

Graphical abstract
Keywords: IVT mRNA, In vitro transcribed mRNA, mRNA structure
Abstract
The accelerated progress and approval of two mRNA-based vaccines to address the SARS-CoV-2 virus were unprecedented. This record-setting feat was made possible through the solid foundation of research on in vitro transcribed mRNA (IVT mRNA) which could be utilized as a therapeutic modality. Through decades of thorough research to overcome barriers to implementation, mRNA-based vaccines or therapeutics offer many advantages to rapidly address a broad range of applications including infectious diseases, cancers, and gene editing. Here, we describe the advances that have supported the adoption of IVT mRNA in the clinics, including optimization of the IVT mRNA structural components, synthesis, and lastly concluding with different classes of IVT RNA. Continuing interest in driving IVT mRNA technology will enable a safer and more efficacious therapeutic modality to address emerging and existing diseases.
1. Introduction
RNAs play diverse functional roles ranging from protein expression, gene regulation, and post-transcriptional modification. One species of RNA is messenger RNA (mRNA) which was first discovered in the 1960s [1]. Although other species of RNAs are also crucial in cellular function, mRNA is a unique entity that is particularly attractive as a therapeutic vector due to its ability to utilize the endogenous translation system to transiently express specific proteins. Despite the appeal of mRNAs as therapeutics, there have been some barriers to the implementation of mRNAs as therapeutic agents. These challenges include difficulty in delivery, immune system activation, and mRNA instability [3]. Due to these challenges, other methods including viral vectors or DNA delivery were initially favored over mRNA for therapeutic applications. However, there are several advantages to mRNA-based therapeutics. Recombinant mRNAs can be easily designed and synthesized in vitro to confer activity in vivo. Compared to viral vectors or DNA delivery, mRNA possesses lower risk of host genome integration and earlier onset of action since mRNAs only need to reach the cytoplasm for translation, rather than transportation into the nucleus [2].
Therefore, harnessing the potential of mRNAs to express proteins of interest has been especially enticing to serve broadly as a therapeutic agent. Advances in lipid nanoparticles mediated delivery has validated mRNA delivery culminating in the FDA approval for use in the SARS-CoV-2 virus vaccines for worldwide administration [5]. Significant effort has been afforded to combat the strong immune stimulatory effects of IVT mRNAs through the incorporation of base-modified nucleosides [6], [7]. Moreover, incorporating base modified nucleosides has also been exhibited to amplify the translational proficiency of IVT mRNAs in vivo [8], [9], [10]. To increase the pharmacokinetic profile of mRNA and stabilize the IVT transcript upon administration, various attempts have been made to design a more efficient mRNA that has increased half-life and translation. These advancements have enabled the investigation of mRNAs across various applications in the clinic, including vaccines, protein replacement, or gene editing [3], [4]. In this article, we focus on the various structural elements of the mRNA that affect its properties to serve as a therapeutic modality and further discuss how the synthesis of IVT mRNA supports this effort. Lastly, we summarize alternative structures of RNAs that may further expand their various therapeutic applications.
2. Structure of mRNA
In vitro transcribed mRNA, or synthetic mRNA, is a single-stranded RNA transcript that has been engineered to mimic naturally occurring mRNAs [11]. The mRNA comprises of five key structural elements: the 5′ cap, 5′ untranslated region (UTR), coding sequence region (CDS), 3′ UTR, and the poly(A) tail. Because of its rapid degradation, fine-tuning and maximizing the activity of synthetically administered mRNAs either by elongating stability or maximizing translation has been crucial to capturing its potential as a therapeutic agent [11]. Researchers have investigated several approaches to optimize whole mRNAs with recent advances in sequencing and computational capacities. Wayment-Steele et al. computed a model to predict average unpaired probability, measurement of secondary structure, and hydrolysis rates to predict and design more stable transcripts of mRNAs by creating highly folded mRNA structures [12]. Taking the outcomes of the highly folded mRNAs, the group further utilized computational calculations incorporating in-cell stability and ribosomal load to predict mRNA sequences that not only sustain high stability but also incorporate high translation capacities [13].
Another strategy to attune the mRNA pharmacokinetic profile has been to assess the impact of specific nucleosides, including the incorporation of modified nucleosides or augmentation of guanine and cytosine (GC) nucleosides. Chemically modified nucleosides were first reported to reduce innate immune stimulatory activation by exogenous mRNA [6]. Interestingly, the substitution of uridine to modified nucleosides, such as pseudouridine (Ψ) or N1-methyl-pseudouridine (m1Ψ), also showed enhanced translation of the encoded protein [14], [15], [16]. Other groups report that rather than a complete replacement, partial replacement with chemically modified nucleosides may induce high protein expression and low immune stimulatory effects [17]. This phenomenon is attributed to reduced RNA sensor engagement by the incorporated modified nucleosides [18]. This potentially begs the question of whether modified nucleosides can also interfere with general protein/RNA interactions, which could decrease the specificity of mRNA interface with RNA binding proteins for translation. Accordingly, Thess et al. evaluated whole sequence engineering with non-chemically modified nucleosides and observed heightened translation and decreased immune stimulation [19]. By relying on optimization of the open reading frame and regulatory sequences within the 5′ and 3′ UTR to maximize GC content, they observed that whole sequence-engineered unmodified RNA exhibited higher luciferase activity compared to modified RNA without sequence engineering in vitro. Complete replacement of UTP with Ψ of the whole sequence mRNA transcript restored luciferase activity and conferred comparable activity between the two transcripts, demonstrating that potent whole sequence engineering is an effective strategy to attain enhanced expression [19]. Additionally, maximizing GC content within the mRNA transcript may yield multiple effects. Uridine can be recognized by toll like receptors 7/8 and may trigger innate immune activation [20]. By increasing GC content, one may minimize the overall number of uridine bases in the transcript to decrease interaction with TLR 7/8. Another important consideration is adenylate/uridylate rich elements (AREs), which are often found in the 3′ UTR of mRNAs that code for proto-oncogenes or cytokines [21]. mRNAs with AREs can be rapidly degraded in response to stress by ARE-binding proteins [22]. Therefore, high GC content within the 3′ UTR may help synthetic mRNAs to evade targeted degradation by ARE-binding proteins. Courel et al. suggest that AU-rich and GC-rich transcripts undergo distinct degradation and translation pathways where GC-rich transcripts are more optimally translated compared to AU-rich transcripts at the 5′ end [23]. Furthermore, increasing the GC content within the mRNA transcript may improve translation by protecting the mRNAs from endoribonuclease digestion [24], [25]. Taken together, nucleoside modification and GC content within mRNAs may improve protein expression ability and reduce immunogenicity of the mRNA.
2.1. 5′ Cap
Eukaryotic mRNAs contain an evolutionarily conserved 5′ m7G cap, which is needed for mediating several biological functions [26]. mRNA cap structures modulate nuclear export, splicing, RNA stability, and efficient translation [27]. In the cytoplasm, the cap functions as an anchor for the recruitment of initiation factors eukaryotic translation initiation factor 4E (EIF4E), which is crucial for translation [28]. The cap can also serve a protective function against 5′ to 3′ exonuclease degradation of mRNA [29]. The capping event is mediated by three separate enzymes including RNA triphosphatase, RNA guanylyltransferase, and RNA (guanine-7)-methyltransferase [30]. Pre-mRNAs are processed by enzymes to generate a 7-methyl guanosine affixed by an inverted 5′-5′ triphosphate bridge to the first nucleoside [31]. The Cap-0 structure is one of the earliest mRNA processing events, which occurs co-transcriptionally with RNA Polymerase II [32]. The attachment of methyl groups to the first and/or second transcribed nucleosides produces the Cap-1 or the Cap-2 structure [33]. Importantly, cap 2′O methylation distinguish self RNA against foreign RNA [34].
2.2. 5′ UTR
5′ Untranslated Region (UTR) is the section of the mRNA upstream of the coding region. While this sequence is not translated, it is a critical region as it serves as a binding site for ribosomes to initiate translation. Therefore, various endeavors have been attempted to elongate stability or increase translation efficiency by optimizing the 5′ UTR region of the mRNA (Table 1 ) [35]. Most eukaryotic 5′ UTR region includes the Kozak consensus sequence (ACCAUGG) which contains the start codon (AUG) to initiate translation [36]. Moreover, the 5′ UTR region tends to be AU sequences rich, which promotes the formation of secondary structures to either impede or engage binding of ribosomes and other regulatory factors to control gene expression [36]. To investigate the relationship between ribosome binding and 5′ UTR sequences, Sample et al. generated a synthetic library of 280,000 randomized 5′ UTR. Combining deep learning algorithms, these sequences were assessed for ribosomal loading capacity. Sample et al. exposed that the predicted models matched the actual measured ribosomal loading of over 30,000 transcripts. By combining the technology of deep learning algorithms, an exogenous optimized 5′ UTR sequence could be identified for various levels of ribosomal loading to finetune control of gene expression following the synthetic 5′ UTR sequence [37]. Linares-Fernandez et al. applied the findings from this earlier study to design an mRNA template with higher protein expression by evaluating several synthetic 5′ UTR sequences with high ribosomal loading. Compared to the native α-globin 5′ UTR sequence, synthetic UTR4 induced greater than 30% more eGFP production in both HeLa and DC2.4 cells [38]. In another work, Zeng et al. performed both comprehensive analysis of endogenous gene expression and de novo design of exogenous sequences to compile a unique combination of 5′ and 3′ UTR sequences. Several rounds of sequence optimization resulted in up to a ten-fold increase in protein translation compared to other tested endogenous UTRs [39]. A separate study by Gebre et al. devised two SARS-CoV-2 mRNA vaccines and compared the efficacy of the vaccines in eliciting a potent immune response [40]. One major difference of these two mRNA vaccine candidates was the inclusion of a 5′ UTR region based on the human hydroxysteroid 17-beta dehydrogenase 4 gene (GSD17B4). The mRNA vaccine with the 5′ UTR region (CV2CoV) was able to elicit a more robust protection compared to the one without the 5′ UTR, which suggests that optimization of non-coding regions can enrich mRNA potency by improving the translation capacity of the target protein.
Table 1.
Selected studies on approaches to 5′ UTR identification and optimization.
| UTR Sequence Origin | Key Findings | Source |
|---|---|---|
| Endogenous sequences | Human α-globin or β-globin based UTRs due to highly stable expression | [41] |
| UTRs based on Cytochrome b-245 alpha chain (CYBA) combinations | [42] | |
| 5′ UTR based on human hydroxysteroid 17-beta dehydrogenase 4 gene | [40] | |
| Endogenous 5′ and 3′ UTR selection and combination for de novo UTR design | [39] | |
| Exogenous sequences | Library of over one million 5′ UTR variants with random 10-nucleotide sequence variation reveals dynamic sequence feature of 5′ UTRs that control mRNA translatability | [43] |
| Library of 280,000 randomized 5′ UTR to build predictive models for translation by directing specific levels of ribosome loading | [37] | |
| 5′ UTR based on synthetic sequences with high ribosomal loading calculations | [38] |
2.3. Coding Sequence
The coding sequence region (CDS), or open reading frame (ORF), is especially important for the development of mRNA therapeutics to encode for an antigen or a protein of interest. The region typically constitutes much of the mRNA transcript; thus, it is unsurprising that modulating this region, specifically in the context of codon optimization, affects overall mRNA stability and translation capacity [44], [45]. The overall translation rate is governed by tRNA species, so optimizing codon usage to reduce the usage of rare tRNA species by replacing codons with more common tRNA species can affect not just protein production, but also the fidelity of translation [46], [47]. The incorporation of modified nucleosides has been widely implemented to reduce innate immune stimulation, but doing so might affect overall translation in both beneficial and disadvantageous ways. Choi et al. reported that (m6A) modification may function as an obstacle to tRNA association and impede translation [48]. Similarly, Eyler et al. illustrated that incorporating pseudouridine alters tRNA binding and reduces translation and increases amino acid substitutions [49]. Contrastingly, Mao et al. incorporated N6-methyladenosine (m6A) within the coding region, which enhanced translation by alleviating mRNA secondary structure to promote translation [50]. Other groups have also investigated the role of secondary structures in protein translation through computational sequence design [13], [25]. Zhang et al. applied an algorithm to balance between stability and codon optimality to design mRNAs with superior protein expression [51]. Therefore, careful consideration of overall codon optimization and base-modified nucleosides should be evaluated to assess the dynamic translation mechanisms for different sequences.
2.4. 3′ UTR
The regulatory elements of mRNAs are situated within the 5′ and 3′ UTR that interact with protein complexes to generate ribonucleoparticles for mRNA transport, stability, and translation [52], [53]. 3′ UTR of mRNAs is vital for targeting transcripts to specific cellular compartments, which is essential for mRNA localization and protein synthesis especially in highly polarized or differentiated cells [54]. 3′ UTR is an especially important regulatory element because microRNA (miRNA) binds to these regions to silence mRNA expression [55], [56]. Sandberg et al. described that rapidly proliferating cells display a shorter 3′ UTR region which in return reduced miRNA binding sites to enable a more proliferative expression of proteins [57]. This property may also be harnessed to design mRNAs that have a specific activity in different cell types by artificially including miRNA binding sites within the 3′ UTR for cell-specific translation [58]. For example, Li et al. included a neuron-specific miRNA targeting site in the 3′ UTR to limit expression of the mRNA in neurons [59]. Furthermore, translation may be enhanced by reducing unstructured sequences within the 3′ UTR to facilitate the binding of poly(A) tail to translation machinery [60]. UTR performance can vary across different cell types, therefore deciding the cell population and optimizing the 3′ UTR for the target cell types should be considered.
2.5. Poly(A) Tail
The last structural element of an mRNA is the poly(A) tail. Poly(A) tail is characterized by the long stretch of repetitive adenosine nucleosides at the 3′ end. In general, a longer poly(A) tail results in a more stable mRNA since the long sequence protects the coding mRNA region from deadenylating and degrading enzymes [61]. However, the dynamic deadenylation and regulatory mechanism of the poly(A) tail reveals that this is not always correct. There are stable mRNAs with a short poly(A) tail [62], [63]. The complex interaction between the poly(A) binding proteins (PABPs) and other translation-initiating proteins work synergistically to regulate the stability and the translation efficiency of the mRNAs [64]. A full-length mRNA sequencing capacity that accurately sequences transcripts of mRNA including the poly(A) tail revealed that there are other nucleosides incorporated into the tail other than just adenosines [65]. Li et al. reported that including cytidine nucleoside into the tails shields synthetic mRNAs from deadenylation by the CCR4-NOT transcription complex, as the CNOT proteins have lower efficiency in removing cytidine [66]. Cytosine incorporation enhanced protein translation and elongated stability of the synthetic mRNA both in vitro and in vivo [66].
There are two strategies to synthesize the poly(A) structure in mRNAs. One way is to extend the IVT mRNA through enzymatic reaction with recombinant poly(A) polymerase [67]. Although this enables the incorporation of modified nucleosides into the poly(A) tail, it lacks the control and precision of generating a fixed length of the tail, which is often desired for clinical applications [11]. The second possibility is to encode a long stretch of A sequences in the DNA template vector. This ensures a highly controlled length of the resulting poly(A) tail. To generate the poly(A) tail, a long adenosine sequence can be destabilizing for DNA plasmids, so one way to bypass this is to incorporate a short UGC linker between the adenosine stretch. The current Pfizer/BioNTech SARS-CoV-2 mRNA vaccine incorporates this strategy to generate a long stable poly(A) tail [68].
3. Synthesis of IVT mRNA
In vitro transcribed mRNA produced by RNA Polymerases (RNAP) differ from chemically synthesized RNA species. Chemical synthesis of RNA is still limited in the length of the sequence to less than 200 base pairs and relies on adding a subsequent nucleoside to the growing chain of RNA polymer through chemical protecting groups. Overall yield decreases with each nucleoside addition [69]. Although innovations in DNA and RNA chemical synthesis have enabled the synthesis of longer strands of RNA molecules up to 200 base pairs, mRNAs are commonly longer than 200 base pairs and require an alternative method to synthesize the transcript. The discovery of RNAP from bacteriophages has allowed for this process which captures the native ability of these enzymes to synthesize long RNA transcripts from a DNA source, whether that is PCR-amplified linearized templates or plasmids [70]. Currently, there have been several RNA Polymerases identified, including T7, T3, and SP6 Polymerases which have been exerted for the IVT process. IVT mRNA by RNA polymerases can achieve long transcripts, more than many kilobases in length with high yield [71], [72].
3.1. RNA Polymerase
A frequently used RNA polymerase is the T7 RNA Polymerase derived from the T7 bacteriophage. T7 RNAP is ubiquitously used because of its high specificity and activity of RNA synthesis [73]. The T7 promoter is a conserved DNA sequence recognized by the T7 RNAP to initiate transcription. Even partially single-stranded DNA templates as long as they are base-paired within −17 to + 1 nucleoside of the promoter sequence will get transcribed into full RNA transcripts [73]. Through the careful dissection of the T7 RNAP, the most widely utilized promoter sequence is TAATACGACTCACTATAGGGAGA starting from −17 position [74]. However, the sequence downstream of the promoter may affect T7 promoter activity, therefore optimization of the promoter sequence might augment transcription based on the downstream sequences [75]. The mRNA sequence, starting from the 5′ UTR region should be placed after this conserved T7 promoter. Although IVT with T7 RNAP yields high production of the desired sequence, there are some limitations. The first limitation is the lower transcription efficiency of sequences that lack G-rich regions in the initial 5′ sequence [76]. A second limitation is the 3′ end heterogeneity due to the synthesis of one or two added nucleosides resulting from incomplete transcription termination [77]. Another limitation is the sly addition of a nucleoside at the 5′ end upon translation initiation given certain 5′ sequences downstream of the promoter [78]. A major downside of IVT mRNA with T7 RNAP is the nonspecific synthesis of double-stranded RNA (dsRNA) byproducts through erroneous 3′ extension [79]. dsRNA is often a trademark of viral infection. Therefore, dsRNA binding proteins actively censor the presence of dsRNA and trigger an innate immune and inflammatory response in presence of dsRNA [80]. Although dsRNA can be purified through chromatography steps after IVT, another way to minimize the presence of dsRNA in the final IVT mRNA product would be to use a thermostable T7 RNAP. Wu et al. generated a thermostable T7 RNAP to reduce the production of dsRNA by diminishing 3′ extension. IVT mRNA transcripts produced by thermostable T7 RNAP displayed reduced innate immune system activation when tested in vitro compared to IVT mRNA produced by nonmodified T7 RNAP [81]. Dousis et al. designed and computationally engineered T7 RNAP with two mutations that transcribed highly pure mRNA transcripts, which lacked byproducts such as dsRNA and increased the fidelity of the IVT mRNA transcripts [82]. Limitations have been addressed by investigating the functional properties of T7 RNAP in detail, especially by analyzing mutants of T7 with altered promoter recognitions [83], [84]. By improving translation initiation sequences, the lack of G-rich 5′ end or the addition of an erroneous nucleoside can be bypassed. To address the addition at the 3′ end, a highly specific purification scheme that can resolve the single-nucleoside difference in size can be used to ensure a homogeneous transcript.
T3 RNAP is derived from the T3 bacteriophage. Like T7 RNAP, T3 RNAP is a DNA-dependent RNA Polymerase. The promoter sequence for T3 is close to T7 but differs in the −10 and −11 positions. The consensus sequence T3 promoter is AATTAACCCTCACTAAAGGGAGA [74]. Due to the similarity in the promoter sequence, T3 RNAP can synthesize mRNA with a T7 Promoter, and vice versa. Nonetheless, transcription efficiency is greatly diminished by up to 90%, which showcases the high specificity and dependence of the promoter sequence for efficient transcription for the different RNAPs [85], [86]. SP6 RNAP is derived from SP6 bacteriophage, which is similar to T7 bacteriophage but is from a distinctly unique lineage [87]. Although the promoter sequence of SP6 RNAP shares similarity with T7 RNAP, SP6 RNAP transcription efficiency is highly specific to the SP6 promoter sequence [88]. The benefit of SP6 RNAP is the ease of production and isolation of these enzymes over comparable T7 or T3 RNAPs [87]. Nonetheless, different RNAPs can similarly produce mRNA transcript if the proper promoters are used.
Over hundred naturally occurring post-transcriptional RNA nucleosides have been identified [89]. Endogenous mRNAs will have modified nucleosides in specific positions within the mRNA [90]. The stratagem to incorporate base-modified nucleosides to decrease innate immunogenicity of exogenous mRNA upon administration begs the question of whether RNAPs can assemble transcripts using chemically modified nucleosides during in vitro transcription. Currently, most synthetic mRNAs utilize complete substitution with modified nucleosides to generate a uniform mRNA product. Milisavljevic et al. discuss that modified pyrimidine nucleosides and ATP with small modifications were good substrates for T7 RNAPs, but modified GTP analogs required the presence of GMP to initiate RNA synthesis [91]. Potapov et al. compared several modified bases including methylated, hydroxymethylated, and isomeric bases (pseudouridine) to equivalent unmodified RNA bases to find that RNAP fidelity decreases with modified bases [92]. The same group later published that N 1-methyl-pseudouridine is integrated with higher fidelity than pseudouridine by T7, T3, and SP6 RNAPs [93]. More investigations into the consistency and efficiency of RNAPs to transcribe mRNA with different chemically modified nucleosides are necessitated to accelerate the robust production of IVT mRNAs.
3.2. mRNA capping
In vitro transcription of mRNA by RNAPs can produce all the structural elements of a native mRNA, except for the 5′ cap structure. The inclusion of the 5′ cap structure has been documented to be necessary for efficient translation initiation [94]. To produce a fully biomimetic mRNA, the 5′ cap must be produced either concurrently during IVT for a one-step process, or enzymatically after the IVT step for a two-step process (Fig. 1 ). The co-transcriptional method works by replacing part of the GTP substrate for a dinucleotide cap analog, commercially available as CleanCap® reagent, which results in a highly efficient cap structure comparable to caps produced by enzymes [95]. However, a portion of the m7GpppG analog can get incorporated in a reverse direction, which is not recognized by the translation machinery for translation initiation. Therefore, anti-reverse cap analogs (ARCAs) were developed, which feature an m27,3’-OGpppG analog to ensure all the cap analogs are incorporated in the correct orientation [96]. ARCA-capped mRNAs displayed better translational capacity than traditionally capped mRNAs [61]. Another synthetic 5′ cap analog based on phosphorothioate cap enhanced RNA stability and translational efficiency in mice in vivo [97]. The one-step capping method is beneficial because a second enzymatic reaction and purification steps are not necessary, but the cost of using cap analogs can be more expensive than performing two-step capping with enzymes [98]. Another downfall of one-step capping is the use of a modified T7 promoter for the incorporation of the cap analog.
Fig. 1.

IVT mRNA capping strategies and cap structures. One-step or co-transcriptional capping uses synthetic GTP Cap analogs during the IVT step. Two-step or post-transcriptional capping uses enzymes and substrates to produce the Cap-0 or Cap-1 structure. B denotes nucleobases. Figure created with BioRender.com.
A two-step process of capping harnesses the function of virus-derived capping enzymes, most commonly vaccinia virus, to create the m7GpppN or Cap-0 structure. A second enzyme, Cap 2′-O-Methyltransferase, derived from the vaccinia virus is required to produce the Cap-1 structure. Cap-1 structure may reduce the innate immune response to the IVT mRNA [99]. Cap 2′-O-Methyltransferase uses S-adenosylmethionine (SAM) to add a methyl group at the 2′-O position at the end of the 5′ end of Cap-0 structures [100]. Enzymatic capping processes are usually very efficient and can achieve up to 100% capping but could have a batch-to-batch variability [101]. A downstream purification step to remove all capping enzymes and reagents from pure mRNA transcript is needed.
3.3. Large-scale manufacturing of IVT mRNA
The benefit of mRNAs as a therapeutic is that they can be purely synthesized cell-free. This negates the necessity to validate the absence of cell-derived impurities or adventitious contaminations [102]. This is especially enticing for manufacturing scale since validation of products from biological systems, such as proteins, is costly and requires stringent regulation and development to guarantee a robust and reproducible manufacturing process [103]. However, because mRNAs can be synthesized in vitro in a relatively well-defined procedure, the manufacturing capacity can be more flexible to adapt to other platforms. Additionally, manufacturing of mRNAs is in a modular process, which can be very quickly and easily switched to synthesize various mRNAs encoding proteins of interest, with very minor modifications in the process. This modularity is especially enticing from a manufacturing perspective since process development, characterization, and scale-up to manufacturing can be standardized in a cell-free system across diverse mRNA transcripts [104].
Currently, the process for mRNA production can be separated into two main steps: upstream and downstream [105]. The upstream process is the production of the IVT mRNA, including the capped structure. The downstream process is further purification, concentration, and sterile filtration to yield a highly homogenous IVT mRNA drug product [98]. A crucial step for the manufacturing of mRNA would be the purification steps to remove any contaminants, such as unreactive nucleosides, enzymes, and most importantly dsRNA. HPLC-purified mRNA reduced immune activation and improved translation by removing contaminants such as dsRNA and RNA fragments [106]. To this end, multiple modes of chromatography have been evaluated, including affinity, size exclusion, ion exchange, and reverse phase separation [105]. An orthogonal purification step that removes specific impurities should be coupled to ensure an effective depletion of all sources of contaminants from IVT mRNA [104].
To be released as a drug substance, IVT mRNA must be fully characterized for its potency, integrity, and purity. One concern would be the stability of RNA. The entire manufacturing operation must be conducted in a very stringent RNase-free environment since the degradation of RNA through ubiquitous RNases is rapid and is a key determinant of mRNA integrity. Key quality control attributes include 5′ capping efficiency, mRNA sequence confirmation, total length, and absence of process and product mediated impurities, such as capping enzymes, DNases, truncated fragments, or dsRNA [107]. A robust characterization assay that meets the release specifications for IVT mRNA will enhance its safety profile to ensure that batch-to-batch variability is minimal.
4. Different structures of translatable RNA
The most common structure of IVT mRNA is the linear form that resembles endogenous mRNA. modRNA is another terminology that describes linear mRNA synthesized by incorporating modified nucleosides, such as pseudouridine or N1-methyl-pseudouridine, during the IVT process. Structurally, there is no difference between linear mRNA and modRNA [108]. While various structural engineering of each part of the mRNA has yielded stable and potent mRNAs, linear mRNAs still suffer from rapid degradation from ubiquitous RNases upon systemic administration. Despite the barriers, interest in linear mRNA has exploded, as demonstrated by not just the number of publications on using mRNA, but also the number of clinical trials that utilize RNA for various therapeutic applications [109].
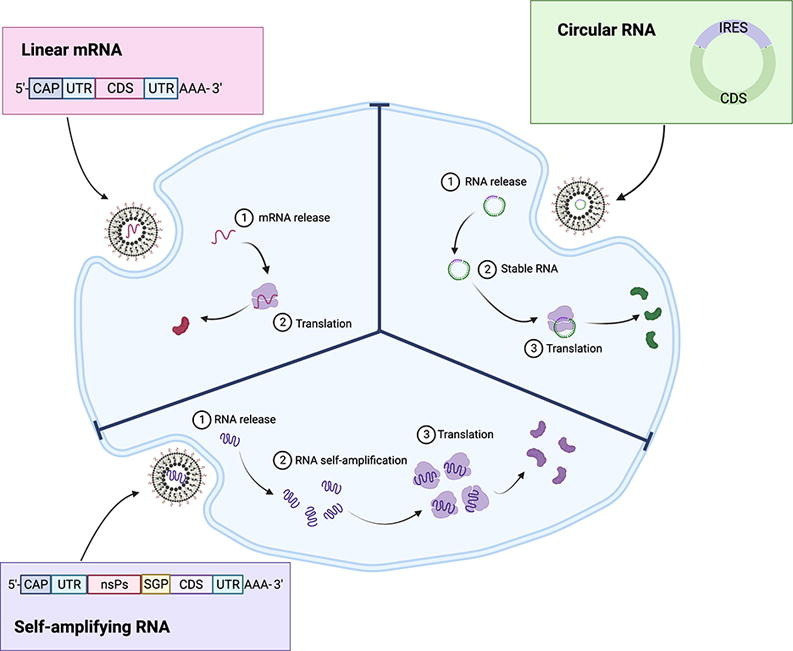
Due to their transient nature, linear mRNAs can be especially enticing as a prophylactic vaccine platform which entail acute expression of the antigen of interest [110], [111]. The mRNA can be directly administered for endogenous translation of the antigen to elicit a humoral immune response [68]. Linear mRNAs have been evaluated as vaccines across multiple infectious diseases or for cancer, culminating in the two FDA approvals of mRNA vaccines for the SARS-CoV-2 virus [112], [113]. Nonetheless, the short half-life of mRNAs can be considered as a barrier to adoption of mRNAs for other applications, such as protein replacement which generally require long duration of protein expression [11]. Structural engineering and optimization of linear mRNA has increased its stability and expression for therapeutic applications. For example, mRNA has been used to encode deficient human factor protein as a hemophilia treatment strategy [114], [115]. mRNA is also under investigation for gene editing applications by encoding for Cas9 protein co-delivered with single guide RNA (sgRNA) in vivo to treat transthyretin amyloidosis in vivo [4]. Although linear mRNA has been demonstrated for protein replacement or gene editing therapeutic applications, it is intrinsically unstable, which is a limitation for chronic therapeutics which require long sustained protein expression. Repeated administration can potentially address the transient duration, but this might cause potential toxicities with repetitive administration of mRNA or its delivery systems [3]. Furthermore, frequent dosing will necessitate a higher production demand for the mRNA therapeutics. To address these issues, researchers actively explore other forms of translatable RNA, such as self-amplifying RNA or circular RNA which exhibit comparatively extended expression (Fig. 2 ).
Fig. 2.

Translational potential of various RNA structures. The RNAs can be broadly divided into two categories, cap dependent translation or cap independent translation. Figure created with biorender.com.
4.1. Self-amplifying RNA
Self-amplifying RNA (saRNA) or replicon RNA addresses the transient and limited translation capacities of linear mRNAs. saRNA is based on the genome of RNA alphavirus but lacks the essential genes required for viral survival. Replicon RNAs include a nonstructural proteins (nsPs) region, which encodes RNA dependent RNA Polymerase (RdRP), which is necessary for self-amplification in the cytoplasm, followed by a subgenomic promoter (SGP). Structural protein genes can be replaced to encode any protein or antigen of interest downstream of the SGP. Thus, the RNA serves two functions. The first is as an mRNA transcript for RdRP. The second function is as a genomic template for amplification by the translated RdRP. The negative-strand RNA operates as a template for continuous translation and replication, thus creating a system for self-amplification [116]. Therefore, one-time administration of saRNA can produce exponential amplification of the desired protein by self-mediated synthesis of the RNA transcripts. Combining the elements from other RNA viruses, Li et al. identified six mutations in the nsPs from the Venezuelan equine encephalitis (VEE) replicon to enhance the expression of the protein that follows the subgenomic promoter [117]. Another strategy to harbor the self-amplification capacity of saRNA but reduce overall mRNA size is to utilize a trans-amplifying RNA strategy. A trans-amplifying RNA will have two separate transcripts, one encoding for a nonreplicating mRNA that encodes for the nsPs, and another that encodes for the protein of interest [118]. One limitation of this strategy is that both RNA transcripts must be delivered into the same cell for self-amplifying capability.
In this way, saRNAs embody the beneficial features of live viruses to express elevated levels of the target protein without the associated risks of viral immunogenicity or integration [119]. This feature is especially desired for vaccine applications because a low dose of the saRNA can elicit a lasting protective immune response [120], [121]. Additionally, since this process creates dsRNA products, replicon RNAs can dually serve as a potent stimulator of the innate immune system, thereby creating an adjuvant effect in the process [122]. Conversely, this stimulation of the innate immune system can be a blessing in disguise. Pepini et al. indicate that to achieve a stronger sustained vaccine potency to the encoded antigen, minimizing the early activation of type I IFN response is needed to increase the initial antigen expression of self-amplifying mRNAs [123]. One potential strategy to minimize type I and III IFN response to the dsRNA formed by saRNAs is to express innate inhibiting proteins (IIPs) [124], [125]. Blackney et al. characterized a library of cis-encoded IIPs in saRNA constructs to demonstrate that cis IIP expression can intensify protein expression by more than 500-fold in vitro and can augment the generation of antigen-specific IgG titer and neutralization in rabbits in vivo [126].
saRNA possesses many strengths to not only serve as a vaccine platform but also work for other therapeutic strategies. The self-amplification property has clear merits for protein replacement therapies because a one-time administration can result in long-sustained protein expression, negating the need for high doses or repeated administration. Li et al. demonstrated that intratumoral administration of LNP-replicons encoding for IL-12 cytokine stimulated the immune system for immune cell-mediated tumor cell killing not just in localized tumor sites, but also in distal metastatic tumors [127]. Du et al. compared modRNA to saRNA for the expression of DNA meiotic recombinase 1 (Dmc1) in spermatocytes to treat male infertility caused by mutated Dmc1. Although delivery of both modRNA and saRNA were able to express the correct Dmc1, a single injection of saRNA was able to extend the expression of the Dmc1 protein to rescue and restore spermatogenesis in the Dmc1 gene knockout mouse model [128]. In some situations, prolonged expression of protein may be undesired after a certain timepoint, or concentration has been met. Wagner et al. constructed an saRNA circuit with RNA binding protein that is responsive to specific small molecules to control expression of proteins [129]. This system can then be induced on or off with small molecules to control translation levels. Nonetheless, the immune stimulatory response to saRNA, especially across different administration routes and delivery vehicles, should be further elucidated to galvanize the adoption of saRNA for various applications.
4.2. Circular RNA
Circular RNA (circRNA) is a natively occurring single-stranded RNA molecule without an exposed 5′ or 3′ end. Due to the lack of any exposed ends, circular RNAs can evade degradation by exonucleases or hydrolysis. This feature equips circRNAs to exhibit high biostability and therefore extended expression [130]. A growing interest in circRNA has uncovered a diverse functional role from cellular activity regulators, protein and gene sponges, or even protein-coding translation templates [131]. The exact mechanism of endogenous circRNA production is still not fully elucidated but is theorized that circRNA is generated by RNA back-splicing events to form exonic circRNA or forward splicing to form intronic circRNA [132], [133]. Therefore, it is possible to have a circRNA that shares the same sequence with the related linear mRNA excluding the back-splicing junction sites. A precursor to circRNA is a linear RNA precursor, which can then be enzymatically or chemically ligated to close and circularize the vector. This feature can be exploited to design mRNA to promote back-splicing to synthesize a synthetic circRNA that encodes for a protein of interest.
circRNA can be generated through two distinct approaches. The first strategy uses an RNA synthesizer with phosphoramidites, which are specialized nucleoside derivatives. These derivatives can form 3′-to-5′ phosphodiester linkage to join and close the loop [134]. Another chemical strategy is an artificial chemical ligation of the linear RNA precursor with cyanogen bromide or 1-Ethyl-3-(3-dimethyl aminopropyl)carbodiimide (EDC) to undergo a condensation reaction, which forms a 3′-5′ phosphodiester bond to join the two ends together [134]. Although chemical synthesis of circRNA is possible, chemical ligation of RNA is inefficient compared to DNA analogs because of competition between intra and inter molecular forces. Therefore, enzymatic strategies to form circRNAs from IVT mRNA are more commonly employed due to their higher ligation efficiency.
The enzymatic strategy uses DNA or RNA ligases from T4 bacteriophage. Using DNA splints to compile the ends of the RNA into proximity, T4 Ligase can then facilitate site-specific ligation [135]. There are different variants of T4 ligase, which have varied preferences at the ligation site, so the linear precursor should be designed in regard to the specific enzyme that will be used to ultimately ligate and form the circRNA moiety [136], [137]. Nonetheless, all these enzymes only perform when the nucleosides have monophosphates, which means that dephosphorylation and re-phosphorylation through phosphatase and kinase or pyrophosphohydrolase treatment is a necessary precursor to enzymatic ligation [134]. Other enzymatic strategies use ribozymes, which are RNA enzymes that can catalyze the ligation of linear RNA to form circRNAs. The ribozymes contain a transposed group I intron precursor RNAs, where the introns are fused with exons. Harnessing the Anabaena pre-tRNAleu gene, a permuted intron–exon (PIE) system was designed. This system transfers 5′ initiating group I intron to the 3′ tail of the exon. The intron is self-cleaved and the exon is ligated to generate a circularized vector [138]. Wesselhoeft et al. engineered the PIE system to construct a wide range of circRNAs of various lengths [130].
The apparent merit of circRNA is its extended stability over linear mRNA. Consequently, synthetic circRNAs can be applied to mimic the broad range of endogenous circRNA functions to serve both as a non-coding and coding vector. For therapeutic applications, utilizing coding circRNA can be especially promising for both vaccines or protein replacement strategies because of the more persistent expression of the protein [142]. Not only is expression elongated due to the stability of the circRNA, but Abe et al. also discussed how the circular structure of the RNA enables a continuous translation where the ribosome can easily re-engage with the RNA after the termination of translation to increase overall translation efficiency of the RNA [143]. A challenge of circRNAs is that it lacks the 5′ cap and the poly(A) tail which recruits translation initiation machinery. Therefore, coding circRNAs should include an internal ribosome entry site (IRES) region to ease translation. Chen et al. performed a systematic analysis and screening of various elements to design a circRNA that displays improved protein translation compared to linear mRNA in vitro and in vivo [144]. Furthermore, Wesselhoeft et al. indicate that nonmodified circRNA is less immunogenic compared to linear mRNA with m1Ψ in part due to reduced TLR sensing which decreases non-specific innate immune system activation [139].
For clinical translation of circRNA, researchers need to work carefully on the manufacturing and characterization of circRNA such as differentiation from its linear cognates. Li et al. reported a programmable RNA-guided, RNA-targeting CRISPR-Cas13 system to differentiate between circRNAs and linear mRNAs by using guide RNAs that target sequences across the back-splicing junction sites [145]. This system can be used to conduct large-scale functional screening of circRNAs across various applications. Nonetheless, the purification of circRNA from the reaction system, such as ribozymes or linear RNAs, can pose a challenge. Large scale synthesis of circRNAs may offer some cost reductions due to absence of capping structures or modified nucleosides, but the circularization process, subsequent purification, and analytical characterization of circRNAs has not yet been fully validated at large scale [140], [141]. The growing interest in coding circRNA necessitates the standardized practice of synthesizing, isolating, and characterizing circRNAs to enable a more robust implementation of circRNAs for therapeutic purposes [146].
5. Conclusion
Since the inception of mRNAs as a therapeutic modality, multiple impediments to the adoption of mRNAs into the clinic have been tackled including its immunogenicity and stability through comprehensive optimization of the IVT mRNA transcript. Structural and sequence engineering from multiple angles have created mRNAs that feature high translational capacity, which is especially propitious as a therapeutic modality. mRNA-based therapeutics including vaccines, protein replacement, or gene editing all benefit from a flexible platform that enables the rapid development and production of the mRNA transcript. An advantage of mRNAs is the transient nature of protein expression, which can be especially promising as a vaccine candidate. However, for some applications, such as protein replacement strategies, this transient property is not always desired. Therefore, modulation of translation dynamics by elongating stability through structural optimizations or utilizing other modalities of RNA such as circular or self-amplifying RNAs can potentially address this shortcoming. Consequently, concurrent research on the development of mRNA structural elements should continue to fulfill the immense potential of mRNAs as drugs. Nonetheless, the cell type specificity of translation machinery complicates the generalization of some of the mRNA optimization strategies. Therefore, more extensive evaluations of the specific potency of the mRNA in representative models are vital to fully characterize the effects of the specific optimization in biorelevant systems. This is further complicated because mRNAs require a delivery system for efficient uptake in vivo. Although recent advances in lipid nanoparticle-based delivery of mRNA permitted the adoption of two mRNA-based vaccines for SARS-CoV-2, more efficient and finetuned systemic delivery still has opportunities for improvement. For therapeutic applications, it can be valuable to achieve specific delivery to target organs and cell types. Therefore, parallel improvements on more safe and efficacious mRNA delivery strategies will also unravel the impending adoption of mRNAs for various applications in clinics.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
Y.D. acknowledges the support from the Maximizing Investigators’ Research Award R35GM144117 from the National Institute of General Medical Sciences. The authors acknowledge the usage of bioRender.com to generate the figures.
References
- 1.Brenner S., Jacob F., Meselson M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature. 1961;190:576–581. doi: 10.1038/190576a0. [DOI] [PubMed] [Google Scholar]
- 2.Yan J., Kang D.D., Dong Y. Harnessing lipid nanoparticles for efficient CRISPR delivery. Biomater. Sci. 2021;9(18):6001–6011. doi: 10.1039/d1bm00537e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbier A.J., Jiang A.Y., Zhang P., Wooster R., Anderson D.G. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 2022;40:840–854. doi: 10.1038/s41587-022-01294-2. [DOI] [PubMed] [Google Scholar]
- 4.Gillmore J.D., Gane E.d., Taubel J., Kao J., Fontana M., Maitland M.L., Seitzer J., O’Connell D., Walsh K.R., Wood K., Phillips J., Xu Y., Amaral A., Boyd A.P., Cehelsky J.E., McKee M.D., Schiermeier A., Harari O., Murphy A., Kyratsous C.A., Zambrowicz B., Soltys R., Gutstein D.E., Leonard J., Sepp-Lorenzino L., Lebwohl D. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 2021;385(6):493–502. doi: 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- 5.Hou X., Zaks T., Langer R., Dong Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021;6(12):1078–1094. doi: 10.1038/s41578-021-00358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kariko K., Buckstein M., Ni H., Weissman D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 7.Nallagatla S.R., Bevilacqua P.C. Nucleoside modifications modulate activation of the protein kinase PKR in an RNA structure-specific manner. RNA. 2008;14:1201–1213. doi: 10.1261/rna.1007408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Svitkin Y.V., et al. N1-methyl-pseudouridine in mRNA enhances translation through eIF2alpha-dependent and independent mechanisms by increasing ribosome density. Nucl. Acids Res. 2017;45:6023–6036. doi: 10.1093/nar/gkx135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bornewasser L., Domnick C., Kath-Schorr S. Stronger together for in-cell translation: natural and unnatural base modified mRNA. Chem. Sci. 2022;13:4753–4761. doi: 10.1039/d2sc00670g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andries O., Mc Cafferty S., De Smedt S.C., Weiss R., Sanders N.N., Kitada T. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Release. 2015;217:337–344. doi: 10.1016/j.jconrel.2015.08.051. [DOI] [PubMed] [Google Scholar]
- 11.Sahin U., Kariko K., Tureci O. mRNA-based therapeutics–developing a new class of drugs. Nat. Rev. Drug Discov. 2014;13:759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 12.Wayment-Steele H.K., Kim D.S., Choe C.A., Nicol J.J., Wellington-Oguri R., Watkins A.M., Parra Sperberg R.A., Huang P.S., Participants E., Das R. Theoretical basis for stabilizing messenger RNA through secondary structure design. Nucl. Acids Res. 2021;49(18):10604–10617. doi: 10.1093/nar/gkab764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leppek K., Byeon G.W., Kladwang W., Wayment-Steele H.K., Kerr C.H., Xu A.F., Kim D.S., Topkar V.V., Choe C., Rothschild D., Tiu G.C., Wellington-Oguri R., Fujii K., Sharma E., Watkins A.M., Nicol J.J., Romano J., Tunguz B., Diaz F., Cai H., Guo P., Wu J., Meng F., Shi S., Participants E., Dormitzer P.R., Solórzano A., Barna M., Das R. Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics. Nat. Commun. 2022;13(1) doi: 10.1038/s41467-022-28776-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karikó K., Muramatsu H., Welsh F.A., Ludwig J., Kato H., Akira S., Weissman D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008;16(11):1833–1840. doi: 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson B.R., Muramatsu H., Nallagatla S.R., Bevilacqua P.C., Sansing L.H., Weissman D., Karikó K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucl. Acids Res. 2010;38(17):5884–5892. doi: 10.1093/nar/gkq347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li B., Luo X., Dong Y. Effects of Chemically Modified Messenger RNA on Protein Expression. Bioconjug Chem. 2016;27:849–853. doi: 10.1021/acs.bioconjchem.6b00090. [DOI] [PubMed] [Google Scholar]
- 17.Kormann M.S.D., Hasenpusch G., Aneja M.K., Nica G., Flemmer A.W., Herber-Jonat S., Huppmann M., Mays L.E., Illenyi M., Schams A., Griese M., Bittmann I., Handgretinger R., Hartl D., Rosenecker J., Rudolph C. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol. 2011;29(2):154–157. doi: 10.1038/nbt.1733. [DOI] [PubMed] [Google Scholar]
- 18.Anderson B.R., Muramatsu H., Jha B.K., Silverman R.H., Weissman D., Kariko K. Nucleoside modifications in RNA limit activation of 2'-5'-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011;39(21):9329–9338. doi: 10.1093/nar/gkr586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thess A., Grund S., Mui B.L., Hope M.J., Baumhof P., Fotin-Mleczek M., Schlake T. Sequence-engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol Ther. 2015;23(9):1456–1464. doi: 10.1038/mt.2015.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verbeke R., Hogan M.J., Lore K., Pardi N. Innate immune mechanisms of mRNA vaccines. Immunity. 2022;55:1993–2005. doi: 10.1016/j.immuni.2022.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen C.Y., Shyu A.B. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 22.von Roretz C., Di Marco S., Mazroui R., Gallouzi I.E. Turnover of AU-rich-containing mRNAs during stress: a matter of survival. Wires Rna. 2011;2:336–347. doi: 10.1002/wrna.55. [DOI] [PubMed] [Google Scholar]
- 23.Courel M., Clément Y., Bossevain C., Foretek D., Vidal Cruchez O., Yi Z., Bénard M., Benassy M.-N., Kress M., Vindry C., Ernoult-Lange M., Antoniewski C., Morillon A., Brest P., Hubstenberger A., Roest Crollius H., Standart N., Weil D. GC content shapes mRNA storage and decay in human cells. Elife. 2019;8 doi: 10.7554/eLife.49708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kudla G., Lipinski L., Caffin F., Helwak A., Zylicz M., Hurst L.D. High guanine and cytosine content increases mRNA levels in mammalian cells. PLoS Biol. 2006;4(6):e180. doi: 10.1371/journal.pbio.0040180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mauger D.M., Cabral B.J., Presnyak V., Su S.V., Reid D.W., Goodman B., Link K., Khatwani N., Reynders J., Moore M.J., McFadyen I.J. mRNA structure regulates protein expression through changes in functional half-life. Proc Natl Acad Sci U S A. 2019;116(48):24075–24083. doi: 10.1073/pnas.1908052116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramanathan A., Robb G.B., Chan S.H. mRNA capping: biological functions and applications. Nucleic Acids Res. 2016;44:7511–7526. doi: 10.1093/nar/gkw551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwer B., Mao X., Shuman S. Accelerated mRNA decay in conditional mutants of yeast mRNA capping enzyme. Nucleic Acids Res. 1998;26:2050–2057. doi: 10.1093/nar/26.9.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y., Kiledjian M. Regulation of mRNA decapping. Wiley Interdiscip Rev RNA. 2010;1:253–265. doi: 10.1002/wrna.15. [DOI] [PubMed] [Google Scholar]
- 29.Galloway A., Cowling V.H. mRNA cap regulation in mammalian cell function and fate. Biochim Biophys Acta Gene Regul Mech. 2019;1862(3):270–279. doi: 10.1016/j.bbagrm.2018.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang S.P., Deng L., Ho C.K., Shuman S. Phylogeny of mRNA capping enzymes. Proc Natl Acad Sci U S A. 1997;94:9573–9578. doi: 10.1073/pnas.94.18.9573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shuman S. The mRNA capping apparatus as drug target and guide to eukaryotic phylogeny. Cold Spring Harb Symp Quant Biol. 2001;66:301–312. doi: 10.1101/sqb.2001.66.301. [DOI] [PubMed] [Google Scholar]
- 32.McCracken S., et al. 5'-Capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev. 1997;11:3306–3318. doi: 10.1101/gad.11.24.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Furuichi Y., Shatkin A.J. Viral and cellular mRNA capping: past and prospects. Adv Virus Res. 2000;55:135–184. doi: 10.1016/s0065-3527(00)55003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daffis S., Szretter K.J., Schriewer J., Li J., Youn S., Errett J., Lin T.-Y., Schneller S., Zust R., Dong H., Thiel V., Sen G.C., Fensterl V., Klimstra W.B., Pierson T.C., Buller R.M., Gale Jr M., Shi P.-Y., Diamond M.S. 2'-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468(7322):452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leppek K., Das R., Barna M. Functional 5' UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat Rev Mol Cell Biol. 2018;19:158–174. doi: 10.1038/nrm.2017.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Araujo P.R., Yoon K., Ko D., Smith A.D., Qiao M., Suresh U., Burns S.C., Penalva L.O.F. Before It Gets Started: Regulating Translation at the 5' UTR. Comp Funct Genomics. 2012;2012 doi: 10.1155/2012/475731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sample P.J., Wang B., Reid D.W., Presnyak V., McFadyen I.J., Morris D.R., Seelig G. Human 5' UTR design and variant effect prediction from a massively parallel translation assay. Nat Biotechnol. 2019;37(7):803–809. doi: 10.1038/s41587-019-0164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linares-Fernandez S., et al. Combining an optimized mRNA template with a double purification process allows strong expression of in vitro transcribed mRNA. Mol Ther Nucleic Acids. 2021;26:945–956. doi: 10.1016/j.omtn.2021.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zeng C., Hou X., Yan J., Zhang C., Li W., Zhao W., Du S., Dong Y. Leveraging mRNA Sequences and Nanoparticles to Deliver SARS-CoV-2 Antigens In Vivo. Adv Mater. 2020;32(40):e2004452. doi: 10.1002/adma.202004452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gebre M.S., Rauch S., Roth N., Yu J., Chandrashekar A., Mercado N.B., He X., Liu J., McMahan K., Martinot A., Martinez D.R., Giffin V., Hope D., Patel S., Sellers D., Sanborn O., Barrett J., Liu X., Cole A.C., Pessaint L., Valentin D., Flinchbaugh Z., Yalley-Ogunro J., Muench J., Brown R., Cook A., Teow E., Andersen H., Lewis M.G., Boon A.C.M., Baric R.S., Mueller S.O., Petsch B., Barouch D.H. Optimization of non-coding regions for a non-modified mRNA COVID-19 vaccine. Nature. 2022;601(7893):410–414. doi: 10.1038/s41586-021-04231-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Z., Day N., Trifillis P., Kiledjian M. An mRNA stability complex functions with poly(A)-binding protein to stabilize mRNA in vitro. Mol Cell Biol. 1999;19:4552–4560. doi: 10.1128/MCB.19.7.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferizi M., Aneja M.K., Balmayor E.R., Badieyan Z.S., Mykhaylyk O., Rudolph C., Plank C. Human cellular CYBA UTR sequences increase mRNA translation without affecting the half-life of recombinant RNA transcripts. Sci Rep. 2016;6(1) doi: 10.1038/srep39149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jia L., Mao Y., Ji Q., Dersh D., Yewdell J.W., Qian S.-B. Decoding mRNA translatability and stability from the 5' UTR. Nat Struct Mol Biol. 2020;27(9):814–821. doi: 10.1038/s41594-020-0465-x. [DOI] [PubMed] [Google Scholar]
- 44.Cannarozzi G., Schraudolph N.N., Faty M., von Rohr P., Friberg M.T., Roth A.C., Gonnet P., Gonnet G., Barral Y. A role for codon order in translation dynamics. Cell. 2010;141(2):355–367. doi: 10.1016/j.cell.2010.02.036. [DOI] [PubMed] [Google Scholar]
- 45.Presnyak V., Alhusaini N., Chen Y.-H., Martin S., Morris N., Kline N., Olson S., Weinberg D., Baker K., Graveley B., Coller J. Codon optimality is a major determinant of mRNA stability. Cell. 2015;160(6):1111–1124. doi: 10.1016/j.cell.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hanson G., Coller J. Codon optimality, bias and usage in translation and mRNA decay. Nat Rev Mol Cell Biol. 2018;19:20–30. doi: 10.1038/nrm.2017.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narula A., Ellis J., Taliaferro J.M., Rissland O.S. Coding regions affect mRNA stability in human cells. RNA. 2019;25:1751–1764. doi: 10.1261/rna.073239.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi J., Ieong K.-W., Demirci H., Chen J., Petrov A., Prabhakar A., O'Leary S.E., Dominissini D., Rechavi G., Soltis S.M., Ehrenberg M., Puglisi J.D. N(6)-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat Struct Mol Biol. 2016;23(2):110–115. doi: 10.1038/nsmb.3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eyler D.E., Franco M.K., Batool Z., Wu M.Z., Dubuke M.L., Dobosz-Bartoszek M., Jones J.D., Polikanov Y.S., Roy B., Koutmou K.S. Pseudouridinylation of mRNA coding sequences alters translation. Proc Natl Acad Sci U S A. 2019;116(46):23068–23074. doi: 10.1073/pnas.1821754116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mao Y., Dong L., Liu X.-M., Guo J., Ma H., Shen B., Qian S.-B. m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat Commun. 2019;10(1) doi: 10.1038/s41467-019-13317-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.H.e. Zhang L. Zhang A. Lin C. Xu Z. Li K. Liu B. Liu X. Ma F. Zhao H. Jiang C. Chen H. Shen H. Li D.H. Mathews Y. Zhang L. Huang Algorithm for Optimized mRNA Design Improves Stability and Immunogenicity 10.1038/s41586-023-06127-z (2023). [DOI] [PMC free article] [PubMed]
- 52.Kislauskis E.H., Singer R.H. Determinants of mRNA localization. Curr Opin Cell Biol. 1992;4:975–978. doi: 10.1016/0955-0674(92)90128-y. [DOI] [PubMed] [Google Scholar]
- 53.Mayr C. What Are 3' UTRs Doing? Cold Spring Harb Perspect Biol. 2019;11(10):a034728. doi: 10.1101/cshperspect.a034728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andreassi C., Riccio A. To localize or not to localize: mRNA fate is in 3'UTR ends. Trends Cell Biol. 2009;19:465–474. doi: 10.1016/j.tcb.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 55.Lytle J.R., Yario T.A., Steitz J.A. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5' UTR as in the 3' UTR. Proc Natl Acad Sci U S A. 2007;104:9667–9672. doi: 10.1073/pnas.0703820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown B.D., Gentner B., Cantore A., Colleoni S., Amendola M., Zingale A., Baccarini A., Lazzari G., Galli C., Naldini L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat Biotechnol. 2007;25(12):1457–1467. doi: 10.1038/nbt1372. [DOI] [PubMed] [Google Scholar]
- 57.Sandberg R., Neilson J.R., Sarma A., Sharp P.A., Burge C.B. Proliferating cells express mRNAs with shortened 3' untranslated regions and fewer microRNA target sites. Science. 2008;320:1643–1647. doi: 10.1126/science.1155390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gillies J.K., Lorimer I.A. Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle. 2007;6:2005–2009. doi: 10.4161/cc.6.16.4526. [DOI] [PubMed] [Google Scholar]
- 59.Li H., Cao Y., Ye J., Yang Z., Chen Q., Liu X., Zhang B., Qiao J., Tang Q., Yang H., Li J., Shi Z., Mao Y. Engineering brain-derived neurotrophic factor mRNA delivery for the treatment of Alzheimer’s disease. Chemical Engineering Journal. 2023;466:143152. [Google Scholar]
- 60.Lai W.C., et al. Intrinsically Unstructured Sequences in the mRNA 3' UTR Reduce the Ability of Poly(A) Tail to Enhance Translation. J Mol Biol. 2022;434 doi: 10.1016/j.jmb.2022.167877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mockey M., Gonçalves C., Dupuy F.P., Lemoine F.M., Pichon C., Midoux P. mRNA transfection of dendritic cells: synergistic effect of ARCA mRNA capping with Poly(A) chains in cis and in trans for a high protein expression level. Biochem Biophys Res Commun. 2006;340(4):1062–1068. doi: 10.1016/j.bbrc.2005.12.105. [DOI] [PubMed] [Google Scholar]
- 62.Meijer H.A., Bushell M., Hill K., Gant T.W., Willis A.E., Jones P., de Moor C.H. A novel method for poly(A) fractionation reveals a large population of mRNAs with a short poly(A) tail in mammalian cells. Nucleic Acids Res. 2007;35(19):e132. doi: 10.1093/nar/gkm830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park J., Kim M., Yi H., Baeg K., Choi Y., Lee Y.-S., Lim J., Kim V.N. Short poly(A) tails are protected from deadenylation by the LARP1-PABP complex. Nat Struct Mol Biol. 2023;30(3):330–338. doi: 10.1038/s41594-023-00930-y. [DOI] [PubMed] [Google Scholar]
- 64.Passmore L.A., Coller J. Roles of mRNA poly(A) tails in regulation of eukaryotic gene expression. Nat Rev Mol Cell Biol. 2022;23:93–106. doi: 10.1038/s41580-021-00417-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Legnini I., Alles J., Karaiskos N., Ayoub S., Rajewsky N. FLAM-seq: full-length mRNA sequencing reveals principles of poly(A) tail length control. Nat Methods. 2019;16:879–886. doi: 10.1038/s41592-019-0503-y. [DOI] [PubMed] [Google Scholar]
- 66.Li C.Y., et al. Cytidine-containing tails robustly enhance and prolong protein production of synthetic mRNA in cell and in vivo. Mol Ther Nucleic Acids. 2022;30:300–310. doi: 10.1016/j.omtn.2022.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao J., Hyman L., Moore C. Formation of mRNA 3' ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev. 1999;63:405–445. doi: 10.1128/MMBR.63.2.405-445.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chaudhary N., Weissman D., Whitehead K.A. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat Rev Drug Discov. 2021;20:817–838. doi: 10.1038/s41573-021-00283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caruthers M.H. A brief review of DNA and RNA chemical synthesis. Biochem Soc Trans. 2011;39:575–580. doi: 10.1042/BST0390575. [DOI] [PubMed] [Google Scholar]
- 70.Tabor S., Richardson C.C. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc Natl Acad Sci U S A. 1985;82:1074–1078. doi: 10.1073/pnas.82.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beckert B., Masquida B. Synthesis of RNA by in vitro transcription. Methods Mol Biol. 2011;703:29–41. doi: 10.1007/978-1-59745-248-9_3. [DOI] [PubMed] [Google Scholar]
- 72.Thiel V., Herold J., Schelle B., Siddell S.G. Infectious RNA transcribed in vitro from a cDNA copy of the human coronavirus genome cloned in vaccinia virus. J Gen Virol. 2001;82:1273–1281. doi: 10.1099/0022-1317-82-6-1273. [DOI] [PubMed] [Google Scholar]
- 73.Milligan J.F., Groebe D.R., Witherell G.W., Uhlenbeck O.C. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 1987;15:8783–8798. doi: 10.1093/nar/15.21.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rong M., He B., McAllister W.T., Durbin R.K. Promoter specificity determinants of T7 RNA polymerase. Proc Natl Acad Sci U S A. 1998;95:515–519. doi: 10.1073/pnas.95.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Conrad T., Plumbom I., Alcobendas M., Vidal R., Sauer S. Maximizing transcription of nucleic acids with efficient T7 promoters. Commun Biol. 2020;3:439. doi: 10.1038/s42003-020-01167-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dunn J.J., Studier F.W., Gottesman M. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J Mol Biol. 1983;166(4):477–535. doi: 10.1016/s0022-2836(83)80282-4. [DOI] [PubMed] [Google Scholar]
- 77.Kholod N., Vassilenko K., Shlyapnikov M., Ksenzenko V., Kisselev L. Preparation of active tRNA gene transcripts devoid of 3'-extended products and dimers. Nucleic Acids Res. 1998;26:2500–2501. doi: 10.1093/nar/26.10.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Helm M., Brule H., Giege R., Florentz C. More mistakes by T7 RNA polymerase at the 5' ends of in vitro-transcribed RNAs. RNA. 1999;5:618–621. doi: 10.1017/s1355838299982328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gholamalipour Y., Karunanayake Mudiyanselage A., Martin C.T. 3' end additions by T7 RNA polymerase are RNA self-templated, distributive and diverse in character-RNA-Seq analyses. Nucleic Acids Res. 2018;46:9253–9263. doi: 10.1093/nar/gky796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen Y.G., Hur S. Cellular origins of dsRNA, their recognition and consequences. Nat Rev Mol Cell Biol. 2022;23:286–301. doi: 10.1038/s41580-021-00430-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu M.Z., Asahara H., Tzertzinis G., Roy B. Synthesis of low immunogenicity RNA with high-temperature in vitro transcription. RNA. 2020;26:345–360. doi: 10.1261/rna.073858.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dousis A., Ravichandran K., Hobert E.M., Moore M.J., Rabideau A.E. An engineered T7 RNA polymerase that produces mRNA free of immunostimulatory byproducts. Nat Biotechnol. 2023;41(4):560–568. doi: 10.1038/s41587-022-01525-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Raskin C.A., Diaz G.A., McAllister W.T. T7 RNA polymerase mutants with altered promoter specificities. Proc Natl Acad Sci U S A. 1993;90:3147–3151. doi: 10.1073/pnas.90.8.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Diaz G.A., Raskin C.A., McAllister W.T. Hierarchy of base-pair preference in the binding domain of the bacteriophage T7 promoter. J Mol Biol. 1993;229:805–811. doi: 10.1006/jmbi.1993.1086. [DOI] [PubMed] [Google Scholar]
- 85.Beier H., Hausmann R. T3 times T7 phage crosses leading to recombinant RNA polymerases. Nature. 1974;251:538–540. doi: 10.1038/251538a0. [DOI] [PubMed] [Google Scholar]
- 86.Klement J.F., Moorefiedl M.B., Jorgensen E., Brown J.E., Risman S., McAllister W.T. Discrimination between bacteriophage T3 and T7 promoters by the T3 and T7 RNA polymerases depends primarily upon a three base-pair region located 10 to 12 base-pairs upstream from the start site. J Mol Biol. 1990;215(1):21–29. doi: 10.1016/s0022-2836(05)80091-9. [DOI] [PubMed] [Google Scholar]
- 87.Butler E.T., Chamberlin M.J. Bacteriophage SP6-specific RNA polymerase. I. Isolation and characterization of the enzyme. J Biol Chem. 1982;257(10):5772–5778. [PubMed] [Google Scholar]
- 88.Krieg P.A., Melton D.A. In vitro RNA synthesis with SP6 RNA polymerase. Methods Enzymol. 1987;155:397–415. doi: 10.1016/0076-6879(87)55027-3. [DOI] [PubMed] [Google Scholar]
- 89.Frye M., Jaffrey S.R., Pan T., Rechavi G., Suzuki T. RNA modifications: what have we learned and where are we headed? Nat Rev Genet. 2016;17:365–372. doi: 10.1038/nrg.2016.47. [DOI] [PubMed] [Google Scholar]
- 90.Roundtree I.A., Evans M.E., Pan T., He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169:1187–1200. doi: 10.1016/j.cell.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Milisavljevic N., Perlikova P., Pohl R., Hocek M. Enzymatic synthesis of base-modified RNA by T7 RNA polymerase. A systematic study and comparison of 5-substituted pyrimidine and 7-substituted 7-deazapurine nucleoside triphosphates as substrates. Org Biomol Chem. 2018;16:5800–5807. doi: 10.1039/c8ob01498a. [DOI] [PubMed] [Google Scholar]
- 92.Potapov V., Fu X., Dai N., Corrêa I.R., Tanner N.A., Ong J.L. Base modifications affecting RNA polymerase and reverse transcriptase fidelity. Nucleic Acids Res. 2018;46(11):5753–5763. doi: 10.1093/nar/gky341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen T.H., Potapov V., Dai N., Ong J.L., Roy B. N(1)-methyl-pseudouridine is incorporated with higher fidelity than pseudouridine in synthetic RNAs. Sci Rep. 2022;12:13017. doi: 10.1038/s41598-022-17249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fabrega C., Hausmann S., Shen V., Shuman S., Lima C.D. Structure and mechanism of mRNA cap (guanine-N7) methyltransferase. Mol Cell. 2004;13:77–89. doi: 10.1016/s1097-2765(03)00522-7. [DOI] [PubMed] [Google Scholar]
- 95.Henderson J.M., et al. Cap 1 Messenger RNA Synthesis with Co-transcriptional CleanCap((R)) Analog by In Vitro Transcription. Curr Protoc. 2021;1:e39. doi: 10.1002/cpz1.39. [DOI] [PubMed] [Google Scholar]
- 96.Stepinski J., Waddell C., Stolarski R., Darzynkiewicz E., Rhoads R.E. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl(3'-O-methyl)GpppG and 7-methyl (3'-deoxy)GpppG. RNA. 2001;7:1486–1495. [PMC free article] [PubMed] [Google Scholar]
- 97.Kuhn A.N., Diken M., Kreiter S., Selmi A., Kowalska J., Jemielity J., Darzynkiewicz E., Huber C., Türeci Ö., Sahin U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010;17(8):961–971. doi: 10.1038/gt.2010.52. [DOI] [PubMed] [Google Scholar]
- 98.Kis Z., Kontoravdi C., Shattock R., Shah N. Resources, Production Scales and Time Required for Producing RNA Vaccines for the Global Pandemic Demand. Vaccines (Basel) 2020;9(1):3. doi: 10.3390/vaccines9010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Williams G.D., Gokhale N.S., Snider D.L., Horner S.M., Schoggins J. The mRNA Cap 2′- O -Methyltransferase CMTR1 Regulates the Expression of Certain Interferon-Stimulated Genes. mSphere. 2020;5(3) doi: 10.1128/mSphere.00202-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Egloff M.P., Benarroch D., Selisko B., Romette J.L., Canard B. An RNA cap (nucleoside-2'-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 2002;21:2757–2768. doi: 10.1093/emboj/21.11.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fuchs A.L., Neu A., Sprangers R. A general method for rapid and cost-efficient large-scale production of 5' capped RNA. RNA. 2016;22:1454–1466. doi: 10.1261/rna.056614.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pardi N., Hogan M.J., Porter F.W., Weissman D. mRNA vaccines - a new era in vaccinology. Nat Rev Drug Discov. 2018;17:261–279. doi: 10.1038/nrd.2017.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Plotkin S., Robinson J.M., Cunningham G., Iqbal R., Larsen S. The complexity and cost of vaccine manufacturing - An overview. Vaccine. 2017;35:4064–4071. doi: 10.1016/j.vaccine.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Whitley J., Zwolinski C., Denis C., Maughan M., Hayles L., Clarke D., Snare M., Liao H., Chiou S., Marmura T., Zoeller H., Hudson B., Peart J., Johnson M., Karlsson A., Wang Y., Nagle C., Harris C., Tonkin D., Fraser S., Capiz L., Zeno C.L., Meli Y., Martik D., Ozaki D.A., Caparoni A., Dickens J.E., Weissman D., Saunders K.O., Haynes B.F., Sempowski G.D., Denny T.N., Johnson M.R. Development of mRNA manufacturing for vaccines and therapeutics: mRNA platform requirements and development of a scalable production process to support early phase clinical trials. Transl Res. 2022;242:38–55. doi: 10.1016/j.trsl.2021.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rosa S.S., Prazeres D.M.F., Azevedo A.M., Marques M.P.C. mRNA vaccines manufacturing: Challenges and bottlenecks. Vaccine. 2021;39:2190–2200. doi: 10.1016/j.vaccine.2021.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Karikó K., Muramatsu H., Ludwig J., Weissman D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011;39(21):e142. doi: 10.1093/nar/gkr695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Meyer, R. A., Trabulo, S., Douthwaite, J. A. & Santos, J. L. in Messenger RNA Therapeutics (eds Stefan Jurga & Jan Barciszewski) 1-16 (Springer International Publishing, 2022).
- 108.Chien K.R., Zangi L., Lui K.O. Synthetic chemically modified mRNA (modRNA): toward a new technology platform for cardiovascular biology and medicine. Cold Spring Harb Perspect Med. 2015;5(1) doi: 10.1101/cshperspect.a014035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lorentzen C.L., Haanen J.B., Met O., Svane I.M. Clinical advances and ongoing trials on mRNA vaccines for cancer treatment. Lancet Oncol. 2022;23:e450–e458. doi: 10.1016/S1470-2045(22)00372-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wolff J.A., Malone R.W., Williams P., Chong W., Acsadi G., Jani A., Felgner P.L. Direct gene transfer into mouse muscle in vivo. Science. 1990;247(4949):1465–1468. doi: 10.1126/science.1690918. [DOI] [PubMed] [Google Scholar]
- 111.Zhang M., Hussain A., Yang H., Zhang J., Liang X.-J., Huang Y. mRNA-based modalities for infectious disease management. Nano Res. 2023;16(1):672–691. doi: 10.1007/s12274-022-4627-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Baden L.R., El Sahly H.M., Essink B., Kotloff K., Frey S., Novak R., Diemert D., Spector S.A., Rouphael N., Creech C.B., McGettigan J., Khetan S., Segall N., Solis J., Brosz A., Fierro C., Schwartz H., Neuzil K., Corey L., Gilbert P., Janes H., Follmann D., Marovich M., Mascola J., Polakowski L., Ledgerwood J., Graham B.S., Bennett H., Pajon R., Knightly C., Leav B., Deng W., Zhou H., Han S., Ivarsson M., Miller J., Zaks T. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med. 2021;384(5):403–416. doi: 10.1056/NEJMoa2035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Polack F.P., Thomas S.J., Kitchin N., Absalon J., Gurtman A., Lockhart S., Perez J.L., Pérez Marc G., Moreira E.D., Zerbini C., Bailey R., Swanson K.A., Roychoudhury S., Koury K., Li P., Kalina W.V., Cooper D., Frenck R.W., Hammitt L.L., Türeci Ö., Nell H., Schaefer A., Ünal S., Tresnan D.B., Mather S., Dormitzer P.R., Şahin U., Jansen K.U., Gruber W.C. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med. 2020;383(27):2603–2615. doi: 10.1056/NEJMoa2034577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang X., Zhao W., Nguyen G.N., Zhang C., Zeng C., Yan J., Du S., Hou X., Li W., Jiang J., Deng B., McComb D.W., Dorkin R., Shah A., Barrera L., Gregoire F., Singh M., Chen D., Sabatino D.E., Dong Y. Functionalized lipid-like nanoparticles for in vivo mRNA delivery and base editing. Sci Adv. 2020;6(34) doi: 10.1126/sciadv.abc2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ramaswamy S., Tonnu N., Tachikawa K., Limphong P., Vega J.B., Karmali P.P., Chivukula P., Verma I.M. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc Natl Acad Sci U S A. 2017;114(10) doi: 10.1073/pnas.1619653114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Phimister E.G., Fuller D.H., Berglund P. Amplifying RNA Vaccine Development. N Engl J Med. 2020;382(25):2469–2471. doi: 10.1056/NEJMcibr2009737. [DOI] [PubMed] [Google Scholar]
- 117.Li Y., Teague B., Zhang Y., Su Z., Porter E., Dobosh B., Wagner T., Irvine D.J., Weiss R. In vitro evolution of enhanced RNA replicons for immunotherapy. Sci Rep. 2019;9(1) doi: 10.1038/s41598-019-43422-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Beissert T., Perkovic M., Vogel A., Erbar S., Walzer K.C., Hempel T., Brill S., Haefner E., Becker R., Türeci Ö., Sahin U. A Trans-amplifying RNA Vaccine Strategy for Induction of Potent Protective Immunity. Mol Ther. 2020;28(1):119–128. doi: 10.1016/j.ymthe.2019.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Maruggi G., Ulmer J.B., Rappuoli R., Yu D. Self-amplifying mRNA-Based Vaccine Technology and Its Mode of Action. Curr Top Microbiol Immunol. 2021 doi: 10.1007/82_2021_233. [DOI] [PubMed] [Google Scholar]
- 120.Geall A.J., Verma A., Otten G.R., Shaw C.A., Hekele A., Banerjee K., Cu Y., Beard C.W., Brito L.A., Krucker T., O’Hagan D.T., Singh M., Mason P.W., Valiante N.M., Dormitzer P.R., Barnett S.W., Rappuoli R., Ulmer J.B., Mandl C.W. Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci U S A. 2012;109(36):14604–14609. doi: 10.1073/pnas.1209367109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mandl C.W., Aberle J.H., Aberle S.W., Holzmann H., Allison S.L., Heinz F.X. In vitro-synthesized infectious RNA as an attenuated live vaccine in a flavivirus model. Nat Med. 1998;4(12):1438–1440. doi: 10.1038/4031. [DOI] [PubMed] [Google Scholar]
- 122.Ulmer J.B., Mason P.W., Geall A., Mandl C.W. RNA-based vaccines. Vaccine. 2012;30:4414–4418. doi: 10.1016/j.vaccine.2012.04.060. [DOI] [PubMed] [Google Scholar]
- 123.Pepini T., Pulichino A.-M., Carsillo T., Carlson A.L., Sari-Sarraf F., Debasitis K.J.C., Maruggi G., Otten G.R., Geall A.J., Yu D., Ulmer J.B., Iavarone C. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J Immunol. 2017;198(10):4012–4024. doi: 10.4049/jimmunol.1601877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu Y., Chin J.M., Choo E.L., Phua K.K.L. Messenger RNA translation enhancement by immune evasion proteins: a comparative study between EKB (vaccinia virus) and NS1 (influenza A virus) Sci Rep. 2019;9:11972. doi: 10.1038/s41598-019-48559-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Beissert T., Koste L., Perkovic M., Walzer K.C., Erbar S., Selmi A., Diken M., Kreiter S., Türeci Ö., Sahin U. Improvement of In Vivo Expression of Genes Delivered by Self-Amplifying RNA Using Vaccinia Virus Immune Evasion Proteins. Hum Gene Ther. 2017;28(12):1138–1146. doi: 10.1089/hum.2017.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Blakney A.K., McKay P.F., Bouton C.R., Hu K., Samnuan K., Shattock R.J. Innate Inhibiting Proteins Enhance Expression and Immunogenicity of Self-Amplifying RNA. Mol Ther. 2021;29(3):1174–1185. doi: 10.1016/j.ymthe.2020.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li Y., Su Z., Zhao W., Zhang X., Momin N., Zhang C., Wittrup K.D., Dong Y., Irvine D.J., Weiss R. Multifunctional oncolytic nanoparticles deliver self-replicating IL-12 RNA to eliminate established tumors and prime systemic immunity. Nat Cancer. 2020;1(9):882–893. doi: 10.1038/s43018-020-0095-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Du S., Li W., Zhang Y., Xue Y., Hou X., Yan J., Cheng J., Deng B., McComb D.W., Lin J., Zeng H., Cheng X., Irvine D.J., Weiss R., Dong Y. Cholesterol‐Amino‐Phosphate (CAP) Derived Lipid Nanoparticles for Delivery of Self‐Amplifying RNA and Restoration of Spermatogenesis in Infertile Mice. Advanced Science. 2023;10(11) doi: 10.1002/advs.202300188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wagner T.E., Becraft J.R., Bodner K., Teague B., Zhang X., Woo A., Porter E., Alburquerque B., Dobosh B., Andries O., Sanders N.N., Beal J., Densmore D., Kitada T., Weiss R. Small-molecule-based regulation of RNA-delivered circuits in mammalian cells. Nat Chem Biol. 2018;14(11):1043–1050. doi: 10.1038/s41589-018-0146-9. [DOI] [PubMed] [Google Scholar]
- 130.Wesselhoeft R.A., Kowalski P.S., Anderson D.G. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat Commun. 2018;9:2629. doi: 10.1038/s41467-018-05096-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen L.L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat Rev Mol Cell Biol. 2020;21:475–490. doi: 10.1038/s41580-020-0243-y. [DOI] [PubMed] [Google Scholar]
- 132.Qian L., Vu M.N., Carter M., Wilkinson M.F. A spliced intron accumulates as a lariat in the nucleus of T cells. Nucleic Acids Res. 1992;20:5345–5350. doi: 10.1093/nar/20.20.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Barrett S.P., Wang P.L., Salzman J. Circular RNA biogenesis can proceed through an exon-containing lariat precursor. Elife. 2015;4:e07540. doi: 10.7554/eLife.07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Obi P., Chen Y.G. The design and synthesis of circular RNAs. Methods. 2021;196:85–103. doi: 10.1016/j.ymeth.2021.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu X., Zhang Y.u., Zhou S., Dain L., Mei L., Zhu G. Circular RNA: An emerging frontier in RNA therapeutic targets, RNA therapeutics, and mRNA vaccines. J Control Release. 2022;348:84–94. doi: 10.1016/j.jconrel.2022.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Nandakumar J., Ho C.K., Lima C.D., Shuman S. RNA substrate specificity and structure-guided mutational analysis of bacteriophage T4 RNA ligase 2. J Biol Chem. 2004;279:31337–31347. doi: 10.1074/jbc.M402394200. [DOI] [PubMed] [Google Scholar]
- 137.Lang K., Micura R. The preparation of site-specifically modified riboswitch domains as an example for enzymatic ligation of chemically synthesized RNA fragments. Nat. Protoc. 2008;3:1457–1466. doi: 10.1038/nprot.2008.135. [DOI] [PubMed] [Google Scholar]
- 138.Puttaraju M., Been M.D. Group I permuted intron-exon (PIE) sequences self-splice to produce circular exons. Nucleic Acids Res. 1992;20:5357–5364. doi: 10.1093/nar/20.20.5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.R. A. Wesselhoeft et al. RNA Circularization Diminishes Immunogenicity and Can Extend Translation Duration In Vivo. Mol Cell 74, 508-520 e504, doi:10.1016/j.molcel.2019.02.015 (2019). [DOI] [PMC free article] [PubMed]
- 140.Loan Young T., Chang Wang K., James Varley A., Li B. Clinical delivery of circular RNA: Lessons learned from RNA drug development. Advanced Drug Delivery Reviews. 2023;197 doi: 10.1016/j.addr.2023.114826. [DOI] [PubMed] [Google Scholar]
- 141.Lee K.H., Kim S., Lee S.-W. Pros and cons of in vitro methods for circular RNA Preparation. Int. J. Mol. Sci. 2022;23(21):13247. doi: 10.3390/ijms232113247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Garber K. Orna Therapeutics: circular logic. Nat. Biotechnol. 2022 doi: 10.1038/d41587-022-00005-1. [DOI] [PubMed] [Google Scholar]
- 143.Abe N., Matsumoto K., Nishihara M., Nakano Y., Shibata A., Maruyama H., Shuto S., Matsuda A., Yoshida M., Ito Y., Abe H. Rolling circle translation of circular RNA in Living Human Cells. Sci. Rep. 2015;5(1) doi: 10.1038/srep16435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chen R., Wang S.K., Belk J.A., Amaya L., Li Z., Cardenas A., Abe B.T., Chen C.-K., Wender P.A., Chang H.Y. Engineering circular RNA for enhanced protein production. Nat. Biotechnol. 2023;41(2):262–272. doi: 10.1038/s41587-022-01393-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li S., Wu H., Chen L.L. Screening circular RNAs with functional potential using the RfxCas13d/BSJ-gRNA system. Nat. Protoc. 2022;17:2085–2107. doi: 10.1038/s41596-022-00715-5. [DOI] [PubMed] [Google Scholar]
- 146.Nielsen A.F., Bindereif A., Bozzoni I., Hanan M., Hansen T.B., Irimia M., Kadener S., Kristensen L.S., Legnini I., Morlando M., Jarlstad Olesen M.T., Pasterkamp R.J., Preibisch S., Rajewsky N., Suenkel C., Kjems J. Best practice standards for circular RNA research. Nat. Methods. 2022;19(10):1208–1220. doi: 10.1038/s41592-022-01487-2. [DOI] [PMC free article] [PubMed] [Google Scholar]