

Abstract
The brain is a highly energy-demanding organ and requires bioenergetics adaptability to balance normal activity with pathophysiologic fueling of spontaneous recurrent seizures, the hallmark feature of the epilepsies. For decades, it has been accepted that recurrent or prolonged seizures can permanently alter neuronal circuitry and cause excitotoxic injury and aberrant inflammation. Further, pathological changes in bioenergetics and metabolism have been considered downstream consequences of epileptic seizures that may begin at the synaptic level. Less appreciated is the fact that primary derangements in cellular or mitochondrial metabolism, in reverse fashion, can result in seizure genesis, and lead to spontaneous recurrent seizures. Increasingly, however, basic-translational research has demonstrated that the relationships between brain metabolism and epileptic seizures are both complex, bidirectional and produce a vicious cycle that compounds the deleterious consequences. From a therapeutic standpoint, metabolism-based treatments such as the high-fat, anti-seizure ketogenic diet have become mainstream, and metabolic substrates and enzymes (such as ketone bodies, certain fatty acids and adenosine) have become attractive molecular targets to prevent seizures and to aid in recovery processes. Moreover, given that metabolism is critical for epigenetic as well as inflammatory changes, the thesis that epileptogenesis – the molecular and cellular processes that produce a chronic epileptic brain – can be both negatively and positively influenced by metabolic changes is rapidly gaining ground. Here, we present the current evidence supporting the view that brain metabolism is both a pathophysiological and therapeutic paradigm for epilepsy.
Keywords: epilepsy, seizure, metabolism, astrocyte, ketogenic diet, fatty acids, adenosine, glucose, ketone bodies, mitochondria, epileptogenesis
Epilepsy is a common neurological disorder that occurs throughout the age-span and has a worldwide prevalence of approximately 1% of the general population1. Spontaneous recurrent seizures (SRS), the defining hallmark of epilepsy, have been linked to increased neuronal excitability and hypersynchrony from various causes – genetic and secondary brain insults, modified further by environmental influences1. Irrespective of the etiology, there is a high energy need to prevent the generation of seizures (ictogenesis), to provide energy to sustain prolonged seizure activity, and to recover from seizures and repair damage. Indeed, without sufficient ATP – the universal currency of life and mostly produced by mitochondria2 – it would be impossible to regenerate membrane potentials needed to enable epileptiform activity 3. Therefore, metabolic treatments such as a ketogenic diet, which increase fuel to the brain in the form of ketone bodies are uniquely suited to prevent seizures and to aid in recovery after a seizure.
In this light, it has long been understood that SRS and prolonged seizures such as status epilepticus induce secondary aberrations in cellular bioenergetics and metabolism4,5. These include glutamate- and free radical-mediated injury, neuroinflammation6–8, and widespread disruptions in brain homeostatic mechanisms. However, less appreciated is the fact that inherent defects in brain energy metabolism, whether through mutations in genes encoding mitochondrial proteins or metabolic substrates and enzymes constituting major biochemical pathways5,9, redox instability10 or epigenetic changes11, can independently precipitate seizures. Indeed, irrespective of the actual inciting events or causes, seizure activity is characterized by a vicious cycle of metabolic derangements and excitotoxic damage to the brain12,13, further compounding the fundamental mechanisms that generate aberrant network excitability and instability.
The mainstay of epilepsy therapeutics is the growing armamentarium of anti-seizure medications (ASMs). For the most part, ASMs are believed to target cellular membrane-bound ion channels and transporters to reduce excitation and/or increase inhibitory neurotransmission at the synaptic level14. While ASMs have been shown to control SRS in most patients with epilepsy (but not without potentially significant side-effects), approximately one-third remain medically intractable1. The sobering reality is that despite decades of basic-translational epilepsy research and the advent of dozens of new drugs approved for clinical use, this refractory population remains unchanged15. This may be due in part because traditional drug development for epilepsy has historically utilized acutely provoked seizure models that may not faithfully recapitulate the pathophysiologic changes seen in chronic epileptic brain and has focused mostly on molecular targets localized to the neuronal cell membrane16. That said, some of the current pharmacological agents in clinical use and under development appear to modulate novel mechanisms such as binding to the synaptic vesicle protein 2A (SV2A) and inhibition of cholesterol 24-hydroxylase17,18. And preclinical development of ASMs has steadily included chronic epilepsy models in the screening process16. Importantly though, it is only recently that metabolic targets have drawn significant attention in the field of epilepsy experimental therapeutics18.
The century-old high-fat, low-carbohydrate ketogenic diet, the most established metabolism-based treatment for epilepsy, provides the strongest evidence that targeting brain bioenergetics and metabolism can mitigate seizure activity – notably, in patients who fail to respond to ASMs19–21. Successful variations of the ketogenic diet, such as the medium-chain triglyceride diet22, modified Atkins diet23, and the low-glycemic index treatment24, have provided further evidence-based validation of metabolic and dietary therapies for the spectrum of epilepsies encountered in clinical practice25. And while the fundamental mechanisms underlying the anti-seizure efficacy of such treatments remain unclear, there is increasing evidence that metabolic approaches abstracted from these diets can afford neuroprotective (and perhaps even disease-modifying) effects26. In this review, we summarize the literature supporting the growing recognition that disruptions in cellular metabolism can be both a cause and consequence of epileptic seizures, hence affording a broad opportunity to exploit this growing science for innovative therapeutic strategies.
Brain Energy Metabolism
The brain is a highly energy-dependent organ. At rest, it requires approximately 20% of oxygen and 25% of glucose relative to the rest of the body, although the brain comprises only 2–3% of total body weight27. Neurons have an exceedingly high energy demand due to many cellular housekeeping functions such as synthesis and degradation of macromolecules, maintenance of cytoskeletal dynamics and axoplasmic transport, as well as other costly bioenergetic functions related to the high level of action potential signaling, synaptic activity, and plasticity changes2,28,29. Although the brain is clearly energy demanding, it does not have an adequate energy reserve; hence, it must rely on a variety of exogenous energy sources to maintain normal function – mostly via transport of substrates through the blood-brain-barrier30,31. That said, while astrocytes are known to store glycogen as a source of glucose-6-phosphate, a main substrate for glycolytic ATP production32, this is wholly insufficient to meet the immediate energy needs of the brain and brain glycogen may act more as an emergency energy reserve and may also serve unique signaling functions between neurons and glia32. Glucose is an obligate source of energy for the brain33, but other fuels such as lactate, ketone bodies and medium-chain fatty acids can be used when the availability of glucose is restricted34–36.
Astrocytes rely on both glycolysis and oxidative metabolism through the tricarboxylic acid (TCA) cycle to produce ATP37. In contrast, neurons are much more dependent on the immediate availability of glucose that is extracted from the capillaries via glucose transporters (GLUT3, in particular)33,38. And while neuronal oxidative metabolism can generate much needed ATP for their high energy needs, neurons might maintain viability and long-term function by accessing some metabolic fuel from astrocytes. Specifically, there is some evidence that astrocytes and neurons control neurometabolic coupling by furnishing lactate according to the astrocyte-neuron lactate shuttle (ANLS) hypothesis39–41 (Fig. 1). Lactate cannot passively diffuse across the blood brain barrier30, and since it cannot be directly utilized for energy production, it must be first transported via monocarboxylic transporters (MCTs) into cells42, and then be subsequently converted enzymatically to pyruvate by lactate dehydrogenase (LDH) which exists in multiple isoforms – LDH1 being primarily expressed in neurons and LDH5 in astrocytes (Fig. 1). However, the ANLS hypothesis is not without some controversy, as there is evidence that oxidative metabolism of lactate in neurons may not always be important for synaptic neurotransmission33,43,44 and the directionalities of lactate transport under different physiological conditions remain unclear. Moreover, when abnormally increased neuronal activity occurs during epileptic seizures, neurons may rely more on their own aerobic glycolysis than astrocyte-derived lactate44. Nevertheless, to better understand cerebral blood flow, metabolism, and metabolic coupling among different cell types, it is important to examine the interdependent relationships between neurons, astrocytes and the microcirculation – elements that comprise at a unitary level the neurovascular unit (Fig. 1). Finally, whether in neurons, glial cells or the vascular endothelium, mitochondria are the most important determinant of bioenergetics capacity, without which there can be no normal – or even pathological – function2,45. Any compromise of mitochondrial function can induce dysregulation of intracellular calcium, increased oxidative stress, and ultimately apoptotic cell death.
Figure 1: Astrocyte-Neuron Lactate Shuttle.
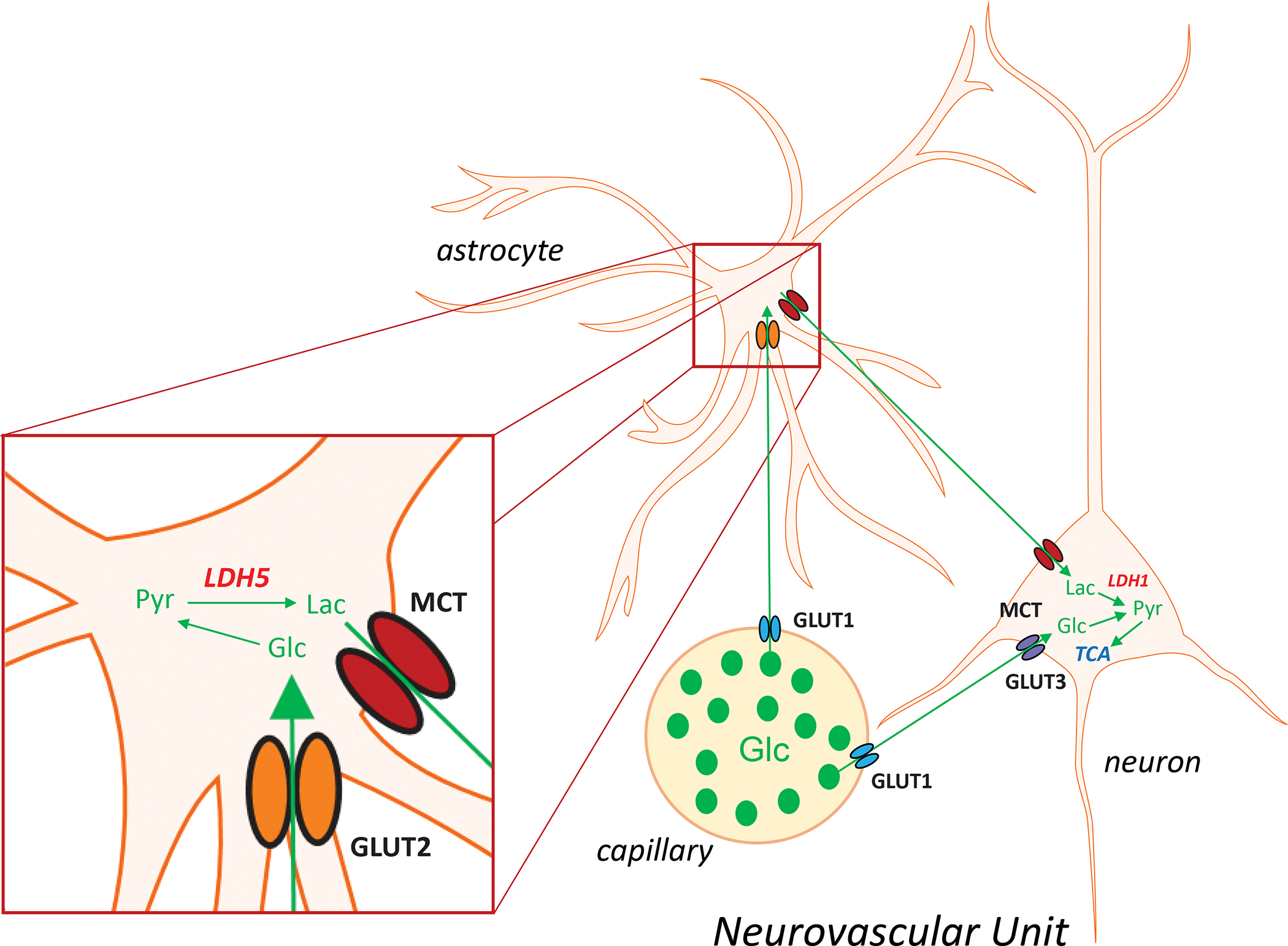
Astrocytes control neurometabolic coupling. Glucose is taken up from the peripheral circulation by glucose transporters (GLUTs) differentially localized to different elements of the tri-partite neurovascular unit. GLUT1 is the main astrocyte glucose transporter, whereas in neurons the principal isoforms are GLUT2 and GLUT3. Glucose is metabolized to pyruvate within the glycolytic pathway, and then converted to lactate via lactate dehydrogenase (LDH) which exists as distinct isoforms as well, with LDH1 and LDH5 being prominently expressed in neurons and astrocytes, respectively. Lactate cannot passively diffuse down its concentration gradient through the blood-brain-barrier; hence, it must be transported via monocarboxylic acid transporters (MCTs). Our current conceptual understanding is that the lactate generated in astrocytes is transported into neurons, is subsequently converted to pyruvate, which then enters the tricarboxylic acid (TCA) cycle to generate energy.
Often neglected in the epilepsy field, glia clearly play a major role in seizure prevention, but also seizure genesis, controlling the ionic balance between intracellular and extracellular compartments, and modulating synaptic neurotransmission37. During normal brain activity astrocytes play an important role in preventing neuronal hyperexcitability and seizures by buffering K+ and by regulating glutamate uptake37. Astrocytes can restore ionic and neurotransmitter homeostasis that is disrupted during seizure activity through various mechanisms, the most important of which are re-uptake of glutamate from the synaptic cleft and buffering of extracellular potassium through glial end-feet processes that link to the brain microvasculature30,37. Importantly, in an in vitro stem cell model, re-uptake of glutamate into astrocytes triggered enhanced glycolysis, which in turn generated more pyruvate that was converted to lactate and subsequently transported to neurons via the ANLS46. Moreover, it is increasingly recognized that astrocytes comprise large networks of highly inter-connected cells, mostly through gap junction channels (connexin43 and Cx30) which enable the bi-directional exchange of ions, nutritional metabolites, second messengers, amino acids, peptides, nucleotides, and even RNA37. Hence, astrocytes are uniquely poised to modulate network activity – electrically and metabolically. In short, astrocytes, being able to use glycogen as fuel, play a critical role in maintaining energy metabolism but also excessive neuronal firing as seen during epileptic seizures. Indeed, there are growing lines of evidence implicating astrocytes in seizure genesis and epileptogenesis37,47–49.
Pathophysiology of Epileptic Seizures
Epileptic seizures are generally divided into two clinical groups according to their presumed site of origin and pattern of spread1. Focal onset seizures are believed to arise from a specific locus in the brain, and the clinical manifestations are a consequence of perturbations in the function(s) ordinarily ascribed to that area. These can at times secondarily generalize to the entire cortex via adjacent spread or through commissural pathways. In contrast, primary generalized seizures manifest at their onset through disruption and enhancement of bilateral and reciprocal thalamocortical connections that are modulated by subcortical structures via multiple ascending pathways50. While this framework is at times viewed as overly simplistic and not reflective of the complex brain dynamics and connectivity required to generate seizures, it is a clinically useful dichotomy that has endured the test of time.
There are numerous mechanisms that have been implicated in ictogenesis. Although much is understood about the electro-clinical phenomenology in epilepsy, fundamentally we know very little about the mechanisms that directly initiate seizure activity and that terminate seizures after variable durations51, and in response to circadian and environmental factors. Where do focal onset seizures originate and are the pathways and structures traditionally implicated in propagation necessary and sufficient? Why does seizure activity eventually stop – even without pharmacological intervention? Are endogenous control mechanisms recruited in response to seizures adequate to do so? The answers to these fundamental questions continue to remain elusive. However, what is clear from the above discussion is that the energy required to sustain epileptic seizure activity is wholly dependent on adaptations in cerebral blood flow and metabolic coupling within the tri-partite neurovascular unit.
For decades, the simple conceptual notion has been that overall central excitatory-inhibitory imbalance precipitates seizure activity. While this makes intuitive sense, the mechanisms that induce network instability cannot all be reduced to a matter of increased excitation and/or reduced inhibition. As an example, increased GABAergic interneuron function has been shown to ‘paradoxically’ induce network excitability and seizures52–54, and there are ephaptic (i.e., non-synaptic) mechanisms55–58 that can regulate neuronal synchrony in either direction. The broad pathophysiological mechanisms elucidated over the past quarter century include ion channel dysfunction (primarily due to genetic defects in various subunit proteins resulting in either loss-of-function or gain-of-function)59, structural brain pathologies such as focal cortical dysplasia and other malformations of cortical development60,61 or secondary changes to post-natal brain insults such as hypoxic-ischemic encephalopathy or traumatic brain injury, cellular loss and synaptic re-organization, neurogenesis, and BBB dysfunction30,31,62, among many others. Molecular disruptions in cell signaling pathways such as mTOR (mammalian target of rapamycin)63, have also been implicated, as well as neuroinflammation from intrinsic and extrinsic causes6,7, stress, and hormones. Additionally, developmental changes in neurotransmitter subunit expression, immaturity of homeostatic mechanisms affecting ionic gradients and transport all have been linked to increased seizure propensity, especially in the developing brain64. More recently, the gut microbiota has been implicated in both the pathogenesis of seizures and the therapeutic efficacy of the ketogenic diet65, further strengthening the notion that a systems biology and whole organismal approach is necessary to better understand the pathogenesis of epilepsy66,67 – particularly in the context of systemic metabolism and inflammation. Finally, relevant to the current review, inborn errors of metabolism, particularly those that impair normal mitochondrial physiology and respiratory chain function, can manifest prominently with epileptic seizures9. Given the ever-expanding list of seizure-modifying factors, we are learning that virtually any pathophysiological mechanism involving the brain – and which induces stress – can elevate the risk of seizures68,69. In support of this, there are an increasing number of studies reporting seizures or epilepsy arising from unexpected mechanisms or genes59,70,71, and clinically, epilepsy is recognized as a fairly common co-morbidity of many neurological disorders. Conversely, there are innovative molecular approaches that are being taken to mitigate epileptogenesis64,72 and identify relevant biomarkers73.
What is the human evidence that physiological and metabolic changes are potentially a cause and/or consequence of seizure activity? Previous studies, utilizing functional and biochemical imaging techniques such as positron emission tomography (PET), functional magnetic resonance imaging (fMRI) and magnetic resonance spectroscopy (MRS) have revealed significant alterations in cerebral blood flow and metabolism in the epileptic brain74–76. Human PET studies utilizing 2-deoxy-2[18F]fluoro-D-glucose (18F-DG) have shown significant increases in focal or regional blood glucose utilization during seizure activity77, and although this has traditionally been interpreted as evidence of a seizure focus, hypermetabolism per se may reflect other pathologies such as brain changes associated with brain plasticity77. Concomitant EEG monitoring during 18F-DG uptake can be used to distinguish between local seizure foci and more general plasticity changes. Interestingly, during the time-span of epileptogenesis, children affected with Sturge-Weber syndrome were characterized by interictal hypermetabolism as opposed to ictal hypermetabolism78. In line with those findings, a follow-up MR-spectroscopy-based study demonstrated in vivo evidence that interictal hypermetabolism is likely based on increased glutamate release in the affected brain regions79. However, 18F-DG studies have also shown interictal hypometabolism in human TLE subjects80, which might indicate an energy crisis as a functional basis for impaired astrocytic K+-buffering and glutamate uptake, and hence a lowering of seizure thresholds. Animal studies, by contrast, have consistently shown increased metabolism during epileptic seizures. Complementing these observations, single-photon computed emission tomography (SPECT) studies have shown dynamic changes in cerebral perfusion, during both interictal and ictal periods76. Functional MRI (fMRI), which measures blood-oxygenation levels as a surrogate for neuronal function and connectivity during cognitive tasks has also proved useful in studying both normal and epileptic brain76. While fMRI has mostly been used to conduct language mapping for epilepsy surgery, there is a growing list of applications, including a broader assessment of neuronal functioning (information processing), brain network connectivity and activity, and even treatment outcomes. fMRI itself is not a technique to directly interrogate brain metabolism, but it can be a valuable tool to dissect functional brain network properties in complementary fashion with MRS, PET, SPECT, and of course, the gold standard electrocephalography (EEG), which is utilized both non-invasively and invasively. Each of these techniques play distinct roles in the clinical armamentarium and given their intrinsic differences in spatial and temporal resolution – and ultimately what the data generated signify, they are often used in combination to delineate the functional and biochemical properties of the epileptic brain.
Despite the numerous scientific and technological advances that have been made over the past few decades, there is an important caveat with respect to an accurate mechanistic understanding of the epileptic brain. As is the case throughout much of science and medicine, it is often tempting to assign causality to any identifiable alteration in the epileptic brain that could theoretically enhance neuronal excitation toward seizure genesis. However, researchers often discover that it is not straightforward to identify the critical mediators and pathways that are directly responsible for ictogenesis and epileptogenesis. Ultimately, it should be emphasized that seizures reflect a complex array of perturbations occurring at multiple hierarchical levels of biochemical, molecular, cellular structure and function, and remain unpredictable consequences of neuronal and glial network activity.
Seizure-Induced Impairment of Metabolic Homeostasis
To truly appreciate the crucial role of metabolism in epilepsy and its pathophysiology, it is important to acknowledge the existence of bi- and multi-directional molecular interactions, which add layers of complexity to the many vicious cycles implicated thus far in the processes of epileptogenesis62,64,81. Broadly stated, seizures by themselves cause an impairment in metabolic homeostasis, and a derailment of key metabolic and biochemical functions contributes to the increased likelihood of seizure generation. In this section, we discuss how epileptic seizures impact cellular metabolism. Acute seizures utilize glucose to fuel heightened energy demand and lead to the preferential formation of lactate rather than acetyl-coA82. Given the availability of glucose, glycolytic flux increases during seizure activity but decreases during the interictal periods – changes which are believed to underlie the phenomena of ictal hypermetabolism and interictal hypometabolism, both long considered the metabolic hallmarks of human and experimental epilepsies83,84. Whereas glycolysis is a less efficient process for ATP production, in comparison to the mitochondrial metabolism of acetyl-CoA, an increase in the glycolytic rate by a factor of 10 to 30 is adequate to generate immediate energy to sustain seizure activity. A shift toward glycolysis under aerobic conditions during seizures is supported by the increased production of lactate,85 mimicking the Warburg effect described in cancer cells which are intriguingly characterized by metabolic derangements mirrored in the epileptic brain86. Increased ictal glucose metabolism may support plasticity processes in the brain, which include gliosis, neurogenesis, and axonal spouting87. Mechanistically, glucose is not only used as a substrate for the generation of ATP, but is also a metabolic precursor for neurotransmitters and neuromodulators including acetylcholine, glutamate, GABA, D-serine, glycine, and D-aspartate87. Together, alterations in key energy metabolites such as glucose, neurotransmitters, and neuromodulators are likely responsible for profound plasticity changes in the brain. A major metabolic consequence of increased ictal glycogen and glucose utilization is the formation of lactate from pyruvate through LDH (Fig. 1). The hypothesis that seizure-induced lactate formation enables epileptic activity is supported by findings that LDH inhibitors provide robust anti-ictogenic effects88. LDH inhibition is likely to interfere with glycolysis by limiting the availability of NAD+ and supporting the oxidative metabolism of pyruvate in mitochondria.
Given the alternative metabolic fates of glycolysis-derived pyruvate (lactate vs. mitochondrial oxidation), the impact of seizures on mitochondrial function is of paramount importance. Several enzymes involved in the mitochondrial TCA cycle, which converts pyruvate into carbon dioxide, are known to be compromised by acute seizures (e.g., status epilepticus or SE) or chronic seizure activity in epilepsy. Reduced activity of pyruvate dehydrogenase, alpha ketoglutarate dehydrogenase, 2-oxoglutarate dehydrogenase, and aconitase89–93, combine to reduce the flux of metabolites through the TCA cycle in the epileptic brain94. In addition, the multimeric protein complexes of the electron transport chain (ETC) which enable oxidative phosphorylation – such as complex I – are known to be impaired in human temporal lobe (TLE) epilepsy and rodent models of epilepsy95–97. Mitochondrial dysfunction based on those alterations is directly supported by findings from experimental SE-induced epileptogenesis models, in which oxygen consumption rates transiently increase within minutes of SE onset, return to baseline during the seizure-free latent period and finally decrease during the chronic epilepsy phase98. Those experimental data on the role of mitochondria in epilepsy are directly supported by clinical findings based on rare inherited mitochondrial disorders associated with epilepsy99.
High levels of oxygen and reactive oxygen species (ROS) are known to be detrimental to mitochondrial health. NADPH oxidase 2, also known as cytochrome b(558) subunit beta or Cytochrome b-245 heavy chain, is a superoxide-generating enzyme expressed in mitochondria and the plasma membrane, which has been shown to play a major role in the generation of SE-induced ROS92,100,101. Seizure-induced increases in mitochondrial O2 and H2O2 production can be attributed to higher substrate utilization and electron transfer to O2 during interictal periods, to an overload of calcium, and to the inhibition of ETC complexes, all of which combine to transfer electrons to oxygen. Seizures also cause inactivation of the mitochondrial antioxidant superoxide dismutase 2 via decreased activity of Sirtuin 3 or altered activities of peroxide detoxification as additional mechanisms contributing to seizure-induced oxidative stress. The generation of ROS can result in the inhibition of complex I of the ETC, leading to ROS-induced ROS production, thus forming a pathologically regenerative cycle driving increased oxidative stress, which results in further free radical damage to cellular macromolecules – a major cause and consequence of extended seizure activity102. Seizure-induced neuronal death can directly be attributed to oxidative stress and the formation of reactive aldehydes, hydroxyl radicals, and redox active iron, which induce damage to mitochondrial DNA, proteins, and lipids10,103. In line with increased oxidative stress as a pathological hallmark of epilepsy, a depletion of the endogenous antioxidant glutathione and the formation of oxidized glutathione disulfide has been demonstrated in both human epilepsy and in rodent models of acquired epilepsy94,104–106. Notwithstanding the above observations and our traditional understanding of the pathological effects of ROS, it is increasingly understood that ROS may possess signaling properties at low concentrations and can modulate GABAergic neurotransmission through both pre- and post-synaptic mechanisms107.
While ROS promote neurotoxicity, seizure-induced disruption of calcium homeostasis may also contribute to the detrimental effects of seizures. Excitotoxic neuronal death is primarily driven by excessive activation of N-methyl-D-aspartate (NMDA) receptors and the subsequent intraneuronal accumulation of toxic levels of calcium. This calcium overload in turn promotes the generation of ROS or reactive nitrogen species, which as outlined above compromise mitochondrial function, cause metabolic impairment and activate necrotic and apoptotic pathways, which are all calcium-dependent processes. Calcium imaging during an ictal event consistently demonstrates increased calcium influx in almost all neurons and astrocytes108. Mitochondria are also crucially involved in the control of calcium homeostasis. Mitochondrial calcium homeostasis depends on mitochondrial calcium uniporter, rapid mitochondrial calcium uptake, and mitochondrial ryanodine receptors109. Changes in calcium concentration in the mitochondrial matrix in turn regulate the activity of the mitochondrial ETC which is required for ATP production110,111. Through this mechanism, an increase in seizure-induced calcium fluxes can directly compromise mitochondrial function.
Metabolic and Epigenetic Changes in Epilepsy
From a metabolic viewpoint, the regulation of energy homeostasis has been crucial for the evolution of all living systems. A rheostat is needed to conserve energy in case of excessive energy consumption or depletion of energy supplies. A relatively simple system to regulate energy homeostasis is based on the ATP-adenosine balance with adenosine acting as a retaliatory metabolite112. From this evolutionary perspective, an epileptic seizure represents excessive energy consumption entailing the degradation of ATP into adenosine, with adenosine being used as an innate feedback rheostat to conserve energy. Therefore, a massive seizure-induced release of adenosine113 acts as the brain’s endogenous anticonvulsant and seizure terminator51,114. Because adenosine is also a building block of RNA and regulator of the S-adenosylmethionine-dependent transmethylation pathway115,116, an energy crisis would also affect RNA synthesis by lowering ATP, and DNA methylation by increasing adenosine. Both processes would combine to reduce gene transcription globally and to conserve energy.
In contrast to the organization of living systems, with a metabolic base and subsequent layers of added complexity (Fig. 2), conventional pharmacotherapy development starts with ‘druggable’ targets at the top of the pyramid. For example, benzodiazepines were almost discovered by chance in 1957 leading to the subsequent characterization of the ‘benzodiazepine receptor’ in the CNS in 1977117. It was later revealed that the benzodiazepine binding site was in fact an integral part of the GABAA receptor complex117. Drug-driven therapy development therefore led to a major focus on G-protein coupled receptors (GPCRs), ion channels, and protein kinases, which still form the mainstay of CNS therapeutics today, and which do not target mechanisms at the base of the evolutionary pyramid. Methods to exploit gene regulation therapeutically are still in its infancy and the therapeutic potential of epigenetic, biochemical, and metabolomics approaches constitutes a new and lesser explored frontier in therapy development. If the metabolic base of the pyramid depicted in Fig. 2 is disrupted, it becomes obvious that a traditional pharmacological treatment approach would have limitations.
Figure 2: Epilepsy Treatment Targets in the Pyramid of Life.
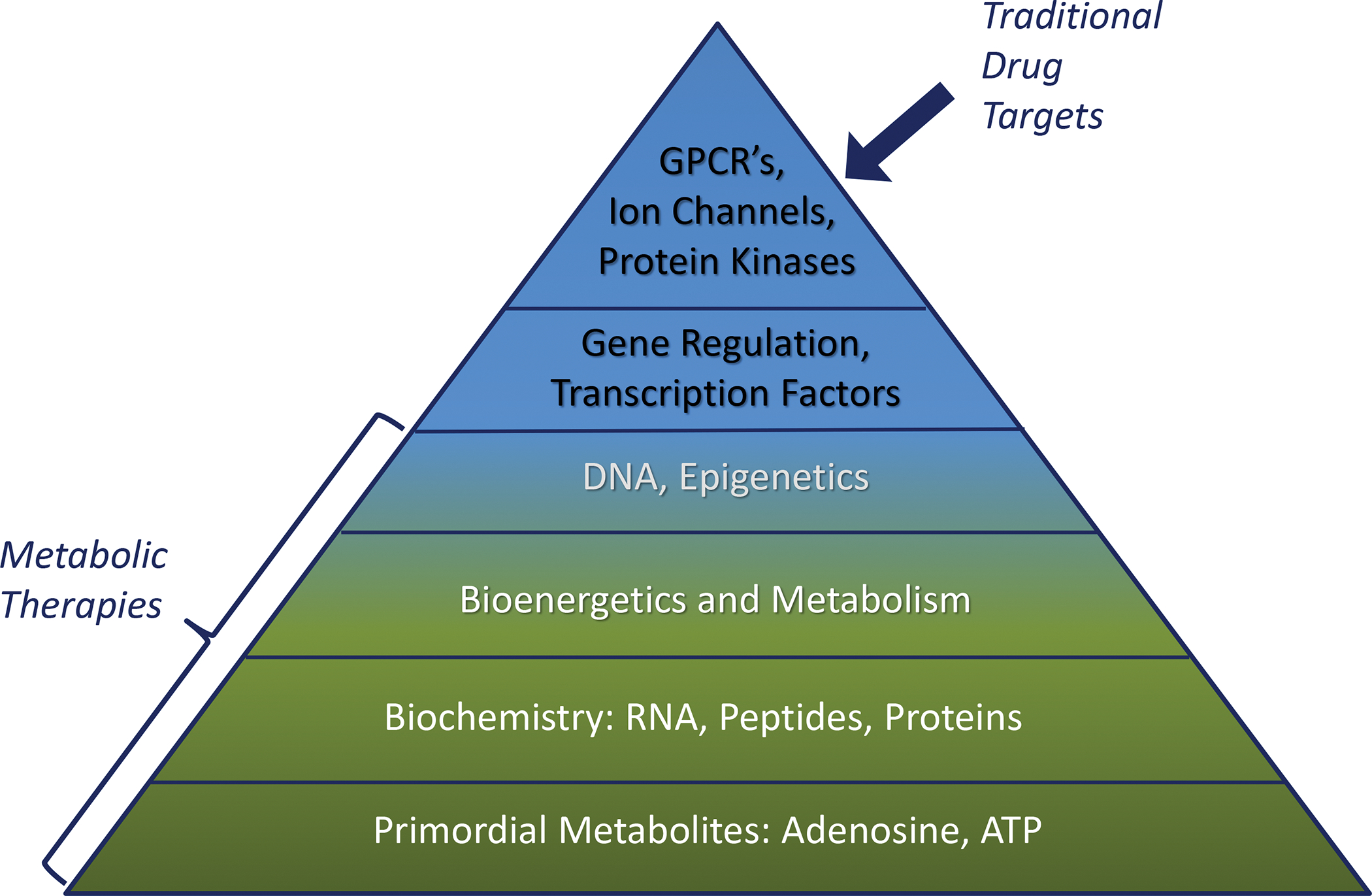
Traditional drug targets are only found at the top of the pyramid, whereas metabolic therapies are uniquely positioned to reset fundamental self-regulatory mechanisms that form functional homeostatic systems. Primordial metabolites such as adenosine and ATP are essential components of complex biochemical networks and represent the foundation for simple metabolism-based regulatory systems to enable energy homeostasis. The evolution of life required the advent of RNA, peptides and proteins, which established the framework for bioenergetics and metabolism. This was followed later by DNA and gene regulatory mechanisms such as epigenetic modifications or use of non-coding RNAs. Transcription factors provided further refinement to these mechanisms. Finally, regulatory systems based on G protein-coupled receptors (GPCRs), ion channels, and protein kinases evolved. From this schema, it becomes evident that if fundamental mechanisms at the base of the pyramid are disrupted, disease can result. Modified from Boison D, The Biochemistry and Epigenetics of Epilepsy: Focus on Adenosine and Glycine, Frontiers in Molecular Neuroscience 2016; 9:26. doi: 10.3389/fnmol.2016.00026.
It is now abundantly clear that metabolic, biochemical, and epigenetic alterations play a major role in the pathophysiology of acquired epilepsies118. Among those alterations, maladaptive changes in adenosine metabolism constitute a central mechanism, linking metabolism with neuronal excitability and gene expression. As a neuromodulator, adenosine exerts a wide range of well-characterized functions based on the activation of a class of four G-protein coupled adenosine receptors (A1, A2A, A2B, A3)119–121. Together, those receptors control neuronal excitability, neuroprotection, synaptic plasticity, and inflammatory responses. Adenosine is under metabolic control of adenosine kinase (ADK), which exists in a cytoplasmic form ADK-S (regulation of tissue levels of adenosine) and a nuclear form (ADK-L), which controls adenosine metabolism in the cell nucleus122. ADK-L drives the flux of methyl groups through the transmethylation pathway, whereby increased ADK-L drives global hypermethylation of DNA (Fig. 3). Increasing adenosine through various methods leads to reduced global 5-methylcytosine (5mC), an effect that is independent of adenosine receptors. Importantly, cultured cell lines engineered to overexpress ADK-L have increased global 5mC levels as compared to ADK-S cells or ADK-deficient cells123.
Figure 3: Adenosine Metabolism and Epileptogenesis.
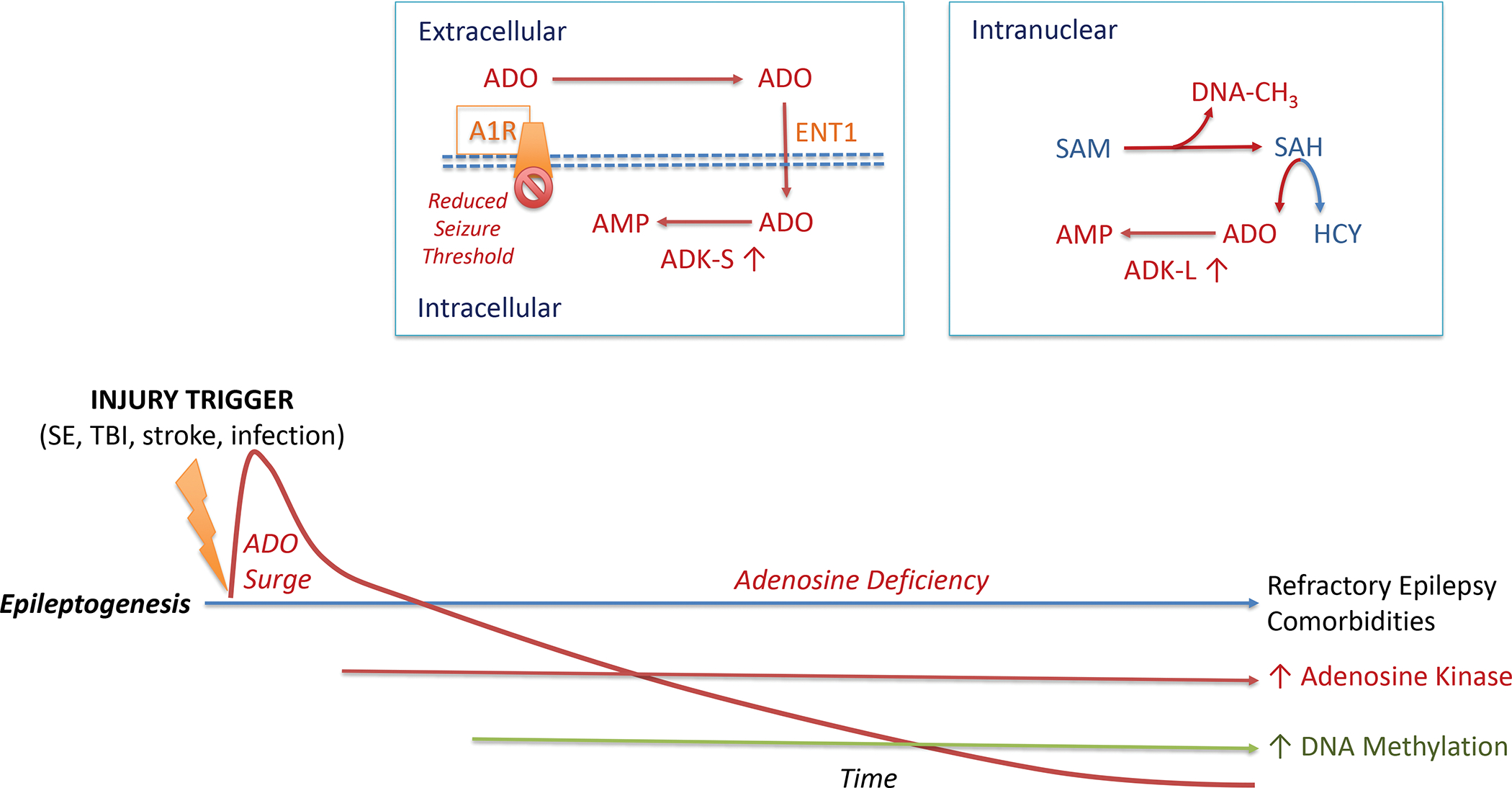
The epileptogenic process leading to acquired epilepsy can be initiated through a variety of injurious triggers including status epilepticus (SE), traumatic brain injury (TBI), stroke, or an infection of the brain. Such precipitating events lead to a biphasic response of the adenosine system: an acute neuroprotective adenosine (ADO) surge followed by progressive adenosine deficiency during epileptogenesis. Chronic adenosine deficiency is driven by maladaptive overexpression of the key adenosine metabolizing enzyme (ADK), which also drives an increase in DNA methylation, an epigenetic hallmark of acquired epilepsies. Extracellular adenosine is regulated by intracellular metabolism of the cytoplasmic isoform of adenosine kinase (ADK-S), which drives the uptake of adenosine through the equilibrative nucleoside transporter 1 (ENT1)). Metabolism-driven cellular uptake of adenosine leads to reduced adenosine A1 receptor (A1R) binding, which is a mechanistic explanation why a pathological increase in ADK-S reduces seizure thresholds. In the cell nucleus, adenosine is coupled to the S-adenosylmethionine (SAM)-dependent transmethylation pathway, which determines DNA methylation (DNA-CH3). The reaction product S-adenosylhomocysteine (SAH) is hydrolized into homocysteine (HCY) and adenosine. Metabolism of adenosine to AMP via the nuclear isoform of adenosine kinase (ADK-L) drives the flux of methylation reactions through this pathway. Thereby, increased ADK-L drives increased DNA methylation. It is important to note that this figure is not drawn to scale. The acute adenosine surge takes place during the first 24 hours after an injurious event, whereas all subsequent steps of the epileptogenic cascade occur over a time-span of days to weeks to months (or longer) after a precipitating event.
Metabolic and Mitochondrial Epilepsies
The mechanisms described above strongly support a metabolic and mitochondrial basis of epilepsy and consequently, metabolic and mitochondrial epilepsies have received increasing attention. Metabolic epilepsy in humans can usually be linked to inborn errors of metabolism, which typically present during infancy or childhood. Those genetically defined aberrations in metabolism provide valuable insights into the etiology of seizures and epilepsy.
A classic example is pyridoxine-dependent epilepsy which presents with recurrent, drug-refractory neonatal seizures, and arises from an inborn error of lysine catabolism. Notably, pyridoxine-dependent epilepsy responds to therapeutic pyridoxine supplementation. Pyridoxine deficiency compromises the function of pyridoxal 5′-phosphate, the active form of vitamin B6, a coenzyme required for the synthesis and metabolism of amino acids. A deficiency in pyridoxal 5′-phosphate thereby leads to decreased GABA concentrations in the brain, as one of the underlying ictogenic mechanisms124.
Of note, too much GABA can also trigger seizures as exemplified by mutations in succinic semialdehyde dehydrogenase deficiency, which is an enzyme of the GABA degradation pathway. Succinic semialdehyde dehydrogenase deficiency causes developmental delay, autism, epilepsy, hypotonia, and extrapyramidal movement disorders125. In addition, the disruption of lysine catabolism results in the accumulation of toxic intermediary substrates such as α-aminoadipic semialdehyde, which increases oxidative stress.
Pyridoxine dependent epilepsy can also be caused by mutations in the PROSC gene, which encodes a pyridoxal 5′-phosphate binding protein126. In a related but distinct condition affecting pyridoxine metabolism, pyridox(am)ine 5′-phosphate oxidase deficiency affects the key enzyme responsible for the conversion of pyridoxine to its active metabolite pyridoxal phosphate127. Clinically, neonatal seizures result, which are unresponsive to pyridoxine, and which are associated with hypoglycemia, lactic acidosis, and encephalopathy128.
Cerebral folate deficiency can have multiple etiologies and is characterized by low levels of 5-methyltetrahydrofolate (the active metabolite of folate) in the brain, but normal folate metabolism in peripheral organs129. The homocysteine-to-methionine conversion requires the enzyme methionine synthetase, methyl-tetrahydrofolate as a methyl donor, and vitamin B12 as a co-factor. Folate deficiency thereby leads to a disruption of methylation reactions and reduced methionine synthesis, resulting in DNA hypomethylation as a possible epigenetic mechanism triggered by a metabolic defect130.
Nonketotic hyperglycinemia results in glycine encephalopathy, which is a rare, inborn defect in the glycine cleavage enzyme complex, and which is characterised by profound accumulation of glycine in the CSF131. Glycine acts as obligatory co-agonist of the NMDA receptor by binding to its strychnine-insensitive glycine binding site132. Because this recognition site is normally not saturated133, an increase in extracellular glycine can potentiate impulse-dependent NMDA receptor activation, which might be the underlying mechanism for seizure generation in this condition.
Glucose transporter 1 deficiency syndrome is characterized by impaired glucose transport into the brain134, which results in infantile-onset refractory seizures, developmental delay, intellectual impairment, and movement disorders134. Interestingly, there is an increased frequency of seizures before meals and providing ketones as an alternative energy fuel via the ketogenic diet can be an effective treatment strategy.
A direct link between metabolism and cellular excitability is exemplified in patients with mutations in ATP-sensitive potassium (KATP) channels, a type of potassium channel involved in the regulation of neuronal excitability of neurons and the physiology pancreatic beta cells135–137. Those channels normally open under conditions of low glucose or low ATP/ADP ratios, and through K+ efflux keeps the post-synaptic neuron hyperpolarized. Mutations in this channel directly impair the brain’s ability to respond to a metabolic challenge by shutting down brain activity – in this scenario clinically, diabetes and seizures result. Metabolic epilepsies can also arise from mutations that lead to the accumulation of toxic compounds such ammonia in urea cycle disorders. Hyperammonemia can cause irreversible neurologic damage138 and increases neuronal excitability likely through excessive NMDA receptor activation139.
Mitochondrial dysfunction is known to precipitate a wide spectrum of neurological manifestations140, whereas ‘mitochondrial epilepsy’ is a unique subset of epilepsy characterized by refractoriness to treatment and poor prognosis in some patients141. Patients with mitochondrial epilepsy are characterized by distinct neuropathological changes, which include a loss of expression of mitochondrial complex I and complex IV subunits in neurons and reactive astrocytes142,143. Based on these observations, a brain slice model of mitochondrial epilepsy was recently developed, one that relies on the pharmacological inhibition of mitochondrial complexes I and IV as well as specific inhibition of the astrocytic TCA cycle enzyme, aconitase142. In this model, astrocytes play a key role in the generation of epileptiform discharges – specifically, the astrocytic GABA-glutamate-glutamine cycle is affected, thus compromising the regulation of GABAergic mediated inhibition142. Findings from this study suggest that glutamine is a critical mediator of the interactions between astrocytic and neuronal compartments controlling the epileptiform network in this in vitro model of mitochondrial epilepsy. In addition, mutations that perturb mitochondrial tricarboxylic acid cycle function, such as nonsense and missense mutations in the citrate transporter SLC13A5, can lead to early infantile epileptic encephalopathies93 Together, the metabolic and mitochondrial epilepsies demonstrate that altered metabolism and energy homoeostasis can have a direct impact on neuronal excitability and provide a rationale for the development of metabolism-based treatments. The myriad metabolic factors altered in many inborn errors of metabolism resulting in synaptic dysfunction are broadly grouped and depicted in Fig. 4.
Figure 4: Principal Mechanisms Involved in the Pathogenesis of Metabolic Epilepsies.
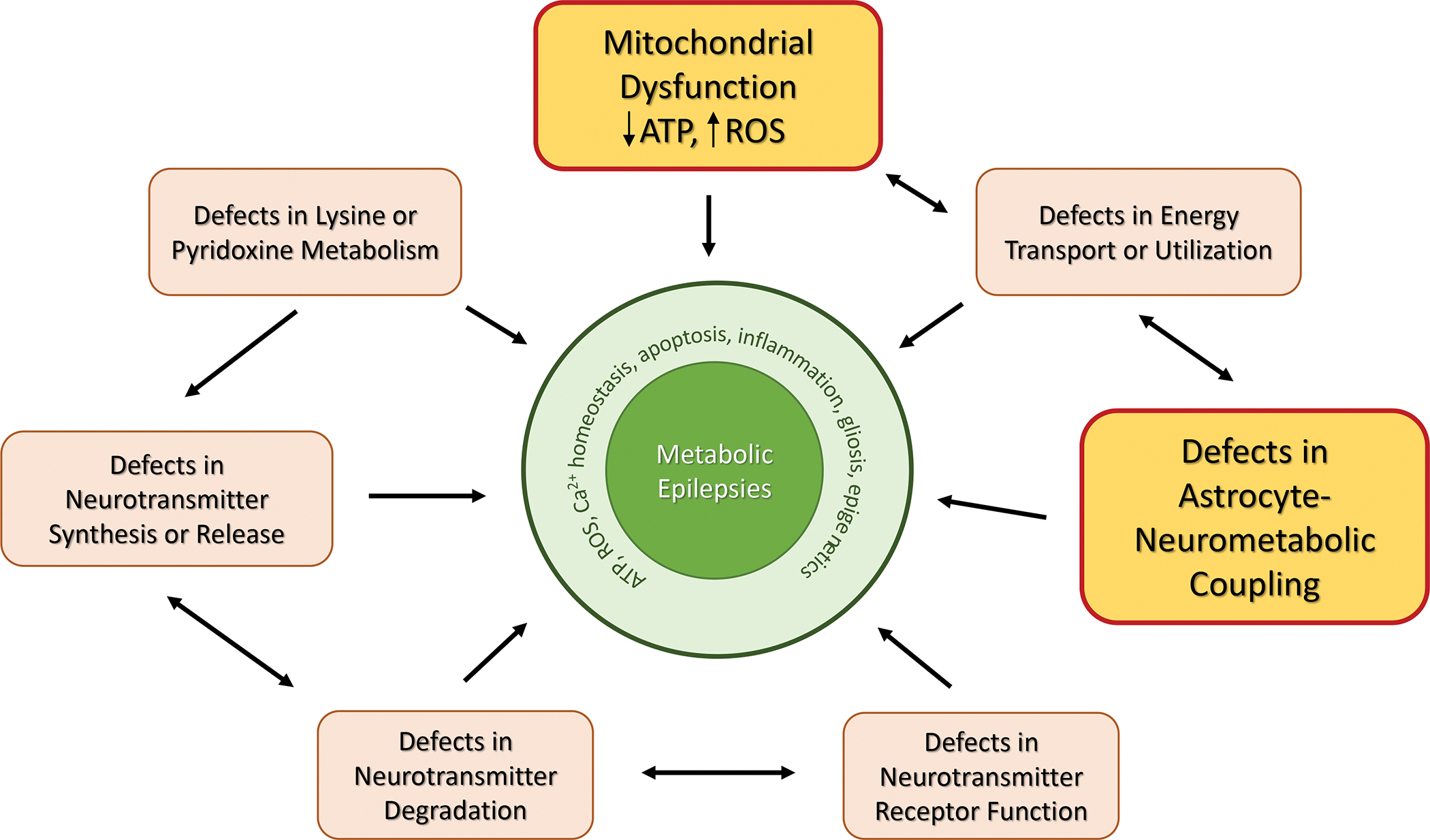
Metabolic epilepsies are complex syndromes characterized by seizures, co-morbidities and developmental impairment. The complex pathology of metabolic epilepsies can be explained by congenital defects leading to an imbalance of neurotransmitter function on the levels of synthesis, degradation, transport, and receptor binding. More importantly, congenital or acquired defects in mitochondrial function and astrocyte-neurometabolic coupling combine to trigger major defects in energy transport or utilization as a potential and preventable (e.g., through metabolic therapies) cause for metabolic epilepsies.
Metabolism-Based Treatments
The signature metabolic therapy for medically intractable epilepsy is the ketogenic diet, which was formulated a century ago to mirror the key biochemical changes associated with fasting – something anecdotally observed over millenia to help control seizures in individuals with epilepsy, but without significant caloric deprivation19. The ketogenic diet is a high-fat, low-carbohydrate and adequate protein diet that has been shown in prospective controlled studies to be effective against medically intractable epilepsy. As the name implies, the ketogenic diet – like fasting – induces prominent ketonemia through enhanced fatty acid oxidation which elevates blood levels of the principal ketone bodies beta-hydroxybutyate and acetoacetate19,34,35. In clinical practice, patients are treated with dietary formulations adhering to ketogenic/anti-ketogenic ratios of 3:1 to 4:1 ratio by weight of fats to carbohydrate plus protein, which results in serum beta-hydroxybutyate levels in the very low millimolar range (typically 2–5 mM, but this is often variable – as low as 0.4 mM and as high as 10 mM)19,144.
Over the years, clinicians have used variations of the ketogenic diet to address common side-effects and theoretically increase ketosis (the medium-chain triglyceride diet)22, to enhance palatability (the modified Atkins diet)23, or have employed diets that capitalize on another seminal biochemical change induced by the ketogenic diet – i.e., relative hypoglycemia (the low-glycemic index treatment)145. Clinicians have reported that these variations of the ketogenic diet are all effective in a small majority of patients with treatment-resistant epilepsy, yielding significantly improved seizure control (>50% reduction in up to 60% of individuals, especially in very young infants20,25,146,147). There is also growing evidence that these diets can be successfully implemented in adult patients148. The traditional ketogenic diet and the medium-chain triglyceride diet appear comparable in terms of clinical effectiveness21, but it is unclear whether the modified Atkins diet and low-glycemic index treatment show similar response rates as there are yet no rigorously controlled prospective head-to-head comparisons25,149,150.
Given the remarkable clinical efficacy of the ketogenic diet and its variants, there has been growing scientific interest in elucidating the mechanisms of these metabolism-based treatments14,151. At present, despite a multiplicity of innovative mechanisms that have been advanced in the literature, and the surprising finding that specific metabolic substrates and enzymes can act like anti-seizure medications or modulate neuronal excitability in unexpected ways (Fig. 5), how diets render such clinical effects remains unclear. Dietary therapies are not without their challenges with respect to compliance and tolerability, so efforts continue to better understand the basic mechanisms of ketogenic diet action, such that insights gleaned from basic-translational studies can uncover novel targets – or indeed paradigms – for experimental therapeutics. In this regard, use of specific fuels such as ketone bodies (as readily administered esters) or medium-chain fatty acids are paving the way for novel therapeutic approaches based on the study of ketogenic diet mechanisms152,153.
Figure 5: Metabolic Substrates and Enzymes Regulating Synaptic Neurotransmission and Neuronal Excitability.
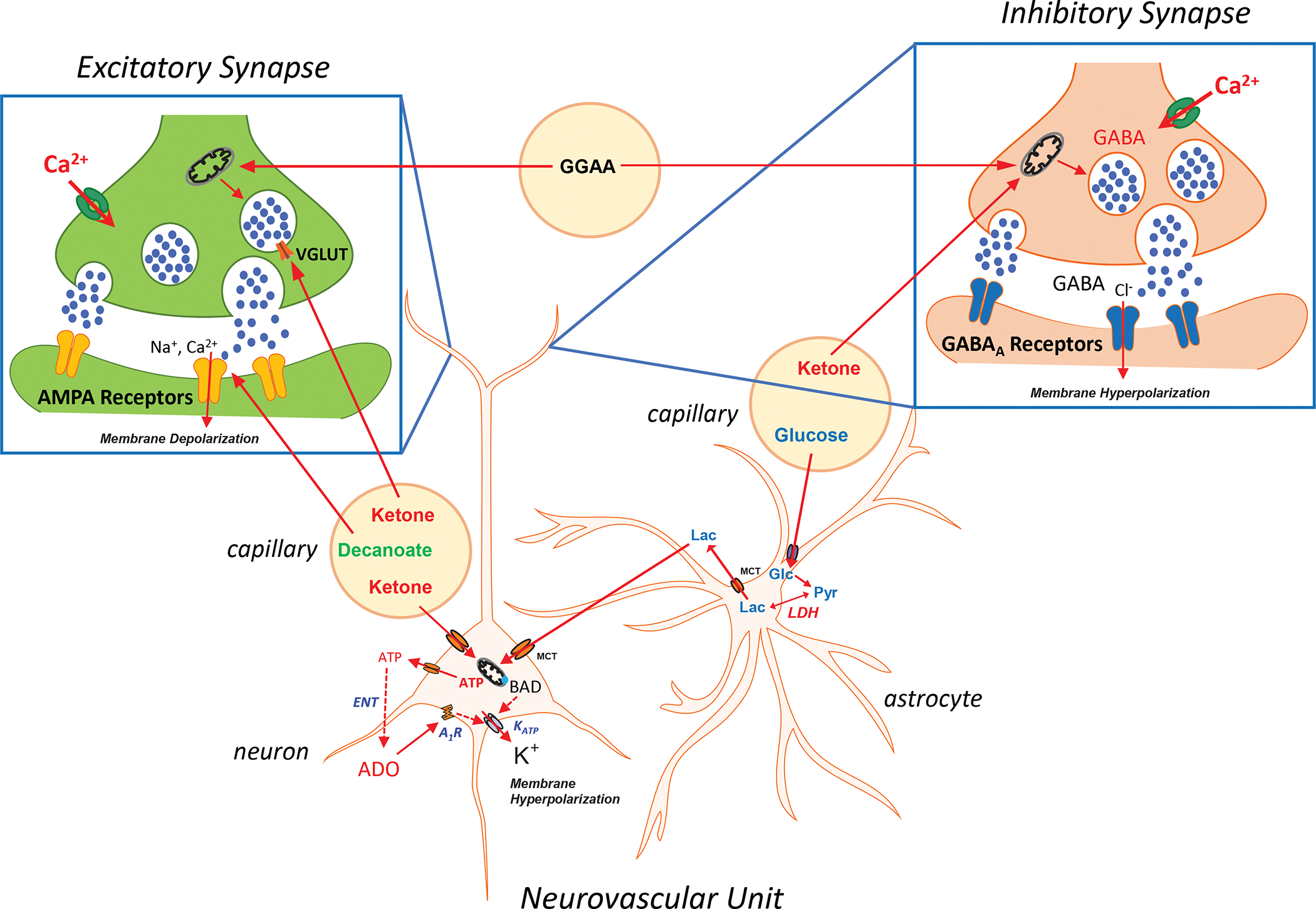
Schematic of the neurovascular unit comprising the neuron, astrocyte and microvasculature, and expanded views of prototypic excitatory and inhibitory central synapses – showing presynaptic and postsynaptic terminals. Ketone bodies can directly compete with the chloride anion regulatory site of vesicular glutamate transporters (VGLUTs) to prevent presynaptic release of glutamate. Decanoate, a C10 medium chain triglyercide, can inhibit post-synaptic AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors much like the anti-seizure medication perampanel. Ketones can also serve as a substrate for glutamate production in inhibitory presynaptic terminals, which increases the synthesis of γ-aminobutyric acid (GABA) via glutamic acid decarboxylase, thereby enhancing inhibitory neurotransmission. Additionally, ketones enhance mitochondrial ATP production. When ATP is released into the extracellular space via pannexin channels, it is degraded to adenosine through an equilibrative nucleotidase (ENT). Adenosine in turn can bind to inhibitory adenosine type 1 receptors (A1Rs) in an autocrine fashion to limit membrane excitability. Ketones, likely through this mechanism, can indirectly activate ATP-sensitive potassium (KATP) channels which induce membrane hyperpolarization. Moreover, increased membrane activity may deplete ATP levels in proximity or subjacent to KATP channels, which would decrease the ATP/ADP ratio and activate these inhibitory channels. KATP channels can also be indirectly regulated by BAD (Bcl2 associated agonist of cell death), a protein that serves a dual function as a pro-apoptotic factor and modulator of glycolysis. Glucose (Glc) transported into astrocytes produces pyruvate (Pyr) which can be converted to lactate (Lac) via lactate dehydrogenase (LDH); inhibition of LDH – as reported using the anti-seizure medication stiripentol – through yet unclear mechanisms affords anti-seizure effects (see Fig. 1 which details some aspects of the astrocyte-neuron lactate shuttle). Finally, ketogenic diet-induced changes in the gut microbiome has been shown to decrease gamma glutamyl amino acids (GGAA) in the blood metabolome, resulting in secondary changes in glutamate and GABA production – specifically, an increase in the GABA/glutamate ratio. Abbreviations: MCT, monocarboxylic acid transporter; ADO, adenosine.
The major proposed mechanisms of metabolic therapies can currently be grouped into a few broad categories (Fig. 6): 1) restoration of impaired bioenergetics and mitochondrial function, notably modulation of TCA cycle and respiratory chain activity154–156, as well as improved redox regulation and antioxidant capacity102,103,106,157; 2) synaptic regulation of both excitatory and inhibitory neurotransmission via metabolic substrates and enzymes88, at both pre-synaptic and post-synaptic levels158,159, and involving γ-aminobutyric acid type A (GABAA), glutamate, and adenosine receptors159 as well as KATP channels160; 3) glycolytic restriction with 2-deoxy-D-glucose161 and regulation of glycolysis by apoptotic factors162; or augmentation of the pentose phosphate pathway by fructose-1,6-bisphosphate (a glycolytic intermediate)163; 4) signaling pathways involved in cellular metabolism such as the mTOR pathway164,165; 5) direct and indirect anti-inflammatory actions on neurons and BBB permeability31,166–168; and 6) epigenetic effects on DNA169 and histones166 which form the basis for disease-modifying and antiepileptogenic effects of the diet. A summary of proposed mechanisms underlying neuroprotective effects of various dietary treatments is depicted in Fig. 7. Beyond this, an exhaustive discussion of proposed mechanisms of dietary therapies is beyond the scope of the present review, and the reader is referred to recent references for additional details14,26,151,170,171. What should be emphasized at the outset though is that the mechanisms underlying the clinical effects of the ketogenic diet are multiple, parallel, overlapping and potentially synergistic. There is no single mechanism that can fully explain these effects. In this regard, the same can be said for the majority of ASMs14,18.
Figure 6: Biochemical Pathways and Mitochondria.
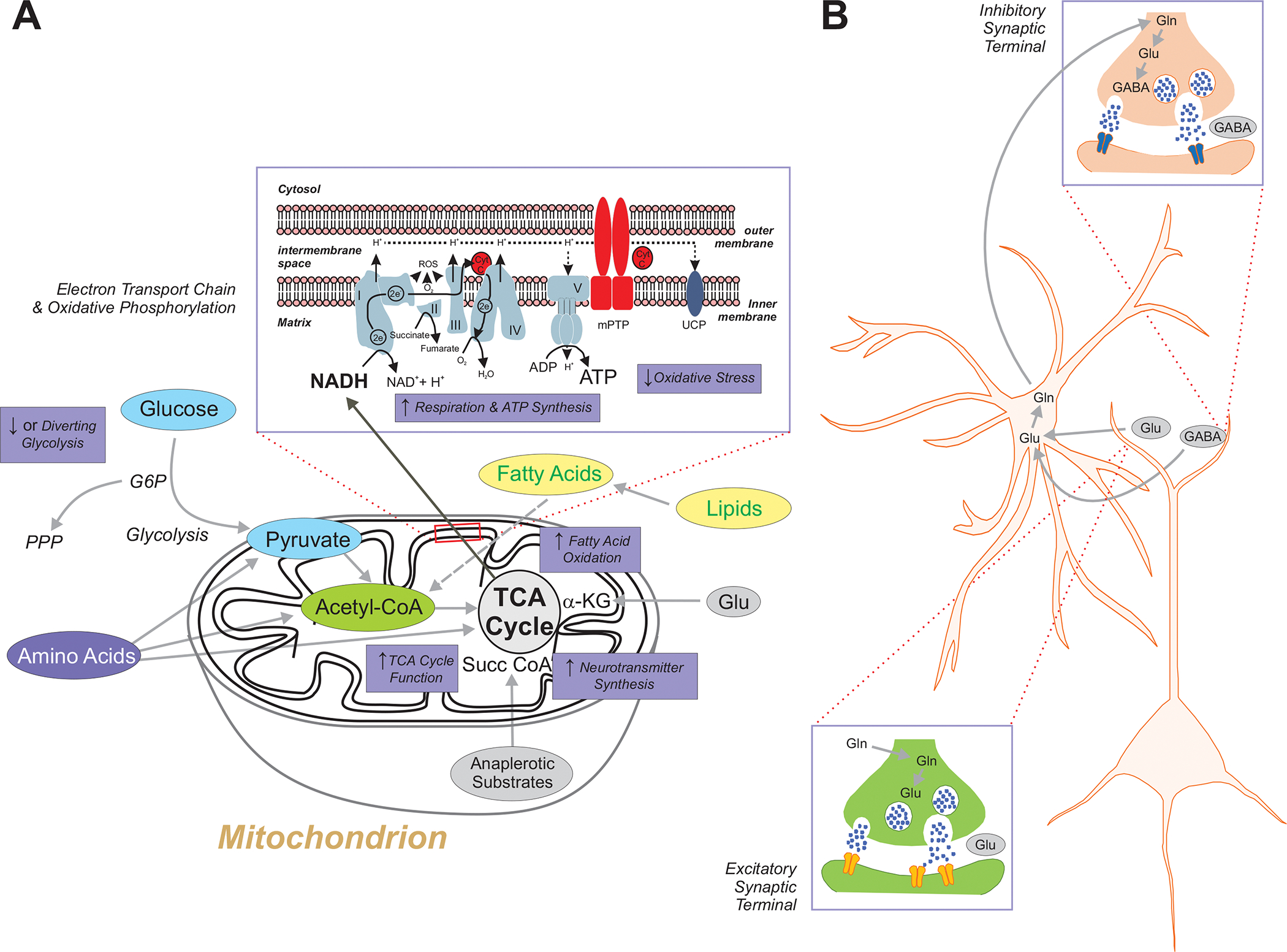
A. The biochemical effects of metabolic anti-seizure treatments have been linked in one way or another to the control of seizures or epilepsy in various experimental models. Inhibition of glycolysis with 2-deoxy-d-glucose results in seizure control in multiple animal models. Additionally, the glycolytic intermediate fructose-1,6-bisphosphate has been shown to exert broad anti-seizure effects, possibly by augmenting the pentose phosphate pathway. All amino acids can be converted for energy utilization if required, and can be glucogenic, ketogenic or both depending on where they enter certain biochemical pathways. For example, alanine, glycine and tryptophan can be converted to pyruvate, leucine and tryptophan to acetyl-coA, and histidine and glutamate into alpha-ketoglutarate of the tricarboxylic acid (TCA) cycle. Fatty acids derived from lipids can undergo fatty acid oxidation within the mitochondrial matrix and generate acetyl-CoA which can be condensed into acetoacetyl-CoA which is a precursor for the synthesis of acetoacetate and beta-hydroxybutyrate. Ketones are not only utilized for ATP production, but they can also be used for the synthesis of neurotransmitters such as glutamate and GABA. Certain fatty acids also enhance mitochondrial uncoupling protein (UCP) activity which partially dissipates the proton gradient across the inner mitochondrial membrane and as a result reduces reactive oxygen species (ROS) and reactive nitrogen species (RNS) production. Triglycerides such as triheptanoin provide anaplerotic substrates that when subsequently metabolized to succinyl CoA in mitochondria can also render anti-seizure effects. Finally, whether through the ketogenic diet (ketogenic diet) or ketone bodies, or through oxidative metabolism, mitochondrial respiratory chain function is enhanced – with resultant increases in oxygen consumption and adenosine triphosphate (ATP) production, and a reduction in oxidative stress. Inset: detailed view of the mitochondrial respiratory chain, and the five respiratory chain complexes denoted in Roman numerals. B. The glutamine-glutamate cycle between neurons and astrocytes is critical for the presynaptic biosynthesis of GABA, as well as providing glutamate that is converted by glutamate dehydrogenase to α-ketoglutarate which enters the TCA cycle as an intermediate87. Abbreviations: NAD+/NADH, oxidized/reduced forms of nicotinamide adenine dinucleotide; CytC, cytochrome C; G6P. glucose 6-phosphate; mPTP, mitochondrial permeability transition pore; TCA, tricarboxylic acid cycle; UCP, mitochondrial uncoupling protein; Gln, glutamine; Glu, glutamate; GABA, γ-aminobutyric acid;
Figure 7: Mechanisms Underlying the Neuroprotective Effects of Ketogenic and Related Diets.
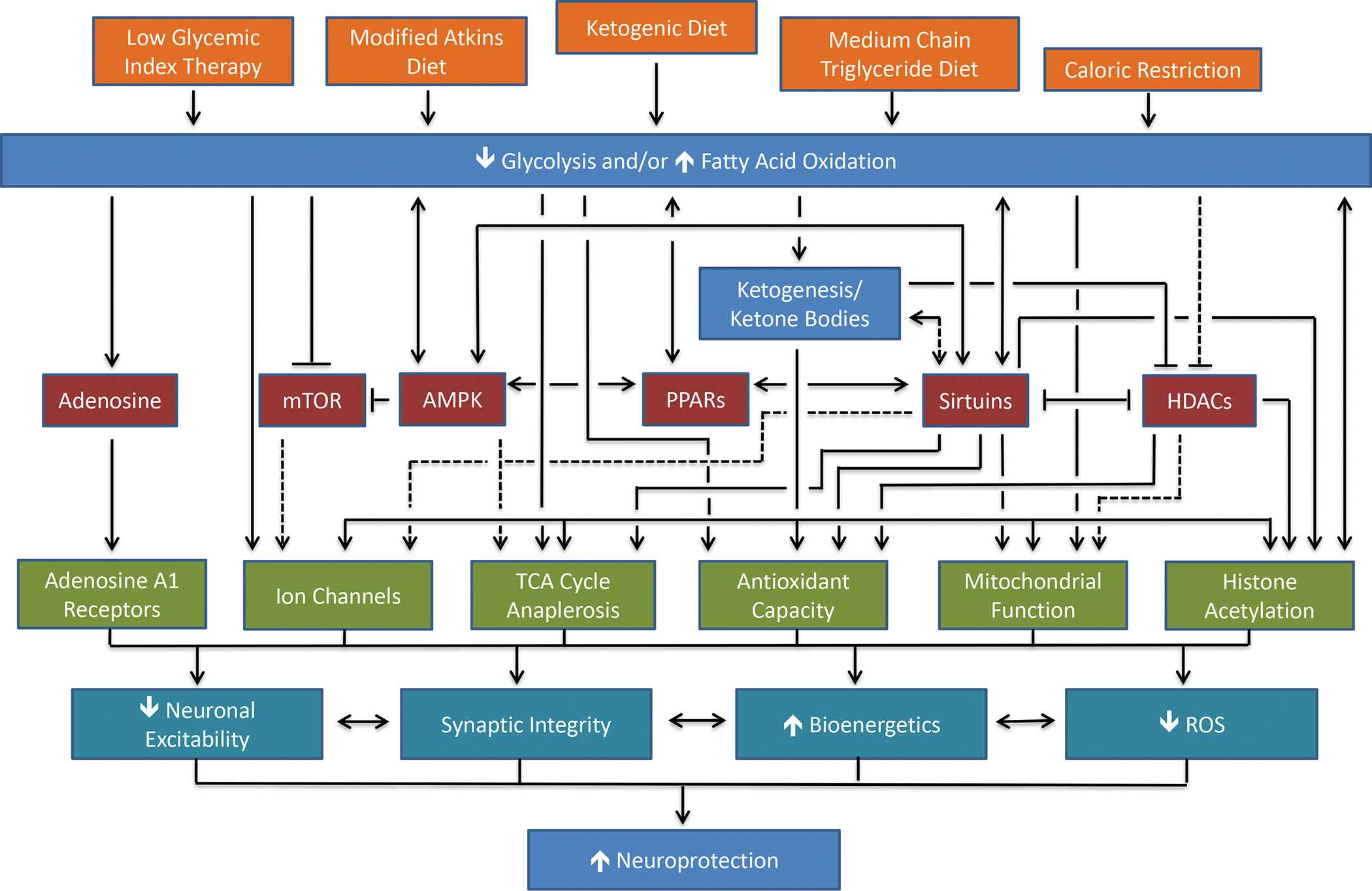
Proposed mechanisms for the neuroprotective effects of the ketogenic diet and its variants. The major dietary interventions are shown in orange, including caloric restriction which mirrors many but not all the biochemical or physiological effects of the exogenous diets. The metabolic effects of the various diets are shown in blue; the energy-sensing pathways that may mediate the effects of the dietary alterations are shown in red; the cellular effects resulting from the diets and/or the energy-sensing pathways are shown in green; and the broad protective effects of the diets and the resulting cellular effects are in shown cyan. Solid black lines indicate linkages detailed in the literature; dashed black lines represent possible, but as yet unproven links. Abbreviations: mTOR, mammalian target of rapamycin; AMPK, adenosine monophosphate-activated kinase; PPARs, peroxisome proliferator-activated receptors; HDACs, histone deacetylases; TCA, tricarboxylic acid cycle; ROS, reactive oxygen species. Modified with permission from Gano et al., Ketogenic Diets, Mitochondria and Neurological Diseases, Journal of Lipid Research, 2014. 55: 2211–2228.
From a therapeutic standpoint, detailed basic-translational studies of dietary therapies for epilepsy have not only strengthened the view that epilepsy in many ways can be considered a metabolic disease, but research has presented a few novel approaches that can theoretically be exploited for clinical development. A few examples illustrating the potential of certain metabolic substrates and enzymes as treatments for epilepsy are provided here. One example is 2-deoxy-D-glucose, which inhibits phosphoglucose isomerase, a key glycolytic enzyme, and which has been shown to possess relatively broad anti-seizure properties, to retard kindling (a model of epileptogenesis)172, and mitigate post-traumatic epilepsy173. However, the protective effects of 2-deoxy-D-glucose may not be simply due to inhibition of glycolysis as other mechanisms have been implicated161. Current clinical efforts – in part encouraged by strong safety data – are being directed toward the treatment of acute repetitive seizures and status epilepticus18. And more recently, a medium-chain triglyceride product has been shown to be effective in pediatric patients with drug-resistant epilepsy174.
As further proof of principle, the anti-epileptogenic potential of transient focal adenosine delivery was first tested in a rat model of systemic kainic acid induced epilepsy123. Young male rats were treated with the excitotoxin kainic acid to trigger epilepsy, and adenosine-releasing silk was implanted 9 weeks later into the lateral brain ventricles. Strikingly, epilepsy progression in the adenosine-group was completely suppressed and this effect was maintained far beyond the expiration of adenosine-release from the polymer – for at least 12 weeks with a reduction in seizure frequency by more than 70%. In addition, the progression of mossy fiber sprouting was also attenuated in the adenosine-treated rats. Mechanistically, the transient delivery of adenosine restored normal 5mC in the hippocampus long-term. A DNA methylation array analysis identified genes whose methylation status changed during epilepsy development and was restored back to normal after adenosine therapy. Among the target genes identified was the polymerase PolD and various DNA interacting proteins including zinc finger proteins123. Taken together, these findings suggest that transient therapeutic adenosine augmentation can restore normal DNA methylation patterns, thereby preventing epilepsy progression (Fig. 3). In this regard, it is important to note that adenosine has been strongly implicated in the mechanism of ketogenic diet action, and that the ketogenic diet has been shown to reverse DNA methylation changes in a chronic animal model of epilepsy169.
Subsequently, it was shown that the transient and systemic use of the ADK inhibitor 5-iodotuber-cidin (to increase adenosine) is antiepileptogenic in the intrahippocampal kainic acid mouse model of temporal lobe epilepsy175. Three days after kainic acid induced status epilepticus in mice, 5-iodotubercidin or vehicle was injected for a restricted time-span of 5 days. Six weeks after the status epilepticus, 94% of mice in the kainic acid/vehicle-injected control group developed robust seizures. In marked contrast, 59% of kainic acid/5-iodotubercidin-treated mice showed almost no seizure activity. These findings are in line with the anti-epileptogenic activity of adenosine that is mediated through an epigenetic mechanism123. Importantly, histopathological analysis demonstrated characteristic granule cell dispersion in KA/vehicle-injected control animals, which was prevented by 5-ITU175. Thus, the transient inhibition of ADK holds promise as a therapeutic strategy for epilepsy prevention.
Specifically, the nuclear and epigenetically active isoform ADK-L is a unique molecular and therapeutic target that links metabolism with developmental and epigenetic functions. It was previously demonstrated that a ketogenic diet leads to a reduction of ADK-L176, a potential mechanism which could explain the epigenetic and disease modifying properties of metabolic treatments169,177. Consistent with this function and therapeutic significance of ADK-L, efforts are currently under way to develop novel small molecule compounds to target ADK-L as potential therapeutic agents for epilepsy prevention178.
Additional strategies are currently pursued for the prevention of human epilepsy based on network pharmacology and the repurposing of FDA-approved drugs179,180. Those approaches include the antibiotic ceftriaxone180, which increases astroglial glutamate uptake via the glutamate transporter GLT-1. Interestingly, through interaction with the adenosine A2A receptor, adenosine has been shown to regulate GLT-1 expression and function181,182 as a possible mechanism through which maladaptive changes in adenosine metabolism during epileptogenesis could influence a downstream target amenable to correction by an existing FDA-approved drug.
In a different vein, ever since the 1930’s, investigators have long been interested in examining whether ketone bodies exert direct anti-seizure effects 183,184. Despite numerous studies in preclinical models, whether ketones are directly responsible for the anti-seizure effects of the ketogenic diet remains unclear, since peripheral ketone levels have not consistently correlated well with seizure control in patients with epilepsy183,185. Nevertheless, the growing evidence that beta-hydroxybutyrate can indirectly modulate KATP channels, act as endogenous ligands for free fatty acid receptors186, inhibits histone deacetylases166, regulate the mitochondrial permeability transition pore187, prevent NLRP3 inflammasome assembly167 – among many other actions188,189 – provides a compelling scientific rationale for exploring ways to induce ketosis without dietary manipulation, such as administrating ketone esters which obviate the challenges inherent in administering ketone salts to achieve low millimolar concentrations in the blood152.
Fatty acids have also drawn great attention in the study of ketogenic diet mechanisms given their important role as the key constituent of ketogenic diets described above. It has long been known that polyunsaturated fatty acids can directly inhibit voltage-gated sodium and calcium ion channels190, but perhaps more importantly they can activate the transcription factor peroxisome proliferator-activated receptor (PPAR) alpha which regulates genes involved in the beta-oxidation of fatty acids and more generally energy homeostasis191. Moreover, the ketogenic diet has been shown to increase PPAR gamma expression, which then leads to anti-inflammatory and anti-oxidant actions192. However, despite strong pre-clinical data, whether polyunsaturated fatty acid ingestion affords anti-seizure effects remain unclear193,194. More recently, medium-chain triglycerides, notably decanoic acid (a C10 medium-chain triglyceride) was shown to inhibit post-synaptic AMPA receptors195 much like the ASM perampanel196, but also increased mitochondrial content in neurons and inhibited mTORC1 activity independent of glucose and insulin signaling165. Indeed, medium chain triglycerides may represent a potential new metabolic treatment for epilepsy across several species, including humans197. For example, a prospective open-label feasibility study of a medical food containing a specific ratio of decanoic acid and octanoic acid revealed beneficial effects on seizure frequency in children with drug-resistant epilepsy174. Furthermore, triheptanoin – the triglyceride of heptanoic acid which promotes anaplerosis (the replenishment of substrates involved in the TCA cycle) – induced anti-seizure effects in both animal models198 and patients199 with epilepsy. Finally, ketogenic diet therapy led to an increase in adenosine176,177, which not only reduced seizure thresholds200 but also exerted potent disease-modifying and antiepileptogenic effects116,175. Collectively, whether certain ketone preparations, adenosine-laden delivery vehicles or fatty acid formulations ultimately prove feasible and effective for epilepsy therapeutics remains to be determined.
An intriguing question that has emerged from the epilepsy and metabolism literature is whether any current ASMs used in clinical practice affect metabolic pathways, actions that may in part explain their efficacy against epileptic seizures. Metabolic targets are not believed to be relevant to ASM mechanisms, with one notable exception. Stiripentol, an ASM that is believed primarily to allosterically modulate GABAA receptors, appears to exert anti-seizure effects via inhibition of lactate dehydrogenase88 (Fig. 5). This important intersection between traditional ASMs and brain metabolism (notably, the ANLS hypothesis summarized in Fig. 1) presents a broad opportunity to integrate concepts in both these areas for therapeutic innovation. Related to this is the notion that ASMs may act in part by reducing the energy needs of the epileptic brain, such that the spared energy can be redirected for normal neuronal signaling. Given that synaptic activity is highly energy demanding, the direct inhibitory effects of ASMs on neurotransmission may yield an important secondary benefit.
A more recent paradigm-shifting discovery has been the observation that the ketogenic diet may provide anti-seizure effects by modulating the gut microbiome – i.e., through specific changes in the abundance and composition of bacterial species that secondarily affect the blood-brain metabolome (e.g., decreasing gamma glutamyl amino acids) and influences brain levels of key neurotransmitters such as GABA and glutamate65. The gut microbiome, which has been increasingly linked to neurological and psychiatric disorders, now stands as a powerful integrator of metabolism and inflammation through the brain-gut axis.
Conclusions/Perspective
From both teleological and evolutionary perspectives, it is clear that cellular metabolism is one of the most important biological frameworks within which epileptic seizures arise. Indeed, without the fundamental substrates, enzymes and biochemical pathways that collectively constitute metabolic homeostasis or disruption in the brain, both normal and abnormal neuronal and glial activity would not be possible. In this sense and in a reductionist manner, epilepsy can be considered a metabolic disease, or more accurately, the heterogeneous epilepsies may represent electroclinical manifestations of complex perturbations in physiological networks fueled by basic biochemical processes. In recent years, this notion has been amplified by basic-translational as well as human studies revealing the metabolic underpinnings of seizure genesis and epileptogenesis. Moreover, the field of metabolism-based treatments for epilepsy has exploited our growing knowledge of brain metabolism to advance both empiric and highly reasoned approaches toward expanding therapeutic options. Additionally, paradigms for novel drug discovery have begun to explore the utility of metabolic readouts in various model systems142,201–204, and technical advances have made it possible for investigators to interrogate cellular metabolism in unprecedented ways – whether through the development of molecular biosensors of specific molecules or biochemical flux, or molecular imaging techniques that provide new insights into the complex inner workings of cells, with ramifications for cellular membrane physiology, epigenetic changes and inflammation3,205. As drug discovery for epilepsy has been hampered for decades by a true lack of understanding of the mechanisms responsible for drug refractoriness206 and given the sobering reality of unaltered treatment outcomes over many decades15, renewed and expanded attention to the metabolic basis of epilepsy might yield new avenues for greater therapeutic gain.
KEY POINTS.
Epileptic seizures induce widespread derangements in cellular and mitochondrial metabolism, as well as cerebral flood flow
Primary defects in genes encoding mitochondrial proteins and/or metabolic substrates and enzymes can result in neuronal and glial network excitability
Brain metabolic homeostasis and function should be viewed as an interplay between the cerebral circulation, glia and neurons
Epilepsy can be viewed as a metabolic disease, and primordial mechanisms evoked by compounds such as adenosine may be highly relevant to seizures and epileptogenesis
Metabolism-based treatments such as the high-fat ketogenic diet and its variants can help restore metabolic homeostasis and lead to seizure control
The metabolic underpinnings of epilepsy provide a rational explanation for its syndromic nature and plethora of symptoms, and explain why metabolic therapies can treat epilepsy comprehensively
As dietary therapies for epilepsy often control seizures in the medically intractable population, experimental therapeutics based on metabolic targets should be increasingly explored
Acknowledgements
JMR has been funded by the Canadian Institutes of Health Research and the NIH R21 NS104513, and DB by the NIH (R01NS103740, R01NS065957) and a CURE Epilepsy Catalyst Award. The authors wish to thank Rose Tobias for editorial assistance.
Footnotes
Competing interests
JMR has been a paid consultant to Aquestive Pharmaceuticals, Danone Nutricia, Mallinckrodt, Eisai Pharma, and Zogenix. JMR has also served on the Scientific Advisory Board of The Charlie Foundation for Ketogenic Therapies (Santa Monica, California, USA). DB is a co-founder of PrevEp Inc. and JMR is the Chief Medical Officer for Path Therapeutics, Inc.
Peer review information
Nature Reviews Neurology thanks C. Juhasz and K. Borges for their contribution to the peer review of this work.
References
- 1.Devinsky O et al. Epilepsy. Nat Rev Dis Primers 4, 18024, doi: 10.1038/nrdp.2018.24 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Hall CN, Klein-Flugge MC, Howarth C & Attwell D Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J Neurosci 32, 8940–8951, doi: 10.1523/JNEUROSCI.0026-12.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rangaraju V, Calloway N & Ryan TA Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835, doi: 10.1016/j.cell.2013.12.042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Otahal J, Folbergrova J, Kovacs R, Kunz WS & Maggio N Epileptic focus and alteration of metabolism. Int Rev Neurobiol 114, 209–243, doi: 10.1016/B978-0-12-418693-4.00009-1 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Zsurka G & Kunz WS Mitochondrial dysfunction and seizures: the neuronal energy crisis. The Lancet Neurology 14, 956–966, doi: 10.1016/s1474-4422(15)00148-9 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Vezzani A, French J, Bartfai T & Baram TZ The role of inflammation in epilepsy. Nat Rev Neurol 7, 31–40, doi:nrneurol.2010.178 [pii] 10.1038/nrneurol.2010.178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vezzani A, Pascente R & Ravizza T Biomarkers of Epileptogenesis: The Focus on Glia and Cognitive Dysfunctions. Neurochemical research 42, 2089–2098, doi: 10.1007/s11064-017-2271-3 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Eyo UB, Murugan M & Wu LJ Microglia-Neuron Communication in Epilepsy. Glia 65, 5–18, doi: 10.1002/glia.23006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lim A & Thomas RH The mitochondrial epilepsies. Eur J Paediatr Neurol 24, 47–52, doi: 10.1016/j.ejpn.2019.12.021 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Pearson-Smith JN & Patel M Metabolic Dysfunction and Oxidative Stress in Epilepsy. Int J Mol Sci 18, doi: 10.3390/ijms18112365 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conboy K, Henshall DC & Brennan GP Epigenetic principles underlying epileptogenesis and epilepsy syndromes. Neurobiol Dis 148, 105179, doi: 10.1016/j.nbd.2020.105179 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Pan JW et al. Neurometabolism in human epilepsy. Epilepsia 49 Suppl 3, 31–41, doi: 10.1111/j.1528-1167.2008.01508.x (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh S et al. Reviving mitochondrial bioenergetics: A relevant approach in epilepsy. Mitochondrion 58, 213–226, doi: 10.1016/j.mito.2021.03.009 (2021). [DOI] [PubMed] [Google Scholar]
- 14.Rogawski MA, Loscher W & Rho JM Mechanisms of Action of Antiseizure Drugs and the Ketogenic Diet. Cold Spring Harbor perspectives in medicine 6, doi: 10.1101/cshperspect.a022780 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Z, Brodie MJ, Liew D & Kwan P Treatment Outcomes in Patients With Newly Diagnosed Epilepsy Treated With Established and New Antiepileptic Drugs: A 30-Year Longitudinal Cohort Study. JAMA Neurol 75, 279–286, doi: 10.1001/jamaneurol.2017.3949 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rho JM & White HS Brief history of anti-seizure drug development. Epilepsia Open 3, 114–119, doi: 10.1002/epi4.12268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lynch BA et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A 101, 9861–9866, doi: 10.1073/pnas.0308208101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bialer M et al. Progress report on new antiepileptic drugs: A summary of the Fifteenth Eilat Conference on New Antiepileptic Drugs and Devices (EILAT XV). II. Drugs in more advanced clinical development. Epilepsia 61, 2365–2385, doi: 10.1111/epi.16726 (2020). [DOI] [PubMed] [Google Scholar]
- 19.Freeman J, Veggiotti P, Lanzi G, Tagliabue A & Perucca E The ketogenic diet: from molecular mechanisms to clinical effects. Epilepsy research 68, 145–180 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Neal EG et al. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. Lancet Neurol 7, 500–506 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Neal EG et al. A randomized trial of classical and medium-chain triglyceride ketogenic diets in the treatment of childhood epilepsy. Epilepsia 50, 1109–1117, doi: 10.1111/j.1528-1167.2008.01870.x (2009). [DOI] [PubMed] [Google Scholar]
- 22.Huttenlocher PR Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatric research 10, 536–540, doi: 10.1203/00006450-197605000-00006 (1976). [DOI] [PubMed] [Google Scholar]
- 23.Kossoff E et al. A Modified Atkins Diet Is Effective for the Treatment of Intractable Pediatric Epilepsy. Epilepsia 47, 421–424 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Muzykewicz DA et al. Efficacy, safety, and tolerability of the low glycemic index treatment in pediatric epilepsy. Epilepsia 50, 1118–1126, doi: 10.1111/j.1528-1167.2008.01959.x (2009). [DOI] [PubMed] [Google Scholar]
- 25.Martin-McGill KJ, Bresnahan R, Levy RG & Cooper PN Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst Rev 6, CD001903, doi: 10.1002/14651858.CD001903.pub5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gano LB, Patel M & Rho JM Ketogenic diets, mitochondria, and neurological diseases. Journal of lipid research 55, 2211–2228, doi: 10.1194/jlr.R048975 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sokoloff L et al. The [14C) Deoxyglucose method for the Measurement of local cerebral glucose utilization: Theory, Procedure, and Normal Values in the Concscious and Anesthetized Albino Rat. . Journal of Neurochemistry 28, 897–916 (1977). [DOI] [PubMed] [Google Scholar]
- 28.Belanger M, Allaman I & Magistretti PJ Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell metabolism 14, 724–738, doi: 10.1016/j.cmet.2011.08.016 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Engl E & Attwell D Non-signalling energy use in the brain. J Physiol 593, 3417–3429, doi: 10.1113/jphysiol.2014.282517 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Profaci CP, Munji RN, Pulido RS & Daneman R The blood-brain barrier in health and disease: Important unanswered questions. J Exp Med 217, doi: 10.1084/jem.20190062 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loscher W & Friedman A Structural, Molecular, and Functional Alterations of the Blood-Brain Barrier during Epileptogenesis and Epilepsy: A Cause, Consequence, or Both? Int J Mol Sci 21, doi: 10.3390/ijms21020591 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dienel GA & Carlson GM Major Advances in Brain Glycogen Research: Understanding of the Roles of Glycogen Have Evolved from Emergency Fuel Reserve to Dynamic, Regulated Participant in Diverse Brain Functions. Adv Neurobiol 23, 1–16, doi: 10.1007/978-3-030-27480-1_1 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Dienel GA Fueling and imaging brain activation. ASN Neuro 4, doi: 10.1042/AN20120021 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Owen OE et al. Brain metabolism during fasting. J Clin Invest 46, 1589–1595, doi: 10.1172/JCI105650 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hasselbalch SG et al. Brain Metabolism During Short-Term Starvation in Humans. Journal of Cerebral Blood Flow and Metabolism 14, 125–131 (1994). [DOI] [PubMed] [Google Scholar]
- 36.Bordone MP et al. The energetic brain - A review from students to students. J Neurochem 151, 139–165, doi: 10.1111/jnc.14829 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Boison D & Steinhauser C Epilepsy and astrocyte energy metabolism. Glia 66, 1235–1243, doi: 10.1002/glia.23247 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maher F, Vannucci SJ & Simpson IA Glucose transporter proteins in brain. FASEB 8, 1003–1011 (1994). [DOI] [PubMed] [Google Scholar]
- 39.Pellerin L & Magistretti PJ Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 91, 10625–10629 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Magistretti PJ & Allaman I Lactate in the brain: from metabolic end-product to signalling molecule. Nature reviews. Neuroscience 19, 235–249, doi: 10.1038/nrn.2018.19 (2018). [DOI] [PubMed] [Google Scholar]
- 41.Machler P et al. In Vivo Evidence for a Lactate Gradient from Astrocytes to Neurons. Cell metabolism 23, 94–102, doi: 10.1016/j.cmet.2015.10.010 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Felmlee MA, Jones RS, Rodriguez-Cruz V, Follman KE & Morris ME Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharmacol Rev 72, 466–485, doi: 10.1124/pr.119.018762 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bak LK et al. Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J Neurochem 109 Suppl 1, 87–93, doi: 10.1111/j.1471-4159.2009.05943.x (2009). [DOI] [PubMed] [Google Scholar]
- 44.Diaz-Garcia CM & Yellen G Neurons rely on glucose rather than astrocytic lactate during stimulation. J Neurosci Res 97, 883–889, doi: 10.1002/jnr.24374 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin AL, Fox PT, Hardies J, Duong TQ & Gao JH Nonlinear coupling between cerebral blood flow, oxygen consumption, and ATP production in human visual cortex. Proc Natl Acad Sci U S A 107, 8446–8451, doi: 10.1073/pnas.0909711107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tarczyluk MA et al. Functional astrocyte-neuron lactate shuttle in a human stem cell-derived neuronal network. J Cereb Blood Flow Metab 33, 1386–1393, doi: 10.1038/jcbfm.2013.81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Devinsky O, Vezzani A, Najjar S, De Lanerolle NC & Rogawski MA Glia and epilepsy: excitability and inflammation. Trends Neurosci 36, 174–184, doi: 10.1016/j.tins.2012.11.008 S0166-2236(12)00205-6 [pii] (2013). [DOI] [PubMed] [Google Scholar]
- 48.Robel S & Sontheimer H Glia as drivers of abnormal neuronal activity. Nature neuroscience 19, 28–33, doi: 10.1038/nn.4184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verhoog QP, Holtman L, Aronica E & van Vliet EA Astrocytes as Guardians of Neuronal Excitability: Mechanisms Underlying Epileptogenesis. Front Neurol 11, 591690, doi: 10.3389/fneur.2020.591690 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huguenard JR & McCormick DA Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci 30, 350–356, doi: 10.1016/j.tins.2007.05.007 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Lado FA & Moshe SL How do seizures stop? Epilepsia 49, 1651–1664 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yekhlef L, Breschi GL, Lagostena L, Russo G & Taverna S Selective activation of parvalbumin- or somatostatin-expressing interneurons triggers epileptic seizurelike activity in mouse medial entorhinal cortex. J Neurophysiol 113, 1616–1630, doi: 10.1152/jn.00841.2014 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Toyoda I, Fujita S, Thamattoor AK & Buckmaster PS Unit Activity of Hippocampal Interneurons before Spontaneous Seizures in an Animal Model of Temporal Lobe Epilepsy. J Neurosci 35, 6600–6618, doi: 10.1523/jneurosci.4786-14.2015 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Righes Marafiga J, Vendramin Pasquetti M & Calcagnotto ME GABAergic interneurons in epilepsy: More than a simple change in inhibition. Epilepsy Behav, 106935, doi: 10.1016/j.yebeh.2020.106935 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Dudek FE, Yasumura T & Rash JE Non-Synaptic Mechanisms in Seizures and Epileptogenesis. Cell Biology International 22, 793–805 (1998). [DOI] [PubMed] [Google Scholar]
- 56.Timofeev I, Bazhenov M, Seigneur J & Sejnowski T Neocortical synchronization. Epilepsia 51, 18, doi: 10.1111/j.1528-1167.2010.02804.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jefferys JG et al. Mechanisms of physiological and epileptic HFO generation. Prog Neurobiol 98, 250–264, doi: 10.1016/j.pneurobio.2012.02.005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shivacharan RS et al. Neural recruitment by ephaptic coupling in epilepsy. Epilepsia 62, 1505–1517, doi: 10.1111/epi.16903 (2021). [DOI] [PubMed] [Google Scholar]
- 59.Noebels J Pathway-driven discovery of epilepsy genes. Nature neuroscience 18, 344–350, doi: 10.1038/nn.3933 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blumcke I et al. Toward a refined genotype-phenotype classification scheme for the international consensus classification of Focal Cortical Dysplasia. Brain Pathol 31, e12956, doi: 10.1111/bpa.12956 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Barkovich AJ, Dobyns WB & Guerrini R Malformations of cortical development and epilepsy. Cold Spring Harbor perspectives in medicine 5, a022392, doi: 10.1101/cshperspect.a022392 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Becker AJ Review: Animal models of acquired epilepsy: insights into mechanisms of human epileptogenesis. Neuropathol Appl Neurobiol 44, 112–129, doi: 10.1111/nan.12451 (2018). [DOI] [PubMed] [Google Scholar]
- 63.Crino PB The mTOR signalling cascade: paving new roads to cure neurological disease. Nat Rev Neurol 12, 379–392, doi: 10.1038/nrneurol.2016.81 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Pitkanen A, Lukasiuk K, Dudek FE & Staley KJ Epileptogenesis. Cold Spring Harbor perspectives in medicine 5, doi: 10.1101/cshperspect.a022822 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Olson CA et al. The Gut Microbiota Mediates the Anti-Seizure Effects of the Ketogenic Diet. Cell 173, 1728–1741 e1713, doi: 10.1016/j.cell.2018.04.027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Loeb JA Identifying targets for preventing epilepsy using systems biology. Neurosci Lett 497, 205–212, doi: 10.1016/j.neulet.2011.02.041 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kirchner A, Dachet F & Loeb JA Identifying targets for preventing epilepsy using systems biology of the human brain. Neuropharmacology 168, 107757, doi: 10.1016/j.neuropharm.2019.107757 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McKee HR & Privitera MD Stress as a seizure precipitant: Identification, associated factors, and treatment options. Seizure 44, 21–26, doi: 10.1016/j.seizure.2016.12.009 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Walsh S et al. A systematic review of the risks factors associated with the onset and natural progression of epilepsy. Neurotoxicology 61, 64–77, doi: 10.1016/j.neuro.2016.03.011 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Chen T, Giri M, Xia Z, Subedi YN & Li Y Genetic and epigenetic mechanisms of epilepsy: a review. Neuropsychiatric disease and treatment 13, 1841–1859, doi: 10.2147/NDT.S142032 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qaiser F, Yuen RKC & Andrade DM Genetics of Epileptic Networks: from Focal to Generalized Genetic Epilepsies. Curr Neurol Neurosci Rep 20, 46, doi: 10.1007/s11910-020-01059-x (2020). [DOI] [PubMed] [Google Scholar]
- 72.Kobow K et al. Finding a better drug for epilepsy: antiepileptogenesis targets. Epilepsia 53, 1868–1876, doi: 10.1111/j.1528-1167.2012.03716.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Engel J Jr. & Pitkanen A Biomarkers for epileptogenesis and its treatment. Neuropharmacology 167, 107735, doi: 10.1016/j.neuropharm.2019.107735 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Duncan R Epilepsy, cerebral blood flow, and cerebral metabolic rate. Cerebrovasc Brain Metabob Rev 4, 105–121 (1992). [PubMed] [Google Scholar]
- 75.Sidhu MK, Duncan JS & Sander JW Neuroimaging in epilepsy. Curr Opin Neurol 31, 371–378, doi: 10.1097/WCO.0000000000000568 (2018). [DOI] [PubMed] [Google Scholar]
- 76.Goodman AM & Szaflarski JP Recent Advances in Neuroimaging of Epilepsy. Neurotherapeutics, doi: 10.1007/s13311-021-01049-y (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chugani HT Hypermetabolism on Pediatric Positron Emission Tomography Scans of Brai Glucose Metabolism: What does it signify? J Nucl Med, doi: 10.2967/jnumed.120.256081 (2021). [DOI] [PubMed] [Google Scholar]
- 78.Alkonyi B, Chugani HT & Juhasz C Transient focal cortical increase of interictal glucose metabolism in Sturge-Weber syndrome: implications for epileptogenesis. Epilepsia 52, 1265–1272, doi: 10.1111/j.1528-1167.2011.03066.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Juhasz C, Hu J, Xuan Y & Chugani HT Imaging increased glutamate in children with SturgeWeber syndrome: Association with epilepsy severity. Epilepsy research 122, 66–72, doi: 10.1016/j.eplepsyres.2016.02.010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nelissen N et al. Correlations of interictal FDG-PET metabolism and ictal SPECT perfusion changes in human temporal lobe epilepsy with hippocampal sclerosis. Neuroimage 32, 684–695, doi: 10.1016/j.neuroimage.2006.04.185 (2006). [DOI] [PubMed] [Google Scholar]
- 81.Rakhade SN & Jensen FE Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol 5, 380–391, doi: 10.1038/nrneurol.2009.80 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ingvar M & Siesjo BK Local blood flow and glucose consumption in the rat brain during sustained bicuculline-induced seizures. Acta Neurol Scand 68, 129–144, doi: 10.1111/j.1600-0404.1983.tb05339.x (1983). [DOI] [PubMed] [Google Scholar]
- 83.Theodore WH Cerebral blood flow and glucose metabolism in human epilepsy. Adv Neurol 79, 873–881 (1999). [PubMed] [Google Scholar]
- 84.Shultz SR, O’Brien TJ, Stefanidou M & Kuzniecky RI Neuroimaging the epileptogenic process. Neurotherapeutics 11, 347–357, doi: 10.1007/s13311-014-0258-1 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.During MJ, Fried I, Leone P, Katz A & Spencer DD Direct measurement of extracellular lactate in the human hippocampus during spontaneous seizures. J Neurochem 62, 2356–2361, doi: 10.1046/j.1471-4159.1994.62062356.x (1994). [DOI] [PubMed] [Google Scholar]
- 86.Boison D & Yegutkin GG Adenosine Metabolism: Emerging Concepts for Cancer Therapy. Cancer Cell 36, 582–596, doi: 10.1016/j.ccell.2019.10.007 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dienel GA Brain Glucose Metabolism: Integration of Energetics with Function. Physiol Rev 99, 949–1045, doi: 10.1152/physrev.00062.2017 (2019). [DOI] [PubMed] [Google Scholar]
- 88.Sada N, Lee S, Katsu T, Otsuki T & Inoue T Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 347, 1362–1367, doi: 10.1126/science.aaa1299 (2015). [DOI] [PubMed] [Google Scholar]
- 89.McDonald TS & Borges K Impaired hippocampal glucose metabolism during and after flurothyl-induced seizures in mice: Reduced phosphorylation coincides with reduced activity of pyruvate dehydrogenase. Epilepsia 58, 1172–1180, doi: 10.1111/epi.13796 (2017). [DOI] [PubMed] [Google Scholar]
- 90.Bhandary S & Aguan K Pyruvate dehydrogenase complex deficiency and its relationship with epilepsy frequency--An overview. Epilepsy research 116, 40–52, doi: 10.1016/j.eplepsyres.2015.07.002 (2015). [DOI] [PubMed] [Google Scholar]
- 91.McDonald TS, Carrasco-Pozo C, Hodson MP & Borges K Alterations in Cytosolic and Mitochondrial [U-(13)C]Glucose Metabolism in a Chronic Epilepsy Mouse Model. eNeuro 4, doi: 10.1523/ENEURO.0341-16.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liang LP, Ho YS & Patel M Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience 101, 563–570, doi: 10.1016/s0306-4522(00)00397-3 (2000). [DOI] [PubMed] [Google Scholar]
- 93.Bainbridge MN et al. Analyses of SLC13A5-epilepsy patients reveal perturbations of TCA cycle. Mol Genet Metab 121, 314–319, doi: 10.1016/j.ymgme.2017.06.009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Smeland OB, Hadera MG, McDonald TS, Sonnewald U & Borges K Brain mitochondrial metabolic dysfunction and glutamate level reduction in the pilocarpine model of temporal lobe epilepsy in mice. J Cereb Blood Flow Metab 33, 1090–1097, doi: 10.1038/jcbfm.2013.54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kunz WS, Goussakov IV, Beck H & Elger CE Altered mitochondrial oxidative phosphorylation in hippocampal slices of kainate-treated rats. Brain Res 826, 236–242, doi:S0006-8993(99)01279-2 [pii] (1999). [DOI] [PubMed] [Google Scholar]
- 96.Folbergrova J, Jesina P, Haugvicova R, Lisy V & Houstek J Sustained deficiency of mitochondrial complex I activity during long periods of survival after seizures induced in immature rats by homocysteic acid. Neurochem Int 56, 394–403, doi: 10.1016/j.neuint.2009.11.011 (2010). [DOI] [PubMed] [Google Scholar]
- 97.Ryan K, Backos DS, Reigan P & Patel M Post-translational oxidative modification and inactivation of mitochondrial complex I in epileptogenesis. J Neurosci 32, 11250–11258, doi: 10.1523/JNEUROSCI.0907-12.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rowley S & Patel M Mitochondrial involvement and oxidative stress in temporal lobe epilepsy. Free radical biology & medicine 62, 121–131, doi: 10.1016/j.freeradbiomed.2013.02.002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rahman S Mitochondrial disease and epilepsy. Dev Med Child Neurol 54, 397–406, doi: 10.1111/j.1469-8749.2011.04214.x (2012). [DOI] [PubMed] [Google Scholar]
- 100.Williams S, Hamil N, Abramov AY, Walker MC & Kovac S Status epilepticus results in persistent overproduction of reactive oxygen species, inhibition of which is neuroprotective. Neuroscience 303, 160–165, doi: 10.1016/j.neuroscience.2015.07.005 (2015). [DOI] [PubMed] [Google Scholar]
- 101.Patel M, Li QY, Chang LY, Crapo J & Liang LP Activation of NADPH oxidase and extracellular superoxide production in seizure-induced hippocampal damage. J Neurochem 92, 123–131, doi: 10.1111/j.1471-4159.2004.02838.x (2005). [DOI] [PubMed] [Google Scholar]
- 102.Patel M Mitochondrial dysfunction and oxidative stress: cause and consequence of epileptic seizures. Free radical biology & medicine 37, 1951–1962, doi: 10.1016/j.freeradbiomed.2004.08.021 (2004). [DOI] [PubMed] [Google Scholar]
- 103.Jarrett SG, Liang LP, Hellier JL, Staley KJ & Patel M Mitochondrial DNA damage and impaired base excision repair during epileptogenesis. Neurobiol Dis 30, 130–138, doi: 10.1016/j.nbd.2007.12.009 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Patsoukis N et al. Thiol redox state and lipid and protein oxidation in the mouse striatum after pentylenetetrazol-induced epileptic seizure. Epilepsia 46, 1205–1211, doi: 10.1111/j.1528-1167.2005.63704.x (2005). [DOI] [PubMed] [Google Scholar]
- 105.Mueller SG, Trabesinger AH, Boesiger P & Wieser HG Brain glutathione levels in patients with epilepsy measured by in vivo (1)H-MRS. Neurology 57, 1422–1427, doi: 10.1212/wnl.57.8.1422 (2001). [DOI] [PubMed] [Google Scholar]
- 106.Liang LP & Patel M Seizure-induced changes in mitochondrial redox status. Free radical biology & medicine 40, 316–322, doi: 10.1016/j.freeradbiomed.2005.08.026 (2006). [DOI] [PubMed] [Google Scholar]
- 107.Beltran Gonzalez AN, Lopez Pazos MI & Calvo DJ Reactive Oxygen Species in the Regulation of the GABA Mediated Inhibitory Neurotransmission. Neuroscience 439, 137–145, doi: 10.1016/j.neuroscience.2019.05.064 (2020). [DOI] [PubMed] [Google Scholar]
- 108.Trevelyan AJ, Sussillo D & Yuste R Feedforward inhibition contributes to the control of epileptiform propagation speed. J Neurosci 27, 3383–3387, doi: 10.1523/JNEUROSCI.0145-07.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Pallafacchina G, Zanin S & Rizzuto R Recent advances in the molecular mechanism of mitochondrial calcium uptake. F1000Res 7, doi: 10.12688/f1000research.15723.1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Giorgi C et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 12, 77–85, doi: 10.1016/j.mito.2011.07.004 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pathak T & Trebak M Mitochondrial Ca(2+) signaling. Pharmacol Ther 192, 112–123, doi: 10.1016/j.pharmthera.2018.07.001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Newby AC Adenosine and the concept of ‘retaliatory metabolites’. Trends Biochem Sci 9, 42–44 (1984). [Google Scholar]
- 113.During MJ & Spencer DD Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann Neurol 32, 618–624 (1992). [DOI] [PubMed] [Google Scholar]
- 114.Dragunow M, Goddard GV & Laverty R Is adenosine an endogenous anticonvulsant? Epilepsia 26, 480–487 (1985). [DOI] [PubMed] [Google Scholar]
- 115.Boison D et al. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci USA 99, 6985–6990 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Williams-Karnesky RL et al. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Inv 123, 3552–3563 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mohler H & Okada T Benzodiazepine receptor: demonstration in the central nervous system. Science 198, 849–851 (1977). [DOI] [PubMed] [Google Scholar]
- 118.Klein P et al. Commonalities in epileptogenic processes from different acute brain insults: Do they translate? Epilepsia 59, 37–66, doi: 10.1111/epi.13965 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fredholm BB, Chen JF, Cunha RA, Svenningsson P & Vaugeois JM Adenosine and brain function. Int Rev Neurobiol 63, 191–270 (2005). [DOI] [PubMed] [Google Scholar]
- 120.Fredholm BB, Chen JF, Masino SA & Vaugeois JM Actions of adenosine at its receptors in the CNS: Insights from knockouts and drugs. Annu Rev Pharmacol Toxicol 45, 385–412 (2005). [DOI] [PubMed] [Google Scholar]
- 121.Fredholm BB, Ijzerman AP, Jacobson KA, Linden J & Muller CE International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors--an update. Pharmacol Rev 63, 1–34, doi:pr.110.003285 [pii] 10.1124/pr.110.003285 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Boison D Adenosine kinase: exploitation for therapeutic gain. Pharmacological Reviews 65, 906–943 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Williams-Karnesky RL et al. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Invest 123, 3552–3563, doi: 10.1172/JCI65636 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kluger G et al. Pyridoxine-dependent epilepsy: normal outcome in a patient with late diagnosis after prolonged status epilepticus causing cortical blindness. Neuropediatrics 39, 276–279, doi: 10.1055/s-0029-1202833 (2008). [DOI] [PubMed] [Google Scholar]
- 125.Malaspina P et al. Succinic semialdehyde dehydrogenase deficiency (SSADHD): Pathophysiological complexity and multifactorial trait associations in a rare monogenic disorder of GABA metabolism. Neurochem Int 99, 72–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tremino L, Forcada-Nadal A & Rubio V Insight into vitamin B6 -dependent epilepsy due to PLPBP (previously PROSC) missense mutations. Hum Mutat 39, 1002–1013, doi: 10.1002/humu.23540 (2018). [DOI] [PubMed] [Google Scholar]
- 127.Mills PB et al. Epilepsy due to PNPO mutations: genotype, environment and treatment affect presentation and outcome. Brain 137, 1350–1360, doi: 10.1093/brain/awu051 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mills PB et al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5’-phosphate oxidase. Hum Mol Genet 14, 1077–1086, doi: 10.1093/hmg/ddi120 (2005). [DOI] [PubMed] [Google Scholar]
- 129.Pope S, Artuch R, Heales S & Rahman S Cerebral folate deficiency: Analytical tests and differential diagnosis. J Inherit Metab Dis 42, 655–672, doi: 10.1002/jimd.12092 (2019). [DOI] [PubMed] [Google Scholar]
- 130.Alonso-Aperte E, Ubeda N, Achon M, Perez-Miguelsanz J & Varela-Moreiras G Impaired methionine synthesis and hypomethylation in rats exposed to valproate during gestation. Neurology 52, 750–756, doi: 10.1212/wnl.52.4.750 (1999). [DOI] [PubMed] [Google Scholar]
- 131.Applegarth DA & Toone JR Glycine encephalopathy (nonketotic hyperglycinaemia) : review and update. J Inherit Metab Dis 27, 417–422, doi: 10.1023/b:boli.0000031222.38328.59 (2004). [DOI] [PubMed] [Google Scholar]
- 132.Labrie V & Roder JC The involvement of the NMDA receptor D-serine/glycine site in the pathophysiology and treatment of schizophrenia. Neurosci Biobehav Rev 34, 351–372 (2010). [DOI] [PubMed] [Google Scholar]
- 133.Bergeron R, Meyer TM, Coyle JT & Greene RW Modulation of N-methyl-D-aspartate receptor function by glycine transport. Proc Natl Acad Sci U S A 95, 15730–15734. (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gramer G et al. Glucose transporter-1 (GLUT1) deficiency syndrome: diagnosis and treatment in late childhood. Neuropediatrics 43, 168–171, doi: 10.1055/s-0032-1315433 (2012). [DOI] [PubMed] [Google Scholar]
- 135.Gloyn AL et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 350, 1838–1849 (2004). [DOI] [PubMed] [Google Scholar]
- 136.Shimomura K et al. A novel mutation causing DEND syndrome: A treatable channelopathy of pancreas and brain. Neurology 69, 1342–1349 (2007). [DOI] [PubMed] [Google Scholar]
- 137.Olson TM & Terzic A Human KATP channelopathies: diseases of metabolic homeostasis. Pflugers Arch 460, 295–306 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Braissant O, McLin VA & Cudalbu C Ammonia toxicity to the brain. J Inherit Metab Dis 36, 595–612 (2013). [DOI] [PubMed] [Google Scholar]
- 139.Llansola M et al. NMDA receptors in hyperammonemia and hepatic encephalopathy. Metab Brain Dis 22, 321–335, doi: 10.1007/s11011-007-9067-0 (2007). [DOI] [PubMed] [Google Scholar]
- 140.Lax NZ, Gorman GS & Turnbull DM Review: Central nervous system involvement in mitochondrial disease. Neuropathol Appl Neurobiol 43, 102–118, doi: 10.1111/nan.12333 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bindoff LA & Engelsen BA Mitochondrial diseases and epilepsy. Epilepsia 53 Suppl 4, 92–97, doi: 10.1111/j.1528-1167.2012.03618.x (2012). [DOI] [PubMed] [Google Scholar]
- 142.Chan F et al. The role of astrocytes in seizure generation: insights from a novel in vitro seizure model based on mitochondrial dysfunction. Brain 142, 391–411, doi: 10.1093/brain/awy320 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lax NZ et al. Extensive respiratory chain defects in inhibitory interneurones in patients with mitochondrial disease. Neuropathol Appl Neurobiol 42, 180–193, doi: 10.1111/nan.12238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kossoff EH et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 3, 175–192, doi: 10.1002/epi4.12225 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pfeifer HH & Thiele EA Low-glycemic-index treatment: a liberalized ketogenic diet for treatment of intractable epilepsy. Neurology 65, 1810–1812, doi: 10.1212/01.wnl.0000187071.24292.9e (2005). [DOI] [PubMed] [Google Scholar]
- 146.Kim SH et al. The ketogenic diet in children 3 years of age or younger: a 10-year single-center experience. Sci Rep 9, 8736, doi: 10.1038/s41598-019-45147-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lyons L, Schoeler NE, Langan D & Cross JH Use of ketogenic diet therapy in infants with epilepsy: A systematic review and meta-analysis. Epilepsia 61, 1261–1281, doi: 10.1111/epi.16543 (2020). [DOI] [PubMed] [Google Scholar]
- 148.Husari KS & Cervenka MC The ketogenic diet all grown up-Ketogenic diet therapies for adults. Epilepsy research 162, 106319, doi: 10.1016/j.eplepsyres.2020.106319 (2020). [DOI] [PubMed] [Google Scholar]
- 149.Sondhi V & Gulati S Efficacy of 3 Major Ketogenic Diet Therapies in Children With Drug-Resistant Epilepsy-Reply. JAMA Pediatr 175, 434–435, doi: 10.1001/jamapediatrics.2020.5451 (2021). [DOI] [PubMed] [Google Scholar]
- 150.Sourbron J et al. Ketogenic diet for the treatment of pediatric epilepsy: review and meta-analysis. Childs Nerv Syst 36, 1099–1109, doi: 10.1007/s00381-020-04578-7 (2020). [DOI] [PubMed] [Google Scholar]
- 151.Boison D New insights into the mechanisms of the ketogenic diet. Curr Opin Neurol, doi: 10.1097/wco.0000000000000432 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Poff AM, Rho JM & D’Agostino DP Ketone Administration for Seizure Disorders: History and Rationale for Ketone Esters and Metabolic Alternatives. Front Neurosci 13, 1041, doi: 10.3389/fnins.2019.01041 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.McDonald T, Puchowicz M & Borges K Impairments in Oxidative Glucose Metabolism in Epilepsy and Metabolic Treatments Thereof. Front Cell Neurosci 12, 274, doi: 10.3389/fncel.2018.00274 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.DeVivo DC, Leckie MP, Ferrendelli JS & McDougal DB Jr. Chronic ketosis and cerebral metabolism. Ann Neurol 3, 331–337, doi: 10.1002/ana.410030410 (1978). [DOI] [PubMed] [Google Scholar]
- 155.Bough KJ et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann Neurol 60, 223–235 (2006). [DOI] [PubMed] [Google Scholar]
- 156.Kim DY, Vallejo J & Rho JM Ketones prevent synaptic dysfunction induced by mitochondrial respiratory complex inhibitors. J Neurochem 114, 130–141, doi: 10.1111/j.1471-4159.2010.06728.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Milder JB, Liang LP & Patel M Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol Dis 40, 238–244, doi: 10.1016/j.nbd.2010.05.030 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Juge N et al. Metabolic control of vesicular glutamate transport and release. Neuron 68, 99–112, doi:S0896-6273(10)00719-1 [pii] 10.1016/j.neuron.2010.09.002 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kawamura M Jr., Ruskin DN & Masino SA Metabolic autocrine regulation of neurons involves cooperation among pannexin hemichannels, adenosine receptors, and KATP channels. J Neurosci 30, 3886–3895 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Ma W, Berg J & Yellen G Ketogenic diet metabolites reduce firing in central neurons by opening K(ATP) channels. J Neurosci 27, 3618–3625, doi:27/14/3618 [pii] 10.1523/JNEUROSCI.0132-07.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shao LR, Rho JM & Stafstrom CE Glycolytic inhibition: A novel approach toward controlling neuronal excitability and seizures. Epilepsia Open 3, 191–197, doi: 10.1002/epi4.12251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gimenez-Cassina A et al. BAD-dependent regulation of fuel metabolism and K(ATP) channel activity confers resistance to epileptic seizures. Neuron 74, 719–730, doi: 10.1016/j.neuron.2012.03.032 S0896-6273(12)00343-1 [pii] (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lian XY, Khan FA & Stringer JL Fructose-1,6-bisphosphate has anticonvulsant activity in models of acute seizures in adult rats. J Neurosci 27, 12007–12011, doi: 10.1523/JNEUROSCI.3163-07.2007 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.McDaniel SS, Rensing NR, Thio LL, Yamada KA & Wong M The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia 52, e7–11, doi: 10.1111/j.1528-1167.2011.02981.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Warren EC et al. Decanoic acid inhibits mTORC1 activity independent of glucose and insulin signaling. Proc Natl Acad Sci U S A 117, 23617–23625, doi: 10.1073/pnas.2008980117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shimazu T et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 339, 211–214, doi: 10.1126/science.1227166 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Youm YH et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nature medicine 21, 263–269, doi: 10.1038/nm.3804 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Koh MY, Lim KS, Fong SL, Khor SB & Tan CT Impact of COVID-19 on quality of life in people with epilepsy, and a multinational comparison of clinical and psychological impacts. Epilepsy Behav 117, 107849, doi: 10.1016/j.yebeh.2021.107849 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kobow K et al. Deep sequencing reveals increased DNA methylation in chronic rat epilepsy. Acta neuropathologica 126, 741–756, doi: 10.1007/s00401-013-1168-8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Augustin K et al. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol 17, 84–93, doi: 10.1016/S1474-4422(17)30408-8 (2018). [DOI] [PubMed] [Google Scholar]
- 171.Elamin M, Ruskin DN, Sacchetti P & Masino SA A unifying mechanism of ketogenic diet action: The multiple roles of nicotinamide adenine dinucleotide. Epilepsy research 167, 106469, doi: 10.1016/j.eplepsyres.2020.106469 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Garriga-Canut M et al. 2-Deoxy-D-glucose reduces epilepsy progression by NRSF-CtBP-dependent metabolic regulation of chromatin structure. Nature neuroscience 9, 1382–1387 (2006). [DOI] [PubMed] [Google Scholar]
- 173.Koenig JB et al. Glycolytic inhibitor 2-deoxyglucose prevents cortical hyperexcitability after traumatic brain injury. JCI Insight 5, doi: 10.1172/jci.insight.126506 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Schoeler NE et al. K.Vita: a feasibility study of a blend of medium chain triglycerides to manage drug-resistant epilepsy. Brain Commun in press: 10.1093/braincomms/fcab160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sandau US et al. Transient use of a systemic adenosine kinase inhibitor attenuates epilepsy development in mice. Epilepsia 60, 615–625, doi: 10.1111/epi.14674 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Masino SA et al. A ketogenic diet suppresses seizures in mice through adenosine A1 receptors. J Clin Inv 121, 2679–2683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Lusardi TA et al. Ketogenic diet prevents epileptogenesis and disease progression in adult mice and rats. Neuropharmacology 99, 500–509, doi: 10.1016/j.neuropharm.2015.08.007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Toti KS, Osborne D, Ciancetta A, Boison D & Jacobson KA South (S)- and North (N)-Methanocarba-7-Deazaadenosine Analogues as Inhibitors of Human Adenosine Kinase. Journal of Medicinal Chemistry 59, 6860–6877 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Schidlitzki A et al. Proof-of-concept that network pharmacology is effective to modify development of acquired temporal lobe epilepsy. Neurobiol Dis 134, 104664, doi: 10.1016/j.nbd.2019.104664 (2020). [DOI] [PubMed] [Google Scholar]
- 180.Welzel L et al. Network pharmacology for antiepileptogenesis: Tolerability and neuroprotective effects of novel multitargeted combination treatments in nonepileptic vs. post-status epilepticus mice. Epilepsy research 151, 48–66, doi: 10.1016/j.eplepsyres.2019.02.010 (2019). [DOI] [PubMed] [Google Scholar]
- 181.Matos M et al. Deletion of adenosine A2A receptors from astrocytes disrupts glutamate homeostasis leading to psychomotor and cognitive impairment: relevance to schizophrenia. Biological psychiatry, doi: 10.1016/j.biopsych.2015.02.026 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Matos M et al. Adenosine A2A receptors modulate glutamate uptake in cultured astrocytes and gliosomes. Glia 60, 702–716, doi: 10.1002/glia.22290 (2012). [DOI] [PubMed] [Google Scholar]
- 183.Simeone TA, Simeone KA, Stafstrom CE & Rho JM Do ketone bodies mediate the anti-seizure effects of the ketogenic diet? Neuropharmacology 133, 233–241, doi: 10.1016/j.neuropharm.2018.01.011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Erecinska M, Nelson D, Daikhin Y & Yudkoff M Regulation of GABA level in rat brain synaptosomes: fluxes through enzymes of the GABA shunt and effects of glutamate, calcium, and ketone bodies. J Neurochem 67, 2325–2334 (1996). [DOI] [PubMed] [Google Scholar]
- 185.Buchhalter JR et al. The relationship between d-beta-hydroxybutyrate blood concentrations and seizure control in children treated with the ketogenic diet for medically intractable epilepsy. Epilepsia Open 2, 317–321, doi: 10.1002/epi4.12058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Won YJ, Lu VB, Puhl HL 3rd & Ikeda SR beta-Hydroxybutyrate modulates N-type calcium channels in rat sympathetic neurons by acting as an agonist for the G-protein-coupled receptor FFA3. J Neurosci 33, 19314–19325, doi: 10.1523/jneurosci.3102-13.2013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kim DY et al. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann Neurol 78, 77–87, doi: 10.1002/ana.24424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Puchalska P & Crawford PA Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell metabolism 25, 262–284, doi: 10.1016/j.cmet.2016.12.022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Moller N Ketone Body, 3-Hydroxybutyrate: Minor Metabolite - Major Medical Manifestations. J Clin Endocrinol Metab 105, doi: 10.1210/clinem/dgaa370 (2020). [DOI] [PubMed] [Google Scholar]
- 190.Elinder F & Liin SI Actions and Mechanisms of Polyunsaturated Fatty Acids on Voltage-Gated Ion Channels. Front Physiol 8, 43, doi: 10.3389/fphys.2017.00043 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bordoni A, Di Nunzio M, Danesi F & Biagi PL Poly unsatured fatty acids: From Diet to Binding to Ppars and other Nuclear Receptors. . Genes & Nutrition 1, 95–106 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Simeone TA, Matthews SA, Samson KK & Simeone KA Regulation of brain PPARgamma2 contributes to ketogenic diet anti-seizure efficacy. Exp Neurol 287, 54–64, doi: 10.1016/j.expneurol.2016.08.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bromfield E et al. A randomized trial of polyunsaturated fatty acids for refractory epilepsy. Epilepsy Behav 12, 187–190, doi: 10.1016/j.yebeh.2007.09.011 (2008). [DOI] [PubMed] [Google Scholar]
- 194.Sarmento Vasconcelos V et al. Polyunsaturated fatty acid supplementation for drug-resistant epilepsy. Cochrane Database Syst Rev, CD011014, doi: 10.1002/14651858.CD011014.pub2 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chang P et al. Seizure control by decanoic acid through direct AMPA receptor inhibition. Brain 139, 431–443, doi: 10.1093/brain/awv325 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Loscher W & Schmidt D Epilepsy: perampanel-new promise for refractory epilepsy? Nat Rev Neurol 8, 661–662, doi: 10.1038/nrneurol.2012.222 (2012). [DOI] [PubMed] [Google Scholar]
- 197.Han FY et al. Dietary medium chain triglycerides for management of epilepsy: New data from human, dog, and rodent studies. Epilepsia 62, 1790–1806, doi: 10.1111/epi.16972 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Willis S, Stoll J, Sweetman L & Borges K Anticonvulsant effects of a triheptanoin diet in two mouse chronic seizure models. Neurobiol Dis 40, 565–572, doi: 10.1016/j.nbd.2010.07.017 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Calvert S, Barwick K, Par M, Ni Tan K & Borges K A pilot study of add-on oral triheptanoin treatment for children with medically refractory epilepsy. Eur J Paediatr Neurol, doi: 10.1016/j.ejpn.2018.07.014 (2018). [DOI] [PubMed] [Google Scholar]
- 200.Boison D Methylxanthines, seizures and excitotoxicity. Handb Exp Pharmacol 200, 251–266 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kumar MG et al. Altered Glycolysis and Mitochondrial Respiration in a Zebrafish Model of Dravet Syndrome. eNeuro 3, doi: 10.1523/ENEURO.0008-16.2016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ibhazehiebo K et al. A novel metabolism-based phenotypic drug discovery platform in zebrafish uncovers HDACs 1 and 3 as a potential combined anti-seizure drug target. Brain 141, 744–761, doi: 10.1093/brain/awx364 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ibhazehiebo K, Rho JM & Kurrasch DM Metabolism-based drug discovery in zebrafish: An emerging strategy to uncover new anti-seizure therapies. Neuropharmacology 167, 107988, doi: 10.1016/j.neuropharm.2020.107988 (2020). [DOI] [PubMed] [Google Scholar]
- 204.Banerji R et al. Enhancing glucose metabolism via gluconeogenesis is therapeutic in a zebrafish model of Dravet syndrome. Brain Commun 3, fcab004, doi: 10.1093/braincomms/fcab004 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Koveal D, Diaz-Garcia CM & Yellen G Fluorescent Biosensors for Neuronal Metabolism and the Challenges of Quantitation. Curr Opin Neurobiol 63, 111–121, doi: 10.1016/j.conb.2020.02.011 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Loscher W & Schmidt D Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia 52, 657–678, doi: 10.1111/j.1528-1167.2011.03024.x (2011). [DOI] [PubMed] [Google Scholar]