

Abstract
Enhancers play a vital role in gene regulation and are critical in mediating the impact of noncoding genetic variants associated with complex traits. Enhancer activity is a cell‐type‐specific process regulated by transcription factors (TFs), epigenetic mechanisms and genetic variants. Despite the strong mechanistic link between TFs and enhancers, we currently lack a framework for jointly analysing them in cell‐type‐specific gene regulatory networks (GRN). Equally important, we lack an unbiased way of assessing the biological significance of inferred GRNs since no complete ground truth exists. To address these gaps, we present GRaNIE (Gene Regulatory Network Inference including Enhancers) and GRaNPA (Gene Regulatory Network Performance Analysis). GRaNIE (https://git.embl.de/grp‐zaugg/GRaNIE) builds enhancer‐mediated GRNs based on covariation of chromatin accessibility and RNA‐seq across samples (e.g. individuals), while GRaNPA (https://git.embl.de/grp‐zaugg/GRaNPA) assesses the performance of GRNs for predicting cell‐type‐specific differential expression. We demonstrate their power by investigating gene regulatory mechanisms underlying the response of macrophages to infection, cancer and common genetic traits including autoimmune diseases. Finally, our methods identify the TF PURA as a putative regulator of pro‐inflammatory macrophage polarisation.
Keywords: enhancers, gene regulatory networks, macrophage biology, multiomics data integration, transcriptional regulation
Subject Categories: Chromatin, Transcription & Genomics; Computational Biology
GRaNIE builds enhancer‐based gene regulatory networks (GRNs) using chromatin accessibility and RNA‐seq data. GRaNPA assesses the biological significance of GRNs and transcription factors. Together, they provide insights into cell‐type‐specific gene regulation.
Introduction
Enhancers are genomic locations that play an important role in cell‐type‐specific gene regulation, and impaired enhancer function has been linked to an increasing number of diseases (Karnuta & Scacheri, 2018; Claringbould & Zaugg, 2021). In particular, genome‐wide association studies (GWAS) have linked over 200,000 common genetic variants with over 40,000 traits and diseases. Since the vast majority of these disease‐associated genetic variants lie in noncoding regions far from promoters (Claringbould & Zaugg, 2021), they are likely affecting enhancers and having a regulatory role.
A big challenge in the post‐GWAS era is the interpretation of these disease‐associated genetic variants in noncoding genomic regions because it is often still unclear what genes they target, and in what cell types. The cell‐type‐specific activity of gene regulatory elements is likely conferred by transcription factors (TFs). And indeed, a recent study points at the importance of studying TFs for understanding genetic variants associated with autoimmune diseases (Freimer et al, 2022). The importance of TFs was confirmed by another study, which found that trans‐expression quantitative trait loci (eQTLs), which likely act via TFs, are more enriched in disease‐associated genes than cis‐eQTLs (Võsa et al, 2021). However, to predict the function of TFs, for example in cell‐fate determination, it is crucial to include putative enhancers (preprint: Janssens et al, 2021; Xu et al, 2021). Enhancer‐mediated gene regulatory networks reconstructed from single‐cell RNA and ATAC‐seq profiling in the fly brain have led to a better understanding of the regulatory diversity across different neuronal cell types (preprint: Janssens et al, 2021). We previously used enhancer‐based analyses to understand disease mechanisms in pulmonary arterial hypertension (Reyes‐Palomares et al, 2020). Thus, for interpreting disease‐associated genetic variants, or enhancers in general, it is crucial to jointly investigate TF activity, enhancers and gene expression in a cell‐type‐specific manner.
Several approaches have been proposed to infer bipartite TF‐gene networks, for example based on co‐expression in bulk (Huynh‐Thu et al, 2010; Haynes et al, 2013), or single‐cell expression data (Aibar et al, 2017; Moerman et al, 2019; preprint: Kamimoto et al, 2020), based on partial information decomposition (Chan et al, 2017), time‐course data (Huynh‐Thu & Geurts, 2018) or data curation (Liu et al, 2015; Han et al, 2018; Garcia‐Alonso et al, 2019; Keenan et al, 2019). At the same time, methods for inferring enhancer‐gene links exist, for example using co‐variation of peaks (Pliner et al, 2018; Fulco et al, 2019), or targeted perturbations of enhancers followed by sequencing (Schraivogel et al, 2020). Only few approaches jointly infer TF‐enhancer and enhancer‐gene links (Marbach et al, 2016) mostly from single‐cell data (preprint: González‐Blas et al, 2022; Fleck et al, 2022) and we currently lack methods to infer cell‐type‐specific networks that enable the study of context‐specific interaction between TFs, regulatory elements and genes.
An important step in regulatory network reconstruction is to evaluate their biological significance. Common approaches for assessing regulatory interactions include benchmarking against networks from simulated data or against known biological networks (Chen & Mar, 2018; Pratapa et al, 2020). Each of these has their own drawback: simulated networks are based on many assumptions about the network structure which may not reflect the “true” biological network while known biological networks typically suffer from a strong literature bias (Weidemüller et al, 2021), low complexity and a limited range of connections and cell types, and are thus not well‐suited for an unbiased evaluation of GRNs. In general, each network inference method will have its own bias and shortcomings, and performance will depend on the benchmarking data set (Chen & Mar, 2018; Pratapa et al, 2020). Thus, there is a need for an unbiased approach to assess the biological relevance of inferred and curated regulatory interactions as well as individual TF regulons (defined as all genes connected to a TF).
Here, we present a tool‐suite for building and evaluating enhancer‐based gene regulatory networks (eGRNs) called GRaNIE (Gene Regulatory Network Inference including Enhancers ‐ https://grp‐zaugg.embl‐community.io/GRaNIE and https://bioconductor.org/packages/GRaNIE) and GRaNPA (Gene Regulatory Network Performance Analysis ‐ https://git.embl.de/grp‐zaugg/GRaNPA), respectively. GRaNIE jointly infers TF‐enhancer and enhancer‐gene interactions based on covariation of bulk RNA‐seq expression and chromatin accessibility (ATAC‐seq) or ChIP‐seq for active histone marks (e.g. H3K27ac) across biological samples. GRaNPA assesses the biological relevance—of any TF‐gene‐based GRN—using a machine learning framework, and identifies TFs that predict cell‐type‐specific expression response to perturbations. We demonstrate that GRaNIE infers biologically meaningful eGRNs using macrophages as example, and validate TF‐enhancer links with ChIP‐seq and enhancer‐gene links with eQTL data. We further demonstrate the cell‐type‐specific nature of GRaNIE‐inferred eGRNs for macrophages, T‐cells and acute myeloid leukaemia (AML) cells using GRaNPA evaluation, and by predicting cell‐type‐specific TF knockout (K/O) data. Using GRaNIE followed by GRaNPA, we identify PURA as putative TF driving the pro‐inflammatory polarisation of macrophages, which we corroborate with orthogonal phosphoproteomics data, and we confirm earlier observations from mice that MBD2 drives the anti‐inflammatory program in macrophages. Furthermore, we find enhancers in the macrophage eGRNs enriched for autoimmune disease variants, which GRaNIE links to upstream TFs and putative target genes. Finally, we provide a comprehensive resource of cell‐type‐specific GRNs for three other cell types (https://apps.embl.de/grn/).
Results
Overview and conceptual description of the GRaNIE algorithm
We developed GRaNIE to interpret genetic and epigenetic variation in regulatory (enhancer and promoter) regions, here defined by ATAC‐seq peaks and hereafter referred to as “peaks”. GRaNIE is an R/Bioconductor package and jointly infers TF‐enhancer/promoter and enhancer/promoter‐gene interactions from the same data in a context‐specific manner. Conceptually, the software is based on an approach we have devised for a recent study in which we investigated enhancer‐mediated disease mechanisms of pulmonary arterial hypertension (Reyes‐Palomares et al, 2020). Briefly, GRaNIE separately identifies TF‐peak and peak‐gene links, and then integrates them into an eGRN.
The TF‐peak links are based on statistically significant co‐variation of TF expression and peak accessibility across samples (e.g. individuals, recommended minimum number ~10–15), taking into account predicted TF binding sites. To obtain them, GRaNIE calculates all pairwise correlations between TF expression levels (RNA‐seq) and peak signal (ATAC/ChIP‐seq), stratified by whether the peak overlaps a predicted binding site of the TF. For each TF, it then uses the distribution of all peaks that do not contain its predicted binding site as background to calculate an empirical FDR for assessing the significance of TF‐peak links (Appendix Fig S1). In our previous work, we have demonstrated that negative TF‐peak correlations indicate the TF acts as transcriptional repressor while positive correlations indicate an activator role, thus allowing the classification of TFs into activators and repressors (Berest et al, 2019). As a quality control (QC), we recommend comparing the number of real TF‐peak links to those obtained from a background set of links inferred from randomised data (see Materials and Methods).
Peak‐gene links are based on significant co‐variation of peak accessibility and gene expression across samples (Fig 1A, Appendix Fig S1). For this, GRaNIE calculates the correlation between the expression of a gene and signals of all peaks within an adjustable, defined distance of its transcription start site (TSS, default is 250 kb). GRaNIE also allows the use of chromatin conformation data, such as Hi‐C, to define which peak‐gene pairs will be tested within a 3D proximity (Appendix Fig S1). Since chromatin accessibility in regulatory elements is generally associated with active gene regulation and transcription, we only expect positive correlations for functional peak‐gene links. Notably, this is still true for repressor‐bound elements, where binding of most repressors leads to loss of both accessibility and transcription (Berest et al, 2019). Negative peak‐gene correlations have no clear biological meaning and may indicate remaining batch effects or random noise. Therefore, one can judge the signal‐to‐noise ratio by assessing positive versus negative peak‐gene correlations and we implemented this as a QC metric in GRaNIE. We recommend comparing these QC metrics with a corresponding background set (implemented in GRaNIE, see Materials and Methods).
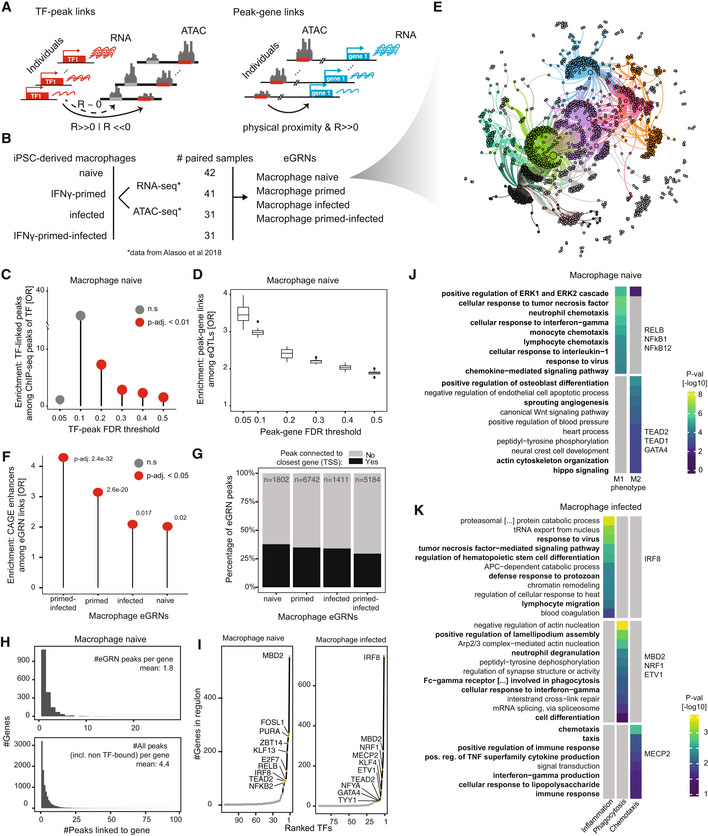
Figure 1. Overview, application and validation of GRaNIE.
-
ASchematic of the eGRN construction by GRaNIE, including the TF to peak (left) and peak to gene (right) links (detailed workflow in Appendix Fig S1).
-
BDatasets used for macrophage eGRN construction and evaluation.
-
CValidation of the eGRN TF‐peak links with ChIP‐seq data. Enrichment of ChIP‐seq peaks overlapping a GRaNIE‐inferred TF‐bound peak (same TF) are shown for different TF‐peak FDRs in the naive macrophage eGRN. Statistical significance was determined using Fishers Exact test; test set: all TF‐peak pairs where the peak contains the motif for the respective TF (n = 25,205, 39,408, 78,971, 109,228, 142,548, 147,226 for TF‐peak FDR 0.05, 0.1, 0.2, 0.3, 0.4, 0.5 respectively), categories: overlap with ChIP‐seq signal, part of GRaNIE‐infer network. Only TFs for which ChIP‐seq data was available are considered (see Appendix Fig S2 for other eGRNs).
-
DValidation of the eGRN peak‐gene links with macrophage eQTLs. Plots show the enrichment of eGRN links overlapping an eQTL over randomly sampled distance‐matched peak‐gene links for different peak‐gene FDRs in the naive macrophage eGRN (see Appendix Fig S3 for other eGRNs). Boxplots: central band: 50% quantile, box: interquartile range (25–75%); whiskers: max/min are 1.5 IQR above/below the box.
-
EForce‐directed visualisation of the naive macrophage eGRN (see Appendix Fig S4 for the other eGRNs). The colours correspond to the identified communities.
-
FEnrichment of macrophage‐specific FANTOM5 CAGE enhancers among the macrophage eGRN peaks. Statistical significance was determined with Fisher's exact test; test set: all peaks that were considered for peak‐gene connections (ATAC consensus peaks located within 250 kb of a TSS of a gene with mean normalised expression across samples > 1) in each eGRN (n = 210,083, 227,035, 227,120 and 219,823 peaks for the naive, infected, primed and primed‐infected eGRN, respectively), categories: overlap with CAGE enhancer, part of GRaNIE network.
-
GFraction of eGRN peaks connected to the closest gene (black) versus other (grey) genes for the macrophage eGRNs.
-
HNumber of peaks linked to a gene shown as histogram for eGRN peaks (top) and all peaks (including non‐TF bound; bottom) for the naive macrophage data (see Appendix Fig S7 for other eGRNs). Mean number of peaks indicated in the panels.
-
INumber of genes connected to each TF for the naive macrophage eGRN (top 10 TFs are labelled).
-
J, KGO enrichment and associated P‐values for selected communities from the naive (J) and infected (K) macrophage eGRN (see Dataset EV7 for the full table of enrichments across communities for all macrophage eGRNs).
GRaNIE then combines TF‐peak and peak‐gene links to create a tripartite TF‐enhancer‐gene eGRN, based on three user‐defined thresholds: FDR of TF‐enhancer edges, FDR of enhancer‐gene edges and maximum distance between enhancer and TSS.
The whole framework is implemented in a user‐friendly R/Bioconductor package. In addition to the eGRN reconstruction function, GRaNIE comprises a set of easy‐to‐use functions for generating and visualising network statistics, identifying network communities, performing subnetwork‐specific Gene Ontology (GO) enrichment analysis and various QC plots. An extensive documentation is available at (https://grp‐zaugg.embl‐community.io/GRaNIE).
Application of GRaNIE for generating cell‐type‐specific eGRN in macrophages
Macrophages are large white blood cells of the innate immune system that can be found in essentially all tissues. They play a role in inflammatory disorders and genetic variants associated with several autoimmune and other diseases are enriched for enhancers active in macrophages (Alasoo et al, 2018; Novikova et al, 2021). Given that inflammatory conditions are underlying many common diseases, macrophages are an important cell type within which disease‐associated variants manifest their effect. Thus, macrophages present an ideal cell type to apply and test the eGRN framework.
We obtained paired RNA‐ and ATAC‐seq data for induced pluripotent stem cell (iPSC) derived macrophages from 31 to 45 individuals in four conditions (naive, interferon gamma (IFN‐γ)‐primed, infected (with Salmonella), and IFN‐γ‐primed‐infected) (Alasoo et al, 2018) (Fig 1B). For each of these, we run GRaNIE using TF‐binding sites predictions based on HOCOMOCO v11 and PWMScan as described previously (Berest et al, 2019).
We next assessed the GRaNIE‐inferred eGRNs using independent molecular evidence, and used this to define reasonable default values for the TF‐peak and peak‐gene FDR thresholds. Since molecular ground truth data for TF‐peak‐gene links does not exist, we evaluated each type of link independently using cell‐type‐specific ChIP‐seq data for the TF‐peak links and cell‐type‐specific eQTL data for the peak‐gene links. Specifically, we obtained macrophage‐specific ChIP‐seq data from ReMap 2022 (Hammal et al, 2022), and quantified the enrichment of GRaNIE‐inferred TF‐bound peaks among ChIP‐seq peaks using ATAC‐peaks that contain the TF motif as background (see Materials and Methods). For the naive, the primed and the infected macrophage eGRNs, this revealed a significant enrichment for ChIP‐seq signal at FDR 0.2 that steadily decreased with increasing FDR (Fig 1C, Appendix Fig S2). The primed‐infected eGRN did not show any significant enrichment, so we excluded this eGRN from further analyses. For the peak‐gene links, we used macrophage‐specific cis‐eQTLs to assess the enrichment of eQTL links in the GRaNIE links over a distance‐matched set of control links at various FDRs. This revealed a steady decrease in the odds ratio with increasing FDR for all three remaining eGRNs (Fig 1D, Appendix Fig S3). Based on these results, we chose 0.2 as default for TF‐peak FDR and 0.1 as default for peak‐gene FDR.
Using these default parameters, we obtained an eGRN for each of the three conditions comprising 92–126 TFs, 1,411–6,742 enhancers, and 1,454–3,869 genes (Fig 1E, Appendix Fig S4A–C; Table 1 and Datasets [Link], [Link]). For all eGRNs, we observed much fewer significant connections when running GRaNIE on randomised data (permuted sample labels, peak labels and motif labels; Appendix Fig S5A–D), signifying that their TF‐peak links pass QC. Similarly, for the peak‐gene links, we find that all eGRNs show more signal for positive (expected signal) than negative (noise) correlations, and that the signal‐to‐noise ratio decreases with peak‐gene distance until no signal is left for random peak‐gene pairs (Appendix Fig S6A–D). In addition, we observed a significant enrichment for the TF‐peak‐gene links among cell‐type‐specific active enhancers based on CAGE data (Andersson et al, 2014) (Fig 1F), further corroborating that GRaNIE infers biologically meaningful eGRNs.
Table 1.
Summary of the eGRNs described in the main text.
| eGRN | # TFs (activators, repressors) | # peaks | # genes | # connections TF‐enhancer‐genes |
|---|---|---|---|---|
| Macrophage naive (naive) | 114 (52, 28) | 1,802 | 1,793 | 3,209 |
| Macrophage IFN‐γ primed (primed) | 126 (65, 31) | 6,742 | 3,869 | 22,082 |
| Macrophage infected with Salmonella (infected) | 92 (35, 26) | 1,411 | 1,454 | 2,128 |
| Macrophage IFN‐γ‐primed and infected (primed‐infected) | 78 (30, 22) | 5,184 | 2,732 | 14,697 |
| AML | 53 (30, 4) | 2,896 | 2,525 | 5,466 |
| Primary CD4+ T‐cells | 94 (20, 16) | 3,469 | 3,258 | 8,920 |
Network statistics for the various eGRNs based on default parameters (TF‐peak FDR = 0.2, peak‐gene FDR = 0.1, peak‐gene distance ≤ 250 kb, activator/repressor stringency threshold based on the 10th percentile).
We observed a slightly larger number of TFs classified as activators than as repressors (1.5–2‐fold), yet activators were connected with more peaks resulting in over 10‐fold more peaks being linked to an activator than to a repressor (Appendix Fig S5A–D; Table 1). Notably, in all eGRNs, only about 20–30% of the peaks are linked to their closest gene TSS (Fig 1G), an observation that is consistent with previous observations in pulmonary arterial endothelial cells (Reyes‐Palomares et al, 2020) and iPSC‐derived cardiomyocytes (preprint: Bunina et al, 2021).
On average, a gene is linked to 4.4 (naive), 2.9 (infected) and 5.9 (primed) peaks, of which 1.8 (naive), 1.5 (infected) and 5.7 (primed) are TF‐bound and thus part of a GRaNIE eGRN (Fig 1H, Appendix Fig S7). This discrepancy suggests that we are still missing some TF‐peak interactions (see Box 1). The majority of TFs are connected to very few genes, yet a handful of TFs are connected to over 50 genes, as exemplified for the eGRN from naive and the infected macrophages (Fig 1I), which is in line with the typically scale‐free structure of GRNs (Ouma et al, 2018). The most connected TFs in the infected and the naive eGRNs include many well‐established macrophage TFs such as IRF8, NFKB2 and RELB (Grigoriadis et al, 1996; Langlais et al, 2016), as well as noncanonical macrophage TFs MBD2, FOSL1 and NRF1. The latter have only recently been implicated in macrophage biology in mouse studies (Morishita et al, 2009; An et al, 2020; Jones et al, 2020).
Box 1. Limitations of GRaNIE.
As with all network inference tools, it is important to keep in mind what an edge means. In the case of GRaNIE, the TF‐peak and peak‐gene links are based on co‐variation across biological samples (in this study variation across individuals). Therefore, it will miss links when either of the nodes (TF expression, peak accessibility, or gene expression) is not variable across samples. For instance, if samples are individuals, GRaNIE may miss house‐keeping and dosage‐sensitive genes, TFs, and enhancers if they are equally active between individuals.
If GRaNIE is run with ATAC‐seq data, the limitations of ATAC‐seq apply: i.e. accessibility may not always reflect activity. Specifically, promoter accessibility is not necessarily correlated with gene expression. Therefore, GRaNIE will likely miss some promoter‐gene connections. Furthermore, it will not detect TFs that do not affect accessibility.
As with most TF‐inference based tools, GRaNIE relies on the availability of a TF binding site within a peak. Therefore, it will miss TFs for which binding sites are unknown, and TFs binding events that do not rely on the TF motif (e.g. cooperative binding).
TF expression is not always predictive of a TF's role in transcriptional regulation. To circumvent this, GRaNIE offers the option of using TF motif accessibility as an estimate of TF activity. This in turn has the caveat that connections will be based on TF motifs, which can be very similar across TFs.
Since GRaNIE is association‐based, it cannot per se distinguish direct from indirect effects. This is particularly important when running it on samples that are very different (e.g. different cell types). It may then become difficult to assess whether the variation in peak accessibility is driven by the TF for which it has a motif, or by some other mechanism. We refer the users to the QC implemented in GRaNIE to judge the extent of such an existing batch effect.
To dive into the biological processes captured by eGRNs, GRaNIE provides functionalities for identifying subnetworks, or communities (using Louvain clustering by default, as implemented in the igraph package in R; Blondel et al, 2008), and performing GO term enrichment on them. In line with a scale‐free architecture of the networks, we typically observe a few large communities and a long tail of very small and isolated nodes for each eGRN (Appendix Fig S8). Among the communities (Appendix Fig S8A) of the naive macrophage eGRN, one is enriched for GO terms related to pro‐inflammatory processes (response to IL‐1, chemotaxis, response to IFN‐γ) and one for anti‐inflammatory processes (angiogenesis, cytoskeleton reorganisation, positive regulation of osteoblast differentiation; Fig 1J), recapitulating the potential of naive macrophages to polarise into either M1 (pro‐inflammatory) or M2 (anti‐inflammatory) cell states (Murray, 2017). We find the M1‐phenotype cluster regulated by NFKB1/2 and REL, while the M2‐phenotype cluster is regulated by TEAD1/2 and GATA4. Among the communities of the infected macrophage eGRN (Appendix Fig S8D), one was enriched for pro‐inflammatory processes, one for phagocytosis‐related processes, and one for chemotaxis (Fig 1K), thus recapitulating the most important facets of macrophage function (Nathan et al, 1983; Parameswaran & Patial, 2010; Meng et al, 2014). Notably, each of these functional communities was regulated by a specific set of TFs: IRF8 for the pro‐inflammatory community, MBD2, NFR1 and ETV1 for the phagocytosis, and MECP2 for the chemotaxis.
As utility evaluation of the GRaNIE eGRNs, we compared real versus permuted eGRNs in terms of number and biological specificity of GO terms enriched in the TF regulons. Notably, the regulons of the permuted networks had the same degree distribution and thus the size distribution of the regulons (see Materials and Methods). The regulons from the permuted networks were enriched in less specific GO terms unrelated to macrophage biology compared with the regulons of the real eGRN (Appendix Fig S9).
In summary, these results demonstrate that GRaNIE‐inferred eGRNs capture molecular evidence from eQTLs, ChIP‐seq and CAGE data, and are useful for investigating TF‐driven biological processes in a cell type/state‐specific manner. Limitations of GRaNIE are outlined in Box 1.
Conceptual description of GRaNPA, an approach for evaluating the biological relevance of GRNs and TFs
The premise of our proposed GRN evaluation framework is that cell‐type‐specific GRNs should capture cell‐type‐specific changes in gene expression patterns that are driven by TFs. For this, we devised a machine learning approach, GRaNPA (Gene Regulatory Network Performance Analysis), which evaluates how well the bipartite TF‐gene connections of an eGRN can predict cell‐type‐specific differential gene expression. At the same time, this framework identifies the TFs that are important for the prediction.
GRaNPA requires differential RNA expression data for a perturbation in the cell type for which the GRN was constructed, and that is independent from the data used to generate the GRN. It then trains a random forest regression model to predict a differential expression value per gene, based on the TF‐target gene connections from the GRN in a 10‐fold cross validation setting (see Materials and Methods; Fig 2A), using R 2, area under the precision‐recall, and receiver operating curves (AUPRC and AUROC) to measure performance. To ensure the prediction is specific to the real GRN, it also trains a separate random forest model based on a permuted GRN, constructed from the same TFs and genes by permuting the edges (thus conserving the degree distribution of the real GRN). A good performance of the permuted GRN indicates that even unspecific TF‐gene connections can predict differential expression, invalidating the real network's specificity. Lastly, to assess overfitting, GRaNPA trains the same permuted network on completely random differential expression data (uniform distribution between −10 and 10; see Materials and Methods). If GRaNPA performs well on this random data, the model is likely overfitting. Notably, GRaNPA can be applied to assess any GRN that contains TF‐gene connections and may be used to benchmark GRNs constructed using various methods (see below). Furthermore, given a predictive GRN and specific differential expression data, GRaNPA estimates the importance of each TF towards the prediction, which provides candidate driver TFs for a specific expression response. The calculation of TF importance is based on the built‐in importance function in the R package ranger that quantifies importance of features in random forest models based on how much their exclusion affects prediction accuracy.
Figure 2. Overview and application of GRaNPA.
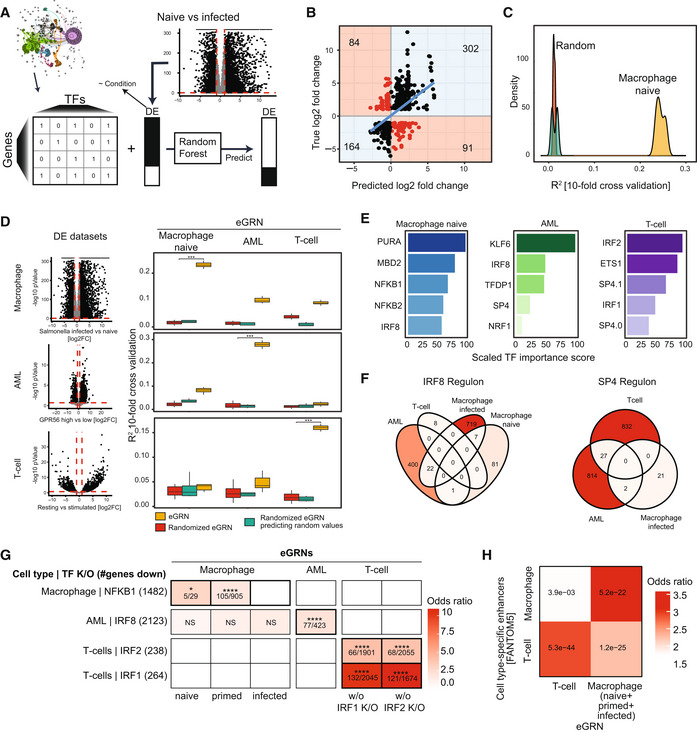
- Schematic of the general GRN evaluation approach GRaNPA.
- Output of GRaNPA is shown as true versus predicted log2 fold‐changes for the macrophage expression response to Salmonella infection. Predictions are based on the naive macrophage eGRN (see Appendix Fig S10 for the other macrophage eGRNs).
- Output of GRaNPA is shown as density distribution of R 2 for 10 random forest runs for the naive macrophage eGRN predicting differential expression upon Salmonella infection, along with the two permuted controls.
- GRaNPA evaluation of eGRNs for naive macrophages (left), AML (middle) and T‐Cells (right) of differential expression from macrophages infected with Salmonella versus naive (top), two subtypes of AML (middle), and resting versus stimulated T‐cells (bottom). Red lines indicate the log2 fold‐change (vertical line) and P‐value (horizontal line) thresholds for genes included in the GRaNPA analysis. Distributions of R 2 from distinct random forest runs (n = 10) are shown as boxplots; t‐tests were performed to compare GRaNPA performance between the permuted and real networks (***P < 0.001). Boxplots: central band: 50% quantile, box: interquartile range (25–75%); whiskers: max/min are 1.5 IQR above/below the box.
- Top 5 most important TFs (0.0 and 0.1 indicate distinct TF motifs as defined by the HOCOMOCO database) for each of the eGRNs in (D) based on prediction in the same cell‐type.
- Overlap of SP4 (left) and IRF8 (right) regulons between eGRNs from different cell types (only eGRNs with at least one connection to the respective TF are shown).
- Enrichment (odds ratio ‐ OR) of NFKB1, IRF8, IRF1 and IRF2 target genes identified in cell‐type specific knockouts (K/O, rows) in the matching macrophage, AML and T‐cell eGRN regulons (columns). Numbers in cells indicate: (# genes in regulon and down in TF K/O)/(# genes in regulon). Asterisks indicate significance using Fisher's exact test; test set: all protein‐coding genes; categories: gene in regulon, gene down in TF K/O (NS: non‐significant, *P‐adj. < 0.05, ****: < 0.001). White squares indicate empty regulons.
- Enrichment of T‐cell and macrophage‐specific FANTOM5 CAGE enhancers among the T‐cell and macrophage eGRN peaks. The numbers inside the tiles are BH‐adjusted P‐values based on Fisher's exact test; test set: all peaks in the respective cell types (102,141 and 248,844 for T‐cells and macrophage eGRNs, respectively); categories: peak in eGRN, peak overlap with CAGE enhancer. The macrophage eGRN is the union between the infected, naive and primed eGRNs.
In short, the GRaNPA strategy is based on the following steps:
Obtain differential expression data for the cell type matching the GRN.
For each cell type, train a random forest regression model (10‐fold cross‐validation) to predict a differential expression value per gene based on TF‐gene links from the GRN.
Compare the performance of models learned on real and permuted TF‐gene links, and TF‐gene links from other cell types (cross‐validation R 2).
Identify important TFs for the given differential expression response.
GRaNPA is implemented as a user‐friendly R‐package (https://git.embl.de/grp‐zaugg/GRaNPA) and documentation is available at (https://grp‐zaugg.embl‐community.io/GRaNPA/). Limitations of GRaNPA are outlined in Box 2.
Box 2. Limitations of GRaNPA.
GRaNPA is based on the assumption that differential gene expression, which is always based on steady‐state RNA expression levels, is explained solely by the action of TFs. This is a simplification and other processes, such as RNA stability, also affect RNA expression levels.
The performance values from GRaNPA are often low, even if they are better than those for permuted networks, suggesting that the GRNs are not picking up all the signal in the data. Adding gene‐specific features e.g. from (Sigalova et al, 2020) may substantially improve performance if desired.
GRaNPA cannot resolve cooperative TF binding.
GRaNPA fails for datasets in which only a small number of differentially expressed genes overlap with the tested GRN.
TFs with few connections in the GRN are less likely to be identified as important TFs with GRaNPA, simply because they do not affect many genes.
GRaNPA evaluation of the macrophage eGRNs
To evaluate the predictive power of the macrophage eGRNs and identify the TFs driving a specific expression response, we obtained RNA‐seq data for naive and Salmonella‐infected macrophages from (Alasoo et al, 2018), and calculated the differential expression using DESeq2 (Love et al, 2014; Materials and Methods). For evaluations, we excluded samples that were used for the eGRN reconstruction.
The three macrophage eGRNs performed well with GRaNPA, predicting differential expression values (random forest regression) with R 2 of 0.15–0.25 (Fig 2B and C, Appendix Figs S10 and S11) and direction of change (classification) with AUPRC of 0.71–0.88 and AUROC of 0.65–75 (Appendix Figs S12 and S13). The performance of the corresponding permuted networks was significantly lower (t‐test P‐value < 1e‐6 for all; Fig 2B and C, Appendix Figs S11–S13). Notably, the eGRN for primed‐infected macrophages that we excluded above due to failed ChIP‐seq validation (Appendix Fig S2) was unable to predict any differential expression (Appendix Figs S10–S13), which highlights the concordance of GRaNPA evaluation with molecular evidence. The significant difference between the permuted and the actual networks shows that the eGRNs indeed capture biologically relevant links between TFs and genes.
eGRNs built from single cell types show cell‐type‐specific predictions
We next assessed the cell‐type specificity of GRaNIE‐inferred eGRNs. To this end, we obtained data sets in different cell types with matched RNA and chromatin accessibility data for primary human CD4+ T‐cells (Freimer et al, 2022) and from AML (Garg et al, 2019) and (He et al, 2022). We ran GRaNIE using the same parameters as described above and obtained additional eGRNs for primary CD4+ T‐cells (Dataset EV5) and AML (Dataset EV6).
To assess their cell‐type‐specific prediction power, we ran GRaNPA on the naive macrophage, T‐cell, and AML eGRNs, and compared their performance to predict differential expression in each of the three cell types. Specifically, we quantified differential expression between resting and lipopolysaccharide (LPS) stimulated follicular CD4+ T‐cells (data from Calderon et al, 2019), between two subtypes of AML (GPR56‐high vs. GPR56‐low; data from Garg et al, 2019), and between naive and Salmonella‐infected macrophages (data from Alasoo et al, 2018). We found that the eGRN that matches the respective cell type led to the best prediction (Fig 2D). While T‐cells and macrophages were only predictive in their own cell type, the AML eGRN was to a smaller extent also predictive for the macrophage response. Since AML cells and macrophages are both from the myeloid lineage, this could indicate some shared regulatory architecture between them. Notably, we found that the R 2 values can be boosted by adding gene specific features, such as expression variation across individuals, in line with our previous work (Sigalova et al, 2020) (Appendix Fig S14). We are primarily interested in evaluating TF‐gene links and eGRN cell‐type specificity, so GRaNPA does not use these gene‐specific features by default.
Using the TF‐importance estimation implemented in GRaNPA, we observed that among the top five important TFs, most are unique for one cell type (Fig 2E) with the exceptions of IRF8, which was important in AML and macrophages, and SP4, important for AML and T‐cells. Notably, the IRF8 regulons in AML and macrophages had only 22 genes (and no single enhancer) in common, while each cell‐type specific regulon included hundreds of nonoverlapping genes (Fig 2F). Similarly, the SP4 regulons of T‐cells and AML were almost mutually exclusive. This suggests a highly cell‐type‐specific regulon composition of IRF8 and SP4.
As an orthogonal validation of the cell‐type specificity of the TF regulons from GRaNIE eGRNs, we compared the regulons with differential expression data upon TF knockout (K/O) in the same cell type. We obtained data for one or two of the top five important TFs in each cell type: NFKB1 in macrophages (Somma et al, 2021), IRF8 in AML (Liss et al, 2021) and IRF1 and IRF2 in T‐cells (Freimer et al, 2022). The genes downregulated upon TF K/O were significantly enriched in the TF regulons of the respective cell types (Fig 2G). Notably, genes downregulated upon IRF8 K/O in AML were specifically enriched for the IRF8 regulon in AML, despite the fact that IRF8 also has a large regulon and is an important TF in macrophages (Fig 2E). This suggests that the cell‐type specificity of the GRaNPA predictions is not only dependent on distinct sets of TFs driving the response, but also on the genes the TF regulates in that cell type, highlighting the importance of cell type‐specific eGRNs.
To validate the cell‐type specificity of enhancers in GRaNIE, we obtained cell‐type‐specific enhancer maps from FANTOM5 using CAGE data for T‐cells and macrophages (Andersson et al, 2014). Quantifying their overlap with enhancers from T‐cell and macrophage eGRNs revealed a stronger significant enrichment among the enhancers from the same cell type as compared with opposite cell types (Fig 2H).
The eGRNs connect TFs to genes through active regulatory regions, comprising both promoters and enhancers. The predictive evaluation set‐up allowed us to compare the relative importance of promoter (i.e. < 10 kb from TSS) and enhancer links (> 10 kb from TSS) in different eGRNs. To do so, we divided the gene‐peak pairs into 10 groups based on their distance to the TSS and ran GRaNPA for each group separately. The promoter‐only eGRNs from infected and primed macrophages showed limited or no predictive power (Appendix Fig S15). This highlights the importance of enhancers and is in line with a recent study that demonstrated the importance of considering enhancers for predicting the cell‐fate potential of TFs (Xu et al, 2021).
Application of GRaNPA to compare GRaNIE eGRNs with other GRN methods
Notably, GRaNPA is applicable to assess any type of bipartite TF‐gene network and can be used more generally to assess the utility of a GRN for understanding cellular response to specific perturbations. Here, we used it to evaluate the performance of several previously published TF‐gene GRNs that draw links between TFs and genes based on different approaches: DoRothEA, which uses manual curation combined with a data‐driven approach including co‐expression to draw TF‐gene links (Garcia‐Alonso et al, 2019; Holland et al, 2020a, 2020b), ChEA3, which uses ChIP‐seq experiments from ENCODE, ReMap, or literature to draw TF‐gene links (Keenan et al, 2019), RegNet, a curated network integrating TFs and miRNAs (Liu et al, 2015), and TRRUST, which is a curation of TF‐gene links based on PubMed indexed articles (Han et al, 2018). We also included an enhancer‐based GRN inferred with ANANSE (Xu et al, 2021) from macrophage data.
The cell‐type‐matched GRaNIE eGRNs and DoRothEA ABC showed good prediction for all datasets tested. The TRRUST, RegNet and ChEA3 networks showed slight predictive power for macrophages, while the only other enhancer‐based network ANANSE showed very poor performance across all cell types (Fig 3A). Thus, GRaNIE networks outperformed most other networks and was on par with the highly curated DoRothEA.
Figure 3. Evaluation of GRaNIE eGRNs and other GRN approaches.
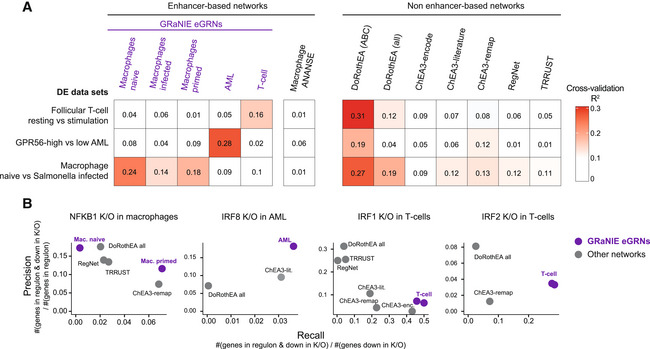
- GRaNPA evaluation of five GRaNIE eGRNs (macrophage naive/primed/infected, AML, and T‐cells), another enhancer‐based eGRN inferred with ANANSE (Xu et al, 2021), and publicly available TF‐gene networks based on data curation (DoRothEA ABC and all (Holland et al, 2020a)), ChIP‐seq data (ChEA3 encode, literature, and ReMap (Keenan et al, 2019)), manual curation (TRRUST (Han et al, 2018) and REGNET (Liu et al, 2015)). GRNs are evaluated by GRaNPA for their performance in predicting the differential expression of resting versus stimulated follicular T‐cells, GPR56 high versus low AML, and naive versus Salmonella‐infected macrophages. Numbers in squares indicate R 2 values.
- Precision‐Recall evaluation of the NFKB1, IRF8, IRF1 and IRF2 regulon from the networks in (A) for identifying genes down‐regulated upon K/O of the respective TF. For GRaNIE eGRNs (purple), the performance of cell‐type matching networks is shown, other networks are the same across all analyses.
To further compare cell‐type specificity, we assessed the overlap between the TF‐regulons identified in the networks with reasonable predictive power and the genes downregulated upon K/O of the same TF (data introduced in Fig 2G). Overall, the cell‐type‐matched GRaNIE eGRNs outperformed all other networks in terms of recall (Fig 3B). Of note, the absolute recall was rather small, likely owing to the fact that TF K/O induces many indirect downstream effects that are not captured by the direct mechanistic links of eGRNs. GRaNIE also outperformed all other networks in terms of precision in AML. While DoRoThEA achieved the highest precision for IRF1 and IRF2 K/O in T‐cells, the recall was smaller. Overall, this cell‐type‐specific TF K/O evaluation highlights the importance of unbiased and cell‐type‐specific eGRNs.
Macrophage eGRNs reveal distinct set of TFs driving response to different types of infection
GRaNIE and GRaNPA can also provide biological insights. Specifically, we employed them for studying different types of pro‐inflammatory M1‐like responses of macrophages to bacterial infections as well as the anti‐inflammatory M2‐like response of breast cancer associated macrophages. We obtained data from previously published studies (Table 2) that measured the expression response of macrophages infected with Mycobacterium Tuberculosis (MTB) (Giraud‐Gatineau et al, 2020), Listeria monocytogenes (Pai et al, 2016), Salmonella Typhimurium (Pai et al, 2016; Alasoo et al, 2018), stimulation with IFN‐γ (Alasoo et al, 2018) and a study that compared tumour associated macrophages with tissue‐resident macrophages from breast cancer tissue (Cassetta et al, 2019).
Table 2.
Differential expression experiments for the different infection settings.
| Cell types | Treatment | Comparison | Reference |
|---|---|---|---|
| Monocyte‐derived macrophages from healthy donors | Listeria monocytogenes | Uninfected versus 2 h post infection | Pai et al (2016) |
| Salmonella Typhimurium | |||
| Mycobacterium Tuberculosis strain resistant to BDQ treatment | Uninfected versus 18 h post infection | Giraud‐Gatineau et al (2020) | |
| Uninfected versus 36 h post infection | |||
| iPSC‐derived macrophages | Salmonella typhimurium | Uninfected versus 5 h post infection | Alasoo et al (2018) |
| 18 h IFN‐γ‐primed versus 18 h IFN‐γ‐primed +5 h post infection | |||
| Interferon‐gamma stimulation | Naive versus 18 h IFN‐γ treatment | ||
| Tumour‐associated and tissue resident macrophages from human breast tissue | Tumour versus tissue‐resident | Tumour versus respective tissue resident macrophages | Cassetta et al (2019) |
Differential expression summary for the different infection datasets/cell types, their treatments and the comparisons used for the differential expression analyses.
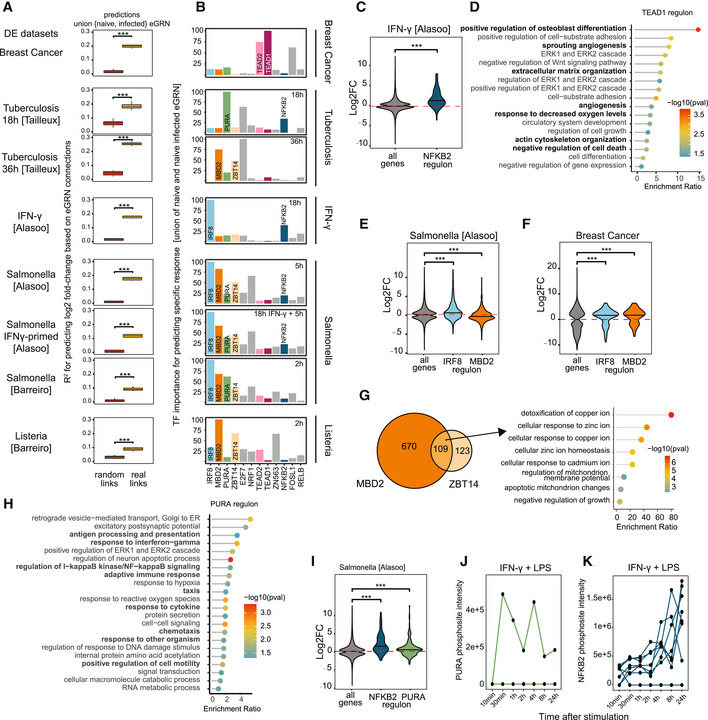
To understand how macrophages respond to these distinct perturbations, we employed GRaNPA using the union of the naive and infected eGRNs (Appendix Fig S16; Dataset EV4), which showed good predictions for all conditions (Fig 4A), and determined the important TFs for each response. One of the well‐understood responses of macrophages is the IFN‐γ‐mediated activation of the NFKB family of TFs (Medzhitov & Horng, 2009). In line with this, we find NFKB2 as one of the most important TFs upon IFN‐γ stimulation (Fig 4B). The NFKB2 regulon was enriched for GO terms related to chemokine signalling and taxis (Appendix Fig S17) and strongly upregulated in response to IFN‐γ (Fig 4C). This demonstrates the ability of GRaNPA to identify biologically meaningful TFs. To assess the robustness of GRaNPA, we compared the TF importance predictions across two independent data sets from Salmonella‐infected macrophages, which revealed very similar profiles despite differences in the experimental set‐up (Fig 4B; iPSC‐derived vs. monocyte‐derived macrophages) and time points (5h and 2h post infection, respectively), thus highlighting the robustness of GRaNPA and the biological congruence between the experiments.
Figure 4. Application of GRaNIE and GRaNPA to investigate macrophage biology.
-
AGRaNPA evaluation of the union of the naive and infected macrophage eGRNs (naive+infected eGRN; real links) and the corresponding permuted control network (random links) across eight experimental settings of macrophage perturbations. Distributions of R 2 from distinct random forest runs (n = 10) are shown as boxplots and two sided t‐tests were performed to compare GRaNPA performance between the permuted and real networks (***P < 0.001). Boxplots: central band: 50% quantile, box: interquartile range (25–75%); whiskers: max/min are 1.5 IQR above/below the box.
-
BTF importance profiles for each of the eight infection settings from (A). The top 5 most predictive TFs in any of the settings are displayed. TFs discussed in the text are individually labelled and coloured.
-
CDistribution of log2 fold‐changes for genes in the NFKB2 regulons from the naive+infected eGRN (n = 85; dark blue) are shown for IFN‐γ stimulation versus naive macrophages alongside the response of all genes (n = 2,976; grey). Central band of the violin plot: median.
-
DGO enrichment of the TEAD1 regulon.
-
E, FDistribution of log2 fold‐changes of Salmonella infection versus naive macrophages (E) and for breast‐cancer associated macrophages (F) are shown for genes in the IRF8 (n = 830; blue) and MBD2 (n = 779; orange) regulons alongside the response of all genes (n = 2,976; grey). Central band of the violin plot: median.
-
GThe overlap between the MBD2 and ZBT14 regulons are shown as Venn Diagram (left). Enriched GO terms for the genes in the intersection are shown as a lollipop plot (right).
-
HGO enrichment of the PURA regulon.
-
IDistribution of log2 fold‐changes of Salmonella infection versus naive macrophages for genes in the NFKB2 (n = 85; blue) and PURA (n = 258; green) regulon alongside the response of all genes (n = 2,976; grey). Central band of the violin plot: median.
-
J, KNormalised mass spectrometry intensity values (y‐axis) for phosphosites detected on PURA (green, J) and NFKB2 (blue, K) in macrophages cultured in the presence of M1 polarising stimuli (IFN‐γ and LPS) for indicated time points (x‐axis). Lines show individual phosphosites detected on each respective TF.
Data information: Two‐sided t‐test was used to determine statistical significance in (C, E, F, and I); data points correspond to genes in tested regulons (numbers given in panels). ***P‐value < 0.001.
In contrast, across conditions, TF‐importance profiles were highly variable (Fig 4B), likely reflecting different roles of macrophages (M1 vs. M2) and their variable defence mechanisms triggered by the pathogens (Leseigneur et al, 2020). Breast‐cancer associated macrophages showed the most distinct profile with TEAD1/TEAD2 as important TFs. GO analysis of the TEAD1/2 regulon revealed a strong enrichment for angiogenesis, osteoblast differentiation and ERK signalling among others (Fig 4D), in line with a more M2‐like phenotype (Corliss et al, 2016; Chen et al, 2020).
The most important TF for predicting the response to Salmonella infection was IRF8, followed by MBD2 and ZBT14 (Fig 4B). IRF8 is a known pro‐inflammatory interferon response factor, associated with the pro‐inflammatory (M1) polarisation of macrophages (Chistiakov et al, 2018), which we confirmed in our data using gene set enrichment analysis (GSEA) of the IRF8 regulon (Appendix Fig S18). Less is known about MBD2 and ZBT14 in macrophages, although MBD2 has been linked to intestinal inflammation in mice (Jones et al, 2020) and with an M2 macrophage programme in pulmonary fibrosis (Wang et al, 2021). In line with this, the MBD2 regulon was downregulated in response to infection (Fig 4E, Appendix Fig S13) but upregulated in breast cancer‐associated macrophages (Fig 4F), showing the opposite pattern to the IRF8 regulon (Fig 4E and F). We further find an enrichment of the M2 gene set among the MBD2 regulon in breast cancer associated macrophages (Appendix Fig S18). The MBD2 and ZBT14 regulons show significant overlap (Fig 4F, P = 3.3e‐13, hypergeometric test) and genes jointly regulated by them are enriched for terms related to response to metal ions (Fig 4G). The use of zinc and copper ions in macrophage defence strategies is well‐documented (Festa & Thiele, 2012; Stafford et al, 2013). Given that ZBT14 and MBD2 are important for predicting response to pathogens, but not to IFN‐γ stimulation (Fig 4B), we speculate that ZBT14 and MBD2 may jointly induce a macrophage‐intrinsic mechanism to counteract toxic metal ions, potentially aimed at overcoming the toxic effects of its own weapons.
GRaNPA identifies PURA as putative proinflammatory TF in macrophages
Among the TFs that are less well known for their role in macrophages, we find PURA for many of the infection settings. In line with a pro‐inflammatory role of PURA, we found GO terms associated with chemotaxis and IFN‐γ response enriched among genes in its regulon (Fig 4H). GSEA found the M1 gene set significantly enriched among the PURA‐regulated differentially expressed genes upon Salmonella infection (Appendix Fig S18). Furthermore, the expression of genes in the PURA regulon were upregulated upon Salmonella infection to a similar extent as the genes in the NFKB2 regulon, which is a known pro‐inflammatory TF (Fig 4I).
To follow‐up on a potential role of PURA in M1 polarisation, we obtained phosphoproteomics data that were collected upon stimulating macrophages with LPS and IFN‐γ towards the M1 phenotype (He et al, 2021). This revealed a specific increase in phosphorylation of Thr187 upon LPS/IFN‐γ stimulation (Fig 4J), following a similar pattern of increasing phosphorylation over time as for phosphosites on NFKB2 (Fig 4K). Notably, this stimulation‐induced phosphosite in PURA is located in the Purα repeats region, which is implicated in DNA binding and crucial for PURA function (Weber et al, 2016). Phosphorylation of DNA‐binding regions has been associated with activation of other TFs (Hirata et al, 1993), suggesting that activation of PURA is perhaps important for M1 polarisation, providing further evidence for its role in macrophages' pro‐inflammatory response.
Overall, these results highlight the use of GRaNPA in conjunction with cell‐type‐specific eGRNs for investigating the biological functions that are regulated by a TF in a specific cell type.
Macrophage‐specific eGRNs are enriched in fine‐mapped GWAS variants and immune‐related traits
The strength of the eGRN framework is that we can specifically investigate the role of gene regulatory elements such as enhancers, which are enriched for disease‐associated genetic variants (Claringbould & Zaugg, 2021). We therefore sought to explore the macrophage eGRNs to learn about gene regulatory mechanisms underlying associations of genetic variants with common complex traits and diseases.
First, we tested whether the peak regions specific to the three macrophage eGRNs that GRaNPA identified as predictive in at least one infection setting (naive, primed, infected) were enriched in heritability for 442 GWAS traits (Dataset EV9). We applied stratified linkage disequilibrium score regression (S‐LDSC; Finucane et al, 2015) and compared the eGRN peaks to all peaks identified in macrophages (see Materials and Methods). Notable enrichments include HbA1c measurement (a measure for diabetes severity), large artery stroke and heart failure for the naive eGRN; adolescent idiopathic scoliosis and nonischemic cardiomyopathy for the primed eGRN and rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) for the infected eGRN (Fig 5A). SLE and RA are both driven by activated macrophages (Udalova et al, 2016; Ma et al, 2019) as a result of known (for SLE) or hypothesised (RA) upregulation of IFN‐γ signalling (Harigai et al, 2008; Rönnblom & Leonard, 2019; Kato, 2020). Interestingly, we find enriched heritability for these traits in the peaks for the IFN‐γ primed eGRN, but not for either naive or infected eGRNs. Given this association, we also assessed the heritability enrichment of the regulatory elements and genes connected to the TFs that are particularly important for predicting the response of macrophages to IFN‐γ (NFKB1/2, RELB, IRF8). Inflammatory bowel disease (specifically ulcerative colitis) comes out as the top enriched trait (Appendix Fig S19), which is in line with the known role of IFN‐γ in this disease (Andreou et al, 2020). Literature evidence for other traits is summarised in Dataset EV10.
Figure 5. Application of GRaNIE for investigating trait‐associated SNPs.
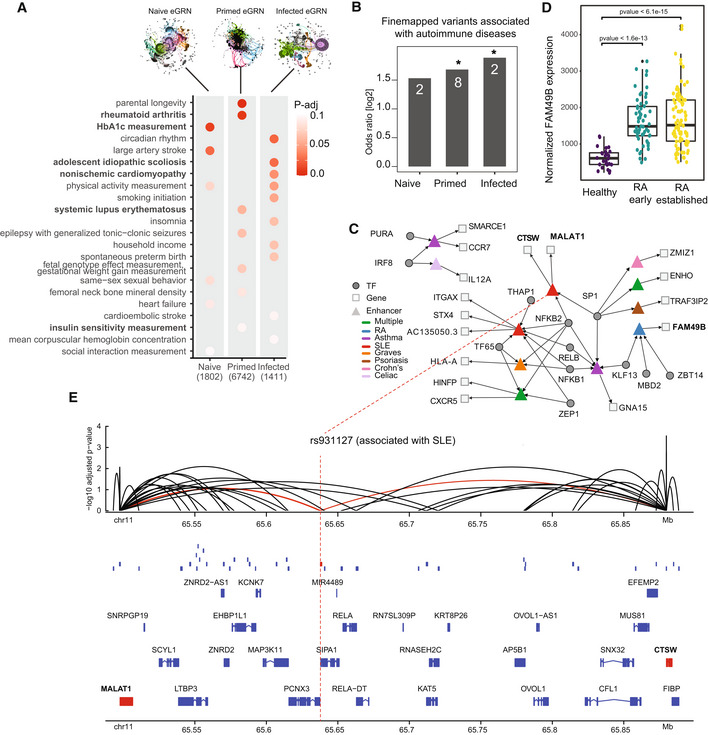
- Heritability enrichment is shown for the naive, primed and infected macrophage eGRNs. The P‐value is adjusted within each trait.
- The enrichment of fine‐mapped GWAS SNPs within the naive, primed, and infected eGRNs is shown as odds ratios; *P‐value < 0.05 (Fisher's exact test, test set: all ATAC‐seq peaks in macrophages – 296,220; categories: peak in eGRN, peak overlap with finemapped SNP); n: number of finemapped SNPs.
- The tripartite TF‐enhancer‐gene network involving all fine‐mapped GWAS variants for autoimmune diseases.
- Normalised expression level of FAM49B is shown as a boxplot for synovial tissue from healthy controls (n = 28) and patients suffering from early (n = 57; green) and established (n = 95; yellow) rheumatoid arthritis (RA). Data from (Guo et al, 2017). Boxplots: central band: 50% quantile, box: interquartile range (25–75%); whiskers: max/min are 1.5 IQR above/below the box. Black dots indicate outliers. Adjusted P‐values were calculated according to the Wald test implemented in DESeq2; replicates are individual donors.
- The genomic context of the fine‐mapped, SLE‐associated variant rs931127 in an ATAC‐seq peak (red box) as gene tracks, including other peaks present in the infected macrophage eGRN (blue boxes), and peak‐gene links from the infected macrophage eGRN (arcs). Genes targeted by the peak overlapping with rs931127 (red) are coloured in red.
Next, we zoomed in to a specific set of fine‐mapped GWAS variants associated with autoimmune diseases with a known link to macrophages. Across all three macrophage eGRNs, we found in total 11 unique fine‐mapped variants that were located in the regulatory regions (2 in the naive, 2 in infected and 8 in the primed eGRN). The infected and primed eGRNs were significantly enriched in fine‐mapped variants (Fig 5B). Investigating the TFs regulating the fine‐mapped autoimmune disease enhancers, we find the known immune response TFs NFKB1/2, RELB and IRF8, but also MBD2, ZBT14 and PURA (Fig 5C), which we identified as important TFs for predicting response to infection. One of the enhancers overlapping with an RA‐associated SNP, regulated by MBD2 and ZBT14, is linked to FAM49B as a target gene. Inspection of FAM49B expression in synovial tissue in a cohort of RA patients compared with healthy controls (Guo et al, 2017) revealed a misregulation upon disease onset, providing additional evidence that FAM49B is indeed the gene targeted by the fine‐mapped SNP (Fig 5D).
One of the fine‐mapped SNPs for SLE, rs9893132, is located in an enhancer regulated by SP1 and NFKB2 and linked to the noncoding RNA MALAT1 and the gene cathepsin W (CTSW) in the primed macrophage eGRN. Both genes are over 50 kb from the fine‐mapped SNP and there are several other genes in the locus that are not linked to the enhancer in question (Fig 5E). MALAT1 has been implicated in SLE through several studies (reviewed in Zhao et al, 2018), suggesting that rs9893132 may target MALAT1. While CTSW expression in lymphocytes has been linked to autoimmune diseases (e.g. Buhling et al, 2002), its role in macrophages is much less studied. Yet, CTSW knockdown in macrophages reportedly increased Mycobacterium tuberculosis survival in macrophages (Pires et al, 2016), suggesting it does play an important function in the pro‐inflammatory macrophage response. Overall, the macrophage eGRNs provide the putative target genes of 11 fine‐mapped GWAS loci, often linking to genes that are over 50 kb away from the SNP (Table 3; Appendix Fig S20).
Table 3.
Predicted target genes of fine‐mapped GWAS autoimmune variants using macrophage eGRNs.
| Peak | TF | Gene name | FM.gwas | rsid | Disease | Network |
|---|---|---|---|---|---|---|
| chr8:129939305‐129940672 | KLF13.0.D, MBD2.0.B, ZBT14.0.C | FAM49B | chr8:129939865 | rs11785995 | Rheumatoid arthritis | Naive |
| chr17:40598595‐40599644 | PURA.0.D | SMARCE1 | chr17:40598769 | rs9893132 | Asthma | Naive |
| chr19:3135922‐3136231 | KLF13.0.D, NFKB1.1.B, NFKB2.0.B, RELB.0.C, SP1.0.A, SP1.1.A | GNA15 | chr19:3136093 | rs117552144 | Asthma | Primed |
| chr16:31265059‐31265802 | NFKB1.1.B, NFKB2.0.B, RELB.0.C, TF65.0.A, THAP1.0.C, ZEP1.0.D | STX4, AC135050.3, ITGAX | chr16:31265490 | rs1143679 | Systemic lupus erythematosus | Primed |
| chr6:30006131‐30007036 | NFKB1.1.B, NFKB2.0.B, TF65.0.A | HLA‐A | chr6:30006148 | rs4313034 | Graves' disease | Primed |
| chr11:65637802‐65638765 | NFKB2.0.B, SP1.0.A, SP1.1.A | MALAT1, CTSW | chr11:65637829 | rs931127 | Systemic lupus erythematosus | Primed |
| chr11:118883323‐118883647 | NFKB2.0.B, TF65.0.A, ZEP1.0.D | CXCR5, HINFP | chr11:118883644 | rs630923 | Multiple sclerosis, Inflammatory bowel disease, Crohn's disease | Primed |
| chr6:111605185‐111606373 | SP1.0.A | TRAF3IP2 | chr6:111605706 | rs7769061 | Psoriasis | Primed |
| chr9:34709959‐34710335 | SP1.0.A, SP1.1.A | ENHO | chr9:34710263 | rs2812378 | Rheumatoid arthritis, Celiac disease | Primed |
| chr10:79285352‐79285717 | SP1.1.A | ZMIZ1 | chr10:79285450 | rs1250569 | Crohn's disease | Primed |
| chr10:79285352‐79285717 | SP1.1.A | ZMIZ1 | chr10:79285523 | rs1250568 | Celiac disease | Primed |
| chr17:40598595‐40599644 | IRF8.0.B | CCR7 | chr17:40598769 | rs9893132 | Asthma | Infected |
| chr3:159929439‐159930124 | IRF8.0.B | IL12A | chr3:159929885 | rs17753641 | Celiac disease | Infected |
Fine‐mapped GWAS variants for autoimmune diseases generated using probabilistic identification of causal SNPs (PICS; see Materials and Methods) for hg38 build overlapping with the peaks of macrophage eGRNs. TF names refer to specific motifs, some TFs have multiple motifs and thus occur multiple times.
Discussion
Phenotypic variation across individuals has two major sources: genetic variation and external influences that can be long‐lived (epigenetics) or short‐lived (signalling). Both can lead to variation in molecular phenotypes that impact on complex traits. Thus, to understand mechanisms underlying phenotypic variation, including disease phenotypes, it is crucial to study the interplay between genetic variants, epigenetic marks and extrinsic cellular signalling. Here, we present GRaNIE and GRaNPA, a tool‐suite that provides a framework for jointly analysing these layers and investigating their biological relevance.
GRaNIE is a flexible and user‐friendly R/Bioconductor package for building enhancer‐based GRNs. It requires RNA‐Seq and open chromatin data such as ATAC‐Seq or ChIP‐Seq for histone modifications (e.g. H3K27ac) across a range of samples (mostly tested in a cohort of at least 10–15 individuals), along with TFBS data (that can either be obtained from the package or provided by the user) to generate cell‐type‐specific eGRNs. It provides a range of quality control plots and functionalities for downstream analyses such as identification of communities within the network, and GO enrichment analyses. A dedicated website accompanies the package and is automatically updated whenever a new package version becomes available.
GRaNPA is an independent R package for evaluating the biological relevance (i.e. predictive power) of any TF‐gene network. It requires a bipartite TF‐gene network and genome‐wide differential expression values as input, and assesses the network's predictive power. In addition, it quantifies the importance of each TF for driving a specific differential expression response. Notably, the prediction performance of GRaNPA can be improved by adding gene‐specific features, for example those shown in (Sigalova et al, 2020); however, this would not help in the assessment of GRNs and is therefore not the main purpose of this study. One attractive use case of GRaNPA is that it can quantitatively compare the performance of different GRNs for predicting a perturbation of interest. It can thus help select the best‐suited network for a given dataset without the need for a “ground truth” network to evaluate their edges and connectivities.
Compared to most of the available GRN reconstruction approaches, GRaNIE infers enhancer‐based regulatory networks that only captures TF‐gene links mediated by enhancers. This has several advantages: first, we showed that for macrophages, parts of their expression response to infection could only be predicted when using enhancer connections. Second, including enhancers in TF‐driven GRNs allows the investigation of mechanisms underlying GWAS traits that are driven by specific TFs, and facilitates interpretation of (fine‐mapped) trait‐associated SNPs. Third, since enhancers tend to be highly cell‐type specific (Roadmap Epigenomics Consortium et al, 2015), eGRNs are likely more cell‐type specific than TF‐gene networks. Finally, by requiring a correlation between the expression level of a TF and the accessibility of the peak, in addition to the motif presence, GRaNIE circumvents the inherent limitation of TF‐binding site predictions, which cannot distinguish between TFs with similar binding motifs (Zeitlinger, 2020). This will exclude many TF‐enhancer links that have the TF motif yet show no correlation with the TF expression level and are thus likely not bound by that TF in the given cell type. GRaNIE bears conceptual similarity with a method published previously (Marbach et al, 2016); however, the data of this work are not available anymore, and the software is neither maintained nor can be downloaded/used.
Enhancer‐based gene regulatory networks consist of TFs and their respective downstream enhancer/promoter and gene targets, which means we can zoom into network communities that capture specific pathways or functions. For example, we showed that when we divide the network into communities, each community is enriched in distinct TF‐driven biological processes. The modularity of the eGRN also showed that NFKB1, NFKB2, RELB and IRF8, the TFs important for predicting the macrophage response to IFN‐γ priming, and their connected regulatory elements and genes were specifically enriched for heritability of GWAS traits that are commonly linked to IFN‐γ signalling. In contrast to other approaches for interpreting trait‐associated variants that are solely based on epigenetics such as the activity by contact model (Nasser et al, 2021), purely based on genetics, such as eQTLs (Võsa et al, 2021) or variable chromatin domains (Waszak et al, 2015), eGRNs by GRaNIE capture TF‐peak‐gene links based on variation due to genetic, epigenetic, or TF‐activity differences across individuals, thus integrating these three layers in one framework. In sum, eGRNs can be used to identify the target genes of individual TFs, to pinpoint the cell‐type‐specific regulatory regions that connect to a TF, and to investigate genetic variation in the tripartite TF‐regulatory element‐gene graphs.
Comparing eGRNs across cell types revealed that for some TFs (e.g. IRF8), the regulons are highly cell‐type specific. Cell‐type‐specific TF functions may be driven by different co‐binding TF partners depending on the cell type. Indeed, in our previous work, we found that TFs regulate distinct biological processes depending on their co‐binding partners (Bunina et al, 2020; Ibarra et al, 2020). An alternative explanation for cell‐type‐specific regulons is that different cell types may differ in their chromatin potential (Ma et al, 2020).
Among the notable observations from applying GRaNIE and GRaNPA to study the gene expression response in macrophages was that some TFs, including MBD2, were specifically important only for predicting response to bacterial infection, and not for IFN‐γ stimulation. Since IRF transcriptional programs are generally more related to a virus response, MBD2 may be required for the bacterial‐specific response, indicating that we can use these networks to identify TFs important for responses to different types of pathogens. The observation that GRaNPA identified distinct sets of important TFs for the different responses may reflect that macrophages use several strategies to fight infections, including phagocytosis followed by degradation mechanisms, starvation of pathogens, and recruiting other players in the immune system (Leseigneur et al, 2020). Another observation is that three TFs important for predicting the response to infection but not to IFN‐γ, are known to bind methylated DNA: MBD2 (Hainer et al, 2016), MECP2 (Lewis et al, 1992) and NRF1 (Domcke et al, 2015). Recent reports provide evidence for a pathogen‐induced global DNA methylation alteration (Qin et al, 2021) downstream of NFKB‐signalling, and it was shown that MBD2 inhibits IFN‐γ by selectively binding to methylated regions in the Stat1 promoter in other cell types (Yue et al, 2021). Our results are consistent with a pathogen‐response mechanism that is partially mediated by DNA methylation, which may modulate the impact of DNA‐methylation sensitive TFs and demonstrates the level of novel biological insights that can be gained with GRaNIE and GRaNPA.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or source | Identifier or catalogue number |
|---|---|---|
| Software | ||
| GRaNIE R package | https://bioconductor.org/packages/GRaNIE/ | |
| GRaNPA R package | https://git.embl.de/grp‐zaugg/GRaNPA | |
| Gephi 0.10.1 | https://gephi.org/ | |
| DESeq2 R package | https://bioconductor.org/packages/DESeq2/ | |
| GeneOverlap R package | https://bioconductor.org/packages/GeneOverlap | |
| fgsea R package | https://bioconductor.org/packages/fgsea | |
| UCSC liftOver web interface | https://genome.ucsc.edu/cgi‐bin/hgLiftOver | |
| External gene regulatory networks | ||
| Dorothea | Garcia‐Alonso et al (2019) | |
| TRRUST | Han et al (2018) | |
| ChEA3 | Keenan et al (2019) | |
| ANANSE | Xu et al (2021) | |
| Other databases and resources | ||
| ReMap 2022 | Hammal et al (2022) | |
| FANTOM5 Human Enhancer Tracks | https://slidebase.binf.ku.dk/human_enhancers/presets Andersson et al (2014). | https://slidebase.binf.ku.dk/human_enhancers/presets/serve/macrophage |
| eQTL catalogue | https://www.ebi.ac.uk/eqtl/ | |
| LDSC Github repository | https://github.com/bulik/ldsc | |
| General genomic features dataset | https://alkesgroup.broadinstitute.org/LDSCORE/ | |
| Fine‐mapped GWAS variants for autoimmune diseases | “PICS2‐GWAScat‐2020‐05‐22.txt.gz” from https://pics2.ucsf.edu | |
Methods and Protocols
Data sets used in this study
For all data sets, we performed PCA along with metadata inspection in the PCA space to evaluate whether samples should be discarded as outliers. If we did, we give details in the respective paragraph.
Expression and chromatin accessibility data for iPSC‐derived macrophages
We used a publicly available data set (ERP020977) for naive and primed macrophages (iPSC‐derived) in two conditions, uninfected and 5‐h infected with Salmonella from (Alasoo et al, 2018). In total, we obtained 304 RNA‐seq profiles from 86 different individuals, of which 145 also had ATACseq data available (https://zenodo.org/record/1188300#.X370PXUzaSN). The samples are split into four groups: primed, primed‐infected, naive and naive‐infected for which 41 (43), 31 (55), 42 (42), 31 (55) paired RNA/ATAC (only RNA‐seq) samples were available, respectively. The data also contained metadata and peak coordinates. The paired samples were used to reconstruct the eGRNs with GRaNIE (see below). The unpaired RNA‐seq data were used for evaluation of the eGRNs with GRaNPA (see below).
Expression data for macrophages infected with Listeria & Salmonella (GEO accession number: GSE73502)
Pai et al (2016) generated expression data on cultured monocytes obtained from PBMCs of healthy donors, for which we downloaded the raw counts data. Matured macrophages were divided into three groups: (i) controls and infected by the (ii) Listeria and (iii) Salmonella bacteria, respectively. We used the RNA‐seq data collected 2 h after infection with Listeria and Salmonella, respectively, for each of the 57 samples.
Expression data for macrophages infected with Tuberculosis (GEO accession numbers: GSE133145, GSE143731)
Giraud‐Gatineau et al (2020) collected two data sets on the effect of bedaquiline (GSE133145) and five other drugs (GSE143731) treatment for Mycobacterium tuberculosis infection in Monocyte‐derived macrophages from healthy donors. The GSE133145 series consists of 16 control and 16 M. tuberculosis‐infected samples, which are later divided into four groups: untreated/DMSO treatment (control)/two variants of bedaquiline treatment (0.5 or 5 μg/ml). Differential expression analysis revealed that differences caused by treatment are not substantial, so we considered the treatment as a controlling variable. The GSE143731 series consists of 28 control and 28 M. tuberculosis‐infected samples, which are later divided into groups corresponding to the treatment with isoniazid (INH), rifampicin (RIF), ethambutol, pyrazinamide (PZA) or amikacin (AMK), and control group. We considered treatment as a controlling variable for the differential expression analysis.
Expression and chromatin accessibility data for CD4 + T‐cells
Paired RNA‐ and ATAC‐seq data were obtained from (Freimer et al, 2022). For RNA‐seq, processed count files were obtained from GSE171737. For ATAC‐seq, raw sequencing files were obtained from GSE171737 and processed and quality‐controlled with an in‐house Snakemake pipeline as previously described (Berest et al, 2019).
Expression data for resting versus LPS‐stimulated CD4 + T‐cells (GSE118165)
RNA‐seq was obtained from (Calderon et al, 2019), which measured expression in resting and stimulated subsets of CD4+ T‐cells. We used the T‐follicular helper cells for differential expression analyses.
Expression and chromatin accessibility for AML
We obtained raw RNA‐seq data for 23 AML patients from (Garg et al, 2019). Processed and quality‐controlled ATAC‐seq data and peaks for the same patients was obtained from (He et al, 2022).
Expression data for TF K/Os
We obtained cell‐type‐specific knockout (K/O) data for THP1‐derived macrophages (NFKB1) (Somma et al, 2021), the human AML cell line MV4‐11 (IRF8) (Liss et al, 2021), and processed differential expression data from primary human CD4+ T cells (IRF1 and IRF2) (Freimer et al, 2022). For the NFKB1 and IRF8 data sets, raw sequencing files were obtained from GSE162015 and GSE163275, respectively, and data processing and quality control was performed with an in‐house Snakemake pipeline as described previously (Berest et al, 2019).
Macrophage phosphoproteomics data
Processed quantitative phosphoproteomics data from polarising THP1‐derived macrophages was obtained from (He et al, 2022).
Differential expression analyses
Differential expression analysis was performed with DESeq2 (Love et al, 2014) for all data sets, typically using the contrast between treatment and no treatment or disease and control (see also Table 2). The design formula generally used was therefore “~condition,” unless otherwise stated. Dataset‐specific details of the differential expression analysis datasets are described below. As input for GRaNPA, we generally used shrunken log2 fold‐changes as implemented in lfcShrink from DESeq2 with the apeglm method (Zhu et al, 2019) unless otherwise indicated, even though it is not a strict requirement of GRaNPA to use any particular transformation.
iPSC‐derived macrophages infected with Salmonella from
Differential expression was calculated using only the RNA‐seq data that were not used for eGRN reconstruction (Alasoo et al 2018). We quantified differential expression for the following contrasts: naive versus infected, naive versus IFN‐γ primed, IFN‐γ primed versus IFN‐γ primed‐infected. The formula used in DESeq2 was “~condition.”
Macrophages infected with Listeria and Salmonella from
We analysed the differential expression between control samples, listeria‐infected samples and salmonella‐infected samples separately (Pai et al 2016). No samples were removed. Information on the donor was used as a covariate, using the design formula: “~patient + condition.”
Macrophages infected with Tuberculosis from
We calculated differential expression between monocyte‐derived macrophages from healthy donors infected with tuberculosis versus control samples (Giraud‐Gatineau et al 2020). Data sets GSE133145 and GSE143731 were analysed separately, but with a common design formula. Although there were also multiple treatments, the expression variance was almost exclusively driven by the difference in disease status. We therefore added the treatment as a covariate to the design formula (“~patient + treatment + condition”), but only investigated differential expression between infected macrophages and controls. One control and one infected sample from the GSE143731 series were removed from the analysis, as they were clear outliers in the PCA plot.
CD4 + follicular T‐cells resting versus LPS‐stimulated
We quantified differential expression between CD4‐positive follicular T‐cells in resting versus stimulated condition (Calderon et al, 2019). The design formula used in DESeq2 is “~condition.”
AML subtypes
Differential expression was calculated using data from (Garg et al, 2019) and comparing samples with high leukaemia stem cell burden (GPR56‐high) versus low leukaemia stem cell burden (GPR56‐low samples) based on immunophenotyping as defined in (Garg et al, 2019). The design formula was: “~GPR56status.” We did not use shrunken log fold‐changes as input for GRaNPA but we verified that results are qualitatively unchanged when doing so.
Tumour‐associated and tissue resident macrophages from human breast tissue
Raw RNA‐sequencing data were obtained from GSE117970 (Cassetta et al, 2019), and processed in the same way as described for expression data for TF K/Os. We obtained differentially expressed genes between tissue resident and tumour associated macrophages using the design formula “~condition.”
GRaNIE: Construction of eGRNs
The following is needed as input for GRaNIE:
Raw or prenormalised chromatin accessibility data (e.g. ATAC‐seq, DNase‐Seq or histone modification ChIP‐seq data such as H3K27ac);
Raw or prenormalised RNA‐seq counts;
Precompiled lists of TFBS predictions per TF (we provide predictions for human and mouse TFBS that were derived as described in Berest et al, 2019); and
TAD domains (optional).
For all data sets in this study, we used the same default parameters when constructing the eGRNs as described below.
GRaNIE is conceptually based on the procedure described in (Reyes‐Palomares et al, 2020) and has the following main steps:
(i) Process chromatin accessibility and RNA‐seq data
Both ATAC‐seq and RNA‐seq may be raw counts or prenormalised counts. If raw counts are provided for RNA‐seq, by default we quantile normalised the RNA‐Seq count data in order to minimise the effects of outlier values that may otherwise have a large influence on the resulting correlations. For chromatin accessibility data, we employ a size factor normalisation as implemented in DESeq2 (Love et al, 2014). However, the user can define which type of normalisation shall be used for either data. Additional filters for excluding particular chromosomes (e.g. sex chromosomes) or genes/peaks with low counts can optionally be used. The latter is implemented by removing genes/peaks if the average counts across all samples are below a specified threshold (5 by default).
(ii) Overlap TF binding sites with ATAC‐Seq peaks
Based on the provided list of putative TFBS per TF (see Berest et al, 2019 for details), we overlap all TFBS from all TF with the open chromatin peaks and record for each peak and TF whether at least one putative TFBS is located within the peak. This binary TF‐peak binding matrix is used in subsequent steps.
(iii) Identify statistically significant TF ‐ peak connections
To identify statistically significant TF‐peak connections, we implement a cell‐type‐specific data‐driven approach. In brief, we first calculate the Pearson's correlation coefficients between the expression level of each TF and the open chromatin signal of each peak across all samples.
We then use an empirical FDR procedure to identify statistically significant TF‐peak connections. For this, for each TF, we split the peaks into two sets: a foreground set containing the peaks with a predicted TFBS and a corresponding background set consisting of peaks without predicted TFBS based on the TF‐peak binding matrix calculated above. We then discretize the TF‐peak correlation r into 40 bins in steps of 0.05 ranging from −1, −0.95, …, 0, …, 1 and calculate a bin‐specific FDR value using two different directions (positive: left to right from −1 to 1, negative: right to left from 1 to −1). For each bin (correlation threshold) k, we calculate the empirical FDR according to the formula , with and denoting the total number of TF‐peaks in the background and foreground, respectively, for which (direction positive) and (direction negative). To make the numbers from foreground and background compatible, we normalise beforehand by their ratio (i.e. , with and denoting the total number of TF‐peaks in the foreground and background, respectively).
(iv) Activator‐repressor TF classification (optional)
Optionally, the TF classification as described in (Berest et al, 2019) can be run and is fully integrated in GRaNIE. It produces a classification of TFs into putative activators, repressors or undetermined. Briefly, it compares the distribution of correlations for peaks with putative binding sites (foreground) against all other peaks (i.e. background) and classifies TFs depending on whether the correlations of putative targets are significantly more positive than (activator), more negative than (repressor) or indistinguishable from (undetermined) the background.
(v) Identify statistically significant peak‐gene connections
Next, we add peak‐gene connections to our network. We identify highly correlated peak‐gene pairs based on their Pearson's correlation and the associated P‐value (using cor.test in R) between the normalised RNA‐seq for the expression of a gene and the corresponding open chromatin peak.
GRaNIE offers two options to decide which peak‐gene pairs to test for correlation: in absence of additional topologically associating domain (TADs) data from Hi‐C or similar approaches it uses a local neighbourhood‐based approach with a custom neighbourhood size (default: 250 kb up‐ and downstream of the peak) to select peak‐gene pairs to test. In the presence of TAD data, only peak‐gene pairs within a TAD are tested. The user has furthermore the choice to specify whether overlapping TADs should be merged or not. We offer two options of where in the gene the overlap with the extended peak may occur: at the 5′ end of the gene (the default) or anywhere in the gene.
GRaNIE also records additional properties for each peak‐gene pair such as their distance as well as gene type & status as annotated by Gencode. By default, only protein‐coding and lincRNA genes are kept in the eGRN, but this can be customised to include other gene types.
(vi) Filter GRN connections and calculate peak‐gene FDR
Lastly, we offer a variety of options to combine and filter TF‐peaks and peak‐genes to derive the full GRN for subsequent analyses. For example, both types of connections can be filtered by their FDRs or by their correlation, peak‐gene links can further be filtered by their distance, gene type, and other criteria. By default, we retain only peak‐gene pairs that are positively correlated. After applying all filters for the peak‐gene links, multiple testing adjustment is performed using Benjamini–Hochberg. The default thresholds for TF‐peak and peak‐gene links are FDR < 0.2 and FDR < 0.1, respectively.
Lastly, we provide heatmaps and boxplots that compare the connectivity for the real and random eGRNs.
GRaNIE quality controls
The package optionally offers PCA plots for both the RNA‐seq and open chromatin data, and upon availability of additional metadata that can be provided, the PCA data can also be coloured accordingly. This facilitates the detection of batch effects and outlier samples that may introduce unwanted variation.
In addition, we implemented a range of quality controls for the different steps to assess both the number as well as signal‐to‐noise for both TF‐enhancer and enhancer‐gene links.
For assessing TF‐enhancer links, we compare the number of links obtained from the real data to a background set of links that we obtain with randomised data using a twofold randomisation scheme (permuting the TF‐peak overlap matrix and sample labels for the RNA counts) by applying the same methods as described before.
For assessing the true enhancer‐gene links, we similarly construct a set of background links. First, after creating the real table for the peak‐gene pairs that fulfill the user‐specified requirements for being tested for correlation, the peaks are shuffled. Notably, this preserves the degree distribution for both peaks and genes. Second, we shuffle the sample labels for the RNA data.
We then base our quality controls on the assumption that peak accessibility and gene expression are positively correlated. Thus, assessing the amount of signal (i.e. small P‐values) of positive versus negative correlations in the real versus the background enhancer‐gene links serves as a proxy for the signal to noise ratio. Specifically, we expect that positive correlations outnumber negative ones for the real enhancer‐gene links, which should not be the case for the background. Lastly, we have a number of additional QC plots that include the enhancer‐gene distance, for which we similarly expect a signal difference for the real but not the background links.
GRaNIE: Downstream analyses implemented in the package
The following functionalities are available within the GRaNIE package:
Descriptive statistics pertaining to the structure of the graph, such as the number of nodes and edges and their types, the distribution of node degrees and the top nodes with regard to degree centrality and eigenvector centrality.
Community identification, for which the package supports multiple algorithms (louvain, walktrap, leading eigenvector, fast greedy and optimal). The communities can be supplemented with descriptive statistics similar to those previously described, but specific to each individual community.
Enrichment analyses in three different flavours; a general enrichment analysis for the whole network, a community‐based enrichment analysis, or a TF‐based enrichment analysis.
Molecular evaluation of GRaNIE‐inferred TF‐peak links using ChIP‐seq
We obtained macrophage‐specific ChIP‐seq data from ReMap 2022 (Hammal et al, 2022) for all TFs that were present in any of the GRaNIE inferred eGRNs (CEBPA, CEBPB, FOS, GABPA, GFI1, IRF8, IRF9, LYL1, MYB, NR1H3, PPARG, RUNX1, STAT1, STAT2, VDR). For these TFs we determined the overlap of GRaNIE inferred TF‐linked peaks (independent of whether they also are linked to a gene) with the respective ChIP‐seq peaks (within 50 bp) and calculated the enrichment over the background set of ATAC‐seq peaks that just contained the TF motif using Fisher's exact test. We excluded two TFs (SPI1 and CTCF) that had more than 10 k connections in GRaNIE at 0.4 or 0.5 FDR thresholds but no connections at lower FDR thresholds.
Molecular evaluation of GRaNIE‐inferred peak‐gene links using eQTL data
We downloaded cis‐eQTL data from the eQTL catalogue (https://www.ebi.ac.uk/eqtl/, accessed on May 5th 2022), selecting all six data sets with eQTLs in monocytes or macrophages. We combined the eQTLs from all data sets and filtered the associations to have a permutation‐based FDR < 0.3. Only GRN peaks that harbour at least one eQTL SNP in these data sets can be evaluated. For every peak, we overlapped the eQTL SNPs, and counted a peak‐gene link as validated if any eQTL SNP affected the same gene as present in the GRN. To test whether the overlap between peak‐gene links and eQTL is enriched as compared to a random background, we also validated links between the GRN peaks and randomly sampled distance‐matched genes based on 50 kb bins. We repeated the random background sampling 20 times and calculated the odds ratio between validated GRN links and validated background links. We calculated the enrichment of validated links by eQTL overlap for a range of peak‐gene FDR thresholds in each of the four macrophage GRNs. We did not add any extra eQTLs from SNPs that were in high linkage disequilibrium with the eQTLs from the six data sets in our link validation. This means we could have missed some eQTL overlaps, but since we used eQTLs from six different datasets, we did include several variants per peak.
Molecular evaluation of GRaNIE‐inferred TF‐peak‐gene links
We obtained the tracks of human enhancers identified using CAGE data from FANTOM5 that were differentially expressed in T‐cells and macrophages (Andersson et al, 2014). Track coordinates were translated to GRCh38 using UCSC liftOver tool (Hinrichs et al, 2006). We quantified the overlap of GRaNIE eGRN enhancers (peaks linked to a TF and a gene) and the respective cell‐type specific (macrophage in Figs 1F and 2H and T‐cell in Fig 2H) CAGE peaks and tested for the association of eGRN peaks (vs. all other peaks within 250 kb of a TSS) and cell‐type‐specific CAGE enhancers using Fisher's exact test.
Utility evaluation of GRaNIE‐inferred eGRN regulons using GO‐term specificity
To assess the specificity of the top GO terms enriched for the predictive TFs (Appendix Fig S9A), we randomised the TF‐gene connections and recalculated the enrichment. Manual inspection suggested that the returned terms for the random network are more general and often unrelated to macrophage biology. To quantify the specificity, we plotted the distribution of the number of genes annotated to each returned GO term for the real network, and for five permuted networks (Appendix Fig S9B), following the rationale that more general terms would have more genes associated with them in the database.
GRaNPA: Prediction model
For the prediction model we do the following:
Compute the differential expression resulting from a perturbation between two different conditions, for which we used DESeq2. We define DE(j) as the differential expression value for the jth Gene.
Define the GRN as a mathematical function:
Based on this, we construct a matrix , a relation matrix between genes and TFs where each row is a gene and each column is a TFs. There is a 1 in the cell for a gene and TF if and only if they are connected through the GRN.
-
iiiRunning a Random Forest (RF) Regression based on the following formula to predict a differential expression value for each gene i based on its connected TFs :
Random Forest has been implemented using the “ranger” package in R. To avoid overfitting we use 10‐fold cross validation; no hyper‐parameter tuning on the random forest was performed. We measure performance by assessing the cross‐validation R 2.
GRaNPA: Construction of the permuted network to assess edge‐specificity
To assess the edge‐specificity of our network during the random forest regression, we constructed a permuted control network based on the structure of the actual GRN. It has the same number of edges with the same degree distribution for TFs with only the gene labels permutated so the connection between TFs and genes are randomised. The differential expression values are unchanged. In case we use any weighing method for the edges, the same method will be applied for each edge of the permuted version to recalculate weight if needed.
GRaNPA: Random signal generation to assess overfitting
To control for over‐fitting in the random forest regression, we used the real network structure and assigned randomly generated values to genes instead of their differential expression value. The random values are chosen from a uniform distribution with minimum and maximum of −5 and 5, respectively. Any prediction for this network is a consequence of overfitting. The reason we used a uniform distribution is that we want to check overfitting and we do not want the distribution to be like any differential expression distribution.
GRaNPA: Calculating variable importance measures
We used the permutation approach to measure variable importance using the “ranger” package in R. This accuracy‐based approach uses the out‐of‐bag sample to calculate the importance of a specific variable. The importance is based on the difference in the prediction accuracy of out‐of‐bag sample and the prediction accuracy of out‐of‐bag sample while its variables have been randomly shuffled while all other variables kept the same.
GRN benchmarking against other networks/tools
Dorothea (Garcia‐Alonso et al, 2019; Holland et al, 2020a, 2020b): a resource containing TF‐target interactions. The connections are tagged by a confidence level based on the number of supporting evidence. The confidence levels range from A: highly reliable to B–D: curated and/or ChIP‐seq interactions to the lowest confidence level E: only computational support.
TRRUST (Han et al, 2018): a database of transcriptional regulatory networks for humans and mice. It has been constructed using text‐mining followed by manual curation. TRRUST v2 regulatory network for humans contains 795 TFs, 2,067 genes and 8,427 regulatory links. We did not use any weighing for the edges as it was not provided by TRRUST v2.
ChEA3 (Keenan et al, 2019): TF‐target gene libraries containing targets determined by ChIP‐seq experiments from ENCODE, ReMap and publications. It also contains co‐expression connections based on RNA‐seq data from resources like GREx and ARCHs4.
ANANSE (Xu et al, 2021): an enhancer‐based cell‐type‐specific network that can predict key transcription factors in cell fate determination. We used ANANSE macrophage‐specific network filtered by 0.8 probability for its links.
Assessing the cell‐type specificity of network models
To assess the cell‐type specificity of network models, we checked their performance on DE data from other cell‐types. In this analysis, we used naive macrophages, naive T‐cell and AML and evaluated their performance for DE data from GPR56‐positive versus GPR56‐negative AML, resting versus stimulated CD4‐positive follicular T‐cell (in 10 different subcell‐types) and naive‐macrophages versus 5‐h infected with Salmonella. We filtered genes using 0.1 adjusted P‐value and 1 absolute log fold change thresholds.
Enrichment of regulons among TF K/O data
We obtained cell‐type‐specific knockout (K/O) data for NFKB1 in THP1‐derived macrophages (Somma et al, 2021), for IRF8 in the human AML cell line MV4‐11 (Liss et al, 2021), and for IRF1 and IRF2 in primary human CD4+ T cells (Freimer et al, 2022). Differential expression analysis was performed using DESeq2 (Love et al, 2014), comparing all K/O conditions to their nontargeting controls. All genes significantly downregulated upon TF K/O (adjusted P‐value < 0.05 and fold‐change < 0), were considered as TF K/O affected genes. For each TF, we then quantified the enrichment of TF K/O affected genes among GRaNIE‐inferred target genes in the respective cell type using Fisher's exact test as implemented in the GeneOverlap package in R. Enrichments with at least five overlapping genes and Fisher's exact P‐value < 0.05 were considered significant.
Visualisation (Shiny App)
We provide a web application based on a Shiny App for interactive visualisation of the eGRNs for different cell types (https://apps.embl.de/grn/).
Gene set enrichment analysis
Preranked gene set enrichment analysis (GSEA, ranking based on log2 fold‐change) was performed using the Bioconductor/R package fgsea (preprint: Korotkevich et al, 2016). The M1 macrophage signature gene set was obtained from (Orecchioni et al, 2019).
GWAS enrichment
We tested whether the macrophage GRNs were enriched for genetic heritability of GWAS traits using stratified linkage disequilibrium score regression (S‐LDSC) (Finucane et al, 2015). 806 GWAS summary statistics that included participants of European descent were downloaded from the GWAS Catalogue (Buniello et al, 2019), harmonised and converted to the LDSC format as described on the LDSC Github repository (https://github.com/bulik/ldsc). We removed summary statistics with fewer than 10,000 individuals and fewer than 100,000 SNPs, because those studies were likely underpowered, leaving 442 traits. We created peak sets for each of the three macrophage GRNs by extracting the enhancer regions that were present in the GRNs after filtering for peak‐gene distance, TF‐peak false discovery rate (FDR) and gene differential expression effect size. We used 54 sets of general genomic features (downloaded from https://alkesgroup.broadinstitute.org/LDSCORE/, following Finucane et al, 2018) and a peak set based on all macrophage enhancers within 250 kb of genes as background regions. Adding these regions as a background ensures that the identified enrichments are not due to the general enrichment of heritability in (macrophage) enhancers near genes, but specifically in the enhancers that are part of the GRNs. We calculated the heritability enrichment P‐values and corrected them for multiple testing within each trait.
GRaNIE: GO enrichment of GRNs
The general enrichment analysis was run using topGO (v2.42.0) as part of the standard GRaNIE workflow, with the foreground being the genes in the filtered GRN, and the background being the genes within a predefined 250 kb neighbourhood of the peaks in the GRN. In more specific enrichment analyses such as those for the top transcription factors, which are ranked by their predictive capacity, the foreground is selected based on the genes a given TF is connected to within the network. Similarly, in community‐based enrichment analyses, the foreground is simply the genes that are classified to a given community. To calculate the enrichment, a Fisher test is used alongside the weight01 algorithm, which is a mixture of the “elim” and the “weight” algorithms introduced by (Alexa et al, 2006), to account for the GO hierarchy. Additionally, terms with less than 4 significantly annotated genes were omitted from the results in the figures. For the sake of better visualising the enriched terms, the figures are limited to the top 10 enriched terms per category. The full list of enriched terms can be found in Datasets EV7 and EV8.
Identifying targets for fine‐mapped GWAS‐SNPs using eGRNs
Fine‐mapped GWAS variants for autoimmune diseases were generated using probabilistic identification of causal SNPs (PICS) algorithm. These variants were downloaded from (i) previously published list and lifted to the hg38 build (Farh et al, 2015) and (ii) data portal under the filename “PICS2‐GWAScat‐2020‐05‐22.txt.gz” from https://pics2.ucsf.edu for the hg38 build. We kept all variants with a PICS probability of greater than 50%. We identified their target genes by overlapping these variants with the peaks from different macrophage eGRNs and then using the peak‐gene links from the respective eGRNs to assign the target genes (Fig 5C). The full list can be found in Table 3.
Author contributions
Aryan Kamal: Conceptualization; data curation; software; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Christian Arnold: Conceptualization; data curation; software; formal analysis; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Annique Claringbould: Conceptualization; data curation; formal analysis; validation; investigation; visualization; writing – original draft; writing – review and editing. Nila H Servaas: Data curation; formal analysis; validation; investigation; visualization. Rim Moussa: Data curation; software; formal analysis; methodology. Maksim Kholmatov: Data curation; formal analysis; validation; visualization. Neha Daga: Data curation; formal analysis; validation; investigation; visualization. Daria Nogina: Data curation; formal analysis. Sophia Mueller‐Dott: Conceptualization; data curation; software; formal analysis; investigation; methodology. Armando Reyes‐Palomares: Conceptualization; methodology. Giovanni Palla: Conceptualization. Olga Sigalova: Data curation; formal analysis. Daria Bunina: Conceptualization. Caroline Pabst: Resources. Judith B Zaugg: Conceptualization; software; supervision; funding acquisition; investigation; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
We declare that none of the authors have any competing interests. JBZ is an editorial advisory board member. This has no bearing on the editorial consideration of this article for publication.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Dataset EV9
Dataset EV10
Acknowledgements
We thank Anna Mathioudaki for proofreading and for naming GRaNIE and GRaNPA, Martina Muckenthaler, Oriana Marques and Christina Mertens for discussions about macrophage biology. MK is funded by the EC H2020 MSCA‐ITN Project ENHPATHY, grant agreement number 860002. AC is funded by an EIPOD fellowship from the EC Horizon 2020 MSCA, grant agreement number 847543. ARP has been granted by the Regional Programme of Research and Technological Innovation for Young Doctors UCM‐CAM (PR65/19‐22460). CA is co‐funded by the European Union (ERC, epiNicheAML, 101044873). Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. Open Access funding enabled and organized by Projekt DEAL.
Mol Syst Biol. (2023) 19: e11627
Data availability
The methods implementation codes are available in the following GitLab repositories: (i) GRaNIE: https://git.embl.de/grp‐zaugg/GRaNIE; (ii) GRaNPA: https://git.embl.de/grp‐zaugg/GRaNPA.
References
- Aibar S, González‐Blas CB, Moerman T, Huynh‐Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J‐C, Geurts P, Aerts J et al (2017) SCENIC: single‐cell regulatory network inference and clustering. Nat Methods 14: 1083–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alasoo K, Rodrigues J, Mukhopadhyay S, Knights AJ, Mann AL, Kundu K, HIPSCI Consortium , Hale C, Dougan G, Gaffney DJ (2018) Shared genetic effects on chromatin and gene expression indicate a role for enhancer priming in immune response. Nat Genet 50: 424–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexa A, Rahnenführer J, Lengauer T (2006) Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22: 1600–1607 [DOI] [PubMed] [Google Scholar]
- An Y, Ni Y, Xu Z, Shi S, He J, Liu Y, Deng K‐Y, Fu M, Jiang M, Xin H‐B (2020) TRIM59 expression is regulated by Sp1 and Nrf1 in LPS‐activated macrophages through JNK signaling pathway. Cell Signal 67: 109522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson R, Gebhard C, Miguel‐Escalada I, Hoof I, Bornholdt J, Boyd M, Chen Y, Zhao X, Schmidl C, Suzuki T et al (2014) An atlas of active enhancers across human cell types and tissues. Nature 507: 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreou N‐P, Legaki E, Gazouli M (2020) Inflammatory bowel disease pathobiology: the role of the interferon signature. Ann Gastroenterol 33: 125–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berest I, Arnold C, Reyes‐Palomares A, Palla G, Rasmussen KD, Giles H, Bruch P‐M, Huber W, Dietrich S, Helin K et al (2019) Quantification of differential transcription factor activity and multiomics‐based classification into activators and repressors: diffTF. Cell Rep 29: 3147–3159 [DOI] [PubMed] [Google Scholar]
- Blondel VD, Guillaume J‐L, Lambiotte R, Lefebvre E (2008) Fast unfolding of communities in large networks. J Stat Mech 2008: P10008 [Google Scholar]
- Buhling F, Kellner U, Guenther D, Kahl S, Brömme D, Weber E, Malfertheiner P, Wex T (2002) Characterization of novel anti‐cathepsin W antibodies and cellular distribution of cathepsin W in the gastrointestinal tract. Biol Chem 383: 1285–1289 [DOI] [PubMed] [Google Scholar]
- Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E et al (2019) The NHGRI‐EBI GWAS catalog of published genome‐wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res 47: D1005–D1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunina D, Abazova N, Diaz N, Noh K‐M, Krijgsveld J, Zaugg JB (2020) Genomic rewiring of SOX2 chromatin interaction network during differentiation of ESCs to postmitotic neurons. Cell Syst 10: 480–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunina D, Germain P‐L, Lopez Tobon A, Fernandez‐Novel Marx N, Arnold C, Ó hEachteirn A, Claringbould A, Lai MC, Rangasamy S, Narayanan V et al (2021) Pathological LSD1 mutations cause HDAC‐mediated aberrant gene repression during early cell differentiation. bioRxiv 10.1101/2021.08.11.455900 [PREPRINT] [DOI]
- Calderon D, Nguyen MLT, Mezger A, Kathiria A, Müller F, Nguyen V, Lescano N, Wu B, Trombetta J, Ribado JV et al (2019) Landscape of stimulation‐responsive chromatin across diverse human immune cells. Nat Genet 51: 1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, Soong DYH, Cotechini T, Anur P, Lin EY et al (2019) Human tumor‐associated macrophage and monocyte transcriptional landscapes reveal cancer‐specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell 35: 588–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TE, Stumpf MPH, Babtie AC (2017) Gene regulatory network inference from single‐cell data using multivariate information measures. Cell Syst 5: 251–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Mar JC (2018) Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinformatics 19: 232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Jiao Y, Liu L, Huang M, He C, He W, Hou J, Yang M, Luo X, Li C (2020) Communications between bone marrow macrophages and bone cells in bone remodeling. Front Cell Dev Biol 8: 598263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN, Bobryshev YV (2018) The impact of interferon‐regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology 223: 101–111 [DOI] [PubMed] [Google Scholar]
- Claringbould A, Zaugg JB (2021) Enhancers in disease: molecular basis and emerging treatment strategies. Trends Mol Med 27: 1060–1073 [DOI] [PubMed] [Google Scholar]
- Corliss BA, Azimi MS, Munson JM, Peirce SM, Murfee WL (2016) Macrophages: an inflammatory link between angiogenesis and lymphangiogenesis. Microcirculation 23: 95–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domcke S, Bardet AF, Adrian Ginno P, Hartl D, Burger L, Schübeler D (2015) Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 528: 575–579 [DOI] [PubMed] [Google Scholar]
- Farh KK‐H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJH, Shishkin AA et al (2015) Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518: 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festa RA, Thiele DJ (2012) Copper at the front line of the host‐pathogen battle. PLoS Pathog 8: e1002887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK, Bulik‐Sullivan B, Gusev A, Trynka G, Reshef Y, Loh P‐R, Anttila V, Xu H, Zang C, Farh K et al (2015) Partitioning heritability by functional annotation using genome‐wide association summary statistics. Nat Genet 47: 1228–1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, Gazal S, Loh P‐R, Lareau C, Shoresh N et al (2018) Heritability enrichment of specifically expressed genes identifies disease‐relevant tissues and cell types. Nat Genet 50: 621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck JS, Jansen SMJ, Wollny D, Zenk F, Seimiya M, Jain A, Okamoto R, Santel M, He Z, Camp JG et al (2022) Inferring and perturbing cell fate regulomes in human brain organoids. Nature 10.1038/s41586-022-05279-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freimer JW, Shaked O, Naqvi S, Sinnott‐Armstrong N, Kathiria A, Garrido CM, Chen AF, Cortez JT, Greenleaf WJ, Pritchard JK et al (2022) Systematic discovery and perturbation of regulatory genes in human T cells reveals the architecture of immune networks. Nat Genet 54: 1133–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Nasser J, Jones TR, Munson G, Bergman DT, Subramanian V, Grossman SR, Anyoha R, Doughty BR, Patwardhan TA et al (2019) Activity‐by‐contact model of enhancer‐promoter regulation from thousands of CRISPR perturbations. Nat Genet 51: 1664–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Alonso L, Holland CH, Ibrahim MM, Turei D, Saez‐Rodriguez J (2019) Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res 29: 1363–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S, Reyes‐Palomares A, He L, Bergeron A, Lavallée V‐P, Lemieux S, Gendron P, Rohde C, Xia J, Jagdhane P (2019) Hepatic leukemia factor is a novel leukemic stem cell regulator in DNMT3A, NPM1, and FLT3‐ITD triple‐mutated AML. Blood 134: 263–276 [DOI] [PubMed] [Google Scholar]
- Giraud‐Gatineau A, Coya JM, Maure A, Biton A, Thomson M, Bernard EM, Marrec J, Gutierrez MG, Larrouy‐Maumus G, Brosch R et al (2020) The antibiotic bedaquiline activates host macrophage innate immune resistance to bacterial infection. Elife 9: e55692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Blas CB, De Winter S, Hulselmans G, Hecker N, Matetovici I, Christiaens V, Poovathingal S, Wouters J, Aibar S, Aerts S (2022) SCENIC+: single‐cell multiomic inference of enhancers and gene regulatory networks. bioRxiv 10.1101/2022.08.19.504505 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriadis G, Zhan Y, Grumont RJ, Metcalf D, Handman E, Cheers C, Gerondakis S (1996) The Rel subunit of NF‐kappaB‐like transcription factors is a positive and negative regulator of macrophage gene expression: distinct roles for Rel in different macrophage populations. EMBO J 15: 7099–7107 [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Walsh AM, Fearon U, Smith MD, Wechalekar MD, Yin X, Cole S, Orr C, McGarry T, Canavan M et al (2017) CD40L‐dependent pathway is active at various stages of rheumatoid arthritis disease progression. J Immunol 198: 4490–4501 [DOI] [PubMed] [Google Scholar]
- Hainer SJ, McCannell KN, Yu J, Ee L‐S, Zhu LJ, Rando OJ, Fazzio TG (2016) DNA methylation directs genomic localization of Mbd2 and Mbd3 in embryonic stem cells. Elife 5: e21964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammal F, de Langen P, Bergon A, Lopez F, Ballester B (2022) ReMap 2022: a database of human, mouse, drosophila and Arabidopsis regulatory regions from an integrative analysis of DNA‐binding sequencing experiments. Nucleic Acids Res 50: D316–D325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Cho J‐W, Lee S, Yun A, Kim H, Bae D, Yang S, Kim CY, Lee M, Kim E et al (2018) TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res 46: D380–D386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harigai M, Kawamoto M, Hara M, Kubota T, Kamatani N, Miyasaka N (2008) Excessive production of IFN‐gamma in patients with systemic lupus erythematosus and its contribution to induction of B lymphocyte stimulator/B cell‐activating factor/TNF ligand superfamily‐13B. J Immunol 181: 2211–2219 [DOI] [PubMed] [Google Scholar]
- Haynes BC, Maier EJ, Kramer MH, Wang PI, Brown H, Brent MR (2013) Mapping functional transcription factor networks from gene expression data. Genome Res 23: 1319–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Jhong J‐H, Chen Q, Huang K‐Y, Strittmatter K, Kreuzer J, Deran M, Wu X, Lee T‐Y, Slavov N et al (2021) Global characterization of macrophage polarization mechanisms and identification of M2‐type polarization inhibitors. Cell Rep 37: 109955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Arnold C, Thoma J, Rohde C, Kholmatov M, Garg S, Hsiao C‐C, Viol L, Zhang K, Sun R et al (2022) CDK7/12/13 inhibition targets an oscillating leukemia stem cell network and synergizes with venetoclax in acute myeloid leukemia. EMBO Mol Med 14: e14990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F et al (2006) The UCSC genome browser database: update 2006. Nucleic Acids Res 34: D590–D598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata Y, Kiuchi K, Chen HC, Milbrandt J, Guroff G (1993) The phosphorylation and DNA binding of the DNA‐binding domain of the orphan nuclear receptor NGFI‐B. J Biol Chem 268: 24808–24812 [PubMed] [Google Scholar]
- Holland CH, Szalai B, Saez‐Rodriguez J (2020a) Transfer of regulatory knowledge from human to mouse for functional genomics analysis. Biochim Biophys Acta Gene Regul Mech 1863: 194431 [DOI] [PubMed] [Google Scholar]
- Holland CH, Tanevski J, Perales‐Patón J, Gleixner J, Kumar MP, Mereu E, Joughin BA, Stegle O, Lauffenburger DA, Heyn H et al (2020b) Robustness and applicability of transcription factor and pathway analysis tools on single‐cell RNA‐seq data. Genome Biol 21: 36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh‐Thu VA, Geurts P (2018) dynGENIE3: dynamical GENIE3 for the inference of gene networks from time series expression data. Sci Rep 8: 3384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh‐Thu VA, Irrthum A, Wehenkel L, Geurts P (2010) Inferring regulatory networks from expression data using tree‐based methods. PLoS One 5: e12776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibarra IL, Hollmann NM, Klaus B, Augsten S, Velten B, Hennig J, Zaugg JB (2020) Mechanistic insights into transcription factor cooperativity and its impact on protein‐phenotype interactions. Nat Commun 11: 124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens J, Aibar S, Taskiran II, Ismail JN, Spanier KI, Bravo Gonzalez‐Blas C, Quan XJ, Papasokrati D, Hulselmans G, Makhzami S et al (2021) Decoding gene regulation in the fly brain. bioRxiv 10.1038/s41586-021-04262-z [PREPRINT] [DOI] [PubMed]
- Jones G‐R, Brown SL, Phythian‐Adams AT, Ivens AC, Cook PC, MacDonald AS (2020) The methyl‐CpG‐binding protein Mbd2 regulates susceptibility to experimental colitis via control of CD11c+ cells and colonic epithelium. Front Immunol 11: 183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimoto K, Hoffmann CM, Morris SA (2020) CellOracle: dissecting cell identity via network inference and in silico gene perturbation. bioRxiv 10.1101/2020.02.17.947416 [PREPRINT] [DOI] [PMC free article] [PubMed]
- Karnuta JM, Scacheri PC (2018) Enhancers: bridging the gap between gene control and human disease. Hum Mol Genet 27: R219–R227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M (2020) New insights into IFN‐γ in rheumatoid arthritis: role in the era of JAK inhibitors. Immunol Med 43: 72–78 [DOI] [PubMed] [Google Scholar]
- Keenan AB, Torre D, Lachmann A, Leong AK, Wojciechowicz ML, Utti V, Jagodnik KM, Kropiwnicki E, Wang Z, Ma'ayan A (2019) ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res 47: W212–W224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, Sergushichev A (2016) Fast gene set enrichment analysis. bioRxiv 10.1101/060012 [PREPRINT] [DOI]
- Langlais D, Barreiro LB, Gros P (2016) The macrophage IRF8/IRF1 regulome is required for protection against infections and is associated with chronic inflammation. J Exp Med 213: 585–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leseigneur C, Lê‐Bury P, Pizarro‐Cerdá J, Dussurget O (2020) Emerging evasion mechanisms of macrophage defenses by pathogenic bacteria. Front Cell Infect Microbiol 10: 577559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer‐Fogy I, Jeppesen P, Klein F, Bird A (1992) Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69: 905–914 [DOI] [PubMed] [Google Scholar]
- Liss F, Frech M, Wang Y, Giel G, Fischer S, Simon C, Weber LM, Nist A, Stiewe T, Neubauer A et al (2021) IRF8 is an AML‐specific susceptibility factor that regulates signaling pathways and proliferation of AML cells. Cancers (Basel) 13: 764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z‐P, Wu C, Miao H, Wu H (2015) RegNetwork: an integrated database of transcriptional and post‐transcriptional regulatory networks in human and mouse. Database (Oxford) 2015: bav095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Xia Y, Yang Q, Zhao Y (2019) The contribution of macrophages to systemic lupus erythematosus. Clin Immunol 207: 1–9 [DOI] [PubMed] [Google Scholar]
- Ma S, Zhang B, LaFave LM, Earl AS, Chiang Z, Hu Y, Ding J, Brack A, Kartha VK, Tay T et al (2020) Chromatin potential identified by shared single‐cell profiling of RNA and chromatin. Cell 183: 1103–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D, Lamparter D, Quon G, Kellis M, Kutalik Z, Bergmann S (2016) Tissue‐specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat Methods 13: 366–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medzhitov R, Horng T (2009) Transcriptional control of the inflammatory response. Nat Rev Immunol 9: 692–703 [DOI] [PubMed] [Google Scholar]
- Meng A, Zhang X, Shi Y (2014) Role of p38 MAPK and STAT3 in lipopolysaccharide‐stimulated mouse alveolar macrophages. Exp Ther Med 8: 1772–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerman T, Aibar Santos S, Bravo González‐Blas C, Simm J, Moreau Y, Aerts J, Aerts S (2019) GRNBoost2 and Arboreto: efficient and scalable inference of gene regulatory networks. Bioinformatics 35: 2159–2161 [DOI] [PubMed] [Google Scholar]
- Morishita H, Saito F, Kayama H, Atarashi K, Kuwata H, Yamamoto M, Takeda K (2009) Fra‐1 negatively regulates lipopolysaccharide‐mediated inflammatory responses. Int Immunol 21: 457–465 [DOI] [PubMed] [Google Scholar]
- Murray PJ (2017) Macrophage polarization. Annu Rev Physiol 79: 541–566 [DOI] [PubMed] [Google Scholar]
- Nasser J, Bergman DT, Fulco CP, Guckelberger P, Doughty BR, Patwardhan TA, Jones TR, Nguyen TH, Ulirsch JC, Lekschas F et al (2021) Genome‐wide enhancer maps link risk variants to disease genes. Nature 593: 238–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan CF, Murray HW, Wiebe ME, Rubin BY (1983) Identification of interferon‐gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med 158: 670–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novikova G, Kapoor M, Tcw J, Abud EM, Efthymiou AG, Chen SX, Cheng H, Fullard JF, Bendl J, Liu Y et al (2021) Integration of Alzheimer's disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat Commun 12: 1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orecchioni M, Ghosheh Y, Pramod AB, Ley K (2019) Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS‐) vs. alternatively activated macrophages. Front Immunol 10: 1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouma WZ, Pogacar K, Grotewold E (2018) Topological and statistical analyses of gene regulatory networks reveal unifying yet quantitatively different emergent properties. PLoS Comput Biol 14: e1006098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai AA, Baharian G, Pagé Sabourin A, Brinkworth JF, Nédélec Y, Foley JW, Grenier J‐C, Siddle KJ, Dumaine A, Yotova V et al (2016) Widespread shortening of 3′ untranslated regions and increased exon inclusion are evolutionarily conserved features of innate immune responses to infection. PLoS Genet 12: e1006338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parameswaran N, Patial S (2010) Tumor necrosis factor‐α signaling in macrophages. Crit Rev Eukaryot Gene Expr 20: 87–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires D, Marques J, Pombo JP, Carmo N, Bettencourt P, Neyrolles O, Lugo‐Villarino G, Anes E (2016) Role of cathepsins in mycobacterium tuberculosis survival in human macrophages. Sci Rep 6: 32247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliner HA, Packer JS, McFaline‐Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, Srivatsan S, Qiu X, Jackson D, Minkina A et al (2018) Cicero predicts cis‐regulatory DNA interactions from single‐cell chromatin accessibility data. Mol Cell 71: 858–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratapa A, Jalihal AP, Law JN, Bharadwaj A, Murali TM (2020) Benchmarking algorithms for gene regulatory network inference from single‐cell transcriptomic data. Nat Methods 17: 147–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Scicluna BP, van der Poll T (2021) The role of host cell DNA methylation in the immune response to bacterial infection. Front Immunol 12: 696280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes‐Palomares A, Gu M, Grubert F, Berest I, Sa S, Kasowski M, Arnold C, Shuai M, Srivas R, Miao S et al (2020) Remodeling of active endothelial enhancers is associated with aberrant gene‐regulatory networks in pulmonary arterial hypertension. Nat Commun 11: 1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium , Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi‐Moussavi A, Kheradpour P, Zhang Z, Wang J et al (2015) Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rönnblom L, Leonard D (2019) Interferon pathway in SLE: one key to unlocking the mystery of the disease. Lupus Sci Med 6: e000270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraivogel D, Gschwind AR, Milbank JH, Leonce DR, Jakob P, Mathur L, Korbel JO, Merten CA, Velten L, Steinmetz LM (2020) Targeted perturb‐seq enables genome‐scale genetic screens in single cells. Nat Methods 17: 629–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigalova OM, Shaeiri A, Forneris M, Furlong EE, Zaugg JB (2020) Predictive features of gene expression variation reveal mechanistic link with differential expression. Mol Syst Biol 16: e9539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somma D, Kok FO, Kerrigan D, Wells CA, Carmody RJ (2021) Defining the role of nuclear factor (NF)‐κB p105 subunit in human macrophage by transcriptomic analysis of NFKB1 knockout THP1 cells. Front Immunol 12: 669906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford SL, Bokil NJ, Achard MES, Kapetanovic R, Schembri MA, McEwan AG, Sweet MJ (2013) Metal ions in macrophage antimicrobial pathways: emerging roles for zinc and copper. Biosci Rep 33: e00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udalova IA, Mantovani A, Feldmann M (2016) Macrophage heterogeneity in the context of rheumatoid arthritis. Nat Rev Rheumatol 12: 472–485 [DOI] [PubMed] [Google Scholar]
- Võsa U, Claringbould A, Westra H‐J, Bonder MJ, Deelen P, Zeng B, Kirsten H, Saha A, Kreuzhuber R, Yazar S et al (2021) Large‐scale cis‐ and trans‐eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat Genet 53: 1300–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang L, Wu G‐R, Zhou Q, Yue H, Rao L‐Z, Yuan T, Mo B, Wang F‐X, Chen L‐M et al (2021) MBD2 serves as a viable target against pulmonary fibrosis by inhibiting macrophage M2 program. Sci Adv 7: eabb6075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waszak SM, Delaneau O, Gschwind AR, Kilpinen H, Raghav SK, Witwicki RM, Orioli A, Wiederkehr M, Panousis NI, Yurovsky A et al (2015) Population variation and genetic control of modular chromatin architecture in humans. Cell 162: 1039–1050 [DOI] [PubMed] [Google Scholar]
- Weber J, Bao H, Hartlmüller C, Wang Z, Windhager A, Janowski R, Madl T, Jin P, Niessing D (2016) Structural basis of nucleic‐acid recognition and double‐strand unwinding by the essential neuronal protein Pur‐alpha. Elife 5: e11297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemüller P, Kholmatov M, Petsalaki E, Zaugg JB (2021) Transcription factors: bridge between cell signaling and gene regulation. Proteomics 21: e2000034 [DOI] [PubMed] [Google Scholar]
- Xu Q, Georgiou G, Frölich S, van der Sande M, Veenstra GJC, Zhou H, van Heeringen SJ (2021) ANANSE: an enhancer network‐based computational approach for predicting key transcription factors in cell fate determination. Nucleic Acids Res 49: 7966–7985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue T, Sun F, Wang F, Yang C, Luo J, Rong S, Zhou H, Xiao J, Wang X, Zhou Q et al (2021) MBD2 acts as a repressor to maintain the homeostasis of the Th1 program in type 1 diabetes by regulating the STAT1‐IFN‐γ axis. Cell Death Differ 29: 218–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J (2020) Seven myths of how transcription factors read the cis‐regulatory code. Curr Opin Syst Biol 23: 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C‐N, Mao Y‐M, Liu L‐N, Li X‐M, Wang D‐G, Pan H‐F (2018) Emerging role of lncRNAs in systemic lupus erythematosus. Biomedicine & Pharmacotherapy 106: 584–592 [DOI] [PubMed] [Google Scholar]
- Zhu A, Ibrahim JG, Love MI (2019) Heavy‐tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35: 2084–2092 [DOI] [PMC free article] [PubMed] [Google Scholar]