevisiae | MutS/MSH | 964 | 19–57 | 295–406 | 620–877 | msh1/2/3/4/5/6, mlh1/2/3, pms1/2 |
| MSH2 | Arabidopsis thaliana | MutS/MSH | 937 | 24–61 | 294–387 | 599–855 | msh1/2/3/6/7, mlh1, pms2, other? |
| MSH2 | Xenopus leavis | MutS/MSII | 933 | 19–57 | 299–393 | 601–849 | msh2 and other? |
| MSH2 | Homo sapiens | MutS/MSH | 934 | 19–57 | 300–394 | 602–849 | msh2/3/4/5/6, mlh1, pms2, other? |
| MSH3 | Saccharomyces cerevisiae | MutS/MSH | 1047 | 164–202 | 447–539 | 757–1005 | msh1/2/3/4/5/6, mlh1/2/3, pms1/2 |
| MSH3 (SWI4) | Saccharomyces pombe | MutSIMSII | 993 | 106–144 | 402–495 | 697–948 | msh3 and other? |
| MSH3 | Homo sapiens | MutS/MSH | 1128 | 232–270 | 535–628 | 831–1091 | msh2/3/4/5/6, mlh1, pms2, other? |
| MSH4 | Saccharomyces cerevisiae | MutS/MSII | 878 | – | 265–356 | 574–813 | msh1/2/3/4/5/6, mlh1/2/3, pms1/2 |
| MSH4 | Homo sapiens | MutS/MSH | 936 | – | 310–402 | 620–858 | msh2/3/4/5/6, mlh1, pms2, other? |
| MSH5 | Saccharomyces cerevisiae | MutSIMSH | 901 | – | 256–348 | 569–843 | msh1/2/3/4/5/6, mlh1/2/3, pms1/2 |
| MSH5 | Homo sapiens | MutS/MSH | 834 | – | 231–319 | 529–776 | msh1/2/3/6/7, mlh1, pms2, other? |
| MSH6 | Saccharomyces cerevisiae | MutS/MSH | 1242 | 314–352 | 614–706 | 905–1164 | msh1/2/3/4/5/6, mlh1/2/3, pms1/2 |
| MSH6 | Arabidopsis thaliana | MutS/MSH | 1338 | 395–433 | 709–802 | 1023–1281 | msh2/3/4/5/6, mlh1, pms2, other? |
| MSH6 | Homo sapiens | MutS/MSH | 1360 | 409–447 | 733–825 | 1057–1321 | msh2/3/4/5/6, mlh1, pms2, other? |
| Archaea | |||||||
| MutS? | Methanobacterium thermoautotrophicum | MutS/MSH | 647 | 186–240 | 402–623 | mutS? | |
aBold, entire genomic sequence currently available bN, N-terminal conserved domain; M, middle conserved domain; C, C-terminal conserved domain; –, absence of domain; coordinates correspond to positions of amino acids in domains..
cGram-positive eubacterium.
dGram-negative eubacterium.
eOctocoral.
Figure 1.

(A) Structure of MutS/MSH and MSP proteins. Light gray bar, N-terminal extension found in MSH3 and MSH6 proteins. Different shaded boxes represent regions of high homology (>50% sequence similarity) within classes; blue, MutS/MSH N-terminal domain; red, MutS/MSH middle domain; green, MutS/MSH C-terminal domain; yellow, MSP C-terminal domain. Domains D1 (ATP-binding) and D2 (‘DELGRG’) retain strict conservation in the MutS/MSH class. I indicates 18 amino acid insertion in the Saccharomyces sp. MSP protein. (B) BOXSHADE output of the MutS/MSH N-terminal domain. Black boxes represent identical amino acids in >80% of the sequences, gray boxes similar amino acids in >50% of the aligned sequences based on Dayhoff’s PAM250 matrix. P (in upper bar), amino acid position; [1], 17 amino acid insertion in the MSH4 protein of S.cerevisiae. (C) MutS/MSH N-terminal conserved domain structure. Aro, (+) and (–) represent aromatic (F/Y/W) and positively (D/E) and negatively (K/R) charged amino acid side chains conserved in MutS/MSH subfamilies, respectively; (n) represents any amino acid. The third position corresponds to the Phe39 (F39) position of T.aquaticus MutS. (D) Middle conserved domain as in (B). The arrow indicates a known dominant negative mutation (R305H) in E.coli MutS. [2], 14 amino acid insertion in the MSH2 protein of S.cerevisiae.
All eubacterial MutS and eukaryotic MSH proteins (MutS/MSH class) appear to function in DNA error correction and/or recombination pathways. These proteins are highly similar with respect to sequence and domain structure (Fig. 1A). Particularly well conserved domains appear in the N-terminal, middle and C-terminal regions (Fig. 1). The N-terminal conserved domain, found only in bacterial MutS and eukaryotic MSH1, MSH2, MSH3 and MSH6 proteins, is predicted to closely interact with DNA mismatches, based on photo-crosslinking and mutagenesis studies of the Phe39 residue in the N-terminal domain of Thermus aquaticus MutS protein (24). MutS/MSH families show different patterns of conservation of residues close to Phe39 (Fig. 1B and C) (24). Proteins that bind base/base mismatches, eubacterial MutS and eukaryotic MSH1, MSH2 and MSH6 proteins, conserve an aromatic (F/Y/W) (F/Y) doublet (e.g. Phe39/Tyr40 in T.aquaticus MutS). In MSH3 proteins, which may participate in binding looped out DNA strands, the two aromatic residues alternate with two positively charged residues: Y (K/R) (Y/F) (R/K). MSH4 and MSH5 proteins, which appear to play no role in error correction, and presumably do not recognize mismatches, show no conservation here.
The middle domain appears in all MutS/MSH proteins and is conserved more or less homogeneously among all MutS/MSH families (Fig. 1D). Three-dimensional structure analysis suggests that this domain lies on the surface of Homo sapiens MSH2 (25). Mutation of a highly conserved arginine in this domain of E.coli MutS (R305H) confers a dominant-negative phenotype (26). Although function cannot be definitively assigned to this domain, it might be involved in protein–protein interactions, such as those between subunits of MutS/MSH protein heterodimers, or in interaction with other components of the mismatch repair apparatus. Two previous studies have focused on mapping interaction domains of human and yeast MSH heterodimers, indicating possible N-terminal and C-terminal heterodimerization domains in MSH2, MSH3 and MSH6 (27,28). These regions do not correspond to the middle domain identified here, thus suggesting that the middle domain is not directly involved in heterodimer formation. However, more definitive studies will be needed to further elucidate MSH interaction domains.
The C-terminal conserved domain (Fig. 1A, alignment not shown; 16) is found in all MutS/MSH sequences and shows the highest conservation of all three domains. It contains helix–turn–helix and nucleotide/magnesium-binding (Walker box) subdomains and is predicted to interact with DNA and to mediate ATP binding and hydrolysis (3,14–16).
No function has been identified for genes of the MSP class, which are found only in certain eubacteria. The predicted MSP protein sequences are relatively conserved over their entire lengths, but differ markedly from those of MutS/MSH protein sequences (Fig. 1A). First, MSP sequences lack both the N-terminal and middle conserved domains of MutS/MSH sequences. Second, MSP sequences contain unique terminal extensions of ~200 amino acids. Third, although certain MSP domains are similar to the conserved C-terminal domains of MutS/MSH sequences, they appear instead near the middle of MSP sequences and do not strictly conserve the spacing of critical functional subdomains seen in MutS/MSH C-terminal domains (Fig. 1A).
Eubacterial MutS proteins have thus far been observed only in conjunction with MutL proteins, and eukaryotic MSH proteins only in conjunction with MLH (PMS) proteins. All eukaryotic MSH proteins, except perhaps MSH1, appear to interact with one or more MLH proteins. MSH2·MSH3 or MSH2·MSH6 heterodimers, plus MLH1·PMS2 heterodimers, are required for mismatch repair functions in yeast and human cells (9,29); direct interactions among these proteins have been demonstrated (30,31). Furthermore, MSH2·MSH3 has recently been shown to interact with a heterodimer of MLH1·MLH3 (4). Genetic epistasis studies indicate that MSH4, MSH5 and MLH1 act in the same meiosis-specific pathway (32), perhaps again interacting with one another in multi-protein complexes. In contrast, MSP genes are found in some eubacteria that lack mutL-like genes. Where both MSP and mutL genes are present, a ‘true’ mutS gene of the mutS/MSH class is also present (see for example 33). MSP proteins thus constitute a class distinct from MutS/MSH proteins.
Among the three complete archeabacterial genome sequences reported, only that of Methanobacterium thermoautotrophicum contains a mutS-like gene (34). This gene predicts a protein, ~150 amino acids shorter than eubacterial MutS proteins, that lacks the N-terminal domain and portions of the middle domain of MutS/MSH sequences, but shows no MSP-like C-terminal extension or other MSP-like characteristics. Methanobacterium thermoautotrophicum encodes no MutL-like protein, so its MutS-like protein might be considered to define a third class.
Optimization of the analysis of the MutS/MSH and MSP phylogeny
In order to rigorously address questions relating to MSH origin and diversification, we initially needed to resolve two general points. First, we needed to define analytical techniques that used as much sequence information as possible, thus maximizing the phylogenetic signal. Second, we needed objective criteria to determine whether MSP sequences, already identified as likely to constitute a distinct class of separate origin (see above), should be included. These points prove to be interrelated: in analysis of complete protein sequences, masking to exclude sequence gaps and regions of ambiguous alignment between the MutS/MSH and MSP groups invariably yielded alignments of only a highly conserved 280 residue region found in the C-terminal regions of MutS/MSH proteins (Fig. 1A), because of the marked structural differences outside this region. We were able to construct neighbor-joining (NJ) minimum evolution and parsimony trees, using (masked) alignment of 28 MutS/MSH and MSP C-terminal protein sequences, representing all classes of MutS-like proteins (Fig. 1). However, we observed several ambiguities in each of a number of trees generated from different sets of sequences based on this alignment (see below).
To quantitatively estimate the amount of phylogenetic signal generated by considering only C-terminal regions, we employed RASA (21; see Materials and Methods). We used taxon-variance ratios to identify long branches (22). In optimizing our analyses in order to increase confidence in the resulting trees (see below), our criteria were: (i) increased values of the tRASA test statistic (a measure of the strength of the phylogenetic signal); (ii) homogeneity of taxon-variance ratios (absence of long-branch attraction), a measure of tree stability; (iii) improved bootstrap support throughout the trees. Figure 2A shows a condensed neighbor-joining tree and taxon-variance ratios, produced by analysis of C-terminal regions of a representative set of all MutS-like proteins (including MutS/MSH and MSP proteins, and the putative archeabacterial MutS from M.thermoautotrophicum). This analysis produced a tRASA value of 9.3 (P << 0.005, see Materials and Methods). The taxon-variance ratio for the M.thermoautotrophicum MutS sequence indicated that it might be problematical here, in the sense of causing long-branch attraction. In the tree, the MSP group branched together with M.thermoautotrophicum MutS within the eubacterial cluster, and the majority of the tree branches showed low bootstrap support. A second analysis, that excluded the M.thermoautotrophicum MutS, produced a higher tRASA value of 11.2 (P << 0.005) and the taxon-variance ratios now suggested that the H.pylori MSP sequence was problematical (data not shown). The tree again showed low bootstrap support for most branches. Strikingly, the MSP group now branched within the eukaryotic MSH cluster, together with the Sarcophytom glaucum mtMutS and close to the MSH4 and MSH5 groups. In both the first (Fig. 2A) and the second case, the MSP group branched closest to the sequence having the longest branch in the respective tree, M.thermoautotrophicum MutS in one case and S.glaucum mtMutS in the other. Confidence in these two trees was thus low, because of the uneven taxon-variance ratios and low bootstrap support. In fact, other analyses using the same C-terminal region of the alignment, but using different subsets of sequences, produced similar outcomes (tree instability), including long-branch attraction and low bootstrap values (data not shown). These examples, representative of tree instabilities observed in ana-lyses of only C-terminal regions, suggest that the C-terminal region alone is not sufficient to resolve critical branching patterns in phylogenetic analyses of MutS-like sequences. The lower phylogenetic signal and instability of trees associated with C-terminal analyses that include both MutS/MSH and MSP protein sequences (Fig. 2A), the gross differences between MutS/MSH and MSP protein sequences (Fig. 1A) and the lack of correlation between the occurrence of MSP and MutL proteins, all suggest that MSP proteins are not closely related to eubacterial MutS proteins. We propose that msp genes arose independently, perhaps through domain shuffling in eubacteria, and have excluded them from the analyses described below.
Figure 2.

Schematic neighbor-joining (NJ) trees and corresponding RASA taxon-variance ratios using different combinations and/or regions of MutS-like protein sequences. For phylogenetic methods see the legend to Figure 3. (A) Representative set of all known MutS-like sequences, aligned as described in the text with a 280 amino acid C-terminal mask. (B) Entire protein regions of MutS/MSH proteins, aligned as described in the text with a complete sequence mask for a core set of 19 sequences. RASA taxon-variance ratios were determined as described in Materials and Methods.
We were now able to include all conserved domains (N-terminal, middle and C-terminal) in alignments (Fig. 1). We conducted RASA and phylogenetic analyses of complete MutS/MSH protein sequences, to determine whether this would increase the overall phylogenetic signal, bootstrap confidence values and more homogeneous taxon-variance ratios. In an analysis of 24 complete MutS/MSH sequences representing all MutS/MSH groups, individual members of the MSH4 and MSH5 groups and the S.glaucum mtMutS now showed problematical taxon-variance ratios. Removal of these sequences produced a ‘core’ tree with no apparent long branches, revealed in the taxon-variance analysis. Figure 2B shows the condensed neighbor-joining distance tree and the taxon-variance ratios for this core set of 19 complete MutS/MSH protein sequences. In comparison to the C-terminal analyses (Fig. 2A), the bootstrap values are significantly increased and show less ambiguity for all branches. All branches are resolved (Fig. 2B) and stable, and the tRASA value increases to 16.7 (P << 0.005) (Fig. 2B). The core tree therefore provides a benchmark reference point when analyses are expanded to include more diverged sequences (e.g. MSH4 and MSH5). Thus, expanded analyses in which branching of the core sequences were significantly altered would a priori be considered questionable.
Reconstruction of the MutS/MSH phylogeny
We next addressed the original MSH origin/diversification questions, using complete protein sequences, excluding the MSP class, and referring back to the core branching pattern. On the basis of protein and DNA sequence data (35,36) and the complementary biochemistry of energy metabolism (37), the eukaryotic cell has been proposed to be the result of endosymbiosis between an archaebacterium (host) and an α-proteobacterium (symbiont) similar to the modern Rickettsia prowazekii (38). Did this eukaryotic DNA mismatch repair gene family evolve from eubacterial and/or archeabacterial precursors subsequent to such an event? Because archaebacteria seem not to possess mismatch repair pathways involving MutS and MutL proteins, eukaryotic MSH genes most likely came from the eubacteria. In our final phylogenetic reconstruction we used 24 complete MutS/MSH sequences, including all groups of MutS/MSH proteins: the core sequences analyzed above (Fig. 2B), the S.glaucum mtMutS and the MSH4 and MSH5 subgroups. The analysis yielded the highest degree of bootstrap support and phylogenetic signal (tRASA = 14.3, P << 0.005) of any analysis that included all groups of MutS/MSH sequences, despite the potentially problematical MSH4/MSH5 and S.glaucum mtmutS taxon-variance ratios. Although the MSH4 and MSH5 subgroups branched together (consistent with the apparent roles of both in meiotic recombination), the mtMutS from S.glaucum did not now branch with (or close to) the MSH4/MSH5 group (Fig. 3), in contrast to the tree shown in Figure 2A and a tree proposed elsewhere (16). Exclusion of any or all of these sequences in this analysis did not significantly change the overall topology of the tree, its bootstrap values or its close similarity to the previous core tree (Fig. 2B). This stable tree thus appears to provide the best estimate of MutS/MSH sequence relationships. The expanded neighbor-joining tree (Fig. 3) and the very similar parsimony tree (not shown) suggest the following postulated scenario for evolution of MSH proteins in eukaryotic cells.
Figure 3.
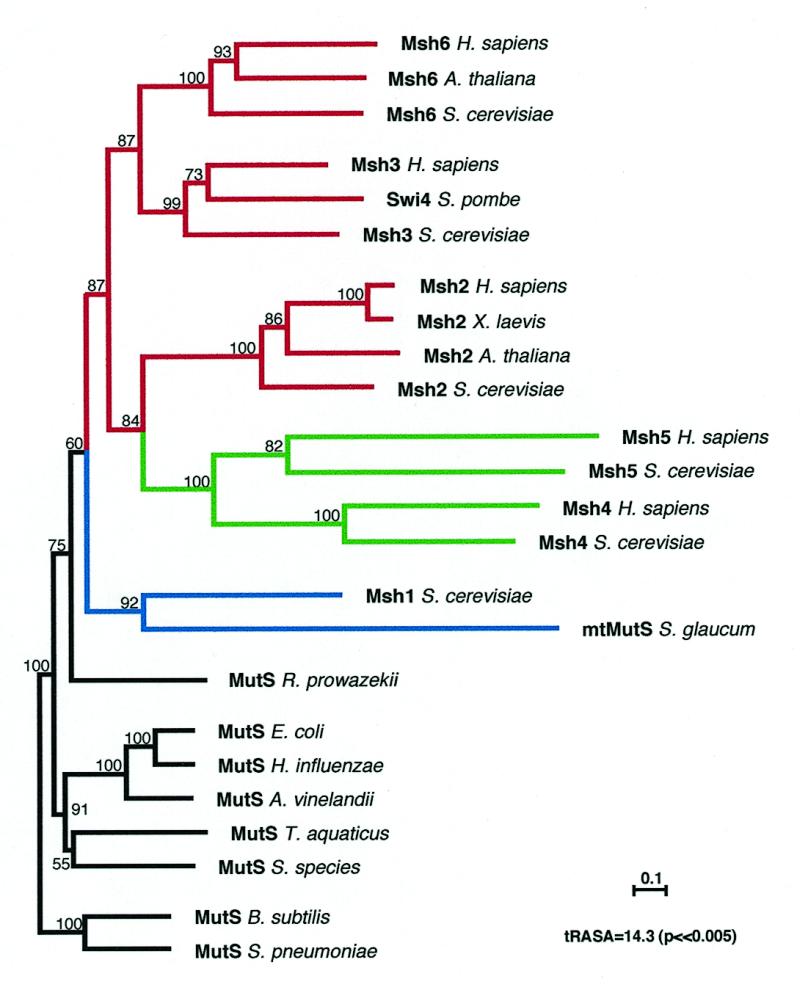
Neighbor-joining (NJ) tree for Dayhoff PAM distances among MutS/MSH protein sequences. Protein parsimony trees were also constructed using PROTPARS (Phylip 3.5), which produced very similar results (not shown). Gaps and regions of ambiguous alignment were excluded from the analysis. The horizontal scale bar indicates evolutionary distance. Numbers above each branch represent the number of times the branch was found in 100 bootstrap replicas. The B.subtilis and S.pneumoniae MutS protein sequences (Gram-positive eubacteria) were used as an outgroup. The masked alignment used to generate this tree (and the tree in Fig. 2B) included the N-terminal, middle and C-terminal regions. Differential shadings reflect the known functional role of each group: black, eubacterial mismatch repair and recombination; blue, mitochondrial mismatch repair in eukaryotes; green, meiotic recombination in eukaryotes; red, nuclear mismatch repair in eukaryotes. All eukaryotic homologs (MSH) are encoded by the nuclear genome, except the mitochondrially encoded mtMutS from S.glaucum.
An engulfed eubacterial (α-proteobacterial) cell, the precursor of the mitochondrion (35,36,39), was the source of the common ancestral MSH gene. The α-proteobacterial mutS gene could have been transferred to the nucleus after the engulfment, as were many other (now) nuclear genes (40,41). Among eukaryotic MSH proteins, the mitochondrial MSH1 protein subfamily is the deepest branching group (Fig. 3) and the yeast MSH1 shows the highest similarity (38%) to the α-proteobacterium R.prowazekii MutS. The R.prowazekii MutS branches within a clade that includes all eukaryotic MSH protein sequences in the final tree, with a bootstrap value of 75% (Fig. 3). In addition, a similar branching pattern is observed in the core tree, here with a bootstrap value of 95%. The R.prowazekii genome does not encode an MSP protein, but it does encode a MutL protein, suggesting the presence of a functional mismatch repair pathway. The close branching of R.prowazekii MutS and eukaryotic MSH1 sequences suggests that eubacterial mutS genes acquired during endosymbiotic events are the direct ancestors of the mitochondrial MSH1 genes, which in turn gave rise to all other MSH genes. Since our analysis here suggests that all members of the eukaryotic MSH gene family are monophyletic (appear to share a common ancestor) with the mitochondrial MSH1 subfamily, any putative former post-replication error correction pathway already present in the protoeukaryote that engulfed the α-proteobacterial mitochondrial ancestor would seem to have disappeared.
Interestingly, the octocoral S.glaucum mtMutS sequence and the yeast MSH1 sequence branch together, although they are encoded respectively by the S.glaucum mitochondrial genome and the yeast nuclear genome (42,43). Others have proposed that the S.glaucum mtmutS is an example of a nuclear gene transferred to the mitochondrial genome (13) or that it is of MSP origin (16). Our analysis is more consistent with two different possibilities: (i) the S.glaucum mtmutS is an MSH1 gene that originated from an α-proteobacterial mutS (not MSP) gene, as did other MSH1 genes, but in this case has remained in its original mitochondrial genome, exemplifying an intermediate step in the transfer and evolution of MSH genes in eukaryotes; (ii) the S.glaucum mtmutS (msh1) has been transferred twice, the second time from the nucleus back into the mitochondrial genome.
A phylogenetic analysis of complete MutS/MSH protein sequences (excluding MSP sequences, but including the M.thermoautotrophicum MutS) places the single archaebacterial MutS within the eubacterial MutS group, branching closest to the T.aquaticus MutS, with a bootstrap value of 78% in the neighbor-joining tree (data not shown). This is in disagreement with organismal and rDNA (5S) phylogenies, which place M.thermoautotrophicum in the Euryachaeota group of the Archaea (44). Methanobacterium thermoautotrophicum MutS may therefore have been acquired through horizontal gene transfer, as previously suggested (16), possibly from another thermophile such as T.aquaticus. Archeabacteria, if capable of post-replication error correction, would seem to use a pathway independent of MutS or MutL proteins.
Rooting the tree
One problem with deep phylogenies (those that encompass highly diverse taxa) is the selection of an appropriate root. This is usually performed using an outgroup (45). In the case of the MutS/MSH phylogeny, we believe the most appropriate root to be within the eubacteria. First, as discussed above, MSH1 genes most likely originated from eubacterial mutS genes following mitochondrial endosymbiotic events, providing a source for the gene duplications that eventually resulted in specialized eukaryotic nuclear MSH genes. Second, there is no evidence that the nuclear MSH genes have an origin independent of MSH1. Archeabacteria do not seem to have a MutS/L pathway, so they are unlikely to be the source of MSH genes. Alternatively, another eubacterium not involved in the original endosymbiotic events might have independently transferred its mismatch repair gene(s) to primitive eukaryotic cells, by the mechanism recently proposed by Doolittle (46) for example. However, no phylogenetic relationships apparent in our analysis suggest that nuclear MSH genes arose from any eubacterial mutS genes other than the α-proteobacterial mutS, via its proposed descendent, the mitochondrial MSH1, regardless of where the root is placed. Our analyses thus suggest that mutS (and mutL) genes appeared early (eubacterial evolution) and were later transferred to eukaryotes as part of the genomes of (Gram-negative) α-proteobacterial endosymbionts. We therefore chose the Gram-positive MutS sequences (here B.subtilis and Streptococcus pneumoniae) as the root for the tree. Indeed, rooted RASA (45) of the rest of the taxa, using a Gram-positive eubacterial root, resulted in the highest tRASA value of any rooted analyses of the entire set of 24 MutS/MSH sequences, indicating that these bacteria are the optimal outgroup (data not shown).
Eukaryotic MSH gene duplication and specialization
The evolution of multiple eukaryotic nuclear MSH gene families may have begun with transfer of a copy of the post-symbiosis mitochondrial MSH1 to the nucleus. The remaining mitochondrial MSH1 would subsequently have been lost, as is typical when nuclear genes encode mitochondrially targeted proteins. Duplication of the nuclear MSH gene would have allowed one to encode an MSH1 protein targeted back to the mitochondrion and the other to give rise to the whole set of nuclear mismatch repair genes, by further duplication and specialization (Fig. 4). The increase in the DNA content of the eukaryotic genome, the development of diploidy, the appearance of multiple chromosomes and the evolution of meiotic recombination may have been concomitant with the evolution of specialized mismatch repair/recombination activities. The first duplication of the nuclear MSH ancestor appears to have yielded the predecessor of the MSH3 and MSH6 genes and the predecessor of the MSH2, MSH4 and MSH5 genes (Figs 3 and 4). This duplication may have been the first step towards mispair recognition by heterodimers. The MSH3 and MSH6 predecessor apparently duplicated again to give rise to the MSH3 and MSH6 subfamilies. MSH3 and MSH6 retained interaction with MSH2, but evolved specialized but overlapping recognition functions. In the MSH2, MSH4 and MSH5 gene family, an MSH4 and MSH5 predecessor with specialized meiotic functions may have diverged from MSH2, giving rise to the individual MSH4 and MSH5 gene families.
Figure 4.

Schematic diagram of MutS/MSH evolution. We suggest that mutS genes arose and evolved in the eubacteria and that a mutS gene was transferred to eukaryotes through mitochondrial endosymbiotic events. This gene (now MSH1) was then transferred to the nucleus and gave rise to all eukaryotic MSH genes.
CONCLUSIONS
To address questions regarding the origin and diversification of eukaryotic MSH proteins, we have systematically optimized our analysis of complete protein sequences. In doing so, we have obtained a comprehensive phylogenetic reconstruction of all known eubacterial, archeabacterial and eukaryotic groups of MutS-like sequences, and identified two broad classes of MutS-like protein sequences, namely MutS/MSH and MSP, consistent with the biochemical function of the former with MutL-like proteins. This approach has established a general framework to accurately classify newly identified MutS-like genes whose functions are unknown. A valuable by-product of this analysis was delineation of three domains that appear in all MutS/MSH proteins thought to be involved in error correction; these domains should provide useful landmarks for establishing alignments and inferring biochemical functions for new sequences.
We suggest that because the conserved C-terminal region of MutS-like proteins does not contain enough phylogenetic information, attempts to employ only this region in comprehensive analyses invariably yield low bootstrap support and unstable trees. The anomalous taxon-variance ratios for certain sequences obtained by C-terminal analyses are indicative of long-branch attraction, which leads to erroneous trees and unlikely hypotheses about the evolution of the MutS/MSH and MSP proteins. We suggest that future phylogenetic reconstructions use the complete amino acid sequences of MutS/MSH proteins, but include only those proteins that do not appear to threaten the accuracy of the tree estimate by long-branch attraction. Our final analysis yields a phylogenetic tree, with high bootstrap support for all branches, that for the first time assigns all known eukaryotic MSH proteins to a single family of proteins having distinct functional subfamilies (Fig. 3), and suggests a eubacterial endosymbiotic origin for all eukaryotic MSH genes.
Preliminary phylogenetic analyses of MutL/MLH protein sequences suggest a similar pattern of evolution for both the MSH and MLH genes, each from a single eukaryotic ancestor (MSH1 and MLH1, respectively; unpublished observations; 15). Interestingly, no gene in the Saccharomyces cerevisiae genome appears to encode a mitochondrially targeted MLH protein and no such genes have yet been identified in other eukaryotes. Further phylogenetic analyses may determine whether both mutS and mutL are likely to have been acquired by eukaryotes at the same time and/or by similar mechanisms. Although S.cerevisiae MSH1 has been shown to bind mismatched DNA substrates with affinities similar to those of other MutS/MSH proteins (47), it and other MSH1 proteins may function via novel error avoidance mechanisms independently of MutL homologs (48).
It would be of great interest to identify organisms with smaller (or larger) sets of nuclear MSH and MLH genes, representing different stages in the duplication–specialization process or gene loss. Plants also encode highly conserved MSH2, MSH3 and MSH6 proteins (12,49); their sequences clearly show conserved biochemical and phylogenetic relationships to their respective MSH subfamilies (K.M.C. and J.B.H., unpublished observations; Fig. 3). This suggests that MSH duplication–specialization events occurred before the evolution of green plants and that plant MSH subfamilies were not acquired from the endosymbiotic bacteria that gave rise to chloroplasts (cyanobacteria). Although no obvious MSH1-like gene is apparent in the C.elegans genome (K.M.C. and J.B.H., unpublished observations) and no MSH1 gene has yet been reported for other animals, an MSH1-like gene is present in the Arabidopsis thaliana genome and shows clear phylogenetic relationships to other MSH1 proteins (G.M.G., K.M.C. and J.B.H, unpublished observations). It remains possible that new subfamilies of MSH genes will be identified in eukaryotes.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs Jonathan Eisen and Jeff Blanchard for discussions. This work was supported by NSF grant 9631048-MCB to J.B.H. and in part by an AP Sloan/DOE post-doctoral fellowship to J.L.-W. and research grants from the NIH to M. Nei. This is contribution 11598 from the Oregon Agricultural Experiment Station.
REFERENCES
- 1.Modrich P. and Lahue,R. (1996) Annu. Rev. Biochem., 65, 101–133. [DOI] [PubMed] [Google Scholar]
- 2.Modrich P. (1991) Annu. Rev. Genet., 25, 229–253. [DOI] [PubMed] [Google Scholar]
- 3.Allen D.J., Makhov,A., Grilley,M., Taylor,J., Thresher,R., Modrich,P. and Griffith,J.D. (1997) EMBO J., 16, 4467–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Flores-Rozas H. and Kolodner,R.D. (1998) Proc. Natl Acad. Sci. USA, 95, 12404–12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reenan R.A. and Kolodner,R.D. (1992) Genetics, 132, 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hollingsworth N.M., Ponte,L. and Halsey,C. (1995) Genes Dev., 9, 1728–1739. [DOI] [PubMed] [Google Scholar]
- 7.Ross-Macdonald P. and Roeder,G.S. (1994) Cell, 79, 1069–1080. [DOI] [PubMed] [Google Scholar]
- 8.Acharya S., Wilson,T., Gradia,S., Kane,M.F., Guerrette,S., Marsischky,G.T., Kolodner,R. and Fishel,R. (1996) Proc. Natl Acad. Sci. USA, 93, 13629–13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marsischky G.T., Filosi,N., Kane,M.F. and Kolodner,R. (1996) Genes Dev., 10, 407–420. [DOI] [PubMed] [Google Scholar]
- 10.Drummond J.T., Li,G.-M., Longley,M.J. and Modrich,P. (1995) Science, 268, 1909–1912. [DOI] [PubMed] [Google Scholar]
- 11.Palombo F., Iaccarino,I., Nakajima,E., Ikejima,M., Shimada,T. and Jiricny,J. (1996) Curr. Biol., 6, 1181–1184. [DOI] [PubMed] [Google Scholar]
- 12.Culligan K.M. and Hays,J.B. (1997) Plant Physiol., 115, 833–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pont-Kingdon G., Okada,N.A., Macfarlane,J.L., Beagley,C.T., Watkins-Sims,C.D., Cavalier-Smith,T., Clark-Walker,G.D. and Wolstenholme,D.R. (1998) J. Mol. Evol., 46, 419–431. [DOI] [PubMed] [Google Scholar]
- 14.Fishel R. and Wilson,T. (1997) Curr. Opin. Genet. Dev., 7, 105–113. [DOI] [PubMed] [Google Scholar]
- 15.Kolodner R. (1996) Genes Dev., 10, 1433–1442. [DOI] [PubMed] [Google Scholar]
- 16.Eisen J.A. (1998) Nucleic Acids Res., 26, 4291–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 18.Hogweg P. and Hesper,B.J. (1984) J. Mol. Evol., 20, 175–186. [DOI] [PubMed] [Google Scholar]
- 19.Smith S.W., Overbeek,R., Woese,C.R., Gilbert,W. and Gillevet,P.M. (1994) CABIOS, 10, 671–675. [DOI] [PubMed] [Google Scholar]
- 20.Felsenstein J. (1989) Cladistics, 5, 164–166. [Google Scholar]
- 21.Lyons-Weiler J., Hoelzer,G.A. and Tausch,R.J. (1996) Mol. Biol. Evol., 13, 749–757. [DOI] [PubMed] [Google Scholar]
- 22.Lyons-Weiler J. and Hoelzer,G.A. (1997) Mol. Phylogenet. Evol., 8, 375–384. [DOI] [PubMed] [Google Scholar]
- 23.Eisen J.A., Kaiser,D. and Meyers,R.M. (1997) Nature Med., 3, 1076–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malkov V.A., Biswas,I., Camerini-Otero,R.D. and Hsieh,P. (1998) J. Biol. Chem., 272, 23811–23817. [DOI] [PubMed] [Google Scholar]
- 25.De Las Alas M.M., De Bruin,R.A.M., Eyck,L.T., Los,G. and Howell,S.B. (1998) FASEB J., 12, 653–663. [DOI] [PubMed] [Google Scholar]
- 26.Wu T.-H. and Marinus,M.G. (1994) J. Bacteriol., 176, 5393–5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alani E. (1996) Mol. Cell. Biol., 16, 5604–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guerrette S., Wilson,T., Gradia,S. and Fishel,R. (1998) Mol. Cell. Biol., 18, 6616–6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li G.-M. and Modrich,P. (1995) Proc. Natl Acad. Sci. USA, 92, 1950–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Habraken Y., Sung,P., Prakash,L. and Prakash,S. (1998) J. Biol. Chem., 273, 9837–9841. [DOI] [PubMed] [Google Scholar]
- 31.Prolla T.A., Pang,Q., Alani,E., Kolodner,R.D. and Liskay,R.M. (1994) Science, 265, 1091–1093. [DOI] [PubMed] [Google Scholar]
- 32.Hunter N. and Borts,R.H. (1997) Genes Dev., 11, 1573–1582. [DOI] [PubMed] [Google Scholar]
- 33.Kaneko T., Sato,S., Kotani,H., Tanaka,A., Asamizu,E., Nakamura,Y., Miyajima N., Hirosawa,M., Sugiura,M., Sasamoto,S. et al. (1996) DNA Res., 3, 109–136. [DOI] [PubMed] [Google Scholar]
- 34.Smith D.R., Doucette-Stamm,L.A., Deloughery,C., Lee,H., Dubois,J., Aldredge,T., Bashirzadeh,R., Blakely,D., Cook,R., Gilbert,K. et al. (1997) J. Bacteriol., 179, 7135–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gray M.W. and Spencer,D.F. (1996) Symp. Soc. Gen. Microbiol., 54, 109–126. [Google Scholar]
- 36.Gupta R.S. and Golding,G.B. (1996) Trends Biochem. Sci., 21, 166–171. [PubMed] [Google Scholar]
- 37.Martin W. and Muller,M. (1998) Nature, 392, 37–41. [DOI] [PubMed] [Google Scholar]
- 38.Andersson S.G., Zomorodipour,A., Andersson,J.O., Sicheritz-Ponten,T., Alsmark,U.C., Podowski,R.M., Naslund,A.K., Eriksson,A.S., Winkler,H.H. and Kurland,C.G (1998) Nature, 396, 133–140. [DOI] [PubMed] [Google Scholar]
- 39.Margulis L. (1970) Origin of Eukaryotic Cells. Yale University Press, New Haven, CT.
- 40.Martin W., Stoebe,B., Goremykin,V., Hansmann,S., Hasegawa,M. and Kowallik,K.V. (1998) Nature, 393, 162–165. [DOI] [PubMed] [Google Scholar]
- 41.Palmer J.D. (1997) Science, 275, 790–791. [DOI] [PubMed] [Google Scholar]
- 42.Pont-Kingdon G.A., Okada,N.A., Macfarlane,J.L., Beagley,C.T., Wolstenholme,D.R., Cavalier-Smith,T. and Clark-Walker,G.D. (1995) Nature, 375, 109–111. [DOI] [PubMed] [Google Scholar]
- 43.Reenan R.A. and Kolodner,R.D. (1992) Genetics, 132, 963–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maidak B.L., Olsen,G.J., Larsen,N., Overbeek,R., McCaughey,M.J. and Woese,C.R. (1997) Nucleic Acids Res., 25, 109–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lyons-Weiler J., Hoelzer,G.A. and Tausch,R.J. (1998) Biol. J. Linn. Soc., 64, 493–511. [Google Scholar]
- 46.Doolittle W.F. (1998) Trends Genet., 14, 307–311. [DOI] [PubMed] [Google Scholar]
- 47.Chi N.-W. and Kolodner,R.D. (1994) J. Biol. Chem., 269, 29984–29992. [PubMed] [Google Scholar]
- 48.Chi N.-W. and Kolodner,R.D. (1994) J. Biol. Chem., 269, 29993–29997. [PubMed] [Google Scholar]
- 49.Bevan M., Bancroft,I., Bent,E., Love,K., Goodman,H., Dean,C., Bergkamp,R., Dirkse,W., Van Staveren,M., Stiekema,W. et al. (1998) Nature, 391, 485–488. [DOI] [PubMed] [Google Scholar]