

Abstract
Plant‐pathogenic fungi are causative agents of the majority of plant diseases and can lead to severe crop loss in infected populations. Fungal colonization is achieved by combining different strategies, such as avoiding and counteracting the plant immune system and manipulating the host metabolome. Of major importance are virulence factors secreted by fungi, which fulfil diverse functions to support the infection process. Most of these proteins are highly specialized, with structural and biochemical information often absent. Here, we present the atomic structures of the cerato‐platanin‐like protein Cpl1 from Ustilago maydis and its homologue Uvi2 from Ustilago hordei. Both proteins adopt a double‐Ψβ‐barrel architecture reminiscent of cerato‐platanin proteins, a class so far not described in smut fungi. Our structure–function analysis shows that Cpl1 binds to soluble chitin fragments via two extended grooves at the dimer interface of the two monomer molecules. This carbohydrate‐binding mode has not been observed previously and expands the repertoire of chitin‐binding proteins. Cpl1 localizes to the cell wall of U. maydis and might synergize with cell wall‐degrading and decorating proteins during maize infection. The architecture of Cpl1 harbouring four surface‐exposed loop regions supports the idea that it might play a role in the spatial coordination of these proteins. While deletion of cpl1 has only mild effects on the virulence of U. maydis, a recent study showed that deletion of uvi2 strongly impairs U. hordei virulence. Our structural comparison between Cpl1 and Uvi2 reveals sequence variations in the loop regions that might explain a diverging function.
Keywords: biotrophy, cerato‐platanin‐like, chitin, Ustilago maydis
Our structure–function analysis of the CP‐like protein 1 (Cpl1) from Ustilago maydis revealed a new mode of chitin binding and suggests synergy with other secreted fungal proteins during infection.
1. INTRODUCTION
Smut fungi constitute a large class of biotrophic plant pathogens that infect mainly grasses, among them many important cereal crops (Zuo et al., 2019). The individual smut fungal species have a narrow host range and establish a tight interaction for their parasitic lifestyle known as biotrophy (Benevenuto et al., 2018). Ustilago maydis infects the maize plant Zea mays and its ancestral form teosinte and forms dark black galls on infected parts of the plants that contain the mature teliospores (Brefort et al., 2009; Dean et al., 2012; Kämper et al., 2006). Plant infection by U. maydis is guided by the secretion of more than 400 effector proteins that allow fungal entry into the plant tissue, suppression of the plant immune system, and metabolic rerouting for resource allocation (Lanver et al., 2017).
Effector proteins are generally classified as small, cysteine‐rich proteins (typically between 200 and 300 amino acids) that harbour an N‐terminal signal peptide for conventional secretion (de Wit et al., 2009; Lanver et al., 2017). Many of these proteins are host‐specific and lack conserved features. Furthermore, computational approaches to predict domains of known function or structure frequently fail to yield reliable results (Jones et al., 2018). In addition, effectors are often encoded in gene clusters or act in concert with other effectors, thus generating functional redundancy, and single deletions might have little influence on the virulence of the deletion strains in infection experiments (Schirawski et al., 2010). The precise molecular functions of most effector proteins therefore remain enigmatic.
An important task of effector proteins from biotrophic fungi is the evasion of the plant immune system, especially during the early steps of host plant colonization. Microbial pathogens are recognized by host cell surface receptors through conserved microbe‐ or pathogen‐associated molecular patterns (MAMPs or PAMPs) (Jones & Dangl, 2006). A prominent PAMP is chitin, an abundant fungal cell wall component (Pusztahelyi, 2018). Specialized receptors in the plant cell wall harbouring LysM domains recognize chitin molecules, which triggers an immune response (Kaku et al., 2006; Kombrink et al., 2011) resulting in a broad range of cellular responses that impair fungal infections.
Consequently, fungal pathogens have evolved a repertoire of effector proteins that protect the fungal cell wall from plant chitinases or serve to scavenge chitin‐derived fragments (Sánchez‐Vallet et al., 2015). These effectors share binding properties towards oligosaccharides but are diverse in structure and function. The most prominent examples are proteins harbouring LysM domains (de Jonge et al., 2010; Hu et al., 2021; Kombrink & Thomma, 2013), proteins harbouring lectin‐like domains (van den Burg et al., 2006), and proteins belonging to the cerato‐platanin (CP)‐like protein family (Pazzagli et al., 2014). CPs comprise a class of chitin‐binding proteins exclusively found in filamentous fungi that exert functions in fungal development, for example, hyphal growth and cell wall remodelling (Chen et al., 2013; Pazzagli et al., 2014).
During the developmental stage associated with plant infection of maize, more than 4500 genes of U. maydis are differentially regulated. Some of those genes have a high transcript abundance in the early stages of maize plant colonization (1–2 days post‐infection) (Lanver et al., 2018). Among the 10 most highly expressed genes is UMAG_01820 (Figure S1a,b). Similarly, UMAG_01820 orthologues from the smut fungi Ustilago hordei and Sporisorium reilianum are highly expressed during infection of their respective hosts, Hordeum vulgare and Z. mays (Figure S1c,d) (Ökmen et al., 2018; Zhang et al., 2021).
Here we present a structural and molecular characterization of the UMAG_01820 and UHOR_02700 gene products. The crystal structures of UMAG_01820 (Cpl1) and UHOR_02700 (Uvi2) uncover a central double‐Ψβ‐barrel (DPBB) characteristic of the CP‐like protein family. Coordination of the chitin oligomers chitobiose and chitotetraose is facilitated at the subunit interface of the homodimers relying on residues provided by the DPBB. The structural comparison between Cpl1 and Uvi2 indicates that subtle changes in the surface‐exposed loops preceding the DPBB might result in diverging functions of these effector proteins during maize and barley infection by U. maydis and U. hordei, respectively. Our study sheds light on the molecular details of Cpl1 and suggests that the CP‐like fold might be more broadly distributed among fungi and functionally more divergent than anticipated so far.
2. RESULTS
2.1. Cpl1 is conserved among related smut fungi
cpl1 encodes a protein of 240 amino acids with a predicted molecular weight of approximately 27 kDa (RefSeq: XP_011387768.1). In silico analyses using the Consensus Constrained TOPology prediction webserver (CCTOP) (Dobson et al., 2015) and SignalP v. 5.0 predicted no transmembrane helices but a signal peptide of 21 amino acids in length at the N‐terminus of the protein. The Basic Local Alignment Search Tool for proteins (BLASTp) (Li et al., 2015) identified homologues of cpl1 only in related smut fungi belonging to the order of Ustilaginales and the class Ustilaginomycetes (Figure S2). These organisms are Pseudozyma hubeiensis SY62, Sporisorium scitamineum, Sporisorium reilianum SRZ2, Sporisorium graminicola, Ustilago trichophora, Melanopsichium pennsylvanicum, Kalmanozyma brasiliensis GHG001, U. hordei, Ustilago bromivora, Moesziomyces antarcticus, Moesziomyces aphidis DSM 70725, and Testicularia cyperi with identities ranging from 72% to 43% (determined by CLUSTAL v. 2.1) (Sievers et al., 2011). Notably, Cpl1 contains four cysteines conserved among all orthologues (Figure S2).
2.2. The crystal structure of Cpl1 reveals a dimeric protein with a DPBB architecture
No structural information on proteins homologous to Cpl1 was available and computational approaches failed to identify structural motifs or domains of known function. Thus, we determined the crystal structure of Cpl1 at 1.8 Å resolution by X‐ray crystallography employing selenomethionine single‐wavelength anomalous dispersion (Se‐SAD; Table S1).
Cpl1 consists of four α‐helices and eight β‐strands (Figure 1a,b) forming a β‐barrel surrounded by flexible loops. The two α‐helices α1 and α2 and a β‐hairpin consisting of β1 and β2 form the N‐terminal domain of the protein (cyan; residues 43 to 105; Figure 1b). The β‐hairpin is stabilized by a highly conserved disulphide bond established by residues Cys77 and Cys96 (Figure S3a). The core of Cpl1 contains a β‐barrel formed by the six β‐strands β3–β8, which is flanked by helices α3 and α4 (blue; residues 106 to 234; Figure 1b). Six‐stranded β‐barrels are found in several proteins; however, the parallel strands rarely form two Ψ‐structures, a fold named DPBB by Castillo et al. (1999). The first Ψ‐structure consists of the loop connecting strands β3 and β4 and strand β7, whereas the second Ψ‐structure consists of the loop connecting strands β7 and β8 and strand β3 (Figure 1b). The second disulphide bond formed by residues Cys124 and Cys157 stabilizes the first Ψ‐structure and the β5/β6 hairpin, which is also part of the β‐barrel (Figure S3a). Four loop regions (LR1–4) protrude from the central structural elements termed loop regions (Figure 1b).
FIGURE 1.
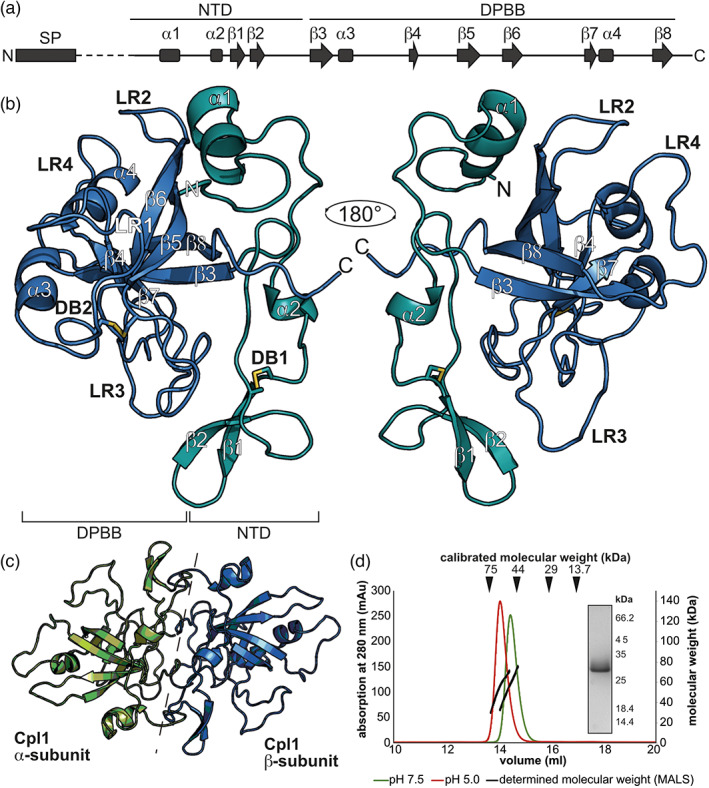
Crystal structure of Cpl1. (a) Secondary structure of Cpl1. The boundaries of the N‐terminal domain (NTD) and the double‐ψβ‐barrel (DPBB) are shown above. (b) Cartoon model of Cpl1 coloured in deep teal (NTD) and blue (DPBB). Secondary structure elements, the two disulphide bridges (DBs), and the four loop regions (LRs) are labelled above the respective elements. (c) Cartoon model of the Cpl1 dimer with the two monomers coloured in green and blue, respectively. (d) Size‐exclusion chromatography‐multi‐angle light scattering (SEC‐MALS) analysis of Cpl1 at pH 7.5 and pH 5.0 shows the presence of a 50‐kDa species (theoretical mass of Cpl1 dimer = 52.4 kDa). The inset shows a Coomassie‐stained SDS‐PAGE of the peak fraction.
The asymmetric unit of the Cpl1 crystals contained six molecules forming three dimers (Figure 1c). The analysis of these dimers using PDBe PISA (Krissinel & Henrick, 2007) shows that the monomers of each dimer cover a buried surface area of approximately 2000 Å2, implicating biological relevance (File S1). Four regions of Cpl1 contribute to the dimer interface between the two molecules. The first region covers residues 43 to 50 and partially aligns with residues 82 to 103 that form the second region (Figure S3b,c) and residues 146 to 160 forming the third region (Figure S3b,d). Finally, the C‐terminal residues 234 to 240 (region 4) are also buried in the dimer interface aligning mainly to residues of the second region (Figure S3b,c). To assess whether Cpl1 forms stable dimers in solution, we employed size‐exclusion chromatography (SEC) coupled with multi‐angle light scattering (MALS). SEC experiments were conducted at pH 7.5 and pH 5.0 to account for the rather acidic milieu of the apoplastic space between fungal and host cells. The MALS experiments determined molecular weights of 53.9 and 54.5 kDa for pH 5.0 and 7.5, respectively, suggesting that the Cpl1 dimer is stable at both pH values (Figure 1d). Notably, Cpl1 also formed SDS‐stable dimers, which could only be disrupted by heating the protein sample prior to SDS‐PAGE analysis.
Our structural and biochemical analyses thus reveal that Cpl1 consists of a central DPBB surrounded by four loop regions stabilized by two conserved disulphide bonds. Two monomers form a stable homodimeric assembly via extensive interactions within two regions of the N‐terminal domain and stretches in the C‐terminal regions of the protein.
2.3. Cpl1 has structural homology to CP‐like proteins and binds to soluble chitin oligomers
With the structure of Cpl1 at hand, we set out to identify homologies to known structures that might allow us to elucidate the potential function of this U. maydis protein. A search with the distance matrix alignment database (DALI) (Holm, 2020) retrieved a high structural similarity to several CP proteins from Moniliophthora perniciosa, a basidiomycete pathogen causing witches' broom disease of the cacao tree (Theobroma cacao). More precisely, the superposition to MpCp5 (Protein Data Bank [PDB] code: 3SUM) yielded a root mean square deviation (r.m.s.d.) of 2.3 Å over 97 Cα‐atoms (Figure 2a). The superposing parts include the six β‐strands β3–β8 as well as helix α4 (Figure S4). Notably, despite some variations in the loop regions, the second disulphide bond (Cys124–Cys157) at Cpl1 superimposes almost perfectly with the first disulphide bond of MpCP5 (Figure S4). The N‐terminus of Cpl1 containing the main dimer interaction interface does not superpose with the corresponding portions in the MpCP5 structures (Figure 2a). However, CPs are known to self‐aggregate under specific conditions and some are also able to form dimers (Gaderer et al., 2014; Seidl et al., 2006). This might indicate a different dimerization mode compared to Cpl1, where monomer formation was not observed.
FIGURE 2.
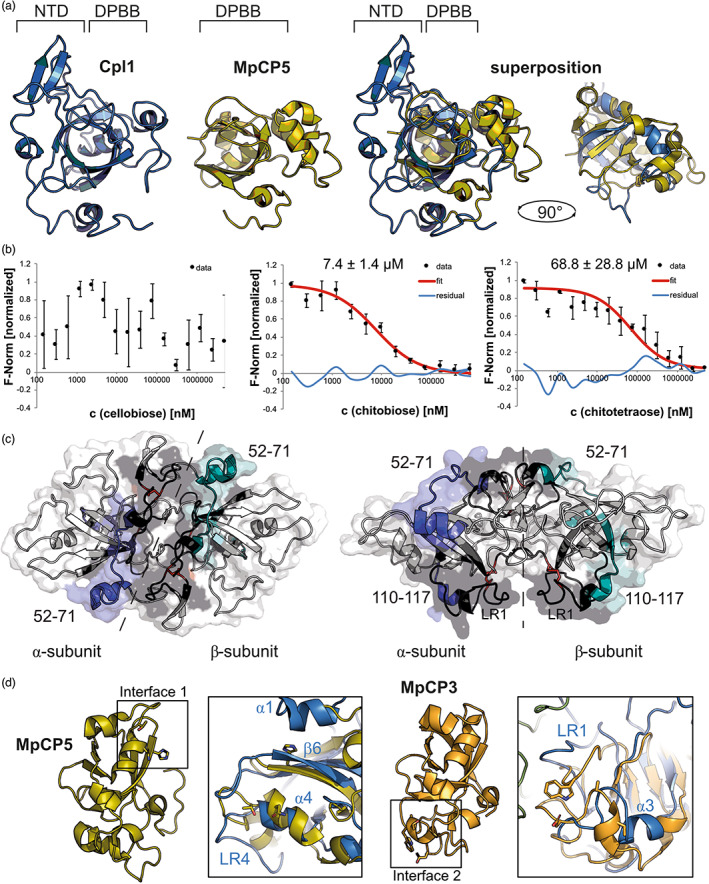
Cpl1 has high structural homology to cerato‐platanin‐like proteins and binds to chitin oligomers. (a) Superposition of a Cpl1 monomer (blue) with MpCP5 (yellow; PDB code: 3SUM). The loop regions of Cpl1 have been truncated for better visibility of the superposed regions with MpCP5 in the right panel. N‐terminal domain (NTD), double‐ψβ‐barrel (DPBB). (b) Microscale thermophoresis (MST) experiments with Cpl1. Shown are normalized fluorescence values (F‐norm, black dots) of Cpl1 titrated against cellobiose, chitobiose, and chitotetraose (concentration in nM). Where possible, a curve was fitted (red) used to calculate the dissociation constant (KD). Data points not included in the fitting are shown as residual data (curve identity, blue). The KD values of Cpl1 binding chitobiose and chitotetraose were 7.4 μM (± 1.4) and 68.8 μM (±28.8), respectively. (c) Areas of Cpl1 that exhibited reduced deuterium incorporation within at least two time points in the presence of chitotetraose (compare to Figure S5) are coloured in blue or dark teal for the α‐ and β‐subunits, respectively, and areas not covered by peptides are shown in black. The side chains of the disulphide bridge‐forming cysteine residues 77/96 and 124/157 are shown as red sticks. (d) The chitin‐binding interfaces of MpCP5 (yellow) and MpCP3 (orange) superposed with Cpl1 (blue) show no structural similarity between the proteins in these regions. LR, loop region.
CPs have been shown to interact with chitin polymers and their monomers commonly found in the fungal cell wall (Pazzagli et al., 2014). Chitin is a polymer of N‐acetyl‐d‐glucosamine monomers linked by β‐1,4‐glycosidic bonds, and based on the high structural homology with CPs, we hypothesized that Cpl1 might also be able to bind chitin. Thus, we performed microscale thermophoresis (MST) experiments to assess whether Cpl1 can bind to soluble chitin monomers, dimers, and tetramers. Cpl1 was labelled with an amine‐reactive dye and titrated with soluble chitin mono‐ and oligomer concentrations ranging from 5 mM to 152 nM. The estimated dissociation constant (KD) for the Cpl1–chitobiose interaction was 7.4 ± 1.4 μM and 68.8 ± 28.8 μM for the Cpl1–chitotetraose interaction, while no interaction of cellobiose with Cpl1 could be observed (Figure 2b).
To identify the binding interface of chitobiose and chitotetraose on Cpl1, we performed hydrogen–deuterium exchange (HDX) coupled with mass spectrometry (MS). Specifically, the degree of deuterium incorporation into Cpl1 in the presence or absence of either ligand was determined. Therefore, the protein was subjected to MS after proteolytic cleavage of the protein into peptides, which allowed the identification of ligand‐dependent differences in HDX (File S2). In total, 79 peptides of Cpl1 were obtained that covered approximately 77% of the Cpl1 amino acid sequence (Figure S5a). Very similar patterns of HDX reduction were apparent for chitobiose (Figure S5b) and chitotetraose (Figure S5c), in particular in peptides spanning residues 52–71 and 110–117 (Figures 2c and S6). In the crystal structure of Cpl1 these residues locate to the upper and lower dimeric interface constituted by the two Cpl1 protomers (Figure 2c, blue and green regions). Unfortunately, peptides covering the regions surrounding the two disulphide bonds could not be retrieved due to incomplete proteolytic digestion (Figure 2c, black regions). This specifically reduced the information on binding events in the larger cleft involving LR1 (Figure 2c, right panel).
We also compared our results on the carbohydrate‐binding interface at Cpl1 with residues of CPs involved in chitin binding obtained by several studies (Barsottini et al., 2013; Pazzagli et al., 2014). In MpCP5, three residues are important for chitin recognition that match to helix α4 and strand β6 on Cpl1 (Figure 2d, left panel). Despite the structural conservation of MpCP5 and CPl1 in this region, our HDX‐MS analysis did not reveal perturbations in this region in the presence of chitin oligomers. However, in MpCP3 a region in close proximity to helix α3 (residues 110–117) was shown to be important for chitin recognition. This binding interface occupies a similar region on CPs and Cpl1 but shows neither structural nor sequence conservation (Figure 2d, right panel). Our data thus suggest two regions within the Cpl1 dimer interface to be involved in binding of soluble chitin oligomers of which one might be distantly related to the binding interface identified in CP proteins.
2.4. A cleft in the dimer interface serves as major chitin‐binding interface
Based on our HDX‐MS analysis, we had a closer view on the two clefts present at the Cpl1 dimer interface. To substantiate our findings, we also performed molecular docking with chitobiose and chitotetraose using AutoDock Vina (Trott & Olson, 2010). The different states of the docked molecules are all located within the cleft formed by LR1 that extends from helix α3 to strand β3 (Figures 2c and 3a). To validate the HDX‐MS and docking results in vitro we chose amino acid residues possibly involved in the chitin interaction and generated single amino acid alanine substitutions to repeat the MST experiments with these mutants. Specifically, we varied D69 and E72, which are both located within or close to helix α2 in the first binding interface identified by HDX‐MS, into alanines. Our MST experiments showed an approximately 10‐fold decrease in affinity towards chitobiose (66.31 ± 3.54 μM in Cpl1D69A/E72A vs. 7.4 ± 1.4 μM in the wild type) (Figure 3b), while the affinity towards chitotetraose was seemingly not affected. However, after mutation of N151 and R155, both of which reside in the cleft on the opposite site of the first interface, into alanines, we could not observe binding of chitobiose anymore (Figure 3b). Furthermore, the affinity towards chitotetraose was reduced roughly twofold (110.54 ± 21.4 μM Cpl1N151A/R155A vs. 68 ± 28.8 μM in the wild type). The two amino acids N151 and R155 are located on both ends of the cleft and the binding interface might therefore accommodate even larger chitin oligomers in the natural context (Figure 3c). In conclusion, we can show that two regions within the interface between two Cpl1 protomers contribute to binding of soluble chitin oligomers, with the one constituted by LR1 being the major one.
FIGURE 3.
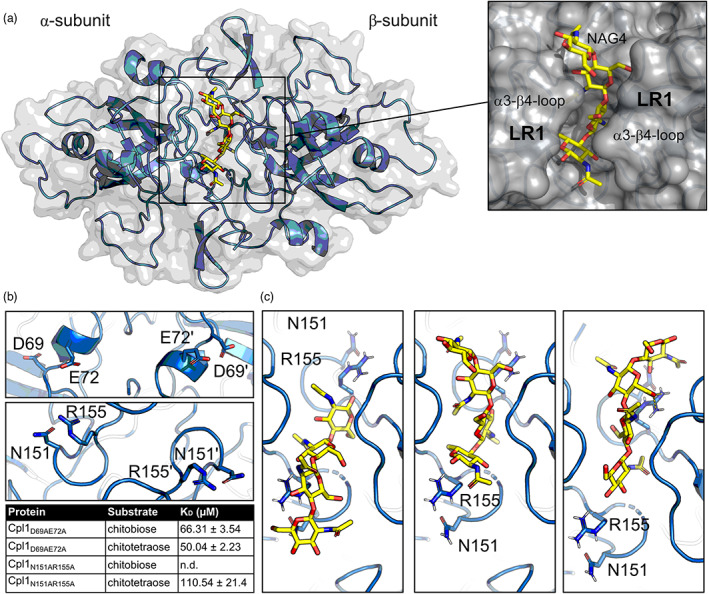
A groove in the dimer interface at Cpl1 is important for binding of soluble chitin oligomers. (a) Cpl1 dimer (blue) with a chitotetraose (yellow) docked into the dimer groove using AutoDock Vina. The inset shows a closeup of the chitotetraose fitted in between the α3–β4–loop (LR1) structures of both protomers. (b) Amino acids that were varied to alanines and the results of microscale thermophoresis (MST) experiments employing Cpl1N151A/R155A and Cpl1D69A/E72A. (c) Closeup of differently docked chitotetraose molecules. N151 and R155 are highlighted and have been mutated to alanines.
2.5. Structural and biochemical comparison of Cpl1 and Uvi2 reveals common principles
Our structural and biochemical analysis revealed that Cpl1 shares the central DPBB with other CP proteins and also binds to soluble chitin oligomers. We next aimed to understand if architecture and biochemical behaviour are shared among Cpl1 homologues from other smut fungi. We chose Uvi2 (the Cpl1 homologue from U. hordei), which has been shown to be an important virulence factor during barley infection (Ökmen et al., 2018). Crystals of Uvi2 diffracted to 1.35 Å resolution and the crystal structure of Uvi2 was solved using Cpl1 as a search model (Table S1, Figure 4a). Superposition of both structures revealed a high structural similarity and an r.m.s.d. of 0.5 Å (Figure 4a). Both proteins form a dimer in the crystal structure that could be confirmed for Uvi2 in solution using SEC‐MALS (Figure 4b,c). We again used two different buffer systems to analyse the oligomeric state at pH 7.5 and pH 5.0. To test if Uvi2 is similarly capable of binding to soluble chitin oligomers, we also performed MST experiments using N‐acetylglucosamine, chitobiose, chitotetraose, and cellulose. We obtained a KD of 14.99 ± 3.72 μM for Uvi2 in the presence of chitobiose, while no KD could be determined for the other carbohydrates (Figure S7). The interaction of Uvi2 with chitobiose is in a similar range as observed for Cpl1 before (7.4 ± 1.4 μM; compare to Figure 2b). Furthermore, both proteins share a sequence identity of 57.5% and regions mapping to the dimer interface are almost identical between Uvi2 and Cpl1 (Figure 4d). The most prominent differences at the sequence level are found in the four loop regions, which also showed a higher local r.m.s.d. than the core of the protein (Figure 4a,d). In conclusion, Uvi2 and Cpl1 share the same biochemical and structural properties except for surface‐exposed loop regions.
FIGURE 4.
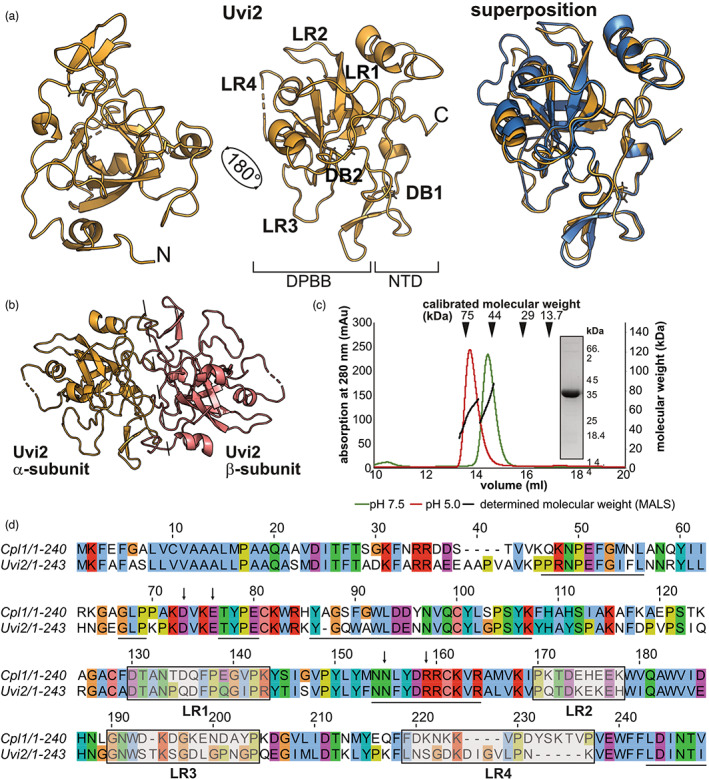
Structural comparison between Cpl1 and Uvi2. (a) Cartoon model of Uvi2 (yellow) alone and superposed with Cpl1 (blue). The loop regions (LRs) and disulphide bonds (DBs) are labelled on the respective elements. N‐terminal domain (NTD), double‐ψβ‐barrel (DPBB). (b) Dimer of Uvi2 as observed in the crystal structure with the two protomers coloured in yellow and salmon. (c) Size‐exclusion chromatography‐multi‐angle light scattering (SEC‐MALS) analysis of Uvi2 at pH 7.5 (green graph) and pH 5.0 (red graph) shows the presence of a 50‐kDa species. The inset shows a Coomassie‐stained SDS‐PAGE gel of the peak fraction. (d) Amino acid sequence alignment of Uvi2 and Cpl1 coloured according to the CLUSTAL colouring scheme (Sievers et al., 2011). The regions involved in dimer formation are underlined in black and residues that were mutated to diminish binding of soluble chitin oligomers are indicated with black arrows. The four LRs showing the highest degree of sequence deviation are indicated with grey boxes.
2.6. Cpl1 is dispensable for sporidial growth of U. maydis and its deletion only mildly affects virulence
Based on the molecular characterization and the high expression of cpl1 during infection, we next aimed to understand the effect of a cpl1 knockout on the growth and the virulence of U. maydis. Therefore, we first generated a cpl1 deletion in wild‐type U. maydis FB1 and FB2 (Banuett & Herskowitz, 1989) by using a CRISPR–Cas9‐dependent approach (Schuster et al., 2016). We could not detect any significant phenotypic differences in disease symptoms between plants infected with FB1 × FB2 and plants infected with FB1Δcpl1 × FB2Δcpl1. Infection with the U. maydis wild‐type strains FB1 × FB2 overall results in stronger infection symptoms on infected plants. Subtle differences in infection behaviour are easily missed as a substantial number of plants died prior to the final examination of the infection experiment (18.6% for FB1 × FB2 vs. 18% FB1Δcpl1 × FB2Δcpl1). We therefore generated strains deleted for cpl1 in the solopathogenic SG200 background. Indeed, we could detect subtle differences in the virulence of strains deleted for cpl1 compared to the respective parental strains (Figure 5a). While the number of plants with small tumours was significantly (p = 0.007) smaller in SG200Δcpl1 compared to SG200 (7% vs. 20%), the total number of larger tumours was higher (normal and heavy tumours 79% in SG200 vs. 89% in SG200Δcpl1; Figure 5a).
FIGURE 5.
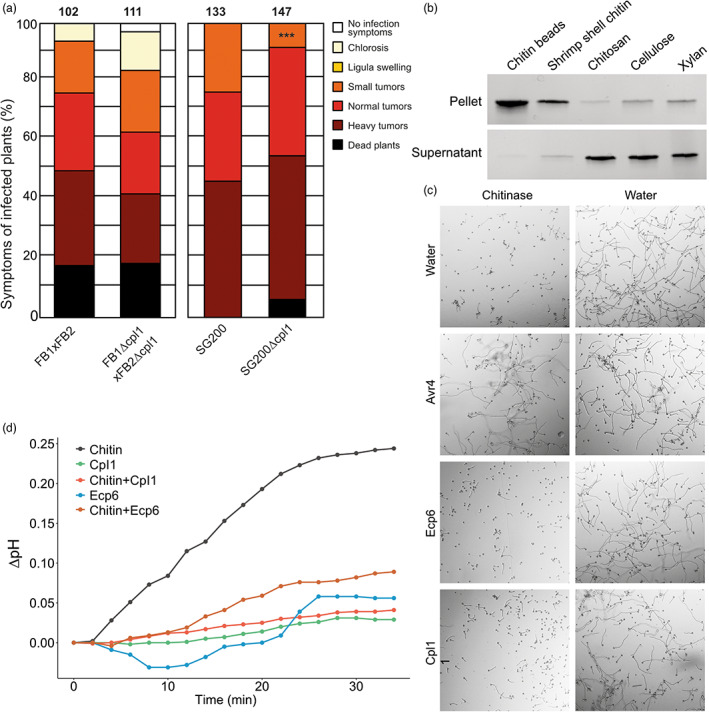
Cpl1 is dispensable for virulence, suppresses chitin‐induced medium alkalization, and binds to chitin, but does not protect hyphal growth against chitinase hydrolysis. (a) Maize infection assays using deletion strains in different genetic backgrounds of Ustilago maydis. cpl1 has been deleted from the genomes of FB1, FB2, and SG200 (from left to right). (b) HA‐tagged Cpl1 was incubated with magnetic chitin beads, shrimp shell chitin, chitosan, cellulose, and xylan for 6 h, and after centrifugation, pellets and supernatants were analysed by protein gel electrophoresis. (c) Microscopic pictures of Trichoderma viride grown in vitro in the presence of Cladosporium fulvum Avr4 or Ecp6 or U. maydis Cpl1 for 2 h, followed by addition of chitinase or water as control. Pictures were taken at 6 h after chitinase addition. (d) The pH of Nicotiana tabacum ‘Bright Yellow‐2’ cell suspensions treated with 100 nM chitin hexamer, 1 μM U. maydis Cpl1, 1 μM C. fulvum Ecp6 (positive control), or mixtures of chitin hexamer and effector protein was measured for 34 min. The assay was performed twice with similar results.
Although the available transcriptional data (Lanver et al., 2018) suggest that cpl1 is not expressed under axenic conditions, we tested whether the growth of sporidial forms of these strains was affected under various stress conditions or if any morphological changes could be detected in sporidia. We could observe neither differences in cell morphology nor differential responses towards the stress conditions tested (Figure S8a,b). In conclusion, our data show that Cpl1 is dispensable for growth of U. maydis in axenic culture and its deletion only mildly affects the virulence of U. maydis.
2.7. Cpl1 binds insoluble chitin fragments, does not protect hyphal growth against chitinase hydrolysis, and has the potential to suppress chitin‐triggered host immunity
Significant branches of plant immunity are targeted towards chitin as a nonself molecule that occurs in microbial pathogens such as fungi. Accordingly, fungal pathogens have evolved various types of effector proteins that protect the fungal cell wall from hydrolytic enzymes to avoid the release of chitin‐derived MAMPs as well as cellular collapse (Sánchez‐Vallet et al., 2015). These include not only particular LysM effectors, but also effectors that carry an invertebrate chitin‐binding domain, both of which have been shown to contribute to fungal virulence and bind to immunogenic chitin oligomers that are typically longer in size, as well as to insoluble chitin fragments (de Jonge et al., 2010; Tian et al., 2022; van den Burg et al., 2006; van Esse et al., 2007). To test whether Cpl1 binds such fragments too, HA‐tagged Cpl1 was subjected to a polysaccharide precipitation assay that included magnetic chitin beads, shrimp shell chitin, and chitosan as chitin‐related substrates and the plant‐derived carbohydrates cellulose and xylan. After incubation and centrifugation, Cpl1 clearly precipitated together with chitin beads and shrimp shell chitin, and only weakly with chitosan, cellulose, and xylan (Figure 5b). Thus, Cpl1 clearly binds to insoluble chitin and only poorly to other carbohydrate substrates.
Next, it was tested whether, based on its chitin‐binding ability, Cpl1 is able to protect hyphae of Trichoderma viride, a fungus that exposes its cell wall chitin in vitro, against chitinase hydrolysis (Mauch et al., 1988). The previously characterized chitin‐binding Cladosporium fulvum effector proteins Avr4 and Ecp6 were used as positive and negative controls, respectively (de Jonge et al., 2010; van den Burg et al., 2006; van Esse et al., 2007). As expected, the addition of chitinases from Clostridium thermocellum drastically inhibited T. viride hyphal growth, and the addition of Avr4, but not of Ecp6, protected the hyphae against chitinase hydrolysis (Figure 5c). However, the addition of Cpl1 did not lead to the protection of hyphae against chitinases (Figure 5c).
Several chitin‐binding effector proteins are known to suppress chitin‐triggered immune responses in their plant hosts, including not only LysM effectors, but also effectors that do not employ LysM domains to bind chitin (de Jonge et al., 2010; Fiorin et al., 2018; Sánchez‐Vallet et al., 2015; Tian et al., 2022; Volk et al., 2019). To test whether Cpl1 can suppress chitin‐triggered immunity in plants, medium alkalinization upon treatment of suspension‐cultured Nicotiana tabacum ‘Bright Yellow‐2’ cells with chitin oligosaccharides was tested in the presence or absence of effector protein (de Jonge et al., 2010). The C. fulvum LysM effector Ecp6, which suppresses such alkalinization, was used as a positive control in this assay (Figure 5d). Remarkably, preincubation of 100 nM chitin with 1 μM Cpl1 prior to addition to the suspension cells led to a significant reduction of the alkalinization response (Figure 5d), demonstrating its ability to suppress chitin‐induced immunity. Collectively, these experiments revealed that Cpl1 can bind insoluble chitin fragments, does not protect hyphae against chitinase hydrolysis, but has the potential to suppress chitin‐triggered immunity.
2.8. Cpl1 localizes to the fungal cell wall and interacts with other cell wall‐associated proteins during infection
To consolidate our findings that Cpl1 binds to soluble chitin oligomers, which suggests that it may localize to the fungal cell wall, we complemented SG200Δcpl1 by constitutively expressing cpl1 fused to a C‐terminal HA‐tag. Strains overexpressing cpl1‐HA were grown in liquid culture and harvested, the culture supernatant was precipitated using trichloroacetic acid, and the cells were subsequently lysed. Only faint amounts of Cpl1‐HA were detected in the concentrated supernatant samples (Figure S9) but prominent amounts of Cpl1 could be detected in the cell samples (Figure S9). Again, overproduction of Cpl1‐HA did not influence the morphology of U. maydis sporidia or influenced their growth under different stress conditions (Figure S8).
To analyse if Cpl1‐HA indeed resides in the cell wall, we stimulated strains with hydroxy‐fatty acids and sprayed them on Parafilm to induce filamentation. The filaments were subjected to immunostaining using an anti‐HA primary antibody and an Alexa Fluor 488 (AF488)‐conjugated secondary antibody. We detected evenly distributed fluorescence on long and branched filaments. However, shorter filaments also showed a homogenous distribution of fluorescence (Figure 6a–d). Interestingly, we did not detect any fluorescence in the areas surrounding the hyphae, indicating that Cpl1‐HA was tightly bound to the fungal cell wall. Taken together, our data support a role of Cpl1 in the fungal cell wall during maize infection.
FIGURE 6.
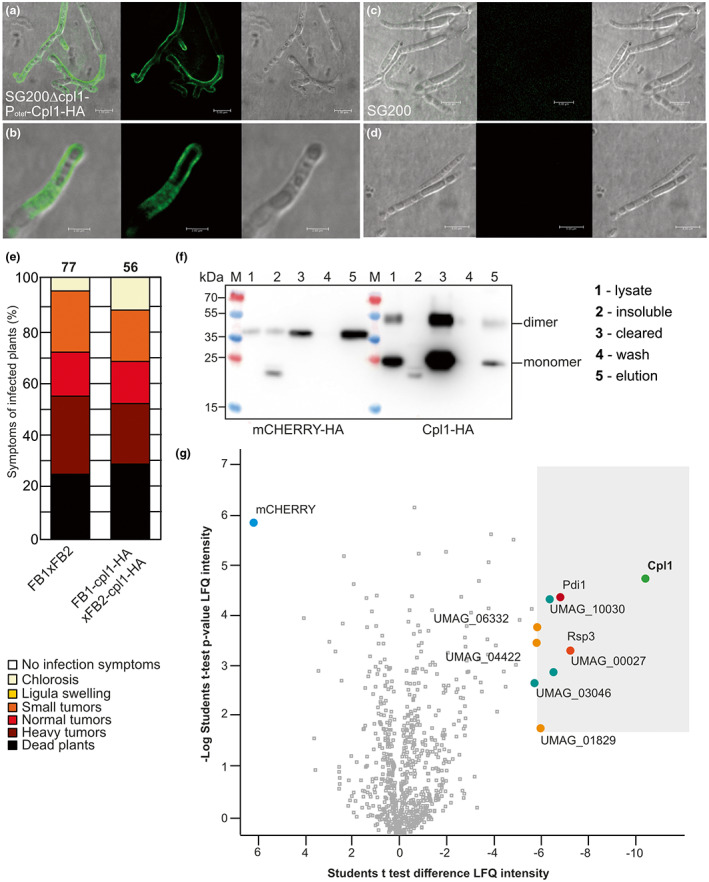
Cpl1 decorates the fungal cell wall of Ustilago maydis hyphae and is copurified with cell wall‐degrading and cell wall‐decorating virulence factors during infection. (a–d) Cpl1‐HA is secreted and binds to the fungal cell wall of U. maydis filaments grown on a Parafilm surface. Confocal microscopy of strains immunostained with an anti‐HA primary antibody and an AF488‐conjugated secondary antibody. Shown are merge, green fluorescent protein (GFP) channel, and differential interference contrast (DIC) images. Panels (a) and (b) show several hyphae and a closeup of SG200Δcpl1 complemented with cpl1‐HA under the control of the constitutive otef promoter, respectively. Panels (c) and (d) show two representative images of SG200 hyphae. (e) Maize infection assay with the indicated U. maydis strains. (f) Western blot analysis of Cpl1‐HA derived from infected maize leaves 3 days post‐infection. For the control bait mCherry‐HA in the ip locus fused to a sequence encoding the signal peptide of Cmu1 under the control of its own promoter was used. (1) Whole lysate of infected maize leaves, (2) insoluble fraction, (3) cleared lysate, (4) wash fraction, and (5) elution fraction. (g) Volcano plot depicting the results of the third of the three co‐immunoprecipitation experiments conducted with FB1 and FB2 containing Cpl1‐HA as bait and mCherry‐HA as the control sample. Shown are normalized label‐free quantification (LFQ) intensities as either the −log(p) value (Student's t test; y axis) or the LFQ intensity difference. Data were acquired by liquid chromatography coupled with mass spectrometry combined with the detection and quantification of peptide intensities. A high −log(p) value (y axis) indicates that many peptides were found. A negative difference (x axis) means that peptides were specific towards the bait sample containing Cpl1‐HA (green and bold) and positive values mean that peptides were specific towards the mCherry‐HA control (blue and bold).
Based on the localization of Cpl1‐HA at U. maydis filaments, we expected to potentially identify secreted maize proteins that might interact with Cpl1 in planta. This observation would be in line with Uvi2 from U. hordei interacting with a barley thaumatin in yeast two‐hybrid experiments (Ökmen et al., 2018). Therefore, we generated cpl1‐HA complementation strains in the FB1 and FB2 deletion background to perform plant infection experiments followed by co‐immunoprecipitation (Co‐IP) coupled with MS (Figure 6e). We used an mCHERRY‐HA gene fused to the sequence encoding the signal peptide of cmu1 under control of the cmu1 promoter as a control. Both proteins were purified from leaves of infected maize seedlings 3 days post‐inoculation (dpi) and detected by western blotting (Figure 6f).
Eight U. maydis proteins were found to be significantly and repeatedly enriched in all three replicates based on their −log(p) value of the label‐free quantification (LFQ) intensity and their LFQ intensity difference (Figure 6g, Table S5). These proteins are UMAG_10030 (uncharacterized), UMAG_10156 (protein disulphide isomerase 1 [Pdi1]; Marín‐Menguiano et al., 2019), UMAG_06332 (endoglucanase 1 [Egl1]; Schauwecker et al., 1995), UMAG_04422 (an endo‐1,4‐β‐xylanase; Moreno‐Sánchez et al., 2021), UMAG_03274 (Rsp3, cell wall‐bound, protects against ZmAFP1/2; Ma et al., 2018), UMAG_00027 (uncharacterized), UMAG_03046 (uncharacterized), and UMAG_01829 (Erc1, a 1,3‐β‐glucanase; Ökmen et al., 2022). Notably, all of them have an N‐terminal signal peptide (as predicted by SignalP v. 5.0), are rich in cysteine residues (an average of 9.5 per protein, UniProt), and have no predicted transmembrane helices (THMHH v. 2.0; DTU Health Tech). We also compared the amount of identified peptides from our Co‐IP experiments with the respective gene expression according to the transcriptome data from Lanver et al. (2018). The overall gene expression of most identified proteins correlated with the peptide abundance (Figure S10). Only in the case of UMAG_10030 and Pdi1, which are both less prominently expressed at 2 dpi, more peptides could be recovered. Tryptic digestion of disulphide bond‐rich effector proteins is often challenging, and we cannot rule out the possibility that UMAG_10030 and Pdi1 are more efficiently digested or be sure that the enrichment in the Co‐IP experiment has biological relevance. In conclusion, our results suggest that Cpl1 localizes in the fungal cell wall in proximity to or directly interacting with cell wall‐remodelling proteins and virulence factors during plant infection.
3. DISCUSSION
3.1. Cpl1—A new type of CP?
In this work, we structurally and biochemically characterized a conserved virulence factor from U. maydis that we termed Cpl1 due to its high structural similarity with proteins of the CP family. CP proteins represent a group of expansin‐related proteins found exclusively in filamentous fungi (Chen et al., 2013) that are highly expressed both during filamentous growth on culture plates and on the surface of host plants (Baccelli, 2015; Luti et al., 2019; Narvaez‐Barragan et al., 2020). These small proteins typically harbour a central DPBB and four conserved cysteine residues, and either reside in the fungal cell wall tightly bound to chitin or are secreted into the apoplast (Luti et al., 2019; Pazzagli et al., 2014). Cpl1 shares the central DPBB, one of the disulphide bonds, and the chitin‐binding properties with other CP‐like proteins and also localizes to the fungal cell wall of U. maydis hyphae. Cpl1 is also exclusively expressed during infectious, and thus filamentous, growth of U. maydis. However, its deletion had no impact on fungal morphology and only mildly influenced virulence. CPs studied during plant colonization in several relevant plant pathogens attenuated virulence in some cases while being dispensable in other (Narvaez‐Barragan et al., 2020). In CPs chitin binding is achieved through a flat and shallow surface groove that is conserved among most members of this protein class (Chen et al., 2013). In contrast, this binding site is not conserved in Cpl1, where chitin oligomers bind in two grooves located within the dimer interface (Figures 2c,d and 3). Cpl1 forms a stable homodimer, while CPs are mainly monomeric, although higher oligomeric states have been observed in some cases (Pazzagli et al., 2014).
CP proteins have only been found in filamentous fungi, and the fungi in which CPs contribute to virulence are characterized by their broad host range and aggressive hemibiotrophic or necrotrophic plant colonization behaviour (Luti et al., 2019). Intriguingly, CP proteins are also linked to plant immunity, with many of them eliciting a defence response that triggers a hypersensitivity response, subsequently resulting in cell death, and were thus regarded as PAMPs or MAMPs (Li et al., 2019; Luti et al., 2019; Narvaez‐Barragan et al., 2020; Pazzagli et al., 2014). A role of Cpl1 in eliciting a hypersensitivity response would be highly unlikely given the different infection strategies of biotrophic and necrotrophic fungi (Glazebrook, 2005). However, future research needs to address the precise role of Cpl1 during plant infection in more detail.
Taken together, we demonstrate that CPs and Cpl1 share common features but also show some prominent differences. Our structure of Cpl1 suggests that Cpl1 is not a bona fide CP but that the CP‐like core fold instead serves as a robust scaffold. The dimer with four loop regions on each Cpl1 protomer might therefore be the basis for new functionalities based on the CP‐like core fold.
3.2. Cpl1 interacts with fungal proteins during infection
Co‐IP experiments using Cpl1 as a bait revealed that several fungal proteins copurify with Cpl1 during maize infection. A more detailed inspection of these proteins identified revealed the presence of three glycoside hydrolases, namely UMAG_04422 (Xyn1, a β‐xylanase), UMAG_01829 (Erc1, a 1,3‐β‐glucanase), and UMAG_06332 (Egl1, an endoglucanase). These cell wall‐degrading enzymes play important roles in cell wall remodelling during hyphal growth and plant cell penetration. Especially in the early stages of infection, the removal of l‐arabinose groups from arabinoxylans incorporated into the plant cell wall as a defence mechanism has been shown to be an important step to increase the accessibility of xylan and penetrate the plant cell wall (de Vries et al., 2000; Doehlemann et al., 2008). The remodelling of fungal and plant cell walls needs to be well orchestrated to prevent host defence signalling. In contrast to necrotrophic and hemibiotrophic pathogens, biotrophic pathogens such as U. maydis have a reduced repertoire of cell wall‐degrading enzymes and might use these enzymes in a more fine‐tuned manner (Spanu et al., 2010). Cpl1 could interact with these cell wall‐degrading enzymes during infection and thereby spatially direct their enzymatic activity.
Furthermore, Cpl1 also enriched the disulphide isomerase Pdi1, which was shown to be important for quality control of cysteine‐rich effectors during infection (Marín‐Menguiano et al., 2019). Pdi1 resides in the endoplasmic reticulum of U. maydis sporidia, but a significant amount is also localized to the cell wall (Marín‐Menguiano et al., 2019). As our Co‐IP experiments do not discriminate between Cpl1 that travels through the secretory pathway and mature cell wall‐bound molecules, we can only speculate where a potential interaction with Pdi1 occurs. In addition, we could identify several peptides corresponding to Rsp3, an important cell wall‐decorating protein shielding infectious hyphae from the activity of two mannose‐binding proteins (Ma et al., 2018). Fungi have evolved several lines of defence to both protect their cell wall against attacking plant enzymes and prevent the generation of MAMPs that would elicit a plant immune response (Tanaka & Kahmann, 2021). Together with Rsp3, Cpl1 might also have a function in either shielding fungal hyphae or scavenging chitin fragments. Our assays could not establish a role in the protection of fungal hyphae against hydrolysis by chitinases in vitro, in contrast to previously characterized chitin‐binding LysM effectors from the wheat pathogen Zymoseptoria tritici (Marshall et al., 2011; Sánchez‐Vallet et al., 2020; Tian et al., 2022) as well as the effector protein Avr4 from the tomato leaf mould fungus C. fulvum (van den Burg et al., 2006; van Esse et al., 2007). However, also the LysM effector Ecp6, which is produced by C. fulvum during host colonization and contributes to virulence during infection, is not able to protect fungal hyphae against chitinase hydrolysis (de Jonge et al., 2010), demonstrating that chitin binding itself is not sufficient to provide protection. Intriguingly, similar to C. fulvum Ecp6, Cpl1 has the capacity to suppress chitin‐triggered host immunity, as Cpl1 application prevented the chitin‐triggered medium alkalinization in a suspension of tobacco cells. Although such suppression of immunity is well known for fungal LysM effectors (de Jonge et al., 2010; Kombrink & Thomma, 2013; Sánchez‐Vallet et al., 2015; Tian et al., 2022), other types of fungal chitin‐binding effectors that do not employ LysM domains for substrate binding have also been shown to display such activity (Fiorin et al., 2018; Volk et al., 2019). However, typically effectors that suppress chitin‐triggered immunity are secreted into the apoplast, where they can accumulate to high concentrations (Kombrink & Thomma, 2013; Sánchez‐Vallet et al., 2015). For example, Ecp6 is one of the most abundant proteins in the apoplast of tomato leaves that are infected by C. fulvum (Bolton et al., 2008). Probably, this release is required for the efficient suppression of immune responses as this may permit efficient scavenging of chitin fragments throughout the apoplast (de Jonge et al., 2010; Kombrink & Thomma, 2013; Sánchez‐Vallet et al., 2015). Although Cpl1 can efficiently suppress chitin‐triggered immunity when applied to a suspension of tobacco cells, it may be questioned whether it is able to do so during host infection by U. maydis, given that the experiments described in this manuscript reveal that Cpl1 mainly localizes in the fungal cell wall, and may therefore not be released (in sufficient amounts) in the apoplast of infected maize tissue. As further effectors of yet unknown function were identified in our Co‐IP experiments, more information on their roles during infection and how they connect to Cpl1 will probably help to understand the precise role of Cpl1 for the virulence of U. maydis.
3.3. Cpl1 and Uvi2 might have diverging functions during plant infection
In a previous study, Ökmen et al. (2018) reported that deletion of uvi2, the U. hordei homologue of cpl1, led to a substantial decrease in fungal biomass during infection of barley. Furthermore, they performed yeast two‐hybrid experiments and identified a barley thaumatin as an interaction partner of Uvi2. To explain the prominent differences of uvi2 and cpl1 deletion strains in plant experiments and apparent differences in the interactome, we solved the crystal structure of Uvi2 and compared it with the structure of Cpl1 (Figure 4). Both proteins form stable dimers and share the ability to bind to soluble chitin oligomers. The central DPBB and both grooves on the upper and lower sides of the protein were also conserved at both the sequence and the structural level. Here, the highest structural deviation could be observed in the loop regions, which also showed the overall lowest sequence similarity (Figure 4d). We therefore consider it likely that sequence variations in these loop regions might explain functional differences between Uvi2 and Cpl1. In our Co‐IP experiments Cpl1 was copurified with several fungal effectors during maize infection (Figure 6g). However, peptides corresponding to, for example, maize homologues of the barley thaumatin, which was suggested to be an interaction partner of Uvi2, could not be identified. It is possible that these loop regions provide a modular binding interface for interaction partners, as has been shown recently for members of the plant protein family of kiwellins, which also share the DPBB core fold with Cpl1 and Uvi2 (Altegoer et al., 2020; Bange & Altegoer, 2019).
The interplay between virulence factors produced by smut fungi during infection is still poorly understood. Often, single deletion mutants are readily compensated by redundancy, which masks the underlying function of the individual proteins. In humans, at least an estimated 22% of all proteins are part of complexes (Giurgiu et al., 2019), and there is evidence that some virulence proteins of U. maydis might be part of larger complexes (Alcantara et al., 2019). Slight variations in the amino acid sequence might not only alter the interactome but also allow for, for example, immune escape, as recently shown for a stem rust effector (Ortiz et al., 2022). Our findings on Cpl1 with Uvi2 shed light on the role of a conserved virulence factor with potentially diverging functions during plant infection by U. maydis and U. hordei due to subtle amino acid variations.
4. EXPERIMENTAL PROCEDURES
4.1. Accession numbers
Protein sequences are available from UniProt (https://uniprot.org) under the following accession numbers: U. maydis Cpl1 (A0A0D1E4Q7 [https://www.uniprot.org/uniprot/A0A0D1E4Q7]), U. hordei Uvi2 (I2G262 [https://www.uniprot.org/uniprot/I2G262]).
4.2. Strains and growth conditions
Escherichia coli DH5α (New England Biolabs) was used for cloning purposes. E. coli SHuffle (DE3) (Novagen) was used to express all produced proteins in this study. E. coli strains were grown under constant shaking in a temperature‐controlled incubator. Z. mays ‘Early Golden Bantam’ (Urban Farmer) was used for infection assays with U. maydis (Kämper et al., 2006). Z. mays was grown in a temperature‐controlled greenhouse (light/dark cycles of 14 h at 28°C/10 h at 20°C). U. maydis strains used in this study are listed in Table S4. U. maydis was grown in YEPSlight medium (1% wt/vol yeast extract, 0.4% wt/vol peptone, and 0.4% wt/vol sucrose) at 28°C with baffled flasks under constant shaking at 250 rpm or on potato dextrose agar at 28°C.
4.3. DNA amplification and molecular cloning
All plasmids and primers used in this study are listed in Tables S2 and S3. The open reading frames of all effectors cloned in this study were amplified from the genomic DNA of U. maydis SG200. All genes were amplified without their predicted signal peptide (SignalP v. 5.0) (Almagro Armenteros et al., 2019) employing specific primers (Table S3). A standard PCR protocol with Phusion High Fidelity DNA polymerase (New England Biolabs) and primer‐specific annealing temperatures was used for DNA amplification. For plasmid construction, standard molecular cloning strategies and techniques were applied. Standard plasmid construction using restriction enzymes BspHI and XhoI (New England Biolabs) was used for UMAG_01820. Modular cloning using the restriction enzyme BsaI and T4 DNA ligase (New England Biolabs) was used to generate recombinant plasmids harbouring UHOR_02700 and all other genes used in this study. Briefly, vector and insert containing BsaI recognition sites were digested with BsaI at 37°C for 4 min and ligated at 16°C for 5 min. This reaction was repeated for five to eight cycles with a final ligation step at 16°C for 10 min. For plasmid amplification, recombinant plasmids were transformed in chemically competent E. coli DH5α (New England Biolabs). The correct sequence of all plasmids was verified using Sanger sequencing (Microsynth, Switzerland) with primers specific for the T7 promoter and terminator regions. For protein production cpl1 was cloned into the pEMGB1 vector containing the solubility tag GB1 with a hexahistidine (6His) tag and a tobacco etch virus (TEV) protease recognition site. UHOR_02700 was inserted into pET24d for protein production.
4.4. Transformation and generation of U. maydis strains
U. maydis protoplasts were transformed as previously described by Bösch et al. (2016). Briefly, 2 μg of donor DNA and 1 μg of plasmid in a volume of 10 μL double‐distilled water were added to U. maydis protoplasts, which were incubated on ice for 10 min. Five hundred microlitres of ice‐cold sterile STC (1 M sorbitol, 10 mM Tris–HCl pH 7.5, 100 mM CaCl2) solution supplemented with 40% vol/vol polyethylene glycol (PEG) 3350 was added to the protoplasts, which were incubated for 15 min on ice. Transformants were plated on double‐layered regeneration agar plates. The first layer was supplemented with 4 μg/mL carboxin topped with a layer of regeneration agar (10 g/L yeast extract, 20 g/L Bacto‐peptone [Difco], 20 g/L sucrose, 1 M sorbitol, 15 g/L agar) without antibiotics. Cells were grown for 5 days and subsequently analysed for successful transformation.
The UMAG_01820 gene was disrupted in U. maydis SG200 using the CRISPR–Cas9 approach described for genetic manipulation of U. maydis (Schuster et al., 2016). Donor DNA was supplied during transformation to delete the respective open reading frame from the genome without further disrupting neighbouring genes. Isolated U. maydis transformants were confirmed by colony PCR using the primers listed in Table S3 and sequencing (Microsynth, Switzerland). For the knockout generation, the plasmid pMS73 was digested with Acc65I to integrate the respective single guide RNA expression cassette via Gibson assembly, according to Schuster et al. (2016). The PCR produced a double‐stranded DNA fragment containing the respective target sequences, scaffold, terminator, and the corresponding overlapping sequences. The fragments were cloned into pMS73 (Table S2). The target sequences (Table S3) were designed using the E‐CRISP tool (Heigwer et al., 2014).
The construction of HA‐tagged cpl1 was done as described for knockouts of cpl1 but with donor DNA encoding an HA tag with flanks designed for the C‐terminus of Cpl1. The inserts in all plasmids and knockouts were validated by sequencing.
4.5. Plant infection assays
U. maydis cpl1 knockout strains (SG200, FB1, and FB2) were grown in YEPSlight medium to an OD600 of 0.7 and subsequently adjusted to an OD600 of 1 using sterile double‐distilled water. For the infection of maize plants, 500 μL of culture was injected into the stem of 7‐day‐old maize seedlings using a syringe as described by Kämper et al. (2006). In the case of FB1 and FB2, cultures of both strains were mixed 1:1 before injection. Disease symptoms of infected plants were scored at 12 dpi as described in Kämper et al. (2006). Disease symptoms were quantified based on three biological replicates and are presented as stacked histograms (File S3).
4.6. Immunolocalization
To localize Cpl1‐HA in budding cells and filamentous hyphae, U. maydis strains constitutively expressing Cpl1‐HA were suspended in 2% YEPSlight containing 0.1 mM 16‐hydroxy hexadecanoic acid at a final OD600 of 0.5 and sprayed onto Parafilm. The Parafilm was placed on top of wetted paper towels inside square Petri dishes and incubated at 28°C for 17 h. The Parafilm was washed with water and blocked with phosphate‐buffered saline (PBS) containing 3% (wt/vol) bovine serum albumen and then incubated in α‐HA antibody (Sigma; 1:1500 dilution) diluted in PBS and 3% wt/vol bovine serum albumen at 4°C overnight. The samples were washed with PBS and incubated with a goat anti‐mouse IgG secondary antibody conjugated with AF488 (Life Technologies; 1:1500 dilution) for 1 h at 4°C. After washing, the samples were analysed using an SP8 LSM confocal microscope equipped with a 100× objective (NA 1.4; Leica). Fluorophores were excited with a pulsed white light laser source at 488 nm. Photon emission was detected with a hybrid detector at the appropriate wavelength (495–530 nm). Images were processed with Leica LAS AF software.
4.7. Fungal stress assays
Fungal stress assays were performed as described previously (Weiland & Altegoer, 2021). Briefly, fungal strains were grown in YEPSlight medium to an OD600 of 1.0. The cells were pelleted and resuspended in sterile double‐distilled water to an OD600 of 0.1. For the stress assays, 5 μL of the culture and indicated serial dilutions was spotted on complete medium (Holliday, 1974) plates supplemented with 50 μg/mL Congo red, 45 μg/mL calcofluor white (Sigma‐Aldrich), 1.5 mM H2O2, 1 M NaCl, or 1 M sorbitol. Images were taken after overnight incubation at 28°C.
4.8. Protein production and purification
E. coli Shuffle (DE3) cells (Novagen) were transformed with pEMGB1‐cpl1 to produce Cpl1 fused to an N‐terminal GB1‐tag including a hexahistidine tag. Transformed cells were grown on Luria–Bertani (LB) agar plates supplemented with 100 μg/mL ampicillin. Colonies from the plate were used as preculture in 100 mL LB medium supplemented with 100 μg/mL ampicillin and grown for 16 h at 37°C under constant shaking at 180 rpm. The main culture was inoculated with the preculture to an OD600 of 0.1 and subsequently grown at 30°C and 180 rpm to an OD600 of 0.6. The cultures were then cooled to 20°C and protein production was induced by adding 0.5 mM isopropyl‐β‐d‐1‐thiogalactopyranoside. The cells continued to grow for 20 h at 20°C and 180 rpm. The cultures were harvested by centrifugation (4000 g, 15 min, 4°C), resuspended in buffer A (20 mM HEPES pH 8, 20 mM KCl, 40 mM imidazole, 250 mM NaCl), and subsequently disrupted using a microfluidizer (M110‐L; Microfluidics). The cell debris was removed by centrifugation (50,000 g, 20 min, 4°C). The supernatant was loaded onto Ni‐NTA FF‐HisTrap columns (GE Healthcare) for affinity purification via the hexahistidine tag. The columns were washed with buffer A (10× column volume) and eluted with buffer B (20 mM HEPES pH 8, 20 mM KCl, 250 mM imidazole, 250 mM NaCl). Prior to SEC, the GB1‐tag was cleaved off by adding 0.4 mg purified TEV directly to the eluate and incubating under constant rotation at 20°C for 3 h. Cleaved His‐tagged GB1 and remaining TEV were removed via a second Ni‐NTA purification step after buffer exchange to buffer A using an Amicon Ultra‐10 K centrifugal filter (Merck Millipore). The tag‐free protein was subjected to SEC using a Superdex S200 Increase 26/600 column equilibrated in HEPES buffer (20 mM HEPES pH 7.5, 20 mM KCl, 200 mM NaCl). The peak fractions were analysed using a standard SDS‐PAGE protocol, pooled, and concentrated with Amicon Ultra‐10 K centrifugal filters.
4.9. Selenomethionine incorporation for anomalous diffraction
E. coli SHuffle T7 cells were transformed with pEMGB1‐cpl1 and grown on LB agar plates supplemented with ampicillin (100 μg/mL) for 16 h at 37°C. Colonies from the plate were used as a preculture for inoculation into 400 mL LB medium (containing 100 μg/mL ampicillin), followed by incubation for 16 h at 37°C under constant shaking at 180 rpm. The cells were harvested at 4000 g for 15 min and resuspended in 10 mL M9 medium (37.25 g/L Na2HPO4, 16.5 g/L KH2PO4, 2.75 g/L NaCl, 5.5 g/L NH4Cl, pH 7.5). The resuspended cells were used to inoculate 5 L of M9 medium (with 100 μg/mL ampicillin) to an OD600 of 0.1. The M9 medium was infused with sterile and freshly made SolX solution (1 g/L l‐lysine, 1 g/L l‐threonine, 1 g/L l‐phenylalanine, 0.5 g/L l‐leucine, 0.5 g/L l‐isoleucine, 0.5 g/L valine, 0.25 g/L selenomethionine, 80 g/L glucose, 100 mM MgCl2, 10 mM CaCl2). The cells were grown to an OD600 of 0.6 at 37°C and 180 rpm. Protein production was induced by adding 1 mM isopropyl‐β‐d‐1‐thiogalactopyranoside. The cultures continued to grow at 37°C and 180 rpm for 20–22 h. The cells were harvested by centrifugation and flash‐frozen in liquid nitrogen to be stored at a temperature of −80°C or immediately used for protein preparation.
4.10. Protein crystallization
Crystallization was performed using the sitting‐drop method at 20°C in 0.5–0.75‐μL drops. The crystallization drops contained the protein and precipitant solutions at a ratio of either 1:1 or 1:2. Crystallization drops were set automatically using a Crystal Gryphon robot (Art Robbins Instruments). NeXtal JCSG suites I–IV and Classics II were used to screen for crystallization conditions. Native Cpl1 crystallized at 0.7 mM within 21 days in 0.8 M LiCl, 0.1 M citrate pH 5.0, and 22.5% wt/vol PEG 6000. Selenomethionine Cpl1 crystallized at 0.7 mM within 1 month in 0.8 M LiCl, 0.1 M citrate pH 5.0, and 20% wt/vol PEG 6000 streak‐seeded with native Cpl1 crystals. Uvi2 crystallized at 0.85 mM within a week in 0.1 M sodium acetate pH 5.0 and 10% vol/vol 2‐methyl‐2,4‐pentanediol at a final pH of 5.0.
4.11. Structural analysis by X‐ray crystallography
Prior to data collection, the crystals were flash‐frozen in liquid nitrogen employing a cryosolution that consisted of crystallization solution supplemented with 15% vol/vol glycerol. The data were collected under cryogenic conditions at the EMBL beamline P13 (Deutsches Elektronen Synchrotron; DESY). The data were integrated and scaled using XDS and merged with XSCALE (Kabsch, 2010). The structure of Cpl1 was determined by isomorphous replacement using data obtained from single‐wavelength anomalous dispersion gathered by incorporating selenomethionine. The structure of Uvi2 was determined by molecular replacement in PHASER (McCoy et al., 2007) using the crystal structure of Cpl1 as a search model. Both structures were manually built in COOT (Emsley & Cowtan, 2004) and refined with PHENIX (Adams et al., 2010). The figures were prepared with PyMOL (Delano, 2002) and Chimera (Pettersen et al., 2021).
4.12. Molecular docking
To virtually identify the putative binding pocket, molecular docking was carried out through AutoDock Vina (Trott & Olson, 2010). The receptor PDB file was prepared with AutoDockTools 4 (Morris et al., 2009) by adding the polar hydrogens and performing the conversion to PDBQT. Likewise, the ligand PDB file was also converted to PDBQT using AutoDockTools 4. The search grid box covered the whole receptor, while the exhaustiveness parameter was set to 10,000. Multiple AutoDock Vina runs with randomized seeds resulted in the same putative binding pocket, indicating an informative prediction (Jaghoori et al., 2016).
4.13. SEC‐MALS
SEC‐MALS was performed using an Äkta PURE system (GE Healthcare) with a Superdex 200 Increase 10/300 column attached to a MALS detector 3609 (Postnova Analytics) and a refractive index detector 3150 (Postnova Analytics). The column was equilibrated with 0.2‐μm‐filtered HEPES buffer for analysis at pH 7.5 or citrate buffer for studies at pH 5.0. The column was calibrated for apparent molecular weight determination using a mix of proteins with known molecular weights (conalbumin, 75 kDa; ovalbumin, 44 kDa; carbonic anhydrase, 29 kDa; ribonuclease A1, 13.7 kDa; aprotinin, 6.5 kDa). The molecular weight was calculated by combining the refraction index and MALS values using a Debye fitting.
4.14. Determination of dissociation constants by MST
Dissociation constants of Cpl1 with different sugars were determined via MST. MST experiments were performed in HEPES buffer containing 0.06% vol/vol Tween 20 using a Monolith NT.115 with red LED power set to 100% and infrared laser power set to 75% (Jerabek‐Willemsen et al., 2011). Tag‐free Cpl1 was labelled according to the supplier's instructions (dye NT 647, Nano‐Temper Technologies). Subsequently, 500 nM of labelled protein was titrated against decreasing amounts of mannose, xylose, arabinose, chitobiose, chitotetraose, or cellobiose starting from 5 mM down to 0.15 μM. MST data were recorded at 680 nm and processed by NanoTemper analysis 1.2.009 and Origin8G (File S4).
4.15. HDX‐MS
Samples for HDX‐MS were prepared by mixing 225 μL of purified Cpl1 (50 μM) with 25 μL of double‐distilled water (apo state) or 25 μL of 50 mM concentrated chitobiose or chitotetraose, yielding a final ligand concentration of 5 mM.
Preparation of the HDX reactions was aided by a two‐arm robotic autosampler (LEAP technologies). A 7.5‐μL aliquot of sample was mixed with 67.5 μL of D2O‐containing SEC buffer (20 mM HEPES‐Na pH 7.5, 20 mM KCl, 20 mM MgCl2, 200 mM NaCl) to start the exchange reaction and incubated for 10, 30, 100, 1000, or 10,000 s at 25°C. Subsequently, 55 μL of the reaction was withdrawn and mixed with an equal volume of quench buffer (400 mM KH2PO4/H3PO4, 2 M guanidine‐HCl, pH 2.2) at 1°C. Ninety‐five microlitres of the resulting mixture was injected into an ACQUITY UPLC M‐Class System with HDX Technology (Waters) (Wales et al., 2008). Undeuterated samples were prepared by a similar procedure (incubation for approximately 10 s) through 10‐fold dilution of the protein samples with H2O‐containing SEC buffer. The injected samples were flushed out of the loop (50 μL) with H2O + 0.1% vol/vol formic acid (100 μL/min) and guided to a protease column (2 mm × 2 cm) kept at 12°C that was filled with a 1:1:1 mixture of the proteases porcine pepsin, protease type XIII from Aspergillus saitoi, and protease type XVIII from Rhizopus sp. immobilized to bead material. The resulting peptides were collected on a trap column (2 mm × 2 cm) filled with POROS 20 R2 material (Thermo Scientific) kept at 0.5°C. After 3 min of digestion and trapping, the trap column was placed in line with an ACQUITY UPLC BEH C18 1.7 μm 1 × 100 mm column (Waters), and the peptides were eluted at 0.5°C using the following gradient of H2O + 0.1% vol/vol formic acid (A) and acetonitrile + 0.1% vol/vol formic acid (B) at a flow rate of 60 μL/min: 0–7 min, 95%–65% A; 7–8 min, 65%–15% A; 8–10 min, 15% A; 10–11 min, 5% A; 11–16 min, 95% A. The eluting proteins were guided to a G2‐Si HDMS mass spectrometer with ion mobility separation (Waters), peptides were ionized with an electrospray ionization source (capillary temperature 250°C, spray voltage 3 kV), and mass spectra were acquired in positive ion mode over a range of 50 to 2000 m/z in enhanced high‐definition MS (HDMSE) or high‐definition MS (HDMS) mode for undeuterated and deuterated samples, respectively (Geromanos et al., 2009; Li et al., 2009). [Glu1]‐Fibrinopeptide B standard (Waters) was employed for lock‐mass correction. During separation of the peptide mixtures on the ACQUITY UPLC BEH C18 column, the protease column was washed three times with 80 μL of wash solution (0.5 M guanidine hydrochloride in 4% vol/vol acetonitrile) and blank injections were performed between each sample to reduce peptide carry‐over. All measurements were carried out in triplicate, that is, separate HDX reactions.
Peptide identification and analysis of deuterium incorporation were carried out with ProteinLynx Global SERVER (PLGS, Waters) and DynamX v. 3.0 software (Waters) as described previously (Osorio‐Valeriano et al., 2019).
4.16. Co‐IP and MS
U. maydis strains FB1 and FB2 harbouring cpl1‐HA in its native locus were used to infect Z. mays plants. The control infection was done using U. maydis SG200 containing HA‐tagged mCherry with the signal peptide and the promoter of UmCmu1 (UMAG_05731) integrated into the ip locus. Infected plant leaves were harvested at 3 dpi and flash‐frozen in liquid nitrogen. Frozen plant material was pulverized using a cryogenic mill (MM400; Retsch) with 50‐mL beakers loaded with a metal sphere of 2 cm diameter. Pulverization took place for 1 min at 30 Hz under cryogenic conditions. The plant powder was transferred to 50‐mL Falcon tubes and stored at −80°C.
For the Co‐IP experiments, 2 g of plant powder was added to 6 mL of HNN buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 50 mM NaF, 5 mM EDTA) freshly infused with 1 mM PMSF, diluted cOmplete protease inhibitor cocktail from Roche (stock solution 1:100), and 1% wt/vol polyvinylpyrrolidone K30. The solution was kept at room temperature. A Dounce homogenizer (Carl Roth CXE1.1) was used to disrupt and dissolve the plant powder entirely before adding 0.1% vol/vol NP‐40, 0.5% wt/vol deoxycholic acid sodium salt, 1% wt/vol dodecyl‐β‐d‐maltosid, 1% wt/vol dodecyldimethylaminoxid, and a cell wall‐degrading enzyme mix (1 U cellulase Onozuka‐R10, Macerozyme R‐10, and cellulase from Aspergillus niger). The solution was rotated at room temperature for 30 min. Subsequently, the cell debris was spun down at 4200 g for 10 min at 4°C. The supernatant was transferred to Eppendorf tubes and split into 1‐mL aliquots, to which 15 μL of magnetic anti‐HA beads (Pierce, Thermo Scientific) was added, followed by incubation while rotating at 4°C for 30 min. The beads were separated from the lysate using a magnetic rack. The supernatant was discarded, and the beads were washed three times with 400 μL HNN buffer (containing all ingredients mentioned above except for the cell wall‐degrading enzymes). The beads were washed five times with 800 μL 100 mM ammonium bicarbonate and subsequently flash‐frozen in liquid nitrogen for the subsequent analyses by MS as described previously (Treuner‐Lange et al., 2020). In short, purified proteins were processed by on‐bead digestion using trypsin, followed by reduction (with 5 mM Tris(2‐carboxyethyl)phosphin [TCEP]) and alkylation (with 10 mM iodoacetamide) of peptides. Peptides were further desalted by solid phase extraction on C18 reverse phase spin columns (Macherey‐Nagel) and subsequently analysed by liquid chromatography coupled with MS. Peptides were first separated by an Ultimate 3000 RSLC nano and a Q‐Exactive Plus mass spectrometer (both Thermo Scientific). Settings were set as described previously (Sander et al., 2019). The gradient length was adopted. Peptides were separated over 40 min from 98% solvent A (0.15% formic acid) and 2% solvent B (99.85% acetonitrile, 0.15% formic acid) to 35% solvent B at a flow rate of 300 nL/min. LFQ was carried out by MaxQuant (Tyanova, Temu, & Cox, 2016) using standard settings with variable (oxidized M, deamidated N and Q) and fixed modification (carbamidomethylated C). The resulting MaxQuant “proteinGroups.txt” output table was loaded into Perseus (v. 1.5.2.6) (Tyanova, Temu, Sinitcyn, et al., 2016). For calculation of enrichment factors in samples versus controls, values for proteins not detected in the control were imputed using the imputation function from normal distribution implemented in Perseus using default settings (width, 0.3; down‐shift, 1.8) (Files S5 and S6).
4.17. Chitin precipitation assay
A solution of HA‐tagged Cpl1 (350 mg/mL) was diluted to a concentration of 40 μg/mL using chitin binding buffer (50 mM Tris pH 8.0, 150 mM NaCl), and 800 μL of protein solution was incubated with 50 μL magnetic chitin beads or 5 mg shrimp shell chitin, chitosan, cellulose, or xylan on a rotary shaker at 4°C for 6 h. The insoluble fraction was pelleted by centrifugation at 13,500 g for 5 min, washed three times with water, and resuspended in 100 μL demineralized water. Supernatants were collected and concentrated to a volume of approximately 100 μL using Microcon Ultracel YM‐10 tubes (Merck). Next, 30 μL of the pellet solution and 30 μL of the concentrated supernatant were mixed with 10 μL of protein loading buffer (200 mM Tris–HCl pH 6.5, 0.4 M dithiothreitol, 8% SDS, 6 mM bromophenol blue, 40% glycerol) and incubated at 95°C for 10 min. All protein samples were subjected to protein electrophoresis using a Mini‐PROTEAN TGX stain‐free protein gel (Bio‐Rad).
4.18. Hyphal protection against chitinase hydrolysis
T. viride conidiospores were collected from 5‐day‐old potato dextrose agar plates and adjusted to a concentration of 5 × 105 spores/mL with a total volume of 1 mL (800 μL demineralized water and 200 μL potato dextrose broth). Conidiospore suspensions were dispensed into a 96‐well microtitre plate in aliquots of 50 μL and incubated at room temperature overnight. Effector proteins were dialysed against 20 mM NaCl and adjusted to a concentration of 100 μM. Of each effector, 9 μL was added to appropriate wells to reach a final concentration of 15 μM. After 2 h of incubation, 3 μL chitinase (25 U/mg) from C. thermocellum (Creative Enzymes) was added to the appropriate wells, while sterile water was added as control. All samples were incubated for 6 h and hyphal growth was monitored with an AE2000 microscope (Motic).
4.19. Chitin‐induced medium alkalinization
Medium alkalinization assays were performed with a 5‐day‐old N. tabacum ‘Bright Yellow‐2’ cell suspension. To measure medium alkalinization, 3‐mL aliquots of the suspension were distributed into a 12‐well culture plate that was equilibrated for 4 h on a rotary shaker at 200 rpm. Upon addition of 100 nM chitin hexamer, 1 μM effector protein, or mixtures of chitin hexamer and effector protein with the same final concentrations, the pH of the medium was continuously measured using a pH electrode (InLab Micro) combined with a SevenDirect SD20 pH meter (Mettler Toledo) for 34 min. Prior to addition, mixtures of chitin hexamer and effector protein were incubated at room temperature for 2 h.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests.
Supporting information
Figure S1 Top 10 most abundant transcripts during early infection stages of three smut fungi.
Figure S2 Multiple sequence alignment of Cpl1 homologues.
Figure S3 The Cpl1 dimer is formed by an extensive interaction network between the two protomers.
Figure S4 Structural conservation between Cpl1 and cerato‐platanins.
Figure S5 Binding of chitobiose and chitotetraose by Cpl1 analysed with hydrogen‐deuterium exchange coupled with mass spectrometry (HDX‐MS).
Figure S6 Representative peptides of Cpl1 exemplifying chitobiose/chitotetraose‐dependent changes in hydrogen‐deuterium exchange coupled with mass spectrometry (HDX‐MS).
Figure S7 Affinity of N‐acetylglucosamine oligomer binding to Uvi2.
Figure S8 Stress assays.
Figure S9 Western blot of SG200 strains overexpressing Cpl1‐HA.
Figure S10 Correlation between transcript abundance and identified peptides of the top eight candidates from the Cpl1‐HA co‐immunoprecipitation.
File S1 Dataset‐PISA analysis. Residues involved in the dimer interaction of the two Cpl1 protomers obtained from PDBePISA analysis of the Cpl1 structure.
File S2 Dataset‐HDX. Hydrogen‐deuterium exchange coupled with mass spectrometry (HDX‐MS) data of Cpl1–apo, Cpl1–chitobiose, and Cpl1–chitotetraose.
File S3 Dataset‐PlantInfections. Symptom scoring from plant infection experiments.
File S4 Dataset‐MST. Data obtained from microscale thermophoresis experiments of Cpl1 with different carbohydrates.
File S5 Dataset‐LFQ‐data‐matrix. Data matrix of mass spectrometry experiments after label‐free quantification.
File S6 Dataset‐MScounts. MS count data from mass spectrometry experiments.
Table S1 Crystallographic data collection and refinement statistics.
Table S2 Plasmids used in this study.
Table S3 Primers used in this study.
Table S4 Ustilago maydis strains used in this study.
Table S5 All eight significantly and repeatedly enriched proteins with their label‐free quantification (LFQ) values.
ACKNOWLEDGEMENTS
We thank Regine Kahmann for her continuous support throughout the work on this manuscript and her group for helpful discussions. We also thank Mariana Schuster for expert help with the CRISPR–Cas9 system and Xiaowei Han for assistance with strain construction and plant infection experiments. We thank Vera Göhre and Michael Feldbrügge for critical reading and fruitful discussions on the manuscript. We thank the EMBL Hamburg at the PETRA III storage ring (DESY, Hamburg, Germany) for support. G.B. thanks the European Research Council for support through the project “KIWIsome” (grant agreement 101019765) and the German Research Foundation (Deutsche Forschungsgemeinschaft [DFG]) through the Core Facility for Interactions, Dynamics and Macromolecular Assembly (project 324652314). We acknowledge the contribution of the Core Facility “Protein Biochemistry and Spectroscopy” of Philipps‐University Marburg. B.P.H.J.T. acknowledges funding by the Alexander von Humboldt Foundation in the framework of an Alexander von Humboldt Professorship endowed by the German Federal Ministry of Education and is furthermore supported by the DFG under Germany's Excellence Strategy EXC 2048/1 (project ID: 390686111). B.P.H.J.T. and F.A. acknowledge funding through the DFG (project 458090666/CRC1535/1). Open access funding enabled and organized by Projekt DEAL.
Weiland, P. , Dempwolff, F. , Steinchen, W. , Freibert, S.‐A. , Tian, H. , Glatter, T. et al. (2023) Structural and functional analysis of the cerato‐platanin‐like protein Cpl1 suggests diverging functions in smut fungi. Molecular Plant Pathology, 24, 768–787. Available from: 10.1111/mpp.13349
Contributor Information
Gert Bange, Email: gert.bange@synmikro.uni-marburg.de.
Florian Altegoer, Email: altegoer@hhu.de.
DATA AVAILABILITY STATEMENT
Coordinates and structure factors have been deposited within the PDB at www.rcsb.org under accession codes 8A14 and 8A4O. The authors declare that all other data supporting the findings of this study are available within the article and its supplementary information files.
REFERENCES
- Adams, P.D. , Afonine, P.V. , Bunkóczi, G. , Chen, V.B. , Davis, I.W. , Echols, N. et al. (2010) PHENIX: a comprehensive python‐based system for macromolecular structure solution. Acta Crystallographica. Section D, Biological Crystallography, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcantara, A. , Bosch, J. , Nazari, F. , Hoffmann, G. , Gallei, M. , Uhse, S. et al. (2019) Systematic Y2H screening reveals extensive effector‐complex formation. Frontiers in Plant Science, 10, 1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almagro Armenteros, J.J. , Tsirigos, K.D. , Sønderby, C.K. , Petersen, T.N. , Winther, O. , Brunak, S. et al. (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks. Nature Biotechnology, 37, 420–423. [DOI] [PubMed] [Google Scholar]
- Altegoer, F. , Weiland, P. , Giammarinaro, P.I. , Freibert, S.‐A. , Binnebesel, L. , Han, X. et al. (2020) The two paralogous kiwellin proteins KWL1 and KWL1‐b from maize are structurally related and have overlapping functions in plant defense. Journal of Biological Chemistry, 295, 7816–7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccelli, I. (2015) Cerato‐platanin family proteins: one function for multiple biological roles? Frontiers in Plant Science, 5, 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bange, G. & Altegoer, F. (2019) Plants strike back: kiwellin proteins as a modular toolbox for plant defense mechanisms. Communicative & Integrative Biology, 12, 31–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banuett, F. & Herskowitz, I. (1989) Different a alleles of Ustilago maydis are necessary for maintenance of filamentous growth but not for meiosis. Proceedings of the National Academy of Sciences of the United States of America, 86, 5878–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsottini, M.R. , De Oliveira, J.F. , Adamoski, D. , Teixeira, P.J.P.L. , Do Prado, P.F.V. , Tiezzi, H.O. et al. (2013) Functional diversification of cerato‐platanins in Moniliophthora perniciosa as seen by differential expression and protein function specialization. Molecular Plant‐Microbe Interactions, 26, 1281–1293. [DOI] [PubMed] [Google Scholar]
- Benevenuto, J. , Teixeira‐Silva, N.S. , Kuramae, E.E. , Croll, D. & Monteiro‐Vitorello, C.B. (2018) Comparative genomics of smut pathogens: insights from orphans and positively selected genes into host specialization. Frontiers in Microbiology, 9, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton, M.D. , van Esse, H.P. , Vossen, J.H. , de Jonge, R. , Stergiopoulos, I. , Stulemeijer, I.J. et al. (2008) The novel Cladosporium fulvum lysin motif effector Ecp6 is a virulence factor with orthologues in other fungal species. Molecular Microbiology, 69, 119–136. [DOI] [PubMed] [Google Scholar]
- Bösch, K. , Frantzeskakis, L. , Vraneš, M. , Kämper, J. , Schipper, K. & Göhre, V. (2016) Genetic manipulation of the plant pathogen Ustilago maydis to study fungal biology and plant microbe interactions. Journal of Visualized Experiments, 115, 54522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brefort, T. , Doehlemann, G. , Mendoza‐Mendoza, A. , Reissmann, S. , Djamei, A. & Kahmann, R. (2009) Ustilago maydis as a pathogen. Annual Review of Phytopathology, 47, 423–445. [DOI] [PubMed] [Google Scholar]
- Castillo, R.M. , Mizuguchi, K. , Dhanaraj, V. , Albert, A. , Blundell, T.L. & Murzin, A.G. (1999) A six‐stranded double‐psi barrel is shared by several protein superfamilies. Structure, 7, 227–236. [DOI] [PubMed] [Google Scholar]
- Chen, H. , Kovalchuk, A. , Keriö, S. & Asiegbu, F.O. (2013) Distribution and bioinformatic analysis of the cerato‐platanin protein family in Dikarya. Mycologia, 105, 1479–1488. [DOI] [PubMed] [Google Scholar]
- de Jonge, R. , van Esse, H.P. , Kombrink, A. , Shinya, T. , Desaki, Y. , Bours, R. et al. (2010) Conserved fungal LysM effector Ecp6 prevents chitin‐triggered immunity in plants. Science, 329, 953–955. [DOI] [PubMed] [Google Scholar]
- de Vries, R.P. , Kester, H.C. , Poulsen, C.H. , Benen, J.A. & Visser, J. (2000) Synergy between enzymes from Aspergillus involved in the degradation of plant cell wall polysaccharides. Carbohydrate Research, 327, 401–410. [DOI] [PubMed] [Google Scholar]
- de Wit, P.J. , Mehrabi, R. , Van den Burg, H.A. & Stergiopoulos, I. (2009) Fungal effector proteins: past, present and future. Molecular Plant Pathology, 10, 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean, R. , Van Kan, J.A.L. , Pretorius, Z.A. , Hammond‐Kosack, K.E. , Di Pietro, A. , Spanu, P.D. et al. (2012) The top 10 fungal pathogens in molecular plant pathology. Molecular Plant Pathology, 13, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano, W.L. (2002) The PyMOL molecular graphics system. San Carlos: DeLano Scientific. [Google Scholar]
- Dobson, L. , Reményi, I. & Tusnády, G.E. (2015) CCTOP: a consensus constrained TOPology prediction web server. Nucleic Acid Research, 43, W408–W412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehlemann, G. , Wahl, R. , Vranes, M. , de Vries, R.P. , Kamper, J. & Kahmann, R. (2008) Establishment of compatibility in the Ustilago maydis/maize pathosystem. Journal of Plant Physiology, 165, 29–40. [DOI] [PubMed] [Google Scholar]
- Emsley, P. & Cowtan, K. (2004) Coot: model‐building tools for molecular graphics. Acta Crystallographica Section D Biological Crystallography, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fiorin, G.L. , Sanchéz‐Vallet, A. , Thomazella, D.P.T. , do Prado, P.F.V. , do Nascimento, L.C. , Figueira, A.V.O. et al. (2018) Suppression of plant immunity by fungal chitinase‐like effectors. Current Biology, 28, 3023–3030.e5. [DOI] [PubMed] [Google Scholar]
- Gaderer, R. , Bonazza, K. & Seidl‐Seiboth, V. (2014) Cerato‐platanins: a fungal protein family with intriguing properties and application potential. Applied Microbiology and Biotechnology, 98, 4795–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geromanos, S.J. , Vissers, J.P.C. , Silva, J.C. , Dorschel, C.A. , Li, G.Z. , Gorenstein, M.V. et al. (2009) The detection, correlation, and comparison of peptide precursor and product ions from data independent LC‐MS with data dependant LC‐MS/MS. Proteomics, 9, 1683–1695. [DOI] [PubMed] [Google Scholar]
- Giurgiu, M. , Reinhard, J. , Brauner, B. , Dunger‐Kaltenbach, I. , Fobo, G. , Frishman, G. et al. (2019) CORUM: the comprehensive resource of mammalian protein complexes‐2019. Nucleic Acids Research, 47(D1), D559–D563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazebrook, J. (2005) Contrasting mechanisms of defense against biotrophic and necrotrophic pathogens. Annual Review of Phytopathology, 43, 205–227. [DOI] [PubMed] [Google Scholar]
- Heigwer, F. , Kerr, G. & Boutros, M. (2014) E‐CRISP: fast CRISPR target site identification. Nature Methods, 11, 122–123. [DOI] [PubMed] [Google Scholar]
- Holliday, R. (1974) Ustilago maydis. In: King, R.C. (Ed.) Handbook of genetics, Vol. 1. New York: Plenum, pp. 575–595. [Google Scholar]
- Holm, L. (2020) Using Dali for protein structure comparison. Methods in Molecular Biology, 2112, 29–42. [DOI] [PubMed] [Google Scholar]
- Hu, S.‐P. , Li, J.‐J. , Dhar, N. , Li, J.‐P. , Chen, J.‐Y. , Jian, W. et al. (2021) Lysin motif (LysM) proteins: interlinking manipulation of plant immunity and fungi. International Journal of Molecular Sciences, 22, 3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaghoori, M.M. , Bleijlevens, B. & Olabarriaga, S.D. (2016) 1001 ways to run AutoDock Vina for virtual screening. Journal of Computer‐Aided Molecular Design, 30, 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerabek‐Willemsen, M. , Wienken, C.J. , Braun, D. , Baaske, P. & Duhr, S. (2011) Molecular interaction studies using microscale thermophoresis. Assay and Drug Development Technologies, 9, 342–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, D.A. , Bertazzoni, S. , Turo, C.J. , Syme, R.A. & Hane, J.K. (2018) Bioinformatic prediction of plant‐pathogenicity effector proteins of fungi. Current Opinion in Microbiology, 46, 43–49. [DOI] [PubMed] [Google Scholar]
- Jones, J.D.G. & Dangl, J.L. (2006) The plant immune system. Nature, 444, 323–329. [DOI] [PubMed] [Google Scholar]
- Kabsch, W. (2010) XDS. Acta Crystallographica. Section D, Biological Crystallography, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaku, H. , Nishizawa, Y. , Ishii‐Minami, N. , Akimoto‐Tomiyama, C. , Dohmae, N. , Takio, K. et al. (2006) Plant cells recognize chitin fragments for defense signaling through a plasma membrane receptor. Proceedings of the National Academy of Sciences of the United States of America, 103, 11086–11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kämper, J. , Kahmann, R. , Bölker, M. , Ma, L.‐J. , Brefort, T. , Saville, B.J. et al. (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis . Nature, 444, 97–101. [DOI] [PubMed] [Google Scholar]
- Kombrink, A. , Sánchez‐Vallet, A. & Thomma, B.P.H.J. (2011) The role of chitin detection in plant–pathogen interactions. Microbes and Infection, 13, 1168–1176. [DOI] [PubMed] [Google Scholar]
- Kombrink, A. & Thomma, B.P.H.J. (2013) LysM effectors: secreted proteins supporting fungal life. PLoS Pathogens, 9, e1003769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel, E. & Henrick, K. (2007) Inference of macromolecular assemblies from crystalline state. Journal of Molecular Biology, 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Lanver, D. , Müller, A.N. , Happel, P. , Schweizer, G. , Haas, F.B. , Franitza, M. et al. (2018) The biotrophic development of Ustilago maydis studied by RNA‐seq analysis. The Plant Cell, 30, 300–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanver, D. , Tollot, M. , Schweizer, G. , Lo Presti, L. , Reissmann, S. , Ma, L.‐S. et al. (2017) Ustilago maydis effectors and their impact on virulence. Nature Reviews Microbiology, 15, 409–421. [DOI] [PubMed] [Google Scholar]
- Li, G.‐Z. , Vissers, J.P.C. , Silva, J.C. , Golick, D. , Gorenstein, M.V. & Geromanos, S.J. (2009) Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics, 9, 1696–1719. [DOI] [PubMed] [Google Scholar]
- Li, S. , Dong, Y. , Li, L. , Zhang, Y. , Yang, X. , Zeng, H. et al. (2019) The novel cerato‐platanin‐like protein FocCP1 from Fusarium oxysporum triggers an immune response in plants. International Journal of Molecular Sciences, 20, 2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, W. , Cowley, A. , Uludag, M. , Gur, T. , McWilliam, H. , Squizzato, S. et al. (2015) The EMBL‐EBI bioinformatics web and programmatic tools framework. Nucleic Acid Research, 43, W580–W584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luti, S. , Sella, L. , Quarantin, A. , Pazzagli, L. & Baccelli, I. (2019) Twenty years of research on cerato‐platanin family proteins: clues, conclusions, and unsolved issues. Fungal Biology Reviews, 34, 13–24. [Google Scholar]
- Ma, L.‐S. , Wang, L. , Trippel, C. , Mendoza‐Mendoza, A. , Ullmann, S. , Moretti, M. et al. (2018) The Ustilago maydis repetitive effector Rsp3 blocks the antifungal activity of mannose‐binding maize proteins. Nature Communications, 9, 1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín‐Menguiano, M. , Moreno‐Sánchez, I. , Barrales, R.R. , Fernández‐Álvarez, A. & Ibeas, J. (2019) N‐glycosylation of the protein disulfide isomerase Pdi1 ensures full Ustilago maydis virulence. PLoS Pathogens, 15, e1007687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, R. , Kombrink, A. , Motteram, J. , Loza‐Reyes, E. , Lucas, J. , Hammond‐Kosack, K.E. et al. (2011) Analysis of two in planta expressed LysM effector homologs from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat. Plant Physiology, 156, 756–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch, F. , Mauch‐Mani, B. & Boller, T. (1988) Antifungal hydrolases in pea tissue: II. Inhibition of fungal growth by combinations of chitinase and β‐1,3‐glucanase. Plant Physiology, 88, 936–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy, A.J. , Grosse‐Kunstleve, R.W. , Adams, P.D. , Winn, M.D. , Storoni, L.C. & Read, R.J. (2007) Phaser crystallographic software. Journal of Applied Crystallography, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Sánchez, I. , Pejenaute‐Ochoa, M.D. , Navarrete, B. , Barrales, R.R. & Ibeas, J.I. (2021) Ustilago maydis secreted endo‐xylanases are involved in fungal filamentation and proliferation on and inside plants. Journal of Fungi, 7, 1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, G.M. , Huey, R. , Lindstrom, W. , Sanner, M.F. , Belew, R.K. , Goodsell, D.S. et al. (2009) AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. Journal of Computational Chemistry, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narvaez‐Barragan, D.A. , Tovar‐Herrera, O.E. , Segovia, L. , Serrano, M. & Martinez‐Anaya, C. (2020) Expansin‐related proteins: biology, microbe–plant interactions and associated plant‐defense responses. Microbiology, 166, 1007–1018. [DOI] [PubMed] [Google Scholar]
- Ökmen, B. , Jaeger, E. , Schilling, L. , Finke, N. , Klemd, A. , Lee, Y.J. et al. (2022) A conserved enzyme of smut fungi facilitates cell‐to‐cell extension in the plant bundle sheath. Nature Communications, 13, 6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ökmen, B. , Mathow, D. , Hof, A. , Lahrmann, U. , Aßmann, D. & Doehlemann, G. (2018) Mining the effector repertoire of the biotrophic fungal pathogen Ustilago hordei during host and non‐host infection. Molecular Plant Pathology, 19, 2603–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz, D. , Chen, J. , Outram, M.A. , Saur, I.M.L. , Upadhyaya, N.M. , Mago, R. et al. (2022) The stem rust effector protein AvrSr50 escapes Sr50 recognition by a substitution in a single surface‐exposed residue. The New Phytologist, 234, 592–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osorio‐Valeriano, M. , Altegoer, F. , Steinchen, W. , Urban, S. , Liu, Y. , Bange, G. et al. (2019) ParB‐type DNA segregation proteins are CTP‐dependent molecular switches. Cell, 179, 1512–1524.e15. [DOI] [PubMed] [Google Scholar]
- Pazzagli, L. , Seidl‐Seiboth, V. , Barsottini, M. , Vargas, W.A.A. , Scala, A. & Mukherjee, P.K.K. (2014) Cerato‐platanins: elicitors and effectors. Plant Science, 228, 79–87. [DOI] [PubMed] [Google Scholar]
- Pettersen, E.F. , Goddard, T.D. , Huang, C.C. , Meng, E.C. , Couch, G.S. , Croll, T.I. et al. (2021) UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Science, 30, 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusztahelyi, T. (2018) Chitin and chitin‐related compounds in plant–fungal interactions. Mycology, 9, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez‐Vallet, A. , Mesters, J.R. & Thomma, B.P.H.J. (2015) The battle for chitin recognition in plant–microbe interactions. FEMS Microbiology Reviews, 39, 171–183. [DOI] [PubMed] [Google Scholar]
- Sánchez‐Vallet, A. , Tian, H. , Rodriguez‐Moreno, L. , Valkenburg, D.J. , Saleem‐Batcha, R. , Wawra, S. et al. (2020) A secreted LysM effector protects fungal hyphae through chitin‐dependent homodimer polymerization. PLoS Pathogens, 16, e1008652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander, T. , Farke, N. , Diehl, C. , Kuntz, M. , Glatter, T. & Link, H. (2019) Allosteric feedback inhibition enables robust amino acid biosynthesis in E. coli by enforcing enzyme overabundance. Cell Systems, 8, 66–75.e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauwecker, F. , Wanner, G. & Kahmann, R. (1995) Filament‐specific expression of a cellulase gene in the dimorphic fungus Ustilago maydis . Biological Chemistry Hoppe‐Seyler, 376, 617–625. [DOI] [PubMed] [Google Scholar]
- Schirawski, J. , Mannhaupt, G. , Munch, K. , Brefort, T. , Schipper, K. , Doehlemann, G. et al. (2010) Pathogenicity determinants in smut fungi revealed by genome comparison. Science, 330, 1546–1548. [DOI] [PubMed] [Google Scholar]
- Schuster, M. , Schweizer, G. , Reissmann, S. & Kahmann, R. (2016) Genome editing in Ustilago maydis using the CRISPR‐Cas system. Fungal Genetics and Biology, 89, 3–9. [DOI] [PubMed] [Google Scholar]
- Seidl, V. , Marchetti, M. , Schandl, R. , Allmaier, G. & Kubicek, C.P. (2006) Epl1, the major secreted protein of Hypocrea atroviridis on glucose, is a member of a strongly conserved protein family comprising plant defense response elicitors. The FEBS Journal, 273, 4346–4359. [DOI] [PubMed] [Google Scholar]
- Sievers, F. , Wilm, A. , Dineen, D. , Gibson, T.J. , Karplus, K. , Li, W. et al. (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology, 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanu, P.D. , Abbott, J.C. , Amselem, J. , Burgis, T.A. , Soanes, D.M. , Stuber, K. et al. (2010) Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science, 330, 1543–1546. [DOI] [PubMed] [Google Scholar]
- Tanaka, S. & Kahmann, R. (2021) Cell wall‐associated effectors of plant‐colonizing fungi. Mycologia, 113, 247–260. [DOI] [PubMed] [Google Scholar]
- Tian, H. , MacKenzie, C.I. , Rodriguez‐Moreno, L. , van den Berg, G.C.M. , Chen, H. , Rudd, J.J. et al. (2022) Three LysM effectors of Zymoseptoria tritici collectively disarm chitin‐triggered plant immunity. Molecular Plant Pathology, 22, 683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treuner‐Lange, A. , Chang, Y.W. , Glatter, T. , Herfurth, M. , Lindow, S. , Chreifi, G. et al. (2020) PilY1 and minor pilins form a complex priming the type IVa pilus in Myxococcus xanthus . Nature Communications, 11, 5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott, O. & Olson, A.J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova, S. , Temu, T. & Cox, J. (2016) The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nature Protocols, 11, 2301–2319. [DOI] [PubMed] [Google Scholar]
- Tyanova, S. , Temu, T. , Sinitcyn, P. , Carlson, A. , Hein, M.Y. , Geiger, T. et al. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods, 13, 731–740. [DOI] [PubMed] [Google Scholar]
- van den Burg, H.A. , Harrison, S.J. , Joosten, M.H. , Vervoort, J. & de Wit, P.J. (2006) Cladosporium fulvum Avr4 protects fungal cell walls against hydrolysis by plant chitinases accumulating during infection. Molecular Plant‐Microbe Interactions, 19, 1420–1430. [DOI] [PubMed] [Google Scholar]
- van Esse, H.P. , Bolton, M.D. , Stergiopoulos, I. , de Wit, P.J.G.M. & Thomma, B.P.H.J. (2007) The chitin‐binding Cladosporium fulvum effector protein Avr4 is a virulence factor. Molecular Plant‐Microbe Interactions, 20, 1092–1101. [DOI] [PubMed] [Google Scholar]
- Volk, H. , Marton, K. , Flajšman, M. , Radišek, S. , Tian, H. , Hein, I. et al. (2019) Chitin‐binding protein of Verticillium nonalfalfae disguises fungus from plant chitinases and suppresses chitin‐triggered host immunity. Molecular Plant‐Microbe Interactions, 32, 1378–1390. [DOI] [PubMed] [Google Scholar]
- Wales, T.E. , Fadgen, K.E. , Gerhardt, G.C. & Engen, J.R. (2008) High‐speed and high‐resolution UPLC separation at zero degrees Celsius. Analytical Chemistry, 80, 6815–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland, P. & Altegoer, F. (2021) Identification and characterization of two transmembrane proteins required for virulence of Ustilago maydis . Frontiers in Plant Sciences, 12, 669835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B. , Zhang, N. , Zhang, Q. , Xu, Q. , Zhong, T. , Zhang, K. et al. (2021) Transcriptome profiles of Sporisorium reilianum during the early infection of resistant and susceptible maize isogenic lines. Journal of Fungi, 7, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo, W. , Ökmen, B. , Depotter, J.R.L. , Ebert, M.K. , Redkar, A. , Villamil, J.M. et al. (2019) Molecular interactions between smut fungi and their host plants. Annual Review of Phytopathology, 57, 411–430. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Top 10 most abundant transcripts during early infection stages of three smut fungi.
Figure S2 Multiple sequence alignment of Cpl1 homologues.
Figure S3 The Cpl1 dimer is formed by an extensive interaction network between the two protomers.
Figure S4 Structural conservation between Cpl1 and cerato‐platanins.
Figure S5 Binding of chitobiose and chitotetraose by Cpl1 analysed with hydrogen‐deuterium exchange coupled with mass spectrometry (HDX‐MS).
Figure S6 Representative peptides of Cpl1 exemplifying chitobiose/chitotetraose‐dependent changes in hydrogen‐deuterium exchange coupled with mass spectrometry (HDX‐MS).
Figure S7 Affinity of N‐acetylglucosamine oligomer binding to Uvi2.
Figure S8 Stress assays.
Figure S9 Western blot of SG200 strains overexpressing Cpl1‐HA.
Figure S10 Correlation between transcript abundance and identified peptides of the top eight candidates from the Cpl1‐HA co‐immunoprecipitation.
File S1 Dataset‐PISA analysis. Residues involved in the dimer interaction of the two Cpl1 protomers obtained from PDBePISA analysis of the Cpl1 structure.
File S2 Dataset‐HDX. Hydrogen‐deuterium exchange coupled with mass spectrometry (HDX‐MS) data of Cpl1–apo, Cpl1–chitobiose, and Cpl1–chitotetraose.
File S3 Dataset‐PlantInfections. Symptom scoring from plant infection experiments.
File S4 Dataset‐MST. Data obtained from microscale thermophoresis experiments of Cpl1 with different carbohydrates.
File S5 Dataset‐LFQ‐data‐matrix. Data matrix of mass spectrometry experiments after label‐free quantification.
File S6 Dataset‐MScounts. MS count data from mass spectrometry experiments.
Table S1 Crystallographic data collection and refinement statistics.
Table S2 Plasmids used in this study.
Table S3 Primers used in this study.
Table S4 Ustilago maydis strains used in this study.
Table S5 All eight significantly and repeatedly enriched proteins with their label‐free quantification (LFQ) values.
Data Availability Statement
Coordinates and structure factors have been deposited within the PDB at www.rcsb.org under accession codes 8A14 and 8A4O. The authors declare that all other data supporting the findings of this study are available within the article and its supplementary information files.