

Summary
Early-life immune development is critical to long-term host health. However, the mechanisms that determine the pace of postnatal immune maturation are not fully resolved. Here, we analyzed mononuclear phagocytes (MNPs) in small intestinal Peyer’s patches (PPs), the primary inductive site of intestinal immunity. Conventional type 1 and 2 dendritic cells (cDC1 and cDC2) and RORgt+ antigen-presenting cells (RORgt+ APC) exhibited significant age-dependent changes in subset composition, tissue distribution, and reduced cell maturation, subsequently resulting in a lack in CD4+ T cell priming during the postnatal period. Microbial cues contributed but could not fully explain the discrepancies in MNP maturation. Type I interferon (IFN) accelerated MNP maturation but IFN signaling did not represent the physiological stimulus. Instead, follicle-associated epithelium (FAE) M cell differentiation was required and sufficient to drive postweaning PP MNP maturation. Together, our results highlight the role of FAE M cell differentiation and MNP maturation in postnatal immune development.
Keywords: neonatal immunology, dendritic cell, Peyer's patch, microbiota, post-natal establishment of intestinal homeostasis, cDC2, RORgt+ APC, M cell
Graphical abstract
Highlights
-
•
Peyer’s patch (PP) CD4+ T cells exhibit delayed maturation after birth
-
•
cDC1 and RORgt+ APC are increased whereas cDC2 are diminished in the neonatal PP
-
•
PP APCs exhibit halted maturation and cDC2 show reduced T cell priming capacity
-
•
M cell development is required and sufficient to drive APC maturation
Despite the critical importance of immune development in early life, our mechanistic understanding of this process is limited. Torow et al. demonstrate that postnatal establishment of intestinal microbe-host interaction is not expressly driven by early microbial colonization, but rather the maturation of Peyer’s patch M cells and antigen-presenting cells.
Introduction
The neonatal immune system differs in many aspects from the adult in both mice and men. This reflects the ongoing tissue development and stepwise processes that ultimately establish the self-controlled adaptive immune system.1,2 However, even though post-natal immune maturation impacts life-long host-microbial homeostasis and health, the mechanisms determining mucosal immune maturation are incompletely understood.
Early-life developmental processes contribute to cell homing and differentiation, tissue microarchitecture, and organ function. Environmental cues mainly represented by microbiota-derived signals stimulate immune maturation and reinforce mucosal barrier integrity.3 They play a protective role in various immune-mediated diseases in mice4,5,6,7 and humans.8,9,10 Microbial proteins represent the main source of immunogenic antigens since breast-milk proteins represent self-antigen. Endogenous mediators such as type I interferon (IFN) alter the basal state of immune cells,11 induce cell maturation,12 augment endocytic and proteolytic activity, increase antigen cross-presentation capacity11,13,14,15 and enhance adaptive immunity.16,17 The role of endogenous mediators and microbial stimuli for post-natal immune maturation and the spectrum of immunogenic antigens at the neonatal inductive sites remain ill-defined.
Small intestinal (SI) Peyer’s patches (PPs) represent a main mucosal inductive site and anatomical place of early T cell homing in the murine mucosa.18 In the adult host, microfold (M) cells as integrative part of the PP follicle-associated epithelium (FAE) sample antigen directly from the SI lumen and transfer it to subepithelial antigen-presenting cells (APCs) that are arrayed in an intricate microarchitecture in the subepithelial dome (SED) and the interfollicular region (IFR) where they prime T cells.19 T cell priming in turn aids B cell activation and IgA production, a fundamental feature of host-microbial homeostasis in the intestine.20,21 The intense and continuous interaction between gut luminal stimuli and the mucosal immune system under homeostatic conditions explains the extraordinarily high frequency of antigen-experienced CD4+ T cells and germinal center B cells in adult PP. The frequency of mature T cells is markedly lower in neonatal PP and this remains unchanged for a prolonged time after birth despite their principal functionality.18 Although post-natal maturation of the PP mononuclear phagocyte (MNP) compartment has not been systematically investigated, recent work highlighted the presence of non-conventional APCs in the murine mesenteric lymph node (MLN). Beside type 3 innate lymphoid cell (ILC) APCs that were required for regulatory T cell maturation in the MLN, distinct clusters of RORγt+MHCII+, so called Thetis cells (TCs), were described, which exhibited a non-redundant functional role during early life.22,23,24
Here, we investigated cellular mechanisms in early life initiating PP homeostatic immune responsiveness. We hypothesized that APCs represent key determinants of this process and analyzed subset composition, anatomical organization, maturation trajectories, tissue environment, and functional profile of neonatal PP MNP. Beside the type 1 and 2 conventional dendritic cells (cDCs) and monocyte-derived MNPs (monocyte-derived cell [MC]), we identified and characterized a population of RORγt+ APC. We also investigated the influence of the early enteric microbiota, the spectrum of available antigens and type I IFN on APC maturation and T cell priming. Finally, we studied the post-natal differentiation of M cells within the FAE overlaying PP and translocation of luminal particulate antigen. Our results suggest that post-natal FAE M cell differentiation determines the onset of APC maturation and functionality as well as intestinal immune homeostasis. A better understanding of immune priming in early life may aid to unravel the functional importance of this critical time window for immune homeostasis and ultimately allow manipulation of immune effector function in the neonatal host.
Results
Reduced number and antigen presentation capacity of cDC2
Thymic output in the murine host is initiated around birth and homing of T cells to the neonatal intestinal lymphoid tissue occurs independent of signals from the enteric microbiota (Figure S1A). To determine the immune reactivity of enteric lymphoid tissues to mucosal antigen exposure in the neonatal setting, we transferred OTII cells into neonatal animals and orally administered OVA in presence or absence of FTY720, an inhibitor of lymphocyte egress. FTY720 treatment enriched OTII cells in PP, suggesting that they represent a primary site of early-life immune reactivity (Figure 1A). Nevertheless, under homeostatic conditions the percentage of CD44hi CD4+ T cells in PP remained low throughout the post-natal period. In contrast, in adult PP, >50% of CD4+ T cells were CD44hi, reminiscent of an activated phenotype. T cell activation increased after 4 weeks of age, concordant with increased FAS+GL7+ germinal center B cells (Figure 1B).
Figure 1.

Neonatal PP MNPs exhibit diminished antimicrobial activity and reduced antigen processing and presentation capacity
(A) OTII cell numbers (PP, Peyer’s patch; MLN, mesenteric lymph node; SPL, spleen) of PND10 mice administered with OVA ± FTY720 on PND 6–8 (n = 9–13, mean, one-way ANOVA/Kruskall-Wallis test).
(B) Percentage of CD44hi of PP CD4+ T cells (left y axis, black line) and GC B cells of B cells (right y axis, yellow green line) (n = 4–20, mean + SD, one-way ANOVA/Kruskal-Wallis test).
(C) Representative immunofluorescent images of PP stained for CD11c (red), phalloidin (white), DAPI (blue); scale bars, 200 μm.
(D) Percentage of PP MNP subsets (cDC1 = XCR1+SIRPα−MHCII+CD11c+, cDC2 = SIRPα+/hiBST2−XCR1−MHCII+CD11c+, Rorγt+ APC = RORγt+SIRPαloXCR1−MHCII+CD11c+, MC = BST2+SIRPα+XCR1−MHCII+CD11c+) quantified by FACS (n = 5–12, mean + SD; one-way ANOVA/Kruskall-Wallis test).
(E) scRNA-seq of PP MNP (n = 2; pooled from PP from 1 L or 3 adult animals per sample). Pie charts depict relative contribution of PND11 and adult cells within the indicated subset.
(F and G) Pseudobulk profile comparison of scRNA-seq clusters from (D) against (F) all ImmGen gene expression profiles and (G) scRNA-seq of RORγt+MHCII+ cells from Akagbosu et al.24 using a cosine similarity metric.
(H) DE genes in CCR7− qDC1, CCR7− qDC2, MC, and RORγt+ APC between PND11 and adult mice.
(I) GO terms overrepresented in qDC2 of PND11 (upper panel, blue circles) or adult (lower panel, red circles). Top 5 GO terms are labeled. See also Figure S1.
Given the central role of MNPs in antigen processing, environmental and microbial signal integration, and T cell priming, we next compared the localization and transcriptional profile of PP MNP between neonatal and adult mice. To avoid problems linked to cell isolation from small sized tissue samples, we first measured the strength and distribution of the pan-phagocyte marker CD11c normalized to the area of the patch. The total phagocyte density was not reduced in neonatal (post-natal day [PND]5 and PND11) vs. weanling (PND21) or adult PP (Figures 1C and S1B). Next, we analyzed PP MNP by flow cytometry to identify age-dependent differences in the subset composition of the various MNP (Figures 1D and S1C).25 Whereas cDC2s were the dominant MNP subset in adult PP in neonatal PP, cDC2 were reduced, while cDC1 were enriched. RORγt+ APC have been described in the neonatal spleen (SPL) and MLNs.24,26,27 We detected RORγt+SIRPαloXCR1−MHCII+CD11c+ APC in PP, which we refer to as RORγt+APC. The frequency of this subset was elevated in early life. The percentage of MCs had similar proportions at PND11 compared with the adult (Figure 1D). Similar age-dependent differences in the MNP subset composition were not observed in lamina propria APCs and no significant numbers of RORγt+ APC were detected (Figure S1D, data not shown).
Additionally, we sorted PP MNP from neonatal (PND11) and adult mice and performed single-cell RNA sequencing (scRNA-seq) to identify age-dependent differences in the transcriptional profile of the various PP MNP subsets. The UMAP revealed six major distinct clusters, of which five were assigned to cDC1, cDC2, or MCs based on known marker genes of cDCs and monocyte-derived APCs (Figures 1E and S1E). The sixth cluster was identified as RORγt+ APC. The scRNA-seq analysis confirmed age-dependent changes in the MNP subset composition observed by flow cytometry with a higher frequency of cDC1 and RORγt+ APC and reduced proportion of cDC2 in the neonatal host (Figure 1E). cDC1 and cDC2 each formed two separate clusters representing immature/quiescent (q) and mature/activated (act) CCR7+ DC.28 The transcriptional profiles of qDC1 and qDC2 were distinct from each other and were identified by the expression of the hallmark genes Sirpa and Xcr (Figures 1E and S1F). As previously described, the homeostatic maturation process was accompanied by substantial transcriptional changes leading to the distinct clustering of actDC from the corresponding qDC counterparts and substantial reduction of the DC lineage-defining genes Sirpa and Xcr1.29,30 In addition, homeostatically activated DCs were characterized by expression of a discrete set of genes that was largely overlapping between actDC1 and actDC2 consistent with their functional commitment. Nevertheless, expression of Cd8a and Cst3 remained detectable in actDC1, albeit at lower levels than in qDC1, whereas S100a4, S100a6, Klrd1, and H2-DMb2 expressed in qDC2 were reduced but remained detectable in actDC2 (Figures 1E, S1E, and S1F). To further confirm our annotation of cDC subsets, we compared the top differentially expressed genes between qDC and actDC identified in Figure S1F with the previously published definition of a core transcriptome of homeostatically quiescent vs. activated cDC from skin, lung, and thymus.30 The majority of differentially expressed genes identified in PP cDC subsets was also found in the respective qDC vs. actDC core transcriptome and the expression profile was almost identical.
To better understand the nature of RORγt+ APC, we first compared the six clusters identified in Figure 1E with the gene expression profiles of cells available on ImmGen (Figure 1F). Of all the queried ImmGen cell types, the RORγt+ APC transcriptional profile showed the greatest similarity to DCs and particularly to the intestinal lamina propria cDC2s. The correlation profile of RORγt+ APC and qDC2 in our dataset to the ImmGen transcriptomes was very similar indicating their transcriptional similarity. RORγt+MHCII+ antigen-presenting TCs were suggested to be enriched in the gut draining lymph node of young mice and to be distinct from type 3 ILCs (ILC3s).24 We compared our six cell clusters to the published scRNA-seq data of RORγt+MHCII+ cells representing Thetis I–IV subsets and ILC3 subsets isolated from PND14 MLN.24 Overall, CCR7+ subsets (actDC1, actDC2, TCIII, and TCIV) showed the greatest similarity to one another reflecting similar general functionality of CCR7+ activated/mature cells. PP RORγt+ APC displayed an intermediate similarity to TCIII and IV and low similarity to antigen-presenting ILC3 cells (Figure 1G).
Comparative analysis of the transcriptional profiles of the MNP subsets revealed that genes involved in (1) antigen presentation and processing (e.g., Cd74, H2-Eb1, H2-Dmb2, H2-Aa, H2-Ab1, and H2-K1), (2) microbicidal activity (e.g., Rnaset2a and Lyz1), and (3) IFN I signaling (e.g., Ifitm1, Ifitm2, and Ifitm3) had decreased expression in neonatal subsets compared with their adult counterparts (Figure 1H). Neonatal MNP were enriched in transcripts associated with stemness (Lgals1 and Birc5). Both actDC1 and actDC2 were transcriptionally similar between PND11 and adult within the subset (Figure S1H). The altered expression profile of CD74, H2-K1, and lysozyme on protein level was confirmed by FACS (Figure S1I). Consistently, gene ontology (GO) enrichment analysis confirmed that GO terms associated with antigen processing and presentation but also response to IFN I and II were underrepresented in neonatal cells, whereas cell division and morphology-associated pathways were overrepresented compared with adult mice (Figure 1I). Together, these results demonstrate an increase of cDC1 and RORγt+ APC and reduction of cDC2 in neonatal PP and indicate an overall lower capacity for antigen processing/presentation.
Enrichment of cDC2 at the PP SED
Functional changes of neonatal PP MNPs could be due to altered tissue organization and cellular interaction. Both, overall anatomical MNP distribution or the T cell-to-phagocyte ratio would influence the stochastic probability of T cells recognizing their cognate antigen and could explain the observed lack of homeostatic T cell activation. Quantitative flow cytometric analysis of the ratio of antigen-presenting MHCII+CD11c+ cells and CD4+ T cells in PP did not reveal any difference between PND11 and adult mice (Figure 2A). Also, the overall MNP tissue distribution showed no age-dependent phenotype. Radial segmentation in adult PP revealed a CD11c signal enriched toward the outer edges of PP that accommodate the SED and IFR, as well as the serosal side of the follicle leading to a negative slope of the line displaying radial (circumferential to central) signal intensity (Figure 2B). The mean slope (Δ) was neutral at PND5 indicating a somewhat more equal distribution of CD11c+ cells in PP during the first week of life but became and remained negative after that, suggesting an overall adult-like anatomical distribution of CD11c+ cells from PND11 onward (Figure 2C). However, MNP subsets display a sophisticated anatomical organization in the adult PP.31 We therefore additionally employed confocal spectral immunofluorescence microscopy to dissect the microarchitecture and tissue distribution of different MNP subtypes. Similar to adults, cDC1 localized mainly to the IFR in the neonatal PP (Figures 2D and S2A). In contrast, marked differences in the MNP distribution between neonatal and adult mice were found in the SED and FAE (Figure 2E). cDC2 were more abundant in the FAE of neonatal mice. Also, they were not restricted to the edge of the SED but present in high numbers at the middle top of the SED in neonatal mice (PND11) interacting intensely with the epithelium (Figures 2E, 2G, S2A, and S2B). Inversely, mococyte-derived lysozyme-expressing (lyso)MNP were more dominant in the adult SED. This resulted in an increased cDC2:lysoMNP ratio in the neonatal SED (Figure 2F). RORγt+ APC in PP of neonatal mice localized both to SED and IFR suggesting a maturation/migration pattern similar to cDC2 (Figures 2G and S2B). Taken together, despite a similar MNP/CD4+ T cell ratio and overall distribution, cDC2 were enriched at the SED and RORγt+ APC in the SED and IFR in the neonate.
Figure 2.

Anatomical distribution of MNPs in neonatal and adult PP
(A) FACS quantification of CD4+ T cells and phagocytic cells in PP. Ratio of mean T cell:phagocyte is indicated.
(B) Sample preparation and signal distribution model of CD11c using radial segmentation of the PP to obtain Δ.
(C) Δ calculated in individual PP (n = 25–36 follicles, Kruskall-Wallis test).
(D, E, and G) Representative spectral confocal imaging projection of PP illustrating anatomical distribution of (D) TLR3+IRF8+CD11c+CX3CR1− cDC1 (TLR3, orange; IRF8, magenta; GFP, green; CD11c, red; CD4, blue [only TLR3+ IRF8+ cells represent cDC1]), (E) SIRPα+CD11c+CX3CR1−lysozyme− cDC2 (SIRPα, magenta; EpCAM, gray; GFP, green; CD11c, red; lysozyme, yellow; CD4, blue [arrowheads point toward cDC2 in the SED]) and (G) RORγt+CD11c+CX3CR1−CD4− APC (RORγt, magenta; GFP, green; CD11c, red; lysozyme, yellow; CD4, blue [boxed areas of individual RORγt+APC shown below]). Scale bars, 20 μm.
(F) Ratio between SIRPα+CX3CR1+Lysozyme+CD11c+ MC and SIRPα+CX3CR1−lysozyme−CD11c+ cDC2 in SED shown in (E). (n = 5–6, median, Mann-Whitney U test.) Each dot represents one animal (A) or one follicle (C and G). See also Figure S2.
Reduced CD4+ T cell priming in neonatal PP
Next, we functionally evaluated priming of CD4+ and CD8+ T cells by neonatal PP MNP.26,28 Therefore, we transferred proliferation dye-labeled TCR-transgenic CD4+ (OTII) or CD8+ (OTI) T cells into neonatal and adult recipients and proliferation was assessed 48 h after oral OVA administration (Figures 3A and 3B). Efficient CD8+ T cell priming was observed both in neonatal and adult PP (Figure 3A). In contrast, CD4+ T cell priming was impaired in neonatal animals (Figure 3B). Also, we assessed the priming capacity of PP MNP toward different T helper subsets in vitro by co-culturing sort-purified CD11c+MHCII+ PP MNP with OTII cells and measuring secreted T helper cytokines in the supernatant. OTII cells co-cultured with neonatal PP MNP secreted enhanced levels of IFNγ and IL-17A consistent with our observation of increased cDC1 and RORγt+ APC numbers in neonatal PP but decreased levels of IL-4 (Figures 3C and S3). These results show that soluble antigen was cross-presented to neonatal PP CD8+ T cells with adult-like efficiency. In contrast, an overall decreased conventional major histocompatibility class (MHC)II presentation of soluble antigen and subsequent CD4+ T cell priming was observed in neonatal mice associated with significant alterations in the spectrum of secreted T helper cytokines.
Figure 3.

Altered T cell priming by neonatal PP MNPs
(A and B) Proliferation of transferred (A) OTI and (B) OTII cells in PP 48 h after oral OVA (n = 3–11, mean, Mann-Whitney U test).
(C) T helper cytokine levels in the supernatant of OTII and PP MNP co-cultures in presence of OVA (n = 3–11, mean + SD, two-way ANOVA/Sidak’s multiple comparison test). See also Figure S3.
Microbiota-derived signals promote cDC2 and RORγt+ APC maturation
Changes in the transcriptional profile, anatomical localization, and function of PP MNP in the neonate may reflect alterations in cellular maturation.25 We analyzed the maturation profile of PP MNP during the post-natal period by generating pseudotime trajectories. The trajectory seed branch was determined by high expression of cell cycling genes and the marker gene of progenitor-like cells Birc5. The relative contribution of neonatal cells to the pseudotime trajectory of cDC1 was comparable to that of adult cells suggesting a similar pace of maturation (Figure S4A). In line with that the proportion of CCR7+ actDC1 determined by FACS, albeit low in general, was not altered (Figure S4B). In contrast, the relative contribution of neonatal cells to the pseudotime trajectory of cDC2 steadily decreased with advancing degree of maturation (Figure 4A). Pseudotime analysis and sub-clustering of the cDC2 identified Birc5, Ccr6, Plet1, Itgam (encoding CD11b), and Ccr7 as key marker genes of cDC2 maturation stages (Figure 4B). We then studied cell maturation by flow cytometry utilizing Plet1 (maturation state [M] 1), CD11b (M2), and CCR7 (M3) as markers for the last three successive developmental stages in the life cycle of PP cDC2 (Figure 4C). Comparing those cDC2 developmental stages between PND11 and adult animals, we detected marked differences with an increased proportion of cells in the M1 state but decreased levels of M2 and M3 in the neonate (Figure 4D). Again, this indicated skewing toward a maturational delay of neonatal cDC2. Pseudotime analysis revealed an early maturational delay of neonatal cells also for RORγt+ APC, with neonatal cells enriched at early differentiation stages (Birc5+) and reduced in the mature/activated subset (Ccr7+) (Figure 4E).
Figure 4.

Maturation delay of PP cDC subsets during the post-natal period
(A) cDC2 pseudotime trajectory; pie charts indicate relative contribution of adult and PND11 cells to specific branch; genes indicate marker genes in and color code is consistent with (B).
(B) Seurat subclusters contributing to cDC2 (upper left, subclustered from Figure 1E), proposed maturation trajectory (upper right) and marker genes for maturational stages of cDC2 (bottom).
(C) Representative FACS plots of PP cDC2 showing the expression of PLET1, CD11b, and CCR7 for maturational stages M1, M2, and M3, respectively.
(D) Percentage of M1–M3 cDC2 (n = 8–10, mean + SD, two-way ANOVA/Bonferroni test between age groups within the same maturational stage).
(E) RORgt+ APC pseudotime trajectory; pie charts indicate relative contribution of adult and PND11 cells to specific branches.
(F) Percentage of M1–M3 of cDC2 in PND11 germ-free (GF) vs. conventional (CV) animals (n = 4–12, mean + SD; two-way ANOVA/Bonferroni test, statistical significance between colonization groups within the same maturational stage) determined by FACS.
(G) Percentage of RORgt+APC in germ-free (GF) vs. conventional (CV) animals (n = 4–11, mean + SD, one-way ANOVA/Kruskall-Wallis test, statistical significance indicated within the same age group). See also Figure S4.
We next asked whether microbial cues contributed to the post-natal MNP maturation and the detected age-related differences.32 The number of CCR7+ migratory cDC1 was independent of the presence of viable microbiota (Figure S4C). In contrast, cDC2 of germ-free (GF) PND11 mice were increased proportionally in the M2 state (CD11b+) but decreased in M3 (CCR7+) suggesting that microbial signals contributed to the final maturation step of cDC2 from M2 to M3 (Figure 4F). cDC2 were also enriched at M2 in animals with myeloid-specific MyD88 deficiency (MyD88ΔCD11c), important for microbiota-dependent innate immune signaling (Figure S4D).33 Taken together our results identify markers of cDC2 maturation and show that microbiota-derived signals promote cDC2 and RORγt+ APC maturation after birth.
Early rise in SI microbiota density, diversity, and antigenicity
Lack of innate immune stimulation or reduced exposure to microbial immunogenic antigens might contribute to the observed delay in MNP maturation. We comparatively analyzed the density and composition of the SI microbiota in neonatal and adult mice by quantitative (q) PCR and metagenomic sequencing, respectively. qPCR for bacterial 16S rRNA gene copies revealed a high bacterial load as early as 24 h post parturition remaining unchanged until PND14 in SI tissue with a further transient increase during weaning but not a reduced bacterial load during early life compared with adult (Figure 5A). Similarly, anaerobic culture of luminal material on nutrient-rich agar plates revealed a comparable number of colony-forming units (CFUs) per gram SI tissue (Figure 5B). Fluorescence in situ hybridization (FISH) staining using a eubacterial probe on cross-sections in neonatal (PND11) and adult mice revealed a patchy distribution of commensal bacteria but overall confirmed a similar bacterial load in PND11 and adult mice (Figure S5). A delayed increase in the bacterial density was detected in the colon (Figure 5C). Sequence reads from metagenomic sequencing of DNA prepared from luminal material of neonatal and adult small intestines were matched against the integrated mouse gut metagenome catalog (iMGMC)34 and 164 metagenome-assembled genomes (MAGs) were identified (Figure 5D; Table S1). The overall species richness was higher in adult than neonatal mice (Figure 5E) but was largely confined to low abundance species. The core microbiome represented by highly abundant MAGs, mostly of the Firmicutes and Bacteroidetes phyla, was present early in life (Figure 5D). By calculating knee points in saturation plots, we could confirm that the number of species to reach saturation was not significantly different between both age groups (Figures 5F and 5G).
Figure 5.

Rapid establishment of a dense and immunogenic small intestinal core microbiome in absence of adaptive immune maturation
(A) Bacterial density determined by quantitative 16S rRNA gene qRCR normalized to murine Gapdh in total small intestinal (SI) tissue (n = 5, mean + SD, Kruskall-Wallis/Dunn’s multiple comparison to adult).
(B) Anaerobic culture of homogenized small intestinal tissue (n = 4, mean, Mann-Whitney U test).
(C) Bacterial density determined by quantitative 16S rDNA quantitative real-time PCR normalized to murine Gapdh in total colonic tissue (n = 5, mean + SD, Kruskall-Wallis/Dunn’s multiple comparison to adult).
(D) Phylogenetic placement, phylum assignment (inner circle), and relative abundance (outer circle) of metagenome-assembled genomes (MAGs) in the SI luminal content by shotgun sequencing.
(E) Molecular species richness determined in (D) (n = 6, mean, Mann-Whitney U test).
(F) Saturation plot depicting number of MAGs required to cover most of the functional potential (n = 6, median + SD). Knee points indicated with vertical lines.
(G) Knee points of the individual saturation curves indicated in (F) (n = 6, mean + SD, Mann-Whitney U test).
(H) BOTA scores of all MAGs from (D) (nPND11 = 59, nAdult = 149 MAGs, violin plot, Mann-Whitney U test).
(I) Cumulative abundance-weighted BOTA scores from (D) (n = 6, mean, Mann-Whitney-U test).
(J) Venn diagram of non-host-derived protein species identified by mass spectrometry in small intestinal luminal material (n = 6).
(K and L) Percentage of M1–M3 of cDC2 in PND11 (K) pIgR+/− offspring born to pIgR−/− dams or pIgR+/+ offspring born to pIgR+/+ dams (n = 4–5) and (L) wildling (WLD) animals (n = 11–12), (mean + SD; two-way ANOVA/Bonferroni test between genetic groups within the same maturational stage) determined by FACS.
(M) Percentage of M1–M3 of cDC2 in PND12 mice after oral administration of adult intestinal content (adultIC) on PND9-11 (n = 5), (mean + SD; two-way ANOVA/Bonferroni test between treatment groups within the same maturational stage).
(N) Percentage of CD44hi cells among total PP CD4+ T cells in 14-day-old mice after oral administration of adult intestinal content (adultIC) on PND1-9 (n = 6–8; median).
(O–Q) Percentage of M1–M3 of cDC2 in PND12 mice that were administered (O) B. fragilis by oral gavage on PND9-11 (n = 5), (P) 8 h after E. coli OMVs oral gavage (n = 6–7), and (Q) weaning intestinal content (weanIC) by oral gavage on PND9-11 (n = 5–9) (mean + SD, two-way ANOVA/Bonferroni test between treatment groups within the same maturational stage). See also Figure S5 and Tables S1 and S2.
Beside the strength of innate immune signals, a reduced spectrum of immunogenic peptides derived from microbiota in the neonatal vs. adult SI might explain the observed delay in immune maturation after birth. Using MAGs identified in neonatal and adult small intestine in combination with a previously reported in silico pipeline bacteria-origin T cell antigen (BOTA) predictor to determine peptides likely to be presented on host MHCII molecules derived from proteins encoded in bacterial genomes.35 Scoring the 164 MAGs revealed a higher mean BOTA score for MAGs present in adult compared with the neonatal consortium (Figure 5H). Consistent with the presence of most highly abundant taxa in the neonate intestine, however, the cumulative weighted BOTA score that incorporates the abundance of detected species showed no significant difference between neonatal (PND11) and adult mice (Figure 5I). Similarly, proteomic analysis of intestinal content reflecting mainly proteins of highly abundant bacterial taxa revealed a large core proteome of 1,060 proteins in both neonatal (PND11) and adult mice and a much smaller number of 388 and 145 age-specific proteins, respectively (Figure 5J; Table S2). Thus, global lack of innate immune stimulation or immunogenic microbial antigens are unlikely factors to explain the delayed MNP maturation in the neonatal host. Instead, specific microbial signals from less abundant taxa might exert an effect.
Next, we analyzed cDC2 maturation (M1–M3) in neonatal mice under various conditions associated with an altered microbiota exposure. Breast milk from pIgR-deficient dams have greatly reduced SIgA-specific for enteric bacteria.21,36 pIgR+/− offspring from pIgR−/− dams, however, exhibited no detectable difference in PP cDC2 maturation (Figure 5K). Mice with a microbiome like in their natural environment (“wildlings”) exhibit a highly diverse microbiota and contain higher numbers of antigen-experienced lymphocytes in the adult.37,38,39 Nevertheless, we did not observe differences in subset or stage composition of cDC2 in neonatal PP (data not shown and Figure 5L). To test whether the adult antigenic repertoire would induce T cell priming in the neonate, we gavaged intestinal content from adult animals (adultIC) to neonatal mice. No detectable influence of adult microbiota on PP cDC2 maturation was detected (Figure 5M) and, consistently, the proportion of CD4+ TEM at PND14 was not influenced (Figure 5N). Bacteroides spp. as well as microbiota-derived outer membrane vesicles (OMVs) exert immune maturation activity.40,41 Oral application of a murine strain of Bacteroides fragilis or OMVs isolated from E. coli did not influence PP cDC2 maturation (Figures 5O and 5P). Finally, we gavaged intestinal content from weaning animals (weanIC) to neonatal mice in order to test the PP maturation promoting effect of bacterial taxa that exhibit a transiently enhanced abundance during the shift from a neonatal to an adult microbiota.42 An enhanced proportion of stage M2 (CD11b+) cDC2 cells was detected upon administration of weanIC (Figure 5Q). Thus, the murine SI is colonized rapidly after birth and global differences in luminal bacterial density, composition and antigenicity appear not to account for the observed delay in DC maturation in the neonatal host. Instead, signals from specific taxa with enhanced abundance during weaning may contribute to PP DC maturation.
IFN I promotes activation of PP DC but does not recapitulate adult to neonatal transition
Endogenous immune stimuli might induce PP DC maturation. IFN-stimulated genes (ISGs) such as Ifitm1, Ifitm2, and Ifitm3 showed the greatest expression reduction in neonatal cDC2 but also cDC1 (Figures 1H and 1I). Tonic-microbiota-induced IFN I signaling was shown to enhance antimicrobial resistance in adult animals.11 Therefore, increased IFN I signaling might also promote maturity and functionality of neonatal PP MNP. To test this hypothesis, we employed the Toll-like receptor (TLR) 7 ligand R848, which activates SI LP and PP DC in adult animals via release of IFN I and TNF-α by plasmacytoid (p)DC.25,43 Neonatal mice exhibited a higher proportion of PP pDC (Figure S6A) and enhanced expression of the co-stimulatory molecule CD86 at 8h after oral administration of R848 (Figure S6B). Proinflammatory serum cytokines, including IFN I, were higher in neonatal animals compared with their adult counterparts following R848 treatment (Figure 6A). R848 treatment also promoted cDC2 progression to M2 and M3 and enhanced the percentage of CCR7+-activated cDC1 and RORγt+ APC (Figures 6B–6D).
Figure 6.

IFN I induces DC activation in neonatal PP and modifies the adaptive immune response
(A) Heatmap of serum cytokines 8 h after R848 (n = 4–5; Kruskall-Wallis test, comparison within age group).
(B) Percentage of M1–M3 of cDC2 in PND11 mice after R848 (n = 4–5), (mean + SD; two-way ANOVA/Bonferroni test between treatment groups within the same maturational stage) determined by FACS.
(C and D) Percentage of CCR7+ among (C) cDC1 and (D) RORγt+ APC in PP after R848 in PND11 mice (mean + SD, Mann-Whitney U test).
(E) Proliferation dye dilution in OTII cells in PP of neonatal mice 48 h after oral gavage of OVA ± R848 normalized to mean PBS (n = 7–8, mean, Mann-Whitney U test).
(F) Neonatal PAA-STm/R848 vaccination experimental setup.
(G) Percentage of IFNγ+ of CD4+ T cells in colonic LP of STm infected mice vaccinated with PAA STm ± R848 as neonates (n = 5–6, mean + SD, Mann-Whitney U test).
(H) scRNA-seq of PP MNP (n = 1; cells pooled from 4 animals per sample).
(I) Top 10 upregulated genes in PND11 (dark blue) and adult (berry) R848 vs. PBS qDC2.
(J) Pseudotime trajectory of cDC2 from PND11 mice ± R848, pie charts represent the relative contribution of cells from PBS and R848 treated animals to indicated branches.
(K) Percentage of M1–M3 of cDC2 in adult IFNARΔCD11c and IFNARfl/fl mice (n = 5), (mean + SD, two-way ANOVA and Bonferroni test between genetic groups within the same maturational stage) determined by FACS. See also Figures S6 and S7.
We addressed how oral R848 affected adaptive immune priming and memory formation in neonates. OTII priming in neonatal PP was diminished compared with adult PP (Figure 3B) but oral co-administration of R848 with OVA partially reversed this phenotype (Figures 6E and S6F). Next, the influence of R848 on T and B cell responses was studied using a vaccination protocol with peracetic acid (PAA)-inactivated, orally administered Salmonella Typhimurium (STm) followed by oral challenge with viable S. Typhimurium in the adult animal as previously described (Figure 6F).44 R848 administration with the first and second vaccine dose at PND7 and 14, respectively, enhanced the percentage of Th1 cells. However, it diminished the percentage of IFNγ+ CD8 cytotoxic T cells (Figures 6G and S6G). Further, R848 abrogated the anti-STm IgA response in the SI mucosa (Figure S6H). The systemic anti-STm IgG and IgM antibody response showed a similar trend (Figures S6I and S6J).
R848 potently activated neonatal PP APC (Figures 6B–6E). To better understand whether the effect of R848 mimicked the physiological PP APC maturation during the neonatal-to-adult transition, sorted PP phagocytes from PND11 and adult mice orally treated with R848 or PBS were subjected to scRNA-seq analysis (Figure 6H). Although neonatal mice exhibited a very potent systemic response to R848 (Figure 6A), the local mucosal response was less pronounced than in the adult, as depicted by the changes in qDC/actDC ratios for cDC1 and cDC2 (Figure 6H). Transcriptional changes after R848 administration were mainly driven by elevated ISG expression, with Isg15, Irf7, Ifi27l2a, and Ifitm3 being among the top 10 increased genes both in adult and neonatal qDC2, qDC1, and RORγt+ APC (Figures 6I and S6C). Pseudotime trajectory analysis of cDC2, cDC1, and RORγt+ APC confirmed an activation pattern of DCs in R848-treated animals. R848-mediated activation was distinct from the homeostatic activation and mainly defined by an enhanced ISG profile with a bimodal splitting of the CCR7+ cells into two different branches representing steady state vs. R848 (Figures 6J, S6D, and S6E). Further, we analyzed genes and corresponding GO terms that were differentially expressed in both, adult vs. neonatal and PND11 PBS vs. R848 comparisons (Figure S7). Very few genes were co-regulated in both datasets. Even within common GO terms, different genes contributed to GO term enrichment, illustrating that the IFN-I-driven activation in the neonate did not recapitulate the neonatal-to-adult transition. Adult mice deficient in the IFN type I receptor in CD11c+ cells (IFNARΔIEC) exhibited no decrease in PP cDC2 maturation (Figure 6K). Thus, the type-I-IFN-inducer R848 stimulated a strong systemic cytokine response in neonates and was able to overcome the maturational arrest of neonatal DC in respect to CD4+ T cell priming. However, despite its potent activity, IFN I appear not to represent the physiological stimulus that drives PP APC maturation in steady state.
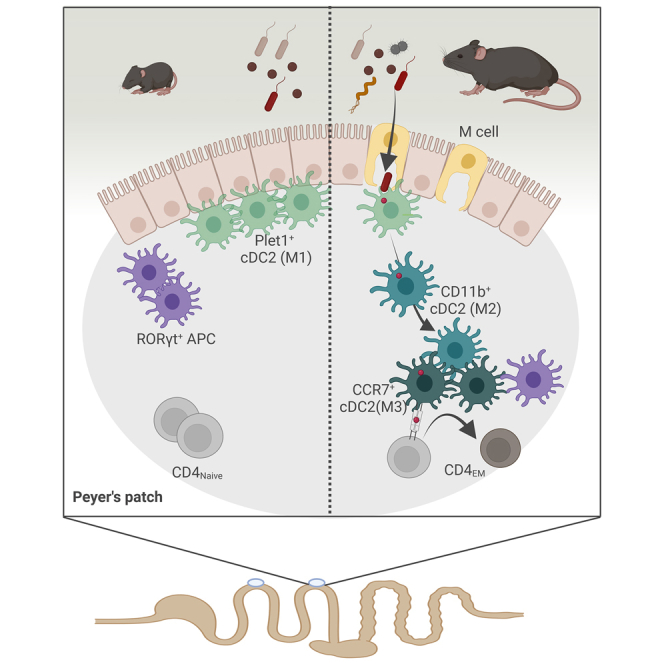
Appearance of M cells determines post-natal cDC2 maturation
M cells as part of the PP FAE represent the primary site of luminal sampling and translocation of particulate antigen, which initiates mucosal IgA production to commensal enteric bacteria.45 M cells are absent from the FAE of neonatal mice and appear only during the third week of life as illustrated by the age-dependent expression of the M cell master transcription factor SpiB and markers Ccl9 and Gp2 (Figure 7A) and staining of GP2+ mature M cells in the FAE (Figure 7B).46,47 The age-dependent appearance of M cells was functionally relevant, illustrated by differences in the uptake of orally administered fluorescent 100-nm latex beads by SED phagocytes between neonatal and adult PP (Figure 7C). Whereas >90% of sections of adult PP contained bead-positive phagocytes in the SED, only approximately 40% of neonatal PP phagocytes had internalized beads at 6 h after oral gavage (Figure 7D). Also, the number of intra-phagocytic beads in the SED was reduced in the neonate (Figure 7E). Thus, the differentiation of M cells and their function as gatekeepers of luminal microbial stimuli might induce PP APC maturation. Indeed, adults lacking M cells due to receptor activator of NF-κB (RANK) deficiency in the intestinal epithelium exhibited impaired maturity of cDC2 in PP tissue (Figure 7F).48 Inversely, accelerated induction of M cells in PND11 mice through administration of RANK ligand enhanced the fraction of M2 PP cDC2 (Figure 7G). In addition to RANK ligand, the TLR5 ligand flagellin will stimulate M cell development.49 Consistently, administration of flagellin induced premature appearance of GP2+ M cells (Figure 7J) and enhanced cDC2 maturity (Figure 7I). STm induces M cell maturation to facilitate its own uptake.50,51 Consistently, infection with STm enhanced the percentage of CCR7+ mature PP cDC2 (Figure 7H). Together, our results suggest a contribution of FAE M cells to the maturation of PP APC. Beside specific microbial signals provided by the microbiota at weaning, the appearance of M cells during the 3rd week after birth critically influences the emergence of fully mature PP APC that in turn regulate the maturation of mucosal T cells, germinal center B cells and the production of secretory IgA.
Figure 7.

M cells are associated with PP DC maturation
(A) Quantitative real-time PCR of Spib, Ccl9, and Gp2 from PP (n = 5–6, mean, Brown, Forsyth, and Welch ANOVA).
(B) Spectral confocal imaging projection representative of PP dome regions. Sections illustrate epithelial cells and M cells (right panels) using EpCAM (cyan) and GP2 (orange). Arrowheads point toward M cells in the FAE. Scale bars, 20 μm.
(C) Immunofluorescence imaging representative of PND11 and adult PP after oral administration of 100-nm fluorescent beads stained for CD11c (red), phalloidin (blue), beads (green); asterisks indicate intraphagocytic beads in the SED. Scale bars, 20 μm.
(D) Percentage of PP with beads in CD11c+ cells within the SED (n = 23–26 follicles from 4 animals, Fisher’s exact test).
(E) Intraphagocytic beads normalized to SED area (n = 23–26 follicles, Mann-Whitney U test).
(F–I) Percentage of M1–M3 of cDC2 in (F) adult RANKΔIEC or RANKfl/fl (n = 4–9) or PND12 mice (G) after RANKL administration (n = 3–6) or (H) S. Typhimurium infection (n = 4–12) or (I) or flagellin administration (n = 4–9); (mean + SD, two-way ANOVA/Bonferroni test between genetic or treatment groups within the same maturational stage) determined by FACS.
(J) Spectral confocal imaging projection representative of dome regions of PP illustrating M cells stained for CCL9 (green) and GP2 (orange) (left and mid panels) as well as SED myeloid cells stained for SIRPa (red) and epithelial cells using EpCAM (cyan) (right panels) arrowheads point toward M cells in FAE. Scale bars, 20 μm.
Discussion
The ontogeny of immune maturation is of major conceptual and functional relevance. Epidemiological studies highlight the early post-natal period as a critical and non-redundant time window for the establishment of a fully functional mucosal immune system.4,6,52,53,54,55,56,57 Also, immune maturation represents a timed succession of events that activate regulatory features prior to effector functions to maintain homeostasis.24,58 Characterization of the post-natal immune maturation advances our understanding of the mechanisms that facilitate life-long immune homeostasis and determine the susceptibility to inflammatory and immune-mediated diseases.
Our results show that delayed post-natal PP CD4+ T cell maturation is paralleled by changes in the APC subset composition, localization, and cell differentiation. Given the principle functionality of neonatal T cells, the underrepresentation and functional impairment of cDC2 function could explain the prolonged low frequency of activated CD4+ T cells and germinal center B cells after birth.59,60,61,62 Additional factors might contribute but the previously identified mechanisms namely maternal immunoglobulins and neonatal T regulatory cells, only accounted for a minor part of this suppression.18,63 In contrast to cDC2, cDC1 were overrepresented, displayed a similar localization and conferred an adult-like activation of transferred OTI cells in the neonatal PP. Also, RORγt+ APC were overrepresented in the neonatal PP and their numbers were enhanced by the presence of a viable microbiota in early but not later life. RORγt+ ILC3-like APCs have recently been described in the MLN and shown to be required for peripheral regulatory T cells (pTregs) induction.22,23 However, the transcriptional profile of RORγt+ APC clearly clustered with DC rather than ILC3 or natural killer (NK) cells and failed to express important ILC3 marker molecules. Another group recently described antigen-presenting RORγt+ TCs in the MLN that emerge during an early time window and also induce pTreg in the colon.24 PP RORγt+ APC displayed an intermediate level of similarity with the CCR7+ fraction of MLN TCs but were present at high levels already in the first vs. second week of life and did not express the gene for cleavage of TGF-β, Itgb8, which was shown by Akagbosu et al.24 to be the mechanism through which pTregs were induced. Thus, the function of RORγt+ APC is likely different from TCs. PP RORγt+ APC might still exert tolerogenic function through other, tissue-environment-specific mechanisms or contribute to type III immune responses. Type III immune responses are driven by IL-17, and we detected increased levels of IL-17 in the PP MNP OTII co-cultures from neonatal mice. RORγt+ APC have also been detected in the SPL of neonatal mice and thus might represent a more global feature of the neonatal immune system.27 Also, the ontogeny of neonatal RORγt+ APC remains unknown. Their dependency on Clec9a-expressing progenitors was ruled out.24,27 However, as neonatal cDC2 unlike their adult counterparts also largely do not derive from Clec9a+ precursors, it remains to be tested whether RORγt+ APC have a common precursor with cDC.27 Taken together, APC T cell interactions in the neonate are characterized by ignorance of microbiota-derived antigen, a DC1 over DC2 bias, and the presence of RORγt+ APC with yet unknown function. APC in the neonatal PP appear to be designated to not engage in homeostatic crosstalk but respond efficiently to pathogenic, mostly viral, challenge in accordance with many previous reports.60,64
The high frequency of activated CD4+ T cells in the healthy adult intestine reflects the continuous and intense interaction between the mucosal immune system and the enteric microbiota. We found that the SI microbiota exhibits a high bacterial density, substantial microbial diversity and broad antigenicity early after birth, similar to what has been described in the oral cavity.65 A delayed increase in bacterial diversity over 2–3 years in humans and 6–8 weeks in mice was described, but these studies lacked a quantitative analysis and investigated fecal samples that reflect the distal intestinal tract.42,66,67,68 A global lack of innate stimuli or limited spectrum of immunogenic antigens therefore appears an unlikely explanation for the delayed T cell maturation in the murine neonatal small intestine.
Nevertheless, a significant difference in the maturation stage and number of cDC2 and RORγt+ APC was observed between SPF and GF animals, respectively. Several attempts failed to identify the underlying stimulus. Only the transfer of a weaning age microbiota exerted a significant stimulatory effect. The subsequent identification of the critical role of M cell development and the differentiation-promoting effect of flagellin hints to a possible role of flagellated taxa with only transient abundance. Flagellated bacterial taxa may bloom during the early post-natal period with loss of the recently discovered TLR 5-mediated counterselection of flagellated bacteria in the small intestine.69
Instead, the age-dependent differentiation of M cells within the FAE was required and sufficient to initiate cDC2 maturation. M cells show a characteristic morphology and provide sufficient luminal antigen to supply underlying PP APC through their high endocytotic and transcytotic capacity.70,71,72 We found that cDC2 accumulated at and interacted tightly with the neonatal FAE prior to M cell maturation possibly awaiting antigen delivery. Consistently, expression of the successive M cell maturation markers SipB, Ccl9, and Gp2 increased steadily during post-natal development in our study and previous reports.46,47,73,74 Receptor activator of nucleic factor-kappa B ligand (RANKL) expressed by subepithelial stromal M cell inducer (MCi) cells stimulates M cell differentiation in adult mice.75 Since parenteral RANKL administration to neonatal mice induced premature M cell differentiation, the pace of MCi cell differentiation and their ability to present RANKL may ultimately determine the onset of M cell differentiation. Alternatively, inhibitory signals like the developmental regulator Blimp1, suppressive factor ONECUT2 or breast-milk constituents such as epidermal growth factor (EGF) may reduce the sensitivity of the FAE to RANKL or reduce RANKL expression or its signal strength.76,77,78 Future work needs to address the influence of developmental and exogenous factors on M cell development.
M cells also serve as scaffold for certain types of APC to directly sample luminal material for subsequent processing and presentation.71 Interestingly, we observed enhanced numbers of cDC2 that interacted with the neonatal FAE and exhibited membrane extensions toward the FAE surface. These epithelium-associated cDC2 might receive the first luminal material translocated by emerging M cells after weaning.79 In wildling mice, cDC2 still exhibited a delayed time course of immune maturation suggesting that exposure to viral or bacterial pathobionts or pathogens might not suffice to accelerate M cell differentiation.
A mechanistic understanding of ontogenetic features of immune maturation and their biological relevance as well as the ability to manipulate immune reactivity might ultimately facilitate age-adapted interventional precision strategies to prevent or treat diseases of the term and preterm human neonate.80 Infections represent the main cause of morbidity and mortality in neonates and young infants worldwide and preventive measures such as vaccines could reduce childhood mortality.74,81,82,83,84,85,86,87,88 The use of vaccines in neonates that carry the largest disease burden, however, is less effective calling for strategies to accelerate immune maturation and improve the immune response to the vaccine antigen in particular at mucosal body surfaces accessible to non-parenteral vaccine administration.80
Limitations of the study
Important questions remain about antigen acquisition by MNPs, both in neonatal and adult PPs. For example, whereas cDC2 reside in both SED and IFR and likely sample antigen directly, IFR-resident cDC1 may rely on antigen transfer from other phagocytes. In contrast, MCs localize next to M cells and take up particulate antigen but migrate and prime T cells inefficiently. Therefore, putative antigen transfer from MCs might be an important mechanism of antigen acquisition that merits further investigation. Likewise, it remains unclear whether PP uptake of soluble and particulate antigens entails different routes.
We identified M cell development as prerequisite for cDC2 maturation; however, cellular and molecular mediators of M cell development largely remain unclear. Finally, we demonstrated selective enrichment of RORgt+ APC in neonatal PPs. Low cell numbers precluded functional ex vivo studies, but future studies utilizing genetic mouse models will address their function in steady state and inflammation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD317 (BSTII) APC (clone 927) | BioLegend | Cat# 127016; RRID:AB_1967127 |
| CD317 (BSTII) BV421 (clone 927) | BioLegend | Cat# 127023; RRID:AB_2687109 |
| CCR7 PE (clone 4B12) | BioLegend | Cat# 120106; RRID:AB_389358 |
| CCR7 PECy5 (clone 4B12) | BioLegend | Cat# 120113; RRID:AB_493571 |
| CD11b BV786 (clone M1/70) | BioLegend | Cat# 101243; RRID:AB_2561373 |
| CD11c PE Cy7(clone N418) | BioLegend | Cat# 117318; RRID:AB_493568 |
| CD11c BUV737 (clone N418) | BD Biosciences | Cat# 749039; RRID:AB_2873433 |
| CD45 APC-R700 (clone 30-F11) | BD Biosciences | Cat# 565478; RRID:AB_2739257 |
| CD45 R718 (clone 30-F11) | BD Biosciences | Cat# 567075 |
| CD64 PE Dazzle 594 (clone X54-5/7.1) | BioLegend | Cat# 139319; RRID:AB_2566558 |
| CD172a (SIRPa) APC (clone P84) | BioLegend | Cat# 144014; RRID:AB_2564061 |
| CD172a (SIRPa) FITC (clone P84) | BioLegend | Cat# 144006; RRID:AB_11204425 |
| I-A/I-E (MHCII) BV510 (clone M5/114.15.2) | BioLegend | Cat# 107635; RRID:AB_2561397 |
| RORgt PE (clone B2D) | Thermo Fisher Scientific | Cat# 12-6981-82; RRID:AB_10807092 |
| XCR1 BV650 (clone ZET) | BioLegend | Cat# 148220; RRID:AB_2566410 |
| XCR1 BV421 (clone ZET) | BioLegend | Cat# 148216; RRID:AB_2565230 |
| CD74 APC (clone In1/CD74) | BioLegend | Cat# 151003; RRID:AB_2632608 |
| CD86 APC (clone GL-1) | BioLegend | Cat# 105011; RRID:AB_493343 |
| IgA PE (clone mA-6E1) | Thermo Fisher Scientific | Cat# 12-4204-81; RRID:AB_465916 |
| IgG1 FITC (clone RG11/39.4) | BD Biosciences | Cat# 553892; RRID:AB_395120 |
| CD4 BV510 (clone RM4-5) | BioLegend | Cat# 100559; RRID:AB_2562608 |
| CD8a BV711 (clone 53-6.7) | BioLegend | Cat# 100748; RRID:AB_2562100 |
| CD44 BV785 (clone IM7) | BioLegend | Cat# 103059; RRID:AB_2571953 |
| CD45.1 BUV 737 (clone A20) | BD Biosciences | Cat# 612811; RRID:AB_2870136 |
| CD45.1 FITC (clone A20) | BioLegend | Cat# 110706; RRID:AB_313495 |
| CD45.2 AF700 (clone 104) | BioLegend | Cat# 109822; RRID:AB_493731 |
| CD69 PE Cy7 (clone H1.2F3) | BioLegend | Cat# 104512; RRID:AB_493564 |
| Va2 BV421 (clone B20.1) | BD Biosciences | Cat# 562944; RRID:AB_2737910 |
| TCRb APC Cy7 (clone H57-597) | BioLegend | Cat# 109220; RRID:AB_893624 |
| GL7 PerCP-Cy5.5 (clone GL7) | BioLegend | Cat# 144609; RRID:AB_2562978 |
| B220 (CD45R) PE/Fire 700 (clone RA3-6B2) | BioLegend | Cat# 103279; RRID:AB_2876408 |
| CD19 Spark NIR 685 (clone 6D5) | BioLegend | Cat# 115568; RRID:AB_2819829 |
| CD3 APC/Fire 810 (clone 17A2) | BioLegend | Cat# 100267; RRID:AB_2876392 |
| IgM eF450 (clone II/41) | Thermo Fisher Scientific | Cat# 48-5790-80; RRID:AB_2574072 |
| IgD BV480 (clone 11-26c.2a) | BD Biosciences | Cat# 566199; RRID:AB_2739590 |
| CD64 (FcγRI) BV605 (clone X54-5/7.1) | BioLegend | Cat# 139323; RRID:AB_2629778 |
| CD45 BUV 395 (clone 30-F11) | BD Biosciences | Cat# 564279; RRID:AB_2651134 |
| CD95 BUV 563 (clone Jo2) | BD OptiBuild | Cat# 741292; RRID:AB_2870823 |
| Chicken anti-GFP unlabeled | Aves Labs | Cat# GFP-1020; RRID:AB_2307313 |
| Hamster anti-mouse CD11c unlabeled (clone N418) | Biolegend | Cat# 117302; RRID:AB_313771 |
| Rabbit anti-mouse IRF8 unlabeled | Bethyl Laboratories | Cat#A304-028A-T; RRID:AB_2620376 |
| Rabbit anti-lysozyme unlabeled | Agilent | Cat# A0099; RRID:AB_2341230 |
| Rat anti-mouse TLR3 unlabeled (clone 11F8) | Bio-Rad | Cat#MCA5891GA |
| Rat anti-mouse SIRPa unlabeled (clone P84) | Biolegend | Cat# 144002; RRID:AB_11203711 |
| Rat anti-ROR gamma (t) unlabeled (clone AFKJS-9) | Thermo Fisher Scientific | Cat# 14-6988-82; RRID:AB_1834475 |
| Rat anti-mouse GP2 unlabeled (clone 2F11-C3) | MBL International | Cat# D278-3; RRID:AB_10598188 |
| Rat anti-mouse CD45 unlabeled (clone 30F11) | Biolegend | Cat# 103101; RRID:AB_312966 |
| Rat anti-mouse CD4-eFluor 450 (clone RM4-5) | Thermo Fisher Scientific | Cat# 48-0042-82; RRID:AB_1272194 |
| Rat anti-mouse Ep-CAM-Alexa Fluor 647 (clone G8.8) | Biolegend | Cat# 118211; RRID:AB_1134104 |
| Donkey anti-chicken IgG- Alexa Fluor 488 | Jackson ImmunoResearch | Cat# 703-545-155; RRID:AB_2340375 |
| Donkey anti-rat IgG-Cy3 | Jackson ImmunoResearch | Cat# 712-165-153; RRID:AB_2340667 |
| Goat anti-Armenian hamster IgG-Alexa Fluor 594 | Jackson ImmunoResearch | Cat# 127-585-160; RRID:AB_2338999 |
| Goat anti-rabbit IgG- Alexa Fluor 514 | Thermo Fisher Scientific | Cat# A-31558; RRID:AB_2536173 |
| Bacterial and virus strains | ||
| Bacteroides fragilis | Clavel Lab | CLA-KB-H83 and CLA-JM-H48 |
| Salmonella Typhiumurium | N/A | SL1344 |
| Biological samples | ||
| E.coli OMVs | Gerkara Lab, Erttmann et al.41 | N/A |
| PAA Salmonella Typhiumurium | Moor et al.44 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| R848 | Invivogen | TLRL-R848 |
| Ovalbumin | Sigma | A5253-500G |
| Ultrapure flagellin from S. typhimurium | Invivogen | tlrl-epstfla-5 |
| RANKL | Biomol | RP0437M-100 |
| Fluoresbrite® YG Microspheres 0.10μm | Polysciences | 17150-10 |
| Liberase TH | Sigma Aldrich | 5401135001 |
| DNAse I | Sigma Aldrich | 11284932001 |
| Critical commercial assays | ||
| LEGENDplex™ Mouse Anti-Virus Response Panel (13-plex) with V-bottom Plate | Biolegend | 740622 |
| LEGENDplex™ MU Th Cytokine Panel (12-plex) w/ VbP V03 | Biolegend | 741044 |
| Chromium Single Cell 3’ v2 kit | 10xGenomics | PN-120267 |
| Deposited data | ||
| scRNAseq data | this manusript | GSE188714 |
| Metagenomic data | this manusript | BioProject # PRJNA794356 |
| Experimental models: Organisms/strains | ||
| C57BL/6J (B6) | N/A | The Jackson Laboratory |
| B6.Cg-Tg(TcraTcrb)425Cbn Rag1tm1Mom/J (OTII) | Barnden et al.89 | The Jackson Laboratory |
| C57BL/6-Tg(TcraTcrb)1100Mjb/J (OTI) | Hogquist et al.90 | The Jackson Laboratory |
| B6.129P2(SJL)-Myd88tm1DefrCg-Tg(Itgax-cre)1-1Reiz/J (MyD88DCD11c) | Caton et al.91 and Hou et al.92 | The Jackson Laboratory |
| B6.SJL-PtprcaPepcb/Boy/J (CD45.1) | Shen et al.93 | The Jackson Laboratory |
| C57BL/6-pIgR™/J (pIgR-/-) | Shimada et al.94 | The Jackson Laboratory |
| B6.Cg-Tnfrsf11atm1.1Irw/J-Tg(Vil1-cre)997Gum/J (RANKDIEC) | Rios et al.45 | The Jackson Laboratory |
| B6(Cg)-Ifnar1™/J-Tg(Itgax-cre)1-1Reiz/J (IFNARDCD11c) | Caton et al.91 and Kamphuis et al.95 | The Jackson Laboratory |
| B6.129P2(Cg)-Cx3cr1tm1Litt/J (CX3CR1-GFP) | Jung et al.96 | The Jackson Laboratory |
| Oligonucleotides | ||
| 5′-GCTGCCTCCCGTAGGAGT-3′ | IDT | Eub338 3′ and 5′ AF647 labeled |
| 5′-CCTACGGGNGGCWGCAG-3′ (16S_341_F) | Klindworth et al.97 | 16S_341_F |
| 5′-GACTACHVGGGTATCTAATCC-3′ (16S_805_R) | Klindworth et al.97 | 16S_805_R |
| Ccl9 | Thermofisher Scientific | Mm00441260_m1 |
| Spib | Thermofisher Scientific | Mm03048233_m1 |
| Gp2 | Thermofisher Scientific | Mm00482557_m1 |
| Intronic GAPDH | Thermofisher Scientific | Mm05724508-g1 |
| Hprt | Thermofisher Scientific | Mm00446968_m1 |
| Software and algorithms | ||
| FlowJo v10 | BD | RRID: SCR_008520 |
| Seurat v3.1.0 | Satija et al.98 | https://satijalab.org/seurat/ |
| Prism v9 | GraphPad Software | RRID: SCR_002798 |
| Cell Ranger v2.1.1 | 10x Genomicx | https://support.10xgenomics.com/single-cell-gene-expression/software/release-notes/2-1 |
| DeqMS v1.8.0 | N/A | https://github.com/yafeng/DEqMS |
| Monocle v2.12.0 | Trapnell et al.99 | https://www.bioconductor.org/packages/release/bioc/html/monocle.html |
| BOTA v0.1.0 | Graham et al.35 | N/A |
| MEGAhit v1.2.9 | Li et al.100 | https://kbase.us/applist/apps/MEGAHIT/run_megahit/release |
| PROKKA v1.13 | Seemann et al.101 | https://github.com/tseemann/prokka/releases |
| Kofamscan v1.3.0 | Aramaki et al.102 | https://github.com/takaram/kofam_scan |
| Trimmomatic v0.36 | Bolger et al.103 | http://www.usadellab.org/cms/?page=trimmomatic |
| BBmap 11-06-2018 | Bushnell104 | https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/bbmap-guide/ |
| BBsplit 11-06-2018 | Bushnell104 | https://github.com/BioInfoTools/BBMap/blob/master/sh/bbsplit.sh |
| iTOL v6.5 | Letunic et al.105 | https://itol.embl.de/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mathias W. Hornef (mhornef@ukaachen.de).
Materials availability
Material transfer agreements with standard academic terms will be established to document reagent sharing by the lead contact’s institution.
Experimental model and subject details
Animals
All animal experiments were performed in compliance with the German animal protection law (TierSchG) and approved by the local animal welfare committees, the Landesamt für Natur, Umwelt und Verbraucherschutz, North Rhine Westfalia (84-02.04.2015.A063 and 84-02.04.2016.A207) and Regierungspräsidium Freiburg, Freiburg (X-20/05F). C57BL/6 wild-type (WT), B6.Cg-Tg(TcraTcrb)425Cbn Rag1tm1Mom/J (OTII), C57BL/6-Tg(TcraTcrb)1100Mjb/J (OTI), B6.129P2(SJL)-Myd88tm1DefrCg-Tg(Itgax-cre)1-1Reiz/J (MyD88ΔCD11c), B6.SJL-Ptprca Pepcb/Boy/J (CD45.1), C57BL/6-pIgR™/J (pIgR-/-), B6.Cg-Tnfrsf11atm1.1Irw/J-Tg(Vil1-cre)997Gum/J (RANKΔIEC), B6(Cg)-Ifnar1™/J-Tg(Itgax-cre)1-1Reiz/J (IFNARΔCD11c) and B6.129P2(Cg)-Cx3cr1tm1Litt/J (CX3CR1-GFP) mice were bred locally and held under specific pathogen–free (SPF) or germ-free (GF) conditions at the Institute of Laboratory Animal Science at RWTH Aachen University Hospital. C57BL/6NTac wildling mice were created through inverse germ-free rederivation as previously desribed,38 housed as well as bred locally at the animal facility of the Medical Center – University of Freiburg, Germany. C57BL/6NTac murine pathogen free (MPF) control mice were originally purchased from Taconic Biosciences, subsequently bred locally and housed under SPF conditions at the animal facility of the Medical Center - University of Freiburg, Germany. Wildlings and C57BL/6NTac MPF mice were age matched for all experiments.
In vivo models
For R848 administration, PND11 and adult mice were administered with 0.4 μg/g body weight (b.w.) R848 by intragastric gavage. For the OTI/OTII transfer, 1-2x105 eF670 marked OTI or OTII cells/g b.w. were transferred into PND11 and adult mice i.p. and OVA was administered 24 h later by intragastric gavage at 2 mg/g body weight 48 h after OVA administration, the proliferation of the transferred T cells was assessed. In experiments involving R848, R848 was orally gavaged 4-5 h after OVA at 0.4 μg/g b.w. For exposure of neonatal mice to the adult/weaning enteric microbiota, small intestine, caecum, and colon of one adult animal or two PND21-22 animals were opened longitudinally and flushed with 3-5 mL sterile PBS per animal for 5 min. under gentle agitation. Tissues were removed and clumps were suspended by vigilant pipetting. Large matter was removed by brief centrifugation at 300g for 30s and the supernatant was collected. Aliquots of adult intestinal content were stored at -20°C for single use and administered daily by intragastric gavage to neonatal mice starting at PND3-10 for T cell activation analysis or PND9-11 for the MNP analysis. For administration of B.fragilis (mix of strains CLA-KB-H83 and CLA-JM-H48) 5x109 bacterial cells from frozen stocks were administered by oral gavage at PND9-11. E.coli OMVs were prepared as described by Erttman et al., and gavaged 25μg were administered by oral gavage. Flagellin (from S.Typhimurium, ultrapure, Invivogen) and recombinant RANKL (Biomol) were injected i.p. at 2μg and 10μg at PND9-11, respectively. For the administration of fluorescent beads, 5x1011 yellow-green fluorescent 100 nm beads/g b.w. (Polysciences) were administered by intragastric gavage to PND11 and adult mice and PP were examined 6 h later.
Vaccination against S.Typhiumrium/ S.Typhimurium infection
The vaccination protocol from Moor et al.44 was adapted. Salmonella Typhimurium (STm) (SL1344) was grown in 500 mL at 37°C and 200 rpm to late stationary phase, harvested by centrifugation (3000 xg, 15 min., 4°C) and resuspended at 109-1010 CFU/mL in PBS. Bacterial suspensions were incubated in 10 mL aliquots in 50 mL tubes with 0.04% peracetic acid (PAA) (Sigma) for 1 h at RT, centrifuged at 3000 xg for 10 min. and resuspended at 1011 particles/ mL. 100 μl of the suspension was plated to verify complete inactivation. Mice were orally gavaged with 4x109 particles PAA STm/g b.w. ± 0.4 μg/b.w. R848 at PND7 and 14 and with PAA STm but no R848 at PND21. Adult mice were pretreated with streptomycing and orally infected with 103 CFU STm. Serum and SI wash and colonic LP cells were collected 24 hours post infection. Neonatal mice were infected orally with 2x103 CFU STm.
Method details
Isolation of PP cells
For neonatal PP a dissection microscope was used. PP were excised, counted and digested using 0.2 mg/mL Liberase TH/DNAse I for 45 min at 37°C and then dissociated by vigorous pipetting. Myeloid cells were positively enriched using CD11c MicroBeads (Miltenyi). OTI and OTII cells were isolated from lymph nodes and digested using 0.2 mg/mL Liberase TH/DNAse I for 45 min at 37°C and then dissociated by vigorous pipetting. OTI cells were then further enriched using a naïve CD8+ T cell isolation kit (Miltenyi).
Flow cytometry and cell sorting
Single cell suspensions were blocked with anti-mouse CD16/32 (clone 93, Biolegend, 1:200 dilution) and stained with fluorescently labeled antibodies for 20-40 min. at 4°C. Antibodies were used at a 1:200 dilution unless stated otherwise. CCR7 surface staining was incubated at room temperature for 45 min. at a dilution of 1:50 prior to surface staining with the other antibodies. To determine coating of Salmonella with specific immunoglobulins from serum and small intestinal washes the protocol published by Moor et al.44 was used. Blood was collected into clotting activating gel tubes (Sarstedt), incubated 15-30 min. at RT and centrifuged at 16000 xg for 15 min.; serum was stored at -20°C until further use. Small intestines were opened longitudinally, suspended in sterile PBS for 5 min., and vortexed. The tissue was removed and the SI wash was centrifuged at 16000 xg for 10 min.; supernatants were stored at -20°C until further use. Serum and SI washes were inactivated at 56°C for 30 min. and centrifuged at 16000 xg for 10 min. Salmonella Typhimurium (SL1344) were grown to late stationary phase in liquid medium, pelleted at 3000 xg at 4°C for 10 min., washed in 0.4 μm filtered PBS, and resuspended at 107 CFU/mL in 0.4 μm filtered 3%FCS/PBS. Sera and SI washes serially diluted (1:2) with an initial dilution of 1:3 and undiluted, respectively. 25 μl of bacteria were incubated with 25 μl of serum/SI wash for 1 h at 4°C, washed twice with 200 μL 0.4 μm filtered 3% FCS/PBS, stained with fluorescently labeled antibodies for 1 h at 4°C and washed before acquisition.
Following antibody clones (Biolegend) were used: anti-CD11b (clone M1/70), anti-CD11c, anti-CD4 (clone RM4-5), anti-CD44 (clone IM7), anti-CD45R/B220 (clone RA3-6B2), anti-CD86 (clone GL1), anti-IA/IE (clone M5/114.15.2, 1:400), anti-XCR1 (clone ZET), anti-BST2 (clone 927), anti-CCR7 (clone 4B12), anti-CD8a (clone 53-6.7), anti-CD19 (clone 6D5), anti-CD45 (clone 30-F11), anti-CD45.1 (clone A30), anti-CD45.2 (clone 104), anti-SIRPa (clone P84), anti-CD74 (clone In1/CD74), anti-mouse IgG1 (clone RMG1-1, 1:40), anti-mouse IgA (clone C10-3, 1:50), anti-mouse IgM (clone RMM-1), anti-RORgt (clone B2D), anti-TCRb (clone H57-597), anti-TCRVa2 (clone 20.1), anti-GL7 (clone GL7), anti-B220 (clone RA3-6B2), anti-CD19 (clone 6D5), anti-CD3 (clone 17A2), anti-CD64 (clone X54-5/7.1), anti-FAS (clone Jo2, BD opti build). Homemade PLET1 antibody was provided by C.Ruedl and conjugated to AF647. Dead cells were stained with 7-AAD (Biolegend) or ZOMBIE NIR (Biolegend). Countbrite counting beads (Biolegend) were added to the FACS samples to assess the absolute cell numbers. Cells were acquired on a BD LSR Fortessa or a Cytek Aurora. Cell sorting was carried on a FACS ARIA II (BD) equipped with a 100 mm nozzle using BD FACS DIVA software. Final analysis was performed using FlowJo (V 10, BD).
DC OTII co-culture and T helper cytokine induction
PP MNP (MHCII+CD11c+CD45+live) from PND11 and adult wild type mice (pooled litter, 4 adult animals per biological replicate) and OTII (CD4+live) were FACS sorted and co-cultured at a ratio of 1:5 in IMDM 10% FCS, 1% penicillin and streptomycin (GIBCO), 1% L-glutamine (GIBCO) and 5 nM 2-mercaptoethanol (Carl-Roth) in the presence of 1 mg/mL OVA for 5 days. Supernatants were analyzed using the Legendplex mouse T helper cytokine panel version 3 (Biolegend) according to the manufacturer’s instructions. Independent experiments were pooled after normalization.
Quantification of 16S rRNA gene copy numbers
The 16S rRNA gene copy numbers were determined in total homogenized small intestinal and colonic tissue samples of C57BL/6J wildtype mice collected at 1, 7, 14, 28, and 56 days after birth (n = 6/age group) and described previously.67 Total DNA was isolated from approximately 200 mg frozen tissue by repeated bead-beating combined with chemical lysis and column-based purification using the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.106 Subsequently, the PCR primers 5′-CCTACGGGNGGCWGCAG-3′ (16S_341_F) and 5′-GACTACHVGGGTATCTAATCC-3′ (16S_805_R) were used for amplification.97 The real-time PCR was performed on a MyiQTM System (BioRad, USA) in a reaction-volume of 25 μl with 12.5 μl iQTM SYBR Green (Biorad), 2 μl template DNA and 0.75 μl primers (10 μM). The cycling conditions were 95 °C for 3 min. followed by 35 cycles of 95 °C for 15 s; 55 °C for 20 s and 72 °C for 30 s. Total 16S gene copy numbers were calculated by comparing the CT value to a standard curve with known concentrations of a plasmid encoding the 16S rDNA gene target sequence of E.coli. Tissues from germ free animals were used as negative controls, in which no specific PCR product was detected.
Quantification of bacteria by anaerobic culture
Small intestines were opened longitudinally, quickly transferred to Hungate tubes containing anaerobic PBS (0.05% L-cysteine, 0.02% DTT; gas mixture of 6% CO2, 4.7% H2 in N2) with ∼10 glass beads and vortexed. To ensure anaerobic conditions, the supernatant was transferred into another Hungate tube containing anaerobic PBS (0.05% L-cysteine, 0.02% DTT) using a sterile syringe fitted with a needle to inoculate through the rubber stopper of the Hungate tube. The suspension was plated onto tryptone soy agar plates with 5% sheep blood (Oxoid) in an anaerobic workstation (MBRAUN, Garching, Germany; same gas mixture as above). All materials were brought into the workstation at least 24h prior to work. CFU were quantified after 24h of incubation at 37 °C.
Immunofluorescence and confocal microscopy
For spectral confocal imaging CX3CR1-GFP+/- reporter mice were used. PP were excised and fixed using Antigenfix for 45 min. at 4°C and incubated in 30% sucrose/PBS until saturated. Then, they were embedded in OCT compound as part of intestinal tube in an upright position, frozen over liquid nitrogen and stored at -80°C. Sections were cut at 10μm thickness and slides were stored at -20°C. After permeabilization and unspecific binding site blocking with PBS containing 0.5% saponin, 2% bovine serum albumin, 1% fetal calf serum, and 1% donkey serum for 30 minutes, sections were labeled overnight at 4°C with primary antibodies followed by washing in PBS before incubation for 1 hour at room temperature with secondary antibodies. Sections were then washed and blocked with sera of primary antibody species for 30 minutes, before staining in the same blocking buffer with fluorochrome-coupled antibodies for 1 hour. Slides were mounted in ProLong Gold and observed with a Zeiss LSM 780 confocal microscope using the spectral imaging mode.107 For FISH staining tissues were fixed using methacarn to ensure the preservation of the mucus structure and subsequently paraffin embedded. Sections were deparaffined using successive xylene washes and ethanol washes (95%, 90%) before being rehydrated with ddH20. Sections were stained for commensal bacteria using the eubacterial FISH probe Eub338 (GCT GCC TCC CGT AGG AGT) labelled with AF647 at 100nM in hybridization buffer (0.9M NaCl, 20-mM Tris-Hcl pH 7.2 0.1%SDS Tris) at 50°C overnight. Slides were washed with prewarmed wash buffer (0.9M NaCl, 20-mM Tris-Hcl pH 7.2) prior to Hoechst 33342 (10ug/mL) nucleic acid staining. Slides were mounted using ProLong Antifade Gold and imaged using a Nikon 90i at the University of Pittsburgh Center for Biological Imaging.
Quantification of the CD11c signal distribution in PP
In a first instance all images were manually segmented by encircling each follicle individually with a polygon. This polygon mask served, together with the image, as basis for further processing steps. To determine the distribution of the CD11c signal with respect to the distance to the surrounding shape the following steps were performed: First, the image was filtered with a Gaussian kernel (σ=9, filter size: 201x201) to achieve a smoother surface and to account for the sparse distribution of positive cells within the image. Next, mean pixel intensities were computed within rings with a specified minimum and maximum distance from the masks' contour. This was obtained using morphological erosion with circular structuring elements of different diameters. To measure the occurrence of the CD11c signal with respect to distance to the contour, we extracted these mean values in non-overlapping, consecutive rings within the whole mask as illustrated in Figure 3B. The thickness (diameter) of each ring was set to 5 pixels to achieve a good trade-off between resolution and smoothness. The center circle was not considered for further processing since it can become arbitrarily small leading to inaccurate measurements. With the obtained mean values, we fit a first order polygon and used the slope Δ as a descriptor for each follicle. The descriptor was normalized for the size of the mask by multiplying the slope with the number of rings.
Single cell RNA sequencing and analysis
Pooled cells from a litter of neonatal mice (5-8 animals) or 3 adult mice were used and samples were analyzed in biological duplicates for the PND11 vs. adult PP MNP experiment and as single samples for the R848 stimulation experiment. MACS enriched cells isolated from PP were subjected to FACS sorting as CD11c+MHCII+CD45+live+IgA-TCRb- and further processed using the Chromium Single Cell 3’ v2 kit (10xGenomics) according to the manufacturer’s protocol. After library generation, sequencing was performed on a NextSeq 550 (IZKF genomics facility of the RWTH Aachen University, Aachen, Germany). Sequencing was performed on Illumina NextSeq 550 (paired-ends, 2×75 bp). With default parameters, we used Cell Ranger (version 2.1.1) to align reads to the mouse genome mm10. Following that, we utilized Seurat (v3.1.0)98 to achieve a high-level analysis of the scRNA-seq data. Cells with high mitochondrial (≧10%) and ribosomal (≧25%) gene content, as well as cells with ≧30.000 UMIs having an increased probability to represent doublets and cells with ≦30 detected genes were excluded from further analysis. We used Seurat to regress out cell cycle, mitochondrial, ribosomal, and UMI counts and performed a log-normalization of read counts. In a second step contaminant cell types (B cells, Cd19; pDCs, Siglec; villus macrophages; Fcgr1 (the latter only for the data set examining naïve PND11 vs. adult MNP)) were removed. After quality control and contaminant removal 3659 and 10756 cells were analyzed and depicted in the UMAPs in Figures 1E and 6H, respectively. Canonical correlation analysis (CCA) based on the first 15 CCs was used to combine samples from R848 and PBS experiments.108 Unsupervised clustering was performed using a shared nearest neighbors graph with a resolution of 0.8 and k = 15. The following parameters were used to build UMAP representations: min dist = 0.3, max.dim = 2, seed.use = 36). To determine cluster specific markers, we implemented the FindMarkers gene function with an adjusted p value of 0.05. Differential gene expression analysis was performed with Seurat using the Wilcoxon test. Next, we used monocle (version 2.12.0)99 in the integrated data to reconstruct cell development trajectories. For the reconstruction of cell development trajectories, we employed the 1000 most relevant genes identified by Monocle's unsupervised feature selection approach (dpFeature). The root of the trajectory was defined choosing the branch with the highest S-score, which also correlated with the expression of stemness-associated genes such as Birc5. For the PND11 PBS vs. PND11 R848 and adult PBS vs. adult R848 scRNA-seq data set data integration was performed by anchors identification based on the results of CCA analysis. The first 20 CCs were used for the anchor identification and first 20 PCs from PCA analysis were used for UMAP representation. For clustering resolution was set to 0.5. For the analysis of commonly regulated genes in PND11 vs. Adult and PND11 PBS vs. R848 qDC2 Gene Set Enrichment Analysis (GSEA) via gseGO function and the compareClusters function from clusterProfiler(cite here) (version 4.6.0) package was performed. Both sets were further sub-divided on average log_FC values - genes with average log_FC > 0 were considered to be Upregulated while those with average log_FC < 0 were considered to be Downregulated. The enrichGO function via the fun parameter of the compareClusters function was used to carry out enrichment analysis of respective upregulated and downregulated genes and the resulting shared set of GO terms and associated genes were depicted using a cnet plot. Comparative GO term enrichment was further carried out using the REVIGO pipeline.109 Pseudobulk analysis against the ImmGen clusters was carried out as previously described.110 Raw data were deposited under GEO accession number GSE188714.
Shotgun metagenomic sequencing
DNA for the metagenomic analysis was isolated using a modified protocol according to Godon et al.111 Snap frozen samples were mixed with 600 μl stool DNA stabilizer (Stratec Biomedical), thawed, and transferred into sterile 2-mL screw-cap tubes containing 500 mg 0.1 mm (diameter) silica/zirconia beads. 250 μl 4 M guanidine thiocyanate in 0.1 M Tris (pH 7.5) and 500 μl 5 % N-lauroyl sarcosine in 0.1 M PBS (pH 8.0) were added. Samples were incubated at 70 °C and 700 rpm for 60 min. A FastPrep® instrument (MP Biomedicals) cooled with dry ice was used for cell disruption (3 x 40 s at 6.5 M/s). Subsequently, 15 mg polyvinylpyrrolidone (PVPP) was added and samples were vortexed, followed by 3 min. centrifugation at 15000 xg and 4°C. Approximately 650 μl of the supernatant was transferred into a new 2 mL tube, which was centrifuged for 3 min. at 15000 xg and 4°C. Afterwards, 500 μl of the supernatant were transferred into a new 2 mL tube and 50 μg of RNase was added. After incubation at 37 °C and 700 rpm for 20 min., the gDNA was isolated using the NucleoSpin® gDNA Clean-up Kit from Macherey-Nagel according to the manufacturer’s protocol. DNA was eluted from columns twice using 40 μl elution buffer and the concentration was determined using a NanoDrop® (Thermo Scientific). Samples were stored at -20 °C.
Sequencing of the metagenomic DNA was performed as follows: mechanical shearing of 600 ng DNA to a size of 200 bp (200 s, peak incident power: 75 W, duty factor: 10%, cycles per burst: 200) was performed using a Covaris M220. Subsequently the libraries were prepared with the NEBNext® Ultra II DNA Library Prep Kit for Illumina® (NEB, USA) according to the manufacturer’s protocol. The PCR enrichment of adaptor-ligated DNA was conducted with 7 cycles and NEBNext® Multiplex Oligos for Illumnina® (NEB, USA) for single-end barcoding. Size selection and clean-up of adaptor ligated DNA was conducted using AMPure beads (Beckman Coulter, USA). The quality (TapeStation, Agilent Technologies, USA) and quantity (Quantus, Promega, USA) of the resulting libraries was checked at the IZKF Core Facility (UKA, RWTH Aachen University) prior to sequencing on a NextSeq500 (Illumina, USA) with a NextSeq500 High Output Kit v2.5 (300 Cycles).
Metagenomic data analysis
For quality control, the metagenomic reads were first trimmed using Trimmomatic103 using a sliding window of 5:20, minimum length of 50, trailing of 3, leading of 3 and illumine clipping values of 2:30:10. Host reads were removed using BBmap104 to map the reads against the genome of Mus musculus GRCm38. Alignment was done using a minimum ratio of 0.9, maximum indel of 3, band width ratio of 0.16, band width of 12, minimum hits of 2 with at least 25 consecutive matches using a kmer length of 14. The first 10bp on each read before mapping and regions with an average score below 10 were also trimmed but undone after mapping (untrim).
The host-filtered reads of the metagenomic DNA were then assembled using MEGAhit100 using a kmer list of 21,27,33,37,43,55,63,77,83,99, with a minimum multiplicity for filtering kmers of 5. PROKKA was used to predict the proteome from the assembled contigs using default options.101 The predicted proteome was functionally annotated against the KEGG database using Kofamscan.102
Host-filtered reads were then aligned to the iMGMC non-redundant collection of metagenome-assembled genomes (MAGs)34 using BBsplit104 with a minimum identity of 90% to produce a coverage statistics file which was converted into TPM via the makeTPMfromCovStats.sh script from the iMGMC best-practices repository. The TPM values were used to calculate the minimal number of species that covered the maximal amount of the data based on the knee point, implemented in python using the Kneed module.112 Only MAGs with a TPM value >100 in >2 samples were retained for further analysis.
A phylogenomic tree of the 153 iMGMC-derived MAGs was generated using their predicted proteomes in Phylophlan3 v3.0.60.113 Diversity was set as high using the ‘supermatrix_aa.cfg’ config file. The tree was visualized in iTOL.105 Raw data were deposited under BioProject accession number PRJNA794356.
Antigenic peptide prediction from metagenomic data using BOTA
The proteins encoded on the 153 MAGs recovered from iMGMC were predicted using PROKKA v1.14.6.101 The genome, predicted proteome and general features were used as input for BOTA v0.1.035 using the H-2-IAb model for MHC class II (allele IA) antigen prediction.
Metaproteomic analysis
For metaproteome analysis, samples were dissolved in 500 μl SDS lysis buffer (0.29 g NaCl, 1M Tris-HCL pH=8, 5M EDTA pH=8, 0.4g SDS in 100 mL water). Protein extraction was done by bead beating (FastPrep-24, MP Biomedicals, Sanra Ana, CA, USA; 5.5 ms, 1 min., 3 cycles) followed by ultra-sonication (UP50H, Hielscher, Teltow, Germany; cycle 0.5, amplitude 60%) and centrifugation (10,000 xg, 10 min.). The protein lysate was loaded on a SDS-gel and run for 10 min. The gel piece was cut, washed and incubated with 25 mM 1,4 dithiothreitol (in 20 mM ammonium bicarbonate) for 1 h and 100 mM iodoacetamide (in 20 mM ammonium bicarbonate) for 30 min., destained, dehydrated and proteolytically cleaved overnight at 37 °C with trypsin (Promega). The proteolytically cleaved peptides were extracted and desalted using ZipTip μC18 tips (Merck Millipore, Darmstadt, Germany). The peptide lysates were re-suspended in 15 μl 0.1% formic acid and analysed by nanoliquid chromatography mass spectrometry (UltiMate 3000 RSLCnano, Dionex, Thermo Fisher Scientific). Mass spectrometric analysis of eluted peptide lysates was performed on a Q Exactive HF mass spectrometer (Thermo Fisher Scientific) coupled with a TriVersa NanoMate (Advion, Ltd., Harlow, UK). Peptide lysates were injected on a trapping column (Acclaim PepMap 100 C18, 3 μm, nanoViper, 75 μm x 2 cm, Thermo Fisher Scientific) with 5 μl/min. using 98% water/2% ACN 0.5% trifluoroacetic acid, and separated on an analytical C18 column (Acclaim PepMap 100 C18, 3 μm, nanoViper, 75 μm x 25 cm, Thermo Fisher Scientific) with a flow rate of 300 nl/min. The mobile phase was 0.1% formic acid in water (A) and 80 % ACN/0.08 % formic acid in water (B). Full MS spectra (350–1,550 m/z) were acquired on the Orbitrap at a resolution of 120,000 with an automatic gain control (AGC) target value of 3×106 ions. Data resulting from LC-MS/MS measurements were analyzed with the Proteome Discoverer (v.2.4, Thermo Fischer Scientific) using SEQUEST HT. Protein identification was performed using a self-built reference database downloaded from UniProt (reference numbers: UP000008827.fasta, UP000019116.fasta, UP000007305.fasta and the predicted proteome from the combined metagenomic assembly). Searches were conducted with the following parameters: Trypsin as enzyme specificity and two missed cleavages allowed. A peptide ion tolerance of 10 ppm and an MS/MS tolerance of 0.05 Da. As modifications, oxidation (methionine) and carbamidomethylation (cysteine) were selected. Peptides that scored a q-value >1% based on a decoy database and with a peptide rank of 1, were considered identified. For the differential analysis of the proteomics data the R package DeqMS 1.8.0 (https://doi.org/10.1074/mcp.TIR119.001646) was used. For each group (PND11 and adult), proteins with valid quantification in at least half of the samples (3/6) were included.
Quanitification and statistical analysis
The Mann-Whitney U test was used to compare two groups, the Kruskall-Wallis with Dunn’s multiple comparison was used to compare multiple groups and one variable, 2-way-ANOVA with Bonferroni’s multiple comparison test was used to compare multiple groups and two variables. Statistical tests were carried out using Graphpad Prism and p<0.05 was considered significant. The number of biological replicates analyzed and the statistical test used are indicated in the respective figure legends. ns, not significant; ∗, p<0.05; ∗∗, p<0.01; ∗∗∗, p<0.001, ∗∗∗∗, p<0.0001.
Acknowledgments
We would like to thank A. Schwentker, D.P.P. Branco, D. Wylensek (RWTH Aachen), and C. Wagner (CIML Marseille) for help with animal experiments/data analysis; V. Cerovic (RWTH Aachen) for critical discussion, E. Slack (ETH Zürich) for advice on Salmonella vaccination, S. Jennings (RWTH Aachen) for gnotobiotic animals, A. Dupont (RWTH Aachen) for mouse breeding management, C. Ruedl (Nanyang Technological University Singapore) for the Plet1 antibody, and Nelson Gekara and Kyaw Min Aung (both Stockholm University) for generation of E. coli MVs and the genomics and flow cytometry core facilities of the Interdisciplinary Center for Clinical Research (IZKF) Aachen within the Faculty of Medicine at RWTH Aachen University. This work was supported by TO1052/1-1 (to N. Torow) and the Collaborative Research Center CRC1382 (project ID 403224013—SFB 1382 projects rising star, A05, B01, and Q02 to N. Torow, M.W.H., M.v.B., and T.C.), Clinical Research Unit 344 (KFO344, project ID 417911533 to N. Torow), CRC/TRR 359 (project ID 491676693 to S.P.R., T.C., and M.W.H.), Emmy Noether-Programm RO 6247/1-1 (project ID 446316360 to S.P.R.) and SFB1160-2 IMPATH (project ID 256073931 to S.P.R.) from the German Research Foundation (DFG) as well as the ERC Advanced Grant EarlyLife (project ID: 101019157 to M.W.H.). N. Torow was supported by the START-Program of the Faculty of Medicine, RWTH Aachen (to N. Torow). H.L. was supported by the French National Research Agency ANR-20-CE15-0016 and used the PICSL imaging facility of the CIML (ImagImm), member of the national infrastructure France-BioImaging, supported by the French National Research Agency (ANR-10-INBS-04). M.v.B. and N.J. were supported by the UFZ for the ProMetheus platform for proteomics and metabolomics.
Author contributions
Conceptualization, N. Torow, M.W.H., and I.G.C.; methodology, N. Torow, R.L., T.C.A.H., M.G., D.M., N.J., M.v.B., H.L., and I.G.C.; software, R.L., T.C.A.H., A.B., M.G., D.M., I.G.C., T.M., and N.M.K.P.G.; validation, N. Torow, M.W.H., and I.G.C.; formal analysis, N. Torow, R.L., T.C.A.H., C.M., S.A.B., N.v.B., E.-L.S., A.B., M.G., I.G.C., and N.M.K.P.G.; investigation, N. Torow, C.M., S.A.B., N.v.B., E.-L.S., S.R., N. Treichel, F.H., H.L., B.S., and D.A.A.; resource, D.M., S.P.R., M.v.B., T.C., F.T., I.G.C., O.P., and T.W.H.; data curation, N.T., R.L., T.C.A.H., and N. Treichel; writing – original draft, N. Torow and M.W.H.; writing – review & editing, N. Torow, M.W.H., T.C., H.L., and O.P.; visualization, N. Torow, R.L., T.C.A.H., C.M., H.L., T.M., and N.M.K.P.G.; supervision, N. Torow, M.W.H., and I.G.C.; project administration, N. Torow and M.W.H.; funding acquisition, N.T., M.W.H., and I.G.C.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: May 1, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.immuni.2023.04.002.
Contributor Information
Natalia Torow, Email: ntorow@ukaachen.de.
Mathias Walter Hornef, Email: mhornef@ukaachen.de.
Supplemental information
Strains, corresponding BOTA scores, and TPM values obtained by shotgun sequencing shown in Figure 5.
DeqMS analysis output table of differential proteomic analysis.
Data and code availability
-
-
scRNAseq data are deposited with the National Center for Biotechnology Information Gene Expression Omnibus (GEO) under the accession numbers GSE188714.
-
-
Metagenomic data are deposited under the NCBI-BioProject accession number PRJNA794356: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA794356.
-
-
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
-
This paper does not report original code.
References
- 1.Herzenberg L.A., Herzenberg L.A. Toward a layered immune system. Cell. 1989;59:953–954. doi: 10.1016/0092-8674(89)90748-4. [DOI] [PubMed] [Google Scholar]
- 2.Hornef M.W., Torow N. 'Layered immunity' and the 'neonatal window of opportunity' - timed succession of non-redundant phases to establish mucosal host-microbial homeostasis after birth. Immunology. 2020;159:15–25. doi: 10.1111/imm.13149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdel-Gadir A., Stephen-Victor E., Gerber G.K., Noval Rivas M., Wang S., Harb H., Wang L., Li N., Crestani E., Spielman S., et al. Microbiota therapy acts via a regulatory T cell MyD88/RORgammat pathway to suppress food allergy. Nat. Med. 2019;25:1164–1174. doi: 10.1038/s41591-019-0461-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cahenzli J., Köller Y., Wyss M., Geuking M.B., McCoy K.D. Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe. 2013;14:559–570. doi: 10.1016/j.chom.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynn M.A., Tumes D.J., Choo J.M., Sribnaia A., Blake S.J., Leong L.E.X., Young G.P., Marshall H.S., Wesselingh S.L., Rogers G.B., et al. Early-life antibiotic-driven dysbiosis leads to dysregulated vaccine immune responses in mice. Cell Host Microbe. 2018;23:653–660.e5. doi: 10.1016/j.chom.2018.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Olszak T., An D., Zeissig S., Vera M.P., Richter J., Franke A., Glickman J.N., Siebert R., Baron R.M., Kasper D.L., Blumberg R.S. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zanvit P., Konkel J.E., Jiao X., Kasagi S., Zhang D., Wu R., Chia C., Ajami N.J., Smith D.P., Petrosino J.F., et al. Antibiotics in neonatal life increase murine susceptibility to experimental psoriasis. Nat. Commun. 2015;6 doi: 10.1038/ncomms9424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kirjavainen P.V., Karvonen A.M., Adams R.I., Täubel M., Roponen M., Tuoresmäki P., Loss G., Jayaprakash B., Depner M., Ege M.J., et al. Farm-like indoor microbiota in non-farm homes protects children from asthma development. Nat. Med. 2019;25:1089–1095. doi: 10.1038/s41591-019-0469-4. [DOI] [PubMed] [Google Scholar]
- 9.Schuijs M.J., Willart M.A., Vergote K., Gras D., Deswarte K., Ege M.J., Madeira F.B., Beyaert R., van Loo G., Bracher F., et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science. 2015;349:1106–1110. doi: 10.1126/science.aac6623. [DOI] [PubMed] [Google Scholar]
- 10.Vatanen T., Kostic A.D., d'Hennezel E., Siljander H., Franzosa E.A., Yassour M., Kolde R., Vlamakis H., Arthur T.D., Hämäläinen A.M., et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. 2016;165:842–853. doi: 10.1016/j.cell.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaupp L., Muth S., Rogell L., Kofoed-Branzk M., Melchior F., Lienenklaus S., Ganal-Vonarburg S.C., Klein M., Guendel F., Hain T., et al. Microbiota-induced Type I interferons instruct a poised basal state of dendritic cells. Cell. 2020;181:1080–1096.e19. doi: 10.1016/j.cell.2020.04.022. [DOI] [PubMed] [Google Scholar]
- 12.Koya T., Yanagisawa R., Higuchi Y., Sano K., Shimodaira S. Interferon-alpha-inducible dendritic cells matured with OK-432 exhibit TRAIL and Fas ligand pathway-mediated killer activity. Sci. Rep. 2017;7 doi: 10.1038/srep42145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Date I., Koya T., Sakamoto T., Togi M., Kawaguchi H., Watanabe A., Kato T., Jr., Shimodaira S. Interferon-alpha-induced dendritic cells generated with human platelet lysate exhibit elevated antigen presenting ability to cytotoxic T lymphocytes. Vaccines (Basel) 2020;9 doi: 10.3390/vaccines9010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spadaro F., Lapenta C., Donati S., Abalsamo L., Barnaba V., Belardelli F., Santini S.M., Ferrantini M. IFN-alpha enhances cross-presentation in human dendritic cells by modulating antigen survival, endocytic routing, and processing. Blood. 2012;119:1407–1417. doi: 10.1182/blood-2011-06-363564. [DOI] [PubMed] [Google Scholar]
- 15.Zietara N., Łyszkiewicz M., Gekara N., Puchałka J., Dos Santos V.A., Hunt C.R., Pandita T.K., Lienenklaus S., Weiss S. Absence of IFN-beta impairs antigen presentation capacity of splenic dendritic cells via down-regulation of heat shock protein 70. J. Immunol. 2009;183:1099–1109. doi: 10.4049/jimmunol.0803214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gough D.J., Messina N.L., Clarke C.J., Johnstone R.W., Levy D.E. Constitutive type I interferon modulates homeostatic balance through tonic signaling. Immunity. 2012;36:166–174. doi: 10.1016/j.immuni.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taniguchi T., Takaoka A. A weak signal for strong responses: interferon-alpha/beta revisited. Nat. Rev. Mol. Cell Biol. 2001;2:378–386. doi: 10.1038/35073080. [DOI] [PubMed] [Google Scholar]
- 18.Torow N., Yu K., Hassani K., Freitag J., Schulz O., Basic M., Brennecke A., Sparwasser T., Wagner N., Bleich A., et al. Active suppression of intestinal CD4(+)TCRalphabeta(+) T-lymphocyte maturation during the postnatal period. Nat. Commun. 2015;6 doi: 10.1038/ncomms8725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luciani C., Hager F.T., Cerovic V., Lelouard H. Dendritic cell functions in the inductive and effector sites of intestinal immunity. Mucosal Immunol. 2022;15:40–50. doi: 10.1038/s41385-021-00448-w. [DOI] [PubMed] [Google Scholar]
- 20.Bunker J.J., Flynn T.M., Koval J.C., Shaw D.G., Meisel M., McDonald B.D., Ishizuka I.E., Dent A.L., Wilson P.C., Jabri B., et al. Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity. 2015;43:541–553. doi: 10.1016/j.immuni.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pabst O., Slack E. IgA and the intestinal microbiota: the importance of being specific. Mucosal Immunol. 2020;13:12–21. doi: 10.1038/s41385-019-0227-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lyu M., Suzuki H., Kang L., Gaspal F., Zhou W., Goc J., Zhou L., Zhou J., Zhang W., et al. JRI Live Cell Bank ILC3s select microbiota-specific regulatory T cells to establish tolerance in the gut. Nature. 2022;610:744–751. doi: 10.1038/s41586-022-05141-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kedmi R., Najar T.A., Mesa K.R., Grayson A., Kroehling L., Hao Y., Hao S., Pokrovskii M., Xu M., Talbot J., et al. A RORgammat(+) cell instructs gut microbiota-specific Treg cell differentiation. Nature. 2022;610:737–743. doi: 10.1038/s41586-022-05089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akagbosu B., Tayyebi Z., Shibu G., Paucar Iza Y.A., Deep D., Parisotto Y.F., Fisher L., Pasolli H.A., Thevin V., Elmentaite R., et al. Novel antigen presenting cell imparts Treg-dependent tolerance to gut microbiota. Nature. 2022;610:752–760. doi: 10.1038/s41586-022-05309-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonnardel J., Da Silva C., Wagner C., Bonifay R., Chasson L., Masse M., Pollet E., Dalod M., Gorvel J.P., Lelouard H. Distribution, location, and transcriptional profile of Peyer's patch conventional DC subsets at steady state and under TLR7 ligand stimulation. Mucosal Immunol. 2017;10:1412–1430. doi: 10.1038/mi.2017.30. [DOI] [PubMed] [Google Scholar]
- 26.Brown C.C., Gudjonson H., Pritykin Y., Deep D., Lavallée V.P., Mendoza A., Fromme R., Mazutis L., Ariyan C., Leslie C., et al. Transcriptional basis of mouse and human dendritic cell heterogeneity. Cell. 2019;179:846–863.e24. doi: 10.1016/j.cell.2019.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papaioannou N.E., Salei N., Rambichler S., Ravi K., Popovic J., Küntzel V., Lehmann C.H.K., Fiancette R., Salvermoser J., Gajdasik D.W., et al. Environmental signals rather than layered ontogeny imprint the function of type 2 conventional dendritic cells in young and adult mice. Nat. Commun. 2021;12 doi: 10.1038/s41467-020-20659-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cabeza-Cabrerizo M., Cardoso A., Minutti C.M., Pereira da Costa M., Reis e Sousa C. Dendritic cells revisited. Annu. Rev. Immunol. 2021;39:131–166. doi: 10.1146/annurev-immunol-061020-053707. [DOI] [PubMed] [Google Scholar]
- 29.Miller J.C., Brown B.D., Shay T., Gautier E.L., Jojic V., Cohain A., Pandey G., Leboeuf M., Elpek K.G., Helft J., et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol. 2012;13:888–899. doi: 10.1038/ni.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ardouin L., Luche H., Chelbi R., Carpentier S., Shawket A., Montanana Sanchis F., Santa Maria C., Grenot P., Alexandre Y., Gregoire C., et al. Broad and largely concordant molecular changes characterize tolerogenic and immunogenic dendritic cell maturation in thymus and periphery. Immunity. 2016;45:305–318. doi: 10.1016/j.immuni.2016.07.019. [DOI] [PubMed] [Google Scholar]
- 31.Da Silva C., Wagner C., Bonnardel J., Gorvel J.P., Lelouard H. The Peyer's patch mononuclear phagocyte system at steady state and during infection. Front. Immunol. 2017;8 doi: 10.3389/fimmu.2017.01254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torow N., Hornef M.W. The neonatal window of opportunity: setting the stage for life-long Host-Microbial Interaction and Immune Homeostasis. J. Immunol. 2017;198:557–563. doi: 10.4049/jimmunol.1601253. [DOI] [PubMed] [Google Scholar]
- 33.Hooper L.V., Littman D.R., Macpherson A.J. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lesker T.R., Durairaj A.C., Gálvez E.J.C., Lagkouvardos I., Baines J.F., Clavel T., Sczyrba A., McHardy A.C., Strowig T. An integrated metagenome catalog reveals new insights into the murine gut microbiome. Cell Rep. 2020;30:2909–2922.e6. doi: 10.1016/j.celrep.2020.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graham D.B., Luo C., O'Connell D.J., Lefkovith A., Brown E.M., Yassour M., Varma M., Abelin J.G., Conway K.L., Jasso G.J., et al. Antigen discovery and specification of immunodominance hierarchies for MHCII-restricted epitopes. Nat. Med. 2018;24:1762–1772. doi: 10.1038/s41591-018-0203-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rogier E.W., Frantz A.L., Bruno M.E., Wedlund L., Cohen D.A., Stromberg A.J., Kaetzel C.S. Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc. Natl. Acad. Sci. USA. 2014;111:3074–3079. doi: 10.1073/pnas.1315792111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beura L.K., Hamilton S.E., Bi K., Schenkel J.M., Odumade O.A., Casey K.A., Thompson E.A., Fraser K.A., Rosato P.C., Filali-Mouhim A., et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 2016;532:512–516. doi: 10.1038/nature17655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosshart S.P., Herz J., Vassallo B.G., Hunter A., Wall M.K., Badger J.H., McCulloch J.A., Anastasakis D.G., Sarshad A.A., Leonardi I., et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science. 2019;365 doi: 10.1126/science.aaw4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosshart S.P., Vassallo B.G., Angeletti D., Hutchinson D.S., Morgan A.P., Takeda K., Hickman H.D., McCulloch J.A., Badger J.H., Ajami N.J., et al. Wild mouse gut microbiota promotes Host Fitness and Improves Disease Resistance. Cell. 2017;171:1015–1028.e13. doi: 10.1016/j.cell.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stefan K.L., Kim M.V., Iwasaki A., Kasper D.L. Commensal microbiota modulation of natural resistance to virus infection. Cell. 2020;183:1312–1324.e10. doi: 10.1016/j.cell.2020.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Erttmann S.F., Swacha P., Aung K.M., Brindefalk B., Jiang H., Härtlova A., Uhlin B.E., Wai S.N., Gekara N.O. The gut microbiota prime systemic antiviral immunity via the cGAS-STING-IFN-I axis. Immunity. 2022;55:847–861.e10. doi: 10.1016/j.immuni.2022.04.006. [DOI] [PubMed] [Google Scholar]
- 42.Jiang H.Q., Bos N.A., Cebra J.J. Timing, localization, and persistence of colonization by segmented filamentous bacteria in the neonatal mouse gut depend on immune status of mothers and pups. Infect. Immun. 2001;69:3611–3617. doi: 10.1128/IAI.69.6.3611-3617.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yrlid U., Milling S.W., Miller J.L., Cartland S., Jenkins C.D., MacPherson G.G. Regulation of intestinal dendritic cell migration and activation by plasmacytoid dendritic cells, TNF-alpha and type 1 IFNs after feeding a TLR7/8 ligand. J. Immunol. 2006;176:5205–5212. doi: 10.4049/jimmunol.176.9.5205. [DOI] [PubMed] [Google Scholar]
- 44.Moor K., Wotzka S.Y., Toska A., Diard M., Hapfelmeier S., Slack E. Peracetic acid treatment generates potent inactivated oral vaccines from a Broad Range of culturable bacterial species. Front. Immunol. 2016;7 doi: 10.3389/fimmu.2016.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rios D., Wood M.B., Li J., Chassaing B., Gewirtz A.T., Williams I.R. Antigen sampling by intestinal M cells is the principal pathway initiating mucosal IgA production to commensal enteric bacteria. Mucosal Immunol. 2016;9:907–916. doi: 10.1038/mi.2015.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanaya T., Hase K., Takahashi D., Fukuda S., Hoshino K., Sasaki I., Hemmi H., Knoop K.A., Kumar N., Sato M., et al. The Ets transcription factor Spi-B is essential for the differentiation of intestinal microfold cells. Nat. Immunol. 2012;13:729–736. doi: 10.1038/ni.2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang K., Dupont A., Torow N., Gohde F., Leschner S., Lienenklaus S., Weiss S., Brinkmann M.M., Kühnel M., Hensel M., et al. Age-dependent enterocyte invasion and microcolony formation by Salmonella. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knoop K.A., Kumar N., Butler B.R., Sakthivel S.K., Taylor R.T., Nochi T., Akiba H., Yagita H., Kiyono H., Williams I.R. RANKL is necessary and sufficient to initiate development of antigen-sampling M cells in the intestinal epithelium. J. Immunol. 2009;183:5738–5747. doi: 10.4049/jimmunol.0901563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Donaldson D.S., Pollock J., Vohra P., Stevens M.P., Mabbott N.A. Microbial stimulation reverses the age-related decline in M cells in aged mice. iScience. 2020;23 doi: 10.1016/j.isci.2020.101147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tahoun A., Mahajan S., Paxton E., Malterer G., Donaldson D.S., Wang D., Tan A., Gillespie T.L., O'Shea M., Roe A.J., et al. Salmonella transforms follicle-associated epithelial cells into M cells to promote intestinal invasion. Cell Host Microbe. 2012;12:645–656. doi: 10.1016/j.chom.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 51.Savidge T.C., Smith M.W., James P.S., Aldred P. Salmonella-induced M-cell formation in germ-free mouse Peyer's patch tissue. Am. J. Pathol. 1991;139:177–184. [PMC free article] [PubMed] [Google Scholar]
- 52.Vatanen T., Kostic A.D., d'Hennezel E., Siljander H., Franzosa E.A., Yassour M., Kolde R., Vlamakis H., Arthur T.D., Hämäläinen A.M., et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. 2016;165 doi: 10.1016/j.cell.2016.05.056. [DOI] [PubMed] [Google Scholar]
- 53.Ege M.J., Mayer M., Normand A.C., Genuneit J., Cookson W.O., Braun-Fahrländer C., Heederik D., Piarroux R., von Mutius E., GABRIELA Transregio 22 Study Group Exposure to environmental microorganisms and childhood asthma. N. Engl. J. Med. 2011;364:701–709. doi: 10.1056/NEJMoa1007302. [DOI] [PubMed] [Google Scholar]
- 54.Depner M., Taft D.H., Kirjavainen P.V., Kalanetra K.M., Karvonen A.M., Peschel S., Schmausser-Hechfellner E., Roduit C., Frei R., Lauener R., et al. Maturation of the gut microbiome during the first year of life contributes to the protective farm effect on childhood asthma. Nat. Med. 2020;26:1766–1775. doi: 10.1038/s41591-020-1095-x. [DOI] [PubMed] [Google Scholar]
- 55.Constantinides M.G., Link V.M., Tamoutounour S., Wong A.C., Perez-Chaparro P.J., Han S.J., Chen Y.E., Li K., Farhat S., Weckel A., et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science. 2019;366 doi: 10.1126/science.aax6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ozkul C., Ruiz V.E., Battaglia T., Xu J., Roubaud-Baudron C., Cadwell K., Perez-Perez G.I., Blaser M.J. A single early-in-life antibiotic course increases susceptibility to DSS-induced colitis. Genome Med. 2020;12 doi: 10.1186/s13073-020-00764-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martínez I., Maldonado-Gomez M.X., Gomes-Neto J.C., Kittana H., Ding H., Schmaltz R., Joglekar P., Cardona R.J., Marsteller N.L., Kembel S.W., et al. Experimental evaluation of the importance of colonization history in early-life gut microbiota assembly. eLife. 2018;7 doi: 10.7554/eLife.36521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Al Nabhani Z., Dulauroy S., Marques R., Cousu C., Al Bounny S., Déjardin F., Sparwasser T., Bérard M., Cerf-Bensussan N., Eberl G. A weaning reaction to microbiota is required for resistance to immunopathologies in the adult. Immunity. 2019;50:1276–1288.e5. doi: 10.1016/j.immuni.2019.02.014. [DOI] [PubMed] [Google Scholar]
- 59.Thapa P., Guyer R.S., Yang A.Y., Parks C.A., Brusko T.M., Brusko M., Connors T.J., Farber D.L. Infant T cells are developmentally adapted for robust lung immune responses through enhanced T cell receptor signaling. Sci. Immunol. 2021;6 doi: 10.1126/sciimmunol.abj0789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sarzotti M., Robbins D.S., Hoffman P.M. Induction of protective CTL responses in newborn mice by a murine retrovirus. Science. 1996;271:1726–1728. doi: 10.1126/science.271.5256.1726. [DOI] [PubMed] [Google Scholar]
- 61.Garcia A.M., Fadel S.A., Cao S., Sarzotti M. T cell immunity in neonates. Immunol. Res. 2000;22:177–190. doi: 10.1385/IR:22:2-3:177. [DOI] [PubMed] [Google Scholar]
- 62.Forsthuber T., Yip H.C., Lehmann P.V. Induction of TH1 and TH2 immunity in neonatal mice. Science. 1996;271:1728–1730. doi: 10.1126/science.271.5256.1728. [DOI] [PubMed] [Google Scholar]
- 63.Koch M.A., Reiner G.L., Lugo K.A., Kreuk L.S., Stanbery A.G., Ansaldo E., Seher T.D., Ludington W.B., Barton G.M. Maternal IgG and IgA antibodies dampen mucosal T helper cell responses in early life. Cell. 2016;165:827–841. doi: 10.1016/j.cell.2016.04.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pott J., Stockinger S., Torow N., Smoczek A., Lindner C., McInerney G., Bäckhed F., Baumann U., Pabst O., Bleich A., Hornef M.W. Age-dependent TLR3 expression of the intestinal epithelium contributes to rotavirus susceptibility. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koren N., Zubeidat K., Saba Y., Horev Y., Barel O., Wilharm A., Heyman O., Wald S., Eli-Berchoer L., Shapiro H., et al. Maturation of the neonatal oral mucosa involves unique epithelium-microbiota interactions. Cell Host Microbe. 2021;29:197–209.e5. doi: 10.1016/j.chom.2020.12.006. [DOI] [PubMed] [Google Scholar]
- 66.Roswall J., Olsson L.M., Kovatcheva-Datchary P., Nilsson S., Tremaroli V., Simon M.C., Kiilerich P., Akrami R., Krämer M., Uhlén M., et al. Developmental trajectory of the healthy human gut microbiota during the first 5 years of life. Cell Host Microbe. 2021;29:765–776.e3. doi: 10.1016/j.chom.2021.02.021. e763. [DOI] [PubMed] [Google Scholar]
- 67.van Best N., Rolle-Kampczyk U., Schaap F.G., Basic M., Olde Damink S.W.M., Bleich A., Savelkoul P.H.M., von Bergen M., Penders J., Hornef M.W. Bile acids drive the newborn's gut microbiota maturation. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-17183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yatsunenko T., Rey F.E., Manary M.J., Trehan I., Dominguez-Bello M.G., Contreras M., Magris M., Hidalgo G., Baldassano R.N., Anokhin A.P., et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fulde M., Sommer F., Chassaing B., van Vorst K., Dupont A., Hensel M., Basic M., Klopfleisch R., Rosenstiel P., Bleich A., et al. Neonatal selection by toll-like receptor 5 influences long-term gut microbiota composition. Nature. 2018;560:489–493. doi: 10.1038/s41586-018-0395-5. [DOI] [PubMed] [Google Scholar]
- 70.Ohno H. Intestinal M cells. J. Biochem. 2016;159:151–160. doi: 10.1093/jb/mvv121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lelouard H., Fallet M., de Bovis B., Méresse S., Gorvel J.P. Peyer's patch dendritic cells sample antigens by extending dendrites through M cell-specific transcellular pores. Gastroenterology. 2012;142:592–601.e3. doi: 10.1053/j.gastro.2011.11.039. [DOI] [PubMed] [Google Scholar]
- 72.de Lau W., Kujala P., Schneeberger K., Middendorp S., Li V.S., Barker N., Martens A., Hofhuis F., DeKoter R.P., Peters P.J., et al. Peyer's patch M cells derived from Lgr5(+) stem cells require SpiB and are induced by RankL in cultured "miniguts". Mol. Cell. Biol. 2012;32:3639–3647. doi: 10.1128/MCB.00434-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Creamer B. The turnover of the epithelium of the small intestine. Br. Med. Bull. 1967;23:226–230. doi: 10.1093/oxfordjournals.bmb.a070561. [DOI] [PubMed] [Google Scholar]
- 74.Kotloff K.L., Nataro J.P., Blackwelder W.C., Nasrin D., Farag T.H., Panchalingam S., Wu Y., Sow S.O., Sur D., Breiman R.F., et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 75.Haber A.L., Biton M., Rogel N., Herbst R.H., Shekhar K., Smillie C., Burgin G., Delorey T.M., Howitt M.R., Katz Y., et al. A single-cell survey of the small intestinal epithelium. Nature. 2017;551:333–339. doi: 10.1038/nature24489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Muncan V., Heijmans J., Krasinski S.D., Büller N.V., Wildenberg M.E., Meisner S., Radonjic M., Stapleton K.A., Lamers W.H., Biemond I., et al. Blimp1 regulates the transition of neonatal to adult intestinal epithelium. Nat. Commun. 2011;2 doi: 10.1038/ncomms1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Knoop K.A., Gustafsson J.K., McDonald K.G., Kulkarni D.H., Coughlin P.E., McCrate S., Kim D., Hsieh C.S., Hogan S.P., Elson C.O., et al. Microbial antigen encounter during a preweaning interval is critical for tolerance to gut bacteria. Sci. Immunol. 2017;2 doi: 10.1126/sciimmunol.aao1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luna Velez M.V.N., H.K., Snabel R.R., Quint Y., Qian C., Martens A., Veenstra G.J.C., Freeman M.R., van Heeringen S.J., Vermeulen M. 2023. ONECUT2 restricts microfold cell numbers in the small intestine; a multi-omics study. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rivera C.A., Randrian V., Richer W., Gerber-Ferder Y., Delgado M.G., Chikina A.S., Frede A., Sorini C., Maurin M., Kammoun-Chaari H., et al. Epithelial colonization by gut dendritic cells promotes their functional diversification. Immunity. 2022;55:129–144.e8. doi: 10.1016/j.immuni.2021.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kollmann T.R., Marchant A., Way S.S. Vaccination strategies to enhance immunity in neonates. Science. 2020;368:612–615. doi: 10.1126/science.aaz9447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.G. B. D. Child Mortality Collaborators Global, regional, national, and selected subnational levels of stillbirths, neonatal, infant, and under-5 mortality, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1725–1774. doi: 10.1016/S0140-6736(16)31575-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Levine M.M., Nasrin D., Acácio S., Bassat Q., Powell H., Tennant S.M., Sow S.O., Sur D., Zaidi A.K.M., Faruque A.S.G., et al. Diarrhoeal disease and subsequent risk of death in infants and children residing in low-income and middle-income countries: analysis of the GEMS case-control study and 12-month GEMS-1A follow-on study. Lancet Glob. Health. 2020;8:e204–e214. doi: 10.1016/S2214-109X(19)30541-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mathers C., Stevens G., Hogan D., Mahanani W.R., Ho J. In: Disease Control Priorities: Improving Health and Reducing Poverty, RD. Jamison D.T., Gelband H., Horton S., Jha P., Laxminarayan R., Mock C.N., Nugent R., editors. The International Bank for Reconstruction and Development/The World Bank; 2017. Global and regional causes of death: patterns and trends; pp. 2000–2015. [DOI] [PubMed] [Google Scholar]
- 84.Oppong T.B., Yang H., Amponsem-Boateng C., Kyere E.K.D., Abdulai T., Duan G., Opolot G. Enteric pathogens associated with gastroenteritis among children under 5 years in sub-Saharan Africa: a systematic review and meta-analysis. Epidemiol. Infect. 2020;148:e64. doi: 10.1017/S0950268820000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Prudden H.J., Hasso-Agopsowicz M., Black R.E., Troeger C., Reiner R.C., Breiman R.F., Jit M., Kang G., Lamberti L., Lanata C.F., et al. Meeting Report: WHO Workshop on modelling global mortality and aetiology estimates of enteric pathogens in children under five. Cape Town, 28-29th November 2018. Vaccine. 2020;38:4792–4800. doi: 10.1016/j.vaccine.2020.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Costello A., Naimy Z. Maternal, newborn, child and adolescent health: challenges for the next decade. Int. Health. 2019;11:349–352. doi: 10.1093/inthealth/ihz051. [DOI] [PubMed] [Google Scholar]
- 87.Boerma T., Requejo J., Victora C.G., Amouzou A., George A., Agyepong I., Barroso C., Barros A.J.D., Bhutta Z.A., Black R.E., et al. Countdown to 2030: tracking progress towards universal coverage for reproductive, maternal, newborn, and child health. Lancet. 2018;391:1538–1548. doi: 10.1016/S0140-6736(18)30104-1. [DOI] [PubMed] [Google Scholar]
- 88.MacLennan C.A., Saul A. Vaccines against poverty. Proc. Natl. Acad. Sci. USA. 2014;111:12307–12312. doi: 10.1073/pnas.1400473111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Barnden M.J., Allison J., Heath W.R., Carbone F.R. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 90.Hogquist K.A., Jameson S.C., Heath W.R., Howard J.L., Bevan M.J., Carbone F.R. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 91.Caton M.L., Smith-Raska M.R., Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J. Exp. Med. 2007;204:1653–1664. doi: 10.1084/jem.20062648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hou B., Reizis B., DeFranco A.L. Toll-like receptors activate innate and adaptive immunity by using dendritic cell-intrinsic and -extrinsic mechanisms. Immunity. 2008;29:272–282. doi: 10.1016/j.immuni.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shen F.W., Saga Y., Litman G., Freeman G., Tung J.S., Cantor H., Boyse E.A. Cloning of Ly-5 cDNA. Proc. Natl. Acad. Sci. USA. 1985;82:7360–7363. doi: 10.1073/pnas.82.21.7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shimada S., Kawaguchi-Miyashita M., Kushiro A., Sato T., Nanno M., Sako T., Matsuoka Y., Sudo K., Tagawa Y., Iwakura Y., et al. Generation of polymeric immunoglobulin receptor-deficient mouse with marked reduction of secretory IgA. J. Immunol. 1999;163:5367–5373. [PubMed] [Google Scholar]
- 95.Kamphuis E., Junt T., Waibler Z., Forster R., Kalinke U. Type I interferons directly regulate lymphocyte recirculation and cause transient blood lymphopenia. Blood. 2006;108:3253–3261. doi: 10.1182/blood-2006-06-027599. [DOI] [PubMed] [Google Scholar]
- 96.Jung S., Aliberti J., Graemmel P., Sunshine M.J., Kreutzberg G.W., Sher A., Littman D.R. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 2000;20:4106–4114. doi: 10.1128/MCB.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Klindworth A., Pruesse E., Schweer T., Peplies J., Quast C., Horn M., Glöckner F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li D., Liu C.M., Luo R., Sadakane K., Lam T.W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–1676. doi: 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 101.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 102.Aramaki T., Blanc-Mathieu R., Endo H., Ohkubo K., Kanehisa M., Goto S., Ogata H. KofamKOALA: KEGG ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2020;36:2251–2252. doi: 10.1093/bioinformatics/btz859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bushnell B. 2014. BBMap: A fast, accurate, splice-aware aligner. 9th Annual Genomics of Energy & Environment Meeting. [Google Scholar]
- 105.Letunic I., Bork P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47:W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Salonen A., Nikkilä J., Jalanka-Tuovinen J., Immonen O., Rajilić-Stojanović M., Kekkonen R.A., Palva A., de Vos W.M. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods. 2010;81:127–134. doi: 10.1016/j.mimet.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 107.Lelouard, H., Mailfert, S., and Fallet, M. (2018). A ten-color spectral imagingstrategy to reveal localization of gut immune cell subsets. https://www.zeiss.fr/content/dam/Microscopy/fr/download/note_application_a_ten_color.pdf.
- 108.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Supek F., Bošnjak M., Škunca N., Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang J., Lareau C.A., Bautista J.L., Gupta A.R., Sandor K., Germino J., Yin Y., Arvedson M.P., Reeder G.C., Cramer N.T., et al. Single-cell multiomics defines tolerogenic extrathymic Aire-expressing populations with unique homology to thymic epithelium. Sci. Immunol. 2021;6 doi: 10.1126/sciimmunol.abl5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Godon J.J., Zumstein E., Dabert P., Habouzit F., Moletta R. Molecular microbial diversity of an anaerobic digestor as determined by small-subunit rDNA sequence analysis. Appl. Environ. Microbiol. 1997;63:2802–2813. doi: 10.1128/aem.63.7.2802-2813.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Satopaa V.A., Albrecht J., Irwin D., Raghavan B. 2011. Finding a “Kneedle” in a Haystack: detecting knee points in system behavior. 2011 31st International Conference on Distributed Computing Systems. [Google Scholar]
- 113.Asnicar F., Thomas A.M., Beghini F., Mengoni C., Manara S., Manghi P., Zhu Q., Bolzan M., Cumbo F., May U., et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-16366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Strains, corresponding BOTA scores, and TPM values obtained by shotgun sequencing shown in Figure 5.
DeqMS analysis output table of differential proteomic analysis.
Data Availability Statement
-
-
scRNAseq data are deposited with the National Center for Biotechnology Information Gene Expression Omnibus (GEO) under the accession numbers GSE188714.
-
-
Metagenomic data are deposited under the NCBI-BioProject accession number PRJNA794356: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA794356.
-
-
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
-
This paper does not report original code.