


Abstract
LR1 is a B cell-specific, sequence-specific duplex DNA binding activity which is induced in B cells carrying out class switch recombination. Here we identify several properties of LR1 which enable it to function in transcriptional regulation. We show that LR1 contributes to transcriptional activation by the Eµ immunoglobulin heavy chain intron enhancer by binding to a site within the enhancer core. We further show that LR1 bends DNA upon binding. In addition, we show that LR1 is itself a bona fide transcriptional activator, as multimerized LR1 sites produce an element which can enhance transcription from a minimal promoter. In order for class switch recombination to occur, an activating signal must be transmitted via the Eµ core, and both S regions targeted for recombination must be actively transcribed. The properties of LR1 that we have identified suggest distinct potential functions of LR1 duplex DNA binding activity in class switch recombination. First, LR1 may contribute to recombinational activation by the Eµ core. Second, there are multiple potential LR1 duplex binding sites in each of the G-rich switch regions, and LR1 bound at contiguous sites may enhance recombination by stimulating transcription of the S regions.
INTRODUCTION
Immunoglobulin heavy chain class switch recombination is a regulated recombination event which depends on both transcriptional and recombinational activation at the heavy chain locus (1). Following antigen stimulation, switch recombination occurs to join an expressed heavy chain variable region to a new heavy chain constant region, deleting the DNA between (Fig. 1). Class switch recombination involves repetitive, G-rich regions of DNA, called switch (S) regions, which are 2–10 kb in length and are located in the intron upstream of each constant region that participates in class switch recombination. Switch recombination is not sequence-specific but region-specific. Examination of sequences of hundreds of switch recombination junctions has shown that both donor and acceptor end points are heterogeneous within the S regions and that junctions are heterogeneous in sequence (reviewed in 2).
Figure 1.

Switch recombination alters the immunoglobulin heavy chain locus. (A) The murine heavy chain locus is diagrammed in the top line, which shows the rearranged variable region [V(D)J], the intron enhancer (Eµ), the switch (S) and constant (C) regions. Recombination from µ (IgM) to γ1 (IgG1) expression will result in joining of the Sµ and Sγ1 switch regions at sites that are heterogeneous in sequence and in position, as diagrammed below. (B) The LR1 site in the core Eµ enhancer. The region of the murine heavy chain intron enhancer (Eµ) which contains the binding sites for LR1, E4 and OCTA (underlined) is shown. (C) CAT expression driven from Eµ reporter constructs carrying wild-type or mutant LR1 sites. The graph shows levels of transcription activation as a percentage of total activation by pIgE-P-CAT. Mock, no DNA control.
Switch recombination is regulated by the Eµ enhancer, which is located in the intron between the JH regions and Sµ–Cµ (Fig. 1) (3–5). The region within Eµ that is both necessary and sufficient for efficient switch recombination has been mapped to a 220 bp HinfI fragment, referred to as the enhancer core. The Eµ core is critical for transcriptional regulation and V(D)J joining as well as class switch recombination (5,6). The Eµ core contains sites for half a dozen or more sequence-specific DNA binding proteins (reviewed in 7). Switch recombination also requires that both S regions targeted for recombination must be actively and simultaneously transcribed (reviewed in 1). Transcription of the S regions is induced by extracellular signals mediated by cytokines and lymphokines, which are produced by activated T cells and bind to receptors on the B cell surface. Binding triggers a series of intracellular events, culminating in induction of transcription at each of the S regions targeted for recombination.
One B cell-specific factor with a binding site in both the Eµ core enhancer and the S regions is LR1. LR1 is a sequence-specific duplex DNA binding activity present only in activated primary B cells and pre-B and B cell lines (8,9). In these lines, the presence of LR1 duplex DNA binding activity correlates with the ability of a cell to support recombination of transfected extrachromosomal switch substrates (10,11). LR1 has been shown to regulate transcription at sites in the c-myc (12) and the Epstein–Barr virus (EBV) EBNA-1 gene (13) promoters. LR1 also binds duplex DNA sites in the immunoglobulin S regions, although its function at these sites is not clearly understood (8,9). LR1 has an unusual composition for a sequence-specific duplex DNA binding protein: it is a heterodimer of the ubiquitous protein, nucleolin, and a specific isoform of hnRNP D (14,15). These two subunits enable LR1 to interact tightly and specifically with G-G paired DNA, a property which may contibute to the interactions between G-rich S regions that are essential to switch recombination (16–18).
The affinity of LR1 for duplex DNA is very high: the KD for binding to one duplex site in the Sγ1 switch region is 1.8 nM (14). In contrast, many mammalian duplex DNA binding factors have affinities in the 20–50 nM range (19 and references therein). This high binding affinity suggests that duplex DNA binding is an important property of LR1. We have therefore examined activities of LR1 that depend on interaction with the DNA duplex and that could contribute to possible function(s) of LR1 in regulation or activation of switch recombination. We report that LR1 can regulate transcription from a site in the core region of Eµ, that LR1 can bend DNA, and that LR1 is itself a bona fide transcriptional activator. In addition to further defining the properties of LR1 in transcriptional regulation, these observations suggest several distinct potential functions for LR1 duplex DNA binding activity in activation of switch recombination: LR1 bound at the Eµ core may regulate recombination; LR1 may enhance recombinational activation by altering DNA structure; and LR1 bound at sites in the S regions may stimulate S region transcription that is essential to recombination.
MATERIALS AND METHODS
Plasmids and constructs
The pIgE-P-CAT reporter construct carries the 682 bp XbaI–EcoRI fragment containing the wild-type murine Eµ enhancer (20) and the VH186.2 heavy chain gene promoter from the B1-48 hybridoma (21). To create pIgE-P-CAT, a XbaI–HindIII fragment carrying EµPH was excised from the Bluescribe vector (pBP-IgEP; 22) and inserted into the corresponding site in the backbone of the pSV2CAT reporter (23), created by excision of the SV40 early enhancer–promoter by AccI and HindIII digestion and conversion of the AccI site to a XbaI site. To generate pIgE29-P-CAT, the Eµ LR1 binding site just upstream of the E4 binding site (see Fig. 1) was mutated by amplifying Eµ in the pBP-IgEP template with mutagenic PCR primers in combination with Bluescribe vector primers T3 and T7 (Stratagene, La Jolla, CA). The mutagenic primers, GATCCCCGAAAGTCCAGATCTAGCAAAACACCACCA and GATCTGGTGTGTGTTTTGCTAGATCTGGACTTTCGGG, were designed to alter a critical portion of the LR1 binding site; in doing so, a DdeI restriction site is replaced by a BglII site (underlined). Following BglII digestion, PCR products were ligated, digested with XbaI and HindIII and reinserted into pBluescribe. The mutation was confirmed initially by diagnostic BglII digestion, and then by sequencing the entire PCR-amplified region. The mutated enhancer region was then used to replace the wild-type sequence in the CAT reporter. To create pIgE33-P-CAT, a similar strategy was followed, except that amplification was carried out with the mutagenic primers GGACAGATCTTTTGGTGGG and CTGAAGATCTCACCACCTGG in combination with vector primers; BglII digestion and religation resulted in deletion of the LR1 site. Multimeric LR1 DNA binding site constructs, p(Sγ1)6-P-CAT and p(G14A)6-P-CAT, were cloned by ligating duplex oligonucleotides carrying BamHI/BglII ends, digesting with BamHI and BglII to destroy all but head-to-tail ligation products, then isolating hexamers on low melting point agarose and cloning them into the BamHI site of pBS/KS+ to create pBS(Sγ1)6 and pBS(G14A)6. The multimers in these plasmids correspond to an LR1 site in the murine Sγ1 region (Sγ1, GATCCTCCTGGGTCAAGGCTGAATAGACGCAGGGGA and GATCTCCCCTGCGTCTATTCAGCCTTGACCCAGGAG) or to a single base G→A mutation in that binding site (G14A, GATCCTCCTGGGTCAAGACTGAATAGACGCAGGGGA and GATCTCCCCTGCGTCTATTCAGTCTTGACCCAGGAG). The resulting plasmids were sequenced and inserts excised with XbaI and KpnI and inserted into a derivative of pIgE-P-CAT from which the enhancer had been excised and replaced with a XbaI–SalI–KpnI polylinker.
Transfections and CAT assays
Conditions for DEAE–dextran transfection were as previously described (24). Graphs presented reflect the average of two transient transfections of PD31 cells with independent DNA preparations and with all data points represented in triplicate.
Protein purification
LR1 was purified 12 000-fold from murine pre-B cells (15). All purified fusion proteins chromatographed as single species on SDS–PAGE. Concentrations of proteins were determined by Bradford microassay (Bio-Rad, Hercules, CA).
DNA mobility shift analysis
Most binding assays used a synthetic duplex deoxyoligonucleotide carrying the LR1 site from the murine Sγ1 switch region (top strand, GATCCTCCTGGGTCAAGGCTGAATAGACGCAGGGGA; bottom strand, GATCTCCCCTGCGTCTATTCAGCCTTGACCCAGGAG; 8). For DNA bending assays, the LR1 Sγ1 site was cloned into the filled SalI site of pBEND2 (25) to create pBEND2(Sγ1). For bending assays, pBEND2(Sγ1) was digested with MluI, NheI, XhoI, PvuII, NruI or XbaI combined with HindIII, to release 164 bp fragments with the LR1 Sγ1 site at permuted positions. Fragments were treated with calf intestinal phosphatase (Boehringer, Indianapolis, IN), end-labeled with [γ-32P]ATP and T4 polynucleotide kinase and gel purified prior to use in bending assays. To create randomly mutated binding sites, the top strand of the LR1 Sγ1 site was synthesized under mutagenic conditions (26) and cloned into the BamHI site of pUC19. DNAs were released by BamHI digestion and labeled by filling in with Klenow fragment for binding assays. Binding to duplex DNA was carried out in 15 µl reactions containing 20 mM HEPES, pH 7.5, 150 mM KCl, 1 mM DTT, 0.1% NP-40, 2.5% glycerol, 2% polyvinylalcohol, 100 µg/ml bovine serum albumin, 4 fmol 32P-labeled duplex DNA (0.1 ng), 10-fold mass excess poly(dI–dC) (1 ng) and 0.2 pmol purified LR1 for 15 min at room temperature. Assays with antibodies used 2 µg rabbit polyclonal anti-nucleolin antibody, purified by HiTrap protein A–Sepharose chromatography (Pharmacia, Piscataway, NJ) and concentrated in a Centricon concentrator (Amicon, Beverly, MA) (14). Quantification of band intensity was carried out using a Bio-Rad phosphorimager (Bio-Rad).
RESULTS
Regulation of the immunoglobulin heavy chain enhancer by LR1
In vitro DNA binding analysis showed that LR1 binds to a site in the immunoglobulin heavy chain intron enhancer (Eµ) (Fig. 1A) (8). This site is just upstream of the E4 and OCTA binding regions (Fig. 1B), within the core region of the Eµ enhancer shown to be necessary and sufficient for switch recombination (5). To assess the contribution of LR1 to Eµ enhancer regulation, Eµ enhancer mutants were generated in which the LR1 binding site was mutated to a BglII restriction site (pIgE29-P-CAT) or deleted (pIgE33-P-CAT). The effect of mutation was assayed by comparing transcriptional activation from a CAT reporter following transfection of PD31 pre-B cells. This cell line was chosen because it had previously been shown to contain LR1 DNA binding activity and to support recombination of extrachromosomal switch substrates (10). As shown in Figure 1C, mutation of the Eµ LR1 binding site reduced reporter gene expression to 50% of wild-type levels; this is comparable to the level of effect observed upon mutation of other binding sites in Eµ (27). Deletion reduced transcriptional activity to 30% of wild-type levels, just slightly above the background evident in mock-transfected cells. These data indicate that LR1 may contribute to regulation of Eµ in vivo.
LR1 bends DNA
In principle, the contribution of LR1 to enhancer function could reflect either the ability of LR1 itself to activate polymerase or the ability of LR1 to alter enhancer architecture, for example by bending the DNA so that transcriptional activators bound to the promoter could interact more effectively with the RNA polymerase complex.
To test the possibility that LR1 alters enhancer architecture, we asked if LR1 can bend duplex DNA. DNA bending can be readily assayed by comparing electrophoretic mobilities of protein bound to sites at permuted positions in fragments of uniform length (25,28,29). While this assay may be affected by protein conformation (30,31), it can nonetheless provide a useful picture of protein–DNA interactions (32,33; for a review see 34). We assayed DNA bending by LR1 by performing gel mobility shift assays of protein binding to labeled fragments, 164 bp in length, carrying an LR1 site at permuted positions (Fig. 2, top). The LR1 site in these constructs was the first LR1 site identified in the Sγ1 switch region (8) and is therefore referred to as the Sγ1 site. This assay showed that relative retardation reflected the position of the LR1 binding site within the fragment (Fig. 2, bottom left). Relative retardation of the fragments was identical in gels run at 4 or 25°C (data not shown), ruling out protein-independent alterations in DNA structure as the source of altered mobility. The degree of bending was determined by comparing the relative gel mobility of the LR1–DNA complexes (35,36) and estimated to be 54°.
Figure 2.
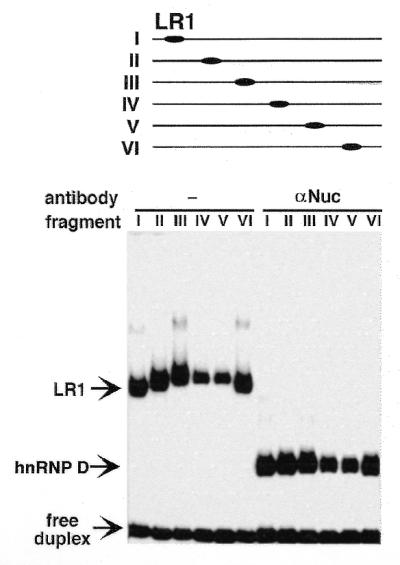
LR1 bends DNA. (Top) Gel mobility shift assay of LR1 binding to 164 bp fragments of pBEND2(Sγ1) with the LR1 site at permuted positions, as indicated above; this corresponds to digestion with: I, MluI; II, NheI; III, XhoI; IV, PvuII; V, NruI; VI, XbaI + HindIII. (Bottom) Binding was carried out in the absence (left) and presence (right) of anti-nucleolin antibodies. Arrows indicate free duplex DNA and the complexes bound by LR1 (nucleolin/hnRNP D) and hnRNP D.
Both nucleolin and hnRNP D contribute to DNA bending by the LR1 heterodimer
Treatment of the LR1–duplex DNA complex with anti-nucleolin antibodies will remove nucleolin from the DNA binding complex, leaving hnRNP D in contact with the DNA (14,15). To determine if the apparent bending was due to interaction of nucleolin or hnRNP D with DNA, relative mobilities were assayed in reactions treated with anti-nucleolin antibodies. Following antibody treatment, the fragments carrying permutated sites did not migrate with identical mobility (Fig. 2, bottom right), although the differences in relative retardation of the hnRNP D–DNA complexes was significantly less than that of the LR1–DNA complexes. The degree of bending by hnRNP D, determined as above, was estimated to be 26°. We conclude that both nucleolin and hnRNP D contribute to the apparent DNA bending by LR1.
The hnRNP D subunit of the LR1 heterodimer contacts the minor groove
Previous experiments had suggested that at least one critical contact for LR1 is in the minor groove of duplex DNA (14,15). As the antibiotic distamycin A fits snugly within the narrow minor groove of B-form DNA, distamycin sensitivity provides a convenient test of minor groove interactions (37–40). We therefore assayed the interaction between purified LR1 and the Sγ1 duplex site in concentrations of distamycin A up to 30 µM. The LR1–DNA interaction proved to be very sensitive to the presence of distamycin A. As shown in Figure 3A, concentrations of distamycin A >10 µM completely inhibited LR1 binding to duplex DNA. The distamycin A concentration at which binding was reduced to 50% of untreated levels (the IC50) was 3 µM (Fig. 3C). This is comparable to the IC50 at which distamycin A inhibits the binding of other proteins to the minor groove of B-form DNA (41,42). Inclusion of anti-nucleolin antibodies in the binding reactions did not alter distamycin sensitivity (Fig. 3B). It is therefore the hnRNP D subunit of the heterodimer that makes intimate contact with the minor groove.
Figure 3.

Distamycin A inhibits LR1 duplex DNA binding. Binding to the 32P-labeled duplex LR1 Sγ1 site was carried out in the presence of 0–30 µM distamycin A, as indicated. (A) Binding by purified LR1. (B) Binding in the presence of anti-nucleolin antibody, which removes nucleolin from the LR1 heterodimer to leave hnRNP D in contact with the DNA. (C) The fraction of DNA bound at each concentration, relative to the untreated sample, is plotted with respect to distamycin A concentration. Proteins assayed were purified LR1 (filled boxes) and hnRNP D (filled diamonds).
Tandemly repeated LR1 sites activate transcription
To ask if LR1 can function as a transcriptional activator, we assayed reporter gene expression from plasmids carrying a hexameric repeat of an LR1 site first identified (8) in the murine Sγ1 switch region [p(Sγ1)6-P-CAT]; a hexameric repeat of the G14A mutation of this LR1 binding site [p(G14A)6-P-CAT]; or the basal IgH promoter alone (pIgP-CAT). Expression was assayed following transfection of the murine pre-B cell line PD31. Previous experiments in our laboratory have shown that PD31 supports recombination of extrachromosomal switch substrates (10). Figure 4 shows that expression of the reporter gene in p(Sγ1)6-P-CAT is ∼9-fold higher than in pIgP-CAT. Expression is reduced ∼2-fold when driven by the construct carrying the G14A mutation in the repeats. The relatively low reduction in transcriptional activation due to mutation probably reflects the fact that while the G14A mutation decreases binding 12-fold (14), the binding constant for the interaction of LR1 with DNA containing this mutated site is 24 nM, still well within the biologically relevant range.
Figure 4.

Activation of transcription from a minimal promoter by tandemly repeated LR1 sites. CAT expression driven from reporter constructs carrying a minimal promoter and hexamers of Sγ1 or G14A mutant LR1 sites. The graph shows levels of expression driven by constructs carrying multimerized binding sites relative to the pIgP-CAT construct, which carries only the minimal promoter.
LR1 has multiple potential binding sites in the S regions
The ability of LR1 to activate transcription from multimerized sites suggested that it would be useful to identify potential LR1 sites within the immunoglobulin heavy chain S regions. Previous DNA binding and footprinting analysis had identified the region bound by LR1 and defined a preliminary duplex DNA binding consensus sequence (8). The consensus was further refined by assaying LR1 binding to duplexes carrying single base substitutions and by inclusion of sites at which LR1 has been shown to regulate transcription, including Eµ, the c-myc promoter (12) and the EBV EBNA-1 gene (13). As summarized in Figure 5A, mutations G8A and G14A have the most severe effect, decreasing binding 7- and 12-fold (see also 14), respectively; mutations G7C, T9A, A12C and G13C diminished binding <2-fold and mutations at several other positions had essentially no effect on binding. We further note that the G14A mutation, which had the most deleterious effect on binding, increased the KD only to 24 nM, which is comparable to the affinity of many mammalian transcription factors for their specific sites, and that this mutation had only a 2-fold effect on transcriptional activation from a 6mer site (Fig. 3). Taken together, these results show that the LR1 recognition consensus is loose rather than stringent.
Figure 5.

Potential LR1 binding sites are dispersed throughout the S regions. (A) Summary of analysis of LR1 binding to switch region and promoter sites previously examined (8,12,13) (left) and to synthetic duplexes carrying single base changes (right). Binding is relative to the Sγ1 site. (B) S region sequences were searched using FINDPATTERNS (GCG program package) for matches to the LR1 consensus, GGNCNAG(G/C)CTG(G/A), allowing two mismatches except in the case of the murine Sγ2b region, where three mismatches were allowed. Only S regions for which at least 1 kb of contiguous sequence was available in GenBank have been included in the tabulation and for this reason the murine Sγ2a region is omitted. Accession numbers of GenBank files containing murine sequences are: Sµ, J00442; Sγ3, M12182; Sγ1, M12389; Sγ2b, U85373; Sɛ, M57385, X53677; Sα, J00474. Accession numbers of files containing human sequences are: Sµ, X54713; Sγ3, U39935; Sγ1, U39737; Sγ2, U39934; Sɛ, X56797; Sα, L19121. (C) Organization of potential LR1 binding sites in the murine Sγ3–Cγ3 region. The top line shows the murine heavy chain locus, with S regions as open boxes and V, D, J and C regions as shaded boxes. The lower lines shows the Sγ3–Cγ3 region, with potential LR1 binding sites indicated by ovals. LR1 binding sites are similarly dispersed throughout other S regions.
The murine and human S regions are from 2 to 10 kb in length. Each S region consists of repeats of a characteristic consensus motif and all these motifs are G-rich on the non-template strand. In order to obtain a rough estimate of the number of potential LR1 sites in duplex S region DNA, we searched human and murine S region sequences for matches to the LR1 consensus, GGNCNAG(G/C)CTG(A/G), allowing two mismatches. This mismatch level was selected because the affinity of LR1 for the DNA duplex is so high (KD = 1.8 nM) that even the most deleterious single base change identified thus far (G14A, KD = 24 nM) would still bind LR1 with an affinity consistent with in vivo function. The search analyzed the long stretches of contiguous S region DNA sequence contained in the human Sµ, Sγ1, Sγ2, Sγ3, Sα and Sɛ and the murine Sµ, Sγ3, Sγ1, Sα and Sɛ sequence files in GenBank. The density of LR1 sites in each S region was calculated by dividing the number of sites by the length of S region sequence searched. This showed that there are from 12 (Sγ1) to 50 (Sα) potential LR1 sites per kb in the murine S regions and from 9 (Sγ1) to 94 (Sα) LR1 sites per kb in the human S regions (Fig. 5B). Assuming that DNA is random in sequence, matches to the LR1 consensus (allowing two mismatches) should occur, on average, once every 4.8 kb. The density of sites in the switch regions is therefore from one to two orders of magnitude higher than predicted as occurring by chance.
Figure 5C illustrates the density of potential LR1 binding sites in the Sγ3–Cγ3 region of the murine immunoglobulin heavy chain locus. Potential LR1 binding sites are densely clustered within the S region and only a few are scattered within the adjacent C region. Similar clustering of LR1 sites is evident in the other S regions.
DISCUSSION
LR1 is a B cell-specific factor which binds tightly to duplex DNA. LR1 regulates transcription at the c-myc P1 promoter (12), at the EBV F promoter (13) and, as shown in this communication, at the heavy chain intron enhancer. We have further shown that LR1 can regulate transcription in two distinct ways, as a bona fide transcriptional activator and by bending DNA and thereby altering DNA architecture. LR1 duplex DNA binding sites are present not only in promoter regions but also within the S regions that participate in class switch recombination. Class switch recombination and transcriptional activation are intimately coupled in vivo and the results reported here also provide insights into how LR1 duplex DNA binding activity may contribute to switch recombination.
Transcriptional activation by LR1
The primary sequence and predicted structural motifs of the LR1 heterodimer provide clues likely to be relevant to the mechanism of transcriptional activation by LR1. LR1 is composed of nucleolin and a specific isoform of hnRNP D. One sequence motif common to transcriptional activators are acidic regions, or ‘acid blobs’ (43). The N-terminus of nucleolin contains long acidic stretches, including three uninterrupted runs of 16, 21 and 38 amino acid residues, which could function as ‘acid blobs’ to regulate transcription. Acidic activators have the potential to synergize with activators contaning glutamine-rich regions (44) and hnRNP D contains a prominent glutamine run (six of seven residues).
The ability of LR1 to bend DNA depends on both the nucleolin and hnRNP D subunits of this factor. DNA bending decreases from 54 to 26° when nucleolin is removed to leave hnRNP D bound to DNA. This is likely to correspond to a decrease of ∼50% in bending, although our experiments do not preclude the possibility that the bending angle is reversed in the absence of nucleolin. While nucleolin may enhance bending indirectly, for example by altering hnRNP D conformation, two observations suggest that during LR1 binding nucleolin forms direct contacts with the DNA: the presence of nucleolin in the LR1 heterodimer has been shown to modulate the sequence specificity of DNA binding (14); nucleolin can be UV crosslinked to a bromodeoxyuridine-substituted LR1 binding site (8,9). The enhanced bending in the presence of nucleolin is therefore probably due to contacts made by nucleolin with the DNA.
Potential roles of LR1 duplex DNA binding activity in class switch recombination
The results reported here suggest specific potential functions of LR1 duplex DNA binding activity in class switch recombination. A 220 bp region within the Eµ intron enhancer is necessary and sufficient for efficient switch recombination (5). We have shown that LR1 can contribute to transcriptional activation by Eµ from its site within the enhancer core and LR1 binding to this same site may also contribute to recombinational activation by this enhancer. Second, LR1 bound at sites within the S regions may enhance recombination by stimulating transcription of the S regions that is known to be essential for class switching. The LR1 DNA binding consensus is G-rich and relatively unconstrained and there are multiple potential LR1 sites in each S region. The fact that multimerization of LR1 sites creates a transcriptional enhancer suggests that occupancy of contiguous or neighboring sites would permit LR1 to function as a transcriptional activator.
Other factors have been identified which have duplex DNA binding sites within the immunoglobulin heavy chain S regions. One category includes a variety of transcription factors which bind the I region enhancers to regulate sterile transcription and promote recombination to specific isotypes (reviewed in 45). Another category includes factors which, like LR1, have been shown to bind to S region sequences. One of these is a factor or factors related to the p50 subunit of NF-κB, which appear to contribute to regulation at the 3′ enhancer (46–48). Like LR1, NF-κB bends DNA (49), but bending is mediated by the p65 subunit and does not involve p50 (50). Another factor, SNAP, has been reported to bind specifically to sites in the Sγ switch regions and to be related to (though distinct from) E47 (51). E47 is an E box binding protein and, since there are E boxes in the Eµ core, SNAP may, like LR1, contribute to the function of this core enhancer.
In addition, a factor referred to as SWAP has been reported to contain three ubiquitous polypeptides, nucleolin, PARP and nucleophosmin, as well as a B cell-specific duplex DNA binding activity SWAP70. In contrast to LR1, the affinity of SWAP70 for DNA is relatively weak (KD >> 60 nM). Moreover, abundant proteins can readily co-purify with the activities of interest and it remains to be shown that nucleolin, PARP, nucleophosmin and SWAP70 form a specific complex. Nucleolin was shown to be an intrinsic component of the LR1 heterodimer by transfecting cells with a construct that expressed nucleolin bearing an HA-tag and then showing that an anti-HA monoclonal antibody recognized LR1 DNA binding activity in gel shift assays (14). A similar approach could be used to verify the composition of the SWAP complex.
DNA bending may contribute to function in both transcription and recombination
A number of DNA binding proteins which function in both recombination and transcription have been shown to bend DNA and also, like LR1, to bind in the minor groove. One of the best studied members of this group of proteins is IHF, the small prokaryotic protein that enhances transcription and recombination. IHF binds DNA with sequence preference, but without sequence specificity, recognizes determinants in the minor groove and bends DNA by >160° upon binding (see 53 and references therein). In the mammalian V(D)J cleavage reaction, the RAG 1 and RAG 2 proteins have been shown to collaborate with the HMG1 and HMG2 proteins that recognize the minor groove of B-form DNA and bend the DNA duplex upon binding (54,55). Other proteins unrelated to IHF and HMG that bind in the minor groove, bend DNA and stimulate both transcription and recombination include the bacteriophage λ Int and Xis proteins, Fis and γδ resolvase (25,35,36,56–58).
Two lines of evidence now demonstrate that the hnRNP D subunit of LR1 is largely responsible for minor groove interactions with duplex DNA. Experiments presented here show that hnRNP D–DNA interactions are sensitive to the antibiotic distamycin A, which binds to the narrow groove of B-form DNA (Fig. 3), and previous experiments assigned to hnRNP D critical minor groove contacts at position G14 within the LR1 binding site (14).
Proteins that bend DNA and bind in the minor groove are thought to act in recombination by inducing alterations in DNA architecture, thereby increasing the lability of the DNA backbone and facilitating cleavage by and interactions between other participating factors. The ability of LR1 to bend DNA could allow synergistic interactions between LR1 molecules (or other factors) bound to physically separated sites and may also distort the phosphodiester backbone to enhance cleavage.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Anna Birkenblit, April Brys, Laurie Dempsey, Angela DePace, Bonnie Gould, Helios Leung, Marna Williams and William P. Russ for valuable discussions and experimental contributions. This research was supported by NIH R01 grant GM39799 to N.M. and a Ford Foundation Postdoctoral Fellowship to L.A.H.
REFERENCES
- 1.Snapper C.M., Marcu,K.B. and Zelazowski,P. (1997) Immunity, 6, 217–223. [DOI] [PubMed] [Google Scholar]
- 2.Dunnick W., Hertz,G.Z., Scappino,L. and Gritzmacher,C. (1993) Nucleic Acids Res., 21, 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bottaro A., Young,F., Chen,J., Serwe,M., Sablitzky,F. and Alt,F.W. (1998) Int. Immunol., 10, 799–806. [DOI] [PubMed] [Google Scholar]
- 4.Gu H., Zou,R.-R. and Rajewsky,K. (1993) Cell, 73, 1155–1164. [DOI] [PubMed] [Google Scholar]
- 5.Sakai E., Bottaro,A. and Alt,F.W. (1999) Int. Immunol., 11, 1709–1713. [DOI] [PubMed] [Google Scholar]
- 6.Sakai E., Bottaro,A., Davidson,L., Sleckman,B.P. and Alt,F.W. (1999) Proc. Natl Acad. Sci. USA, 96, 1526–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ernst P. and Smale,S.T. (1995) Immunity, 2, 427–438. [DOI] [PubMed] [Google Scholar]
- 8.Williams M. and Maizels,N. (1991) Genes Dev., 5, 2353–2361. [DOI] [PubMed] [Google Scholar]
- 9.Williams M., Brys,A., Weiner,A.M. and Maizels,N. (1992) Nucleic Acids Res., 20, 4935–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li M.-J., Chung,W. and Maizels,N. (1997) Mol. Immunol., 34, 201–208. [DOI] [PubMed] [Google Scholar]
- 11.Li M.-J. and Maizels,N. (1999) J. Immunol., 163, 6659–6664. [PubMed] [Google Scholar]
- 12.Brys A. and Maizels,N. (1994) Proc. Natl Acad. Sci. USA, 91, 4915–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulfone-Paus S., Dempsey,L.A. and Maizels,N. (1995) Proc. Natl Acad. Sci. USA, 92, 8293–8297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanakahi L.A., Dempsey,L.A., Li,M.-J. and Maizels,N. (1997) Proc. Natl Acad. Sci. USA, 94, 3605–3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dempsey L.A., Hanakhai,L.A. and Maizels,N. (1998) J. Biol. Chem., 273, 29224–29229. [DOI] [PubMed] [Google Scholar]
- 16.Dempsey L.A., Sun,H., Hanakhai,L.A. and Maizels,N. (1999) J. Biol. Chem., 274, 1066–1071. [DOI] [PubMed] [Google Scholar]
- 17.Hanakahi L.A., Sun,H. and Maizels,N. (1999) J. Biol. Chem., 274, 15908–15912. [DOI] [PubMed] [Google Scholar]
- 18.Maizels N. (1999) Am. J. Hum. Genet., 64, 1270–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhodes D. and Burley,S.K. (2000) Curr. Opin. Struct. Biol., 10, 75–77. [DOI] [PubMed] [Google Scholar]
- 20.Gillies S.D., Morrison,S.L. and Tonegawa,S. (1983) Cell, 33, 717–728. [DOI] [PubMed] [Google Scholar]
- 21.Krawinkel U., Zoebelein,G. and Bothwell,A.L.M. (1986) Nucleic Acids Res., 14, 3871–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leung H. and Maizels,N. (1992) Proc. Natl Acad. Sci. USA, 89, 4154–4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorman C.M., Moffat,L.F. and Howard,B. (1982) Mol. Cell. Biol., 2, 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li M.-J., Leung,H. and Maizels,N. (1996) In Adolph,K.W. (ed.), Methods in Molecular Genetics. Academic Press, Orlando, FL, Vol. 8, pp. 375–387.
- 25.Kim J., Zwieb,C., Wu,C. and Adhya,S. (1989) Gene, 85, 15–23. [DOI] [PubMed] [Google Scholar]
- 26.Derbyshire K.M., Salvo,J.J. and Grindley,N.D.F. (1986) Gene, 46, 145–152. [DOI] [PubMed] [Google Scholar]
- 27.Su L.-K. and Kadesch,T. (1990) Mol. Cell. Biol., 10, 2619–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prentki P., Pham,M.-H. and Galas,D.J. (1987) Nucleic Acids Res., 15, 10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu M., Hammer,R.E., Blasquez,V.C., Jones,S.L. and Garrard,W.T. (1989) J. Biol. Chem., 264, 21190–21195. [PubMed] [Google Scholar]
- 30.Sitlani A. and Crothers,D.M. (1996) Proc. Natl Acad. Sci. USA, 93, 3248–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sitlani A. and Crothers,D.M. (1998) Proc. Natl Acad. Sci. USA, 95, 1404–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kerppola T.K. (1996) Proc. Natl Acad. Sci. USA, 93, 10117–10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerppola T.K. (1997) Biochemistry, 36, 10872–10884. [DOI] [PubMed] [Google Scholar]
- 34.Harrington R.E. and Winicov,I. (1994) Prog. Nucleic Acid Res. Mol. Biol., 47, 195–270. [DOI] [PubMed] [Google Scholar]
- 35.Thompson J.F. and Landy,A. (1988) Nucleic Acids Res., 16, 9687–9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim S. and Landy,A. (1992) Science, 256, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X., Ramakrishnan,B. and Sundaralingam,M. (1997) J. Mol. Biol., 267, 1157–1170. [DOI] [PubMed] [Google Scholar]
- 38.Coll M., Frederick,C.A., Wang,A.H.-J. and Rich,A. (1987) Proc. Natl Acad. Sci. USA, 84, 8385–8389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kopka M.L., Yoon,C., Goodsell,D., Pjura,P. and Dickerson,R.E. (1995) Proc. Natl Acad. Sci. USA, 82, 1376–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pelton J.G. and Wemmer,D.E. (1990) J. Am. Chem. Soc., 112, 1393–1399. [Google Scholar]
- 41.Bellorini M., Moncollin,V., D’Incalci,M., Mongelli,N. and Mantovani,R. (1995) Nucleic Acids Res., 23, 1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geiss G.K., Radebaugh,C.A. and Paule,M.R. (1997) J. Biol. Chem., 272, 29243–29254. [DOI] [PubMed] [Google Scholar]
- 43.Ptashne M. (1988) Nature, 335, 683–689. [DOI] [PubMed] [Google Scholar]
- 44.Escher D., Bodmer-Glavas,M., Barberis,A. and Schaffner,W. (2000) Mol. Cell. Biol., 20, 2774–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henderson A. and Calame,K. (1998) Annu. Rev. Immunol., 16, 163–200. [DOI] [PubMed] [Google Scholar]
- 46.Wuerffel R., Nathan,A.T. and Kenter,A.L. (1990) Mol. Cell. Biol., 10, 1714–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wuerffel R., Jamieson,C.E., Morgan,L., Merkulov,G.V., Sen,R. and Kenter,A.L. (1992) J. Exp. Med., 176, 339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Michaelson J.S., Singh,M., Snapper,C.M., Sha,W.C., Baltimore,D. and Birshtein,B.K. (1996) J. Immunol., 156, 2828–2839. [PubMed] [Google Scholar]
- 49.Schreck R., Zorbas,H., Winnacker,E.L. and Baeuerle,P.A. (1990) Nucleic Acids Res., 18, 6497–6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuprash D.V., Rice,N.R. and Nedospasov,S.A. (1995) Nucleic Acids Res., 23, 427–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma L., Hu,B. and Kenter,A.L. (1997) Int. Immunol., 9, 1021–1029. [DOI] [PubMed] [Google Scholar]
- 52.Borggrefe T., Wabl,M., Akhmedov,A.T. and Jessberger,R. (1998) J. Biol. Chem., 273, 17025–17035. [DOI] [PubMed] [Google Scholar]
- 53.Rice P.A., Yang,S., Mizuuchi,K. and Nash,H.A. (1996) Cell, 87, 1295–1306. [DOI] [PubMed] [Google Scholar]
- 54.Sawchuk D.J., Weis-Garcia,F., Malik,S., Besmer,E., Bustin,M., Nussenzweig,M.C. and Cortes,P. (1997) J. Exp. Med., 185, 2025–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Gent D.C., Hiom,K., Paull,T.T. and Gellert,M. (1997) EMBO J., 16, 2665–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hatfull G.F., Noble,S.M. and Grindley,N.D.F. (1987) Cell, 49, 103–110. [DOI] [PubMed] [Google Scholar]
- 57.Liu-Johnson H.N., Gartenberg,M.R. and Crothers,D.M. (1986) Cell, 47, 995–1005. [DOI] [PubMed] [Google Scholar]
- 58.Salvo J.J. and Grindley,N.D. (1988) EMBO J., 7, 3609–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]