


Abstract
Decoding of the UGA selenocysteine codon for selenoprotein translation requires the SECIS element, a stem–loop motif in the 3′-UTR of the mRNA carrying short or large apical loops. In previous structural studies, we derived a secondary structure model for SECIS RNAs with short apical loops. Work from others proposed that intra-apical loop base pairing can occur in those SECIS that possess large apical loops, yielding form 2 SECIS versus the form 1 with short loops. In this work, SECIS elements arising from eight different selenoprotein mRNAs were assayed by enzymatic and/or chemical probing showing that seven can adopt form 2. Further, database searches led to the discovery in drosophila and zebrafish of SECIS elements in the selenophosphate synthetase 2, type 1 deiodinase and SelW mRNAs. Alignment of SECIS sequences not only highlighted the predominance of form 2 but also made it possible to classify the SECIS elements according to the type of selenoprotein mRNA they belong to. Interestingly, the alignment revealed that an unpaired adenine, previously thought to be invariant, is replaced by a guanine in four SECIS elements. Tested in vivo, neither the A to G nor the A to U changes at this position greatly affected the activity while the most detrimental effect was provided by a C. The putative contribution of the various SECIS motifs to function and ligand binding is discussed.
INTRODUCTION
Selenocysteine is the major biological form of selenium. Only bacteria, archaea and animals can co-translationally incorporate it into selenoproteins at specific positions. Such a translational mechanism requires the participation of a complex molecular machinery, in particular for designating an internal, in-frame UGA as the selenocysteine-encoding and not a stop codon (for reviews see 1–4). The mechanism leading to selenocysteine biosynthesis and incorporation into selenoproteins has been unravelled in Escherichia coli (3). The general translation elongation factor EF-Tu does not intervene in this process where it is replaced by the specialized translation factor SELB. Indeed, the SELB/GTP/Sec-tRNASec complex binds to a stem–loop structure adjacent to the UGA selenocysteine codon in the mRNA, forming a quaternary complex that directs the charged tRNASec to the A site of the ribosome. In eukaryotes, a hairpin structure called SECIS (SElenoCysteine Insertion Sequence) resides in the 3′-UTR of selenoprotein mRNAs. It is mandatory for recognition of UGA as a selenocysteine codon (1). Different SECIS-binding proteins have been described (5–11), but among these only SBP2 was shown to be effectively required for mammalian selenoprotein translation (11). Since SECIS-binding proteins are not the eukaryotic homologs of the bacterial SELB, it looks as if two (SBP2 and the eukaryotic SELB factor) or more proteins act in concert to accomplish this recoding event, in contrast to bacteria (8,11).
The spacing between the UGA codon and the SECIS element, that can reside in some instances as far away as 5 kb, raises several structural and functional issues regarding how the RNA–protein complex formed at the SECIS hairpin can fulfil its role at a distance. In an attempt to better understand the underlying mechanism, we previously undertook the experimental determination of the secondary structure of the SECIS RNA element. Structure probing experiments combined with sequence comparisons enabled us to propose a structural model consisting of a hairpin comprising the two helices I and II separated by an internal loop (12). Most predominantly, helix II contains a quartet of non-Watson–Crick base pairs constituting a motif essential to mediate selenoprotein translation (13). Using sequence comparisons and site-directed mutagenesis, other investigators have proposed a model similar to ours but with additional base pairing in the apical loop of some of the SECIS elements (14,15). Their findings led to the categorization of the SECIS elements into two forms according to the size of the apical loop: form 1 corresponds to the model described in Walczak et al. (12) and Martin et al. (14), and form 2, containing a larger number of nucleotides in the apical loop, enables intra-loop base pairing.
By virtue of a screen searching SECIS secondary structures in databases, we earlier identified the four novel selenoproteins SelN, SelX, SelZf1 and SelZf2 (16). Also discovered by this approach were the SelD and SelY SECIS elements in the human selenophosphate synthetase 2 and type 2 iodothyronine deiodinase mRNAs, respectively (17,18) that had not been reported in the literature at the time of our search. Here, our objective was to determine experimentally the form adopted by these SECIS elements, and also to examine the structures of a few others proposed in the literature to fold into form 2. Arising from this work, structure probing experiments as well as sequence comparisons with SECIS elements in the database support the notion that the form 2 model is representative of a substantial number of SECIS elements. Additionally, both the expansion of the repertoire of SECIS sequences and functional assays led us to establish that replacement of an adenine, considered so far as invariant, is not as detrimental as anticipated.
MATERIALS AND METHODS
EMBL/GenBank/DDBJ data
The accession number for Drosophila melanogaster selenophosphate synthetase 2 homology is AF279253.
Constructs and site-directed mutagenesis
Vectors for in vitro transcription of the various SECIS elements were made as follows. The BclI–KpnI digested fragments of the pSelN, pSPS2 (also called pSelD), pSelX, pSelY, pSelZ and pSel15 plasmids (16) were introduced into BclI–KpnI digested pT7BcK (12) vectors, giving rise to pT7BcKSelN, pT7BcKSelD, pT7BcKSelX, pT7BcKSelY, pT7BcKSelZ and pT7BcKSel15, respectively.
Xenopus laevis type 3 iodothyronine deiodinase (XlDI3) was obtained by PCR of genomic DNA. Primers were designed to amplify the sequence spanning positions 1274–1380 (19). The 5′ and 3′ primers incorporated BamHI+T7 promoter and EcoRI sites, respectively. An EcoRI–BamHI digest containing the SECIS sequence was inserted into the EcoRI–BamHI cleaved pUC119 plasmid, generating pSEX5.
The rat PHGPx SECIS element was constructed by hybridizing the overlapping oligodeoxynucleotides 5′-GATCACCTTCCACCCCG-3′, 5′-GCACTCATGACGGTCTGCCTGAA-3′, 5′-AACCAGCCCGCTGGTGGGGCAGTC-3′, 5′-CGAGGACCTGGCGTGCATCC-3′, 5′-CCGCCGGAGGAAGGGGTAC-3′, 5′-CCCTTCCTCCGGCGGGGATGCACG-3′, 5′-CCAGGTCCTCGGGACTGCCC-3′, 5′-CACCAGCGGGCTGGTTTTCAGGCAGAC-3′ and 5′-CGTCATGAGTGCCGGGGTGGAAGGT-3′. The resulting fragment (positions 52–147; 20), containing BclI and KpnI sites at the 5′ and 3′ ends, respectively, was cloned into the pT7BcK vector to produce pT7BcKPHGPx.
To linearize DNAs for further transcription by T7 RNA polymerase and to allow reverse transcription of the RNAs, an XhoI site was introduced by site-directed mutagenesis into pT7BcK, 3′ to the universal primer hybridization sequence, yielding pT7BcKX. BclI–KpnI digests of pT7BcKSelX, pT7BcKSelY, pT7BcKSelD and pT7BcKPHGPx were ligated to BclI–KpnI digested pT7BcKX vectors. The BamHI–EcoRI fragment of pSEX5 was inserted into the BamHI–EcoRI cleaved pT7BcKX vector.
An EST (accession no. AA541242) containing the partial sequence of the drosophila cDNA, putatively encoding the selenocysteine-containing selenophosphate synthetase 2 (SelD), was obtained by querying the EST database with TBLASTN at NCBI using the human selenophosphate synthetase 2 amino acid sequence (17). The EST was purchased at Genome Systems (St Louis, MO) and sequenced to identify the SECIS element (accession no. AF279253).
Mutants carrying G, C or T at position 1017 in the SECIS element of the rat glutathione peroxidase were obtained by hybridization of the appropriate oligodeoxynucleotides, following the procedure fully described in Walczak et al. (13). The assembled DNA fragments, containing BclI and KpnI sites at the 5′ and 3′ ends, respectively, were ligated to pRGPx-BcK and pT7BcK vectors according to Walczak et al. (13).
In vitro transcription
Vectors were linearized by EcoRI or XhoI prior to in vitro transcription. Ten micrograms of linearized DNA was incubated for 2 h in 250 µl of a buffer containing 40 mM Tris–HCl pH 8.0, 22 mM MgCl2, 1 mM spermidine, 0.01% Triton X100, 5 mM DTE, 24 U RNasin, 4 mM of each NTP, 15 µl of T7 RNA polymerase. To produce 5′ ApG-ending RNAs for 5′-labeling, 5 mM ApG was added and the GTP concentration decreased to 1 mM. After 1 h incubation at 37°C, 1 mM GTP was added and the mixture incubated for a further 1 h. The RNAs were purified on 10% denaturing polyacrylamide gels and electroeluted.
RNA structure probing
Enzymatic cleavage. 5′ ApG RNAs were 5′ end-labeled with [γ-32P]ATP. Digestions were performed for 5–15 min at 20°C in a buffer containing 20 mM Tris–HCl pH 7.5, 5 mM MgCl2, 100 mM KCl, with either 2 × 10–3 U/µg RNase V1, 5 × 10–3 U/µg RNase T2 or 10–3 U/µg RNase T1 (12). Samples were fractionated on 10% sequencing gels.
Alkaline ladders were performed by incubating 5′ end-labeled RNAs in 50 mM sodium carbonate pH 8.9 for 5 min at 90°C. RNAs were precipitated with 600 µl of 2% LiClO4 in acetone.
For RNase T1 ladders, 105 c.p.m. of 5′ end-labeled RNAs were digested with 5 × 10–4 U/µg RNase T1 for 10 min at 55°C in 25 mM sodium citrate pH 5.5, 1 mM EDTA, 8 M urea, 0.025% bromophenol blue, 0.025% xylene cyanol (12).
Chemical probing. Cleavage of 5′ end-labeled RNAs by Pb++ ions occurred in 25 mM HEPES–NaOH pH 7, 10 mM Mg acetate, 50 mM KCl containing 25 × 104 c.p.m. RNA and 8 µg carrier tRNA. Samples were preincubated for 2 min before adding lead acetate to the final concentrations indicated in Figure 2.
Figure 2.



Enzymatic and Pb(II) probing of the SECIS RNAs shown in Figure 1. (A) hSelD; (B) hSelN; (C) hSelX; (D) hSelY; (E) hSelZ; (F) mSel15; (G) rPHGPx; (H) XlDI3. The RNAs were digested with RNases T1, T2 and V1 for 5 and 10 min. (I) hSelX SECIS was cleaved at 20°C with 4, 7.5 and 10 mM Pb(II) acetate for 2 and 5 min (lanes 2–7, respectively). (J) rPHGPx SECIS was cleaved at 37°C for 2 min with 1.25 (lane 2), 2.5 (lane 3) and 5 mM (lane 4) Pb(II) acetate. Lanes marked C are control reactions; L, alkaline ladder; T1, RNase T1 ladder. The numbering corresponds to that in Figure 1.
Chemical reactions with dimethylsulfate (DMS) and carbodiimide (CMCT) were performed using the standard procedures described in Krol and Carbon (21) and the legend to Figure 3. Detection of the modified bases was accomplished by extension of the 5′ end-labeled 17mer universal primer. The positions of modifications were mapped by dideoxysequencing with reverse transcriptase. The reverse transcripts were separated on 10% sequencing gels.
Figure 3.

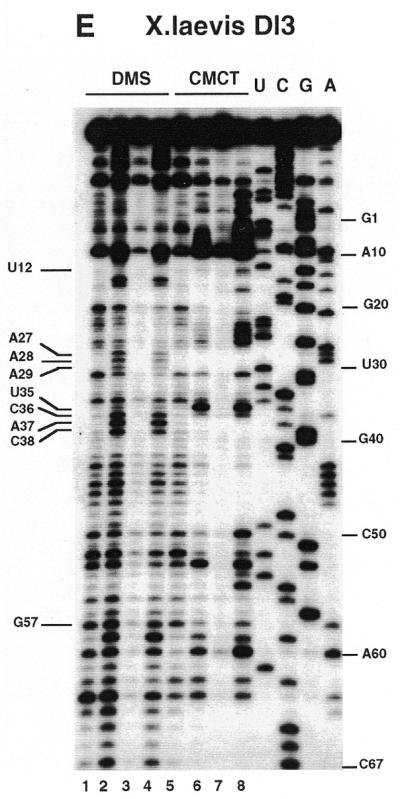
Chemical probing with DMS and CMCT. (A) hSelD; (B) hSelX; (C) hSelY; (D) rPHGPx. A short migration is also displayed. (E) XlDI3. RNAs were treated for 5 min at 20°C with 0.5 µl DMS under native (lanes 2) or semi-denaturing conditions (lanes 4), or with 10 mg/ml CMCT for 10 min at 25°C, under native (lanes 6) or semi-denaturing (lanes 8) conditions. Lanes 1, 3, 5 and 7 are controls. A, G, C, U are sequencing lanes. Numbering is from Figure 1.
Transfection of COS-7 cells and glutathione peroxidase assays
COS-7 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 0.1 mg/ml gentamycin according to standard cell culture procedures. Transient transfections were carried out as described (13,16) with 5 µg of test DNA, 4 µg of tRNASec expression vector and 1 µg of plasmid CMV-LacZ as the transfection standard. Sodium selenite (10 nM) was added to the culture medium. Prior to glutathione peroxidase activity measurements, β-galactosidase activities were assayed with one-tenth volume of crude cell extract to normalize the results (13,16). Assays were done in triplicate.
RESULTS
Structure probing of form 1 and form 2 SECIS RNAs
We carried out the secondary structure determination of eight SECIS RNAs contained in the mRNAs of the human SelN, SelX, SelZ (SelZf1 and SelZf2 mRNAs sharing the same SelZ SECIS element), SelD and SelY (16), mouse Sel15 kDa (22), rat phospholipid hydroperoxide glutathione peroxidase (20) and XlDI3 (19) selenoproteins. In the course of this study, they will be termed hSelN, hSelX, hSelZ, hSelD, hSelY, mSel15, rPHGPx and XlDI3, respectively. Figure 1 (left hairpins except hSelN) shows the predicted secondary structure models. Seven hairpins have the potential to form the short helix 3 giving rise to the SECIS form 2 displayed on the right. In hSelN, helix 3 could form through complementarity between A26UC and G32AU in the apical loop. However, this would imply participation of A26, the 3′ purine in the invariant AAR triplet found unpaired in all SECIS RNAs described so far (12,15). To test the models, the RNA transcripts were submitted to enzymatic probing with RNase T1 (cleaves 3′ to Gs, mostly in single-stranded regions under the experimental conditions), RNase T2 (specific for single strands without pronounced base specificity) and RNase V1 (cleaves in helices and stacked regions), shown in Figure 2A–H. In some instances, lead acetate-induced cleavage was employed to sense single-stranded RNA regions. Representative gels for hSelX and rPHGPx are shown in Figure 2I–J, respectively. Where necessary, chemical probing with DMS (reacts at the N1-A and N3-C positions) and CMCT (reacts at the N1-G and N3-U) was undertaken in an attempt to resolve ambiguities. In such experiments, protection of the chemical groups of the bases against chemical modification is indicative of their participation in base pairing. The reactions were conducted under native or semi-denaturing conditions to provide information about the stability of the base pairs that might be disrupted in the absence of Mg++ and K+ ions (Fig. 3A–E). A compilation of the probing data for each SECIS RNA is represented in Figure 1, arising from the gels in Figures 2 and 3 and data not shown. Interpretation of the results generated by combination of the various probes led to the following conclusions. The eight SECIS RNAs can adopt the overall 2D structure model proposed for the SECIS element, with helices 1 and 2 separated by the internal loop 1 of variable size (12). Because helix 1 varies in length, only the top five base pairs were depicted in Figure 1. Helix 2 falls within a range of 13–15 bp, consistent with the consensus SECIS secondary structure model and other data (12,15 and our unpublished data). Only the mSel15 and XlDI3 helices II are interrupted by the A51 and U54 bulges, respectively. Similar to the human and rat type 1 deiodinases, and rat glutathione peroxidases SECIS (12), the existence of the quartet of non-Watson–Crick base pairs is supported by the weak RNase T1 susceptibilities of the GpA bond in the GA tandem and the lack of strong Pb++ cleavages (Fig. 2A–J). Formation of the quartet is further substantiated by the protection under native conditions of N3-U10, N1-G11, N1-G53 and N1-G55 in hSelD (Fig. 3A), N3-U10, N1-G11, N3-U13, N1-G52, N1-G53 and N3-U55 in hSelY (Fig. 3C), and N3-U12 and N1-G57 in XlDI3 (Fig. 3E). In hSelN, the apical loop is readily cleaved by RNases T1 and T2 (Fig. 2B), thus excluding base pairing of A26UC with G32AU. The hSelN SECIS element therefore adopts the form 1 structure with a 14–15 bp helix 2 and a 10–12 nt long apical loop depending on whether the closing G.U base pair does form. For the hSelY, hSelZ, mSel15, rPHGPx and XlDI3 SECIS, analysis of the RNases and Pb++ cleavages enabled us to propose the formation of the short helix 3 (Fig. 1). Indeed, the five RNAs show high susceptibility towards RNase T2 (and T1) and Pb++ in the purine stretch of internal loop 2 and in the apical loop (Fig. 2D–H, J). The occurrence of helix 3 is further supported by the reactivities under native conditions of N1-A25, N1-A26, N1-A27, N1-A33, N3-U34, N3-C35, N1-A41 in hSelY, of N1-G24, N1-A25, N1-A26, N1-A27, N1-A28 and N1-G36 in rPHGPx, and of N1-A27, N1-A28, N1-A29, N3-U35, N3-C36, N1-A37, N3-C38, N1-A43, N1-A44 in XlDI3 (Fig. 3C–E).
Figure 1.


Experimentally supported secondary structure models for the SECIS RNAs of the human SelN, SelD, SelX, SelY, SelZ, mouse Sel15, rat PHGPx and XlDI3. The structures were manually folded and tested by enzymatic and/or chemical probing. Potential base pairings in the apical loops are drawn. Sequences were taken from the references indicated in the text. Enzymatic and Pb(II) cleavages are indicated by arrowheads: red, RNase T1; green, RNase T2; blue, RNase V1; black, Pb(II). The sizes of the arrowheads are roughly proportional to the intensity of cleavage. Circles indicate bases that are reactive towards DMS or CMCT under native (orange) or semi-denaturing (purple) conditions. Moderate reactivities are highlighted in yellow (native) or light purple (semi-denaturing). The form 2 SECIS models arising from the probing experiments are displayed on the right for each SECIS hairpin. For hSelD and hSelX, the equilibrium arrows schematize the occurrence of the transient form 1 and form 2 hairpins. The non-Watson–Crick quartet is boxed in red. Owing to the variable lengths of helix 1 in the various SECIS, the numbering starts at the fifth base pair below internal loop 1 in each SECIS element.
Additional information could be gained for the rPHGPx helix 1. Figure 3D shows that N1-A6, N3-C7, N1-G64, N3-U65, N1-G66, N1-A68 and N3-U69 are reactive, whereas N1-G4 and N3-C5 are protected under native conditions. These reactivities argue more in favor of the base pairing between CCGGC (positions 1–5) and GCCGG (positions 74–78), than between GCAC (positions 4–7) and GUGC (positions 64–67), as was proposed earlier (20). The base pairing is further supported by the compensatory base pair change C2-G77 to U-A in the porcine PHGPx SECIS (Table 1).
Table 1. Structure-based sequence alignment of SECIS RNA showing conservation of the characteristic sequence and structural features.

Twenty six sequences were aligned to maximize structural homologies. SECIS elements were classified according to form 1 (12) or form 2 (15). Sequence and structural features are highlighted: the conserved A/G purine in internal loop 1 (orange), the base pairing partners of the non-Watson–Crick quartet (grey), the A stretch in the apical loop of form 1 or in the internal loop 2 of form 2 (red), the apical loop (light grey), and the base pairing partners of helix 3 (yellow). SECIS sequences were: hSelN, human SelN (16); O.nDI1a and b, O.niloticus type 1 deiodinase first SECIS and second SECIS (accession no. Y11109); zGPx, zebrafish glutathione peroxidase (accession no. AW232474); cDI1, chicken type 1 deiodinase (accession no. Y11110); hSelD, human selenophosphate synthetase 2 (16); dSelD, drosophila selenophosphate synthetase 2 (accession no. AF270253); mSelD, mouse selenophosphate synthetase 2 (16); hSelX, human SelX (16); hSelT, human SelT (26); hSelZ, human SelZ (16); bTrxR2, bovine mitochondrial thioredoxin reductase (accession no. AB022283); hTR3, human thioredoxin reductase (accession no. AF171054); mTrxR1, mouse cytoplasmic thioredoxin reductase (accession no. AB027565); C.eTrxR, C.elegans thioredoxin reductase (24); zSelW, zebrafish SelW (accession no. AW128765); m and hSel15, mouse and human Sel15 (22); hSelY, human SelY (16); mDI2, mouse type 2 deiodinase (accession no. AF096875); cDI2, chicken type 2 deiodinase (accession no. AF125575); X.lDI3, X.laevis type 3 deiodinase (19); cDI3, chicken type 3 deiodinase (accession no. Y11273); O.n DI3, O.niloticus type 3 deiodinase (accession no. Y11111); rPHGPx, rat phospholipid hydroperoxide glutathione peroxidase (20); and pPHGPx, porcine PHGPx (23). The SECIS RNAs with an asterisk had their structures experimentally determined in this work.
Interpretation of the data is less straightforward for the hSelD and hSelX SECIS. In hSelD, the apical loop is fairly cleaved by RNase T2 between C24 and A28, C32 and A37. In addition, the G38-G39-A40 bonds are readily hydrolysed by RNase T1 (Fig. 2A). Chemical probing indicated reactivities of N3-U25, N1-A26 to A28, N3-C29, N1-A33, N3-U34, N3-U35, N1-A36 and N1-A37, but protection of N1-G38 and N1-G39 under native conditions (Fig. 3A). However, a pause of reverse transcriptase compromised the assignment of the status of C30-C32, the putative base pairing partners of G38 and G39. From the low yield of RNase T2 cleavage between C29-C32, the protection of N1-G38 and N1-G39 towards CMCT and the RNase T1 cleavage after these positions, one can hypothesize that helix 3 effectively forms but is rather unstable. In hSelX, N1-A25 to A27, N3-U31, N3-U32, N3-C33, N3-C34, N1-A35 and N1-A40 are reactive under native conditions (Fig. 3B). RNase T2 and Pb++ cleavages are observed between C24 and C28, and C30 and A35, RNase V1 cuts moderately the C29-C30 bond but RNase T1 cleavages are weak after G36 and G37 (Fig. 2C). Altogether, the data for hSelD and hSelX point to a dynamic structure in equilibrium between an open form of the apical loop and the short helix 3, under our experimental conditions.
In conclusion, hSelY, hSelZ, mSel15, rPHGPx, XlDI3 fold into form 2, whereas hSelN adopts form 1. In the absence of clear-cut data, we propose that the hSelD and hSelX SECIS secondary structures transiently fold into the form 2 model.
Database search and structure-based sequence alignment of SECIS elements
Next, we wished to identify and select sequences in the 3′-UTR that can fold into SECIS elements that have not been reported in a structural alignment yet. To this end, we searched cDNAs or ESTs putatively encoding homologs to previously reported selenoproteins in various organisms. Nucleotide databases were queried with TBLASTN at NCBI, using the amino acid sequences of the human cellular glutathione peroxidase, human selenophosphate synthetase 2, rat SelW, human thioredoxin reductases, and human type 1, 2 and 3 iodothyronine deiodinases. Except those already indexed as SECIS elements in complete cDNA sequences (Table 1), two zebrafish ESTs were obtained (GenBank accession nos AW232474 and AW128765) that putatively encode the GPx and SelW homologs, respectively. A SECIS element could be identified in each of the EST sequences by detecting the invariant RUGA-(13–15 nt)-AAR-(23–26 nt)-GA sequence motif, a characteristic structural feature of the SECIS element (12,24). These sequences were manually examined for their ability to adopt either form 1 or form 2. Also, a drosophila cDNA (GenBank accession no. AF279253) was identified as the putative human selenophosphate synthetase 2 homolog. It differs from the previously reported putative drosophila selenophosphate synthetase gene (25) in that it contains an in-frame UGA codon and a SECIS element.
Twenty six SECIS are listed in Table 1. They include those found in the search, others (15,16,24,26) aligned for comparison, and SECIS RNAs whose structure was probed in this work. The majority can fold into form 2 and a few of them only adopt form 1, corroborating an earlier observation (15). The alignment and the probing data showed that the length of helix 3 is variable, ranging from 2 (hSelD and mTrxR1) to 7 bp in the chicken type 3 deiodinase. The 3 bp helix 3 in the drosophila and mouse SelD is supported by the compensatory base pair change U-A to G-C. No significant sequence conservation could be inferred in the short apical loop of form 2. Interestingly, the form 2 SECIS RNAs expose the invariant adenine stretch in the internal loop 2. This loop was shown to be very accessible to RNases and chemicals in this work. It is exclusively composed of purines in type 2 deiodinases, X.laevis, chicken and Oreochromis niloticus type 3 deiodinases, TrxR2 thioredoxin reductases and hSelZ.
An adenine is found 5′ to the non-Watson–Crick quartet. It has been tacitly considered as invariant because the sample of the examined SECIS elements was too limited to allow the discovery of variants. However, a G was recently discovered in the SECIS element of the Caenorhabditis elegans thioredoxin reductase (24). Table 1 shows that this also happens in the chicken and O.niloticus type 3 deiodinases and in the second SECIS element of the O.niloticus type 1 deiodinase (O.n.DI1b).
The GPx SECIS function can tolerate A to G, C or U changes 5′ to the non-Watson–Crick quartet
The finding that a G can occur in a few SECIS at the position believed to be occupied by an invariant A 5′ to the UGA sequence of the quartet prompted us to examine whether substituting G and also C or U for A1017 in the SECIS element of the cellular GPx mRNA (12) would alter its function. GPx SECIS DNA variants containing either G, C or T at position 1017 were prepared and inserted into the GPx cDNA to replace the residing SECIS element. After transfection of COS-7 cells with the GPx cDNAs, the glutathione peroxidase activity, acting as the reporter since selenocysteine is part of the active site of the enzyme, was measured in cell extracts with the assay described in Walczak et al. (13). Figure 4 indicates that the 1017 position is rather tolerant to mutation. Indeed, only the substitution of C for A led to a substantial drop to 34% residual GPx activity. The effects of the A to G or A to U changes were only moderate with 70 and 78% residual activity, respectively.
Figure 4.

Activities of the A1017G, A1017C and A1017U GPx SECIS mutants in transfected COS-7 cells, determined with the glutathione peroxidase activity assay. The average values above the bars, subtracted from the endogenous GPx level, are given with respect to the wt GPx activity taken as 100%.
In conclusion, the mutational analysis revealed that a G can substitute for A without significantly affecting SECIS function, in accordance with the alignment shown in Table 1. This was also the case for a U, even though no U was found in SECIS sequences so far. Replacement of A by a C, however, was more deleterious but did not abrogate SECIS activity.
DISCUSSION
In this work, we have experimentally analyzed the secondary structures of eight different SECIS elements. We proposed in an earlier work secondary structure models for the mammalian type 1 iodothyronine deiodinase and cellular glutathione peroxidase SECIS RNAs (12). The motivation for investigating the structure of other SECIS RNAs arose from the finding by site-directed mutagenesis that certain SECIS elements with large apical loops could adopt the form 2 structure with intra-apical loop base pairing (15). Using an ensemble of enzymatic and chemical probes, we could show that among the RNAs analyzed, the human SelY and SelZ, rat PHGPx, X.laevis type 3 deiodinase and mouse Sel15 SECIS elements adopt the form 2. The stability of the short helix 3 is apparently too low in the human SelD and SelX to enable detection of form 2, exclusively. Rather, there was likely an equilibrium between forms 1 and 2 under the experimental conditions employed and it may be that it is shifted in vivo toward one form or the other by the presence of SECIS-binding protein(s). Only one SECIS, human SelN, could adopt form 1. A structure-based sequence alignment with SECIS sequences other than those used previously (12,15) clearly argues in favor of form 2 being predominant. Besides, information emanating from Table 1 and Walczak et al. (12) and Grundner-Culeman et al. (15) indicates that SECIS elements can be categorized according to the type of selenoprotein mRNA they belong to. For example, type 1 deiodinases and cellular GPx fall into form 1 whereas type 2 and 3 deiodinases and thioredoxin reductases belong to form 2. Two other interesting findings could be derived from the data that led to Table 1: (i) the existence of a selenocysteine-containing, in addition to the arginine-containing (25), selenophosphate synthetase (SPS2) in drosophila, like in mammals (17) and a few eubacteria and archaea (3); (ii) the adenosine 5′ to the non-Watson–Crick quartet is not invariant but rather semi-conserved since a guanine was discovered in four SECIS elements.
Another interesting example is provided by the O.niloticus type 1 deiodinase. Apart from the SelP mRNA that contains several selenocysteine codons (4), this is the only reported case of a selenoprotein mRNA containing two SECIS elements. There is no evidence yet for the functionality and need for two SECIS to read a single selenocysteine codon in the O.niloticus type 1 deiodinase. However, the compensatory base changes observed in helices 1 of the two SECIS (U-A, G-C and U-A in the first SECIS to G-C, U-A and C-G in the second, respectively), the A to G change below the quartet, the several pyrimidine or purine transitions in the loops and the C.A to A.A change at the top base pair of the quartet, argue in favour of the maintenance of two functional SECIS structures by selective pressure.
The probing data revealed the strong accessibility of the conserved adenine stretch in internal loop 2. Recent data concluded that the SECIS-binding protein SBP2 does not require this region for binding (11). The internal loop 2 and the exposed adenines herein could serve as a signal for the interaction with the specialized translation factor SelB or be part of a more complex, long-range RNA–RNA interaction with upstream regions of the mRNA or the ribosome.
According to the compilation in this work and in Walczak et al. (12) and Grundner-Culeman et al. (15), it appears that form 2 accounts for 66% of the known SECIS elements. The need for the dichotomy in favor of form 2 is unclear at the present time, but several possibilities can be envisaged. Form 1 and form 2 SECIS perform slightly different functions, or form 1 could just represent the relics of evolution, form 2 being fixed because of evolutionary advantages. Among the advantages, one could invoke the binding of different proteins, but SBP2 has been shown to bind to both types of SECIS RNAs (10). Another advantage could be correlated to the stretch of conserved adenines at the apex of the SECIS stem–loop, that occurs in a slightly different conformation in the two forms. The As are accessible in form 1 within the apical loop and well exposed and bulging out in the internal loop 2 of form 2. We hypothesize that this duality can provide a differential affinity towards a protein or RNA ligand perhaps leading to the observed distinct efficiencies of form 1 and form 2 SECIS elements in UGA readthrough (27).
The recent characterization of SBP2 and development of an in vitro assay for selenoprotein translation (11) will certainly help define the contribution of the various SECIS motifs to SBP2 binding and selenoprotein translation.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to N. Hubert for the rat PHGPx construct, J. Fletcher for reading the manuscript and C. Loegler for excellent technical assistance. This work was supported by grants from the Ligue Nationale Contre le Cancer (Comité du Haut-Rhin) and the Centre Volvic pour la Recherche sur les Oligo-Eléments. D.F. was awarded a fellowship from the Ministère de l’Education Nationale, de la Recherche et de la Technologie. R.W. was a recipient of a predoctoral fellowship of the Centre Volvic pour la Recherche sur les Oligo-Eléments.
REFERENCES
- 1.Low S.C. and Berry,M. (1996) Trends Biochem. Sci., 21, 203–208. [PubMed] [Google Scholar]
- 2.Hubert N., Walczak,R., Sturchler,C., Myslinski,E., Schuster,C., Westhof,E., Carbon,P. and Krol,A. (1996) Biochimie, 78, 590–596. [DOI] [PubMed] [Google Scholar]
- 3.Hüttenhofer A. and Böck,A. (1998) In Simons,R.W. and Grunberg-Manago,M. (eds), RNA Structure and Function. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 603–639.
- 4.Burk R.F. and Hill,K.E. (1999) Bioessays, 21, 231–237. [DOI] [PubMed] [Google Scholar]
- 5.Shen Q., McQuilkin,P.A. and Newburger,P.E. (1995) J. Biol. Chem., 270, 30448–30452. [DOI] [PubMed] [Google Scholar]
- 6.Hubert N., Walczak,R., Carbon,P. and Krol,A. (1996) Nucleic Acids Res., 24, 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen Q., Wu,R., Leonard,J.L. and Newburger,P.E. (1998) J. Biol. Chem., 273, 5443–5446. [DOI] [PubMed] [Google Scholar]
- 8.Fujiwara T., Busch,K., Gross,H.J. and Mizutani,T. (1999) Biochimie, 81, 213–218. [DOI] [PubMed] [Google Scholar]
- 9.Fagegaltier D., Hubert,N., Carbon,P. and Krol,A. (2000) Biochimie, 82, 117–122. [DOI] [PubMed] [Google Scholar]
- 10.Copeland P.R. and Driscoll,D.M. (1999) J. Biol. Chem., 274, 25447–25454. [DOI] [PubMed] [Google Scholar]
- 11.Copeland P.R., Fletcher,J.E., Carlson,B.A., Hatfield,D.L. and Driscoll,D.M. (2000) EMBO J., 19, 306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walczak R., Westhof,E., Carbon,P. and Krol,A. (1996) RNA, 2, 367–379. [PMC free article] [PubMed] [Google Scholar]
- 13.Walczak R., Carbon,P. and Krol,A. (1998) RNA, 4, 74–84. [PMC free article] [PubMed] [Google Scholar]
- 14.Martin G.W., Harney,J.W. and Berry,M.J. (1998) RNA, 4, 65–73. [PMC free article] [PubMed] [Google Scholar]
- 15.Grundner-Culeman E., Martin,G.W., Harney,J.W. and Berry,M.J. (1999) RNA, 5, 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lescure A., Gautheret,D., Carbon,P. and Krol,A. (1999) J. Biol. Chem., 274, 38147–38154. [DOI] [PubMed] [Google Scholar]
- 17.Guimaraes M.J., Peterson,D., Vicari,A., Cocks,B.G., Copeland,N.G., Gilbert,D.J., Jenkins,N.A., Ferrick,D.A., Kastelein,R.A., Bazan,J.F. and Zlotnik,A. (1996) Proc. Natl Acad. Sci. USA, 93, 15086–15091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buettner C., Harney,J.W. and Larsen,P.R. (1998) J. Biol. Chem., 273, 33374–33378. [DOI] [PubMed] [Google Scholar]
- 19.St.Germain D.L., Schwartzman,R.A., Croteau,W., Kanamori,A., Wang,Z., Brown,D.D. and Galton,V.A. (1994) Proc. Natl Acad. Sci. USA, 91, 11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lesoon A., Mehta,A., Singh,R., Chisolm,G.M. and Driscoll,D.M. (1997) Mol. Cell. Biol., 17, 1977–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krol A. and Carbon,P. (1989) In Dahlberg,J.E. and Abelson,J.N. (eds), Methods in Enzymology, Volume 180. Academic Press Inc., San Diego, CA, pp. 212–227.
- 22.Gladyshev V.N., Jeang,K.T., Wootton,J.C. and Hatfield,D.L. (1998) J. Biol. Chem., 273, 8910–8915. [DOI] [PubMed] [Google Scholar]
- 23.Brigelius-Flohé R., Aumann,K.-D., Blöcker,H., Gross,G., Kiess,M., Klöppel,K.-D., Maiorino,M., Roveri,A., Schuckelt,R., Ursini,F., Wingender,E. and Flohé,L. (1994) J. Biol. Chem., 269, 7342–7348. [PubMed] [Google Scholar]
- 24.Buettner C., Harney,J.W. and Berry,M.J. (1999) J. Biol. Chem., 274, 21598–21602. [DOI] [PubMed] [Google Scholar]
- 25.Persson B.C., Böck,A., Jäckle,H. and Vorbrüggen,G. (1997) J. Mol. Biol., 274, 174–180. [DOI] [PubMed] [Google Scholar]
- 26.Kryukov G.V., Kryukov,V.M. and Gladyshev,V.N. (1999) J. Biol. Chem., 274, 33888–33897. [DOI] [PubMed] [Google Scholar]
- 27.Kollmus H., Flohé,L. and McCarthy,J.E.G. (1996) Nucleic Acids Res., 24, 1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]