


Abstract
The low and unpredictable uptake and cytosolic transfer of oligonucleotides (ODN) is a major reason for their limited benefit. Improving the ODN potential for therapy and research requires a better understanding of their receptor-mediated endocytosis. We have undertaken to identify a membrane ODN receptor on HepG2 cells by ligand blotting of cell extracts with [125I]ODN and by photolabelling of living cells with a [125I]ODN-benzophenone conjugate. A major band at 66 kDa was identified by the two methods. Its labelling was saturable and competed for by unlabelled ODN of various sequences and irrespective of the presence of a phosphodiester or phosphorothioate backbone. This protein remained sedimentable after carbonate extraction, indicating strong membrane association. About half of the total cell amount resisted extensive surface proteolysis, suggesting a dual localisation at the plasma membrane and cytoplasmic vesicles. The protein was purified using a biotinylated ODN-benzophenone conjugate by photocrosslinking followed by streptavidin affinity purification. A sequence obtained by Edman degradation showed no homology with known proteins. Using anti-peptide antisera, labelling by western blotting revealed at 66 kDa a band with comparable properties as found by ligand blotting. Thus, a new membrane protein acting as an ODN receptor has been demonstrated.
INTRODUCTION
Oligonucleotides (ODNs) are short nucleic acid sequences with important potential for research and therapy (1). ODNs can act in many ways. The original aim of their application to living cells was to inhibit specific gene expression by an antisense mechanism (2). Besides steric arrest of the translational machinery, some ODNs can further recruit RNase H (3) or RNase L (4) that degrade complexed (pre)mRNA. Different ODNs, designed to form triple helices with DNA sequences, have been developed to inhibit expression upstream, at the transcriptional level (5). Successful use of ODNs to correct point mutations (6) or splicing errors (7) has also been reported. Ribozymes are oligonucleotides with intrinsic catalytic activity (8). ODNs referred to as aptamers are selected to bind with high affinity to predefined proteins so as to inhibit their function (9,10).
Unmodified ODNs are very sensitive to nuclease attack. While capping of the 3′- and 5′-ends protects ODNs against exonucleases, backbone modifications such as phosphorothioate modification of DNA further prevent endonuclease attack (11). However, these ODN modifications may alter intracellular trafficking, specificity and mode of action (12–15).
To be effective, intact ODNs must reach the cytosol or the nucleus. Nuclear pores do not represent a barrier, since ODNs microinjected into the cytosol are rapidly found in the nucleus (16). In contrast, lipid bilayers are impermeable to polyanions such as ODNs (12,17–19). Accordingly, ODNs are taken up by endocytosis, a process that is indeed blocked at low temperature (15,17,19–21), except in keratinocytes (22,23). After endocytic uptake, ODNs are sequestered in punctate intracellular structures (13,15) that are sedimentable (24). This represents an intracellular membrane-bound reservoir, from which ODNs are supposed to somehow escape.
Conflicting results in the literature on the mode of ODN endocytosis may be due to variations in the choice of cell line as well as ODN structure and concentration. In most cases, fluid phase pinocytosis or adsorptive endocytosis seem predominant in the micromolar range generally used for in vitro experiments (12,17,19,21). However, non-specific effects are frequent at such high concentrations. These drawbacks can be sequence independent (24,25) or due to a special motif such as a G-tetrad (26,27) and CG-rich sequences (28,29). The non-specific effects of ODNs at high extracellular concentrations and the presumably inefficient cytosolic transfer might explain failures of their use. Specificity problems can be minimised in two ways : (i) avoiding some ODN classes or sequences; (ii) reducing ODN concentration in the medium, provided uptake in target cells remains sufficient or cytosolic translocation can be improved.
The most efficient mechanism of vesicular uptake is receptor-mediated endocytosis (30), which involves two concentration steps. In the first step, a ligand becomes concentrated at the cell surface upon binding to its specific plasma membrane receptor. The level of ligand concentration reflects receptor abundance and affinity. In the second concentration step, ligand–receptor complexes are clustered in and preferentially internalised by clathrin-coated pits, leading to accelerated endocytosis. Receptor-mediated endocytosis of ODN has been suggested (18). Furthermore, specific ODN-binding proteins acting at relatively low concentrations have been found on the surface of various cell lines (17,19,20,23,31), but none has been characterised so far. In addition, multifunctional binding proteins, including albumin (32), the scavenger receptor (24,27,33) and Mac-1 (34), have been implicated in ODN uptake at high concentration.
In this paper, we aimed at the purification of an ODN membrane receptor. We have selected the human hepatocarcinoma cell line HepG2 for several reasons. (i) Hepatocytes are involved in the clearance of injected ODN (24); (ii) the rate of ODN capture is particularly high in the HepG2 cell line (15); and (iii) ODN intracellular routing has been extensively studied in these cells (15). The occurrence of a specific membranous receptor for ODN in these cells was demonstrated using ligand blotting and in situ photolabelling with an ODN-benzophenone conjugate (35). The receptor was purified using a biotinylated ODN-benzophenone conjugate and streptavidin affinity chromatography and found by microsequence analysis to be a novel protein. Partial resistance of the receptor to extensive surface protease digestion indicates a dual localisation at the plasma membrane and on intracellular vesicles, compatible with endocytosis of ligand–receptor complexes and receptor recycling.
MATERIALS AND METHODS
Oligonucleotides
Four different 25mer phosphodiester ODNs, with a sequence complementary to the AUG initiation site of HIV-1 gag, were used in this study (Table 1). The first ODN bore a fluorescein moiety at its 5′-end and its 3′-end was capped with a free amine. In the third and fourth ODNs, biotin was introduced at the 5′-end during synthesis. In addition, cytidine at the 5′ second position of the fourth ODN was replaced by a fluorescein via fluorescein phosphoramidite. All these ODNs were purchased from Eurogentec (Seraing, Belgium). The last three ODNs were further derivatised with benzophenone, by adapting the coupling procedure to ODNs bearing a free amine at the 3′-end (Amino-modifier C6 dT; Eurogentec) as previously described (35). Products were purified by HPLC and the efficiency of biotin coupling was demonstrated by the disappearance of HPLC signal upon incubation with streptavidin-coated beads (Immobilised Streptavidin; Boehringer, Mannheim, Germany) in 20 mM phosphate buffer, pH 7.4, for 30 min at room temperature. Once coupled to benzophenone, ODNs were handled in the dark or under a safe light. In addition, a phosphorothioate ODN of identical sequence was used for competition experiments (a kind gift of Dr A.C. Roche, Orléans, France).
Table 1. ODN derivatisations.

F, fluorescein (Fl); B, benzophenone (BΦ); b, biotin (biot.).
Fluoresceinated ODNs were radiolabelled using IodoBeads (Pierce, Rockford, IL) to a specific radioactivity ranging from 10 000 to 20 000 c.p.m./ng. Briefly, 1 mCi of Na125I (Amersham Pharmacia Biotech, Uppsala, Sweden) was diluted into 400 µl of 10 mM phosphate buffer, pH 6.6, and added to washed IodoBeads. After 5 min, 2–10 nmol of ODN in 100 µl were added. After 30 min of gentle intermittent mixing, the reaction was arrested by the addition of 500 µl of a 30 mM NaI, 25 mM Na2S2O3 solution. Free Na125I was finally removed by gel filtration through a Sephadex G-25 column (Pharmacia).
Cell culture and in situ photocrosslinking
HepG2 cells (kindly donated by Prof. G. Strous, Utrecht, The Netherlands) were propagated in Falcon flasks at 37°C under a humidified atmosphere containing 8% CO2, in RPMI 1640 medium (Gibco BRL, Gaithersburg, MD) supplemented with amino acids (200 µM alanine, 1 mM arginine, 400 µM cysteine, 4 mM glutamine, 130 µM histidine, 500 µM isoleucine, 200 µM serine and 300 µM tyrosine), 60 µg/ml of both penicillin and streptomycin (Sigma, St Louis, MO) and 10% foetal calf serum (PAA Laboratories, Linz, Austria).
For receptor photocrosslinking, cells at near confluency were incubated in Petri dishes for 2 h in the dark at 37°C with 20–100 nM ODN-benzophenone derivatives. When radiolabelled conjugates were used, radioactivity was kept constant at 5 000 000 c.p.m./ml. Petri dishes were then transferred to 4°C, washed eight times with phosphate-buffered saline containing 0.44 mM Ca2+ (PBS-Ca2+) and irradiated without a cover, using three UV lamps (λmax 365 nm, total power 60 W; Vilber Lourmat, Marne-la-Vallée, France) for 15–60 min. Preliminary experiments indicated that cell iodination by this photocrosslinking procedure levels off after ~5 min irradiation. This plateau may reflect a balance between further photocrosslinking with increasing irradiation time and 125I abstraction by UV, as indicated by progressive release of ethanol-soluble material (data not shown). For purification experiments, the irradiation time was extended to 1 h, in order to maximise photocrosslinking yield (~50% as estimated by silver staining; 35).
Protease digestion, cell lysis and carbonate extraction
In some experiments, cell surface proteins were extensively digested at 4°C, using 0.1% (w/v) protease XIV (Sigma) in RPMI culture medium for 1 h. Protein surface digestion also conveniently resulted in cell detachment from the plastic support. After cell harvesting and centrifugation, pellets were washed with 1 ml of PBS-Ca2+, transferred to lysis buffer made up of 10 mM Tris–HCl, pH 7.4, 0.5% (v/v) Nonidet™ P40 (Boehringer) and protease inhibitors (Complete Mini; Boehringer) as recommended by the manufacturer, and briefly sonicated. For carbonate extraction, whole cells or subcellular fractions (see below) were resuspended in 100 mM Na2CO3, pH 11.5, and incubated on melting ice for 30 min (36). After centrifugation at 6 000 000 g × min, the supernatant was removed and the pellet was resuspended in lysis buffer. The slight decrease in electrophoretic mobility on SDS–PAGE of membrane proteins after carbonate extraction is attributed to the residual salt content.
Subcellular fractionation
Cells were photocrosslinked with [125I]ODN-BΦ as described above and harvested in 4 ml of a homogenisation medium containing 0.25 M sucrose, 3 mM imidazole, pH 7.0, and 1 mM EDTA on melting ice. After a first homogenisation by six passages through a tight Dounce homogeniser, the material was centrifuged at 10 000 g × min (GR 4.11 centrifuge; Jouan, St Nazaire, France). The upper third of the supernatant was collected and the remainder was homogenised again by two Dounce passages and centrifuged at 7000 g × min. Again, the upper third was removed before repeating Dounce homogenisation and centrifugation. Then the supernatant was entirely removed. To maximise subcellular particle extraction, 1 ml of the homogenisation medium was again added to the pellet that was passed once through the Dounce homogeniser and pelleted again at 7000 g × min, a procedure repeated twice. The final pellet is denoted the N fraction and the pool of supernatants is denoted the E fraction. This cell extract was fractionated by differential sedimentation using a Ti50 rotor (Beckman, Palo Alto, CA) to yield either the M and LP pellets, obtained successively at 33 000 and 3 000 000 g × min, or ML pellets, obtained by directly centrifuging at 275 000 g × min. The 3 000 000 g × min supernatant is denoted the S fraction. Pellets were directly resuspended in lysis buffer or first submitted to carbonate extraction, as described above.
Streptavidin affinity purification
Lysates from cells incubated with biotin-ODN-BΦ were diluted at a 0.5 mg/ml protein concentration in affinity chromatography buffer made up of 50 mM Tris–HCl, 1% (w/v) SDS, 0.5% (w/v) deoxycholate, 0.5 M NaCl and supplemented with protease inhibitors (Complete; Boehringer) as recommended by the manufacturer. Lysates were mixed with 20 µl of streptavidin-coated beads (Boehringer) at 4°C and incubated under gentle rotation for 2 h or overnight. After three washes of the beads with buffer, pooled supernatants (unbound fraction) were precipitated with 10% (w/v) trichloroacetic acid to reduce volumes and NaCl concentration, then dissolved in SDS–PAGE buffer [2% (w/v) SDS, 0.1 M dithiothreitol, 10% (v/v) glycerol, pH 6.8]. Beads were boiled in SDS–PAGE buffer to release bound material.
Analysis of proteins on gels and blots
After proteins were separated by SDS–PAGE, some gels were revealed by silver staining or reverse-zinc staining for microsequence analysis (37). Radioactive gels were scanned using a PhosphorImager (Molecular Dynamics, Los Alamos, CA). For internal sequencing, the excised band was cleaved in situ with CNBr and the resulting peptides were purified by capillary RP-HPLC (38). The N-terminal amino acid sequences were determined at the picomole level using a LF3400D microsequencer (Beckman). Alternatively, after SDS–PAGE, proteins were electrotransferred to a PVDF membrane (Boehringer), then the membrane was saturated for 3 h to overnight using a development solution containing 50 mM Tris–HCl, pH 8.0, 90 mM NaCl, 2 mM CaCl2 and 0.5% (v/v) Tween-20. This was followed by ligand blotting or western blotting.
For ligand blotting, the membrane was incubated for 1 h at room temperature with 50 ml of the same solution supplemented with 5 nmol of [125I]Fl-ODN (~50 µg ODN in total), 500 µg of dextran sulphate and 500 µg of suramin (both from Sigma). The membrane was washed eight times using the development solution and dried for PhosphorImager analysis. A 100-fold molar excess of the phosphorothioate ODN Gem91™ or the same concentration of an equimolar mixture of 1024 different 21mer phosphodiester ODNs (Gibco BRL) were used for competition experiments.
For western blotting, antibodies were raised in three mice against the peptide CSILTSPDTEQNNYVP coupled to keyhole limpet haemocyanin (Eurogentec). The saturated membrane was incubated overnight at 4°C with pre-immune or immune serum diluted 400-fold, washed five times, further incubated with 0.05 µCi/ml [125I]protein G (Amersham), washed again five times, dried and analysed with the PhosphorImager.
RESULTS
Identification of a Mr 66 kDa band as a cell membrane ODN receptor by ligand blotting of cell extracts
As a first procedure to identify a putative receptor, lysates of naive cells were analysed by ligand blotting using [125I]Fl-ODN in the presence of polysulphated (dextran sulphate) and polysulphonated (suramin) compounds. At the concentrations used, these polyanions did not inhibit [125I]ODN binding to HepG2 cells (data not shown) but strongly decreased background binding to the blots.
Five major bands were labelled at Mr 66, 57, 52, 46 and 39 kDa, respectively (Fig. 1, lane 1). When incubation was performed with a 100-fold molar excess of an unlabelled ODN of the same sequence in the phosphorothioate series (Gem91™, lane 2), all radioactive bands disappeared. With the same excess of a mixture composed of 1024 unrelated 21mer phosphodiester ODNs (lane 3), the three upper bands also disappeared and the two lower ones almost vanished. These experiments indicate specificity of ODN interactions.
Figure 1.
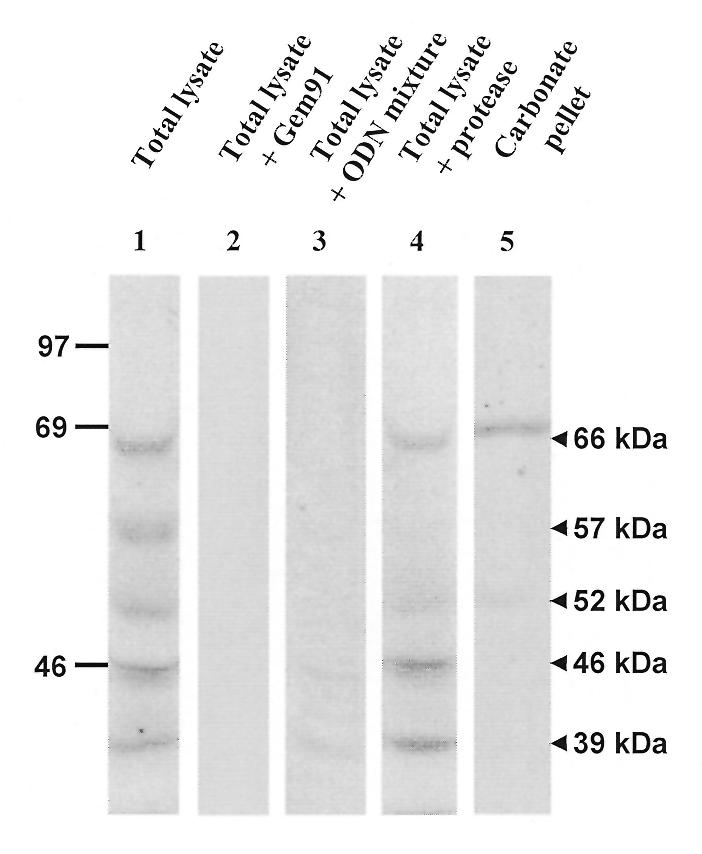
Ligand blotting. Lysates or extracts from naive cells were resolved by SDS–PAGE and transferred onto a PVDF membrane. ODN-binding proteins were revealed using 100 nM [125I]Fl-ODN in the presence of a 10-fold mass excess of suramin and dextran sulphate. Lanes 1–3, total lysates (100 µg protein/lane) without competitor (lane 1), with a 100-fold molar excess of the phosphorothioate Gem91™ (lane 2) or a 100-fold molar excess of a mixture of unrelated 21mer phosphodiesters (lane 3). Lane 4, total lysate (100 µg protein) from surface-digested cells. Lane 5, 50 µg protein of membrane pellet after whole cell carbonate extraction. Positions of molecular weight standards (in kDa) are indicated on the left.
When cells were lysed after extensive proteolytic digestion of the cell surface, the intensity of the bands at 66 and 52 kDa decreased ~2-fold (compare lanes 1 and 4) and the band at 57 kDa vanished, whereas the bands at 46 and 39 kDa were unaffected. These observations indicate that the 57 kDa band occurs at the cell surface, suggest an intracellular localisation of the 46 and 39 kDa bands and are compatible with a dual localisation of the 66 and 52 kDa bands. Only the 66 kDa band remained sedimentable with membranes after carbonate extraction (lane 5), indicating that this is a membranous ODN receptor.
Identification of a saturable pool of the cell membrane ODN receptor by in situ photoaffinity labelling
Specific binding of a [125I]ODN-benzophenone conjugate ([125I]Fl-ODN-BΦ) to a particular protein in a mixture followed by UV light irradiation generates a covalent linkage, resulting in selective stable radiolabelling of that protein (35). When the conjugate was applied to HepG2 cells at 37°C prior to photocrosslinking, a major band was radiolabelled, also at 66 kDa (Berens et al., in preparation; Fig. 2A, lane 2). Progressive extinction of its labelling upon competition by increasing Fl-ODN concentrations demonstrated saturation of the ODN receptor (lanes 3–7) and allowed an estimation of the Ki as ~220 nM (Fig. 2B). Scatchard plot analysis indicated that the total number of photolabellable receptors at ODN saturation was ~21 000 per cell. However, a more reasonable estimation of the accessible receptor population, which takes into account both photocrosslinking efficiency (~50%) and the loss of radioactivity caused by the irradiation damage (estimated at ~50%; our unpublished observations), reaches ~80 000 receptors/cell. Efficient displacement by a competitor ODN devoid of benzophenone also demonstrated that labelling was not due to the benzophenone moiety.
Figure 2.

Photoaffinity labelling of intact cells. (A) PhosphorImager scan of a representative isotopic dilution experiment. Nearly confluent HepG2 cells were incubated with 33 nM [125I]Fl-BΦ-ODN either alone (lane 2) or with the indicated concentrations of Fl-ODN as competitor (in nM), prior to photocrosslinking for 15 min (lanes 3–7). After lysis, 100 µg of protein from each dish were loaded per lane and separated by SDS–PAGE. Lane 1 shows molecular weight standards with indications of molecular masses (in kDa) on the left. (B) Quantification of radioactivity of the 66 kDa band in two separate experiments, represented by different symbols.
Subcellular fractionation
As a first step to purify the 66 kDa ODN receptor, HepG2 cells were photolabelled in situ with [125I]Fl-ODN-BΦ and its subcellular distribution was analysed by differential sedimentation. Fractions N (nuclei contaminated by a few intact cells and by large cell debris), E (post-nuclear extract), M (large post-nuclear particles), LP (small post-nuclear particles) and S (cytosol) were collected. Large post-nuclear particles were enriched ~4.4-fold in the radiolabelled 66 kDa band, as compared with the post-nuclear extract (see Fig. 3, lanes 2 and 4). This band was much less abundant in fractions N and LP (lanes 3 and 7) and was not detected in the high speed supernatant S (lane 10). After carbonate extraction of fractions M and LP, which in both cases resulted in the partition of 22% of particulate protein with sedimentable membranes (Cp) and 78% with the supernatant (Cs), the radioactivity at 66 kDa was almost exclusively recovered in the membrane pellets (lanes 5 and 8). Thus, carbonate extraction confirmed the membrane topology of the ODN receptor in subcellular particles and led to an additional 3-fold enrichment.
Figure 3.

Subcellular fractionation. Nearly confluent HepG2 cells were incubated with 20 nM [125I]Fl-ODN-BΦ prior to photocrosslinking for 15 min and homogenisation. The homogenate was separated by differential centrifugation, successfully yielding a low speed N pellet, large post-nuclear particles (M), smaller particles (LP) and the final supernatant (S). E is the total post-nuclear supernatant. 100 µg of protein from each fraction were resolved by SDS–PAGE. Both the M and LP fractions were further extracted at alkaline pH, yielding a carbonate pellet (Cp, 22 µg protein) and a carbonate supernatant (Cs, 78 µg protein). Lane 1 shows molecular weight standards, with indications of molecular masses (in kDa) on the left.
Receptor purification
To achieve protein purification, we repeated the procedure of photocrosslinking with an ODN-benzophenone conjugate bearing an additional biotin moiety. To first test the validity of the procedure at an analytical scale, cells were incubated with 100 nM [125I]Fl-biotin-ODN-BΦ, photocrosslinked and 100 mg of protein of total lysates were incubated with streptavidin beads. Whereas ~70% of the radioactivity was retained by the beads (Fig. 4A), the bulk of the protein was recovered in the unbound fraction and no protein could be detected by silver staining in the bound fraction (Fig. 4B). This makes it impossible to calculate the purification achieved by affinity isolation. However, based on the high sensitivity of silver staining (~10 ng protein in a faint band), purification can be estimated to exceed 4000-fold.
Figure 4.

Affinity purification on streptavidin beads. (A and B) Analytical scale. Nearly confluent HepG2 cells were incubated with ~50 nM [125I]Fl-biotin-ODN-BΦ followed by photocrosslinking for 15 min. 100 µg of protein from total cell lysate were affinity purified on streptavidin beads. The supernatant was concentrated by TCA precipitation. Bound and unbound fractions were analysed with a PhosphorImager and by silver staining. (C) Preparative scale (600 000 000 cells). After incubation with 100 nM biotin-ODN-BΦ followed by photocrosslinking for 60 min, 20 mg of protein from the membrane pellet after carbonate extraction of the ML fraction were affinity purified with streptavidin beads, eluted upon boiling in SDS–PAGE buffer and resolved by SDS–PAGE. The gel was stained with the reverse-zinc procedure. Positions of molecular weight markers (in kDa) are indicated on the left.
The procedure was then repeated at a preparative scale on 600 000 000 cells incubated with 100 nM biotin-ODN-BΦ and photocrosslinked. After isolation of the carbonate-extracted membrane pellet from the ML fraction and affinity purification on streptavidin beads, bound material was resolved by SDS–PAGE followed by reverse-zinc staining. To monitor purification, a smaller number of cells was incubated in parallel with 100 nM [125I]Fl-biotin-ODN-BΦ and, again, the 66 kDa radiolabelled band was almost exclusively seen in the carbonate-extracted membrane pellet of the ML fraction. Based on its radioactivity, ~5000 copies were photocrosslinked per cell.
For microsequencing, at least 1 pmol of the purified band is needed. Assuming equal labelling efficiency when using the non-radiolabelled conjugate, ~3.5 pmol of receptor were covalently conjugated. After affinity purification, the reverse-zinc procedure stained one major band at 66 kDa and a second band of lesser intensity at 57 kDa. After in situ CNBr cleavage of the excised 66 kDa band and RP-HPLC peptide isolation, microsequence analysis by Edman degradation yielded one sequence of 16 amino acids, (M)SILTSPDTEQN(N/X)YVP. Comparison of this sequence with protein and genomic databases showed no significant identity or similarity.
Western blotting
The 16 amino acid peptide was coupled to keyhole limpet haemocyanin and antisera were raised in three mice. By western blotting of cell lysates, all three antisera revealed several bands, including a major one at 66 kDa that was not detected by pre-immune sera under identical conditions (Fig. 5, compare lanes 1 and 2). As in ligand blotting, the intensity of this band decreased by half if cells had been surface-digested prior to lysis (lane 3), and it was the major one in the membrane pellet after carbonate extraction (lane 4). Interestingly, pre-incubation of the blot with Gem91™ (an ODN which best competes for [125I]Fl-ODN uptake in HepG2 cells; data not shown) strongly decreased labelling of the 66 kDa band by the antibodies, suggesting that the recognised epitope could be close to the ODN-binding site (compare lanes 4 and 5).
Figure 5.
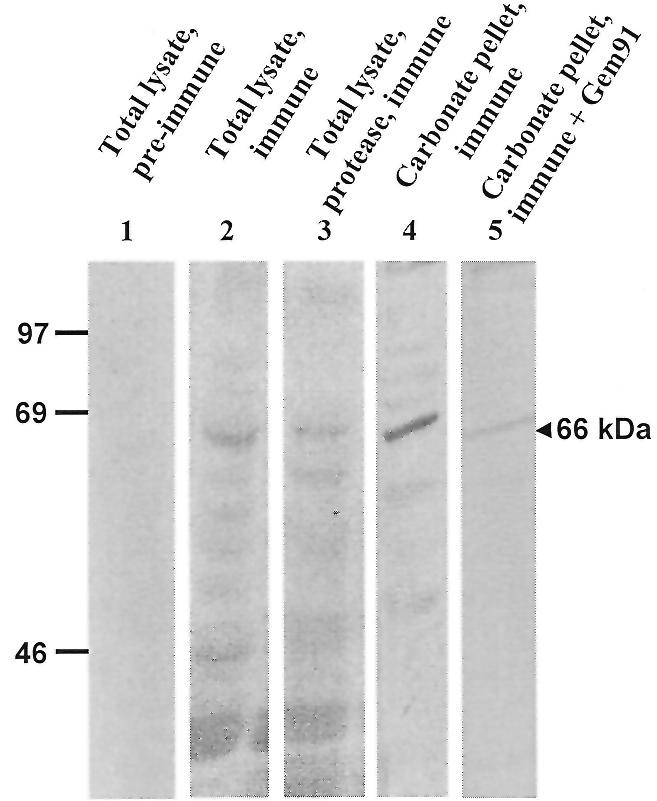
Western blotting. The same lysates and extracts as in Figure 1 were used. Lysates from naive cells (lanes 1 and 2, 100 µg protein/lane), surface-digested cells (lane 3, 100 µg protein) or a membrane pellet after carbonate extraction (lanes 4 and 5, 50 µg protein/lane) were transferred to a PVDF membrane. Lane 5 was saturated using the development solution supplemented with 1 µM Gem91™. Proteins were revealed using pre-immune antiserum (lane 1) or anti-peptide antiserum (lanes 2–5) followed by [125I]protein G. Positions of molecular weight standards (in kDa) are indicated on the left.
DISCUSSION
In this paper, we have identified a major ODN membrane receptor in human HepG2 cells by three different methods: ligand blotting, photocrosslinking and western blotting. To our knowledge, this is also the first report of the purification and partial sequencing of a novel ODN cell membrane receptor.
Ligand blotting experiments on total cell extracts showed five major bands. Because membrane incubation was performed with an excess of polyanionic competitors, these bands are the signature of proteins sharing affinity for ODNs independently of their polyanionic nature. Moreover, these proteins can bind ODNs of different sequence and with either a phosphodiester or a phosphorothioate backbone. Surface protease digestion identified three different classes of ODN-binding proteins. Some proteins are unaffected by this treatment and therefore presumably intracellular, while another disappears after surface proteolysis and is therefore located on the plasma membrane. Two other bands belong to a third class: a decrease by half in labelling after extensive surface digestion suggests a dual localisation at the cell surface and in an intracellular compartment. One such band, the only one to be strongly associated with membranes, is thus the best candidate for an ODN receptor involved in endocytosis.
A similar band was obtained by photocrosslinking on living cells (Berens et al., in preparation; this paper). The affinity constant of this receptor for Fl-ODN was estimated as 220 nM. Comparable affinity values were reported for phosphodiester ODNs in mouse fibroblasts L929 (200 nM; 17,18), in human colorectal adenocarcinoma HCT-15 cells (160 nM; 39) and in human HeLa cells (210 nM; 39), although the Mr of the ODN receptor varied between cell types. By photoaffinity labelling of isolated plasma membrane fractions from HepG2 cells, an ODN-binding protein was demonstrated as a doublet at ~110 kDa, with an affinity of 60 nM, and its labelling was totally displaced by 2 µM dextran sulphate (40). Such properties are clearly distinct from those of the 66 kDa band analysed in the present study. A completely different affinity value was reported in polymorphonuclear leukocytes, where the integrin Mac1 was found to be involved in ODN binding (8.8 µM; 34).
Several purification methods for an ODN receptor can be envisaged. Affinity chromatography on an oligo(dT) matrix has been used (20). Membrane proteins were radiolabelled at the cell surface prior to lysis, then applied to an ODN column. After washing, bound proteins were eluted upon boiling in SDS, resolved by electrophoresis and detected by autoradiography. In this approach, however, the moderate affinity of the receptor for the ODN requires a high protein concentration for sufficient binding to occur and it limits washing times and stringency, thereby increasing the danger of non-specific binding. Subsequent purification steps are thus necessary. Alternatively, ODNs modified with alkylating moieties (17,23) generate covalent labelling, but have several limitations, including auto-alkylation, deactivation by nucleophiles in the medium and possible persistence of the alkylating group leading to interference with subsequent steps.
Benzophenone photocrosslinking offers several advantages. (i) Photolabelling is a more flexible approach, as crosslinking can be performed after removal of non-specifically bound material and initiated when a defined compartment has been reached. (ii) The chemical properties of benzophenone allow for photocrosslinking by near-UV light, therefore inducing less damage than other photocrosslinking agents which require far-UV irradiation, and offer much higher yields, typically ~50% (35). (iii) For purification purposes, the combination of photocrosslinking using a biotinylated ODN with affinity chromatography on streptavidin-coated beads is also clearly superior to affinity chromatography on immobilised ODN, since the extremely high affinity of the biotin–avidin complex (~10 fM) makes it possible to incubate beads at low protein concentrations and to carry out stringent washings. In our experiments, a >4000-fold purification was achieved in one step, with a yield sufficient for microsequencing.
All three mice immunised against the peptide, which was determined by microsequencing of a CNBr fragment of the purified receptor, produced antibodies recognising one major band by western blotting after membrane extraction by carbonate treatment. In addition, the intensity of this band was decreased 2-fold after surface protease digestion. Thus indistinguishable properties in ligand and western blotting, together with competition by the phosphorothioate Gem91™ for recognition of the antibodies, are consistent with the identity of this band as the ODN receptor.
The peptide determined by microsequencing does not resemble any of the known scavenger receptors, a family of multifunctional proteins (33) that have been suggested as candidates for receptor-mediated endocytosis of ODN in endothelial liver cells and in macrophages (24,27). The receptor is instead a novel protein, which we currently aim at defining by molecular cloning. Until its sequence becomes available, it is too early to speculate on the natural function of a plasma membrane protein that presumably uses ODNs as opportunistic ligands.
In conclusion, we have identified a membrane receptor for ODNs on HepG2 cells by three alternative methods: ligand blotting and western blotting of cell lysates or membrane extracts, as well as photoaffinity labelling of intact cells. This receptor interacts with phosphodiester and phosphorothioate ODNs of variable sequence. It is partly exposed on the cell surface (protease-sensitive pool) and partly intracellular (protease-resistant pool). Whether this dual localisation reflects receptor-mediated endocytosis of ODN and receptor recycling to the plasma membrane requires further cell bio-logical experiments that are now made possible thanks to anti-receptor antibodies.
Acknowledgments
ACKNOWLEDGEMENTS
Gels and blots were analysed using NIH Image v.1.59 software. We thank Dr A.C. Roche for providing GEM91 and Drs P. Henriet and Ch. Josenhans for critical reading of the manuscript. P.deD. is a research fellow (SmithKline Beecham grant) of the Fonds National de la Recherche Scientifique (FNRS, Belgium) at which R.W. is a scientific collaborator. C.B. was a research fellow at the FNRS, then recipient of a PhD training grant from UCL. This work was supported by grants from Agence Nationale de Recherche contre le SIDA (ANRS, France), as well as the FNRS, Concerted Research Actions and Interuniversity Attraction Poles (Belgium).
REFERENCES
- 1.Roush W. (1997) Science, 276, 1192–1193. [DOI] [PubMed] [Google Scholar]
- 2.Zamecnik P.C. and Stephenson,M.L. (1978) Proc. Natl Acad. Sci. USA, 75, 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haeuptle M.T., Frank,R. and Dobberstein,B. (1986) Nucleic Acids Res., 14, 1427–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Torrence P.F., Maitra,R.K., Lesiak,K., Khamnei,S., Zhou,A. and Silverman,R.H. (1993) Proc. Natl Acad. Sci. USA, 90, 1300–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kukreti S., Sun,J.-S., Garestier,T. and Hélène,C. (1997) Nucleic Acids Res., 25, 4264–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoon K., Cole-Strauss,A. and Kmiec,E.B. (1996) Proc. Natl Acad. Sci. USA, 93, 2071–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmajuk G., Sierakowska,H. and Kole,R. (1999) J. Biol. Chem., 274, 21783–21789. [DOI] [PubMed] [Google Scholar]
- 8.Lewin A.S., Drenser,K., Hauswirth,W.W., Nishikawa,S., Yasumura,D., Flannery,J.G. and LaVail,M.M. (1998) Nature Med., 4, 967–971. [DOI] [PubMed] [Google Scholar]
- 9.Bridonneau Ph., Chang,Y.-F., O’Connell,D., Gill,S.C., Snyder,D.W., Johnson,L., Goodson,Th., Herron,D.K. and Parma,D.H. (1998) J. Med. Chem., 41, 778–786. [DOI] [PubMed] [Google Scholar]
- 10.Toulmé J.J., Le Tinévez,R. and Brossalina,E. (1996) Biochimie, 78, 663–673. [DOI] [PubMed] [Google Scholar]
- 11.Bennett C.F. (1998) Biochem. Pharmacol., 55, 9–19. [DOI] [PubMed] [Google Scholar]
- 12.Stein C.A. and Cheng,Y.C. (1993) Science, 261, 1004–1012. [DOI] [PubMed] [Google Scholar]
- 13.Tonkinson J.L. and Stein,C.A. (1994) Nucleic Acids Res., 22, 4268–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agrawal S., Jiang,Z., Zaho,Q., Shaw,D., Cai,Q., Roskey,A., Channavajjala,L., Saxinger,C. and Zhang,R. (1997) Proc. Natl Acad. Sci. USA, 94, 2620–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pichon C., Arar,K., Stewart,A.J., Duc Dodon,M., Gazzolo,L., Courtoy,P.J., Mayer,R., Monsigny,M. and Roche,A.-C. (1997) Mol. Pharmacol., 51, 431–438. [PubMed] [Google Scholar]
- 16.Leonetti J.-P., Mechti,N., Degols,G., Gagnor,C. and Lebleu,B. (1991) Proc. Natl Acad. Sci. USA, 88, 2702–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yakubov L.A., Deeva,E.A., Zarytova,V.F., Ivanova,E.M., Ryte,A.S., Yurchenko,L.V. and Vlassov,V.V. (1989) Proc. Natl Acad. Sci. USA, 86, 6454–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vlassov V.V., Balakireva,L.A. and Yakubov,L.A. (1994) Biochim. Biophys. Acta, 1197, 95–108. [DOI] [PubMed] [Google Scholar]
- 19.Beltinger Ch., Saragovi,H.U., Smith,R.M., LeSauteur,L., Shah,N., DeDionisio,L., Christensen,L., Raible,A., Jarett,L. and Gewirtz,A.M. (1995) J. Clin. Invest., 95, 1814–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loke S.L., Stein,C.A., Zhang,X.H., Mori,K., Nakanishi,M., Subasinghe,C., Cohen,J.S. and Neckers,L.M. (1989) Proc. Natl Acad. Sci. USA, 86, 3474–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Temsamani J., Kubert,M., Tang,J., Padmapriya,A. and Agrawal,S. (1994) Antisense Res. Dev., 4, 35–42. [DOI] [PubMed] [Google Scholar]
- 22.Noonberg S.B., Garovoy,M.R. and Hunt,C.A. (1993) J. Invest. Dermatol., 101, 727–731. [DOI] [PubMed] [Google Scholar]
- 23.Laktionov P.P., Dazard,J.-E., Vives,E., Rykova,E.Y., Piette,J., Vlassov,V.V. and Lebleu,B. (1999) Nucleic Acids Res., 27, 2315–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bijsterbosch M.K., Manoharan,M., Rump,E.T., De Vrueh,R.L.A., van Veghel,R., Tivel,K., Biessen,E., Bennett,C.F., Dan Cook,P. and van Berkel,T.J.C. (1997) Nucleic Acids Res., 25, 3290–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaerman J.L., Moureau,P., Deldime,F., Lewalle,P., Lammineur,C., Morschhauser,F. and Martiat,P. (1997) Blood, 90, 331–339. [PubMed] [Google Scholar]
- 26.Pearson A.M., Rich,A. and Krieger,M. (1993) J. Biol. Chem., 268, 3546–3554. [PubMed] [Google Scholar]
- 27.Benimetskaya L., Berton,M., Kolbanovsky,A., Benimetsky,S. and Stein,C.A. (1997) Nucleic Acids Res., 25, 2648–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Agrawal S. and Zhao,Q. (1998) Curr. Opin. Chem. Biol., 2, 519–528. [DOI] [PubMed] [Google Scholar]
- 29.Weiner G.J., Liu,H.-M., Wooldridge,J.E., Dahle,C.E. and Krieg,A.M. (1997) Proc. Natl Acad. Sci. USA, 94, 10833–10837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldstein J.L., Brown,M.S., Anderson,R.G., Russell,D.W. and Schneider,W.J. (1985) Annu. Rev. Cell Biol., 1, 1–39. [DOI] [PubMed] [Google Scholar]
- 31.Hawley P. and Gibson,I. (1996) Antisense Nucleic Acids Drug Dev., 6, 185–195. [DOI] [PubMed] [Google Scholar]
- 32.Geselowitz D.A. and Neckers,L.M. (1995) Antisense Res. Dev., 5, 213–217. [DOI] [PubMed] [Google Scholar]
- 33.Biessen E.A.L., Vietsch,H., Kuiper,J., Bijsterbosch,M.K. and van Berkel,T.J.C. (1998) Mol. Pharmacol., 53, 262–269. [DOI] [PubMed] [Google Scholar]
- 34.Benimetskaya L., Loike,J.D., Khaled,Z., Loike,G., Silverstein,S.C., Cao,L., El Khoury,J., Cai,T.-Q. and Stein,C.A. (1997) Nature Med., 3, 414–420. [DOI] [PubMed] [Google Scholar]
- 35.Berens C., Courtoy,P.J. and Sonveaux,E. (1999) Bioconjugate Chem., 10, 56–61. [DOI] [PubMed] [Google Scholar]
- 36.Fujiki Y., Hubbard,A.L., Fowler,S. and Lazarow,P.B. (1982) J. Cell Biol., 93, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez-Patron C., Calero,M., Collazo,P.R., Garcia,J.R., Madrazo,J., Musacchio,A., Soriano,F., Estrada,R., Frank,R., Castellanos-Serra,L.R. and Mendez,E. (1995) Anal. Biochem., 224, 203–211. [DOI] [PubMed] [Google Scholar]
- 38.Wattiez R., Hermans,C., Bernard,A., Lesur,O. and Falmagne,P. (1999) Electrophoresis, 20, 1634–1645. [DOI] [PubMed] [Google Scholar]
- 39.Nakai D., Seita,T., Terasaki,T., Iwasa,S., Shoji,Y., Mizushima,Y. and Sugiyama,Y. (1996) J. Pharmacol. Exp. Ther., 278, 1362–1372. [PubMed] [Google Scholar]
- 40.Yao G.-Q., Corrias,S. and Cheng,Y.-C. (1996) Biochem. Pharmacol., 51, 431–436. [DOI] [PubMed] [Google Scholar]