

Significance
Dengue places a huge burden to public health systems. More than forty percent of the human population is at risk of developing dengue fever or dengue hemorrhagic fever. Pathogenesis and disease severity of dengue are associated with increased production of proinflammatory cytokines. In this study, we identified a dengue virus mechanism to limit cytokine secretion during infection. Studying viral–host interactions, we found an additional function of the viral protein NS5 in antagonizing immune signaling and limiting proinflammatory cytokine production in a dengue virus serotype-specific manner. This work reveals a mechanism critical for dengue virus pathogenesis and provides important information for improving tetravalent live attenuated vaccines.
Keywords: dengue virus, host–virus interactions, evasion of innate antiviral responses, NS5 viral protein activation, dengue virus pathogenesis
Abstract
Dengue virus (DENV) is the most important human virus transmitted by mosquitos. Dengue pathogenesis is characterized by a large induction of proinflammatory cytokines. This cytokine induction varies among the four DENV serotypes (DENV1 to 4) and poses a challenge for live DENV vaccine design. Here, we identify a viral mechanism to limit NF-κB activation and cytokine secretion by the DENV protein NS5. Using proteomics, we found that NS5 binds and degrades the host protein ERC1 to antagonize NF-κB activation, limit proinflammatory cytokine secretion, and reduce cell migration. We found that ERC1 degradation involves unique properties of the methyltransferase domain of NS5 that are not conserved among the four DENV serotypes. By obtaining chimeric DENV2 and DENV4 viruses, we map the residues in NS5 for ERC1 degradation, and generate recombinant DENVs exchanging serotype properties by single amino acid substitutions. This work uncovers a function of the viral protein NS5 to limit cytokine production, critical to dengue pathogenesis. Importantly, the information provided about the serotype-specific mechanism for counteracting the antiviral response can be applied to improve live attenuated vaccines.
Dengue is the most important viral disease in humans transmitted by mosquitos. Despite its global burden, there are no antiviral agents or universal vaccines available to cope with dengue virus (DENV) infections. Vaccine development has been a great challenge due to the presence of four DENV serotypes (DENV1-DENV4). One important feature of DENV infection is that a previous exposure to any of the four serotypes can lead to a more severe clinical manifestation upon infection with a heterologous serotype (1–3), a phenomenon that requires effective tetravalent vaccines. A great limitation in vaccine development is the different properties of the distinct serotypes. They differ in how they trigger and counteract antiviral responses, which complicates the design of live attenuated tetravalent vaccine formulations. Previous experience has shown that DENV2 is difficult to include in tetravalent formulations due to unbalanced replication, and chimeric viruses with other serotypes were required (4). In this regard, it is important to understand the molecular differences among the four DENV serotypes and how they lead to infection and pathogenesis.
DENV is a member of the Flavivirus genus, together with other emerging and reemerging pathogens such as Zika virus (ZIKV), yellow fever virus (YFV), West Nile virus (WNV), and Japanese encephalitis virus. Flaviviruses are positive sense, single-stranded RNA viruses. Their genome encodes a single polyprotein that is cleaved by viral and cellular proteases into three structural (C, prM, and E) and at least seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (5). NS5 is the largest viral protein and displays multiple enzymatic activities. It contains two domains, the N-terminal methyltransferase (MTase) domain, essential for viral RNA capping, and the C-terminal RNA-dependent RNA-polymerase (RdRp) responsible for viral RNA synthesis (6–8). NS5 also contains an extraordinary set of tools to dismantle the innate host antiviral response. One crucial cellular defense mechanism targeted by NS5 is the interferon (IFN)-signaling pathway. It has been documented that DENV NS5 from all serotypes blocks type I IFN signaling by proteasome-mediated human STAT2 degradation (9, 10). This process requires binding of UBR4 to an in situ proteolytically cleaved DENV NS5 (9, 11). In contrast, ZIKV NS5 mediates STAT2 degradation in a UBR4-independent manner and does not require proteolytic processing of the NS5 N terminus (12). In the case of YFV, NS5 protein also binds STAT2 but only after IFN stimulation, and the binding does not mediate STAT2 degradation (13–15). Also, the NS5 from WNV, tick-borne encephalitis and Langat viruses suppress expression of the IFN receptor, activity that involves the NS5 RdRp domain (13, 14). Therefore, NS5 protein from different flaviviruses uses different strategies to subvert type I IFN signaling, limiting the expression of several antiviral genes and generating a more favorable environment for viral replication.
To dig deeper into the multiple functions of NS5, we previously performed a proteomic study in the context of DENV infection, and constructed a host protein–NS5 interaction map (16). We found an interplay between NS5 and the cellular splicing machinery, which resulted in an increase in intron retention and modulation of IFN-stimulated gene expression. Mechanistically, we found that NS5 directly interacts with core components of the U5 small nuclear ribonucleoprotein complex (snRNP) to alter the splicing process (16). In this regard, alteration of SAT1 mRNA alternative splicing by NS5 was also found to reduce the abundance of this antiviral protein (17). Here, we further explore the NS5 interactome map to validate the binding and relevance of additional host proteins in the context of viral infection. We identified the host protein ERC1 with antiviral activity.
ERC1 is a regulatory subunit of the IKK complex involved in the nuclear factor-κB (NF-κB) activation pathway (18, 19), it participates in the docking and/or fusion of Rab6-positive vesicles at the cell cortex (20) and it forms a functional complex that drives cell motility together with LL5 proteins, LL5α and LL5β, and liprin-α1 (21). We found that ERC1 disappears during DENV2 infection and that protein degradation is mediated by the methyltransferase domain of NS5 in a proteasome-dependent manner. Interestingly, although the four DENV serotypes degrade STAT2, ERC1 was not degraded by DENV4 infection, suggesting mechanistic differences between STAT2 and ERC1 degradation. By designing a battery of recombinant DENVs, we separated the molecular determinants required for ERC1 and STAT2 degradation. In addition, using a DENV2 with a single-residue substitution in NS5, without the ability to degrade ERC1, we found significantly higher levels of NF-κB dependent proinflammatory cytokine expression and secretion during infection, higher IFN-β expression, and increased cell motility compared with the WT DENV2. This work provides an additional function of NS5 in modulating host innate antiviral responses and uncovers molecular differences between DENV serotypes relevant for vaccine formulations.
Results
ERC1 Has an Antiviral Function and Is Degraded after DENV2 Infection.
We have previously developed a genetic system for mapping physical interactions of NS5 with host proteins using an NS5 tagged DENV2 infectious clone. Employing affinity-purification mass spectroscopy, we constructed an NS5-host protein interactome map using infected human cells (SI Appendix, Fig. S1), and validated the functionality of a number of NS5 ligands (16, 17). To identify relevant binders of NS5, we carried out a knockdown screening assay of a set of NS5-interactors and assessed viral replication after infection with a reporter DENV2 encoding Renilla luciferase gene (RepDENV) (22). We focused on cellular proteins with potential antiviral activity and identified a host protein named ERC1. Knockdown of this protein enhanced viral replication, which was measured by RepDENV luciferase activity reflecting viral RNA replication (Fig. 1A). The increased level of reporter activity was similar to that observed by silencing proteins previously reported to display antiviral functions, STAT2 and CD2BP2 (Fig. 1A). To further confirm this observation, infections with WT DENV2 at multiplicity of infection (MOI) of one were performed in ERC1 silenced human A549 cells. Viral RNA was assessed in cells at 8 and 24 h postinfection. A fourfold increase in viral replication was observed when compared to replication levels in cells silenced with a nonrelated siRNA control (Fig. 1B). These results suggest an antiviral role of the host protein ERC1 during infection.
Fig. 1.
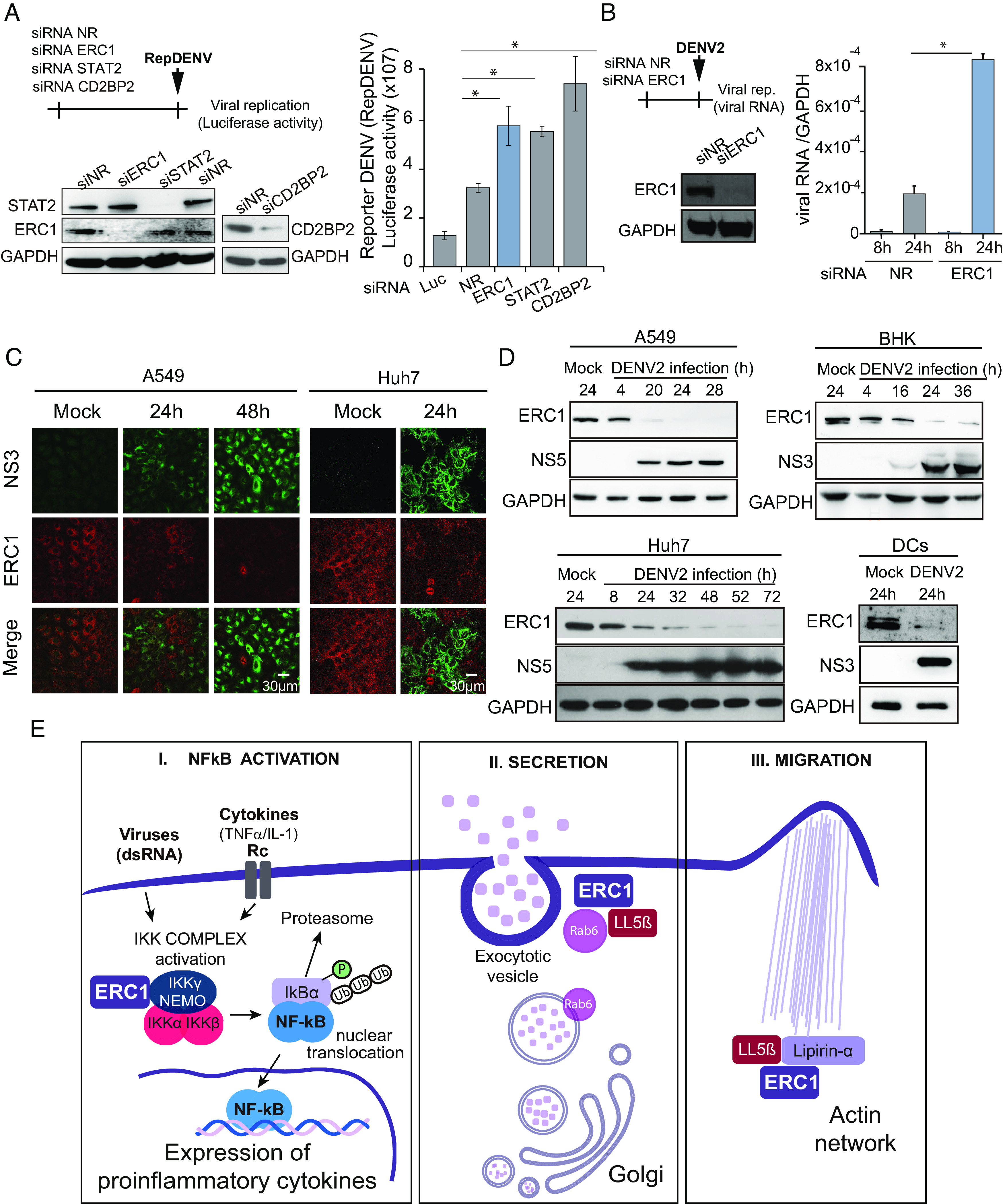
The host ERC1 protein has an antiviral activity and is degraded during DENV2 infection. (A) ERC1 has an antiviral function. Luciferase activity showing the replication of DENV2 reporter (RepDENV) 36 hpi in A549 cells transfected 48 h previously with siRNAs directed to ERC1 (blue) or directed to two antiviral proteins: STAT2 and CD2BP2, as indicated. Two additional controls were included, a nonrelated siRNA (NR) and a positive siRNA control directed to Renilla luciferase (Luc). WB assays indicate the levels of silenced proteins. Statistical significance is shown with the following notation: *P < 0.05. (B) ERC1 silencing reduces DENV replication. Viral replication is shown by qRT PCR (mean ± SD) at 8 and 24 hpi (MOI 1) in A549 cells previously transfected with a nonrelated siRNA (NR), or a siRNA directed to ERC1. WB shows the levels of ERC1 in each condition. Statistical significance is shown with the following notation: *P < 0.05. (C) ERC1 levels are reduced in DENV2-infected human cells. Immunofluorescence (IF) assays against ERC1 (red) and the viral protein NS3 (green, to label infection), are shown in human A549 and Huh7 cells either mock infected or infected with DENV2 (MOI 3). Objective: 40× (D) ERC1 protein is degraded upon DENV2 infection at MOI of five in different cell lines. WB analyses showing the levels of ERC1 as a function of time postinfection, as indicated in each panel. Cytoplasmic extracts from infected A549 (human), BHK (hamster), Huh7 (human), DCs (monocyte-derived human DCs) are shown. (E) Schematic description of the multiple functions of ERC1. (I) ERC1 is involved in NF-kB activation by Toll-like and cytokine receptors (TNF-α, IL1) or by DNA damage. ERC1 interacts with the IKKγ/NEMO regulatory subunit of IκB kinase (IKK) complex and enhances IKK activation. This activation induces IκBα phosphorylation, followed by ubiquitination and proteasome degradation of NF-kB inhibitory protein IκBα. NF-kB complex is then translocated to the nucleus and acts as a transcription factor that induces genes related to immune responses, such as proinflammatory cytokines (TNF-α, IL6). (II) ERC1 is involved in secretion. It interacts with Rab6-dependent vesicles acting as a docking and/or fusion protein in the cell cortex, where it tags the site of arrival of Rab6- vesicles for secretion. (III) ERC1 is involved in cell migration. The host protein forms a functional complex with LL5 proteins LL5a and LL5b and liprin-α1 that drives cell motility.
To study ERC1 localization during the infection process, indirect immunofluorescence (IF) assays were carried out in DENV2 or mock infected human A549 and Huh7 cells. Interestingly, the ERC1 signal was undetectable in infected cells, suggesting viral-induced protein degradation (Fig. 1C). To further explore this possibility, western blot assays (WB) were performed in cytoplasmic extracts obtained as a function of time from infected A549 and Huh7 cells with MOI of 5. The data indicate that ERC1 disappears in the course of infection in both human cells (Fig. 1D). To expand this observation, we used BHK cells and human monocyte-derived dendritic cells (DCs), which are rodent and primary targets for DENV infection, respectively (Fig. 1D). The results showed that ERC1 protein levels greatly decrease, while the viral proteins accumulate in all the infected cell lines analyzed. Together, we identified a protein with possible antiviral activity that is degraded during the course of DENV2 infection.
Multiple Functions of the Cellular ERC1 Protein.
ERC1 is a ubiquitous protein with multiple coiled-coil motifs that enable interaction with many proteins. ERC1 has been previously implicated in the activation of NF-kB through both the canonical (mediated by Toll-like and cytokine receptors) (19) and the atypical pathway (activated by genotoxic stress, DNA damage) (18) (Fig. 1E). ERC1 has also been involved in intracellular membrane trafficking. It participates in the docking and/or fusion of Rab6-positive vesicles at the cell cortex and in secretion by its binding with LL5 proteins, LL5α and LL5β (20, 23). Its interaction with the latter together with lipirin-α has been implicated in the regulation of cell motility (21, 24) (Fig. 1E). In order to further evaluate the roles of ERC1, we either overexpressed or knocked down its expression in HEK 293T and A549 human cell lines.
ERC1 involvement in the NF-kB activation pathway was evaluated by overexpression of the host protein and subsequent measurement of NF-kB reporter activation. Overexpression of ERC1 increased more than 20-fold the luciferase activity of the NF-kB reporter compared to that of the control vector (Fig. 2A). On the other hand, silencing ERC1 in A549 cells stimulated with 5 ng/mL TNF-α, significantly reduced IL6 and TNF-α mRNA, as well as IL6 secretion (Fig. 2B). Given that NF-kB is involved in the IFN-β pathway (25), we activated cells by 10 μg/mL poly I:C treatment (double-stranded RNA analog) in ERC1 knockdown or control conditions, and evaluated cytokine and IFN-β expression. In the presence of poly I:C, induction of IL6, TNF-α and IFN-β mRNA expression was observed, which was greatly reduced upon ERC1 silencing (Fig. 2C). The cytokines were also detected at significantly lower levels in the supernatant of ERC1-depleted cells (Fig. 2 C, Lower). These results support a role of ERC1 on expression of NF-kB dependent proinflammatory genes and IFN-β. To evaluate the role of ERC1 on cell motility, we determined the impact of ERC1 knockdown in a wound-healing assay. Wounds were made on a monolayer of A549 cells following ERC1 silencing or treatment with a nonrelated siRNA control. Images of the gaps were recorded immediately after the wound (0 h) and 24 h postwound. Measurement of the distance migrated by cells within 24 h demonstrated that cell invasion into the cell-free region was significantly promoted in the presence of ERC1, supporting a role of this protein in cell migration in A549 cells (Fig. 2D). Taken together, our observations confirmed previous reports regarding the ERC1 role in the NF-kB pathway and in cell migration.
Fig. 2.

ERC1 enhances NF-kB-dependent cytokine production and is necessary for cell motility in human cells (A) ERC1 overexpression activates NF-kB reporter activity. The luciferase activity of the NF-kB reporter was measured in cells expressing or not ERC1. On the top, schematic representation of the experiment. 293T cells were cotransfected with 10 ng of the NF-kB reporter plasmid (pFLucNFkB Rep), 4 ng Renilla luciferase expression plasmid (pRLuc) as control to standardize transfection and either, 26 ng ERC1 plasmid (pERC1) or 26 ng of an empty control plasmid (pControl). Relative luciferase activity (FLuc/RLuc) is shown at 16 h posttransfection. (B) ERC1 is involved in proinflammatory cytokine expression and secretion. On the top, schematic representation of the protocol used and WB showing the efficiency of ERC1 silencing. At the bottom, cytokine expression (mRNA of TNF-α and IL6) relative to GAPDH is shown measured by RT-qPCR, and secreted IL6 protein measured by ELISA, in ERC1 silenced or control A549 human cells treated with 5 ng/mL TNF-α during 2, 4, and 6 h. (C) TNF-α, IL6, and IFN-β mRNA expression and protein secretion after 16 and 24 h of 10 μg/mL poly I:C treatment in ERC1 silenced or control A549 cells. On the Top panel, schematic representation of the protocol used and WB showing the efficiency of ERC1 silencing. (D) ERC1 role on A549 cell motility. On the top, schematic representation of the protocol used and WB showing the efficiency of ERC1 silencing. Wound-healing assays are shown in ERC1 silenced or control A549 cells. Representative images of scratched monolayer at 0 and 24 h are shown for each condition. Objective: 5×. The wound closure was quantified by measuring the distance covered by the migrated leading edge using Zen 2.3 lite software. Statistical significance is shown in the different panels with the following notations: ****P < 0.0001; *P < 0.05.
The Methyltransferase Domain of NS5 Is Responsible for ERC1 Degradation Mediated by UBR4.
Because ERC1 was found to be a binder of NS5, we hypothesized that this viral protein was responsible for ERC1 degradation during infection. It has been previously reported that NS5 was necessary but not sufficient for STAT2 degradation. In that case, a precursor of NS5 was required (9). We thus evaluated whether NS5 was sufficient for ERC1 fate during infection. To this end, NS5/GFP, which contained a self-cleaving 2A peptide to release the reporter protein, was overexpressed in A549 cells, and ERC1 was examined by immunofluorescence (Fig. 3A). As a control, a plasmid expressing NS3/GFP was used.
Fig. 3.

Mechanism of ERC1 degradation during DENV infection. (A) NS5 protein is sufficient for ERC1 degradation. IF and WB assays showing ERC1 levels in cells expressing NS5-GFP or NS3-GFP control, as indicated in each case. IF assays were carried out 24 h posttransfection of A549 cells with either NS5 or NS3 expression plasmids. Objective: 40×. For WB analysis, 293T cells were transfected during 24 h with NS5-GFP plasmid and sorted by FACS to separate GFP-positive (NS5 +) from GFP-negative (NS5 −) cells. (B) The methyltransferase domain (MTase) of NS5 is responsible for ERC1 degradation. IF and WB assays showing ERC1 levels in cells expressing RdRp or MTase domains of NS5. For IF, A549 cells were transfected with 250 ng of each plasmid. Images were acquired with the objective 63× 24 h posttransfection. On the right, WB showing endogenous ERC1 levels in 293T cells expressing different amounts of the viral proteins or control plasmid (pCL), as indicated in each case. (C) ERC1 degradation is proteasome-dependent. WB showing expression of ERC1 or the viral NS5 protein in DENV2-infected A549 cells with an MOI of 3 during 12 h and subsequently treated with 20 uM MG132 or DMSO as control during 2, 6, and 10 h. (D) UBR4 E3 ubiquitin ligase mediates ERC1 degradation. The levels of ERC1 and viral proteins are shown in UBR4-silenced A549 cells infected or not with DENV2. Samples at 24 h postinfection with an MOI of 5 are shown. On the Right panel, levels of UBR4 mRNA relative to GAPDH measured by qPCR are included. (E) DENV2 T2A and G3A mutant viruses are replication competent. On the right, DENV NS5 sequence alignment of the first N-terminal eleven amino acids of the four serotypes. Two N-terminal NS5 amino acid substitutions were introduced independently in the DENV2 infectious clone: T2A or G3A. The amino acid change is shown in red. On the left, IF assays showing that T2A and G3A viruses replicate in A549 cells 24 h post infection with an MOI of 10. The viral NS3 protein is shown in green and DAPI in blue. Objective: 20×. (F) T2A or G3A substitutions in NS5 prevent ERC1 degradation. WB showing ERC1 at 24 h postinfection with DENV2 WT, T2A, or G3A. The viral proteins NS5 and NS3 as well as GAPDH are also shown, as indicated in each case. (G) DENV2 WT degrades mouse ERC1 protein. Quantification of ERC1-positive and -negative melanoma mouse cells (B16-F10) infected during 24 h with an MOI of 10 of WT, T2A, or G3A viruses is shown. The percentage of ERC1-positive cells was calculated for each viral infection.
We observed that transfected cells expressing NS5/GFP displayed almost undetectable ERC1 fluorescent signal when compared with cells expressing NS3/GFP protein control (Fig. 3A). To confirm this observation, sorted GFP-positive (NS5+) from GFP-negative (NS5−) cells were collected and used for ERC1 detection in each population by WB. ERC1 protein levels were greatly reduced in NS5 expressing cells, suggesting that DENV2 NS5 is the only viral component required for ERC1 degradation (Fig. 3A).
NS5 protein contains two defined domains, the methyltransferase (MTase) of 32 kDa and the RNA-dependent RNA polymerase (RdRp) of 75 kDa. To determine which of these domains was responsible for ERC1 degradation, they were independently cloned in expression plasmids with GFP, containing a 2A cleavage site. Overexpression of the two domains of NS5 in A549 cells showed that the MTase was responsible for ERC1 disappearance, while cells expressing RdRp domain retained similar ERC1 levels to that observed in cells expressing GFP alone (Fig. 3B). WBs of cytoplasmic extracts of 293T cells expressing GFP alone, MTase, or RdRp confirmed that ERC1 levels were significantly reduced when the MTase domain was expressed (Fig. 3B). We conclude that the viral protein NS5 is responsible for ERC1 degradation and that this property resides in its MTase domain.
To evaluate whether the ubiquitin proteasome pathway is involved in ERC1 degradation, the proteasome inhibitor MG132 was used during DENV2 infection in a controlled manner since it is a potent inhibitor of viral replication. Either MG132 or DMSO (control treatment) was added 12 h postinfection, once the viral infection was established. Cells were then harvested every two hours to detect NS5 and ERC1 levels by WB (Fig. 3C). MG132 prevented ERC1 degradation during infection between 6 and 10 h after drug addition, supporting proteasome involvement.
Protein degradation by the proteasome pathway requires a E3 ubiquitin ligase. UBR4 was identified as binder of NS5 in our proteomic studies (SI Appendix, Fig. S1). In addition, UBR4 was previously found to interact with DENV NS5 for STAT2 degradation (11). Thus, we hypothesized that UBR4 might be required for ERC1 degradation. We evaluated the ability of DENV2 infection to degrade ERC1 in UBR4 silenced cells. UBR4 was knocked down with specific siRNAs and A549 cells were infected with DENV2 (Fig. 3D). Control cells were treated with a nonrelated siRNA previous infection. UBR4 mRNA levels were greatly reduced upon silencing, thus ERC1 protein levels were assessed 24 h post DENV infection by WB. Viral infection was evaluated by the levels of the viral proteins NS3 and NS5 in the same samples. We observed that UBR4 knockdown prevented ERC1 degradation upon viral infection, suggesting the involvement of this E3 ubiquitin ligase in degradation of the host protein (Fig. 3D).
It has been previously shown that T2A and G3A substitutions in NS5 affect UBR4 binding, while retaining interaction with STAT2 in overexpressed NS5 conditions (11); however, this has not been evaluated in the context of an infectious clone. Thus, we designed recombinant DENV2 viruses carrying the single substitutions, T2A or G3A, in NS5 (Fig. 3E). To characterize the replication of the two mutants, we performed growth curves in mosquito and human cells. While in mosquito cells, the two viruses displayed the same replication ability than the WT virus (SI Appendix, Fig. S2), a lower replication was observed when the mutants were used to infect human cells (SI Appendix, Fig. S3).
We then generated viral stocks of T2A and G3A viruses in mosquito cells and used them to infect A549 cells at high (MOI of 10) (Fig. 3E). In these conditions, in which the complete monolayer of human A549 cells was infected, DENV2 T2A and DENV2 G3A were unable to degrade ERC1 as shown by WB analysis (Fig. 3F). This was also true in B16-F10 mouse cells, in which ERC1 remained present in DENV2 T2A- or G3A-infected cells (Fig. 3G). We thus conclude that T2 and G3 residues of NS5 are required for ERC1 degradation, likely due to their participation in UBR4 interaction as previously reported (11).
DENV Serotypes Differently Degrade ERC1 Protein during Infection.
To determine the extent of ERC1 degradation by different DENV serotypes, IF and WB analyses were carried out in A549 cells infected with each of the four DENV serotypes. IF assays (Fig. 4A) and WB analysis (Fig. 4B) indicated that DENV1, 2, and 3 efficiently degraded the host protein, while DENV4 infections show mock levels of ERC1. In contrast, degradation of STAT2 was observed with all four DENV serotypes (Fig. 4B), as previously reported (11). This result was intriguing and revealed differences among the DENV serotypes in the ability to degrade STAT2 and ERC1 (Fig. 4B). DENV4 NS5 must retain the ability to bind UBR4 for STAT2 degradation (11), but there must be an additional requirement in the viral NS5 for ERC1 association, which appears to be different among the four DENV serotypes (Fig. 4C).
Fig. 4.

DENV serotypes differentially degrade ERC1 (A) ERC1 protein is not degraded during DENV4 infection. IF showing ERC1 or the viral protein NS3 in cells infected with an MOI of 5 with DENV1, DENV2, DENV3, or DENV4, as indicated. Objective: 40× (B) DENV4 infection differentially degrades STAT2 and ERC1. WB assay showing ERC1, STAT2, NS5, and GAPDH control in extracts from cells infected during 24 h with an MOI of 5 with each of the four DENV serotypes, as indicated. (C) STAT2 and ERC1 degradation by the four DENV serotypes. Schematic representation of the ability of NS5 from different serotypes to degrade STAT2 and ERC1. (D) DENV4 NS5 lacks the ability to degrade ERC1. Levels of ERC1 in cytoplasmic extracts of 293T sorted cells expressing or not GFP from cells transfected during 24 h with 15 μg DENV2-NS5-GFP, DENV4-NS5-GFP, or GFP plasmids. Cytoplasmic extracts from equal amounts of cells were loaded in each lane. Levels of ERC1, GFP, and GAPDH are shown. (E) The MTase domain of DENV4 NS5 lacks the ability to degrade ERC1. Levels of ERC1 in cytoplasmic extracts of 293T sorted cells expressing or not GFP from cells 24 h posttransfection with 15 μg DENV2-MTase-GFP, DENV4- MTase-GFP, or GFP plasmids. Cytoplasmic extracts from equal amounts of cells were loaded in each lane. Levels of ERC1 and GAPDH are shown. (F) Schematic representation of the differential degradation of ERC1 by the MTase of DENV2 and DENV4.
To confirm the differential ability of the NS5 protein from DENV2 and DENV4 to degrade ERC1, we cloned and overexpressed NS5 of these two viruses and evaluated their capacity to degrade ERC1. The proteins were expressed in fusions with GFP in 293T cells. Transfected cells were sorted in GFP-positive (NS5+) and -negative (NS5−) cells and protein levels assessed by WB. DENV2 NS5-positive cells showed consistently lower amounts of ERC1- than DENV2 NS5-negative ones, while DENV4 NS5-positive and -negative cells had similar levels of ERC1, suggesting the inability of DENV4 NS5 to degrade ERC1 (Fig. 4D). We also evaluated whether the NS5 MTase domain from DENV2 and DENV4 retained the differential ability to degrade ERC1. To this end, we constructed and cloned the two MTase domains. WB analyses showed that expression of the DENV4 protein did not alter the levels of ERC1 (Fig. 4E). Based on these results, we propose a model in which DENV2 MTase domain is the one responsible for interacting with both UBR4 and ERC1 for protein degradation via proteasome. In contrast, DENV4 protein would bind UBR4 but lacks the ability to degrade ERC1 (Fig. 4F).
Mapping Structural Determinants within DENV NS5 for ERC1 Degradation.
We then searched for the molecular determinants that could explain the serotype-specific degradation of ERC1. We aligned and compared the amino acid sequence of the MTases from the four DENV serotypes. Additionally, we used the crystal structure of DENV2 MTase to identify solvent-exposed regions, which could potentially interact with ERC1 (Fig. 5A). Based on this analysis, we designed and cloned three chimeric DENV2-DENV4 MTases: DENV2 MTase R1 (DENV4 14-27), DENV2 MTase R2 (DENV4 43-52), and DENV2 MTase R3 (DENV4 173-176) (Fig. 5A), which were expressed fused to a self-cleaving 2A peptide and GFP. The ability of these chimeric proteins to degrade ERC1 was tested in transfected cells by IF and WB analyses. MTase R1 and MTase R2 showed no ERC1 degradation, whereas MTase R3 showed similar ERC1 levels as the DENV2 MTase (Fig. 5 B and C).
Fig. 5.

Mapping the molecular determinants in NS5 for ERC1 degradation. (A) Sequence alignment of MTases from DENV1, 2, 3, and 4. Amino acid sequences of the four DENV serotypes are shown. R1, R2, and R3 in green boxes show solvent-exposed regions of the MTase domain with significant sequence differences between DENV4 and the other three serotypes (in gray). Residue numbers corresponding to the R1, R2, and R3 are shown in each case with a representation of the protein secondary structure below in red. Below: MTase chimeras R1, R2, and R3 are shown based on DENV2 protein crystal structure (DENV2 in green and the DENV4 region in red). (B) Chimeric R1 and R2 MTase proteins lack the ability to degrade ERC1. Representative images of A549 cells transfected with 250 ng chimeric MTase-GFP (R1-R3), DENV2 MTase-GFP, DENV4 MTase-GFP, or GFP control plasmids are shown by IF. Detection of GFP and ERC1 is indicated in each case. Objective: 63×. (C) ERC1 protein levels by WB in cytoplasmic extracts of 293T cells expressing the chimeric MTase-GFP is shown. Proteins NS5 and GAPDH for each sample are also shown. (D) Replication and propagation of chimeric DENV2 viruses. Viral RNA from R1 and R2 chimeras were transfected in BHK cells, and viral propagation was evaluated by detecting NS3 protein using IF. DAPI-staining nucleus shows the integrity of the monolayer. Objective: 20×. (E) DENV2 chimeric virus R1 lacks the ability to degrade ERC1. A549 cells were infected during 24 h with DENV2 (MOI:3) and chimeric virus R1 (MOI:20). IF showing NS5 in green indicating DENV2-infected cells and ERC1 in red showing degradation of the host protein in infected cells. Objective: 63× (F) WB showing ERC1 levels in cytoplasmic extracts of A549 cells infected during 24 h with DENV2 (MOI:5), DENV4 (MOI:5), or R1 chimeric virus (MOI:20). (G) Amino acid sequence alignment of the four DENV serotypes and seven recombinant DENV2 viruses. In red, the amino acid substitution is shown in each recombinant virus. (H) ERC1 degradation during infection with each recombinant DENV. WB of cytoplasmic extracts of A549 cells infected during 24 h with the recombinant viruses as indicated on the top of the gel. For each infection, ERC1 levels are shown and viral replication evaluated by NS3. DENV2 WT and mock infections were used as controls. (I) G21D and T2A recombinant DENV2 viruses are unable to degrade ERC1. IF showing that infection with an MOI of 5 of DENV2 WT degrades ERC1, while G21D and T2A do not alter ERC1 staining compared to the mock infection. In red ERC1 and in green NS3. Objective: 63× (J) Infection with DENV2 G21D differentially degrades STAT2 and ERC1. WB of cytoplasmic extracts of A549 cells infected during 24 h with an MOI of 5 of WT, G21D, or T2A DENV2. ERC1, STAT2, NS3, NS5, and GAPDH are indicated. (K) G21D amino acid change in NS5 protein alters ERC1 binding. Immunoprecipitation analyses are shown. A549 cells were infected with DENV2 WT, G21D, or T2A viruses (MOI: 5), as indicated on the top of the gels. Eight hours postinfection cells were treated with 20 μM MG132 for 6 h. Lysates were immunoprecipitated with anti-ERC1 antibody (indicated as IP: ERC1), and WB were performed using anti ERC1 and anti NS5, as shown. Before the IP assay, an aliquot of the whole-cell extract (WCE) was spared as input control of the IP, shown on the left.
To determine whether chimeric R1 and R2 MTases were functional in the context of the infectious DENV clone, we generated recombinant viruses replacing the specific regions of DENV2 MTase. It is important to highlight that viruses in which the complete MTase was exchanged between DENV2 and DENV4 were nonviable (26). Thus, we generated the DENV2 carrying only the R1 and R2 regions of the DENV4 protein. Viral full-length RNA from WT and, R1 and R2 chimeric viruses were transfected in BHK cells and their replication capacity was assessed by IF assay. R1 chimeric virus produced lower levels of progeny detected after 48 h posttransfection, whereas the R2 chimera was replication impaired (Fig. 5D). In order to evaluate possible defects in the enzymatic activities of NS5 in the chimeric viruses, the replication kinetics were evaluated in mosquito C6/36 and human cells (SI Appendix, Figs. S2 and S3). The R2 chimeric virus was replication-impaired in both cell lines while the R1 virus showed 10 and 30 fold reduced replication in mosquito and human cells, respectively, in comparison to the WT virus, suggesting that both chimeras displayed defects in the RNA replication functions of NS5. Chimeric R1 viral stocks were generated in C6/36 cells and used to infect A549 cells at high MOI. NS5 from the WT and R1 chimera showed a preponderant nuclear localization in infected cells (Fig. 5E). Interestingly, the replicating R1 virus lost the ability to degrade ERC1 (Fig. 5 E and F). This observation suggests that amino acids replaced in the R1 region of the MTase could be involved in the different phenotypes observed between serotypes DENV2 and DENV4 regarding ERC1 degradation.
A single Amino Acid Substitution in the DENV2 NS5 Recapitulates the Phenotype of DENV4 regarding ERC1 and STAT2 Degradation.
The chimeric DENV2 R1 virus contains four residues that are unique for DENV4 (Fig. 5G). To narrow down the requirements for ERC1 degradation, we generated a battery of recombinant viruses containing single amino acid changes and combinations of 2 or 3 substitutions in NS5. We evaluated viral replication capacity in mosquito C6/36 and A549 cells (SI Appendix, Figs. S2 and S3), and the ability of the battery of mutated viruses to degrade ERC1 in A549 cells (Fig. 5 G and H). Two recombinant viruses, DENV2 A19S and G21D, showed efficient replication, while the others showed a different degree of impairments (SI Appendix, Figs. S2 and S3, see also levels of NS3 in Fig. 5H). Interestingly, the mutant DENV2 G21D also lost the ability to degrade ERC1 (Fig. 5H). Thus, changing a single amino acid at position 21 of the DENV2 NS5 MTase domain mimicked DENV4 in preventing ERC1 degradation.
DENV2 T2A virus displays a defect in ERC1 and STAT2 degradation, which can be attributed to a reduced UBR4 binding, previously shown by Morrison et al. (11). On the other hand, because DENV4 infection leads to STAT2 degradation, we hypothesize that the DENV2 G21D virus should retain the ability to degrade STAT2. To evaluate this hypothesis, A549 cells were infected with either DENV2 WT, DENV2 G21D, or DENV2 T2A viruses using MOI of 10. Replication and protein levels were assessed by IF and WB. IF assays showed that cells infected with either virus, T2A or G21D DENV2, displayed mock levels of ERC1 (Fig. 5I). In addition, WB analysis of extracts from T2A or G21D DENV2-infected cells indicated that T2A virus showed a reduced ability of degrading STAT2 and ERC1, while G21D virus efficiently degraded STAT2 but not ERC1. The data show a dissociation of the molecular requirements for ERC1 and STAT2 degradation within NS5. Evaluation of the timing of degradation of the two host proteins during DENV2 infection also shows differences between these two processes (SI Appendix, Fig. S4). We observed that while most of STAT2 is degraded 4 hpi, ERC1 degradation is evident after 8 h of infection.
The mutation G21D in NS5 prevents ERC1 degradation, thus we evaluated a possible impact of this mutation on ERC1 binding. To analyze this, we carried out immunoprecipitation (IP) assays of ERC1 in mock and infected A549 cells with WT, G21D and T2A viruses and evaluated complex formation with the different NS5 proteins. Because ERC1 is degraded during infection with the WT virus, we used MG132 to inhibit host protein degradation (20 μM of MG132 added 8 h after infection). In these conditions, while the input levels of ERC1 were comparable (Fig. 5 K, Left) a significant reduction of G21D NS5 protein was observed in the IPs in comparison to WT and T2A-infected cells (Fig. 5 K, Right). These results suggest that mutation G21D and not T2A affects the interaction of NS5 with ERC1.
In summary, by comparing DENV2 and DENV4 NS5 sequences and constructing chimeric viruses, we identified a single residue in NS5 that is necessary for efficient ERC1 binding and degradation, allowing us to design a DENV2 virus that resembles DENV4 regarding the described NS5 properties.
Lack of ERC1 Degradation in DENV2 G21D Results in Higher Expression and Secretion of Cytokines upon Infection.
Infections with DENV2 or DENV4 produce significantly different amounts of proinflammatory cytokines (27). In this regard, DENV4 infections were associated with a higher level of cytokine production. Taking into account the role of NS5 and the relevance of dissecting molecular differences between DENV serotypes, we further investigated the implication of the differential ERC1 degradation in the activation of the NF-kB pathway during infection.
We compared the WT and the G21D viruses assessing replication kinetics, cytokine production, and cell motility of the infected cells. Viral stocks were produced in mosquito C6/36 cells, and one-step replication curves were carried out in both mosquito and human cells. Viral infection and replication in mosquito cells showed indistinguishable RNA replication and infectious viral progeny production when the G21D mutant was compared to the WT virus (Fig. 6A). In contrast, replication attenuation was observed with the G21D virus in human cells (Fig. 6B). A delayed viral RNA synthesis and lower infectious particle production was observed when G21D virus was compared with the WT. In human cells, viral protein accumulation and ERC1 levels were also evaluated as a function of time. The results showed complete ERC1 protein degradation at 16 h postinfection with the WT virus, while with DENV2 G21D ERC1 levels were similar to mock-infected cells even at 48 h postinfection with high MOI (Fig. 6C). In addition, to characterize the replication of the G21D virus, we performed infections in ERC1-silenced cells. The results show that the G21D replicates similar to WT in ERC1 KD A549 cells, confirming the role of ERC1 in the differential replication of the two viruses (Fig. 6D).
Fig. 6.

ERC1 degradation limits cytokine production and cell motility. (A) G21D and WT DENV2 show similar replication kinetics in mosquito C6/36 cells. Viral replication as a function of time is shown in one-step growth curve (MOI: 10). (A, Left) RT qPCR of intracellular viral RNA relative to 18S. (A, Right) Plaque assay of infectious viral particles in the supernatant of infected cells. PFU: plaque-forming units. (B) G21D DENV2 is attenuated in human A549 cells. Viral replication as a function of time is shown in one-step growth curve (MOI: 10). (B, Left) RT qPCR of intracellular viral ARN relative to GAPDH. (B, Right) Plaque assay of infectious viral particles in the supernatant of infected cells. (C) ERC1 degradation upon WT or G21D virus infection. WB analysis of cytoplasmic extracts of A549 cells infected with an MOI of 10 with either WT or G21D DENV2 as a function of time. Mock infections were used as a control of ERC1 levels. (C, Left) WB of cytoplasmic extracts of A549 cells infected with WT DENV2 as a function of time is shown. A sample of G21D at 48 h postinfection is included for comparison. (C, Right) WB of cytoplasmic extracts of A549 cells infected with G21D virus as a function of time is shown. A sample of WT at 48 h postinfection is included for comparison. Viral replication is shown by NS3 protein accumulation. (D) G21D virus replication is enhanced in ERC1 knockdown cells to WT levels. G21D and WT viral replication is shown by qRT PCR (mean ± SD) at 24 hpi (MOI 5) in A549 cells previously transfected with an ERC1 siRNA or a nonrelated siRNA (NR). (E) Expression and secretion of cytokines during infection with an MOI of 1 of WT or G21D DENV2s. (E, Upper) replication curve and cytokine mRNA expression of WT and G21D DENV infections. Viral RNA levels are shown as a function of time after infection and measured by RT qPCR and normalized to GAPDH. IL6, TNF-α, and IFN-β mRNA expression as a function of time of infected cells was measured by RT qPCR and expressed relative to GAPDH, as indicated in each case. (E, Lower) IL6, TNF-α, and IFN-β protein secretion measured by ELISA in the supernatant of infected cells. (F) Increased cell motility by G21D DENV infection. (F, Left) schematic representation of the protocol used. (F, Middle) representative images of the wound-healing assays in A549 cells previously infected with an MOI of 3 with either WT or G21D viruses. Images of the wounds at 0 and 24 h are shown in mock or infected cell monolayers. Objective: 5×. (F, Right) the wound closure was quantified by measuring the distance covered by the migrated leading edge using Zen 2.3 lite software. Additionally, an IF assay to detect NS3 viral protein is shown, which was performed once the wound-healing assay was concluded. Statistical significance is shown with the following notations: ****P < 0.0001; *P < 0.05.
We then compared the cytokine expression and secretion after WT and G21D viral infections in A549 cells. Although DENV2 WT replicated to higher levels than the G21D in human cells, the mutant virus induced significantly higher levels of proinflammatory cytokines (TNF-α and IL6) and IFN-β mRNA than the WT virus (Fig. 6E), supporting a role of ERC1 in this process. The level of secreted cytokines was also assessed in cell supernatants by ELISA assays, showing significantly higher levels in the G21D-infected cells in respect to that with the WT virus. Because the two viruses only differed in a single amino acid and had identical replication in mosquito cells, the differential expression and secretion of cytokines could be attributed to the lack of ERC1 degradation. The results support the idea that DENV2 infection limits the production of proinflammatory cytokines by eliminating ERC1, providing a possible molecular explanation to the different properties of DENV serotypes.
Because ERC1 has been implicated in cell migration (21), we also hypothesized that DENV2 infection would impair this function. To examine this possibility, we performed a wound-healing assay after mock, DENV2 WT, or DENV2 G21D infection (Fig. 6F). The wound size was registered immediately after the wound was generated (0 h) and 24 h postwound. Infection of complete monolayers was verified by IF analysis using anti-NS3 antibodies. While uninfected cells showed complete wound healing at 24 h, the DENV2-infected cells showed significant retarded cell migration (Fig. 6F). In addition, when infected cells with the two viruses WT and G21D were compared, the mutant virus showed significantly higher cell migration than WT. We conclude that ERC1 degradation during DENV2 infection reduces cell migration, phenotype that is mitigated in the D21G mutant.
Discussion
Here, we identified a function of the DENV NS5 protein in modulating NF-κB activation by degradation of the cellular protein ERC1. This mechanism is mediated by the proteasome, requires UBR4, and involves the MTase domain of the viral protein. Importantly, we uncovered molecular details of how different DENV serotypes differentially control ERC1 degradation, and separated this function from STAT2 degradation. Because DENV infection is characterized by induction of large amounts of proinflammatory cytokines, our findings are relevant for understanding viral pathogenesis and defining the molecular bases of how distinct DENV serotypes limit NF-κB activation (28–32).
NS5 is a potent antagonist of type I IFN signaling of the DENV proteins by suppressing the human JAK-STAT signaling. NS5 from different flaviviruses evolved remarkable convergent strategies to interfere with this pathway, but using distinct mechanisms. In the case of DENV, the process is mediated by STAT2 degradation through the ubiquitin-proteasome pathway via UBR4 E3 ubiquitin ligase (9–11). UBR4 binds to the MTase domain of NS5, while STAT2 interacts with the RdRp domain, at residues 202 to 306, leading to STAT2 degradation (33). Here, we found that UBR4 is also involved in ERC1 degradation; however, in this case, the MTase domain is also responsible for ERC1 binding. Thus, NS5 functions as a scaffold protein with different binding sites for the host STAT2 and ERC1 proteins, but sharing a requirement for UBR4.
Proteolytic cleavage of NS5 N terminus in the context of the viral polyprotein was reported to be necessary for STAT2 degradation (9), presumably due to the requirement of determinants at the N terminus of NS5 for UBR4 binding (11). Our data indicate that the matured NS5 is capable of binding in a functional manner to UBR4 for host protein degradation in the absence of in situ N-terminal processing of NS5. Thus, the reported NS5 proteolysis required for STAT2 degradation must have another requisite. Interestingly, while mouse STAT2 is not degraded by DENV NS5 (34), the mouse ERC1 was efficiently degraded during DENV infection. This information is relevant when evaluating animal models for viral infection and pathogenesis, and for further analyses of ERC1 function in in vivo models.
Although the four DENV serotypes degrade STAT2, ERC1 is not degraded by DENV4 infection, indicating differences between the MTase domains of NS5 from DENV2 and DENV4. This observation is particularly important because ERC1 degradation significantly reduced NF-κB dependent cytokine expression and secretion. In this regard, DENV4 replication promotes stronger innate immune responses, with increased dendritic cell activation, expression of migration markers, and cytokine production, compared with DENV2 (27). Epidemiological data also suggest that DENV2 secondary infections are associated with more severe disease than DENV4 infections (30, 31, 35). The pathogenic versus protective roles of cytokines during DENV infection are not completely understood and a “cytokine storm” may result in more severe disease (36). Our findings comparing DENV2 and DENV4 NS5 properties explain, at least in part, the distinct antiviral innate immune induction by distinct serotypes that can be associated with different viral fitness and pathogenesis.
Unveiling differences among serotypes is key for developing effective tetravalent vaccines. Here, we dissected the molecular determinants within the NS5 protein associated to the different abilities of DENV2 and DENV4 to degrade ERC1. Through the generation of NS5 chimeras and recombinant DENVs, we show that UBR4 and ERC1 binding require residues that are both at the N terminus of the viral protein. NS5 with point mutations T2A or G3A have been reported to impair UBR4 interaction, and here we have observed that these mutants in the context of the infection clone affect STAT2/ERC1 degradation. In addition, a DENV2 carrying the D21 residue from DENV4 MTase replicated to WT levels in mosquito cells, while it was attenuated in human cells, inducing increased expression and secretion of proinflammatory cytokines and IFN-β when compared to the WT DENV2. This mutant virus degraded STAT2 but lacked the ability to degrade ERC1. This finding further supports the role of ERC1 in the NF-κB activation pathway. In line with our findings, it has been previously shown that DENV2 can block NF-κB activation to down-regulate cytokine production, although the mechanism of this process was not described (37).
NF-κB regulates the expression of hundreds of genes related to a variety of cellular processes such as apoptosis, inflammation, and immune responses. It is activated within minutes after viral exposure. Therefore, many viruses have evolved different strategies to modulate NF-κB activation cascade (38, 39). For instance, measles virus V protein directly binds to NF-kB to suppress its activity (40); cowpox virus can inhibit NF-kB gene induction by interfering with IkBα degradation process (41). Likewise, HIV type 1 (HIV-1) Vpu protein inhibits IkBα degradation by preventing its ubiquitination (42). Another mechanism involves interaction with IKK complex. HCV NS5B protein binds to IKKα and blocks IKK activation (43), whereas membrane M protein of SARS-CoV interacts with IKKβ and thus precludes NF-κB activation cascade (44). Our data provide an additional mechanism by which DENV prevents NF-κB activation by degrading a factor that enhances the activity of IKK complex (19). We hypothesize that the resulting decrease in cytokine expression may be favorable for DENV2 replication and propagation in in vivo infections. Given the role of ERC1 in cell motility, its degradation might even have a major impact in natural infections. Reduction of the infected cell mobility could be relevant for limiting the migration of DCs to the lymph nodes for T cell activation. Further studies will be necessary to evaluate the impact of ERC1 degradation on dendritic cell migration in infected animals.
One drawback in the formulation of DENV tetravalent vaccines is the inability to properly attenuate DENV2 (45). Therefore, recombinant DENV2/DENV4 viruses are being used in current formulations for clinical trials (4, 46). The generation here of a mutant DENV2 that is attenuated due to its incapacity to degrade ERC1, resembling DENV4, provides a strategy that could be taken into consideration for the design of live-attenuated vaccines.
This work discloses an additional function of the DENV NS5 in limiting the cellular innate antiviral response. Importantly, we defined molecular bases for differences among DENV serotypes, which could be of use for tetravalent vaccine development.
Materials and Methods
Cells and Viruses.
Mammalian cells: A549 cells (human lung adenocarcinoma epithelial cell line, ATCC, CCL- 185) and Huh7 cells (human hepatoma cell line, JCRB Cell Bank #0403) were maintained in Dulbecco’s Modified Eagle’s Medium/ F-12 (Ham’s), HEK 293 cells (human embryonic kidney cell lines, ATCC CRL-1573) were maintained in Dulbecco’s Modified Eagle’s Medium high glucose, BHK-21 cells (baby hamster kidney cell line, ATCC, CCL-10) were maintained in Minimum Essential Medium Alpha. All the media were supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Human DCs were obtained from peripheral blood mononuclear cells (PBMC) from healthy volunteer donors. Samples were deidentified prior to processing. Monocytes were purified by positive selection using anti-CD14 antibodies immobilized on magnetic beads (Miltenyi, USA). DCs were obtained by culture of monocytes with IL-4 + GM-CSF (30 ng/mL) for 5 d. Mosquito cells: C6/36HT (Aedes albopictus cells ATCC, CRL-1660 were adapted to grow at 33 °C) were maintained in Leibovitz’s L-15 Medium, supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, 0.3% tryptose phosphate, 0.02% glutamine, and 1% MEM nonessential amino acid solution. Stocks of wild-type, chimeric, and mutated DENV2 (16681) were prepared in mosquito C6/36 cells and used to infect the different cell lines as indicated in each case. DENV2 D2S10 (47) was kindly provided by Dr. Sujan Shresta and was used to infect DCs.
Recombinant DENVs.
The desired chimeric viruses or mutations were introduced in a DENV2 cDNA clone (48) (GenBank accession number U87411). Chimeric DENV2-DENV4 cDNA clones were obtained by replacing amino acids from 14 to 27 of NS5 (R1) or from 43 to 52 of NS5 (R2) fragment of the WT plasmid with the respective fragment derived from DENV4 (strain 341750) by doing an overlapping PCR (primers listed in SI Appendix, Table S1). Mutations in the MTase domain of DENV2 were also introduced by overlapping PCR using primers AVG 1,320 and 1,323 and the specific primer to introduce the different mutations (listed in SI Appendix, Table S1). In all cases, the mutated fragment was exchanged on the DENV2 cDNA clone by digestion with NheI and AvrII enzymes. Recombinant DENV plasmids were linearized using XbaI and used for transcription with T7 RNA polymerase (Invitrogen) in the presence of cap analog (m7GpppA). RNA transcripts were transfected with Lipofectamine 2,000 (Invitrogen) into BHK-21 or C6/36HT cells. Viral replication was evaluated by indirect immunofluorescence. Supernatants were harvested at different times posttransfection and used to infect C6/36HT cells to produce viral stocks. Infectious viral particles were assessed by plaque assays in BHK-21 cells.
Plasmids.
All DENV proteins were cloned in the pcDNA plasmid fused to a 2A self-cleavage sequence and GFP sequence. NS5 DENV2/GFP plasmid was generated by amplifying GFP with primers AVG2375 and AVG2376 (SI Appendix, Table S2), which included a 2A sequence and cloned with XhoI and ApaI in a pcDNA plasmid carrying NS5 DENV2 protein previously obtained in the laboratory. RdRpGFP DENV2 and MTaseGFP DENV2 plasmids were obtained by introducing the 2A_GFP sequence (which was previously coned in the NS5 DENV2/GFP), in pcDNAs plasmids expressing these NS5 domains by digestion with XhoI and ApaI enzymes. NS5 DENV4/GFP plasmid was obtained by amplifying the NS5 DENV4 sequence from strain 341750 with primers AVG2375 and 2377 and subsequent replacement of the NS5 on the pcDNA NS5 DENV2/GFP plasmid by digestion with Xho enzyme. MTaseGFP DENV4 was generated by amplification with primers AVG2269 and 2270 and this region replaced in MTaseGFP DENV2 plasmid. Finally, chimeric R1, R2, and R3 MTase/GFP plasmids were obtained by overlapping PCR with primers AVG2380 and the specific reverse primer (AVG2384, AVG2385 or AVG2386, respectively) and specific forward primers (AVG2381, AVG2382 or AVG2383, respectively) and primer AVG2387. The amplified products were replaced in the MTaseGFP DENV2 plasmid by digestion with AflII and XhoI enzymes. ERC1 expression plasmid (pCMV6-ELKS) was purchased from Origen (RC213864). NF-kB reporter plasmid [NFkb pGL 4.32 (FFLuc)] was kindly provided by Ricardo Rajsbaum.
Transfection Assays.
A549 cells were used for transfection assays with viral protein expression plasmids and subsequent immunofluorescent assays. HEK 293 cells were used for transfection assays and posterior sorting or western blot assays. In both cases, cells were transfected with the indicated amount of plasmid in a ratio 1:3 with lipofectamine 2,000. Four hours after transfection, cell media were exchanged with fresh media (5% FBS or Fetal Bovine Serum) for 20 h.
Antibodies.
Antibodies against ERC1 [ELKS (E-1)] and STAT2 were purchased from Santa Cruz Biotechnology (sc-365715 and sc-476, respectively). Anti-GAPDH (mouse monoclonal) was purchased from Abcam (ab8245). Antibodies against DENV NS3 and NS5 (rabbit polyclonals) were obtained in our laboratory.
Knockdown Assays.
RNA interference experiments were carried out as previously described (16) using ON-TARGET plus SMART pool siRNA oligonucleotides (Dharmacon RNA Technologies, Lafayette, CO, USA). A nonrelated siRNA (siRNA NR) was used as negative control, and a siRNA against Renilla Luciferase (siRNA Luc) was used as an additional control in infections with reporter DENV2. A hundred thousand A549 cells per well were seeded in a 24 multiwell plate 24 h previous to transfection with the corresponding siRNA using Oligofectamine (Invitrogen). After 48 h of transfection, cells were infected with either reporter DENV or DENV2 or treated with TNF-α or poly I:C. For the reporter DENV containing the Renilla luciferase gene (22), cells were incubated for 36 h prior to measuring luciferase activity. For infection with WT DENV (16,681), cells were infected with a MOI of 5, and intracellular RNA was measured by quantitative real-time PCR using primers: ACAAGTCGAACAACCTGGTCCAT (Forward) and GCCGCACCATTGGTCTTCTC (Reverse). Knockdown was confirmed by WB analyses or by RT qPCR (in the case of UBR4). Primers: GGTGTTCCAGAGGCTAGTGATC (Forward) and CCAACTGCTTCTGCGGTTCCTT (Reverse) (SI Appendix, Table S3).
TNF-α and Poly I:C Treatments.
Knockdown A549 cells were treated with 5 ng/mL human TNF-α (Peprotech) during 2, 4, or 6 h. For poly I:C treatment, silenced A549 cells were transfected with 10 μg/mL poly I:C in a relation 10:1 of poly I:C (Sigma): lipofectamine 2,000 (Invitrogen) and harvested 16 and 24 h posttransfection. After the indicated time, the cell supernatant was harvested to measure cytokine production by ELISA, and the intracellular RNA was extracted with TRIzol to quantify cytokine expression by RT qPCR.
Real-Time Quantitative PCR (qPCR) and Primers.
TRIzol reagent (Invitrogen) was used for total RNA extraction, and cDNA was synthesized with random decaoligonucleotide primer mix by M-MLV Reverse Transcriptase (Promega). qPCRs were performed by using SYBR Green dye and specific primers for different mRNAs. GAPDH mRNA was used as a control housekeeping gene. Sequences of primer used for qPCR assays are listed in SI Appendix, Table S3.
Immunofluorescence Assays.
Immunofluorescence assays were performed as previously described (22). Briefly, 1 × 105 cells per well were seeded into 24-well plates containing glass coverslip. For infection assays, cells were mock infected or infected with DENV. After 24 or 48 h postinfection, coverslips were collected, and the cells were fixed with paraformaldehyde 2%, sucrose 2%, in PBS pH 7.4 at room temperature for 20 min. PFA fixed cells were then permeated with 0.1% Triton X-100 for 4 min at room temperature. For transfection assays, A549 cells were transfected with the indicated expression plasmids and 24 h posttransfection, coverslips collected as previously described. Images were obtained with a Zeiss Axio Observer three inverted fluorescence microscope, with 20× objective. For confocal microscopy, the Carl Zeiss LSM 5 Pascal confocal microscope was used and images taken with 40× or 63× objectives.
NF-kB Reporter Assay.
HEK 293 cells (5 × 104 cells) were transfected with 10 ng NF-kB luciferase reporter plasmid together with either 26 ng ERC1 expression vector or 26 ng of an empty plasmid (pcDNA) using Lipofectamine 2,000. Four ng Renilla luciferase-expressing plasmid was used for normalizing transfection efficiencies. After 16 h of transfection, luciferase activity was determined using Dual Luciferase Reporter assay system according to the manufacturer’s instructions (Promega).
ELISA Assays.
Human IL6 and TNF-α ELISA Set were purchased from BD (555220 and 555212 respectively) and human IFN beta ELISA kit from PBL Assay Science (41410–1). Cytokines were measured in the cell supernatant following the manufacturer’s instructions.
Wound-Healing Assay.
Wound-healing assays were performed in 1.2 × 106 A549 cells knocked down with ERC1 or a nonrelated siRNA and in mock, WT, and G21D DENV2-infected A549 cells. In either assay, the cell monolayer was scratched by a sterile micropipette tip, followed by washes with PBS to remove the floating cells. Images of the wound-healing process were obtained with Zeiss Axio Observer three inverted fluorescence microscope immediately after the scratch and 24 h later (Objective: 5×). Cell migration into the gap was evaluated by measuring the distance covered by the migrated leading edge after scratching using Zen 2.3 software.
Cell Sorting Experiments.
For cell sorting assays, 9 × 106 HEK 293 cells were transfected with 15 μg of the indicated expression plasmid (NS5 DENV2/GFP, GFP, NS5 DENV4/GFP, MTase DENV2/GFP or MTase DENV4/GFP) and 45 μL lipofectamine 2000. Twenty-four hours posttransfection, cells were sorted by fluorescence-activated cell sorter (FACS) analyses (FACS AriaFusion) to obtain GFP-positive and -negative cells. One hundred cells per condition were lysed and subsequently loaded in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) 10× gels for further blotting assays.
Proteasome Inhibition Assay.
A549 cells (2 × 105 cells) were either mock or infected with DENV2 with an MOI of 3 during 12 h to allow viral infection. At 12 h postinfection, cells were treated with 20 uM MG132 or DMSO as control during 2, 6, and 10 h. Cells were lysed with cracking buffer (SDS 4%, Tris-HCl 0.5 M pH 6.8, glycerol 20%, EDTA 20 mM, Bromofenol azul 0.001%) and subsequently analyzed by western blot assays.
Immunoprecipitation Assays.
A549 cells were either mock or infected with DENV2 WT, G21D, and T2A mutants with an MOI of 5 during 8 h to allow viral infection. At 8 h postinfection, cells were treated with 20 uM MG132 during 6 h. Cell were then harvested by scraping, washed with PBS, centrifuged at 100 rcf for 5 min at 4 °C, and the pellet lysed with 1 mL lysis buffer (50 mM Tris-HCl pH 7.4, 150 mMNaCl, 1 mM EDTA, 0.5% NP40, protease and phosphatase inhibitors, +/− DNase I and RNase A) chilled on ice. Cell lysates were incubated in a rotating mixer for 30 min at 4 °C and then clarified by centrifugation at 3,000 rcf for 20 min at 4 °C. An aliquot of clarified supernatant was spared as input control (whole-cell extract). The clarified lysates were overnight incubated in a rotating mixer at 4 °C with 30 μL protein G plus/protein A agarose previously coated with 1 μg antibody against ERC1 [ELKS (E-1)]. Beads were recovered by centrifugation at 400 rcf for 2 min at 4 °C and washed seven times with 1 mL wash buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.05% NP40) chilled on ice. Beads were finally incubated with 80 μL of cracking buffer (SDS 4%, Tris-HCl 0.5 M pH 6.8, glicerol 20%, EDTA 20 mM, Bromofenol azul 0.001%) and heated for 10 min at 99 °C. Twenty microliters of whole-cell extract and of the immunoprecipitated protein were loaded in an SDS-PAGE 10% gel and blotted against NS5 and ERC1 proteins.
Statistical Analysis.
All statistical tests and plots were performed using GraphPad Prism 8.0 software. The p values were calculated using the Mann–Whitney U test to compare the mean values of two groups. Statistical significance is shown in the figure legends with the following notations: *P < 0.05; ****P < 0.0001.
Role of the Funders.
Funding sources played no role in study design; in the collection, analysis, and interpretation of data; in the writing of the report; and in the decision to submit the paper for publication.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
Authors are thankful to Dr. Sujan Shresta for DENV2 D2S10 infectious clone. They are also grateful to Dr. Ana Fernandez-Sesma for helpful discussions. This study has received funding from, NIH (NIAID) (R01.AI095175 and U19AI168631-01 to A.V.G., R01AI166668, R01AI134907, R01AI155466, and P01AI150585-01A1 to R.R., and K12 HD052023 to M.G.), Fondo Nacional para la Investigación Científica y Tecnológica (PICT 2019-02869 to A.V.G. and PICT 2020-01631 and PIBAA 2022-28720210100056CO to M.M.G.L.L.).
Author contributions
M.M.G.L.L., H.M.P., R.R., P.S.S., and A.V.G. designed research; M.M.G.L.L., G.C.N., A.P., F.D.M., N.G.I., L.G., M.G., and A.C. performed research; M.M.G.L.L., S.O.R., H.M.P., F.D.M., N.G.I., L.G., D.S.O., L.d.B., R.R., N.J.K., P.S.S., and A.V.G. analyzed data; and M.M.G.L.L. and A.V.G. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. A.P. is a guest editor invited by the Editorial Board.
Contributor Information
María Mora Gonzalez Lopez Ledesma, Email: mgonzalezlopez@leloir.org.ar.
Andrea V. Gamarnik, Email: agamarnik@leloir.org.ar.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Katzelnick L. C., et al. , Antibody-dependent enhancement of severe dengue disease in humans. Science 358, 929–932 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halstead S. B., O’Rourke E. J., Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J. Exp. Med. 146, 201–217 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Halstead S. B., Mahalingam S., Marovich M. A., Ubol S., Mosser D. M., Intrinsic antibody-dependent enhancement of microbial infection in macrophages: Disease regulation by immune complexes. Lancet. Infect. Dis. 10, 712 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Durbin A. P., Historical discourse on the development of the live attenuated tetravalent dengue vaccine candidate TV003/TV005. Curr. Opin. Virol. 43, 79 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murugesan A., Manoharan M., Dengue virus. Emerg. Reemerging Viral Pathog. 2020, 281–359 (2019). [Google Scholar]
- 6.Egloff M. P., Benarroch D., Selisko B., Romette J. L., Canard B., An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: Crystal structure and functional characterization. EMBO J. 21, 2757 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yap T. L., et al. , Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J. Virol. 81, 4753–4765 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartholomeusz A. I., Wright P. J., Synthesis of dengue virus RNA in vitro: initiation and the involvement of proteins NS3 and NS5. Arch. Virol. 128, 111–121 (1993). [DOI] [PubMed] [Google Scholar]
- 9.Ashour J., Laurent-Rolle M., Shi P.-Y., García-Sastre A., NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 83, 5408–5418 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazzon M., Jones M., Davidson A., Chain B., Jacobs M., Dengue virus NS5 inhibits interferon-α signaling by blocking signal transducer and activator of transcription 2 phosphorylation. J. Infect. Dis. 200, 1261–1270 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Morrison J., et al. , Dengue virus Co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 9, e1003265 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grant A., et al. , Zika virus targets human STAT2 to inhibit type i interferon signaling. Cell Host Microbe 19, 882–890 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sm B., The many faces of the flavivirus NS5 protein in antagonism of type I interferon signaling. J. Virol. 91, e01970-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thurmond S., Wang B., Song J., Hai R., Suppression of type I interferon signaling by flavivirus NS5. Viruses 10, 712 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laurent-Rolle M., et al. , The interferon signaling antagonist function of yellow fever virus NS5 protein is activated by type I interferon. Cell Host Microbe 16, 314–327 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Maio F. A., et al. , The dengue virus NS5 protein intrudes in the cellular spliceosome and modulates splicing. PLoS Pathog. 12, 1–29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pozzi B., et al. , Dengue virus targets RBM10 deregulating host cell splicing and innate immune response. Nucleic Acids Res. 48, 6824 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu Z. H., et al. , ATM- and NEMO-dependent ELKS ubiquitination coordinates TAK1-Mediated IKK activation in response to genotoxic stress. Mol. Cell 40, 75–86 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sigala J. L. D., Activation of transcription factor NF-kB requires ELKS, an IkB kinase regulatory subunit. Science 304, 1963–1967 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Held R. G., Kaeser P. S., ELKS active zone proteins as multitasking scaffolds for secretion. Open Biol. 8, 1–12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sala K., et al. , The ERC1 scaffold protein implicated in cell motility drives the assembly of a liquid phase. Sci. Rep. 9, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Samsa M. M., et al. , Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 5, e1000632 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fourriere L., et al. , RAB6 and microtubules restrict protein secretion to focal adhesions. J. Cell Biol. 218, 2215–2231 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramella M., Maria Ribolla L., de Curtis I., Liquid–liquid phase separation at the plasma membrane–cytosol interface: common players in adhesion, motility, and synaptic function. J. Mol. Biol. 434, 1–14 (2022). [DOI] [PubMed] [Google Scholar]
- 25.Pfeffer L. M., The role of nuclear factor κb in the interferon response. J. Interf. Cytokine Res. 31, 553–559 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teramoto T., Boonyasuppayakorn S., Handley M., Choi K. H., Padmanabhan R., Substitution of NS5 N-terminal domain of dengue virus type 2 RNA with type 4 domain caused impaired replication and emergence of adaptive mutants with enhanced fitness. J. Biol. Chem. 289, 22385–22400 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamlin R. E., et al. , High-dimensional CyTOF analysis of dengue virus-infected human DCs reveals distinct viral signatures. JCI Insight 2, 1–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soo K. M., Khalid B., Ching S. M., Chee H. Y., Meta-analysis of dengue severity during infection by different dengue virus serotypes in primary and secondary infections. PLoS One 11, e0154760 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsang T. K., et al. , Effects of infection history on dengue virus infection and pathogenicity. Nat. Commun. 10, 1–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaughn D. W., et al. , Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J. Infect. Dis. 181, 2–9 (2000). [DOI] [PubMed] [Google Scholar]
- 31.Thomas L., et al. , Influence of the dengue serotype, previous dengue infection, and plasma viral load on clinical presentation and outcome during a dengue-2 and dengue-4 co-epidemic. Am. J. Trop. Med. Hyg. 78, 990–998 (2008). [PubMed] [Google Scholar]
- 32.Thomas L., et al. , Clinical presentation of dengue by serotype and year of epidemic in martinique. Am. J. Trop. Med. Hyg. 91, 138–145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ashour J., Laurent-Rolle M., Shi P.-Y., García-Sastre A., NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 83, 5408–18 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ashour J., et al. , Mouse STAT2 restricts early dengue virus replication. Cell Host Microbe 8, 410–421 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fried J. R., et al. , Serotype-specific differences in the risk of dengue hemorrhagic fever: An analysis of data collected in Bangkok, Thailand from 1994 to 2006. PLoS Negl. Trop. Dis. 4, e617 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothman A. L., Immunity to dengue virus: A tale of original antigenic sin and tropical cytokine storms. Nat. Rev. Immunol. 11, 532–543 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Chang T. H., et al. , Dengue virus serotype 2 blocks extracellular signal-regulated kinase and nuclear factor-κB activation to downregulate cytokine production. PLoS One 7, 4–11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmitz M. L., Kracht M., Saul V. V., The intricate interplay between RNA viruses and NF-κB. Biochim. Biophys. Acta Mol. Cell Res. 1843, 2754–2764 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rahman M. M., McFadden G., Modulation of NF-κB signalling by microbial pathogens. Nat. Rev. Microbiol. 94, 291–306 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schuhmann K. M., Pfaller C. K., Conzelmann K.-K., The measles virus V protein binds to p65 (RelA) to suppress NF-kappaB activity. J. Virol. 85, 3162–3171 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oie K. L., Pickup D. J., Cowpox virus and other members of the orthopoxvirus genus interfere with the regulation of NF-kappaB activation. Virology 288, 175–187 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Bour S., Perrin C., Akari H., Strebel K., The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B. J. Biol. Chem. 276, 15920–15928 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Choi S.-H., et al. , Hepatitis C virus nonstructural 5B protein regulates tumor necrosis factor alpha signaling through effects on cellular IkappaB kinase. Mol. Cell. Biol. 26, 3048–3059 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fang X., et al. , The membrane protein of SARS-CoV suppresses NF-kappaB activation. J. Med. Virol. 79, 1431–1439 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durbin A. P., Kirkpatrick B. D., Pierce K. K., Schmidt A. C., Whitehead S. S., Development and clinical evaluation of multiple investigational monovalent DENV vaccines to identify components for inclusion in a live attenuated tetravalent DENV vaccine. Vaccine 29, 7242–7250 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitehead S. S., Development of TV003/TV005, a single dose, highly immunogenic live attenuated dengue vaccine; what makes this vaccine different from the Sanofi-Pasteur CYDTM vaccine? Expert Rev. Vaccines 15, 509 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prestwood T. R., Prigozhin D. M., Sharar K. L., Zellweger R. M., Shresta S., A mouse-passaged dengue virus strain with reduced affinity for heparan sulfate causes severe disease in mice by establishing increased systemic viral loads. J. Virol. 82, 8411–8421 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kinney R. M., et al. , Construction of infectious cDNA clones for dengue 2 virus: Strain 16681 and its attenuated vaccine derivative, strain PDK-53. Virology 230, 300–308 (1997). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.