

Summary
Genome-wide association studies (GWASs) have identified thousands of variants for disease risk. These studies have predominantly been conducted in individuals of European ancestries, which raises questions about their transferability to individuals of other ancestries. Of particular interest are admixed populations, usually defined as populations with recent ancestry from two or more continental sources. Admixed genomes contain segments of distinct ancestries that vary in composition across individuals in the population, allowing for the same allele to induce risk for disease on different ancestral backgrounds. This mosaicism raises unique challenges for GWASs in admixed populations, such as the need to correctly adjust for population stratification. In this work we quantify the impact of differences in estimated allelic effect sizes for risk variants between ancestry backgrounds on association statistics. Specifically, while the possibility of estimated allelic effect-size heterogeneity by ancestry (HetLanc) can be modeled when performing a GWAS in admixed populations, the extent of HetLanc needed to overcome the penalty from an additional degree of freedom in the association statistic has not been thoroughly quantified. Using extensive simulations of admixed genotypes and phenotypes, we find that controlling for and conditioning effect sizes on local ancestry can reduce statistical power by up to 72%. This finding is especially pronounced in the presence of allele frequency differentiation. We replicate simulation results using 4,327 African-European admixed genomes from the UK Biobank for 12 traits to find that for most significant SNPs, HetLanc is not large enough for GWASs to benefit from modeling heterogeneity in this way.
Keywords: admixture, GWAS, Tractor, local ancestry, heterogeneity, effect sizes, simulation, genetic correlation, complex trait, genetic ancestry
Graphical abstract
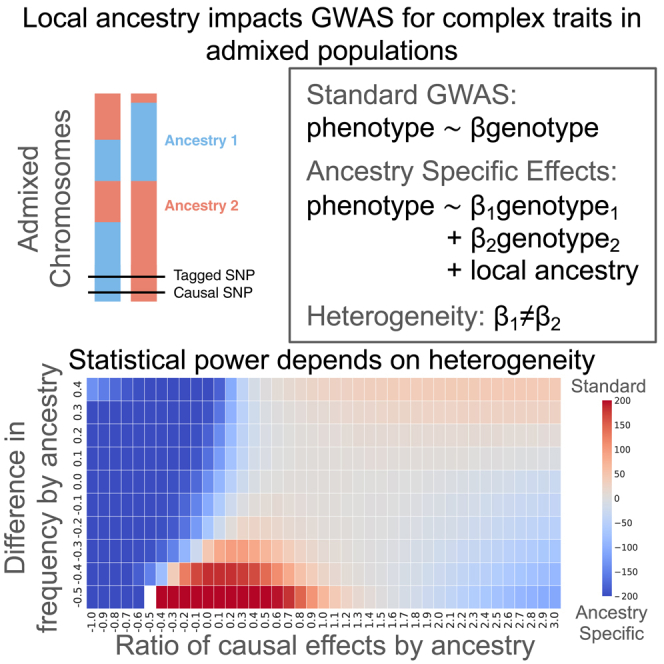
Genome-wide association studies (GWASs) are an important tool for disease mapping but are under-explored for populations of individuals with mixed genetic ancestries (admixed populations). We provide considerations for method selection and show that differences in allele frequency and/or causal effects by ancestry impact statistical power for GWASs in admixed populations.
Introduction
The success of genomics in disease studies depends on our ability to incorporate diverse populations into large-scale genome-wide association studies (GWASs).1,2,3,4 Cohort and biobank studies are growing to reflect this diversity,5,6,7 and a variety of techniques exist which incorporate populations of different continental ancestries into GWASs.8 However, while admixture has been an important factor in other steps in the disease mapping process, such as fine-mapping9 and estimating heritability,10,11 individuals of mixed ancestries (admixed individuals) have largely been left out of traditional association studies. GWASs performed in admixed populations have greater power for discovery compared to similar sized GWASs in homogeneous populations.12,13 Thus, excluding admixed individuals from association studies will not only increase health disparities, but will also disadvantage other populations. To prevent this exclusion, approaches to association studies have been developed specifically for admixed populations.14,15,16,17 However, the impact of HetLanc (differences in estimated allelic effect sizes for risk variants between ancestry backgrounds) on GWAS methods remains under-explored. Of particular interest are recently admixed populations, defined as fewer than 20 generations of mixture between two ancestrally distinct populations. In such populations, the admixture process creates mosaic genomes comprised of chromosomal segments originating from each of the ancestral populations (i.e., local ancestry segments). Local ancestry segments are much larger than linkage disequilibrium (LD) blocks18; thus, LD patterns within each local ancestry block of an admixed genome reflect LD patterns of the ancestral population. Similarly, allele frequency estimates from segments of a particular local ancestry are expected to reflect allele frequencies of the ancestral population. Variation in local ancestry across the genome leads to variability in global ancestry (the average of all local ancestries within a given individual). Such variability in local and global ancestries could pose a problem to GWASs in admixed populations as genetic ancestries are often correlated with socio-economic factors that also impact disease risk, thus yielding false positives in studies that do not properly correct for genetic ancestries. Because local and global ancestry are only weakly correlated,19 complete control of confounding due to admixture requires conditioning on both local and global ancestry.20 However, the success of admixture mapping indicates that the possibility of losing power due to over-correction for local ancestry differences is serious.21,22
GWASs in admixed populations are typically performed either using a statistical test that ignores local ancestry altogether (referred to in this work as “standard GWASs” and defined in Table 1) or using a test that explicitly allows for HetLanc (e.g., Tractor). The former provides superior power in the absence of HetLanc with the latter having great potential for discovery in its presence. However, these methods’ relative statistical power for discovery depends on the cross-ancestry genetic architecture of the trait, i.e., which variants are causal and what are those variants’ ancestry-specific frequencies, causal effects, and linkage disequilibrium patterns. For example, existing studies have found that standard GWASs can yield a 25% increase in power over Tractor13 in the absence of HetLanc while Tractor has higher power when causal effects are different by more than 60%.15 However, the full impact of cross-ancestry genetic architecture on GWAS power in admixed populations remains under-explored.
Table 1.
Summary of GWAS association statistics
| Association statistic | Statistical test | Assumptions on | Ancestry-related covariates | Degrees of freedom |
|---|---|---|---|---|
| ADM | – | 1 | ||
| Standard GWAS | 1 | |||
| SNP1 | 1 | |||
| MIX | 1 | |||
| SUM | and | 2 | ||
| Tractor | and | – | 2 |
All tests adjust for global ancestry and can be used on binary traits, and all tests except MIX can be implemented with adjustment for additional covariates and use on quantitative traits. For more information on the comparison of standard GWAS, ADM, SUM, and MIX, see Pasaniuc et al.14 and Seldin et al.22 We note that while additional methods exist,36,37,38,39 we do not focus on them in this work because they do not directly relate to Equation 1.
In this work, we use simulations to perform a comprehensive evaluation quantifying the impact of these factors on the power of GWAS approaches in admixed populations. We provide guidelines for when to use each test as a function of cross-ancestry genetic architecture. Elements of cross-ancestry genetic architecture such as allele frequencies, global ancestry ratios, and LD are known or can be calculated in advance of a GWAS to determine which of our simulation results apply in each case. Using extensive simulations, we find that standard GWASs should be preferred when HetLanc is small or non-existent. We quantify the extent of HetLanc and the ancestry-specific allele frequency differences required for Tractor to overcome the extra degree of freedom penalty. We further validate our results using the African-European admixed population in the UK Biobank (UKBB). By examining the HetLanc of significant SNPs in the UKBB, we can understand how often it rises to a level that impacts the power of traditional GWASs.
Subjects and methods
Simulated genotypes
We simulate genotypes using the following procedure, which produces a set of genotypes made up of independent SNPs from admixed genotypes with two ancestries.
-
(1)
Draw the individual global ancestry proportion of ancestry 2, for 10,000 individuals where is the expected global ancestry proportion of ancestry 2, and is the variance of global ancestry in the population (we use to reflect the variance of global ancestry found in the UK Biobank admixed population). is coerced between [0,1].
-
(2)
For each individual, draw a local ancestry count , where represents the local ancestry count of ancestry 2.
-
(3)
For each local ancestry, draw a genotype , where represents the allele frequency at local ancestry . Allele frequencies were specified for each simulation scenario according to the figure legends.
Simulated quantitative phenotypes with a single causal SNP
We simulate quantitative phenotypes with a single causal SNP (used in Figures 2C, 2D, 3A, and S1–S8) using the following procedure.
-
(1)
Standardize genotypes so that they have a mean 0 and variance 1.
-
(2)
Given some effect sizes , calculate , where the variance is taken over all individuals, and represents the genetic variance component of the phenotypes.
-
(3)
Given some heritability , calculate , where is the environmental variance component of the phenotypes. This comes from the equation .
-
(4)
For each individual, draw where is the random noise to add to the phenotype to represent environmental variables.
-
(5)
Repeat for 1,000 replicates.
Figure 2.

Association statistics in the absence of HetLanc
(A) Type I error for association statistics. Type I error calculated as the percent of null SNPs with a significant association detected. 95% confidence interval too narrow for display.
(B) Power for association statistics. Power calculated as the percent of simulations to successfully recover the causal variant. Odds ratios OR1 = OR2 = 1.2. 95% confidence interval too narrow for display.
(C) Power for a standard GWAS, SNP1, and Tractor as CAF2 is varied between 0.0 and 1.0 and CAF1 is fixed at 0.5. Power for all three methods varies as CAF difference varies. 95% confidence interval too narrow for display.
(D) Heatmap of percent increase in power of a standard GWAS over Tractor when . Causal allele frequencies CAF1 and CAF2 varied from 0.0 to 0.5 in increments of 0.1.
All simulations are for case-control (A and B) or quantitative (C and D) traits simulated 1,000 times for a population of 10,000 individuals with 100 genotypes each with global ancestry proportion 50/50. Power calculated using (A) nominal threshold p value < 0.05, (B) Bonferroni-corrected threshold p value < 1 × 10−5, or (C and D) standard threshold p value < 5 × 10−8. (A and B) Case-control traits have case-control ratio 1:1, 10% case prevalence, and CAF1 = CAF2 = 0.5. (C and D) Quantitative traits have heritability = 0.005. Heritability, global ancestry, causal effect size , and overall CAF do not qualitatively impact these results (Figures S1–S3).
Figure 3.

Impact of HetLanc on percent difference in power depends on CAF difference
(A) Heatmap of percent difference in power for a standard GWAS versus Tractor. The “∗” indicates the center with no HetLanc or CAF difference. The solid line represents the boundary between when a standard GWAS and Tractor have higher power. The dashed line represents the region in which a standard GWAS always has higher power than Tractor. Quantitative trait simulated 1,000 times for a population of 10,000 individuals on a trait with effect size ranging from −1.0 to 3.0 in increments of 0.1, and effect size = 1.0. Global ancestry proportion 50/50, heritability at = 0.005, and causal allele frequencies CAF1 = 0.5 and CAF2 ranging from 0.1 to 1.0 in increments of 0.1. Power calculated using a standard threshold p value < 5 × 10−8.
(B) Histogram of empirical Rhet for significant SNPs found for 12 phenotypes in the UKBB. estimated using Tractor.
Simulated quantitative phenotypes with multiple causal SNPs
We simulate quantitative phenotypes with multiple causal SNPs using real genotypes (used in Figures 4, S9, and S10) with the following procedure.
-
(1)
Use chromosome 1 of the UK Biobank admixed African-European genotypes.
-
(2)
Given some polygenicity p (p = 100 used in Figures 4 and S9, p = 1, 10, 100 used in Figure S10), randomly choose p SNPs to be causal.
-
(3)
Given some genetic correlation, draw effect sizes for causal SNPs chosen in step 2. Genetic correlations equal to 1.0, 0.5, and −1.0 used in Figures 4 and S9, genetic correlation equal to 1.0 for Figure S10. For more on genetic correlation, see Hou et al.23
-
(4)
Calculate , where the variance is taken over all individuals, and represents the genetic variance component of the phenotypes.
-
(5)
Given some heritability , calculate , where is the environmental variance component of the phenotypes. This comes from the equation . used in Figures 4, S9, and S10.
-
(6)
For each individual, draw where is the random noise to add to the phenotype to represent environmental variables.
-
(7)
Repeat for 100 replicates.
Figure 4.

Effect size heterogeneity in the context of polygenicity
(A) Boxplot of type I error for Tractor and a standard GWAS split by non-differentiated (MAF difference 0.2) and differentiated (MAF difference > 0.2) SNPs.
(B) Boxplot of power for Tractor and a standard GWAS in the case of no effect size heterogeneity split by non-differentiated and differentiated SNPs.
(C) Boxplot of power for Tractor and a standard GWAS in the case of effect size heterogeneity split by non-differentiated and differentiated SNPs.
(D) Boxplot of power for Tractor and a standard GWAS in the case of opposite effect sizes split by non-differentiated and differentiated SNPs.
All simulations used real UKBB admixed genotypes and simulated phenotypes with 100 causal SNPs and a total additive genetic heritability of = 0.5 (see subjects and methods). “∗” indicates a nominally significant p value (<0.05). “∗∗” indicates a Bonferroni-corrected significant p value (<1.28 × 10−3). The boxes show the inter-quartile range while the whiskers show the rest of the distribution (not including outliers).
Simulated case-control phenotypes
We simulate case-control phenotypes (used in Figures 2A and 2B) using the following procedure.
-
(1)Given some SNP, ancestry-specific odds ratios , and a case prevalence c, case-control phenotypes were simulated under the logistic model as in Atkinson et al.15
-
(a)Calculate the genetic component of the phenotype for each individual as .
-
(b)Find some intercept such that , where the bar refers to the mean over all individuals j.
-
(c)For each individual , draw case status from a distribution.
-
(d)Randomly discard control phenotypes until the case:control ratio is 1:1.
-
(a)
-
(2)
Repeat for 100 iterates of 1,000 replicates.
Real genotypes and phenotypes
For our real data analysis, we used genotypes from the UK Biobank. We limited our study to participants with admixed African-European ancestry. Overall, we had 4,327 individuals with an average of 58.9% African and 41.1% European ancestry. We used the imputed genotypes for these individuals with a total of 16,584,433 SNPs. The genotypes were mapped to the GRCh38 build and imputed to the TOPMed reference panel. We calculated the top 10 PCs for these genotypes and added these PCs as covariates to all analyses as our global ancestry component. The phenotypes we used are also from the UK Biobank and include aspartate transferase enzyme (AST), BMI, cholesterol, erythrocyte count, HDL, height, LDL, leukocyte count, lymphocyte count, monocyte count, platelet count, and triglycerides. We log transformed AST, BMI, HDL, leukocyte count, lymphocyte count, monocyte count, platelet count, and triglycerides to analyze all 12 traits as quantitative, continuous traits. We standardized all genotypes and phenotypes to be mean centered at 0.0 and have a variance of 1. This research complies with all relevant ethical regulations. The ethics committee/IRB of UKBB gave ethical approval for collection of UKBB data (https://www.ukbiobank.ac.uk/learn-more-about-uk-biobank/about-us/ethics). Participants signed a written consent form to be a part of the UKBB (https://www.ukbiobank.ac.uk/media/05ldg1ez/consent-form-uk-biobank.pdf). Approval to use UKBB individual-level data in this work was obtained under application 33127 at http://www.ukbiobank.ac.uk.
Association testing on simulated genotypes
We calculate the standard GWAS and Tractor association tests on simulated data using scripts that can be found on https://github.com/rachelmester/AdmixedAssociation. A standard GWAS (referred to as “ATT” in this software package) is a one degree of freedom association test that uses the model to test for against a null hypothesis that includes global ancestry (). Tractor (referred to as “TRACTOR” in this software package) is a two degree of freedom association test that uses the model to test for and against a null hypothesis that includes local ancestry () and global ancestry (). They can both be adapted to be used on case-control phenotypes by substituting logistic regression and odds ratios for linear regression and effect sizes. Additionally, they can both be adjusted for additional covariates such as age and sex. For our simulations, we used global ancestry proportions as our measure of global ancestry () and did not need to adjust for any additional covariates such as age and sex as we did not model those factors in our simulations. For power calculations, we use a standard significance threshold of p value < 5 × 10−8.
Association testing on real genotypes
We used admix-kit (https://kangchenghou.github.io/admix-kit/index.html) to perform the standard GWAS (referred to as “ATT” in this software package) and Tractor (referred to as “TRACTOR” in this software package) association tests on these data and extracted the p values. To determine significant SNPs, we filtered for SNPs with a standard p value of < . For the Manhattan plots, we plot all SNPs with a p value < 10−2 in Figure 5B and a p value < 10−4 in Figure S11 for computational plotting purposes. For Tables S1 and S2 and Figure 5A, to determine whether SNPs were part of the same locus, we grouped SNPs within a 500 kB radius and kept the most significant SNP from each test (standard GWAS and Tractor) in that locus.
Figure 5.

Comparing significant SNPs found with a standard GWAS and Tractor
(A) Venn diagram of independent significant loci found using a standard GWAS and Tractor in the UKBB across 12 quantitative traits.
(B) Manhattan plot of erythrocyte count in the UKBB. Significant SNPs found with a standard GWAS shown in red and significant SNPs found with Tractor shown in blue. Manhattan plot SNPs shown filtered for p value < 0.01 and SNPs are plotted based on post-filter indices.
Measures used to compare our results
In this work, we introduce several key measures that we use to compare our results. The formal definitions of these are the following.
Percent difference in power
Adjusted chi square
We take the p value from a statistic and convert it back to a statistic, regardless of the original degrees of freedom. The adjusted chi square score for a is itself.
Results
Heterogeneity by local ancestry impacts association statistics in admixed populations
HetLanc occurs when a SNP exhibits different estimated allelic effect sizes depending on its local ancestry background. HetLanc can manifest itself at causal SNPs due to genetic interactions between multiple causal variants or differential environments, although recent work suggests that the magnitude and frequency of these types of epistatic effects between causal variants is limited.23 A more common form of HetLanc is observed at non-causal SNPs that tag the causal effect in a differential manner across ancestries. Differential linkage disequilibrium by local ancestry at these non-causal SNPs (tagged SNPs) can cause HetLanc even when allele frequencies and causal effect sizes are the same across ancestries. The extent to which HetLanc exists and the magnitude of these differences in effect sizes are yet uncertain.22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38 However, the existence of HetLanc plays an important role in the power of GWAS methods to detect associations. Consider the example in Figure 1 in which the allelic effect size for a tagged SNP is estimated for a phenotype in an admixed population. In this population, both the tagged SNP and the true causal SNP may exist in regions attributed to both local ancestries present in the population (Figure 1A). Since LD patterns differ by local ancestry, the correlation between the tagged and causal SNPs will also depend on local ancestry (Figure 1B). This differential correlation between tagged and causal SNPs will cause the estimated allelic effect size for the tagged SNP to depend on local ancestry (Figure 1C). Thus, even for cases in which true causal effect sizes are the same across ancestries, allelic effect sizes estimated for the tagged SNP may be heterogeneous. Since GWASs cannot determine true causal effect sizes, we introduce Rhet, a measure of HetLanc which allows for both true causal effect-size heterogeneity and LD- and allele frequency-induced estimated allelic effect-size heterogeneity.
Figure 1.

Toy example of how differential LD by local ancestry can induce HetLanc
(A) Admixed populations contain haplotypes with different local ancestry at the causal or tagged SNP.
(B) The correlation between tagged and causal SNPs depends on their local ancestry due to differential LD by local ancestry.
(C) In a GWAS, the estimated marginal SNP effect size is proportional to the true causal effect size and the correlation between the tagged and causal SNPs (, where refers to the ancestry).
Methods for association testing in admixed populations
We start with a formal definition for a full model relating genotype, phenotype, and ancestry for a single causal SNP:
| (Equation 1) |
where is a phenotype, and are vectors that represent the number of alternate alleles with local ancestry 1 and 2 (such that , the full genotype regardless of ancestry), and are ancestry-specific marginal effect sizes of the SNP, is the vector of local ancestry counts at the locus, is the effect size of is a vector of global ancestry proportions, is the effect size of , is a matrix of additional covariates (such as age and sex), is a vector of effect sizes for these covariates, b is the intercept term multiplied by the column vector 1, and is random environmental noise.
Variability across local and global ancestries has been leveraged in various statistical approaches for disease mapping in admixed populations. One of the first methods developed for association was admixture mapping (ADM).30,36 ADM tests for association between local ancestry and disease status in affected individuals and control subjects or in a case-only fashion. This association is achieved by contrasting local ancestry deviation with expectations from per-individual global ancestry proportions. Therefore, ADM is often under-powered especially in situations in which allele frequency at the causal variant is similar across ancestral populations.31 Genotype association testing is traditionally performed using a linear or logistic regression with some standard covariates. This type of association test, referred to in this work as a standard GWAS, tests for association between genotypes and disease status while correcting for global ancestry to account for stratification.17,32 However, neither ADM nor standard GWASs take advantage of the full disease association signal in admixed individuals. SNP1, SUM, and MIX are examples of association tests that combine local ancestry and genotype information. SNP1 regresses out local ancestry in addition to global ancestry to control for fine-scale population structure. This approach helps control for fine-scale population stratification but may remove the signal contained in local ancestry information.33 SUM34 combines the SNP114 and ADM statistics into a two degree of freedom test. MIX14 is a case-control test that incorporates SNP and local ancestry information into a single degree of freedom test. Most recently Tractor15 conditions the effect size of each SNP on its local ancestry followed by a joint test allowing for different effects on different ancestral backgrounds. This step builds the possibility of HetLanc explicitly into the model, which may be particularly important when SNPs are negatively correlated across ancestries.35 Other varieties of tests have also been developed using different types of frameworks, most notably BMIX35 which leverages a Bayesian approach to reduce multiple testing burden. These statistics have been compared at length.3,14,22 However, existing comparisons do not consider HetLanc, nor do they thoroughly discuss allele frequency differences across ancestries.
Standard GWASs have more power than Tractor in the absence of heterogeneity by ancestry
First, we use simulations to compare type I error and power for each association statistic in Table 1. Starting with 10,000 simulated admixed individuals based on a 50/50 admixture proportion, we simulate 1,000 case-control phenotypes with a single causal SNP (see subjects and methods). We define type I error as the percent of non-causal SNPs found to have significant associations (p value < 0.05) for each score (see subjects and methods). Type I error is well controlled by Tractor (5.01%), SNP1 (5.01%), MIX (5.00%), and standard GWASs (5.01%) (Figure 2A). However, we find that type I error is not as well controlled for ADM (9.15%) and SUM (7.84%). We next calculate power to detect causal SNPs for an odds ratio of OR1 = OR2 = 1.2 (see subjects and methods). We find that SNP1 had the highest power at 42.14%. However, SNP1 was not significantly more powerful than either MIX (power 42.12%, p value 0.878) or a standard GWAS (power 42.05%, p value 0.325, Figure 2B). The power of all three of these tests was significantly higher (p value 1 × 10−16) than for SUM (power = 33.4%), ADM (power = 0.039%), or Tractor (power = 31.9%). Since Tractor is a statistical test specifically designed to find SNP-trait associations with effects that are heterogeneous by local ancestry,15 this loss of power is expected for Tractor when effect sizes are the same across ancestries, which is not the genetic architecture for which Tractor was designed. We find that while these association statistics are all well controlled, power does substantially differ between them. In the absence of both HetLanc and allele frequency difference, one degree of freedom SNP association tests outperform two degree of freedom tests.
We next investigate how differences in causal allele frequency (CAF) impact the power of a standard GWAS and Tractor in the case where true causal effect sizes are the same. We investigate the impact of varying CAF in each ancestry independently. Using our 10,000 simulated admixed individuals from the previous experiment, we simulate 1,000 quantitative phenotypes with a single causal SNP (see subjects and methods). We calculate the power of both Tractor and a standard GWAS to find these causal SNPs and then average that power over 100 simulated genotypes with specific allele frequencies. First, we let CAF1 = 0.5 and CAF2 range from 0.0 to 1.0 with a 0.1 increment and plot power over CAF2 (Figure 2C). We find that a standard GWAS and SNP1 have higher power than Tractor at all levels of CAF difference. Since Tractor has an extra degree of freedom compared to a standard GWAS and SNP1, Tractor is disadvantaged when . Additionally, we see that while SNP1 has (insignificantly) higher power than a standard GWAS when CAF1 = CAF2, the power of SNP1 deteriorates as causal allele frequency difference increases. This behavior is qualitatively the same as Tractor. When CAF1 = CAF2, a standard GWAS has 94.7% power, with Tractor at 91.1% power. However, as CAF2 becomes more different from CAF1, a standard GWAS maintains its power at 93.0%. By contrast, Tractor loses much of its power, with only 45.3% power when the causal allele is fixed at 100% in population 2 and only 48.1% power when the causal allele is absent in population 2. A standard GWAS maintains higher power than Tractor even at varying levels of heritability (Figures S1–S3), global ancestry (Figure S1), effect size (Figure S2), and CAF1 (Figure S3). However, the difference in power has a large range depending on the CAF difference between local ancestries.
Next we introduce percent difference in power, a one-dimensional metric to compare between these association statistics (see subjects and methods). We use this metric to visualize how varying CAF1 and CAF2 independently impacts the power of a standard GWAS and Tractor (Figure 2D). The percent increase in power when using a standard GWAS over Tractor when the causal SNP is absent in population 2 is 68%. The power difference between a standard GWAS and Tractor increases as CAF difference increases. Furthermore, the lower the CAF starts out in population 1, the larger the power difference between these two statistics. Specifically, when CAF1 = 0.5 and CAF2 = 0.1, the difference in CAF is 0.4 and a standard GWAS has a 25% power increase over Tractor. However, when CAF1 = 0.4 and CAF2 = 0.0, the difference in CAF is still 0.4 but a standard GWAS has a 43% increase in power over Tractor. While these differences in power do depend on both CAF differences and absolute CAF values in both ancestries, it is worth noting that differences in power along the diagonal axis are not significant. For example, while the increase in power of a standard GWAS over Tractor is 25% when CAF1 = 0.5 and CAF2 = 0.1 and the increase in power of a standard GWAS over Tractor is 26% when CAF1 = 0.1 and CAF2 = 0.5, the difference that occurs when switching causal allele frequencies between ancestries only has a p value of 0.345 in this case.
While this result corroborates previous studies,40,41,42 the relationship between Tractor and admixture mapping provides insight into the mechanism behind this dynamic. Mainly, as allele frequency differentiation by local ancestry increases, so does the power of the admixture mapping test statistic. In fact, ADM has no power when causal allele frequencies do not differ by ancestry but achieves up to 6.7% power when CAF1 = 0.0 and CAF2 = 0.5 (Figure S4A). However, the Tractor method uses the admixture mapping statistic as its null hypothesis. A stronger null hypothesis will be rejected less often than a weaker one even when the alternative hypothesis is the same, causing any test utilizing a strong null hypothesis to have less power. Thus, Tractor will have less power when its null hypothesis (ADM) has more power, which occurs in situations with high allele frequency differentiation. When allele frequencies do not differ by ancestry, Tractor achieves 91% power in our simulations. However, when CAF1 = 0.0 and CAF2 = 0.5, Tractor power plummets to 44% (Figure S4B). SNP1, which also uses ADM as its null hypothesis, suffers from the same deterioration in power as causal allele frequency differentiation increases (Figure S4C). When the causal allele frequencies are the same, SNP1’s power matches that of a standard GWAS, but as causal allele frequency differentiation increases, SNP1 loses power in the same pattern as Tractor. This indicates that Tractor loses power compared to a standard GWAS due to both its additional degree of freedom and due to its choice of null hypothesis.
While high levels of allele frequency differentiation drastically decrease the power of Tractor, a standard GWAS also has a smaller decrease in power at high levels of allele frequency differentiation, from 95% at equal allele frequencies to 93% when CAF1 = 0.0 and CAF2 = 0.5 (Figure S4D). This decrease in power is not as large as that suffered by Tractor, but it is also due to increased power of the null hypothesis at higher frequency differentiation across populations. The null hypothesis of the standard GWAS test statistic only includes global ancestry, but the power of global ancestry alone to predict a trait increases as allele frequency differentiation increases.32 The idea that including global ancestry as a covariate in these analyses reduces power for SNPs with large CAF differences raises the question of how much attenuation can be expected when more exact measures of global ancestry (such as principal components) are included in the analysis. However, the overall power attenuation due to the inclusion of global ancestry is small compared to that due to local ancestry; thus, we shift our focus back to considering local ancestry-specific effects on power.
Impact of HetLanc on power depends on allele frequency differences
Next, we investigate the impact of CAF differences and HetLanc on power differences between a standard GWAS and Tractor. The exact relationship between HetLanc (measured as Rhet), CAF difference, and percent difference in power is complex (Figure 3A). First, there is a window when 0.5 < Rhet < 1.5 in which, regardless of CAF difference, HetLanc is not enough to increase the power of Tractor relative to a standard GWAS. Thus, at these “low” levels of HetLanc, a standard GWAS will reliably have more power than Tractor across the allele frequency spectrum. Similarly, when Rhet < −0.5, there is no allele frequency difference which would increase the power of a standard GWAS relative to Tractor. This corroborates our findings that when effect sizes are in opposite directions, Tractor is expected to have improved power over standard GWASs regardless of CAF difference. We can see that it is characteristics of both standard GWASs and Tractor that drive this trend (Figure S5). The power of a standard GWAS depends most strongly on the magnitude of Rhet and is diminished the most when effect sizes are in opposite directions. By contrast, the power of Tractor depends strongly on both CAF difference and Rhet. These two factors combine to create an asymmetric shape for the percent difference in power (Figure 3A). This asymmetry in power observed for the Tractor method is likely due to correlations between effective sample size, allele frequency, global ancestry, and local ancestry that can occur in an asymmetric manner when causal effect sizes and causal allele frequencies differ between local ancestries.32 In these figures, we must consider that CAF1 is held constant at 0.5 and β2 is held constant at 1.0. For example, Rhet = 0.5 corresponds to β1 = 0.5 and β2 = 1.0. When CAF1 = 0.5 and CAF2 = 0.9, most of the genetic variance from the individuals in the study will come from ancestry 2 due to its larger causal allele frequency and larger effect size. This leaves the association for ancestry 1 with much less genetic variance to work with, and thus will lead Tractor’s ability to detect an association in ancestry 1 to be under-powered. However, when CAF2 = 0.1, much less of the total genetic variation in the population will come from ancestry 2, leading Tractor’s power to detect association in both populations to be more balanced.
We also find that SNP1 power suffers not only when causal allele frequency differences increase but also when HetLanc increases. We additionally investigate similar scenarios for standard GWASs and Tractor with varied global ancestry proportions (Figure S6), population-level CAF (Figure S7), and heritability (Figure S8). While the exact boundaries of these regions do differ, the overall shape of this heatmap and the conclusions mentioned above do not qualitatively change.
Polygenic trait simulations follow the same pattern as single causal variant simulations
We next investigate how HetLanc impacts power in polygenic traits. We consider the genotypes of individuals with African-European admixture in the UK Biobank. These individuals have an average of 58.9% African and 41.1% European ancestry over the population of 4,327 individuals. We simulate phenotypes using 100 causal SNPs along chromosome 1 and compare the power of a standard GWAS and Tractor over 100 simulations. Using real genotypes allows us to consider polygenic traits in the context of more realistic linkage disequilibrium and admixture. We now use genetic correlation23 instead of Rhet to measure HetLanc in the case of polygenic traits and separate our findings by whether or not the causal SNPs are differentiated (MAF difference > 0.2) or non-differentiated (MAF difference 0.2).
First, we find that both standard GWASs and Tractor have relatively well-calibrated type I error rates (Figure 4A). At an expected false positive rate of 5%, a standard GWAS has a 5.06% false positive rate for differentiated SNPs and a 5.00% false positive rate for non-differentiated SNPs. In this situation, in which genetic correlation = 1.0 (which corresponds to zero effect size heterogeneity), Tractor has a well-calibrated false positive rate of 4.99% for differentiated SNPs, but a false positive rate of 3.35% for non-differentiated SNPs, which is significantly deflated (p value < 10−16).
Similar to our simulations with only a single causal SNP, a standard GWAS and Tractor each have higher power in different combinations of genetic correlation and MAF differences. When genetic correlation remains 1.0 (Figure 4B), a standard GWAS has 23.0% power for differentiated SNPs and 25.5% power for non-differentiated SNPs, in contrast to Tractor’s 19.5% power for differentiated SNPs and 23.3% power for non-differentiated SNPs. The difference in power between differentiated SNPs and non-differentiated SNPs is significant (p values 3.53 × 10−3 for a standard GWAS and 1.73 × 10−6 for Tractor). The difference in power between a standard GWAS and Tractor is significant as well (p values 4.84 × 10−4 for differentiated SNPs and 3.22 × 10−4 for non-differentiated SNPs).
After we introduce HetLanc, its direction and magnitude impact which method has the most power, a result which resembles our previous findings. When effect sizes vary by ancestry but are in the same direction (genetic correlation = 0.5, Figure 4C), a standard GWAS has more power for differentiated SNPs (18.7% for a standard GWAS and 16.8% for Tractor, p value 0.04), whereas Tractor has more power than a standard GWAS for non-differentiated SNPs (18.0% for standard GWAS and 20.1% for Tractor, p value 5.44 × 10−4). When effect sizes are in opposite directions however (genetic correlation = −1.0, Figure 4D), Tractor has more power than standard GWASs for both differentiated SNPs (4.90% for a standard GWAS and 11.5% for Tractor, p value = 3.50 × 10−11) and non-differentiated SNPs (1.93% for a standard GWAS and 14.7% for Tractor, p value < 10−16).
We also consider the SNP1 test for these polygenic analyses. As expected, SNP1 remains well calibrated in the polygenic case but falls between Tractor and standard GWASs in terms of power when effect sizes are the same (genetic correlation = 1.0). However, when effect sizes are different (genetic correlation = 0.5 or −1.0), SNP1 performs less well than either standard GWASs or Tractor (Figure S9). We also consider how the level of polygenicity impacts power in the case with genetic correlation = 1.0 (Figure S10). We find that while a standard GWAS remains more powerful than Tractor when polygenicity is reduced to 10, the differences in power between a standard GWAS and Tractor do not remain significant in either the differentiated or non-differentiated case. This is likely due to the high heritability in this case since for the polygenic simulations we held = 0.5. Thus, in the case of 100 causal SNPs, each SNP had a = 0.005, which is identical to the heritability in the single causal SNP simulations. In the case of 10 causal SNPs, however, each SNP had = 0.05, which increased overall power, causing a necessary decrease in power difference between methods.
A standard GWAS finds more significant loci across 12 traits in the UK Biobank
We next seek to understand the impact of correcting for local ancestry in genetic analyses in real data. We investigate both Tractor and a standard GWAS in the same population of African-European admixed individuals from the UK Biobank. In real data, we investigate MAF (minor allele frequency) differences in lieu of CAF differences, since it is common practice to test minor alleles in real GWASs. First, we investigate MAF differences between segments of African and European local ancestry over 16,584,433 imputed SNPs. We find that the mean absolute minor allele frequency difference of these SNPs is 0.0959, with a standard deviation of 0.115. 85.2% of them have an absolute allele frequency difference of <0.2 across local ancestry (Figure S11).
Next, we investigate empirically derived values of Rhet to determine in which region of the heatmap estimated effect sizes are likely to be found in real data (Figure 3B). We ran the Tractor method on 12 quantitative traits to find the actual values of Rhet for the estimated effect sizes and . These traits were aspartate transferase enzyme (AST), BMI, cholesterol, erythrocyte count, HDL, height, LDL, leukocyte count, lymphocyte count, monocyte count, platelet count, and triglycerides. Then, we line up the histogram of these empirically derived values of Rhet with the heatmap. We find that for 69.3% of all SNPs found to be significant using the Tractor test statistic, the empirical value for Rhet is within this [−0.5, 1.5] window. While this is an estimate, we predict the true difference between estimated marginal effect sizes might be smaller than indicated by these empirical values because Tractor is more powerful in identifying SNPs with heterogeneous effect sizes. This result reflects previous findings that causal effects are similar across ancestries within admixed populations.22 Due to this similarity in effect size, most of the significant SNPs sit in the center of the heatmap. This region of this heatmap predicts that standard GWASs will have more power than Tractor. While we cannot directly compare the standard GWAS score with the Tractor score due to their differing degrees of freedom, we can compare the mean adjusted statistics. To calculate the adjusted statistic, we take the p value from a statistic and convert it back to a statistic, regardless of the original degrees of freedom. In this way, we can compare the mean adjusted statistic of the SNPs found to be significant in this case. We find that this statistic is significantly larger for the standard GWAS method than the Tractor method (Figure S12). For significant SNPs, the mean standard GWAS is 42.9, the mean adjusted Tractor is 37.5, and the p value for the difference is 2.11 × 10−4.
In addition to assessing HetLanc directly, we can also compare the number of independent significant SNPs found by a standard GWAS and Tractor for these phenotypes. We find that while the number of independent significant SNPs varies across all traits, including when grouped by independent loci (Table S1), overall a standard GWAS finds more significant independent signals than Tractor (Figure 5A). We find 22 independent significant loci, with 19 loci found in a standard GWAS and 10 found in Tractor. This trend is most pronounced in HDL, in which 5 independent loci were determined to be significant by a standard GWAS compared to none for Tractor. Similarly, BMI, leukocyte count, and monocyte count also only had independent significant loci when testing using a standard GWAS as opposed to Tractor. Cholesterol and LDL had significant loci found by both standard GWAS and Tractor, with a larger number found by the standard GWAS. Height is the only trait for which Tractor identified one significant locus but not the standard GWAS. Unfortunately, our sample sizes were not large enough to detect any significant loci for platelet count, triglycerides, or lymphocyte count. All independent significant loci for these 12 phenotypes are detailed in Table S2.
Additionally, we find that while a standard GWAS often finds more significant independent loci than Tractor, the two methods do not always find the same loci. Erythrocyte count is one phenotype in which we find an equal number of independent significant loci using both a standard GWAS and Tractor. However, not all loci overlap. Investigating the Manhattan plot of erythrocyte count specifically (Figure 5B), we see that loci on chromosome 16 are found by both a standard GWAS and Tractor. But outside of the main locus, both the standard GWAS and Tractor find separate additional significant regions. At the main locus, this Manhattan plot clearly shows that a standard GWAS has significantly smaller p values for the same locus. Thus, in a smaller sample size only a standard GWAS would have found this important region. This example highlights the importance of choosing the most highly powered association statistic for any given situation. Manhattan plots for other phenotypes can be found in Figure S13.
Discussion
In this work, we seek to understand the impact that estimated allelic effect-size heterogeneity by ancestry (HetLanc) has on the power of a GWAS in admixed populations. Our main goal is to find whether conditioning disease mapping on local ancestry leads to an increase or decrease in power. We find that HetLanc and CAF differences are the two most important factors when considering various methods for disease mapping in admixed populations. We focus on two association statistics: a standard GWAS, which ignores local ancestry, and Tractor, which conditions effect sizes on local ancestry. We find that in cases with small or absent levels of HetLanc, a standard GWAS is more powerful than Tractor in simulations of quantitative traits. This conclusion holds across a variety of global ancestry proportions and levels of SNP heritability. We find that as CAF differentiation between ancestries increases, so does the improvement of power of a standard GWAS compared to Tractor. At high HetLanc (Rhet >1.5) or when effect sizes are in opposing directions (Rhet < −0.5), we find that Tractor out-performs a standard GWAS. For African-European admixed individuals in the UKBB, most significant loci have both small measured HetLanc and MAF differences. We find that across 12 quantitative traits, a standard GWAS finds more significant independent loci than Tractor. Furthermore, a standard GWAS has smaller p values for the loci that it shares with Tractor. This suggests that on smaller datasets, more of the shared loci would be found by a standard GWAS than by Tractor.
This work has several implications for GWASs in admixed populations. Our results suggest that usually, a standard GWAS adjusted for global ancestry is the most powerful way to perform a GWAS in an admixed population. However, it may be possible to predict the comparative power of a standard GWAS and Tractor using the allele frequencies and linkage disequilibria of a specific sample. Additionally, since in real analyses a standard GWAS and Tractor often find different loci, it is important to keep both methods in mind when performing analyses. These methods prioritize different types of loci, with standard GWASs likely prioritizing loci with higher MAF differences and Tractor prioritizing loci with higher levels of HetLanc. Furthermore, our findings suggest that conditioning on local ancestry is a major factor in Tractor’s loss of power in situations in which causal allele frequencies differ. Thus, the performance of a method which includes effect size heterogeneity could potentially be considerably improved if local ancestry were not included in the null hypothesis. We leave assessment of the power and calibration of this type of hybrid method for future work.
We conclude with caveats and limitations of our work. When hoping to understand these patterns of power for association statistics, there are many combinations of different elements of genetic architecture to consider. These include phenotypic factors such as environmental variance and polygenicity, as well as elements of admixture such as the number of generations of admixture and the strength of linkage disequilibrium. We could not consider them all, and thus it is likely that additional nuances to our findings exist when other factors are considered. One major element not considered in this work is case-control traits. While we chose to focus on quantitative traits in this analysis due to their simplicity and ubiquity, case-control traits are also important in medicine. It is possible that the behavior of these phenotypes will vary compared to the quantitative traits that we analyze here, both in simulations and real data. We suggest case-control traits as an interesting avenue of research for future works. Lastly, we chose to focus our analyses on standard GWASs and Tractor due to their popularity and ease of use. We compare how these methods work “out of the box” to provide simple and usable guidance for others. However, as discussed in the introduction to this work, a variety of other association tests exist. It is likely that in certain circumstances one of these existing methods would outperform both a standard GWAS and Tractor.
From both scientific and social perspectives, it is important that admixed populations are incorporated more effectively into genetic studies. By providing insight into the strengths and limitations of these methods, we hope to enable studies to maximize their power in admixed populations.
Acknowledgments
The authors would like to acknowledge Ella Petter, Ruth Johnson, and Vidhya Venkateswaran for their insightful feedback. This research was funded in part by National Institutes for Health (NIH) under awards U01-HG011715 (B.P.) and R01-HG009120 (B.P.), R.M. was supported in part by NIH award no. T32HG002536 and B.M.H. and G.M. were supported in part by NIH grant R35GM133531 to B.M.H. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. This research was conducted using the UKBB Resource under application 33127.
Declaration of interests
The authors declare no competing interests.
Published: May 23, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.05.001.
Contributor Information
Rachel Mester, Email: rmester@ucla.edu.
Bogdan Pasaniuc, Email: pasaniuc@ucla.edu.
Supplemental information
Data and code availability
Code for this project including simulation experiments and data processing pipeline are available on github: https://github.com/rachelmester/AdmixedAssociation. An application for UK Biobank individual-level genotype and phenotype data can be made at the UK Biobank: http://www.ukbiobank.ac.uk.
References
- 1.Tian C., Gregersen P.K., Seldin M.F. Accounting for ancestry: population substructure and genome-wide association studies. Hum. Mol. Genet. 2008;17:R143–R150. doi: 10.1093/hmg/ddn268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mills M.C., Rahal C. The GWAS Diversity Monitor tracks diversity by disease in real time. Nat. Genet. 2020;52:242–243. doi: 10.1038/s41588-020-0580-y. [DOI] [PubMed] [Google Scholar]
- 3.Hou K., Bhattacharya A., Mester R., Burch K.S., Pasaniuc B. On powerful GWAS in admixed populations. Nat. Genet. 2021;53:1631–1633. doi: 10.1038/s41588-021-00953-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin A.R., Gignoux C.R., Walters R.K., Wojcik G.L., Neale B.M., Gravel S., Daly M.J., Bustamante C.D., Kenny E.E. Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet. 2017;100:635–649. doi: 10.1016/j.ajhg.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramirez A.H., Sulieman L., Schlueter D.J., Halvorson A., Qian J., Ratsimbazafy F., Loperena R., Mayo K., Basford M., Deflaux N., et al. The All of Us Research Program: data quality, utility, and diversity. Patterns. 2022;3:100570. doi: 10.1016/j.patter.2022.100570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou W., Kanai M., Wu K.H.H., Rasheed H., Tsuo K., Hirbo J.B., Wang Y., Bhattacharya A., Zhao H., Namba S., et al. Global Biobank Meta-Analysis Initiative: Powering genetic discovery across human disease. Cell Genom. 2022;2:100192. doi: 10.1016/j.xgen.2022.100192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg N.A., Huang L., Jewett E.M., Szpiech Z.A., Jankovic I., Boehnke M. Genome-wide association studies in diverse populations. Nat. Rev. Genet. 2010;11:356–366. doi: 10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin H., Morris N., Kang S.J., Li M., Tayo B., Lyon H., Hirschhorn J., Cooper R.S., Zhu X. Interrogating local population structure for fine mapping in genome-wide association studies. Bioinformatics. 2010;26:2961–2968. doi: 10.1093/bioinformatics/btq560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zaitlen N., Pasaniuc B., Sankararaman S., Bhatia G., Zhang J., Gusev A., Young T., Tandon A., Pollack S., Vilhjálmsson B.J., et al. Leveraging population admixture to characterize the heritability of complex traits. Nat. Genet. 2014;46:1356–1362. doi: 10.1038/ng.3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong Y., Perera M.A., Gamazon E.R. On using local ancestry to characterize the genetic architecture of human traits: genetic regulation of gene expression in multiethnic or admixed populations. Am. J. Hum. Genet. 2019;104:1097–1115. doi: 10.1016/j.ajhg.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin M., Park D.S., Zaitlen N.A., Henn B.M., Gignoux C.R. Admixed populations improve power for variant discovery and portability in genome-wide association studies. Front. Genet. 2021;12:673167. doi: 10.3389/fgene.2021.673167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wojcik G.L., Graff M., Nishimura K.K., Tao R., Haessler J., Gignoux C.R., Highland H.M., Patel Y.M., Sorokin E.P., Avery C.L., et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570:514–518. doi: 10.1038/s41586-019-1310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasaniuc B., Zaitlen N., Lettre G., Chen G.K., Tandon A., Kao W.H.L., Ruczinski I., Fornage M., Siscovick D.S., Zhu X., et al. Enhanced statistical tests for GWAS in admixed populations: assessment using African Americans from CARe and a Breast Cancer Consortium. PLoS Genet. 2011;7:e1001371. doi: 10.1371/journal.pgen.1001371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Atkinson E.G., Maihofer A.X., Kanai M., Martin A.R., Karczewski K.J., Santoro M.L., Ulirsch J.C., Kamatani Y., Okada Y., Finucane H.K., et al. Tractor uses local ancestry to enable the inclusion of admixed individuals in GWAS and to boost power. Nat. Genet. 2021;53:195–204. doi: 10.1038/s41588-020-00766-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith M.W., O'Brien S.J. Mapping by admixture linkage disequilibrium: advances, limitations, and guidelines. Nat. Rev. Genet. 2005;6:623–632. doi: 10.1038/nrg1657. [DOI] [PubMed] [Google Scholar]
- 17.Price A.L., Zaitlen N.A., Reich D., Patterson N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 2010;11:459–463. doi: 10.1038/nrg2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korunes K.L., Goldberg A. Human genetic admixture. PLoS Genet. 2021;17:e1009374. doi: 10.1371/journal.pgen.1009374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kang S.J., Larkin E.K., Song Y., Barnholtz-Sloan J., Baechle D., Feng T., Zhu X. Assessing the impact of global versus local ancestry in association studies. BMC Proc. 2009;3:1077–S116. doi: 10.1186/1753-6561-3-s7-s107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shriner D., Adeyemo A., Ramos E., Chen G., Rotimi C.N. Mapping of disease-associated variants in admixed populations. Genome Biol. 2011;12:223–228. doi: 10.1186/gb-2011-12-5-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peterson R.E., Kuchenbaecker K., Walters R.K., Chen C.Y., Popejoy A.B., Periyasamy S., Lam M., Iyegbe C., Strawbridge R.J., Brick L., et al. Genome-wide association studies in ancestrally diverse populations: opportunities, methods, pitfalls, and recommendations. Cell. 2019;179:589–603. doi: 10.1016/j.cell.2019.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seldin M.F., Pasaniuc B., Price A.L. New approaches to disease mapping in admixed populations. Nat. Rev. Genet. 2011;12:523–528. doi: 10.1038/nrg3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hou K., Ding Y., Xu Z., Wu Y., Bhattacharya A., Mester R., Belbin G.M., Buyske S., Conti D.V., Darst B.F., et al. Causal effects on complex traits are similar for common variants across segments of different continental ancestries within admixed individuals. Nat. Genet. 2023;55:549–558. doi: 10.1038/s41588-023-01338-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patel R.A., Musharoff S.A., Spence J.P., Pimentel H., Tcheandjieu C., Mostafavi H., Sinnott-Armstrong N., Clarke S.L., Smith C.J., et al. VA Million Veteran Program Genetic interactions drive heterogeneity in causal variant effect sizes for gene expression and complex traits. Am. J. Hum. Genet. 2022;109:1286–1297. doi: 10.1016/j.ajhg.2022.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marigorta U.M., Navarro A. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet. 2013;9:e1003566. doi: 10.1371/journal.pgen.1003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi H., Gazal S., Kanai M., Koch E.M., Schoech A.P., Siewert K.M., Kim S.S., Luo Y., Amariuta T., Huang H., et al. Population-specific causal disease effect sizes in functionally important regions impacted by selection. Nat. Commun. 2021;12:1098–1105. doi: 10.1038/s41467-021-21286-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown B.C., Asian Genetic Epidemiology Network Type 2 Diabetes Consortium. Ye C.J., Price A.L., Zaitlen N. Transethnic genetic correlation estimates from summary statistics. Am. J. Hum. Genet. 2016;99:76–88. doi: 10.1016/j.ajhg.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galinsky K.J., Reshef Y.A., Finucane H.K., Loh P.R., Zaitlen N., Patterson N.J., Brown B.C., Price A.L. Estimating cross-population genetic correlations of causal effect sizes. Genet. Epidemiol. 2019;43:180–188. doi: 10.1002/gepi.22173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi H., Burch K.S., Johnson R., Freund M.K., Kichaev G., Mancuso N., Manuel A.M., Dong N., Pasaniuc B. Localizing components of shared transethnic genetic architecture of complex traits from GWAS summary data. Am. J. Hum. Genet. 2020;106:805–817. doi: 10.1016/j.ajhg.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKeigue P.M. Mapping genes that underlie ethnic differences in disease risk: methods for detecting linkage in admixed populations, by conditioning on parental admixture. Am. J. Hum. Genet. 1998;63:241–251. doi: 10.1086/301908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mani A. Local ancestry association, admixture mapping, and ongoing challenges. Circ. Cardiovasc. Genet. 2017;10:e001747. doi: 10.1161/CIRCGENETICS.117.001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 33.Liu J., Lewinger J.P., Gilliland F.D., Gauderman W.J., Conti D.V. Confounding and heterogeneity in genetic association studies with admixed populations. Am. J. Epidemiol. 2013;177:351–360. doi: 10.1093/aje/kws234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tang H., Siegmund D.O., Johnson N.A., Romieu I., London S.J. Joint testing of genotype and ancestry association in admixed families. Genet. Epidemiol. 2010;34:783–791. doi: 10.1002/gepi.20520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shriner D., Adeyemo A., Rotimi C.N. Joint ancestry and association testing in admixed individuals. PLoS Comput. Biol. 2011;7:e1002325. doi: 10.1371/journal.pcbi.1002325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X., Zhu X., Qin H., Cooper R.S., Ewens W.J., Li C., Li M. Adjustment for local ancestry in genetic association analysis of admixed populations. Bioinformatics. 2011;27:670–677. doi: 10.1093/bioinformatics/btq709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J., Stram D.O. The role of local ancestry adjustment in association studies using admixed populations. Genet. Epidemiol. 2014;38:502–515. doi: 10.1002/gepi.21835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duan Q., Xu Z., Raffield L.M., Chang S., Wu D., Lange E.M., Reiner A.P., Li Y. A robust and powerful two-step testing procedure for local ancestry adjusted allelic association analysis in admixed populations. Genet. Epidemiol. 2018;42:288–302. doi: 10.1002/gepi.22104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen W., Ren C., Qin H., Archer K.J., Ouyang W., Liu N., Chen X., Luo X., Zhu X., Sun S., Gao G. A generalized sequential Bonferroni procedure for GWAS in admixed populations incorporating admixture mapping information into association tests. Hum. Hered. 2015;79:80–92. doi: 10.1159/000381474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simonin-Wilmer I., Orozco-Del-Pino P., Bishop D.T., Iles M.M., Robles-Espinoza C.D. An overview of strategies for detecting genotype-phenotype associations across ancestrally diverse populations. Front. Genet. 2021;12:703901. doi: 10.3389/fgene.2021.703901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin E.R., Tunc I., Liu Z., Slifer S.H., Beecham A.H., Beecham G.W. Properties of global-and local-ancestry adjustments in genetic association tests in admixed populations. Genet. Epidemiol. 2018;42:214–229. doi: 10.1002/gepi.22103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin H., Zhu X. Power comparison of admixture mapping and direct association analysis in genome-wide association studies. Genet. Epidemiol. 2012;36:235–243. doi: 10.1002/gepi.21616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code for this project including simulation experiments and data processing pipeline are available on github: https://github.com/rachelmester/AdmixedAssociation. An application for UK Biobank individual-level genotype and phenotype data can be made at the UK Biobank: http://www.ukbiobank.ac.uk.