


Abstract
Most small nuclear RNAs (snRNAs) are synthesized by RNA polymerase II, but U6 snRNA is synthesized by RNA polymerase III. In the fruit fly Drosophila melanogaster the RNA polymerase specificity of the snRNA genes is determined by a few nucleotide differences within the proximal sequence element (PSE), a conserved sequence located ∼40–65 bp upstream of the transcription start site. The PSE is essential for transcription of both RNA polymerase II-transcribed and RNA polymerase III-transcribed snRNA genes and is recognized in Drosophila by a multi-subunit protein factor termed DmPBP. Previous studies that employed site-specific protein–DNA photocrosslinking indicated that the conformation of the DNA–protein complex is different depending upon whether DmPBP is bound to a U1 or U6 PSE sequence. These conformational differences of the complex probably represent an early step in determining the selection of the correct RNA polymerase. We have now obtained evidence that DmPBP modestly bends the DNA upon interacting with the PSE and that the direction of DNA bending is similar for both the U1 and U6 PSEs. Under the assumption that DmPBP does not significantly twist the DNA, the direction of the bend in both cases is toward the face of the DNA helix contacted by the 45 kDa subunit of DmPBP. Together with data from partial proteolysis assays, these results indicate that the conformational differences in the complexes of DmPBP with the U1 and U6 PSEs more likely occur at the protein level rather than at the DNA level.
INTRODUCTION
Transcription of genes encoding the spliceosomal small nuclear RNAs (snRNAs) in higher eukaryotes is dependent upon a unique and essential proximal sequence element (PSE) at a conserved location ∼40–65 bp upstream of the transcription start site (1–9). The PSE is required for transcription of U1, U2, U4 and U5 snRNA genes by RNA polymerase II as well as U6 snRNA genes by RNA polymerase III (1,2,10–12). In vertebrates the promoters of snRNA genes transcribed by RNA polymerase II lack TATA boxes, whereas U6 snRNA gene promoters contain TATA boxes. In the presence of the upstream PSE, the TATA box acts as a dominant element for determining the RNA polymerase III specificity of vertebrate U6 gene promoters (2,13,14).
In other organisms, however, the RNA polymerase specificity of snRNA genes can be determined by different mechanisms. For example, plant snRNA genes transcribed by either RNA polymerase II or RNA polymerase III have TATA boxes, but the spacing between the PSE (specifically termed the USE in plants) and the TATA box determines RNA polymerase specificity (15–17). Significantly, in vertebrates, sea urchins and plants, experiments have indicated that the PSEs of U1 and U2 genes are functionally interchangeable with the PSEs of U6 genes (14,16–19).
Surprisingly, in the fruit fly Drosophila melanogaster, RNA polymerase specificity is determined by the actual sequence of the U1 or U6 PSE itself (20). Thus the U1 and U6 PSEs (termed more specifically PSEAs in Drosophila) are not interchangeable (20), even though the Drosophila U1 and U6 PSEAs have considerable sequence similarity that encompasses a 21 bp region. Indeed, the substitution of as few as 3 nt within the 21 bp region that constitutes the PSEAs of Drosophila U1 and U6 genes is sufficient to switch the specificity of these promoters from one RNA polymerase to the other (20).
In the human system, the PSE-binding protein has been named PBP, PTF or SNAPc (21–26). SNAPc consists of five subunits with molecular weights of approximately 19, 43, 45, 50 and 190 kDa (24,26–31), but little is known about the architectural arrangement of these subunits relative to the DNA.
As part of our studies in the Drosophila system, we recently employed a powerful site-specific protein–DNA photocrosslinking technique that identified three subunits (45, 49 and 95 kDa in size) of the D.melanogaster PSEA-binding protein (DmPBP) that are in close proximity to the DNA (32). The photocrosslinking data revealed the relative translational and rotational positions of the three protein subunits relative to the PSEA sequence and therefore relative to each other when bound to the DNA (32).
Importantly, the photocrosslinking pattern of the three DmPBP polypeptides to the DNA was different depending upon whether DmPBP was bound to a U1 or U6 PSEA sequence (32). This indicated that the proximity of the polypeptides to the DNA was altered depending upon the PSEA sequence bound to the protein. Together with the fact that the PSEA acts as the dominant element to determine RNA polymerase specificity in Drosophila, these studies indicated that conformational differences exist in the overall DNA–protein complex that are determined by whether the protein is interacting with a U1 or U6 PSEA.
To gain a better understanding of the nature of the conformational differences involved at U1 and U6 promoters, we have carried out DNA bending analysis and partial proteolysis assays to detect potential differences in DNA and/or protein conformation. Our results suggest that DmPBP bends DNA modestly and that the direction of the DNA bend is similar in the U1 and U6 complexes; the partial proteolysis assays, on the other hand, are consistent with a model for conformational differences at the protein level.
MATERIALS AND METHODS
Source of DmPBP
The D.melanogaster PSEA-binding protein (DmPBP) was partially purified from the soluble nuclear fraction prepared from 0–12 h Drosophila embryos as previously described (33). Briefly, fractionation involved chromatography steps on DEAE–cellulose, heparin–agarose, Affi-Gel Blue and Resource Q columns. The Resource Q fraction was used for all the experiments reported in this paper except for the minicircle binding assays. For those assays, the Resource Q fraction was further subjected to Sephacryl S-300 chromatography as previously described (33), then fractions containing DmPBP activity were pooled and applied to a 1 ml bed volume Ni-NTA–agarose (Qiagen) column in BCZ-100 buffer (20 mM HEPES–KOH, pH 7.9, 5 mM MgCl2, 0.01 mM ZnCl2, 0.2 mM EDTA, 20% v/v glycerol, 3 mM dithiothreitol, 0.5 mM PMSF, 100 mM KCl) containing 0.5 mM imidazole. The column was washed with BCZ-100 buffer containing 5 mM imidazole and DmPBP was eluted with BCZ-100 containing 20 mM imidazole. This fraction was used directly in the minicircle binding assays.
Circular permutation assays
DNA fragments 242 bp in length that contained either the U1 PSEA or the U6 PSEA at various positions relative to the fragment ends (Fig. 1A) were generated via PCR using the constructs U1-8-PB and U6-8-PB (20) as templates. These are synthetic constructs that contain a U1 PSEA or U6 PSEA inserted between the EcoRI and KpnI sites of pUC18, a PSEB (7) sequence 8 bp downstream of the PSEA between the KpnI and BamHI sites of pUC18 and 35 bp of additional synthetic sequence between the BamHI and SalI sites of pUC18 (20,34). The U1 and U6 fragments were identical to each other except at five nucleotide positions within the PSEAs (Fig. 1B). In these constructs the U1 and U6 PSEAs promoted transcription exclusively by RNA polymerase II or III, respectively, in a transcription assay in vitro (20). All PCR fragments used throughout this study were internally labeled by including [α-32P]dCTP and [α-32P]dTTP in the PCR reactions.
Figure 1.
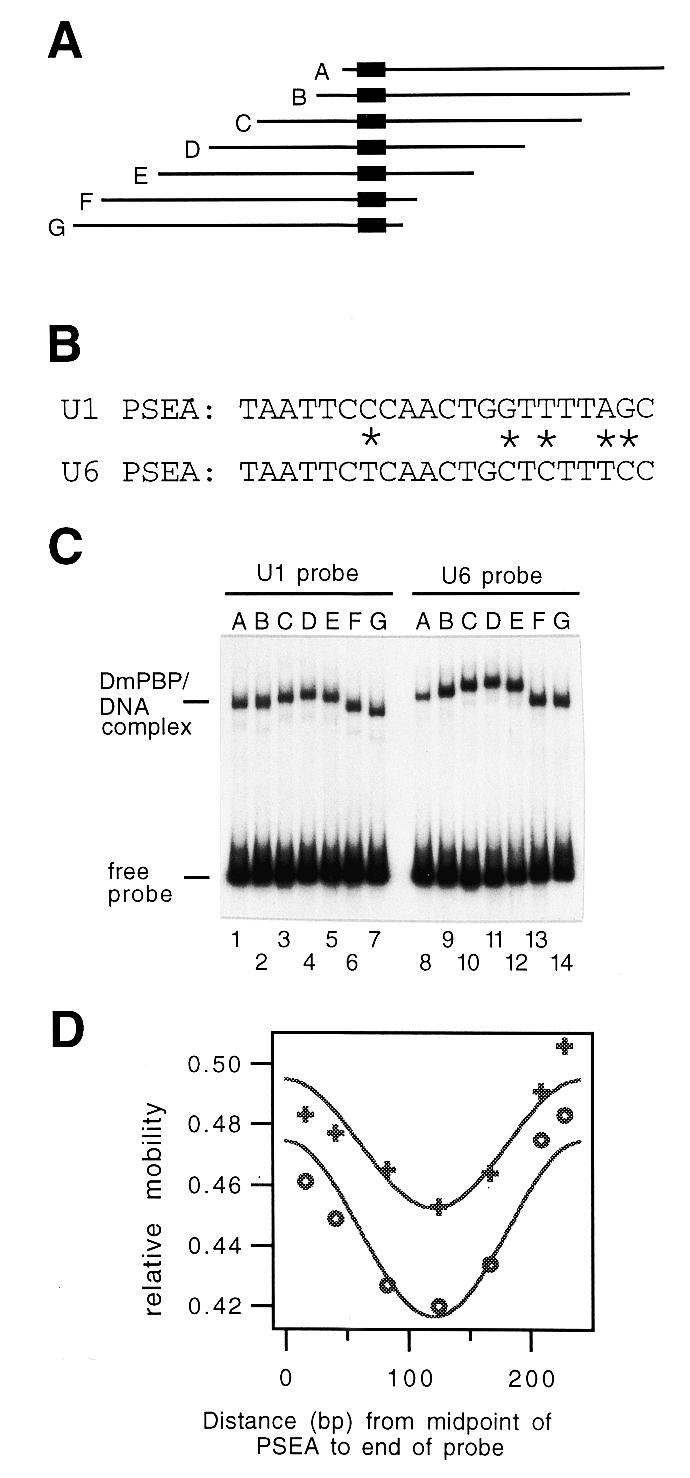
Circular permutation assay with DNA fragments that contain a Drosophila U1 or U6 PSEA. (A) Diagram of a set of seven DNA fragments that contain the U1 or U6 PSEA at various positions relative to the ends of the fragments. The location of the PSEA, which is the binding site for DmPBP, within each 242 bp fragment is indicated by the black rectangle. (B) Sequences of the Drosophila U1 and U6 PSEAs with the differences indicated by asterisks. (C) Autoradiogram following native gel electrophoresis of DmPBP–DNA complexes formed with the U1 (lanes 1–7) or the U6 (lanes 8–14) circularly permuted fragments diagrammed in (A). (D) Plot of the relative mobility of DmPBP–DNA complexes versus the position of the PSEA within the fragment. Plus symbols, fragments containing the U1 PSEA; open circles, fragments containing the U6 PSEA.
The radiolabeled PCR fragments were incubated with DmPBP (Resource Q fraction) at 25°C for 30 min and then separated by electrophoresis through a 4% (29:1 acrylamide:bisacrylamide) gel containing 25 mM Tris–borate, 0.56 mM EDTA. DNA bands were detected by autoradiography. The distance migrated by the protein-bound fragment was divided by the distance migrated by the unbound fragment. Averages were obtained from three experiments and the values were plotted versus the distance in base pairs from the center of the PSEA to the end of the probe. Curve fitting to the circular permutation function µ = µmax[(ACP/2)(cos{2π[(D – CD)/PCP]} – 1) + 1], where µ is the mobility of the complex, µmax is the theoretical maximum mobility, ACP is the circular permutation amplitude, D is the distance from the center of the PSEA to one end of the probe, CD is the center of distortion and PCP is the circular permutation period (35), was carried out using the program IGOR (WaveMetrics, Lake Oswego, OR). Values for ACP were determined from the fitted curve and used to calculate a nominal DNA bend angle from the relationship ACP = 1 – cos(α/2), where α represents the DNA bend angle (35).
PCR templates with an intrinsic DNA bend at varying positions downstream of the U1 or U6 PSEA
The U1-8-PB and U6-8-PB plasmid constructs described above were cut with BamHI and SalI to remove a 41 bp section. Then synthetic double-stranded oligodeoxyribonucleotides containing three phased A tracts with spacers of variable lengths were cloned between the BamHI and SalI sites. The sequences of the five non-template (relative to the PSEA) strand oligonucleotides used for cloning were as follows: d(GATCCTGCAAAACGGGCAAAAACGGGGCAAAAACGGG); d(GATCCTCGGCAAAAACGGGCAAAAACGGGCAAAAACGGG); d(GATCCTCGACGCAAAAACGGGCAAAAACGGGCAAAAACGGG); d(GATCCTCGACACGCAAAAACGGGCAAAAACGGGCAAAAACGGG); d(GATCCTCGACTGACGCAAAAACGGGCAAAAACGGGCAAAAACGGG). In these constructs the middle adenosine residue of the central A tract is separated by 54, 56, 58, 60 or 62 bp from the midpoint of the U1 or U6 PSEA.
The U1 gene PSEA present in these constructs contained a run of four T residues on the non-template strand (Fig. 1B) that would be expected to result in an additional intrinsic bend in the DNA. To minimize this effect, new constructs were prepared that had the sequence of the U1 PSEA changed from a T to a C at position 17. This particular change was chosen because the most common nucleotide in Drosophila U1, U2 and U4 PSEAs at position 17 is a C. The RNA polymerase II specificity of the U1 PSEA with this T→C substitution was confirmed by transcription in vitro (unpublished data). All of the U1 constructs used to generate PCR fragments for the minicircle binding and ligase-catalyzed circularization reactions contained this change in the U1 PSEA.
Minicircle binding assays
Internally labeled DNA fragments were generated by PCR from plasmid templates that contained the midpoint of the U1 or U6 PSEA separated by 54, 56, 58, 60 or 62 bp from the midpoint of an intrinsic bend. Six different primers were synthesized that contained ClaI restriction sites staggered at 2 bp intervals. These primers were used for PCR in appropriate pair-wise combinations so that after digestion of the PCR products with ClaI all DNA fragments were exactly 169 bp in length. By generating the substrates in this manner, torsional effects during ring closure from misaligned ends were avoided. Following gel purification of the ClaI-digested fragments, each was ligated overnight with T4 DNA ligase and the minicircles were purified from linear molecules by gel electrophoresis.
To examine the relative binding affinity of DmPBP for the various minicircles and corresponding linear DNA probes, electrophoretic mobility shift assays were performed under three different experimental conditions. In one instance, the minicircle and linear probes were incubated separately with DmPBP and subsequently loaded onto separate lanes of a gel. In the second instance, the minicircles were incubated with DmPBP followed 5 min later by the addition of the corresponding linear probe to each reaction. In the third case, the order of the addition of the probes was reversed. These order-of-addition experiments were performed to ensure that two conditions were fulfilled: (i) equilibrium was attained in the reactions; (ii) the concentration of free protein was identical in reactions with linear and circular probes (since the two probes were incubated together in the same reaction). After 30–35 min incubation, the samples were subjected to electrophoresis through a native gel. Bands corresponding to unbound and protein-bound minicircular and linear DNA fragments were quantified by phosphorimager analysis.
The relative affinity of DmPBP for the different minicircles relative to the linear DNA was estimated from the following relationship (36): Krel = Kcircle/Klinear = ([unbound linear][bound minicircle])/([bound linear][unbound minicircle]). Relative affinities were normalized to an average affinity of 1 and then values from three experiments were averaged and plotted versus the distance from the midpoint of the PSEA to the midpoint of the intrinsic bend. Curve fitting to the phasing function µ = µavg[(APH/2)(cos{2π[(S – ST)/PPH]}) + 1] (35) was then carried out by substituting relative affinity for relative mobility as the y-axis variable: Krel = Kavg[(APH/2)(cos{2π[(S – ST)/PPH]}) + 1], where Kavg = 1 after normalization, APH is the phasing amplitude, S is the distance from the center of the binding site to the center of the intrinsic bend, ST is the center-to-center distance when the protein-induced and intrinsic bends are in opposite directions and PPH is the phasing period. Curve fitting was then carried out to obtain the values of the constants APH, ST and PPH.
Ligase-catalyzed circularization assays
Linear DNA fragments that contained the U1 or U6 PSEA phased at variable positions relative to the A tracts were prepared exactly as described above for the minicircle binding assays. Each fragment was 169 bp in length with ClaI cohesive ends. In addition, similar fragments that contained a mutant PSEA sequence (CCTGATAGGTGACCAGGACTA) were generated. The linear fragments were incubated with DmPBP for 20 min at 25°C. Five units of T4 DNA ligase were added and incubation continued for another 30 min. The tubes were placed in a 65°C water bath for 15 min to inactivate the ligase. After reducing the temperature to 37°C, 30 U exonuclease III were added to each reaction and incubation was continued at 37°C for 45 min to digest the linear DNA. This was followed by the addition of SDS and proteinase K to final concentrations of 0.5% and 0.2 µg/µl, respectively, and incubation was continued at 37°C for 20 min. The samples were separated by electrophoresis through a 5% native polyacrylamide gel and bands corresponding to minicircles were quantified by phosphorimager analysis.
The relative efficiency of minicircle formation was calculated from the relative intensities of the individual bands. (The linear range of the experiment was confirmed by reactions carried out without exonuclease digestion, which indicated that <20% of the DNA was converted to minicircles during the allotted incubation period.) Relative efficiencies were normalized by setting the average efficiency of minicircle formation for each set of data to the value 1. Normalized data from three experiments were averaged and plotted versus the distance from the midpoint of the PSEA to the midpoint of the intrinsic bend. Curve fitting to the phasing function (35), employing relative efficiency of minicircle formation as the y-axis variable (rather than relative mobility), was carried out in a similar manner as described above for the minicircle binding experiments.
Partial proteolysis assays
Internally labeled PCR fragments that contained a U1 or U6 PSEA (but no A tract intrinsic bends) were incubated with a DmPBP Resource Q fraction as described above for the circular permutation assays. After 30 min incubation, various amounts of endoproteinase Lys-C, Glu-C, chymotrypsin or Asp-N (sequencing grade; Roche Molecular Biochemicals, Indianapolis, IN) were added as described in the legend to Figure 4 and incubation was continued for 10–30 min depending upon the proteinase. The samples were then immediately loaded onto a native polyacrylamide gel and electrophoresed as described above. After drying the gel, bands corresponding to free probe and to protein–DNA complexes were detected by autoradiography.
Figure 4.

Partial proteolysis assays of DmPBP bound to U1 or U6 PSEAs. Labeled DNA fragments that contained the U1 or U6 PSEAs (as indicated by a 1 or 6 above each individual lane) were incubated with DmPBP followed by the addition of increasing amounts of an endoproteinase to partially digest the DmPBP. The free and protein-bound fragments were then separated by native gel electrophoresis and the bands detected by autoradiography. (A) Lanes 3–8, 0.26, 1.3 or 6.4 µg/ml Lys-C; lanes 1, 2, 9 and 10, no proteinase. (B) Lanes 3–12, 0.037, 0.18, 0.91, 4.6 or 23 µg/ml Glu-C; lanes 1, 2, 13 and 14, no proteinase. (C) Lanes 3 and 5, 5.9 µg/ml Lys-C; lanes 4 and 6, 31 µg/ml Glu-C; lanes 1, 2, 7 and 8, no proteinase. (D) Lanes 3–12, 0.17, 0.34, 0.69, 1.4 or 2.8 µg/ml chymotrypsin; lanes 1 and 2, no proteinase. (E) Lanes 3–12, 0.10, 0.31, 0.92, 2.8 or 8.3 µg/ml Asp-N; lanes 1 and 2, no proteinase.
RESULTS
DNA–protein complexes assembled with DmPBP and U1 or U6 PSEAs have anomalous mobilities in a circular permutation assay
To obtain an initial indication of whether DNA is bent when complexed with DmPBP, internally labeled DNA fragments were generated that contained either a U1 or U6 PSEA at various locations relative to the ends of the fragments (Fig. 1A). All fragments were the same length and were prepared by PCR from ‘U1’ or ‘U6’ synthetic templates that were identical in sequence except at five nucleotides within the PSEA (differences indicated by asterisks in Fig. 1B).
These DNA probes were incubated with DmPBP and separated by electrophoresis through a native polyacrylamide gel. All 14 probes migrated with similar mobilities as free DNA (Fig. 1C). However, the shifted U1 and U6 DNA–protein complexes migrated significantly more slowly when the PSEA was located near the center of the fragment than when the PSEA was positioned near to either end of the fragment (Fig. 1C). These data are consistent with DNA bending upon interacting with DmPBP. It is also worth noting that the protein–DNA complexes formed with the U6 fragments exhibited somewhat slower mobilities overall than the corresponding complexes with DNA fragments that contained the U1 PSEA.
Data from three experiments were averaged and fitted to a circular permutation cosine function (35). Figure 1D shows a plot of the results. An apparent or nominal DNA bend angle was calculated from the relationship ACP = 1 – cos(α/2) (35), where ACP is the amplitude of the circular permutation function determined from the fitted curves (Fig. 1D). This formula predicts a nominal bend in the DNA of 48° for the DmPBP-bound U1 PSEA and 57° for the U6 PSEA. However, circular permutation analysis does not differentiate between induction of a directional bend in the DNA versus a point of flexure. More importantly, the protein itself (particularly if elongated in shape rather than globular in structure) can alter mobilities in circular permutation assays in ways that mimic the effect of bent DNA (37,38). Therefore, solution-based assays that involved minicircle binding and circularization were performed to more thoroughly assess the shape and conformation of the DNA when complexed with DmPBP.
Binding of pre-bent DNA to DmPBP
Minicircle binding has been used as a solution-based technique to examine protein-induced bending of DNA (36,38–40). The minicircle binding assay is based upon the principle that a DNA-bending protein will bind with higher affinity to DNA that is pre-bent in the direction of the protein-induced bend but will bind with lower affinity to DNA pre-bent in the opposite direction. To examine the behavior of DmPBP in a minicircle binding assay, two separate series of constructs were prepared in which the PSEA (U1 or U6) was spaced at varying distances from an intrinsic DNA bend provided by three phased A tracts (Fig. 2A). In these constructs, the midpoint of the (U1 or U6) PSEA varied from 54 to 62 bp from the midpoint of the central A tract. DNA fragments were amplified from these templates by PCR using combinations of primers that contained ClaI restriction sites at appropriately staggered positions such that each of the PCR products would have ClaI sites separated by exactly 169 bp (Fig. 2A). After digestion with ClaI and purification, the fragments were circularized by treatment with T4 DNA ligase. This produced 169 bp long minicircles that contained the PSEA pre-bent in various directions that ranged over nearly a turn of the helix.
Figure 2.

Binding of DmPBP to pre-bent (minicircle) DNA. (A) The linear fragments that were used to generate the 169 bp minicircles in which the U1 or U6 PSEA would be bent in various directions are diagrammed. The sequences separating the PSEA and phased A tracts in each fragment are shown explicitly, with the distance in base pairs from the midpoint of the PSEA to the midpoint of the A tracts given to the left. (B) Autoradiogram following native gel electrophoresis after incubation of DmPBP with linear fragments or the corresponding minicircular DNA that contained the U1 PSEA (lanes 1–10) or the U6 PSEA (lanes 11–20). bL, bound linear; fL, free linear; bMC, bound minicircle; fMC, free minicircle. (C) Phasing plot of normalized binding affinity versus the distance between the PSEA and the intrinsic bend. Plus symbols, fragments containing the U1 PSEA; open circles, fragments containing the U6 PSEA.
The minicircles, as well as the corresponding linear DNA fragments, were incubated with DmPBP and subjected to electrophoresis through a native polyacrylamide gel to separate protein-bound DNA from free DNA. Initial experiments were performed in which the minicircle and linear fragments were incubated separately with the DmPBP fraction. However, other experiments were performed in which the protein was first incubated with the minicircles followed by addition of the linear fragments or, conversely, in the opposite order of DNA addition (see Materials and Methods). These order-of-addition experiments were done to ensure that equilibrium was attained in the binding reactions. Furthermore, this method ensured that the concentration of free protein was identical in the reactions with linear and circular fragments since the binding reactions were carried out in the same tube. Each protocol yielded results that were essentially identical. However, for simplicity, only the results from the gels in which the incubations were carried out separately are shown in Figure 2B.
The linear U1 and U6 DNA fragments were bound by DmPBP with affinities that were unaffected by the phasing between the PSEA and the intrinsic bend (Fig. 2B, lanes 1–5 and 11–15). However, when minicircle DNA was used in the binding assay, the protein bound more tightly when the center-to-center distance between the PSEA and the intrinsic bend was 54 or 62 bp, as opposed to the intermediate separations of 56, 58 or 60 bp (Fig. 2B, lanes 6–10 and 16–20). This is most clearly evident in the fact that the bands that correspond to free probe had a notably diminished intensity in lanes 6, 10, 16 and 20 in comparison to the other lanes. Those lanes also contained an increased amount of DNA smearing between the bands corresponding to free and fully shifted probes which was not observed in the absence of DmPBP (not shown). It is possible that the protein–minicircle complexes have an unusual topology that contributes to their partial dissociation during passage through the gel matrix.
To estimate the relative binding affinities of DmPBP for the different pre-bent DNAs in comparison to the linear fragments, bands corresponding to bound and free circular as well as bound and free linear fragments were quantified by phosphorimager analysis. Relative binding affinities were calculated as described by Sitlani and Crothers (41; see Materials and Methods). The average of the relative affinities was normalized to a value of 1 and the corresponding relative affinities were plotted as a function of the separation between the PSEA and the intrinsic bend. The data points were then used to curve fit a cosine phasing function (35) as described in Materials and Methods.
Significantly, the curves for the DNAs that contained the U1 and U6 PSEAs were in phase with each other and essentially overlapped (Fig. 2C). Each was at a minimum when the center-to-center spacing between the PSEA and the intrinsic bend was 58 bp. Moreover, the phasing period (PPH) determined from both the U1 and U6 fitted curves was 10.4 bp, which is the value expected for two independent bends (i.e. the intrinsic and protein-induced bends) separated by B-form DNA. Finally, the amplitudes of the fitted curves are a reflection of the relative affinity of the protein for the PSEA when the DNA is pre-bent in the most favorable versus least favorable direction. The values determined from the curve fitting suggest that there is up to an ∼20-fold difference in affinity of DmPBP for either PSEA that is dependent upon the direction of DNA pre-bending.
Ligase-catalyzed circularization of U1 and U6 protein–DNA complexes
As a complementary and independent method to confirm the DNA-bending properties of DmPBP in solution, we next performed ligase-catalyzed circularization reactions (38,41–43). In these assays minicircles are generated most efficiently when a protein-induced bend and an intrinsic DNA bend cooperate with each other to bend the DNA in the same direction (producing a C-shaped ligation substrate), as opposed to bending the DNA in opposite directions (forming an S-shaped molecule). Linear DNA fragments (each 169 bp in length to avoid torsional effects from misaligned ends) were prepared that contained the U1 or U6 PSEA phased at 2 bp intervals relative to an intrinsic DNA bend (the same fragments as shown in Fig. 2A). Each fragment was incubated with an excess of DmPBP followed by the addition of T4 DNA ligase. The formation of minicircles in the reaction was monitored by autoradiography following exonuclease digestion and gel electrophoresis of the reaction products.
In the presence of DmPBP, DNA fragments that contained either the wild-type U1 or U6 PSEA were circularized most rapidly when the center-to-center separation of the PSEA from the intrinsic DNA bend was 54 or 62 bp, whereas the rate of minicircle appearance was depressed when the separation was 58 bp (Fig. 3A, lanes 1–10). In contrast, a fragment that contained a mutant PSEA to which DmPBP could not bind did not exhibit this pattern (Fig. 3A, lanes 11–15). Additional control reactions were carried out in the absence of DmPBP (Fig. 3B); under these circumstances the fragments that contained the wild-type U1 or U6 PSEA did not exhibit the decrease in minicircle formation at the 58 bp separation relative to the 54 and 62 bp separations (Fig. 3B). Together these results indicate that the reduction in minicircle formation at the 58 bp separation is not intrinsic to the DNA and requires the binding of DmPBP to a non-mutant PSEA.
Figure 3.
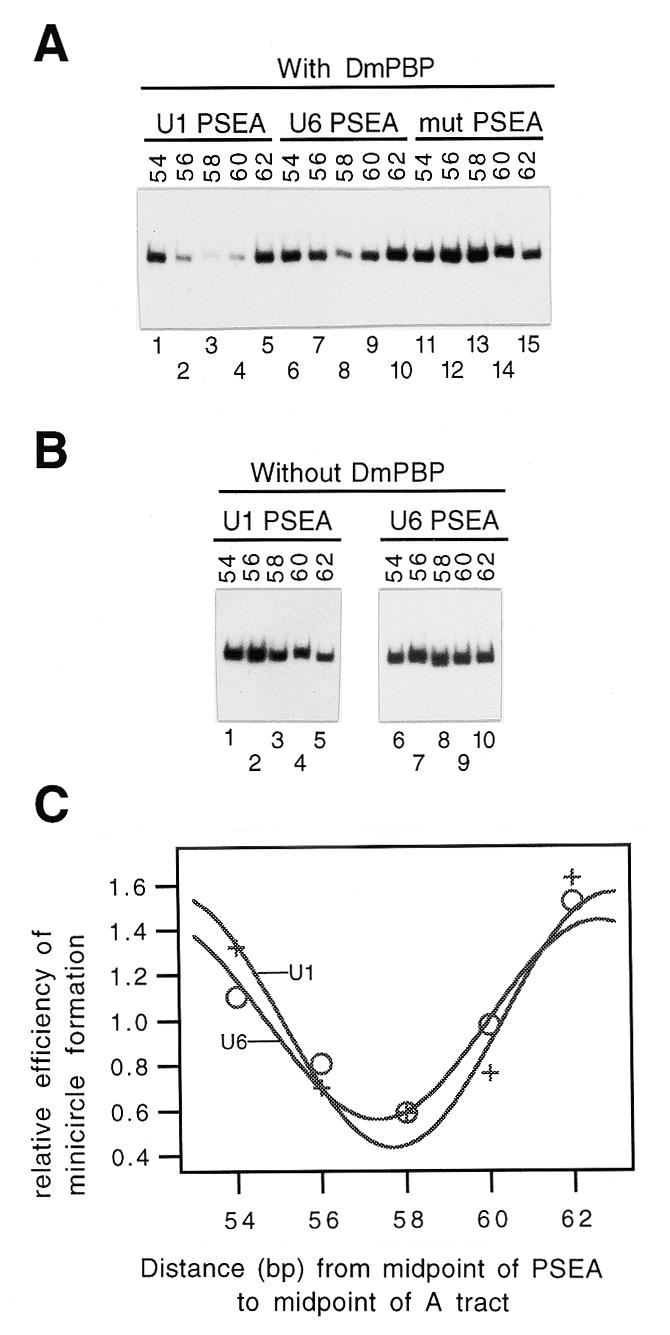
Ligase-catalyzed circularization assays. (A) Following the binding of DmPBP to the DNA fragments diagrammed in Figure 2A, DNA ligase was added and the appearance of circular DNA was monitored by gel electrophoresis following exonuclease digestion. Bands corresponding to 169 bp closed circular DNA are shown. Reactions were performed with DNA fragments that contained a U1 PSEA (lanes 1–5), a U6 PSEA (lanes 6–10) or a mutant PSEA to which DmPBP could not bind (lanes 11–15). (B) Ligation reactions carried out as in (A) but in the absence of the DmPBP fraction. (C) Phasing plot of the normalized efficiency of minicircle formation versus the distance between the PSEA and the intrinsic bend. Plus symbols, fragments containing the U1 PSEA; open circles, fragments containing the U6 PSEA.
The relative efficiencies of minicircle formation were quantified by phosphorimager analysis of the minicircle bands and the average of the efficiencies was normalized to a value of 1. The normalized efficiencies were averaged from three experiments and plotted versus the distance separating the midpoint of the PSEA from the midpoint of the intrinsic bend. A curve was then fitted to the data by applying the phasing function. The results (Fig. 3C) indicated that the curves for the U1 and U6 PSEAs were phased very similarly, with minimum circularization efficiency at a 57–58 bp spacing (ST) and phasing periods (PPH) of ∼10.5 bp. These results imply that DmPBP bends both PSEA sequences in essentially the same direction. Moreover, when ∼58 bp separate the midpoints of the PSEA and intrinsic DNA bend, the intrinsic bend and the protein-induced bend must be in opposite directions in three-dimensional space. Significantly, these results are entirely consistent with the results of the minicircle binding experiments (Fig. 2) and support the notion that DmPBP bends the U1 and U6 PSEAs in similar directions.
Differential sensitivity of DmPBP to proteases as a result of DmPBP interaction with a U1 or U6 PSEA
The minicircle binding and circularization assays suggested that the U1 and U6 PSEAs have a similar conformation when complexed with DmPBP. To detect possible differences in protein conformation, we next carried out experiments to determine whether the protein might be differentially susceptible to proteolysis when bound to a U1 or U6 PSEA (Fig. 4). Following incubation of DmPBP with DNA fragments that contained the U1 or U6 PSEA, the complexes were subjected to partial proteolysis with one of several different proteases prior to native gel electrophoresis. Figure 4A shows the effects of partial proteolysis of these complexes by the endoproteinase Lys-C. As previously noted, complexes of DmPBP with DNA fragments that contained the U6 PSEA migrated more slowly through native polyacrylamide gels than complexes with DNA fragments that contained the U1 PSEA (e.g. Fig. 4A, lanes 1, 2, 9 and 10). Digestion of these complexes with increasing amounts of Lys-C resulted in conversion of both the U1 and U6 complexes to forms that had identical mobilities but which ran slightly ahead of the native U1 complex (Fig. 4A, lanes 3–8).
A similar but distinct partial digestion pattern was observed with the endoproteinase Glu-C (Fig. 4B). The U1 and U6 complexes were converted to faster migrating products that had mobilities nearly identical to each other, but these partial digestion products migrated significantly ahead of the native U1 and U6 complexes (Fig. 4B, compare lanes 11–14). Figure 4C portrays a side-by-side comparison of the native U1 and U6 complexes in relation to those that resulted from the most stringent digestions with endoproteinase Lys-C or Glu-C. Although each enzyme individually digested the U1 and U6 complexes to products having similar mobilities, the products of the Lys-C and Glu-C digestions were clearly distinct.
Partial digestion with chymotrypsin initially converted the DmPBP–U6 DNA complex to a form that had a mobility similar to that of the native U1 complex (Fig. 4D, lanes 3–8). More complete chymotrypsin digestion resulted in the appearance of more rapidly migrating complexes in the case of both U1 and U6 (Fig. 4D, lanes 9–12).
A unique digestion pattern was obtained by treatment of the U1 and U6 DNA–DmPBP complexes with the endoproteinase Asp-N (Fig. 4E). Initial digestion of the U6 complexes resulted in the appearance of an intermediate band that ran with approximately the same mobility as the native U1 complexes (Fig. 4E, lanes 3–6). However, digestion with higher amounts of Asp-N eventually eliminated the signal arising from the U6 complexes, but left a portion of the U1 complexes undigested (Fig. 4E, lanes 9–12). Thus, the U6 complexes appeared to be more sensitive to digestion by Asp-N than the U1 complexes.
DISCUSSION
Previous studies from our laboratory indicated that the DNA–protein complexes formed between DmPBP and the U1 or U6 PSEAs on snRNA promoters have distinct functional and structural properties. First, the sequence of the PSEA itself was sufficient to functionally determine the RNA polymerase specificity of the Drosophila U1 and U6 promoters (20). Second, although identical subunits of DmPBP were photocrosslinked to the phosphodiester backbone of the U1 and U6 PSEAs, the crosslinking patterns of these three subunits exhibited significant differences depending upon which PSEA (U1 or U6) was involved in the interaction. At the extreme, two contrasting models could account for the differences observed in the PSEA-specific DNA–protein interactions. In one model, the conformation of the protein could remain constant but the route of the DNA and its contacts with the protein could vary, i.e. U1 and U6 DNA could be bent or twisted to different degrees or in different directions depending upon its differential interaction with the protein. At the other extreme, the DNA of the U1 and U6 PSEAs could follow identical paths, but the conformation of the protein could be altered as a result of the DNA sequence to which it binds. In this case the PSEA sequences would be acting as differential allosteric effectors of protein conformation. Models that combine either of these two extremes are equally plausible.
The experiments reported in this paper were undertaken to assess the relative contribution of protein or DNA conformational differences in the functionally distinct protein–DNA complexes formed between DmPBP and the U1 or U6 PSEAs. Results of circular permutation experiments were consistent with the possibility that DmPBP might be a DNA-bending protein. Minicircle binding and ligase-catalyzed circularization assays confirmed the DNA-bending properties of DmPBP. These two solution-based assays are believed to be more reliable than assays that rely on relative mobilities in polyacrylamide gels since the shape of the protein, gel conditions and other factors can significantly affect mobilities in gel-based assays (36–38,41). Due to the uncertainties and problems inherent in all types of DNA-bending assays, however, we believe that an accurate and reliable quantification of the DNA bend angle is not feasible under the current circumstances. Nonetheless, our results provide strong evidence that the U1 and U6 PSEAs are bent when complexed with DmPBP and that the direction of bending is similar in each case.
If we further assume that the bending of the DNA is planar and that DmPBP does not twist the DNA, the results of our assays make it is possible to specify the direction of the DmPBP-induced DNA bend relative to the PSEA sequence and relative to the DmPBP subunits that bind to it. The three phased A tracts together produce a bend that is equivalent to a net bend toward the minor groove at the central A of the middle A tract. The protein-induced bend and the intrinsic DNA bend are maximally out-of-phase with each other when the separation between their midpoints is ~58 bp (Figs 2 and 3). Based upon a theoretical and experimentally determined phasing period of 10.5 bp/turn of the DNA helix (Figs 2 and 3), the 58 bp separation constitutes 5.5 turns of the helix. Thus, for a bend in the PSEA to be in the opposite direction in space compared to that of the intrinsic DNA bend, a bend at position 11 in the PSEA would also have to be toward the minor groove. Thus, although we do not know the exact location of the bend within the PSEA (it may be distributed throughout the PSEA), the overall global direction of the protein-induced bend is equivalent to a bend toward the minor groove at the midpoint of the PSEA (position 11 in Fig. 1B).
Previous work identified the faces of the DNA helix contacted by each of the three DNA-binding subunits of DmPBP (32). By combining that knowledge with the results of the DNA-bending assays, the direction of the DNA bend relative to the rotational position of these subunits on the PSEA can be determined. The bend toward the minor groove at position 11 (or any equivalent directional bend, e.g. a bend toward the major groove at position 16) will result in a bend toward the face of the helix that is contacted by the 45 kDa subunit (32). This result is depicted schematically in Figure 5. As mentioned in the preceding paragraph, however, this interpretation is subject to the caveat that DmPBP does not significantly writhe or twist the DNA.
Figure 5.
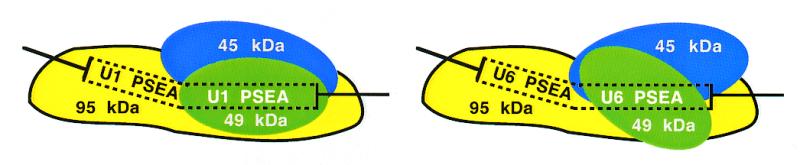
Model of the interaction of DmPBP with Drosophila U1 and U6 gene proximal sequence elements. According to the model, the protein adopts alternative conformations depending upon whether it is bound to a U1 or U6 PSEA sequence. In both cases the DNA is modestly bent upon interacting with DmPBP and the direction of the bend is similar for both the U1 and U6 PSEAs. The figure shows the direction of the DNA bend toward the face of the helix contacted by the 45 kDa subunit under the assumption that DmPBP does not significantly writhe or twist the DNA.
The fact that the U1 and U6 PSEAs behave very similarly in both minicircle binding and circularization experiments suggests that the global trajectory of the DNA as it enters and exits DmPBP is similar for DNA fragments that contain U1 and U6 PSEA sequences. These findings suggest that the predominant conformational differences in the DmPBP–DNA complexes are at the protein rather than the DNA level (Fig. 5). Data from the partial proteolysis assays are also consistent with this hypothesis while providing further evidence that the core structure of the U1 and U6 complexes are very similar (Fig. 4). Although the native U1 and U6 complexes migrated at different rates, partial digestion with Lys-C, Glu-C or chymotrypsin resulted in their reduction to complexes that had similar mobilities. Although differences in the local DNA structure could contribute to this phenomenon, we favor a model in which DmPBP exists in a more extended or strained conformation when bound to the U6 (versus the U1) PSEA and that after proteolytic clipping DmPBP adopts a U1-like conformation even on the U6 PSEA. A more extended or strained conformation of native DmPBP on the U6 PSEA is also consistent with the greater sensitivity of the U6 complex to digestion by endoproteinase Asp-N (Fig. 4E).
Our data do not definitively rule out the possibility that an additional factor in the DmPBP fraction can associate with the U6–DmPBP complex and that this factor is preferentially removed as a result of partial proteolysis. In our experiments, however, we believe that scenario is less likely since the difference in mobility of the U1 and U6 complexes is observed with all fractions of DmPBP obtained throughout the purification procedure (unpublished data). Moreover, recent photocrosslinking studies (32) provided no evidence for the presence of an additional factor in the U6 complexes, even with a less pure DmPBP fraction than that used in the current experiments. However, in a more complete transcription preinitiation complex our model hypothesizes that allosteric conformational differences in DmPBP induced by the PSEA are likely to play an important role in recruiting RNA polymerase II- or RNA polymerase III-specific factors to the U1 and U6 snRNA gene promoters, respectively.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Monica Neuberger and David Pullman for providing access to the computer program IGOR and for assistance with curve fitting. This work was supported by NSF grant MCB-9818000 to W.E.S., by the California Metabolic Research Foundation and by the San Diego State University Department of Chemistry, College of Sciences and Office of Faculty Affairs.
REFERENCES
- 1.Hernandez N. (1992) In McKnight,S.L. and Yamamoto,K.R. (eds), Transcriptional Regulation. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 281–313.
- 2.Lobo S.M. and Hernandez,N.T. (1994) In Conaway,R.C. and Conaway,J.W. (eds), Transcription: Mechanisms and Regulation. Raven Press, New York, NY, pp. 127–159.
- 3.Stefanovic B. and Marzluff,W.F. (1992) Mol. Cell. Biol., 12, 650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wendelburg B.J. and Marzluff,W.F. (1992) Nucleic Acids Res., 20, 3743–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J.-M., Parsons,R.A. and Marzluff,W.F. (1994) Mol. Cell. Biol., 14, 2191–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Das G., Henning,D. and Reddy,R. (1987) J. Biol. Chem., 262, 1187–1193. [PubMed] [Google Scholar]
- 7.Zamrod Z., Tyree,C.M., Song,Y. and Stumph,W.E. (1993) Mol. Cell. Biol., 13, 5918–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vankan P. and Filipowicz,W. (1989) EMBO J., 8, 3875–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waibel F. and Filipowicz,W. (1990) Nucleic Acids Res., 18, 3451–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dahlberg J.E. and Lund,E. (1988) In Birnstiel,M.L. (ed.), Structure and Function of Major and Minor Small Nuclear Ribonucleoprotein Particles. Springer Verlag, Heidelberg, Germany, pp. 38–70.
- 11.Parry H.D., Scherly,D. and Mattaj,I.W. (1989) Trends Biochem. Sci., 14, 15–19. [Google Scholar]
- 12.Dahlberg J.E. and Lund,E. (1991) Science, 254, 1462–1463. [DOI] [PubMed] [Google Scholar]
- 13.Mattaj I.W., Dathan,N.A., Parry,H.D., Carbon,P. and Krol,A. (1988) Cell, 55, 435–442. [DOI] [PubMed] [Google Scholar]
- 14.Lobo S.M. and Hernandez,N. (1989) Cell, 58, 55–67. [DOI] [PubMed] [Google Scholar]
- 15.Goodall G.J., Kiss,T. and Filipowicz,W. (1991) Oxf. Surv. Plant Mol. Cell Biol., 7, 255–296. [Google Scholar]
- 16.Waibel F. and Filipowicz,W. (1990) Nature, 346, 199–202. [DOI] [PubMed] [Google Scholar]
- 17.Kiss T., Marshallsay,C. and Filipowicz,W. (1991) Cell, 65, 517–526. [DOI] [PubMed] [Google Scholar]
- 18.Parry H.D., Tebb,G. and Mattaj,I.W. (1989) Nucleic Acids Res., 17, 3633–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J.-M., Haberman,R.P. and Marzluff,W.F. (1996) Mol. Cell. Biol., 16, 1275–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jensen R.C., Wang,Y., Hardin,S.B. and Stumph,W.E. (1998) Nucleic Acids Res., 26, 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waldschmidt R., Wanandi,I. and Seifart,K.H. (1991) EMBO J., 10, 2595–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wanandi I., Waldschmidt,R. and Seifart,K.H. (1993) J. Biol. Chem., 268, 6629–6640. [PubMed] [Google Scholar]
- 23.Murphy S., Yoon,J.-B., Gerster,T. and Roeder,R.G. (1992) Mol. Cell. Biol., 12, 3247–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoon J.-B., Murphy,S., Bai,L., Wang,Z. and Roeder,R.G. (1995) Mol. Cell. Biol., 15, 2019–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sadowski C.L., Henry,R.W., Lobo,S.M. and Hernandez,N. (1993) Genes Dev., 7, 1535–1548. [DOI] [PubMed] [Google Scholar]
- 26.Henry R.W., Sadowski,C.L., Kobayashi,R. and Hernandez,N. (1995) Nature, 374, 653–656. [DOI] [PubMed] [Google Scholar]
- 27.Yoon J.B. and Roeder,R.G. (1996) Mol. Cell. Biol., 16, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadowski C.L., Henry,R.W., Kobayashi,R. and Hernandez,N. (1996) Proc. Natl Acad. Sci. USA, 93, 4289–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bai L., Wang,Z.X., Yoon,J.B. and Roeder,R.G. (1996) Mol. Cell. Biol., 16, 5419–5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henry R.W., Ma,B.C., Sadowski,C.L., Kobayashi,R. and Hernandez,N. (1996) EMBO J., 15, 7129–7136. [PMC free article] [PubMed] [Google Scholar]
- 31.Wong M.W., Henry,R.W., Ma,B.C., Kobayashi,R., Klages,N., Matthias,P., Strubin,M. and Hernandez,N. (1998) Mol. Cell. Biol., 18, 368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y. and Stumph,W.E. (1998) Mol. Cell. Biol., 18, 1570–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Su Y., Song,Y., Wang,Y., Jessop,L., Zhan,L.C. and Stumph,W.E. (1997) Eur. J. Biochem., 248, 231–237. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y. and Stumph,W.E. (1995) Proc. Natl Acad. Sci. USA, 92, 8606–8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kerppola T.K. and Curran,T. (1993) Mol. Cell. Biol., 13, 5479–5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sitlani A. and Crothers,D.M. (1998) Proc. Natl Acad. Sci. USA, 95, 1404–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gartenberg M.R., Ampe,C., Steitz,T.A. and Crothers,D.M. (1990) Proc. Natl Acad. Sci. USA, 87, 6034–6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCormick R.J., Badalian,T. and Fisher,D.E. (1996) Proc. Natl Acad. Sci. USA, 93, 14434–14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parvin J.D., McCormick,R.J., Sharp,P.A. and Fisher,D.E. (1995) Nature, 373, 724–727. [DOI] [PubMed] [Google Scholar]
- 40.Kim J., de Haan,G., Nardulli,A.M. and Shapiro,D.J. (1997) Mol. Cell. Biol., 17, 3173–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sitlani A. and Crothers,D.M. (1996) Proc. Natl Acad. Sci. USA, 93, 3248–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor W.H. and Hagerman,P.J. (1990) J. Mol. Biol., 212, 363–378. [DOI] [PubMed] [Google Scholar]
- 43.Crothers D.M., Drak,J., Kahn,J.D. and Levene,S.D. (1992) Methods Enzymol., 212, 3–29. [DOI] [PubMed] [Google Scholar]